
Enantioselective allylic amination of MBH carbonates catalyzed by novel chiral 4-dialkylaminopyridine catalysts†‡
Gaoyuan
Ma
and
Mukund P.
Sibi
*
Department of Chemistry and Biochemistry, North Dakota State University, Fargo, ND 58108, USA. E-mail: mukund.sibi@ndsu.edu
First published on 10th September 2014
Abstract
We have investigated the asymmetric allylic amination of Morita–Baylis–Hillman (MBH) carbonates with nitrogen nucleophiles in the presence of newly designed chiral DMAP catalysts. A series of α-methylene β-amino esters were obtained with good yield and selectivities.
The super nucleophilic base, 4-dimethylaminopyridine (DMAP), and its derivatives have general applicability for the nucleophilic catalysis of a wide variety of reactions.1 The range of applications using DMAP as a catalyst in organic synthesis has increased dramatically. In view of the versatility of DMAP, a significant step was to devise a chiral variant that could achieve asymmetric catalysis, and the first process toward attaining this objective was reported by Vedejs in 1996.2 Following this pioneering study, several research groups successively developed chiral DMAP catalysts with a stereogenic center or a chiral axis and planar–chiral DMAP catalysts.3 These catalysts have been successfully used in kinetic resolution of secondary alcohols and amines, desymmetrization of meso diols, asymmetric [3 + 2] cycloadditions, enantioselective protonation of ketenes, O-to-C rearrangements of acyl groups, etc.4
We are interested in investigating DMAP catalysts that incorporate fluxional groups to provide steric control of reactivity. In the past few years, our group has examined a concept called “chiral relay” which utilizes fluxional chirality in achiral templates, ligands, and additives to provide a general method for the enhancement of enantioselectivity in a variety of transformations.5 A body of work has demonstrated that stereochemical information can be conveyed from fluxional stereogenic centers to reaction sites to control diastereo- or enantioselectivities.5,6
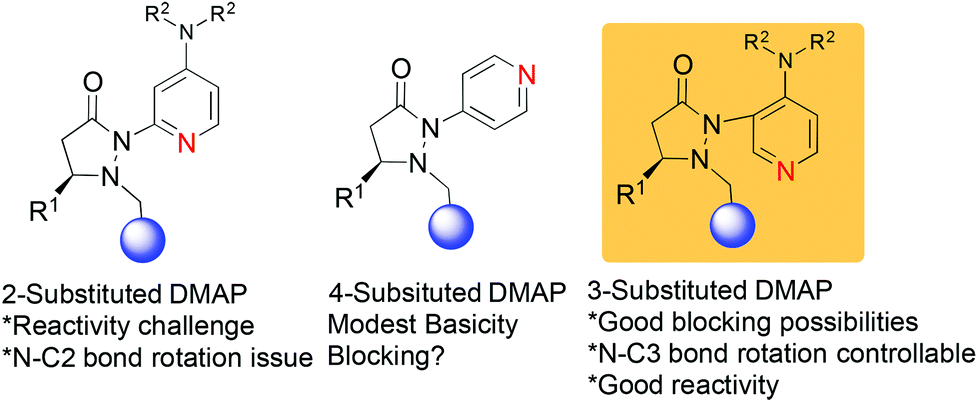
We have recently reported on the development of novel chiral DMAP catalysts that incorporate a fluxional group to control stereoselectivity in kinetic resolutions.7 The design criteria for the new chiral DMAP catalysts were based on well-established principles in the literature (Scheme 1). The initial evaluation involved the proper placement of the fluxional group on the pyridine ring. Early investigations showed that even a small group (methyl) adjacent to the pyridine nitrogen markedly erodes their activity as nucleophilic catalysts. Thus, 2-substituents in DMAP catalyst design are generally not desirable (structure A, Scheme 1).8 Furthermore, 4-substituted pyridines are likely to provide minimal steric shielding due to the remote architecture between the catalytic center and the stereocontrol element (structure B, Scheme 1). Based on these considerations, we connected the 3-position of pyridine with the chiral pyrazolidinone template containing the fluxional group (structure C, Scheme 1). We expected that reactivity should be reasonable, and that the proximity between the fluxional group and the pyridine might afford conformational definition.
 | ||
| Scheme 1 Design of novel chiral DMAP catalysts with fluxional chirality. | ||
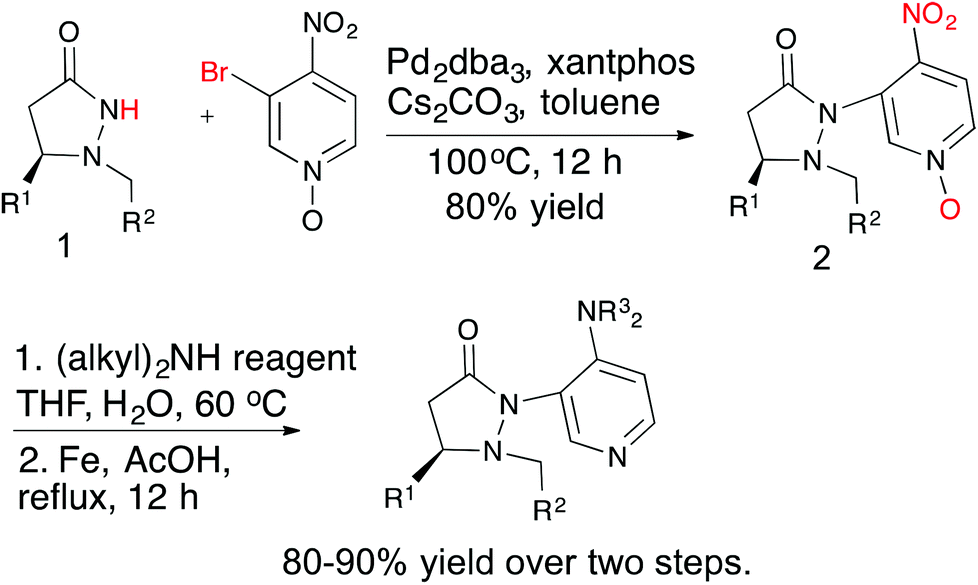
Several chiral DMAP catalysts containing fluxional groups have been prepared from readily available pyrazolidinones 1 (Scheme 2).7 The preparation of new DMAP catalysts started from enantiomerically pure pyrazolidinones 1 and required three steps, which afforded good overall yields. The methodology is amenable for scale up and diversity can be introduced in the fluxional group as well as the pyridine 4-substituent without much difficulty. Five catalysts were synthesized with different R1, R2 and R3 substituents for evaluation.
 | ||
| Scheme 2 Synthesis of chiral DMAP catalysts. | ||
We are interested in evaluation of the newly designed DMAP catalysts in enantioselective transformations. To evaluate the nucleophilic characteristics of the new DMAP catalysts we have chosen the allylic amination of Morita–Baylis–Hillman (MBH) carbonates. The transformation of MBH adducts has attracted a lot of attention since they are synthetically important synthons.9 Although there are a number of reports which utilize chiral palladium complexes, chiral tertiary amines and phosphines for the transformation of MBH adducts,10 to the best of our knowledge there is no example of using chiral DMAP for such a transformation.
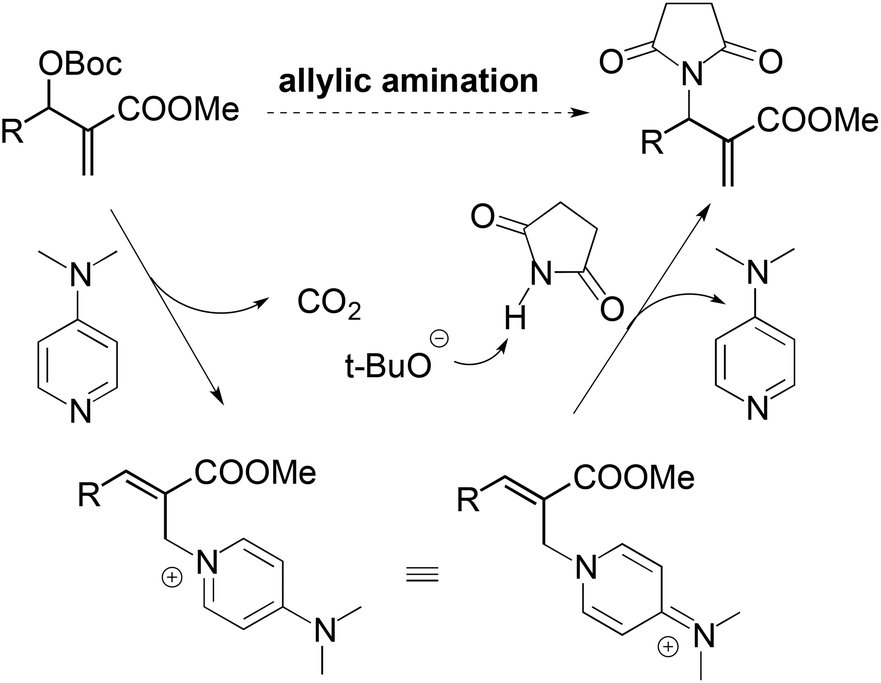
For the nucleophilic allylic amination of MBH adducts, a commonly accepted tandem SN2′–SN2′ mechanism is illustrated in Scheme 3. The nucleophilic catalyst (DMAP) first adds to the MBH adduct in an SN2′ fashion, followed by elimination of the tert-butyl carbonate anion. After elimination of carbon dioxide, the tert-butoxy anion deprotonates the pronucleophile. Then the in situ formed anionic nucleophile attacks the olefinic bond of the cation intermediate to afford the observed amination products.11
 | ||
| Scheme 3 Allylic amination of MBH carbonates catalyzed by DMAP. | ||
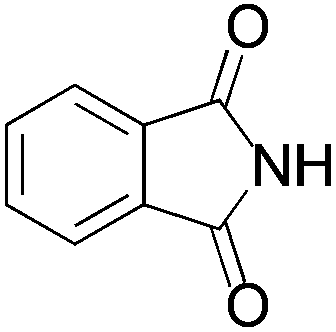
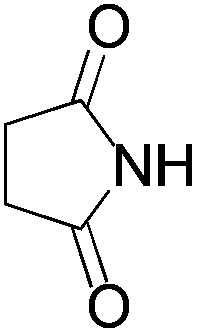
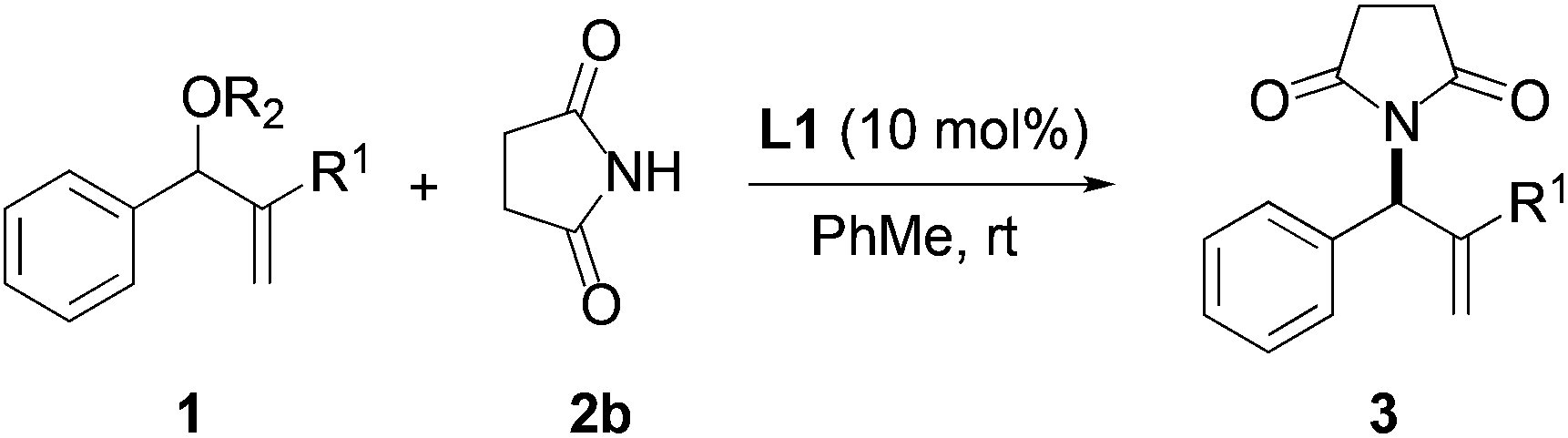
We began our investigation by screening MBH carbonates 1a with a variety of nitrogen nucleophiles 2 in toluene in the presence of 10 mol% L1 at room temperature to furnish the amination product. The results are summarized in Table 1. In general, better yield as well as enantioselectivity was obtained when cyclic imide pronucleophiles were used (Table 1, entries 1, 2). Succinimide was slightly better than phthalimide (compare entry 2 with 1). Both the yield and enantioselectivity in reactions with acyclic benzoylbenzamide and acetylacetamide were lower than cyclic nitrogen nucleophiles (entries 3 and 4). Benzenesulfonamide as a nucleophile was better than acyclic pronucleophiles but inferior to cyclic imides (entry 5). A variety of solvents were screened for the reaction and of these toluene gave the best result (see ESI‡).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Nitrogen Nu | Product | Time | Yieldb (%) | eec (%) |
| a Reaction conditions: the reactions were performed with 1 equiv. of 1, 1.5 equiv. of 2 and 10 mol% of catalyst in toluene at room temperature. b Isolated yield. c Determined by chiral HPLC analysis. | |||||
| 1 |
|
3a | 24 h | 76 | 31 |
| 2 |
|
3b | 24 h | 75 | 39 |
| 3 |
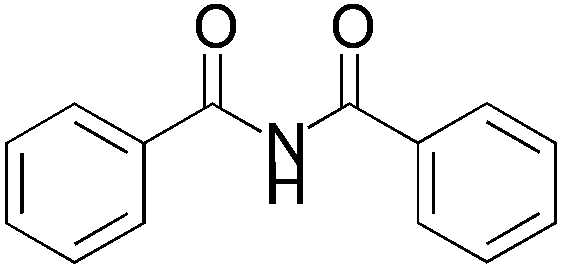
|
3c | 36 h | 26 | 3 |
| 4 |
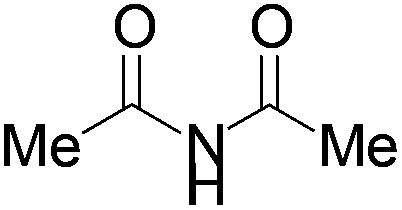
|
3d | 7 d | 16 | 11 |
| 5 |
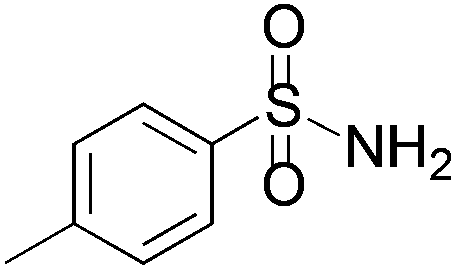
|
3e | 3 d | 17 | 25 |

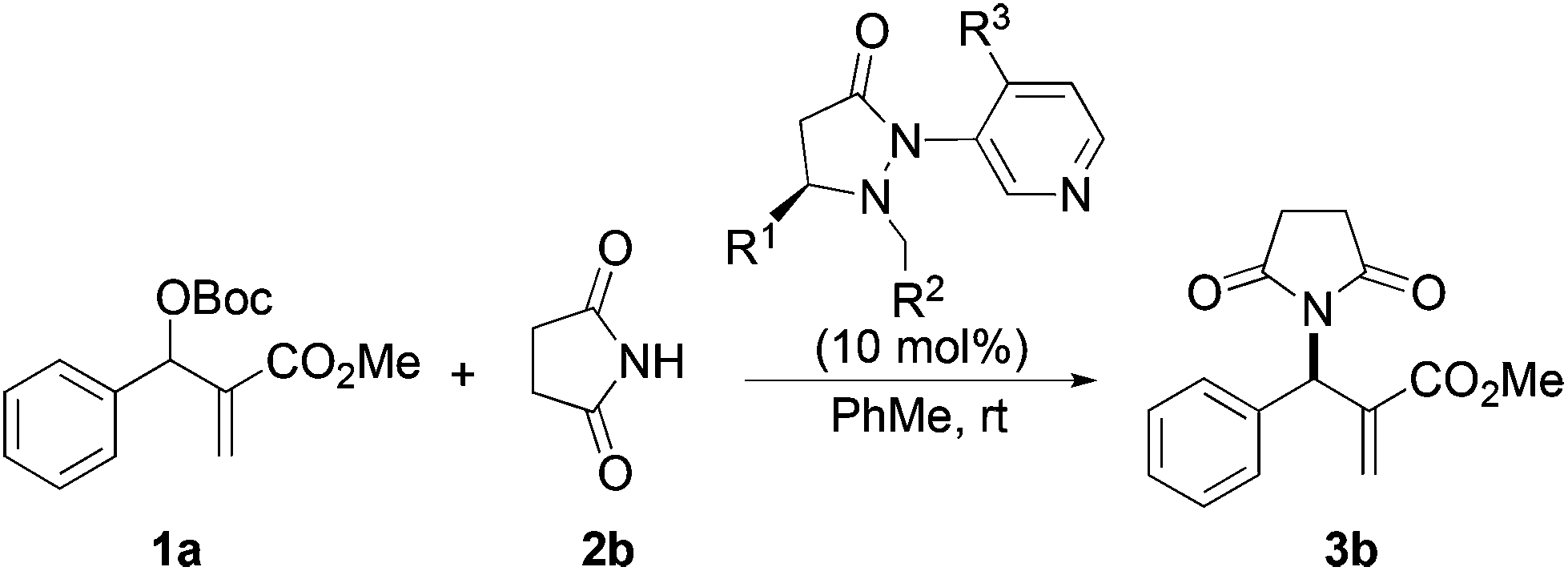
Next we examined MBH adducts with different leaving groups (OR2) and different electron withdrawing substituents (OR1) using succinimide as an aminating agent and L1 as the catalyst (Table 2). We found that the tert-butoxycarbonyloxy group was optimal as a leaving group compared to an acetate or a benzoate group (compare entry 1 with entries 2 and 3). The t-BOC group which produces a tert-butoxide anion in situ generates a nucleophile from the pronucleophile more effectively, leading to the amination product in good yield (75%). Of the different electron withdrawing groups evaluated, the methyl ester gave the product with the highest selectivity (compare entry 1 with entries 4, 5, and 6). In contrast, ethyl and benzyl esters as well as the methyl ketone gave adduct 3 in high yields but with only modest enantioselectivity (entries 4–6).
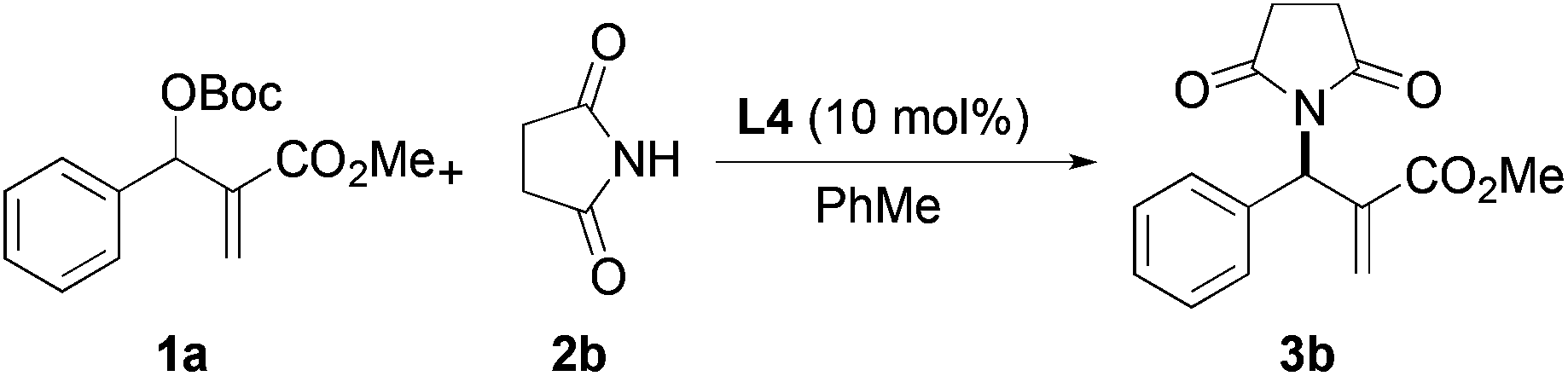
Based on the above results, reaction between MBH carbonates 1a and succinimide 2b was chosen as a model reaction. A number of chiral relay DMAP catalysts were examined next, and these results are summarized in Table 3. In general, all the DMAP catalysts examined in this study were able to catalyze the enantioselective amination of MBH carbonates and provided the product in yields ranging from 75 to 95% (entries 1–6). The catalysts examined in this study varied in the size of the fluxional group and the nature of the dialkylamino substituent on the pyridine ring (catalysts L1–L5). Generally, catalysts with a dimethylamino substituent at pyridine-4 gave higher selectivity (compare L1 with L2 and L4 with L5). Interestingly, catalyst L1 with a bulkier 1-naphthyl fluxional group was less effective than a smaller phenyl group (L4) with respect to enantioselectivity. Catalyst L3 with a smaller chiral i-Pr group gave the product in a lower selectivity than L4 with a bigger t-Bu chiral group (33% vs. 59%). Based on these results, catalyst L4 was chosen for further studies.
|
|
|||
|---|---|---|---|
| Catalysts | Time (h) | Yieldb (%) | eec (%) |
| a b c See Table 1. | |||
| L1 R1 = t-Bu, R2 = 1-naphthyl, R3 = dimethylamino | 24 | 75 | 39 |
| L2 R1 = t-Bu, R2 = 1-naphthyl, R3 = pyrrolidine | 40 | 95 | 29 |
| L3 R1 = i-Pr, R2 = phenyl, R3 = dimethylamino | 48 | 95 | 33 |
| L4 R1 = t-Bu, R2 = phenyl, R3 = dimethylamino | 40 | 85 | 59 |
| L5 R1 = t-Bu, R2 = phenyl, R3 = pyrrolidine | 36 | 90 | 43 |
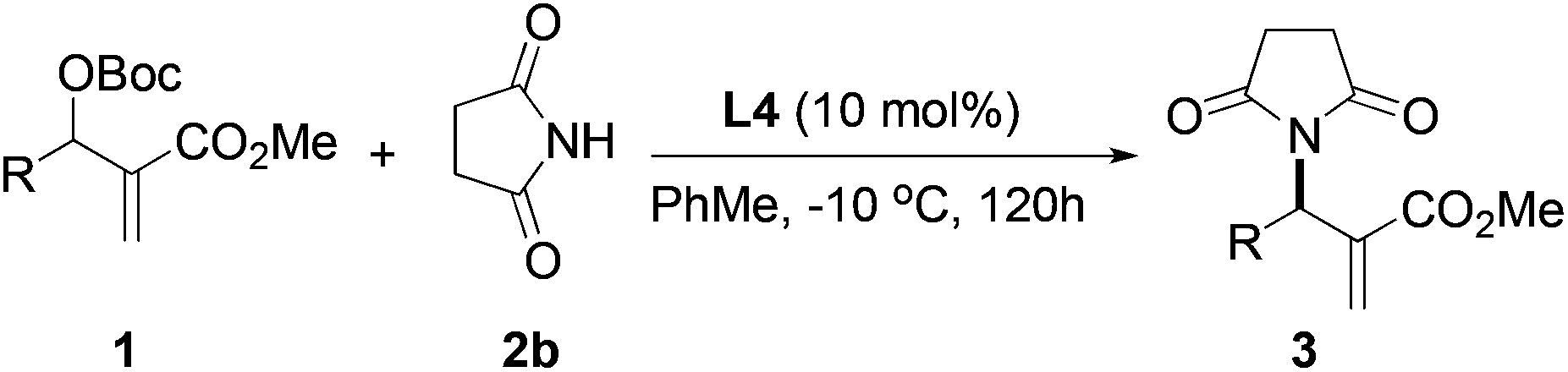
After identifying catalyst L4 that gave high yield and good selectivity, we evaluated the effect of reaction temperature and catalyst loading on the amination reaction. Results from these experiments are tabulated in Table 4. Reactions with 5, 10, and 25 mol% L4 gave the product with similar enantioselectivities and yields (entries 1–3). However, using 5 mol% catalyst, a relatively longer reaction time was required for good conversion (entry 3). Lowering the temperature to −10 °C led to improvement in enantioselectivity up to 67% vs. 59%, although the yield suffered slightly (63% vs. 85%, entry 4).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Temp. (°C) | Cat. loading (mol%) | Time (h) | Yieldb (%) | eec (%) |
| a Reaction conditions: the reactions were performed with 1 equiv. of 1a, 1.5 equiv. of 2b and L4 in toluene at a given temp. b Isolated yield. c Determined by chiral HPLC analysis. | |||||
| 1 | rt | 10% | 40 | 85 | 59 |
| 2 | rt | 25% | 48 | 88 | 57 |
| 3 | rt | 5% | 72 | 80 | 57 |
| 4 | −10 | 10% | 120 | 62 | 67 |
Having established the optimal reaction conditions for the asymmetric allylic amination with succinimide, a variety of MBH carbonates were synthesized to study the substrate scope. Results from these experiments are shown in Table 5. MBH carbonates with electron-deficient aromatics were facile giving the products in excellent yield and good selectivity (entries 2 and 3). Reaction with an electron-rich aromatic proceeded with comparable enantioselectivity (entry 4). An MBH carbonate with a disubstituted aromatic produced the product with good yield and the highest enantioselectivity was observed in this series (entry 5, 72% ee). A substrate containing an ortho substituent was less efficient, giving the product with low enantioselectivity (entry 6). Allylic amination of an alkyl substituted MBH substrate gave low yield and low enantioselectivity (entry 7).
|
|
||||
|---|---|---|---|---|
| Entry | R | Product | Yieldb (%) | eec (%) |
| a Reaction conditions: the reactions were performed with 1 equiv. of 1, 1.5 equiv. of 2b and 10 mol% of L4 in toluene at −10 °C. b Isolated yield. c Determined by chiral HPLC analysis. | ||||
| 1 | C6H5 | 3b | 62 | 67 |
| 2 | 4-NO2C6H4 | 3i | 90 | 62 |
| 3 | 4-CNC6H4 | 3j | 81 | 61 |
| 4 | 3-MeOC6H4 | 3k | 70 | 63 |
| 5 | 3,5-(MeO)2C6H4 | 3l | 78 | 72 |
| 6 | 2-BrC6H4 | 3m | 61 | 39 |
| 7 | n-C4H9 | 3n | 27 | 16 |
Several groups have developed chiral amines and phosphines as efficient organocatalysts for asymmetric substitutions of MBH adducts. High yields and enantioselectivities were achieved with a wide range of MBH adducts.11c,d Similar to cinchona alkaloid-based tertiary amine or chiral tertiary phosphine catalysts, the chiral DMAP catalysts are also effective for the allylic amination of MBH carbonates. Although the selectivities for the DMAP catalyzed reactions are slightly lower, it offers new insights into chiral DMAP catalysis, especially organo-DMAPs with fluxional chirality.
The structure and absolute configuration of L4 were ascertained by single crystal X-ray analysis (Fig. 1). The X-ray crystal structure unambiguously shows that the tert-butyl group could control the stereochemistry of the adjacent benzyl group because of its fluxionality resulting in an anti orientation. The fluxional benzyl group appears to block the back as well as the bottom side of the DMAP pyridine ring, which facilitates enantiocontrol.
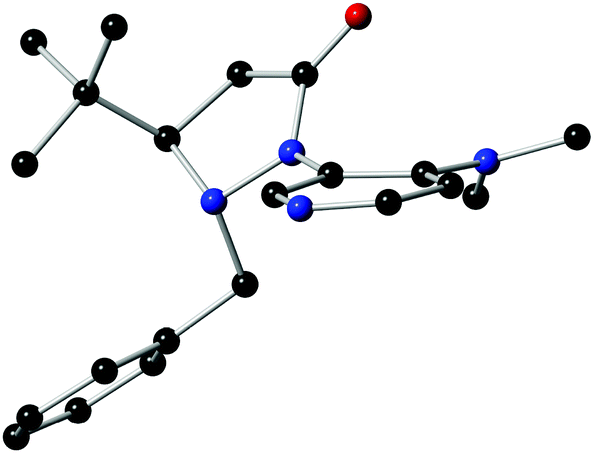 | ||
| Fig. 1 Single crystal X-ray structure of L4. | ||
Conclusions
In conclusion, we have developed the first DMAP catalyzed enantioselective allylic substitution reaction of MBH carbonates. We have also shown that the new DMAP catalysts containing fluxional groups as stereocontrol units can be effectively applied as nucleophilic catalysts in asymmetric allylic aminations. The catalyst preparation is highly tunable and up to 72% ee was achieved using L4 and a range of α-methylene β-amino esters were obtained with good yield and selectivities. Higher selectivity obtained with catalyst L4 with a bulky chiral C-5 substituent in comparison with a reaction with L3 with a smaller C-5 substituent demonstrates that the ‘chiral relay’ concept is operative. Further applications of chiral relay DMAP catalysts in asymmetric catalysis are in progress.Acknowledgements
We thank NSF-CHM-0709061 and the AC Cope Scholar award for financial support. We also thank NSF-CRIF (CHE-0946990) for the purchase of the departmental X-ray diffractometer and Dr Angel Ugrinov for solving the single crystal XRD structures.Notes and references
- (a) G. Höfle, W. Steglich and H. Vorbrüggen, Angew. Chem., Int. Ed. Engl., 1978, 17, 569 CrossRef; (b) E. F. V. Scriven, Chem. Soc. Rev., 1983, 12, 129 RSC; (c) U. Ragnarsson and L. Grehn, Acc. Chem. Res., 1998, 31, 494 CrossRef CAS; (d) R. Murugan and E. F. V. Scriven, Aldrichimica Acta, 2003, 36, 21 CAS; (e) A. C. Spivey and S. Arseniyadis, Angew. Chem., Int. Ed., 2004, 43, 5436 CrossRef CAS PubMed; (f) S. Xu, I. Held, B. Kempf, H. Mayr, W. Steglich and H. Zipse, Chem. – Eur. J., 2005, 11, 4751 CrossRef CAS PubMed.
- E. Vedejs and X. Chen, J. Am. Chem. Soc., 1996, 118, 1809 CrossRef CAS.
- (a) T. Kawabata, M. Nagato, K. Takasu and K. Fuji, J. Am. Chem. Soc., 1997, 119, 3169 CrossRef CAS; (b) J. C. Ruble, H. A. Latham and G. C. Fu, J. Am. Chem. Soc., 1997, 119, 1492 CrossRef CAS; (c) A. C. Spivey, T. Fekner, S. E. Spey and H. Adams, J. Org. Chem., 1999, 64, 9430 CrossRef CAS; (d) S. Arai, S. Bellemin-Laponnaz and G. C. Fu, Angew. Chem., Int. Ed., 2001, 40, 234 CrossRef CAS; (e) S. Yamada, T. Misono, Y. Iwai, A. Masumizu and Y. Akiyama, J. Org. Chem., 2006, 71, 6872 CrossRef CAS PubMed; (f) C. O. Dálaigh and S. J. Connon, J. Org. Chem., 2007, 72, 7066 CrossRef PubMed; (g) C. K. De, E. G. Klauber and D. Seidel, J. Am. Chem. Soc., 2009, 131, 17060 CrossRef CAS PubMed; (h) M. R. Crittall, H. S. Rzepa and D. R. Carbery, Org. Lett., 2011, 13, 1250 CrossRef CAS PubMed; (i) E. Larionov, M. Mahesh, A. C. Spivey, Y. Wei and H. Zipse, J. Am. Chem. Soc., 2012, 134, 9390 CrossRef CAS PubMed.
- (a) G. C. Fu, Pure Appl. Chem., 2001, 73, 347 CAS; (b) S. France, D. J. Guerin, S. J. Miller and T. Lectka, Chem. Rev., 2003, 103, 2985 CrossRef CAS PubMed; (c) G. C. Fu, Acc. Chem. Res., 2004, 37, 542 CrossRef CAS PubMed; (d) S. J. Connon, Lett. Org. Chem., 2006, 3, 333 CrossRef CAS; (e) R. P. Wurz, Chem. Rev., 2007, 107, 5570 CrossRef CAS PubMed.
- (a) M. P. Sibi, L. Venkatraman, M. Liu and C. P. Jasperse, J. Am. Chem. Soc., 2001, 123, 8444 CrossRef CAS; (b) M. P. Sibi, R. Zhang and S. Manyem, J. Am. Chem. Soc., 2003, 125, 9306 CrossRef CAS PubMed; (c) M. P. Sibi, S. Manyem and H. Palencia, J. Am. Chem. Soc., 2006, 128, 13660 CrossRef CAS PubMed; (d) M. P. Sibi, L. M. Stanley, X. Nie, L. Venkatraman, M. Liu and C. P. Jasperse, J. Am. Chem. Soc., 2007, 129, 395 CrossRef CAS PubMed; (e) M. P. Sibi, K. Kawashima and L. Stanley, Org. Lett., 2009, 11, 3894 CrossRef CAS PubMed.
- For selected contributions from other laboratories see: (a) S. D. Bull, S. G. Davis, D. J. Fox, A. C. Garner and T. G. R. Sellers, Pure Appl. Chem., 1998, 70, 1501 CrossRef CAS; (b) E. E. Sugg, J. F. Griffin and P. S. Portoghese, J. Org. Chem., 1985, 50, 5032 CAS; (c) J. Clayden, Chem. Soc. Rev., 2009, 38, 817 RSC; (d) R. W. Parrott II and S. R. Hitchcock, Tetrahedron: Asymmetry, 2007, 18, 377 CrossRef PubMed; (e) H. Suga, Y. Furihata, A. Sakamoto, K. Itoh, Y. Okumura, T. Tsuchida, A. Kakehi and T. Baba, J. Org. Chem., 2011, 76, 7377 CrossRef CAS PubMed.
- G. Ma, J. Deng and M. P. Sibi, Angew. Chem., Int. Ed., 2014 DOI:10.1002/anie.201406684 DOI:10.1002/ange.201406684 , in press.
- T. Sammakia and T. B. Hurley, J. Org. Chem., 1999, 64, 4652 CrossRef CAS PubMed.
- (a) D. Basavaiah, A. J. Rao and T. Satyanarayana, Chem. Rev., 2003, 103, 811 CrossRef CAS PubMed; (b) K. Y. Lee, S. Gowrisankar and J. N. Kim, Bull. Korean Chem. Soc., 2005, 26, 1481 CrossRef CAS; (c) J. N. Kim, H. J. Lee and J. H. Gong, Tetrahedron Lett., 2002, 43, 9141 CrossRef CAS; (d) C.-W. Cho, J.-R. Kong and M. J. Krische, Org. Lett., 2004, 6, 1337 CrossRef CAS PubMed; (e) Y. Du, X. Han and X. Lu, Tetrahedron Lett., 2004, 45, 4967 CrossRef CAS PubMed; (f) T.-Z. Zhang, L.-X. Dai and X.-L. Hou, Tetrahedron: Asymmetry, 2007, 18, 1990 CrossRef CAS PubMed; (g) D. J. V. C. van Steenis, T. Marcelli, M. Lutz, A. L. Spek, J. H. van Maarseveen and H. Hiemstra, Adv. Synth. Catal., 2007, 349, 281 CrossRef CAS; (h) H. Park, C.-W. Cho and M. J. Krische, J. Org. Chem., 2006, 71, 7892 CrossRef CAS PubMed; (i) C.-L. Cao, Y.-Y. Zhou, J. Zhou, X.-L. Sun, Y. Tang, Y.-X. Li, G.-Y. Li and J. Sun, Chem. – Eur. J., 2009, 15, 11384 CrossRef CAS PubMed; (j) S. Xu, S. Zhu, J. Shang, J. Zhang, Y. Tang and J. Dou, J. Org. Chem., 2014, 79, 3696 CrossRef CAS PubMed.
- (a) B. M. Trost, M. R. Machacek and H.-C. Tsui, J. Am. Chem. Soc., 2005, 127, 7014 CrossRef CAS PubMed; (b) B. M. Trost, O. R. Thiel and H.-C. Tsui, J. Am. Chem. Soc., 2003, 125, 13155 CrossRef CAS PubMed; (c) D. Wang, Y.-L. Yang, J.-J. Jiang and M. Shi, Org. Biomol. Chem., 2012, 10, 7158 RSC; (d) J. Peng, X. Huang, H.-L. Cui and Y.-C. Chen, Org. Lett., 2010, 12, 4260 CrossRef CAS PubMed; (e) X. Huang, J. Peng, L. Dong and Y.-C. Chen, Chem. Commun., 2012, 48, 2439 RSC; (f) F.-L. Hu and M. Shi, Org. Chem. Front., 2014, 1, 587 RSC.
- (a) Y. Du, X. Han and X. Lu, Tetrahedron Lett., 2004, 45, 4967 CrossRef CAS PubMed; (b) S.-J. Zhang, H.-L. Cui, K. Jiang, R. Li, Z.-Y. Ding and Y.-C. Chen, Eur. J. Org. Chem., 2009, 5804 CrossRef CAS; (c) T.-Y. Liu, M. Xie and Y.-C. Chen, Chem. Soc. Rev., 2012, 41, 4101 RSC; (d) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659 CrossRef CAS PubMed.
Footnotes |
| † This article is dedicated to Max Malacria on his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1016533. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00210e |
| This journal is © the Partner Organisations 2014 |