
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stabilization of [poly(allylamine)–tannic acid]n multilayer films in acidic and basic conditions after crosslinking with NaIO4†
Vincent
Ball
*ab
aUniversité de Strasbourg, Faculté de Chirurgie Dentaire, 8 Rue Sainte Elisabeth, 67000 Strasbourg, France. E-mail: vball@unistra.fr
bInstitut National de la Santé et de la Recherche Médicale, Unité Mixte de Recherche 1121, 11 Rue Humann, 67085 Strasbourg Cedex, France
First published on 17th June 2015
Abstract
Thin films containing polyphenols offer great perspectives in terms of biomedical applications and as nano-reactors owing to the ability of polyphenols to reduce metallic cations or to form coordination complexes with them. In this report it is shown that films made from the alternated adsorption of a weak polyelectrolyte, poly(allylamine), and tannic acid which are intrinsically instable at low and high pH values can be made pH insensitive, at least from pH = 1 to pH = 13 by a single contact with a 10 mM sodium periodate solution. IO4− anions oxidize tannic acid which then reacts with the amino groups of poly(allylamine) to yield covalent bonds between them. In addition, the permeability of the (PAH–TA)n films for the anionic redox probe hexacyanoferrate is dramatically reduced upon reaction with IO4− without affecting the film morphology.
Introduction
Tannic acid (TA), decagalloyl glucose, is a hydrolysable polyphenol with well known biological properties such as antioxidant and antibacterial activities.1 TA as well as other polyphenols also form spontaneous films at solid liquid interfaces as exemplified in tea cups.2 Films can also be deposited on almost all kinds of surfaces from tannic acid–Fe3+ containing mixtures using the strong metal cation affinity of TA.3 These films decompose spontaneously in acidic conditions owing to the competition between H3O+ cations to protonate TA and Fe3+ cations to coordinate with deprotonated sites on TA. TA can also react with polymers4 and proteins5,6via hydrogen bonds and with polycations allowing to produce pH sensitive films using the step-by-step deposition method.7,8 When using uncharged polymers like poly(N-vinyl caprolactam), poly(N-vinylpyrrolidone) or poly(ethylene oxide), the films are stable in acidic solutions owing to the formation of hydrogen bonds and decompose in basic conditions after protonation of TA7 (with an average pKa value of 8.5 (ref. 7 and 9)). On the other hand when deposited in alternation with polycations the films containing TA are stable in basic conditions and decompose when TA is fully protonated and hence uncharged.8Hence the films made from TA acidic are intrinsically responsive to pH changes and can dissolve in the charged or uncharged state of TA depending on the nature of the interaction partners. It could also be interesting to obtain extremely stable coatings able to remain unchanged in a very broad range of pH values as well as in a very broad range of ionic strength in order to get robust films, for instance in the protection against corrosion. It has been shown previously that polyelectrolyte multilayer films deposited in a step-by-step manner can be crosslinked using a vast repertoire of chemical reactions.10,11 Herein we will exploit the ability of TA to be oxidized to yield a quinone rich structure able to react with polyamines. This will allow to stabilize films made from the alternated deposition of poly(allylamine) (PAH) and TA, hence (PAH–TA)n films where n is the number of deposition cycles against large pH variations as well as large variations in ionic strength. The feasibility of this concept is also extended to other polycations, like poly(-L-lysine). It is also show that the reaction between TA and PAH, triggered by the addition of sodium periodate will affect the permeability of the (PAH–TA)n films for potassium hexacyanoferrate without affecting their morphology.
Results and discussion
The deposition of (PAH–TA)n films was followed in situ and in real time by means of quartz crystal microbalance with dissipation monitoring (QCM-D). PAH and TA were dissolved at 1 mg mL−1 in the presence of 50 mM sodium acetate buffer at pH = 5.0. TA was found to be stable against hydrolysis in these conditions.9 The film deposition experiments were performed on gold coated quartz crystals because of the stability of this substrate in a wide pH window. This is not the case (a priori) for silica coated quartz which undergoes some dissolution at pH values above 10. The stability of the substrate itself was mandatory in order to investigate the pH stability of the deposited coatings. QCM-D allows to show that the frequency changes of the quartz crystal reach a steady state (less than 0.2 Hz frequency changes per min) after 5 min of injection (constant flow of 0.25 mL min−1) of either PAH or TA. The same holds true for the rinsing steps with sodium acetate buffer (Fig. 1). This adsorption and rinsing time duration of 5 min is hence used in all the forthcoming experiments. QCM-D shows also that the frequency changes of the quartz crystal monitored at different overtones (ν = 3, 5 and 7) overlap within 5% suggesting that the (PAH–TA)n films are rigid and homogeneous.12 The growth regime of the (PAH–TA)n films was of the linear type, as found by others,8 meaning a constant increment in film mass per unit area for each deposition cycle.13 | ||
Fig. 1 Deposition of (PAH–TA)4 films on gold modified quartz crystals in the presence of 50 mM sodium acetate buffer at pH = 5.0. The film was further exposed to 10 mM NaIO4 and to HCl at 0.1 M as indicated with black vertical lines. The final rinse with sodium acetate buffer is also indicated whereas the buffer rinsing steps after the reaction with NaIO4 and HCl are omitted for clarity. Each injection step of PAH and TA is indicated with a red and purple line respectively. The reduced frequency changes of the quartz crystal were followed at the third ( ), the fifth ( ), the fifth ( ) and seventh overtone ( ) and seventh overtone ( ) of the fundamental resonance frequency by means of QCM-D. ) of the fundamental resonance frequency by means of QCM-D. | ||
When such films were put in the presence of 0.1 M HCl or 0.1 M NaOH solutions they underwent a rapid (less than 2 min) and almost quantitative desorption from the substrate (see Fig. 2, the corresponding QCM-D experiments are shown in Fig. 1 of the ESI†). This can be easily explained on the basis of the weak polyelectrolyte behaviour of TA and PAH. Under acidic conditions TA is fully protonated and uncharged, interacting weakly or not at all with the fully protonated and positively charged PAH. At high enough pH, typically above the average pKa of PAH, TA is fully deprotonated and negatively charged whereas PAH is unprotonated and non charged. Hence in these very acidic (pH ∼ 1) and very basic solutions (pH ∼ 13) the electrostatic interactions between PAH and TA, which allowed for the deposition of the film at pH = 5, are disrupted and the films disintegrate. By means of UV visible spectroscopy, we showed that the (PAH–TA)4 film deposited at pH = 5.0 remain stable when subsequently immersed in solutions of pH between 4 and 9 (data not shown) but decompose rapidly at pH < 4 and at pH > 9. However, when the (PAH–TA)4 films are put in the presence of a sodium periodate solution at 10 mM during 5 min and further exposed to HCl or NaOH at 0.1 M (with pH close to 1 and 13 respectively), no film erosion is observed (Fig. 1 and 2 in the ESI†). Hence the (PAH–TA)n films become stable in a very broad range of pH values. The stability of the thicker (PAH–TA)8 films was further confirmed by means of ellipsometry measurements for films deposited on Si wafers: no film dissolution was found after 5 min of contact with 10 mM NaIO4 and further exposure to 0.1 M HCl (Fig. 2). However complete film dissolution was observed for untreated films (Fig. 2) after exposure to a 0.1 M HCl solution.
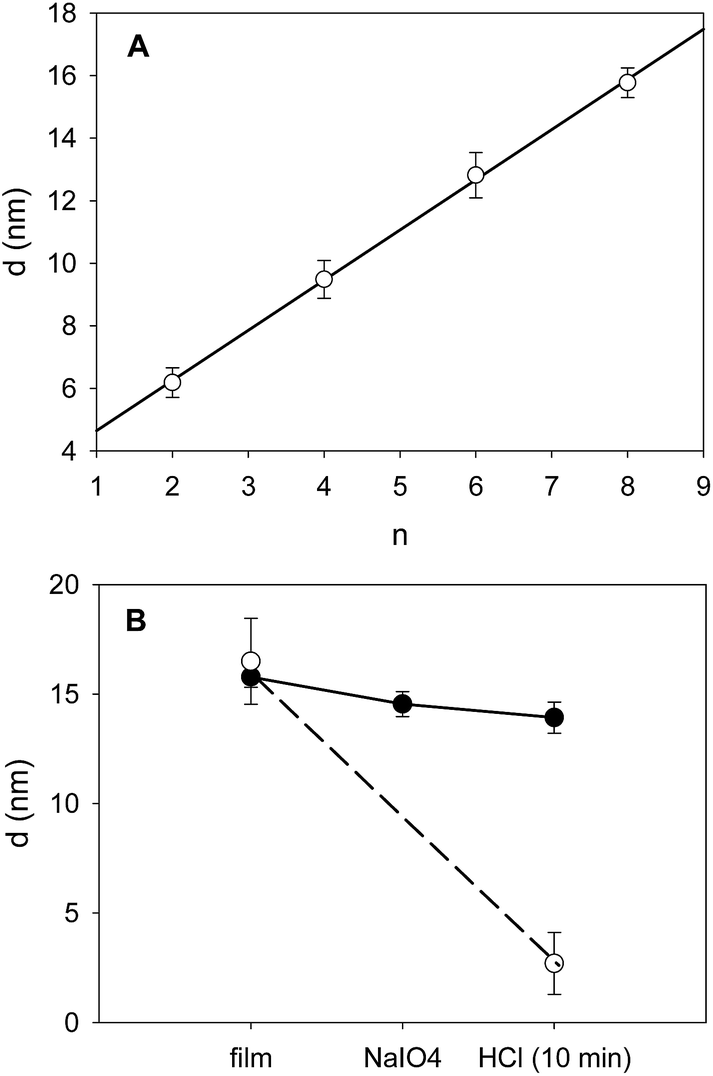 | ||
| Fig. 2 (A) Evolution of the film thickness of a (PAH–TA)n film deposited on a silicon wafer as monitored as a function of the number of deposition cycles by means of ellipsometry. The deposition of the film was performed in the presence of 50 mM sodium acetate buffer at pH = 5. The slope of the straight line obtained by means of linear regression was of 1.60 nm per deposition cycle (r2 = 0.998). (B) Evolution of the film thickness of a (PAH–TA)8 film deposited on Si in the case of an uncrosslinked film (○) and a film crosslinked with 10 mM NaIO4 (5 min) after exposure to 0.1 M NaCl during 10 min (●). | ||
From now on the (PAH–TA)n films treated with NaIO4 at 10 mM will be denoted as (PAH–TA)n + NaIO4 films. When the (PAH–TA)4 + NaIO4 films were exposed to 0.1 M NaOH solution, the absorbance due to the film remained unchanged on the substrate. In the absence of NaIO4 treatment, the film desorption was quantitative after exposure to 0.1 M NaOH (Fig. 3 in the ESI†). The integrity of the (PAH–TA)4 + NaIO4 film after treatment with 0.1 M NaOH even suggests that the modified film protects the underlying quartz from NaOH induced leaching.
The stability of (PAH–TA)n + NaIO4 films deposited on quartz slides in the presence of solutions having a very low pH was also confirmed by means of UV-visible spectroscopy. This characterization method provided further information about the mechanism of film stabilization: the characteristic absorption band of deprotonated TA (at λmax between 300 and 320 nm) in the (PAH–TA)8 film totally disappeared after 5 min of contact with a 10 mM NaIO4 solution (Fig. 3A). When TA was dissolved at 0.05 mg mL−1 in a 50 mM sodium acetate solution at pH = 5.0, the maximal absorbance was detected at λmax = 278 nm in agreement with published data.9 This peak, corresponding to the fully protonated form of TA, disappeared when NaIO4 was added to the TA solution and the absorption maximum is now shifted to about 248 nm (Fig. 3B).
 | ||
Fig. 3 (A) UV-vis spectra of the (PAH–TA)8 film before ( ) and after reaction with 10 mM NaIO4 ( ) and after reaction with 10 mM NaIO4 ( ) and after a final exposure to 0.1 M HCl during 10 min ( ) and after a final exposure to 0.1 M HCl during 10 min ( ). (B) Absorption spectra of a 0.05 mg mL−1 TA solution in the presence of 50 mM sodium acetate buffer at pH = 5.0 before ( ). (B) Absorption spectra of a 0.05 mg mL−1 TA solution in the presence of 50 mM sodium acetate buffer at pH = 5.0 before ( ) and after addition of NaIO4 ( ) and after addition of NaIO4 ( ) to reach a final concentration of 10 mM in the oxidant. The position of the peaks are indicated in both parts (A) and (B). ) to reach a final concentration of 10 mM in the oxidant. The position of the peaks are indicated in both parts (A) and (B). | ||
NaIO4 is a strong enough oxidant (E° = 1.55 V vs. NHE for the IO4−/IO3− couple) to oxidize TA to yield quinone groups. In turn these reactive functionalities are able to form covalent bonds with amino groups14 from PAH to yield a very robust, pH insensitive coating. In confirmation to this assumption, IR spectroscopy shows changes in the spectrum of PAH–TA mixtures when put in contact with 10 mM NaIO4 (Fig. 4).
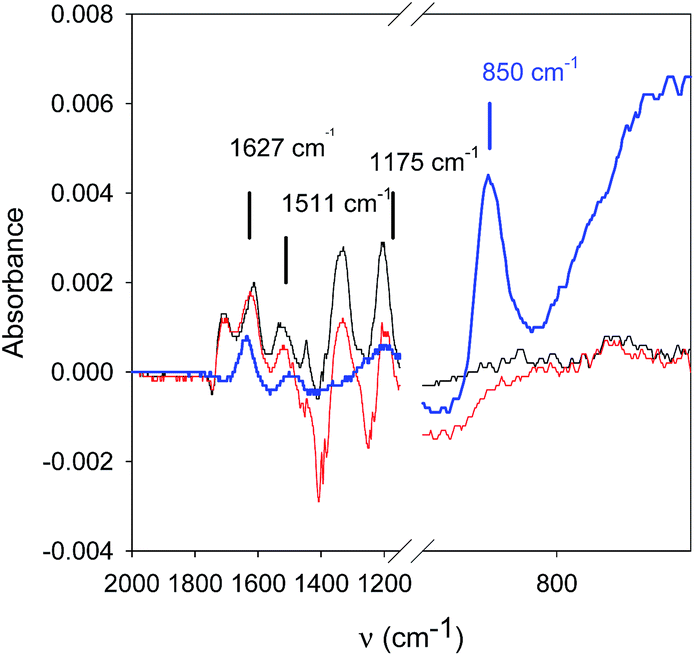 | ||
| Fig. 4 IR spectra in the ATR mode of a PAH + TA mixture (both components at 5 mg mL−1 in the presence of 50 mM sodium acetate buffer), before (black line) and after addition of NaIO4 (to reach a concentration of 10 mM) (red line). The spectrum of a 10 mM NaIO4 solution in 50 mM sodium acetate is also displayed (blue line). The baseline was taken in the presence of the buffer. The main peaks of TA and IO4− anions are indicated with vertical black and blue lines respectively. | ||
The peak due to IO4− anions (850 cm−1 (ref. 15)) disappears in contact with the PAH–TA mixture and the peaks at 1511 and 1172 cm−1 of the PAH–TA mixture are considerably decreased in intensity after addition of NaIO4. Tannic acid displays peaks in its IR spectrum at 860, 1172, 1511 and 1627 cm−1.16
The best way to characterize the formation of covalent bonds between the amino groups of PAH and the quinone groups of oxidized TA would be to perform NMR experiments on PAH–TA mixtures before and after oxidation with NaIO4. However owing to the multiple possible reaction sites and owing to the chemical heterogeneity of TA,17 an easier much qualitative experiment was performed. A turbid mixture of TA (at 1 mg mL−1) and of PAH at 0.5 mg mL−1 was characterized by means of dynamic light scattering after a 5 fold appropriate dilution. This mixture was also diluted in a 50 mM sodium acetate buffer (pH = 5.0) containing 1 M NaCl and 1 M urea in order to respectively weaken the electrostatic interactions and the hydrogen bonds between PAH and TA. Indeed, such a treatment with the NaCl–urea solution allowed for a size reduction from (574 ± 260) nm to (250 ± 120) nm and for a marked decrease in the solution turbidity (Fig. 4 in the ESI†). However, when the PAH–TA mixture was subjected to NaIO4 treatment, a marked color change occurred (due to the oxidation of TA) as well as phase separation. This phase separation remained even after 5 dilution with the same NaCl–urea containing solution. This experiments show that the addition of NaIO4 to the PAH–TA aggregates strengthens their mutual interactions most probably via the formation of covalent bonds between amino and quinone groups.
Even if the crosslinking of the (PAH–TA)n films with NaIO4 had a dramatic influence on the stability of the film at pH values of about 1 and 13, it had a negligible effect on the film morphology as demonstrated by means of contact mode AFM imaging in the dry state (Fig. 5). The root mean squared roughness, determined over a 10 μm × 10 μm surface area of the (PAH–TA)8 films even slightly decreased from to (7.0 ± 0.8) nm to (4.8 ± 0.5) nm after the contact with the 10 mM NaIO4 solution during 5 min. The scratched (PAH–TA)8 films displayed a height change of about 20–25 nm corresponding to the film thickness. This value was slightly larger than the thickness calculated from the ellipsometry experiments, namely 14 nm (Fig. 2). This could originate from an oversimplified modelization of the film as a homogeneous and isotropic coating with a refractive index of 1.50 at λ = 632.8 nm.
 | ||
| Fig. 5 Surface topographies acquired in air and in the contact mode of a (PAH–TA)8 (A) and a (PAH–TA)8 + NaIO4 film (B) and a line profile over a scratched (PAH–TA)8 + NaIO4 film. The average root mean squared roughness of the (PAH–TA)8 and a (PAH–TA)8 + NaIO4 films were of (7.0 ± 0.8) nm and (4.8 ± 0.5) nm respectively. | ||
The (PAH–TA)n films were also deposited on amorphous carbon working electrodes and cyclic voltammetry scans (at 100 mV s−1 sweep rate between −0.2 and +0.8 V vs. Ag/AgCl) were recorded in the presence of 50 mM sodium acetate buffer (pH = 5.0) but the in the absence of any other redox probe. An irreversible oxidation phenomenon with a maximal oxidation current at Epa ∼ 0.45 V vs. Ag/AgCl was observed during the first scan but it was absent during the second one (Fig. 5 in the ESI†). This clearly shows that TA deposited in a (PAH–TA)n film is electroactive as was previously found in (TA–Fe3+)n films.18 When these films were put in contact with potassium hexacyanoferrate (1 mM freshly dissolved in the 50 mM sodium acetate buffer at pH = 5.0), the oxidation current of the Fe(CN)64− anions only slightly decreased with the number of deposition cycles and hence with the film thickness (Fig. 6A). These films were however continuous an pinhole free (at the resolution of the AFM, namely about 10 nm) as shown by AFM (Fig. 5) meaning that they remain strongly permeable for the anionic redox probe. The oxidation peak potential also shifted to higher potentials emphasizing the increased energetic penalty for Fe(CN)64− anions to be oxidized (Fig. 6A). When putting the (PAH–TA)n films in contact with NaIO4 (10 mM) during 5 min, the film permeability for the redox probe was markedly decreased (Fig. 6B). After 6 deposition cycles the (PAH–TA)n + NaIO4 films are almost totally impermeable to the Fe(CN)64− anions. These experiments demonstrate that the crosslinking step with NaIO4 not only dramatically increases the film stability in the presence of concentrated HCl or NaOH solutions but it also increases its compacity by decreasing the probability for a negatively charged redox probe to have access to the surface of the working electrode at least at pH = 5.0 at which the voltammetry experiments were performed. This marked reduction in the film permeability for hexacyanoferrate anions is not due to an increase in film hydrophobicity, and hence a decrease in hydration, following oxidation. Indeed 5 min of oxidation with 10 mM NaIO4 allows for a reduction in the static contact angle from (21.4 ± 3.2) to (5.1 ± 2.0)° for a (PAH–TA)6 coating.
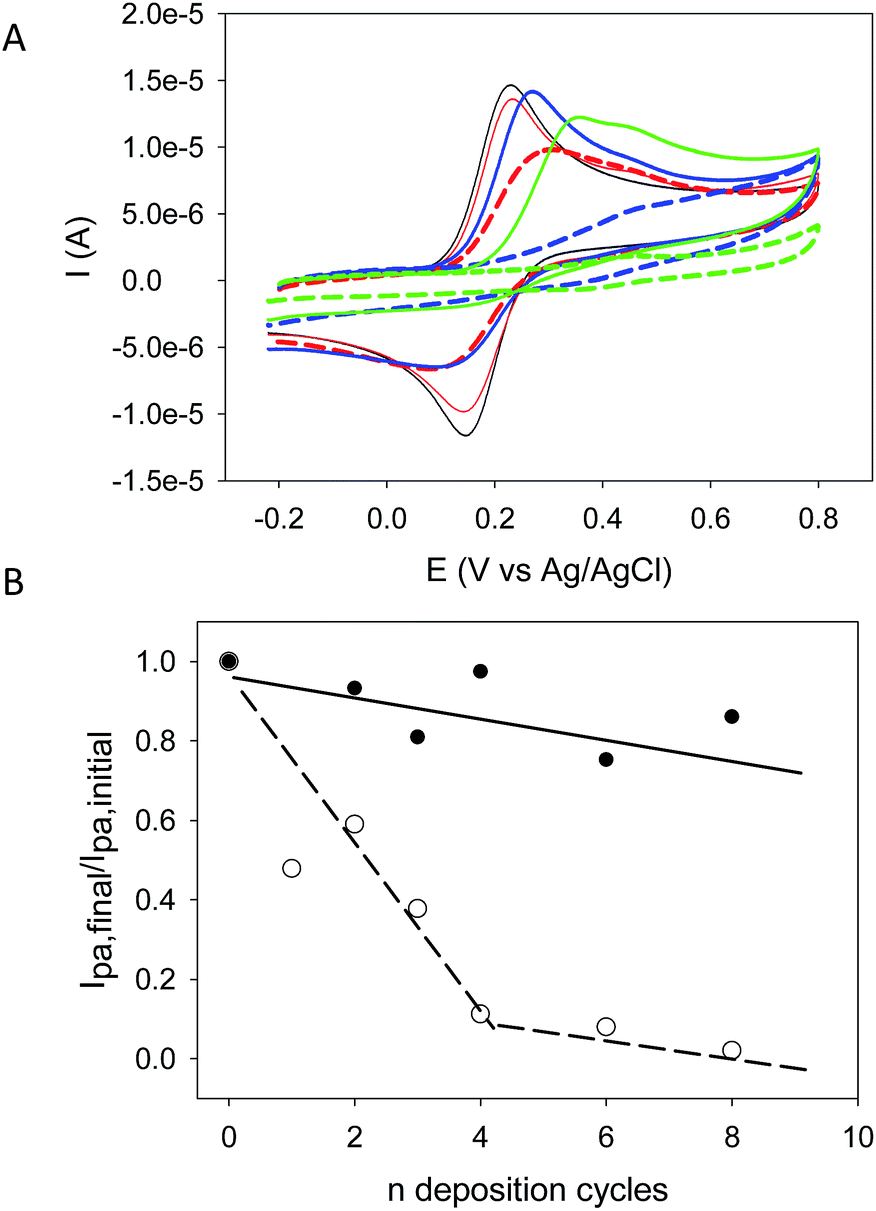 | ||
| Fig. 6 (A) Cyclic voltammograms before (full lines) and after reticulation of (PAH–TA)n films (broken lines) with 10 mM NaIO4 during 5 min in the presence of 1 mM K4Fe(CN)6 dissolved in the 50 mM sodium acetate buffer. The curves correspond to n = 0 (i.e. the pristine amorphous carbon electrode, black curve), and films made from n = 2 (red curves), n = 4 (blue curves) and n = 8 (green curves) deposition cycles. (B) Evolution of the ratio of the maximal oxidation current of Fe(CN)64− anions after (○) and before (●) exposure of the (PAH–TA)n films to 10 mM NaIO4 (5 min). The Ipa,final values were measured either after film deposition (●) or after crosslinking with NaIO4 (○) whereas the Ipa,initial values were measured on the pristine carbon electrode after polishing and before film deposition. | ||
Finally, it was found that the stabilization of (polycation–TA)n films after NaIO4 crosslinking was not limited to PAH as the polycation but films made from poly(-L-lysine) could also be stabilized in strong acidic media as well as at pH values above the average pKa of PLL, namely 10 (data not shown).
Conclusions
The interaction of multi-layered films made from polyamines like PAH and PLL and TA with NaIO4 allows to produce pH resistant coatings even in the presence of 0.1 M HCl and 0.1 M NaOH, conditions in which the untreated films decompose. The oxidation of TA with periodate anions produces quinones which are extremely reactive with the amino groups present on PAH allowing to produce covalent bonds between TA and PAH thus rendering the whole structure of the film pH insensitive. This chemical modification of the film, demonstrated by means of UV-vis and FTIR spectroscopy (but not yet by means of NMR spectroscopy) occurs without a change in the film morphology but affects markedly the film permeability for hexacyanoferrate anions. In a future investigation the interactions between PAH and TA in the presence of NaIO4 will be investigated in solution by means of structural methods like NMR. The NaIO4 treated films will also be investigated for their biological properties.Experimental section
Chemicals
All aqueous solutions were prepared from ultrapure water (Milli Plus, Millipore, Billerica, Massachusetts, USA). Poly(allylamine hydrochloride) (PAH, ref. 283215, Mw = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 as characterized by means of GPC using PEG as a standard) and tannic acid (TA, ref. 403040, Mw = 1701.2 g mol−1) were purchased from Sigma-Aldrich and used without further purification. Poly(-L-lysine hydrobromide) had a molecular mass comprised between 4000 and 15000 g mol−1 as characterized by means of viscosimetry by the furnisher (Sigma-Aldrich, ref. P6516). PAH, PLL and TA were dissolved at 1 mg mL−1 in 50 mM sodium acetate buffer (Merck, ref. 6268) with a pH adjusted to 5.0 using concentrated hydrochloric acid. The polyelectrolyte solutions were freshly prepared before each experiment. NaIO4 (Sigma-Aldrich, ref. 311448) was dissolved at 10 mM in the sodium acetate buffer. These solutions were also freshly prepared before each experiment. Potassium hexacyanoferrate (K4Fe(CN)6, ref. P9387 from Sigma-Aldrich) was dissolved at 1 mM in the sodium acetate buffer to investigate the permeability of (PAH–TA)n or of (PAH–TA)n + NaIO4 films deposited on amorphous carbon electrodes.
000 g mol−1 as characterized by means of GPC using PEG as a standard) and tannic acid (TA, ref. 403040, Mw = 1701.2 g mol−1) were purchased from Sigma-Aldrich and used without further purification. Poly(-L-lysine hydrobromide) had a molecular mass comprised between 4000 and 15000 g mol−1 as characterized by means of viscosimetry by the furnisher (Sigma-Aldrich, ref. P6516). PAH, PLL and TA were dissolved at 1 mg mL−1 in 50 mM sodium acetate buffer (Merck, ref. 6268) with a pH adjusted to 5.0 using concentrated hydrochloric acid. The polyelectrolyte solutions were freshly prepared before each experiment. NaIO4 (Sigma-Aldrich, ref. 311448) was dissolved at 10 mM in the sodium acetate buffer. These solutions were also freshly prepared before each experiment. Potassium hexacyanoferrate (K4Fe(CN)6, ref. P9387 from Sigma-Aldrich) was dissolved at 1 mM in the sodium acetate buffer to investigate the permeability of (PAH–TA)n or of (PAH–TA)n + NaIO4 films deposited on amorphous carbon electrodes.
The adsorption substrates were quartz plates (Thuet, Blodelsheim, France), silicon wafers (Siltronix, Archamps, France) and amorphous carbon electrodes which were used for film characterization by means of UV-vis spectroscopy, AFM + ellipsometry and cyclic voltammetry (CV) respectively.
The quartz and silicon substrates were cleaned with ethanol, Hellmanex (2% v/v in water, Hellma GmbH, Müllheim, Germany); distilled water, 0.1 M HCl, distilled water and finally with an UV–O3 cleaner during 10 min. These cleaning steps were performed before each experiment. The amorphous carbon electrodes (ref. CH 104, CH Instruments, Austin, Texas) used as the working electrodes in the CV experiments were successively polished with 1 μm and 0.1 μm alumina pastes (Escil, France) and sonicated two times in a distilled water bath. The quartz crystal microbalance experiments were performed on gold coated quartz crystals from Q Sense (ref. QSX 303, Q Sense, Sweden). The gold substrates were cleaned in the same manner as the quartz plates and silicon wafers.
The multilayer films were deposited by immersing the quartz plates, silicon wafers or amorphous carbon electrodes in the PAH solution, in the sodium acetate buffer, in the TA solution and again in the rinsing buffer. These four steps constitute an “adsorption cycle” and such cycles were repeated n times to yield (PAH–TA)n films. Each adsorption and rinsing step lasted over t min and the optimal duration was determined from the results of the quartz crystal microbalance with dissipation monitoring experiments (QCM-D). At the end of the deposition and film characterization, the films were immersed during 5 min in a freshly prepared 10 mM NaIO4 solution and finally rinsed with sodium acetate buffer. Finally the resulting (PAH–TA)n + NaIO4 films were immersed either in 0.1 M HCl or 0.1 M NaOH solutions as well as in solutions of lower HCl and NaOH concentrations. Their properties, i.e. their thickness, their morphology, their absorption spectra and their permeability for Fe(CN)64− anions were then measured and compared with the corresponding values before the treatment with NaIO4 and strong acid/base solutions. Similarly, unoxidized (PAH–TA)n films were immersed in either in 0.1 M HCl or 0.1 M NaOH solutions.
The QCM-D experiments were performed with a E1 device from QSense (Göteborg, Sweden). The polyelectrolyte or buffer solutions were injected by means of a peristaltic pump at a constant flow rate of 250 μL min−1. The reduced frequency changes, Δfν/ν, as well as the dissipation changes, ΔDν, at the 3rd (ν = 3), 5th (ν = 5) and 7th (ν = 7) overtone were followed as a function of time. In the case where the reduced frequency changes overlap and if the (dimensionless) dissipation changes are small, the films can be considered as rigid and the Sauerbrey equation19 can be used to calculate the surface coverage in adsorbed molecules.
UV-vis spectroscopy experiments were performed with a single beam Xenius spectrophotometer from Safas, Monaco. The reference spectra were taken with the pristine quartz slide just before the beginning of the coating process.
Infra-red spectra were acquired in the attenuated total reflection mode using a PIKE ATR element fitted with a ZnSe crystal on a Spectrum Two spectrophotometer (Perkin Elmer). The spectra were obtained after averaging 16 interferograms acquired between 700 and 4000 cm−1 with a spectral resolution of 4 cm−1. A PAH–TA mixture with both components at a concentration of 5 mg mL−1 in the presence of 50 mM sodium acetate buffer was put in the liquid circulation cell. The infra-red spectrum was acquired and NaIO4 was subsequently added to this solution to reach a concentration of 10 mM. The spectrum of a 10 mM NaIO4 solution was measured independently. For all the IR spectra, the base line was acquired in the presence of 50 mM sodium acetate buffer.
Ellipsometry experiments were performed with a monochromatic PZ2000 ellipsometer (Horiba, France) at a wavelength of 632.8 nm and a constant angle of incidence of 70°. At least five measurements were performed on dried (PAH–TA)n and (PAH–TA)n + NaIO4 films along the major axis of the rectangular silicon wafers. The obtained ellipsometric angles Δ and ψ were used to calculate the thickness of the coatings assuming a refractive index of 1.50 in the framework of an homogeneous and isotropic film.
The thickness of dried PEI–(TA–Fe3+)n films was measured by means of AFM, which constitutes an absolute measurement provided the piezoelectric ceramic on which the sample is glued is well calibrated. AFM topographic images were obtained in the contact mode and in the dry state using a Nanoscope IV microscope (Bruker, Germany). The used cantilevers were of MSCT type with a nominal spring constant of 0.2 N m−1. Topographies were acquired over 10 μm × 10 μm surface areas after repetitive scans at a frequency of 1 Hz and a resolution of 512 × 512 pixels and the lowest possible deflection set point to ensure minimal damage to the investigated films. The thickness of the films was determined by measuring height changes in the direction perpendicular to lines needle scratched in the film just before imaging. The given thickness corresponds to the average of 20 line profiles ± one standard deviation on images acquired over 20 μm × 20 μm area. The image acquisition was performed using Nascope614r1 as the software.
Dynamic light scattering was performed with a Coulter N4plus device. The autocorrelation function of the light scattered at an angle of 90° with respect to the incident beam was measured and analysed using the Contin algorithm to yield the size distribution in a diluted PAH–TA mixture. Such a mixture was prepared upon injecting a PAH solution (1 mg mL−1) in an equal volume of a TA solution (2 mg mL−1) under vigorous stirring. This solution was then diluted 5 fold either in 50 mM sodium acetate buffer or in 50 mM sodium acetate buffer containing 1 M NaCL and 1 M urea. The strong concentration in NaCl and urea is expected to weaken strongly the electrostatic interactions between PAH and TA. In a final experiment 1 mL of 10 mM NaIO4 solution was added to 5 mL of a PAH–TA mixture and left to react during 5 min before 5 fold dilution with a solution containing 1 M NaCl and 1 M urea.
Static contact angles of water droplets (6 μL) were acquired using a DGD-MCAT goniometer (Digidrop, Bourg de Péage, France).
The cyclic voltammetry (CV) experiments were performed in a three electrode configuration using an amorphous carbon electrode, an Ag/AgCl (ref. 111, CH Instruments) and a Pt wire (ref. 115, CH Instruments) as the working electrode, the reference and the auxiliary electrode respectively. The CV curves were measured by cycling the potential (vs. Ag–AgCl) between −0.20 V and 0.80 V. The as prepared (PAH–TA)n films were incubated with 50 mM sodium acetate buffer and at least 2 CVs at a scan rate of 100 mV s−1 were recorded in the absence of any exogenous redox probe. This experiment was performed in order to investigate the electrochemical behaviour of the TA molecules present in the (PAH–TA)n films. Finally the (PAH–TA)n films were incubated with a 1 mM K4Fe(CN)6 solution in the sodium acetate buffer during 2 min and CV curves at 100 mV s−1 were then acquired to investigate if Fe(CN)64− anions can permeate through the (PAH–TA)n films. Those films were then rinsed with sodium acetate buffer up to the disappearence of any residual current due to Fe(CN)64− anions. The electrode was then immersed for 5 min in a 10 mM NaIO4 solution, its capacitive curve was again measured in the absence of redox probe and finally a last CV curve was acquired in the presence of a 1 mM K4Fe(CN)6 solution. The oxidation current of Fe(CN)64− on the (PAH–TA)n and (PAH–TA)n + NaIO4 films were compared to the oxidation current of the redox probe on the pristine and freshly polished electrode. The ratio of the oxidation current after film oxidation and before its deposition was plotted as a function of the number of deposition cycles.
Acknowledgements
Ch. Ringwald is acknowledged for his help in the QCM-D experiments.Notes and references
- E. Haslam, J. Nat. Prod., 1996, 59, 205–215 CrossRef CAS PubMed.
- H. Ejima, J. J. Richardson, K. Liang, J. P. Best, M. P. van Koeverden, G. K. Such, J. Cui and F. Caruso, Science, 2013, 341, 154–157 CrossRef CAS PubMed.
- D. G. Barrett, T. S. Sileika and P. B. Messersmith, Chem. Commun., 2014, 50, 7265–7268 RSC.
- A. R. Patel, J. Seijen ten-Hoorn, J. Hazekamp, T. B. J. Blijdenstein and K. P. Velikov, Soft Matter, 2013, 9, 1428–1436 RSC.
- M. Liu, Y. Zhang, Q. Yang, Q. Xie and S. Yao, J. Agric. Food Chem., 2006, 54, 4087–4094 CrossRef CAS PubMed.
- K. J. Siebert, N. V. Troukhanova and P. Y. Lynn, J. Agric. Food Chem., 1996, 44, 80–85 CrossRef CAS.
- I. Erel-Unal and S. A. Sukhishvili, Macromolecules, 2008, 41, 3962–3970 CrossRef CAS.
- T. Shutava, M. Prouty, D. Kommireddy and Y. Lvov, Macromolecules, 2005, 38, 2850–2858 CrossRef CAS.
- M. Oćwieja, Z. Adamczyk and M. Morga, J. Colloid Interface Sci., 2015, 438, 249–258 CrossRef PubMed.
- D. E. Bergbreiter and K. S. Liao, Soft Matter, 2009, 5, 23–28 RSC.
- G. Rydzek, P. Schaaf, J.-C. Voegel, L. Jierry and F. Boulmedais, Soft Matter, 2012, 8, 9738–9755 RSC.
- I. Reviakine, D. Johansmann and R. P. Richter, Anal. Chem., 2011, 83, 8838–8848 CrossRef CAS PubMed.
- P. Lavalle, J.-C. Voegel, D. Vautier, B. Senger, P. Schaaf and V. Ball, Adv. Mater., 2011, 23, 1191–1221 CrossRef CAS PubMed.
- B. Liu, L. Burdine and T. Kodadek, J. Am. Chem. Soc., 2006, 128, 15228–15235 CrossRef CAS PubMed.
- F. A. Miller and C. H. Wilkins, Anal. Chem., 1952, 24, 1253–1294 CrossRef CAS.
- J. Iglesias, E. García de Saldaňa and J. A. Jaén, Hyperfine Interact., 2001, 134, 109–114 CrossRef CAS.
- S. Rohn, Food Res. Int., 2014, 65, 13–19 CrossRef CAS.
- C. Ringwald and V. Ball, J. Colloid Interface Sci., 2015, 450, 119–126 CrossRef CAS PubMed.
- G. Sauerbrey, Z. Phys., 1959, 155, 206–222 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra06783a |
| This journal is © The Royal Society of Chemistry 2015 |