
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChelate-free metal ion binding and heat-induced radiolabeling of iron oxide nanoparticles†
Eszter
Boros
ab,
Alice M.
Bowen
c,
Lee
Josephson
d,
Neil
Vasdev
be and
Jason P.
Holland
*be
aThe Athinoula A. Martinos Center for Biomedical Imaging, 149 13th Street, Suite 2301, Charlestown, Massachusetts 02129, USA
bDepartment of Radiology, Massachusetts General Hospital, Harvard Medical School, 55 Fruit Street, Boston, Massachusetts 02114, USA. E-mail: holland.jason@mgh.harvard.edu; jasonpholland@gmail.com; Fax: +1-617-726-6165; Tel: +1-617-726-6107
cCenter for Biomolecular Magnetic Resonance (BMRZ), Institute of Physical and Theoretical Chemistry, Goethe University Frankfurt, Max-von-Laue-Str. 7, Building N140/Ground Floor, 60438 Frankfurt am Main, Germany
dCenter for Advanced Molecular Imaging Sciences, Massachusetts General Hospital, Harvard Medical School, 149 13th Street, Charlestown, Massachusetts 02129, USA
eDivision of Nuclear Medicine and Molecular Imaging, Massachusetts General Hospital, 55 Fruit Street, White 427, Boston, Massachusetts 02114, USA
First published on 29th September 2014
Abstract
A novel reaction for chelate-free, heat-induced metal ion binding and radiolabeling of ultra-small paramagnetic iron oxide nanoparticles (USPIOs) has been established. Radiochemical and non-radioactive labeling studies demonstrated that the reaction has a wide chemical scope and is applicable to p-, d- and f-block metal ions with varying ionic sizes and formal oxidation states from 2+ to 4+. Radiolabeling studies found that 89Zr–Feraheme (89Zr–FH or 89Zr–ferumoxytol) can be isolated in 93 ± 3% radiochemical yield (RCY) and >98% radiochemical purity using size-exclusion chromatography. 89Zr–FH was found to be thermodynamically and kinetically stable in vitro using a series of ligand challenge and plasma stability tests, and in vivo using PET/CT imaging and biodistribution studies in mice. Remarkably, ICP-MS and radiochemistry experiments showed that the same reaction conditions used to produce 89Zr–FH can be employed with different radionuclides to yield 64Cu–FH (66 ± 6% RCY) and 111In–FH (91 ± 2% RCY). Electron magnetic resonance studies support a mechanism of binding involving metal ion association with the surface of the magnetite crystal core. Collectively, these data suggest that chelate-free labeling methods can be employed to facilitate clinical translation of a new class of multimodality PET/MRI radiotracers derived from metal-based nanoparticles. Further, this discovery is likely to have broader implications in drug delivery, metal separation science, ecotoxicology of nanoparticles and beyond.
Introduction
Nanomedicine is the medical application of nanotechnology to diagnose or treat disease.1,2 In the fields of Radiochemistry and Nuclear Medicine, nanomedicines have attracted attention for use in drug delivery and molecular imaging.3,4 As vectors for drug delivery, nanoparticles offer a range of unique advantages including their ability to modify drug pharmacokinetics in vivo and their capacity to be loaded with high concentrations of different drugs for advancing combination therapies and theranostics.5–9 Nanoparticle properties can be tailored to control drug release, modify blood circulation half-lives, improve biodistribution profiles, increase tissue permeability and target specificity, as well as enhanced metabolic stability.10 In molecular imaging research, nanoparticles are frequently used to deliver a diagnostic payload such as a fluorophore for optical imaging with Fluorescence-Mediated Tomography (FMT) or a radionuclide for imaging with single-photon emission computed tomography (SPECT) or positron emission tomography (PET).11,12 Notably, the inherent properties of nanoparticles may also be controlled so that the nanoparticle itself becomes the imaging agent. Prominent examples of using the inherent properties of nanoparticles for imaging include fluorescent emissions from e.g. quantum dots, and the use of ultra-small superparamagnetic iron oxide particles (USPIOs) as magnetic resonance imaging (MRI) contrast agents.13–16Nanoparticles have been labeled with a wide range of radionuclides including 18F (t1/2 = 109.7 min), and radiometals like 64Cu (t1/2 = 12.7 h) and 89Zr (t1/2 = 78.41 h) for PET.15,17–19 However, with current chemical and materials science technology, radiolabeling nanoparticles typically requires significant structural/surface modifications e.g. coating of the particle and loading of the nuclide through the use of a chelate or chemically modifying prosthetic group.12 These chemical transformations can often impinge on the pharmacokinetic properties of nanoparticles leading to suboptimal targeting and distribution in vivo.15 Therefore, development of new radiochemical labeling methods is vital to advance nanomedicine. In particular, a unified strategy for radiolabeling nanoparticles with almost any metallo-radionuclide that avoids challenging, multi-step and nuclide-specific chelation and prosthetic group chemistry has the potential to transform the preclinical and clinical use of radiolabeled nanoparticles by simplifying radiolabeling and quality control (QC) protocols. While recent pioneering reports by Chen et al.,20 Wong et al.,21 Chakravarty et al.,22 and Sun et al.23,24 have disclosed advances in the area of chelate-free radiolabeling of various nanoparticles, to the best of our knowledge, no general method is available for rapid, facile and versatile radiolabeling of nanoparticles using a variety of different metallo-radionuclides.
Here, we report a simple, new reaction that facilitates chelate-free metal ion binding and heat-induced radiolabeling of USPIOs. The chemistry is applicable to a wide range of p-, d- and f-block metal nuclides. We demonstrate that Feraheme (FH; a.k.a. ferumoxytol) – a United States Food and Drug Administration approved USPIO drug for treating anaemia and in multi-center clinical trials as an MRI contrast agent16,25–28 – can be labeled with the metallo-radionuclides 64Cu2+, 111In3+ and 89Zr4+, under the same general reaction conditions and without the use of a chelate, to yield thermodynamically and kinetically stable species. Radiolabeled derivatives of FH and other USPIOs have the potential to be used in hybrid PET/MRI, SPECT/MRI and/or radiotherapeutic applications in vivo. Crucially, our technology allows for radiolabeling of metal-based nanoparticles post-fabrication – an advance that could allow clinical-grade particles conforming to current Good Manufacturing Practice (cGMP) to be employed as reagents in the radiosynthesis.
Experimental methods
Full experimental details are presented in the ESI.†Results
The hypothesis underpinning this work was that simple metal ion salts of for example chloride or oxalate anions, could be reacted thermally with USPIO-based nanoparticles under basic conditions to induce metal ion association to the central iron oxide core without using a chelate. Feraheme (average formula: Fe5874O8752–C11719H18682O9933Na414; MW ∼ 796 kDa) was selected as an appropriate nanoparticle for testing our hypothesis. Feraheme is a USPIO nanoparticle solution composed of a non-stoichiometric Fe3O4 (or Fe2O3:FeO) magnetite core approximately 5–10 nm in diameter, and stabilized with a carboxymethyl dextran coating (total size: ∼17–31 nm diameter; source: http://www.feraheme.com/). The coating increases aqueous solubility and was designed to provide extreme thermal stability of the particles so that terminal sterilization could be achieved by autoclaving at ca. ∼120 °C.26–28 In all reactions, aliquots of FH were used directly from a clinical formulation dose without further purification.Chelate-free, heat-induced radiolabeling of USPIOs
Initial radiochemical studies demonstrated that FH could be labeled with 89Zr4+ ions (using either oxalate or chloride salt formulations)29 under aqueous conditions (pH 8.0) upon heating and stirring the reaction mixture at 120 °C for less than 1 h (Fig. 1A). After cooling to room temperature the reaction was quenched with excess DTPA, EDTA or DFO (50 μL; 10 mM; ∼200–600-fold excess ligand versus total moles of Zr4+ ions). The 89Zr–FH product was isolated by size-exclusion chromatography (SEC; using either 30 kDa spin-column centrifugation or PD-10 column separation; Fig. 1B) in 82–96% decay corrected radiochemical yield (RCY), with an average RCY of 93 ± 3%; (n = 16 separate reactions), and radiochemical purity (RCP) of >98% after formulation in sterile saline (Fig. 1C). On scaling up the reaction to higher initial amounts of 89Zr-oxalate radioactivity (≥3.2 mCi), 89Zr–FH was isolated in 90 ± 2% RCY (n = 3). | ||
| Fig. 1 (A) Reaction of FH with 89Zr4+ ion salts (oxalate or chloride) to give radiolabeled 89Zr–FH. (B) SEC (PD-10) elution profile of the crude 89Zr–FH labeling reactions after stirring for 1 h at either RT (red) or 120 °C (blue) showing specific association of 89Zr-activity with the high MW (>30 kDa) FH fraction. (C) Analytical PD-10 elution profile (40 × 0.2 mL fractions) of purified and formulated 89Zr–FH demonstrating >98% RCP in sterile saline. (D) Representative radio-TLC chromatograms of DTPA-quenched crude 89Zr–FH reactions after 1 h at either RT control (black) or 120 °C (red). Note that in this analytical method, 89Zr–FH remained at the origin (Rf = 0.0) whilst 89Zr-radioactivity coordinated by DTPA during the quenching step migrated with the solvent front (Rf = 1.0). (E) Plot of the percentage RCY of 89Zr–FH versus reaction pH. | ||
89Zr-radiolabeling of the FH nanoparticles was confirmed further by radio-TLC (Fig. 1D; 89Zr–FH and 89Zr–DFO were retained on silica gel plates and remained at the baseline [Rf = 0.0] whereas unreacted 89Zr4+(aq.) ions coordinated as 89Zr–DTPA eluted with the solvent front [Rf = 1.0]). Note that if the 89Zr-radiolabeling reaction is quenched with DFO, radio-TLC on silica gel cannot be used to separate unreacted from nanoparticle-bound 89Zr using an aqueous DTPA (pH 7.5; 50 mM) eluent. The final radiochemical yield of purified 89Zr–FH was typically >95% by radio-TLC and the specific-activity was around 0.915 mCi mg−1 of total Fe (equivalent to 0.3 Ci μmol−1 of FH particles). Radio-TLC data are consistent with previously reported data on 89Zr–DFO–FH in which Thorek et al.15 radiolabeled FH nanoparticles with 89Zr via a DFO chelate covalently bound to the carboxymethyl dextran coating. In general, radio-TLC was found to be less reliable (due to possible instability of the particles on silica gel) than SEC for characterizing the RCP and RCY of radiolabeled FH. Note also that due to the small size of the FH nanoparticles (∼17–31 nm) magnetic separation was not feasible. For this reason, SEC was used as the primary analytical measure in subsequent stability studies (vide infra).
Importantly, control reactions revealed that the observed heat-induced, chelate-free association of the 89Zr-radioactivity to the FH nanoparticles was specific. Control reactions performed by stirring the mixture at room temperature (RT) for 1 h followed by quenching with DTPA showed only 6 ± 2% (n = 4) of the 89Zr-radioactivity co-eluted with FH in SEC (Fig. 1B). Radio-TLC of the crude, DTPA-quenched reaction mixtures analyzed before SEC confirmed the temperature-dependent nature of the radiolabeling reactions. Room temperature reactions gave only ∼7% 89Zr-radioactivity associated with the FH. Further, control studies in which FH was omitted from the 89Zr-mixture found that after DTPA-quenching both the RT and 120 °C reactions, >98% of the 89Zr-radioactivity was measured in the small MW fractions (fractions >2 mL elution volume) from SEC. The absence of 89Zr-radioactivity in the high MW fractions when FH was omitted demonstrates that the activity is bound to the added USPIO and not derived from precipitation, aggregation or agglomeration of 89Zr alone (i.e. experiments show that even under the basic conditions employed the 89Zr-radioactivity does not hydrolyze to form 89Zr-oxide particles).
In order to examine the possibility that the 89Zr-radioactivity may be binding to the carboxymethyl dextran coating of the FH nanoparticles – potentially through coordination by the hydroxyl or carboxylate groups – 89Zr-radiolabeling reactions were conducted using an alternate USPIO nanoparticle (Molday ION) that does not have a surface coating. Reactions of 89Zr-“chloride” with Molday ION nanoparticles (stir; 2 h; 250 μCi; pH 8.0–8.5; 0.2 mg Fe total) at RT gave only low levels of 89Zr-bound nanoparticles (89Zr-NP) at RT (14 ± 3%; n = 3). In comparison, when the reaction was heated at 120 °C higher RCY of 89Zr-NP (68 ± 9%; n = 3) was isolated by SEC. These data confirm that the chelate-free, heat-induced reaction is applicable to both coated and non-coated USPIOs, and suggest that the interaction between 89Zr ions and FH is centered at the iron oxide core rather than the nanoparticle coating.
During optimization studies, the pH dependence of 89Zr–FH radiolabeling was investigated (Fig. 1E). No significant difference in the percentage RCY was observed after reacting 89Zr-oxalate with FH in the pH range 7.1–9.0 (120 °C; 2 h; quenched with DFO at RT for 30 min; n = 3 per data point). Radiochemical yields were consistent with the initial radiolabeling studies (∼90% RCY; vide supra). Acidic pH ranges were not tested because the magnetite crystal core of USPIO nanoparticles, including FH, are known to be thermodynamically unstable with respect to decomposition and Fe3+ ion extraction in the presence of common chelates (e.g. EDTA, citrate etc) in acid.25,30
Stability studies
While the radiolabeling experiments confirmed that 89Zr4+ metal ions specifically associate with the magnetite crystal core of FH, association alone is not sufficient basis for developing radiotracers. Therefore, prior to pursuing detailed studies in vivo, the thermodynamic and kinetic stability of 89Zr–FH was assessed by using ligand challenge and plasma stability studies (Fig. 2).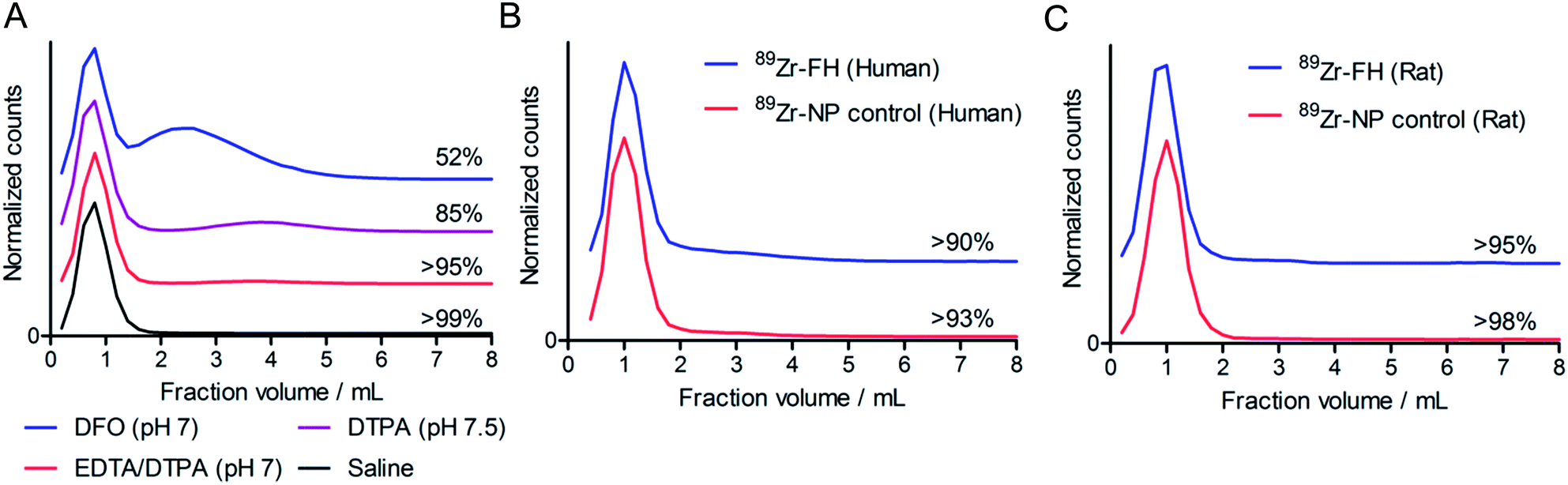 | ||
| Fig. 2 (A) Ligand challenge stability data (from SEC) acquired after 72 h incubation at 37 °C in saline (black), EDTA–DTPA mixture (pH 7; red), DTPA (pH 7.5; purple) and DFO (pH 7; blue). Note that data are offset in the y-axis for clarity. Size-exclusion profiles showing representative plasma challenge data for 89Zr–FH and control particles 89Zr-NP acquired after 72 h incubation at 37 °C in (B) human, and (C) rat plasma. | ||
Ligand challenge experiments were performed by incubating isolated 89Zr–FH (200 μCi; ∼50 μL) in sterile saline, DFO (10 mM; pH 7.0), DTPA (50 mM; pH 7.5); or a DTPA–EDTA mixture (10 mM per each chelate; pH 7.0). Mixtures were incubated at 37 °C for 72 h and analyzed by SEC (Fig. 2A). 89Zr–FH was found to be stable in saline with >99% of the 89Zr-radioactivity remaining bound to the high MW FH fraction after 72 h. When challenged with excess of the poly-carboxylate chelates (200–600-fold versus Zr4+ metal ion concentration) the radiotracer displayed high radiochemical stability with >95% intact 89Zr–FH in the DTPA–EDTA mixture and 85% intact 89Zr–FH in the more concentrated DTPA test after 72 h. In contrast, 52% of the 89Zr-radioactivity remained bound to FH when challenged with DFO.
Several important features of the ligand challenge experiment should be highlighted. First, unlike EDTA and DTPA, DFO is a siderophore with an exceptionally high affinity for Fe3+ ions (formation constant for Fe–DFO, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) β = 30.7). As such, DFO represents a more extreme ligand challenge than either EDTA or DTPA to the stability of USPIOs. The chemical instability of USPIOs to DFO via Fe3+ ion extraction to form Fe–DFO was observed in this experiment. The SEC columns run with DFO challenge samples displayed two intense orange/brown bands. The first band eluted corresponded to the high MW 89Zr–FH fraction while the second band (assigned to Fe–DFO) co-eluted with the small MW species (89Zr–DFO) in the 2.0–6.0 mL fractions. In contrast to the DFO challenge SEC, only one orange/brown band was observed in the ligand challenge experiments with EDTA and DTPA mixtures. Hence, under the conditions employed, EDTA and DTPA are not sufficiently strong chelates to affect Fe3+ ion extraction and/or magnetite core degradation. Second, the small MW fraction peak for the 89Zr–DTPA and 89Zr–EDTA mixtures occurs at fraction volume 3.8–4.0 mL, whereas the equivalent peak for 89Zr–DFO is both broadened and shifted to give a maximum at 2.4–2.6 mL. This peak shift to shorter retention time and band broadening is indicative of association between 89Zr–DFO and the FH (or 89Zr–FH) nanoparticles. Since the FH nanoparticles carry a predominantly negative charge under the conditions employed, it seems likely that due to the 2+ positive charge on the [89Zr–DFO]2+ complex at pH 7, a charge-based equilibrium binding occurs on the SEC column between free cationic 89Zr–DFO (aq.) and 89Zr–DFO bound to FH. Consequently, these ligand challenge studies also indicate that while it is possible to use DFO to quench the 89Zr-radiolabeling reaction with FH, DTPA and/or EDTA represent a more appropriate choice of quenching agent since they facilitate 89Zr–FH purification by SEC and do not induce USPIO degradation.
β = 30.7). As such, DFO represents a more extreme ligand challenge than either EDTA or DTPA to the stability of USPIOs. The chemical instability of USPIOs to DFO via Fe3+ ion extraction to form Fe–DFO was observed in this experiment. The SEC columns run with DFO challenge samples displayed two intense orange/brown bands. The first band eluted corresponded to the high MW 89Zr–FH fraction while the second band (assigned to Fe–DFO) co-eluted with the small MW species (89Zr–DFO) in the 2.0–6.0 mL fractions. In contrast to the DFO challenge SEC, only one orange/brown band was observed in the ligand challenge experiments with EDTA and DTPA mixtures. Hence, under the conditions employed, EDTA and DTPA are not sufficiently strong chelates to affect Fe3+ ion extraction and/or magnetite core degradation. Second, the small MW fraction peak for the 89Zr–DTPA and 89Zr–EDTA mixtures occurs at fraction volume 3.8–4.0 mL, whereas the equivalent peak for 89Zr–DFO is both broadened and shifted to give a maximum at 2.4–2.6 mL. This peak shift to shorter retention time and band broadening is indicative of association between 89Zr–DFO and the FH (or 89Zr–FH) nanoparticles. Since the FH nanoparticles carry a predominantly negative charge under the conditions employed, it seems likely that due to the 2+ positive charge on the [89Zr–DFO]2+ complex at pH 7, a charge-based equilibrium binding occurs on the SEC column between free cationic 89Zr–DFO (aq.) and 89Zr–DFO bound to FH. Consequently, these ligand challenge studies also indicate that while it is possible to use DFO to quench the 89Zr-radiolabeling reaction with FH, DTPA and/or EDTA represent a more appropriate choice of quenching agent since they facilitate 89Zr–FH purification by SEC and do not induce USPIO degradation.
Plasma challenge experiments were conducted by incubating both 89Zr–FH and control Molday ION 89Zr–NP in human (Fig. 2B) or rat (Fig. 2C) plasma for 72 h at 37 °C. Overall, the data demonstrate that radiolabeled nanoparticles are stable with <10% of the 89Zr-radioactivity released as small-molecules from either 89Zr–FH or 89Zr–NP in the presence of human or rat plasma. These data confirm that chelate-free, heat-induced 89Zr-radiolabeling leads to species that are thermodynamically and kinetically stable with respect to loss of the activity and decrease in RCP.
Non-radioactive chemical characterization
After demonstrating that radiolabeled 89Zr–FH was stable, a series of non-radioactive studies were conducted to evaluate the metal ion loading capacity as well as any induced chemical/magnetic changes in the FH properties that occur on reaction with various metal ions. FH was reacted with solutions of non-radioactive Zr4+–chloride in a serial dilution spanning a Zr-to-FH particle mole ratio of 1000 to 0.03 (Fig. 3A; 7-orders of magnitude; mole range n(Zr) = 5 μmol to 8 pmol). Note that in the radiochemical reaction, given the range of specific activity for 89Zr-salts (470–1195 Ci mmol−1),29 the actual amount of Zr used to radiolabel FH was 10−9 to 10−10 moles (equivalent Zr-to-FH ratio of 1.0 to 0.1). These serial dilution experiments (which spanned the radiochemical range) revealed that FH displays a very high capacity for Zr4+ metal ion association. A linear relationship was observed between the initial concentration of Zr4+ and the concentration associated with the high MW FH fraction after reaction at 120 °C for 1 h, quenching with DTPA, and purification by SEC.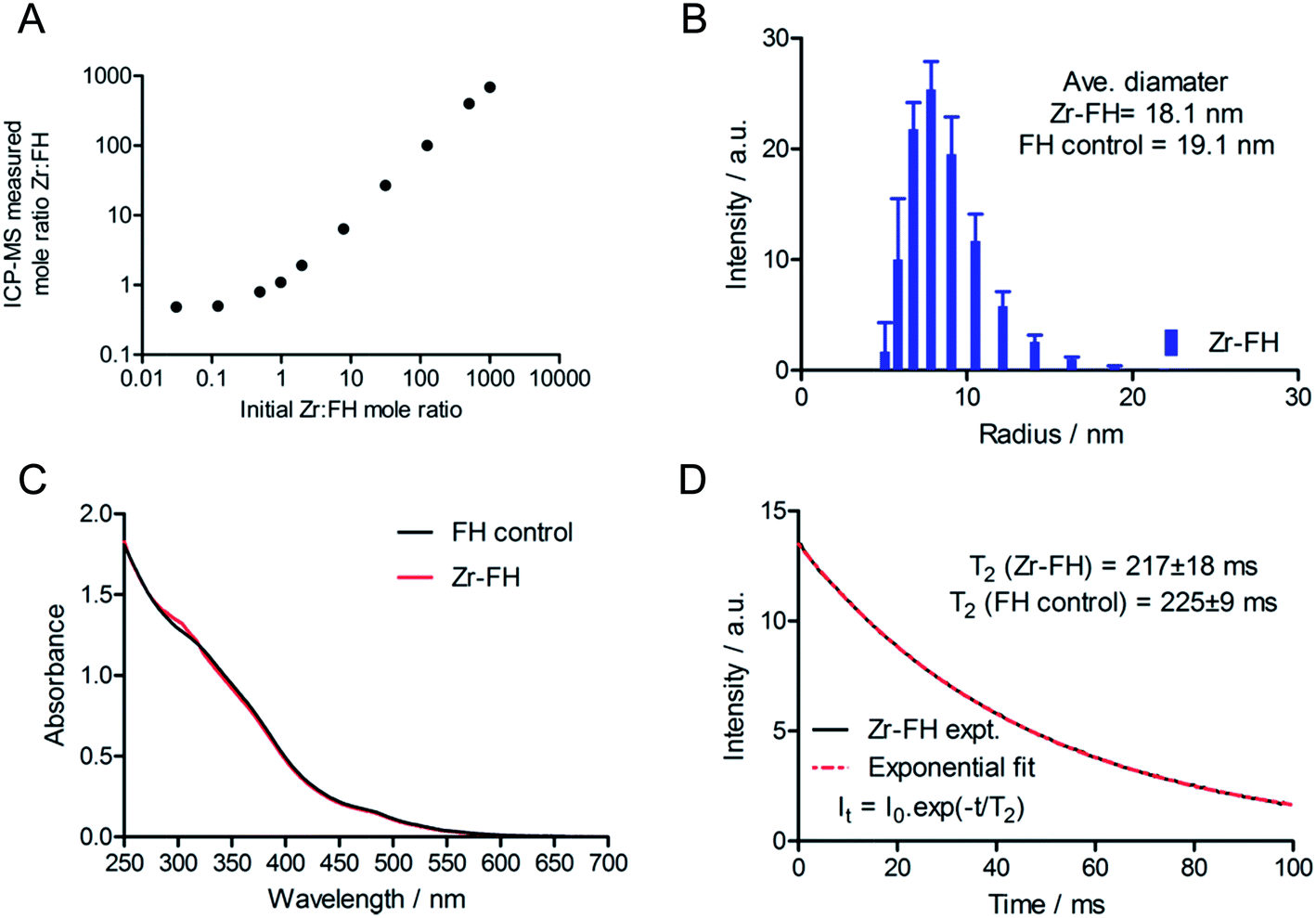 | ||
| Fig. 3 (A) Scatter plot of the measured Zr-to-FH mole ratio (ICP-MS) versus the initial reaction mole ratio showing the linear relationship between increasing Zr-chloride concentration and Zr–FH loading capacity. Linear regression analysis (excluding the two data points at a Zr-to-FH mole ratio below 1.0) gave the relation: measured mole ratio = 0.70 × initial mole ratio + 6.4; correlation coefficient R2 = 0.995. Note that at mole ratios <1.0, the concentration of added Zr lies at the detection limit of our ICP-MS instrumentation. (B) Representative DLS particle size histogram showing the range of Zr–FH particle sizes for a sample loaded with a Zr-to-FH mole ratio ≤125 (data shown for 30-to-1 mole ratio). The data are identical to measurements of control FH samples. (C) Electronic absorption spectra of solutions of Zr–FH (red; Zr-to-FH mole ratio = 30) and FH control in saline at ∼1 mg mL−1 total Fe. (D) T2-relaxation data showing the experiment (black) and modelled curve (red) for Zr–FH (mole ratio = 30) in water at 40 °C. | ||
Next, a combination of dynamic light scattering (DLS), UV/vis spectroscopy and T2 relaxometry was used to investigate the effect of Zr4+ metal ion loading on the physical (particle size and absorption) and magnetic properties of the Zr–FH nanoparticles. DLS studies revealed that for Zr-to-FH mole ratios ≤125, no change in particle size was observed (Fig. 3B). Measurements of Zr–FH particle size at loading mole ratios ≤125 each gave an average particle diameter of ∼18.1 nm compared to ∼19.7 nm for FH control samples. Samples measured at higher Zr-to-FH particle mole ratios (>125) exhibited pronounce aggregation with >90% of the particles in solution ranging from 60–400 nm in diameter (average = 150 nm). These data indicate that as long as the threshold Zr-to-FH mole ratio of approximately 125 is not exceeded (note that radiochemically a mole ratio of 0.1–2.0 is used), the Zr–FH loaded particles remain intact with an equivalent particle size distribution to that observed for clinical FH (diameter of particles in the clinical formulation = 17–31 nm).
Electronic absorption spectroscopy showed that no difference was observed in the absorption profiles of Zr-loaded FH versus FH control samples in saline (Fig. 3C). Magnetic T2 relaxometry measurements also confirmed that the water relaxation properties of Zr–FH were the same as control (reaction blank) FH samples with no difference in T2-relaxation time (Zr–FH: 217 ± 18 ms versus FH: 225 ± 9 ms; Fig. 3D). Interestingly, a modest decrease in T2-relaxation time was observed for both Zr–FH and control FH samples (isolated by PD-10 elution in saline from reactions conducted at RT and 120 °C) when compared to the clinical formulation of FH (experimental t(T2) = 236 ± 2 ms). This minor difference in T2 relaxation time is assigned to a change in particle solvation that occurs upon changing the formulation from mannitol (40 mg mL−1) in the clinical dose to sterile saline used in these studies.
Collectively, the DLS, UV/vis and T2 relaxometry data demonstrate that the FH nanoparticles remain chemically unchanged in terms of their physical size, spectroscopy absorption and magnetic spin–spin relaxation properties after loading with Zr4+ ions to a maximum Zr-to-FH mole ratio of approximately 125-to-1.
PET/CT imaging and biodistribution studies in mice
Based on the encouraging radiochemistry, stability and physical characterization data (vide supra) we next evaluated the pharmacokinetics and distribution profile of 89Zr–FH in wild-type B6C3F1/J mice using PET/CT imaging (Fig. 4) and ex vivo biodistribution studies. Temporal PET/CT imaging studies (n = 5 mice) revealed that 89Zr–FH circulates in the blood during the first ∼6–8 h post-administration (Fig. 4A). Blood pool extraction occurred rapidly with 89Zr-radioactivity accumulating mainly in liver, spleen and lymph nodes (particularly the mesenteric lymph node; Fig. 4B). Whole-body excretion data showed that 89Zr–FH was not eliminated with 95 ± 2% (n = 5) of the 89Zr-radioactivity remaining in the mouse beyond 120 h post-administration (Fig. 4C). Time–activity curves (TACs) derived from volume-of-interest (VOI) analysis of the temporal PET/CT data showed that as the 89Zr-radioactivity clears from the heart/blood pool, a concordant increase in liver uptake occurs (Fig. 4D). Uptake was also observed in the spleen and lymphatic system – consistent with the reported data on 89Zr–DFO–FH15 and other labeled nanoparticles.31–35 Further analysis of the heart TAC revealed that 89Zr–FH clears from the blood pool with an observed extraction half-life of activity in the heart VOI, t1/2(heart) = 2.21 ± 0.55 h (n = 3; correlation coefficient R2 = 0.958; Fig. 4E). This measured half-life in the heart/blood pool is consistent with the known blood half-life36 of ∼40 min for non-labeled FH in mice – differences are assigned to the fact that PET/CT analysis includes the heart tissue and associated blood pool volume in contrast to direct blood sampling methods used previously.36 Overall, the PET/CT data showed that the biodistribution and excretion profile of 89Zr–FH is fully consistent with the known biodistribution and dosimetry data of FDA-approved FH.15,25,31,37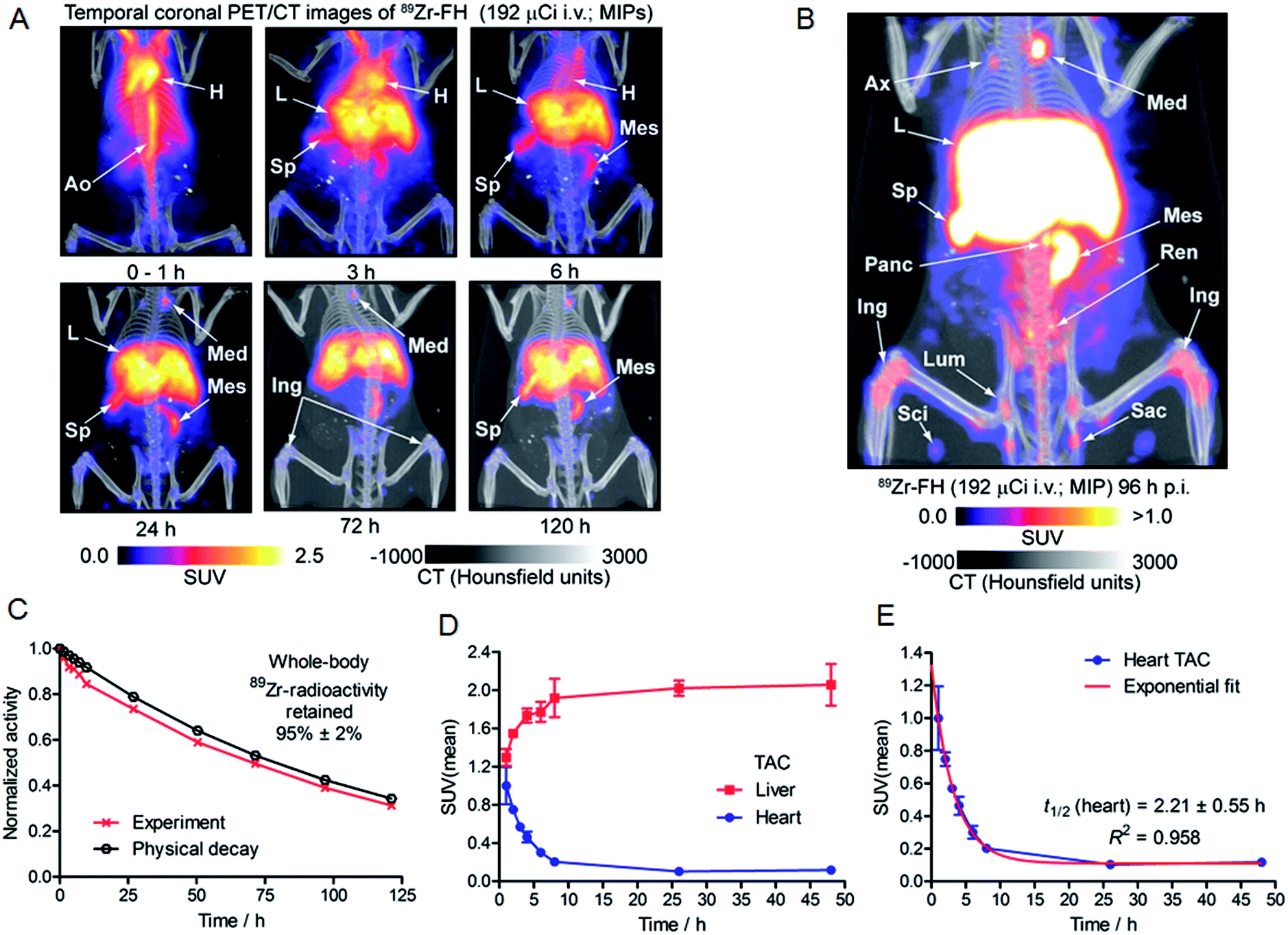 | ||
| Fig. 4 (A) Temporal PET/CT maximum intensity projection (MIP) images recorded between 0–120 h post-i.v. injection of 89Zr–FH in B6C3F1/J wild-type mice. Ao = aorta; H = heart; L = liver; Sp = spleen; Mes = mesenteric lymph node; Ing = inguinal lymph. (B) MIP PET/CT image recorded at 96 h p.i. in a wild-type B6C3F1/J mouse showing uptake of 89Zr–FH in the lymphatic system. Sp = spleen; L = liver; lymph nodes: Ax = axial; Med = mediastinal; Panc = pancreatic; Ren = renal; Sac = sacral; Sci = sciatic; Lum = lumbar. (C) Whole-body measurement of the effective excretion half-life measured by dose calibrator showing that 95 ± 2% (n = 5) of 89Zr–FH remained inside the mice for at least 120 h p.i. (D) Time–activity curves (TACs) derived from VOI analysis of the temporal PET/CT data showing the extraction of 89Zr-radioactivity from the heart and accumulation in the liver. (E) TAC and one-phase exponential decay modeling of the heart TAC data. | ||
Nanoparticles are known to accumulate in activated macrophage cells.10,16,32,38,39 Therefore, we evaluated the uptake of 89Zr–FH in an established subcutaneous (s.c.) chemically induced acute phase response model of inflammation which has been shown to induce infiltration and enrichment of activated macrophage.40–42 Full biodistribution data from tissue dissection and ex vivo counting of groups of mice (n = 5 per group) sacrificed at 1, 4, 24, 48 and 120 h, post-i.v. administration of 89Zr–FH are presented in Table 1 and Fig. 5A. Radiotracer uptake in the inflamed (right hind limb) muscle was found to increase over time from 0.48 ± 0.39 %ID per g at 1 h p.i., to a maximum of 0.95 ± 0.12 %ID per g at 120 h p.i. In contrast, non-specific 89Zr–FH accumulation in the control (left hind limb) muscle showed a maximum value at 1 h (0.27 ± 0.09 %ID per g) decreasing to a minimum of 0.10 ± 0.02 %ID per g at 120 h p.i. A plot of the inflamed-to-control muscle 89Zr–FH uptake revealed that 89Zr-radioactivity continued to accumulate in the inflamed muscle between 1–24 h p.i reaching a ratio of ∼8.3 (Fig. 5B). Beyond 24 h, the inflamed-to-control muscle ratio continued to increase slightly due to wash-out of activity from the control muscle. A representative coronal PET/CT image of 89Zr–FH uptake in the inflamed muscle of the right hind limb is presented in Fig. 5C. Note that the absolute uptake of 89Zr–FH in the inflamed tissue is limited by the comparatively short circulation half-life of these USPIO nanoparticles in mice. Nevertheless, biodistribution data confirm that 89Zr–FH uptake in the macrophage-enriched inflamed tissue is specific. The mechanism of 89Zr–FH delivery and accumulation is consistent with the Enhanced Permeability and Retention (EPR).43 Overall, the PET/CT imaging and biodistribution data in mice are fully consistent with the known distribution of FH (and other USPIO nanoparticles) and with reported data on 89Zr–DFO–FH.15,25,31,37
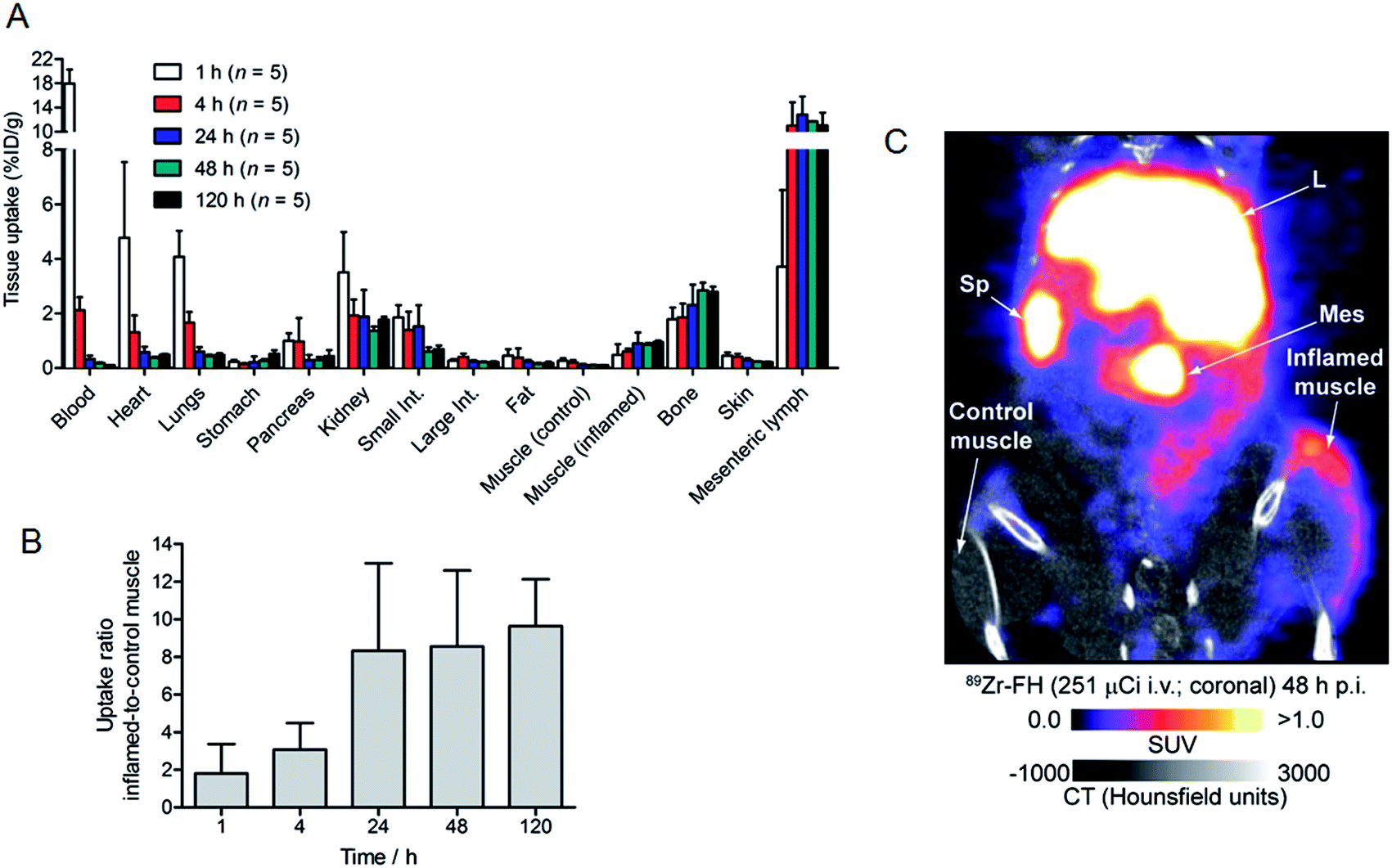 | ||
| Fig. 5 (A) Full biodistribution data (excluding liver and spleen) showing the measured tissue uptake (in units of %ID per g) at 5 time points between 1–120 h post-i.v. injection of 89Zr–FH in B6C3F1/J mice (n = 5 per group). (B) Plot of the change in 89Zr–FH uptake ratio versus time between the inflamed and control muscle. (C) Coronal PET/CT image recorded at 48 h p.i. showing specific 89Zr–FH uptake in the inflamed muscle (right hind limb) versus contralateral control muscle (left hind limb). Sp = spleen; L = liver; Mes = mesenteric lymph node. | ||
| Uptake of 89Zr-radioactivity/%ID/g | |||||
|---|---|---|---|---|---|
| Tissue | 1 h (n = 5) | 4 h (n = 5) | 24 h (n = 5) | 48 h (n = 5) | 120 h (n = 5) |
| Blood | 17.95 ± 2.31 | 2.12 ± 0.48 | 0.33 ± 0.12 | 0.18 ± 0.07 | 0.10 ± 0.02 |
| Heart | 4.78 ± 2.77 | 1.31 ± 0.62 | 0.58 ± 0.20 | 0.38 ± 0.07 | 0.49 ± 0.06 |
| Lungs | 4.08 ± 0.95 | 1.66 ± 0.39 | 0.60 ± 0.16 | 0.44 ± 0.11 | 0.48 ± 0.14 |
| Liver | 75.37 ± 7.86 | 88.10 ± 12.16 | 89.68 ± 16.74 | 89.78 ± 10.28 | 101.93 ± 5.88 |
| Spleen | 106.15 ± 18.10 | 147.16 ± 22.80 | 117.97 ± 24.90 | 112.50 ± 20.58 | 132.96 ± 11.90 |
| Stomach | 0.23 ± 0.07 | 0.14 ± 0.04 | 0.22 ± 0.20 | 0.27 ± 0.13 | 0.52 ± 0.30 |
| Pancreas | 1.00 ± 0.27 | 0.97 ± 0.86 | 0.29 ± 0.19 | 0.30 ± 0.22 | 0.43 ± 0.51 |
| Kidney | 3.50 ± 1.48 | 1.92 ± 0.59 | 1.88 ± 0.98 | 1.37 ± 0.33 | 1.77 ± 0.23 |
| Small intestine | 1.85 ± 0.45 | 1.39 ± 0.67 | 1.53 ± 0.77 | 0.61 ± 0.33 | 0.69 ± 0.29 |
| Large intestine | 0.27 ± 0.05 | 0.40 ± 0.13 | 0.25 ± 0.03 | 0.22 ± 0.04 | 0.22 ± 0.06 |
| Fat | 0.45 ± 0.24 | 0.38 ± 0.34 | 0.23 ± 0.07 | 0.16 ± 0.06 | 0.18 ± 0.11 |
| Muscle (control; left) | 0.27 ± 0.09 | 0.20 ± 0.09 | 0.11 ± 0.04 | 0.10 ± 0.04 | 0.10 ± 0.02 |
| Muscle (inflamed; right) | 0.48 ± 0.39 | 0.61 ± 0.10 | 0.90 ± 0.40 | 0.85 ± 0.15 | 0.95 ± 0.12 |
| Bone | 1.79 ± 0.42 | 1.85 ± 0.51 | 2.31 ± 0.74 | 2.85 ± 0.62 | 2.79 ± 0.42 |
| Skin | 0.45 ± 0.12 | 0.41 ± 0.11 | 0.29 ± 0.07 | 0.23 ± 0.04 | 0.22 ± 0.03 |
| Mesenteric lymph | 3.72 ± 2.80 | 11.01 ± 3.86 | 12.84 ± 2.98 | 11.72 ± 0.03 | 11.05 ± 4.65 |
| Inflamed-to-control muscle ratio | 1.80 ± 1.57 | 3.07 ± 1.41 | 8.33 ± 4.65 | 8.56 ± 4.03 | 9.64 ± 2.49 |
Chemical scope of metal ion binding to USPIOs
Having established that our radiochemical studies in vitro and in vivo on 89Zr–FH provide compelling evidence that chelate-free, heat induced labeling of USPIOs is a practical method for generating novel multi-modality PET/MRI imaging agents, we next investigated the chemical and radiochemical scope of metal ion binding. Temperature dependent FH labeling reactions were performed using non-radioactive chloride ion salts of the p-block ion In3+, first row d-block transition metal ions Mn2+, Co2+, Ni2+, Cu2+ and Zn2+, second row d-block transition metal ion Zr4+, and f-block lanthanide ions Eu3+ and Tb3+ (ESI Table S1†). Competition reactions were also performed in which mixtures of nine metal ions were reacted with FH (Fig. 6A).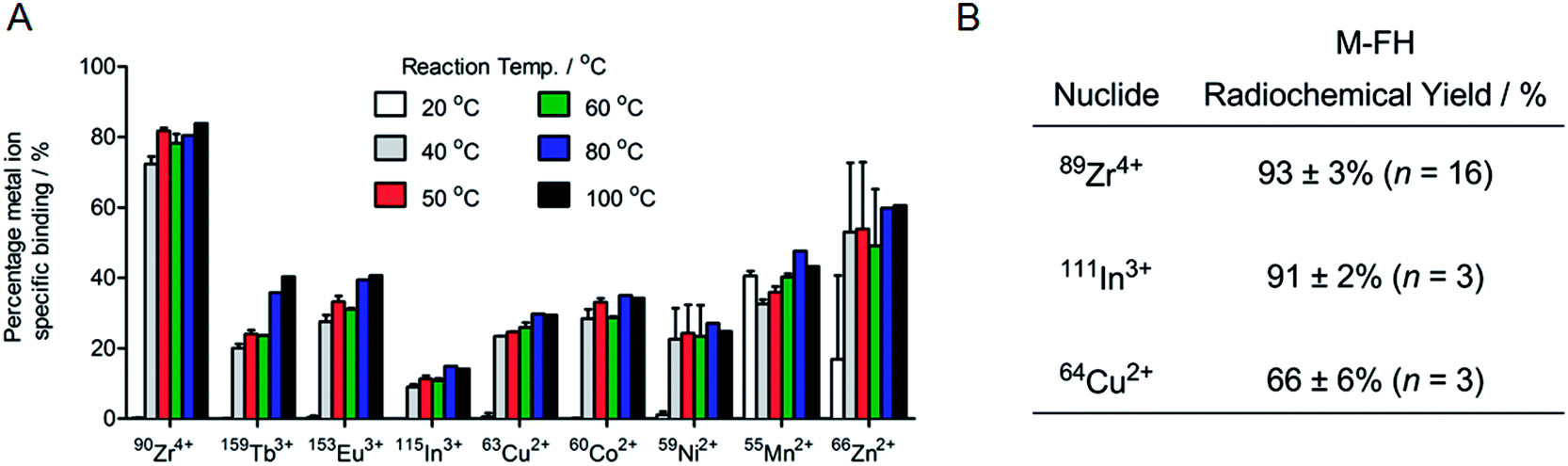 | ||
| Fig. 6 (A) ICP-MS data showing the percentage of specific metal ion binding to FH after heating the reaction mixture containing nine different metal ions for 1 h at temperatures ranging from RT to 100 °C (n = 3). Note the competition reaction was quenched using a DTPA–EDTA (10 mM per each chelate; pH 7.0) solution for 30 min at RT, and purified using SEC (PD-10; eluent: sterile saline). (B) Table showing the average isolated radiochemical yields of M-FH attained after chelate-free, heat induced radiolabeling for 1 h at 120 °C using 89Zr4+, 111In3+ and 64Cu2+ metal ions. | ||
Remarkably, ICP-MS data revealed that all metal ions tested bound to FH. Yields varied with Zr4+ ions displaying the highest specific binding around 75–80% (note that these data are consistent with the radiochemical data and ICP-MS data on the serial dilution loading capacity measurements [vide supra]). In these non-radioactive reactions, In3+ ions displayed the lowest affinity for FH with specific binding yields in the range 8–16%. The lanthanide ions Tb3+ and Eu3+ showed very similar specific binding yields ranging from around 20–40%. Similar binding yields were also observed for Co2+, Cu2+ and Ni2+ ions (20–40%), and a slight increase in binding yield was observed for Mn2+ (∼35–45%) and for Zn2+ ions (16% at RT increasing to 67% at 100 °C). Notably, an equivalent modest temperature-dependence of the labeling yield was observed for all metal ions tested. Almost no specific binding was observed at RT (with the exceptions of Mn2+ and Zn2+ ions). However, upon heating the reactions from 40 °C to 100 °C, specific binding yields increased. In general, labeling yields also tended to increase with increasing temperature, and highest metal ion binding yields were attained at 80 °C to 100 °C for all metal ions tested. Finally, from the metal ion binding competition experiments, Zr4+ ions were found to out-compete the other metal ions tested.
In order to validate the results from the non-radiochemical (ICP-MS) labeling reactions, additional radiolabeling reactions (RT and 120 °C; 1 h, quenching with DTPA–EDTA mixture for 30 min at RT followed by purification by SEC) were performed using chloride salts of the positron-emitting radionuclide 64Cu2+ and the SPECT and Auger electron emitting radionuclide 111In3+ (Fig. 6B). The radiolabeling results with 64Cu2+ and 111In3+ were very similar to those for 89Zr4+ reactions: 64Cu–FH was isolated in 66 ± 6% RCY (n = 3) and 111In–FH was isolated in 91 ± 2% RCY (n = 3) from the reaction at 120 °C (both with >99% RCP). Control reactions at RT revealed low non-specific binding of 11 ± 6% for 64Cu2+ and 9 ± 4% for 111In3+ ions. Ligand challenge and plasma stability studies were not performed on 64Cu–FH and 111In–FH but it is anticipated that these radiotracers will be stable with results similar to those obtained for 89Zr–FH (vide supra), and further studies are underway.
Electron magnetic resonance (EMR) studies
Data presented above suggest that metal ion binding may occur at the magnetite crystal core of the nanoparticles (FH and Molday ION USPIOs). In terms of the mechanism of binding, it is conceivable that the metal ions may bind tightly to the crystal surface. However, in an alternative mechanism we hypothesized that binding could involve thermally-induced diffusion of the metal ions inside the magnetite crystal lattice – akin to thermally-induced “doping” of the nanoparticle core – or inside crystal defects. To test this hypothesis, we performed temperature dependent (5–200 K) high-resolution X-band EMR (also known as Electron Spin Resonance [ESR]) spectroscopy on non-radioactive FH nanoparticles labeled with Zr4+, Cu2+, and In3+ ions, as well as control and drug FH samples (Fig. 7 and ESI Fig. S1–S3†).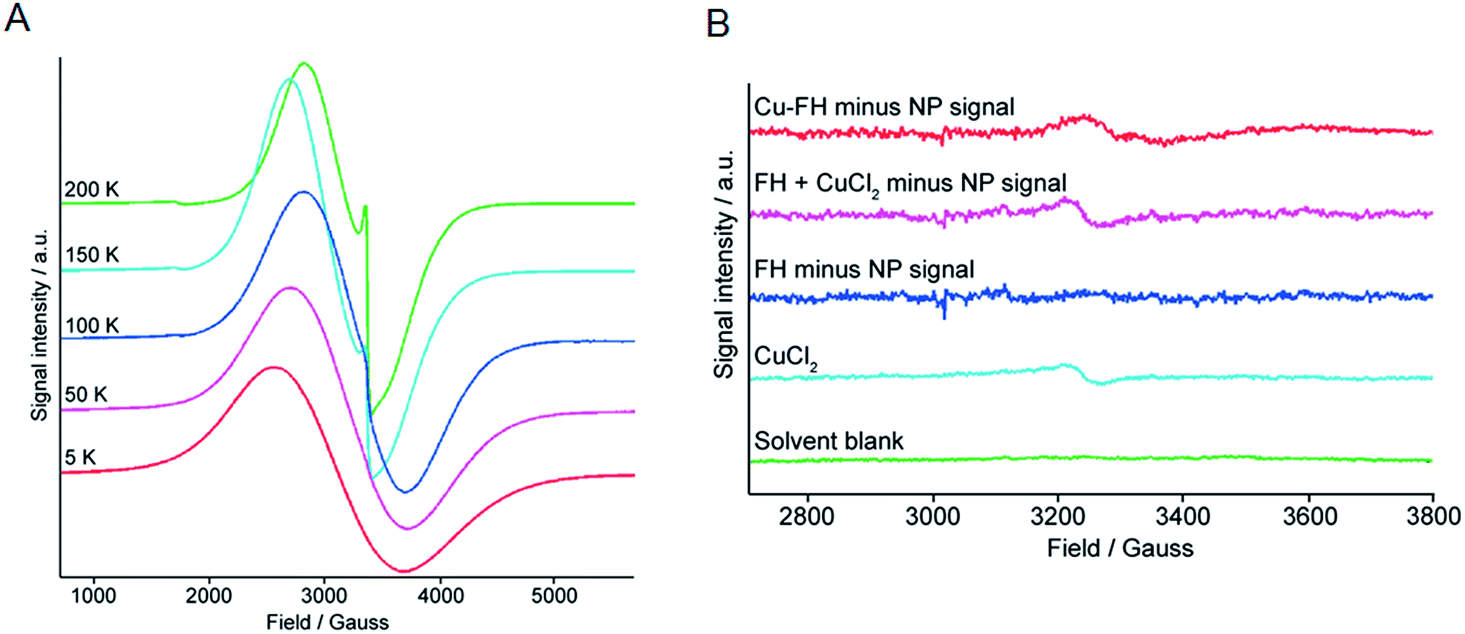 | ||
| Fig. 7 Temperature-dependent X-band ESR spectra of (A) control FH drug nanoparticles, and (B) a stack plot showing the EMR region where the low intensity Cu2+ ions give rise to a paramagnetic signal. The spectra correspond to (from bottom to top): solvent blank (green); CuCl2 control (light blue); the spectrum of FH blank minus the polynomial fitted SPR signature of the nanoparticles showing that no Cu2+ ions are present in the control FH blank samples (dark blue); a control spectrum showing the summed spectrum of FH and CuCl2 minus the SPR nanoparticle signal (purple); and the spectrum of Cu–FH (loaded with ∼60 Cu2+ ions per nanoparticle) with the SPR nanoparticle signal subtracted (red). | ||
Two main features were observed in all cases for the FH nanoparticles. The first feature is a broad line which broadens and moves to lower fields with a decrease in temperature. Second, a sharper feature at g = 2 which is only visible at higher temperatures (Fig. 7A). These spectroscopic features are consistent with previous EMR data recorded on iron oxide nanoparticles.44–46 In addition, two signals were observed in the half-field region. The signal at g = 4.0 is mainly visible at higher temperatures and arises due to multi-quantum transitions.44 The signal at g = 4.3 was only observed at low temperatures and is paramagnetic in origin resulting from Fe3+ ions. It should be noted that the low intensity Fe3+ signal was found to be absent from the FH drug samples but was observed in all metal ion loaded samples (Zr–FH, Cu–FH and In–FH; Fig. S1–S3†) and the FH blank sample that had been treated in the same way as the loaded samples. Thus, the appearance of this impurity signal at g = 4.3 is linked to the synthetic method.
In order to identify if the low intensity paramagnetic Cu2+ signal could be observed under the intense super-paramagnetic resonance (SPR) signature in the Cu–FH spectrum, samples of solvent blank (1:4 v/v ethylene glycol–double distilled H2O), free CuCl2 (prepared at a concentration approximately equivalent to the Cu2+ ion loading in Cu–FH: ∼60 Cu2+ ions per nanoparticle [∼25 μM]), FH reaction blank, and Cu–FH were measured using EMR at 5 K (Fig. 7B). The solvent blank gave no measureable EMR signal, confirming that the signals recorded in other samples arise from the dissolved material (Fig. 7B; green). The sample of CuCl2 gave the expected axial spectrum with a g⊥ ∼ 2.08 (Fig. 7B; light blue. Note the signal-to-noise ratio was not sufficient to determine an accurate g‖ value).
Next, to deduce if a Cu2+ signal was present in the Cu–FH spectrum we first removed the larger SPR nanoparticle signal. Note that simulating the SPR signal from nanoparticles is non-trivial and many different models have been derived.47 For this reason, as a first approximation the nanoparticle SPR signal was fitted using a polynomial function. Prior to applying our method on the Cu–FH spectrum we validated the procedure in two ways. First, the polynomial fitted SPR signal was subtracted from the experimental spectrum of the FH blank (Fig. 7B; dark blue). Second, the experimental signals for the CuCl2 and FH blank samples were summed and then the polynomial fitted SPR signal was subtracted to recover the CuCl2 signal (Fig. 7B; purple). In the first validation test, subtracting the fitted SPR data from the experimental FH spectrum gave no EMR signal. In the second test, subtracting the simulated SPR signal allowed full recovery of the CuCl2 signal, providing confidence in our method. Finally, the polynomial fitted SPR nanoparticle signal was subtracted from the total experimental Cu–FH spectrum to reveal the presence of a low intensity paramagnetic signal (Fig. 7B; red). Data for the Cu–FH sample showed a signal with a g ∼ 2.05, which is consistent with the anticipated range of g⊥ values corresponding to Cu2+ ions. Notably, the signal is shifted from that of free CuCl2 (g⊥ ∼ 2.08) which indicates a change in the Cu2+ ion environment. This shift is consistent with a mechanism of uncoupled/isolated Cu2+ ions binding to the FH nanoparticles and provides direct support indicating that a mechanism of direct metal ion binding to the magnetite crystal surface is plausible.
Discussion
As macromolecular scaffolds for developing drugs and imaging agents, nanoparticles offer unparalleled opportunities in terms of exploiting their chemical and structural features such as enhanced rigidity, controlled shape and size, electromagnetic properties, high surface area, variable porosity, resistance to metabolism in vivo, and tuneable chemical reactivity at the surface, on coatings and inside the particle.1–9 The vast majority of radiochemical methods currently used to label nanoparticles derive from the well-established chemistries of conjugation and radiometal chelation, and from 18F-labeling reactions.48 In this regard, reactions that could be considered specific to particles or surfaces have rarely been exploited as methods for radiolabeling nanoparticles. Goel et al. have recently reviewed the emerging area of intrinsically labeled nanoparticles.49 In their pioneering work on chelate-free radiolabeling, Chen et al.20 noted that arsenic(III) and arsenic(V) ions display high affinity and specific chemical reactivity with the surface of magnetite crystals. Radiolabeling of USPIO with radioarsenic nuclides was enabled through reaction of As(III)O3 trigonal pyramids and As(V)O4 tetrahedra via site-specific occupancy of vacant FeO4 sites on the octahedrally terminated [111] surface. The same group recently followed this work by presenting a method for developing PET/MRI agents using intrinsically labeled 69Ge nanoparticles.22 Inspired by the technology used to produce 68Ge/68Ga generators whereby the parent 68Ge (t1/2 = 279 days) is bound to metal oxide supports including TiO2, ZrO2, CeO2, SnO2, Fe2O3, Fe3O4, Al2O3etc, Chakravarty et al. demonstrated that 89Ge-labeled super-paramagnetic iron oxide nanoparticle (SPION) could be prepared in the absence of a chelate. In a third example, Wong et al.21 reported the synthesis of dextran-coated 64Cu-doped iron oxide nanoparticles. In this work, the 64Cu-radionuclide was added to the reaction mixture prior to synthesis of the nanoparticles. Subsequent standard synthesis of the dextran-coated USPIOs led to facile incorporation of the nuclide inside the nanoparticle magnetite core. Finally, in two recent reports, Sun et al. provided elegant demonstrations of chelate-free 64Cu-radiolabeling of Au nanorods,23 and 64Cu-doped CdSe/ZnS quantum dots24 – data which provide strong support of our assertion of the generality of heat-induced, chelate-free radiolabeling of USPIOs and other metal-based nanoparticles. In these reports, the researchers noted that these agents show promise for dual-modality PET/MRI imaging and these reactions represent key examples of “chelate-free radiometal synthesis”. However, with the exception of the data showing 64Cu-labeling of Au nanorods, these approaches appear to lack generality and/or have drawbacks when labeling pre-fabricated nanoparticles. Labeling with As(III)/As(V) nuclides is specific for the exposed surface defects of magnetite crystals and suffers from a lack of chemical control over the oxidation state of the arsenic species. Production of 69Ge-labeled SPION required an initial 69Ge-labeling step followed by subsequent coating of the nanoparticle by PEGylation to generate the final PET/MRI radiotracer. Although chemical doping during nanoparticle synthesis represents a general way of incorporating different metal radionuclides, this approach requires full batch preparation and QC of the entire nanoparticle for each radiosynthesis. In addition, incorporation of the radiometal pre-nanoparticle fabrication is not feasible for many short-lives nuclides like 68Ga (t1/2 = 67.7 min).19The data presented in this work demonstrates that chelate-free, heat-induced metal ion binding to the magnetite core of USPIO nanoparticles is a facile and robust new reaction. The radiolabeling reaction generates labeled-USPIO nanoparticles which are found to be radiochemically stable with virtually no change in their physical or chemical properties when compared to the native/parent nanoparticle. Notably, minor differences observed in this work are most likely due to differences in the formulation rather than inherent changes to the nanoparticle properties. The fact that the FH nanoparticles retain their physical and chemical characteristics after labeling means that our reaction has the potential to expedite clinical translation of radiolabeled nanoparticles for PET/CT and PET/MRI. The reaction was found to exhibit a broad chemical scope with metal ions of different charge (ranging from 2+ to 4+) undergoing reaction with FH to generate M-FH labeled constructs. Importantly, we also demonstrated the radiochemical versatility of our novel reaction. Different metallo-radionuclides including the positron-emitting radionuclides 89Zr4+ and 64Cu2+, as well as the γ-ray and Auger electron emitting radionuclide 111In3+ were successfully bound to FH producing radiotracers that have potential to be used in PET or SPECT imaging, and in radiotherapy. In addition, as noted by others15,20,21 radiolabeled USPIO nanoparticles (including FH) hold promise for developing dual-modality PET/MRI agents. Indeed, radiolabeled FH shows potential as a PET/MRI imaging agent for characterizing sentinel lymph node metastases and as a potential radiotracer for imaging macrophage activation in e.g. inflammatory diseases, tumors and myocardial infarct etc.15,39
At this juncture, full details of the mechanism of metal ion binding to the magnetite crystal are unknown. Experimental data points toward a specific and very tight interaction which leads to labeled nanoparticles that display excellent stability under extreme chemical and biochemical challenges in vitro and in vivo. Having excluded the possibility of metal ion binding to the nanoparticle coating (see control Molday ION radiolabeling studies and quenching data vide supra), two potential pathways of interaction involve: (i) metal ion binding to the oxide surface layer of the nanoparticle core, and (ii) thermally-induced diffusion of the metal ions inside crystal cavities/defects to generate an equivalent of a “doped” nanoparticle. The ligand challenge experiments using DFO suggest that both mechanisms of binding are plausible. However, the EMR data on Cu–FH point toward the most likely mechanism of metal ion binding involving direct interaction with the oxide surface layer of the magnetite crystal core. If Cu2+ ions penetrate into the FH core such that they became part of the super-paramagnetic system, the Cu2+ signal would be lost because the EMR spectrum would not show coupling of a single unpaired electron to a Cu nucleus. This conclusion is supported by the experimental EMR data that confirms coupling of individual electrons to Fe nuclei is not observed in the FH control sample (Fig. 7A). Thus, observance of the Cu2+ signal in Cu–FH provides direct support for the mechanism of metal ion binding to the crystal surface. However, the absence of a Cu signal does not allow us to exclude the possibility that a thermally-induced “doping” mechanism is occurring such that while some copper interacts primarily on the surface of the nanoparticles other ions diffuse into the crystalline core. Further work using chemical, radiochemical, magnetic spectroscopy (EMR, SQUID magnetometry, Mössbauer Spectroscopy etc) and High-Resolution Transmission Electron Microscopy combined with Energy Dispersive X-ray analysis (HRTEM-EDX) is underway to elucidate the precise location and mechanism of metal ion binding to USPIO nanoparticles. We are also exploring the use of other metal ions (e.g. actinides like 225Ac and heavier p-block metals) to expand the potential of using chelate-free radiolabeling to incorporate other radionuclides including α-particle emitters into various metal-based nanoparticles for therapeutic applications. Given the ICP-MS data showed that only mild heating to ≥40 °C was required to induce metal ion binding to FH, we anticipate that our technology may also be applicable to labeling nanoparticles functionalized with biological targeting vectors including peptides, proteins and immunoglobulin fragments etc. Finally, it is conceivable that our discovery of heat-induced binding of metal ions to metal-based nanoparticles will have implications in areas of science beyond Radiochemistry and Nuclear Medicine including drug development and delivery, separation science and environmental toxicology of nanoparticles.
Conclusions
Experimental studies have established that the novel chelate-free, heat-induced metal ion binding reaction can be used to label USPIO nanoparticles. The reaction has a wide chemical scope and was found to be applicable to p-, d- and f-block metal ions exhibiting a range of ionic sizes and formal oxidation states from 2+ to 4+. Radiolabeling studies found that 89Zr–FH was thermodynamically and kinetically stable in vitro in a series of ligand challenge and plasma stability tests. Further, PET/CT imaging and biodistribution studies showed that 89Zr–FH behaves in an almost identical manner to the parent FH nanoparticle i.e. the radiolabeling reactions and metal ion binding appear to have minimal effect on the physical and biological properties of the FH nanoparticles. Additional imaging and biodistribution studies in mice bearing a subcutaneous acute phase response inflammation model demonstrated the potential of 89Zr–FH to be used for PET/CT (and possibly MRI) detection of tissues enriched with activated macrophage. Subsequent labeling studies combined with ICP-MS and radiochemistry found that the same reaction conditions used to produce 89Zr–FH can be employed to yield 64Cu–FH and 111In–FH. EMR studies found that a low intensity Cu2+ was present in the Cu–FH spectrum and these data provide direct evidence in support of metal ion association with the magnetite crystal core of the FH nanoparticles. Collectively, the data provide support for the hypothesis of tight metal ion binding to the magnetite crystal surface through a likely interaction between the positively charged metal ions and the anionic oxide surface layer. We anticipate that our new reaction will have profound implications in Radiochemistry, Nuclear Medicine and beyond.Acknowledgements
We thank Dr T. Lee Collier, Dr Cory Daignault and David Hill for helpful discussions, and Alicia Leece for technical advice regarding PET/CT. We thank Dr Ron Moore, Dr Jack A. Correia and David Lee and the Massachusetts General Hospital Cyclotron Core facility for target irradiations. We are indebted to Dr Bruce Wieland and Dr Matthew Stokely (BruceTech Targets) for design and production of the custom-made solid metal target for the GE PETtrace cyclotron. We thank Dr Peter Caravan for providing access to the ICP-MS instrumentation. We thank Prof. Thomas Prisner for access to EMR instrumentation. We are grateful to William J. Cupelo and Dr Jack Hoppin (inviCRO, LLC, Boston, MA) for generous support and access to the VivoQuant software. We thank Dr Ivan Greguric and the Australian National Science and Technology Organisation (ANSTO, Sydney, Australia) for providing financial support (JPH). We are grateful to the Swiss National Science Foundation for an advanced Post-doctoral mobility fellowship (EB). AMB gratefully acknowledges the support of a Goethe International Postdoc Programme Fellowship (EU project GO-IN, No. 291776 (COFUND) FP7 Marie Curie Action).Notes and references
- L. Zhang, F. X. Gu, J. M. Chan, A. Z. Wang, R. S. Langer and O. C. Farokhzad, Clin. Pharmacol. Ther., 2007, 83, 761–769 CrossRef PubMed.
- V. Wagner, A. Dullaart, A.-K. Bock and A. Zweck, Nat. Biotechnol., 2006, 24, 1211–1217 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, Adv. Drug Delivery Rev., 2006, 58, 1456–1459 CrossRef CAS PubMed.
- F. X. Gu, R. Karnik, A. Z. Wang, F. Alexis, E. Levy-Nissenbaum, S. Hong, R. S. Langer and O. C. Farokhzad, Nano Today, 2007, 2, 14–21 CrossRef.
- E. S. Kawasaki and A. Player, Nanomed.: Nanotechnol., Biol. Med., 2005, 1, 101–109 CrossRef CAS PubMed.
- O. C. Farokhzad, J. Cheng, B. A. Teply, I. Sherifi, S. Jon, P. W. Kantoff, J. P. Richie and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6315–6320 CrossRef CAS PubMed.
- L. Zhang, A. F. Radovic-Moreno, F. Alexis, F. X. Gu, P. A. Basto, V. Bagalkot, S. Jon, R. S. Langer and O. C. Farokhzad, ChemMedChem, 2007, 2, 1268–1271 CrossRef CAS PubMed.
- R. Jurgons, C. Seliger, A. Hilpert, L. Trahms, S. Odenbach and C. Alexiou, J. Phys.: Condens. Matter, 2006, 18, S2893 CrossRef CAS.
- R. Sinha, G. J. Kim, S. Nie and D. M. Shin, Mol. Cancer Ther., 2006, 5, 1909–1917 CrossRef CAS PubMed.
- S.-D. Li and L. Huang, Mol. Pharm., 2008, 5, 496–504 CrossRef CAS PubMed.
- J. J. Mulvey, C. H. Villa, M. R. McDevitt, F. E. Escorcia, E. Casey and D. A. Scheinberg, Nat. Nanotechnol., 2013, 8, 763–771 CrossRef CAS PubMed.
- A. Ruggiero, C. H. Villa, J. P. Holland, S. R. Sprinkle, C. May, J. S. Lewis, D. A. Scheinberg and M. R. McDevitt, Int. J. Nanomed., 2010, 5, 783–802 CrossRef CAS PubMed.
- A. Jordan, R. Scholz, K. Maier-Hauff, F. H. Landeghem, N. Waldoefner, U. Teichgraeber, J. Pinkernelle, H. Bruhn, F. Neumann, B. Thiesen, A. Deimling and R. Felix, J. Neuro-Oncol., 2006, 78, 7–14 CrossRef CAS PubMed.
- I. Roy, T. Y. Ohulchanskyy, H. E. Pudavar, E. J. Bergey, A. R. Oseroff, J. Morgan, T. J. Dougherty and P. N. Prasad, J. Am. Chem. Soc., 2003, 125, 7860–7865 CrossRef CAS PubMed.
- D. L. Thorek, D. Ulmert, N. F. Diop, M. E. Lupu, M. G. Doran, R. Huang, D. S. Abou, S. M. Larson and J. Grimm, Nat. Commun., 2014, 5, 3097 Search PubMed.
- J. S. Weinstein, C. G. Varallyay, E. Dosa, S. Gahramanov, B. Hamilton, W. D. Rooney, L. L. Muldoon and E. A. Neuwelt, J. Cereb. Blood Flow Metab., 2010, 30, 15–35 CrossRef CAS PubMed.
- S. K. Mouli, P. Tyler, J. L. McDevitt, A. C. Eifler, Y. Guo, J. Nicolai, R. J. Lewandowski, W. Li, D. Procissi, R. K. Ryu, Y. A. Wang, R. Salem, A. C. Larson and R. A. Omary, ACS Nano, 2013, 7, 7724–7733 CrossRef CAS PubMed.
- N. K. Devaraj, E. J. Keliher, G. M. Thurber, M. Nahrendorf and R. Weissleder, Bioconjugate Chem., 2009, 20, 397–401 CrossRef CAS PubMed.
- J. P. Holland, M. J. Williamson and J. S. Lewis, Mol. Imaging, 2010, 9, 1–20 CAS.
- F. Chen, P. A. Ellison, C. M. Lewis, H. Hong, Y. Zhang, S. Shi, R. Hernandez, M. E. Meyerand, T. E. Barnhart and W. Cai, Angew. Chem., Int. Ed., 2013, 52, 13319–13323 CrossRef CAS PubMed.
- R. M. Wong, D. A. Gilbert, K. Liu and A. Y. Louie, ACS Nano, 2012, 6, 3461–3467 CrossRef CAS PubMed.
- R. Chakravarty, H. F. Valdovinos, F. Chen, C. M. Lewis, P. A. Ellison, H. Luo, M. E. Meyerand, R. J. Nickles and W. Cai, Adv. Mater., 2014, 26, 5119–5123 CrossRef CAS PubMed.
- X. Sun, X. Huang, X. Yan, Y. Wang, J. Guo, O. Jacobson, D. Liu, L. P. Szajek, W. Zhu, G. Niu, D. O. Kiesewetter, S. Sun and X. Chen, ACS Nano, 2014, 8, 8438–8446 CrossRef CAS PubMed.
- X. Sun, X. Huang, J. Guo, W. Zhu, Y. Ding, G. Niu, A. Wang, D. O. Kiesewetter, Z. L. Wang, S. Sun and X. Chen, J. Am. Chem. Soc., 2014, 136, 1706–1709 CrossRef CAS PubMed.
- V. S. Balakrishnan, M. Rao, A. T. Kausz, L. Brenner, B. J. Pereira, T. B. Frigo and J. M. Lewis, Eur. J. Clin. Invest., 2009, 39, 489–496 CrossRef CAS PubMed.
- C. W. Jung and P. Jacobs, Magn. Reson. Imaging, 1995, 13, 661–674 CrossRef CAS.
- C. W. Jung, Magn. Reson. Imaging, 1995, 13, 675–691 CrossRef CAS.
- W. Li, S. Tutton, A. T. Vu, L. Pierchala, B. S. Y. Li, J. M. Lewis, P. V. Prasad and R. R. Edelman, J. Magn. Reson. Imag., 2005, 21, 46–52 CrossRef PubMed.
- J. P. Holland, Y. Sheh and J. S. Lewis, Nucl. Med. Biol., 2009, 36, 729–739 CrossRef CAS PubMed.
- M. R. Jahn, H. B. Andreasen, S. Fütterer, T. Nawroth, V. Schünemann, U. Kolb, W. Hofmeister, M. Muñoz, K. Bock, M. Meldal and P. Langguth, Eur. J. Pharm. Biopharm., 2011, 78, 480–491 CrossRef CAS PubMed.
- H. H. Bengele, S. Palmacci, J. Rogers, C. W. Jung, J. Crenshaw and L. Josphson, Magn. Reson. Imaging, 1994, 12, 433–442 CrossRef CAS.
- E. J. Keliher, J. Yoo, M. Nahrendorf, J. S. Lewis, B. Marinelli, A. Newton, M. J. Pittet and R. Weissleder, Bioconjugate Chem., 2011, 22, 2383–2389 CrossRef CAS PubMed.
- M. A. Saksena, A. Saokar and M. G. Harisinghani, Eur. J. Radiol., 2006, 58, 367–374 CrossRef PubMed.
- M. G. Harisinghani, J. Barentsz, P. F. Hahn, W. M. Deserno, S. Tabatabaei, C. H. van de Kaa, J. de la Rosette and R. Weissleder, N. Engl. J. Med., 2003, 348, 2491–2499 CrossRef PubMed.
- M. Harisinghani, R. W. Ross, A. R. Guimaraes and R. Weissleder, Neoplasia, 2007, 9, 1160–1165 CrossRef CAS.
- S. Chen, D. Alcantara and L. Josephson, J. Nanosci. Nanotechnol., 2011, 11, 3058–3064 CrossRef CAS PubMed.
- K. Kanakakorn, I. Cavill and A. Jacobs, Br. J. Haematol., 1973, 25, 637–643 CrossRef CAS PubMed.
- S. Aryal, J. Key, C. Stigliano, M. D. Landis, D. Y. Lee and P. Decuzzi, Small, 2014, 10, 2688–2696 CrossRef CAS PubMed.
- R. Weissleder, M. Nahrendorf and M. J. Pittet, Nat. Mater., 2014, 13, 125–138 CrossRef CAS PubMed.
- J. P. Holland, M. J. Evans, S. L. Rice, J. Wongvipat, C. L. Sawyers and J. S. Lewis, Nat. Med., 2012, 18, 1586–1591 CrossRef CAS PubMed.
- M. J. Evans, J. P. Holland, S. L. Rice, M. G. Doran, S. M. Cheal, C. Campos, S. D. Carlin, I. K. Mellinghoff, C. L. Sawyers and J. S. Lewis, J. Nucl. Med., 2013, 54, 90–95 CrossRef CAS PubMed.
- Y. Ohkubo, H. Kohno, T. Suzuki and A. Kubodera, Radioisotopes, 1985, 34, 7–10 CrossRef CAS.
- C. Heneweer, J. P. Holland, V. Divilov, S. Carlin and J. S. Lewis, J. Nucl. Med., 2011, 52, 625–633 CrossRef CAS PubMed.
- M. M. Noginov, N. Noginova, O. Amponsah, R. Bah, R. Rakhimov and V. A. Atsarkin, J. Magn. Magn. Mater., 2008, 320, 2228–2232 CrossRef CAS PubMed.
- J. Salado, M. Insausti, L. Lezama, I. G. d. Muro, M. Moros, B. Pelaz, V. Grazu, J. M. d. l. Fuente and T. Rojo, Nanotechnology, 2012, 23, 315102 CrossRef CAS PubMed.
- F.-X. Ma, X.-Y. Sun, K. He, J.-T. Jiang, L. Zhen and C.-Y. Xu, J. Magn. Magn. Mater., 2014, 361, 161–165 CrossRef CAS PubMed.
- J. Kliava, in Magnetic Nanoparticles, Wiley-VCH Verlag GmbH & Co. KGaA, 2009, pp. 255–302 Search PubMed.
- R. A. Sperling and W. J. Parak, Philos. Trans. R. Soc., A, 2010, 368, 1333–1383 CrossRef CAS PubMed.
- S. Goel, F. Chen, E. B. Ehlerding and W. Cai, Small, 2014 DOI:10.1002/smll.201401048 , in press.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc02778g |
| This journal is © The Royal Society of Chemistry 2015 |