 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCotA laccase: high-throughput manipulation and analysis of recombinant enzyme libraries expressed in E. coli using droplet-based microfluidics†
Thomas
Beneyton
ab,
Faith
Coldren
a,
Jean-Christophe
Baret
cd,
Andrew D.
Griffiths
ab and
Valérie
Taly
*ae
aLaboratoire de Biologie Chimique, Institut de Science et d'Ingénierie Supramoléculaires (ISIS), CNRS UMR 7006, 8 allée Gaspard Monge, F-67083 Strasbourg, France
bLaboratoire de Biochimie, Ecole Supérieure de Physique et de Chimie Industrielles de la ville de Paris (ESPCI ParisTech), CNRS UMR 7084, 10 rue Vauquelin, F-75231, Paris, France
cDroplets Membranes and Interfaces, Max Planck Institute for Dynamics and Self-Organization, Am Fassberg, 17, D-37077 Goettingen, Germany
dUniversité de Bordeaux, CRPP-CNRS, 115 Avenue Schweitzer, F-33600 Pessac, France
eUniversité Paris Sorbonne Cité, INSERM UMR-S1147, 45 rue des Saints-Pères, F-75270, Paris, France. E-mail: valerie.taly@parisdescartes.fr
First published on 11th March 2014
Abstract
We present a high-throughput droplet-based microfluidic analysis/screening platform for directed evolution of CotA laccase: droplet-based microfluidic modules were combined to develop an efficient system that allows cell detection and sorting based on the enzymatic activity. This platform was run on two different operating modes: the “analysis” mode allowing the analysis of the enzymatic activity in droplets at very high rates (>1000 Hz) and the “screening” mode allowing sorting of active droplets at 400 Hz. The screening mode was validated for the directed evolution of the cytoplasmic CotA laccase from B. subtilis, a potential interesting thermophilic cathodic catalyst for biofuel cells. Single E. coli cells expressing either the active CotA laccase (E. coli CotA) or an inactive frameshifted variant (E. coli ΔCotA) were compartmentalized in aqueous droplets containing expression medium. After cell growth and protein expression within the droplets, a fluorogenic substrate was “picoinjected” in each droplet. Fluorescence-activated droplet sorting was then used to sort the droplets containing the desired activity and the corresponding cells were then recultivated and identified using colorimetric assays. We demonstrated that E. coli CotA cells were enriched 191-fold from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 initial ratio of E. coli CotA to E. coli ΔCotA cells (or 437-fold from a 1:99 initial ratio) using a sorting rate of 400 droplets per s. This system allows screening of 106 cells in only 4 h, compared to 11 days for screening using microtitre plate-based systems. Besides this low error rate sorting mode, the system can also be used at higher throughputs in “enrichment” screening mode to make an initial purification of a library before further steps of selection. Analysis mode, without sorting, was used to rapidly quantify the activity of a CotA library constructed using error-prone PCR. This mode allows analysis of 106 cells in only 1.5 h.
:9 initial ratio of E. coli CotA to E. coli ΔCotA cells (or 437-fold from a 1:99 initial ratio) using a sorting rate of 400 droplets per s. This system allows screening of 106 cells in only 4 h, compared to 11 days for screening using microtitre plate-based systems. Besides this low error rate sorting mode, the system can also be used at higher throughputs in “enrichment” screening mode to make an initial purification of a library before further steps of selection. Analysis mode, without sorting, was used to rapidly quantify the activity of a CotA library constructed using error-prone PCR. This mode allows analysis of 106 cells in only 1.5 h.
1. Introduction
Directed evolution is a powerful biotechnological tool that mimics and accelerates natural evolution at the laboratory scale. It relies on inducing artificial mutations to create populations of new variants and selecting the rare ones which exhibit improved functionality under a user-defined and controlled selection pressure. It has been intensively used this last decade to improve enzymes1 or microorganism strains2 for industrial applications or, for example, to study the fundamentals of evolution itself.3 The impact of directed evolution is however limited by its need for efficient high-throughput screening methods, especially when selection occurs on the enzymatic activity. Indeed, efficient directed evolution requires screening of very large libraries under conditions that closely match the desired activity. As genetic information is encoded in the genotype and the selection is based on phenotypic activities, genotype and phenotype have to be linked. Ideally, they must be physically linked by compartmentalization as it is in cells. Nowadays, the most controlled and flexible high-throughput enzymatic activity screening technique is the robotized compartmentalization of assays in microtitre plates, allowing us to process up to 1 assay per second. This technology has even reached its physical limits as reducing assay volumes to below 1 μL is problematic due to evaporation and capillarity.4 One way to miniaturize reaction vessels exploits microarrays and microfluidic lab-on-chip technology, decreasing the reaction volume to ∼250 pL.5 Nevertheless, these screening techniques are still limiting the applicability of directed evolution.6 Another way is to use in vitro compartmentalization (IVC): genotype and phenotype are linked by compartmentalizing individual components of a library in water-in-oil droplets.7 Initially used with bulk emulsion, IVC is suitable for in vitro evolution, reducing the reaction volume below 1 pL and allowing screening of large libraries (up to 1011).8 However, the range of activities that can be screened and the flexibility of the method are rather limited because of the lack of control of the droplet volume and timing. Moreover, most assays require the addition of new reagents at defined times to initiate or terminate a reaction. As bulk emulsions are hardly manipulated, the applicability of IVC is markedly limited in that case. These limitations can be overcome by using the droplet-based microfluidic technology, which offers the possibility to use IVC in a highly controlled manner.Indeed, droplet-based microfluidics allows the production of highly monodisperse water-in-oil droplets and the manipulation of those at a single level and at very high frequencies.9 During recent years, this technology has been subjected to increasing interest for screening applications, which has led to intensive developments of efficient tools to handle single droplets within an emulsion. Different droplet-based microfluidic modules have been developed to perform controlled droplet manipulation, such as fusion,10–12 picoinjection,13,14 incubation15,16 or sorting based on fluorescence,17 especially in the droplets' volume range of 10 pL to 100 pL, suitable for biological assay and cell handling. This technology has arrived at the point where single modules can be combined to create efficient integrated screening platforms.18 However, very few directed evolution platforms have been described in the literature. An integrated encapsulation/fusion/sorting platform has been developed for complete in vitro directed evolution.19In vivo screening platforms have also been described but their flexibility is limited as they only consist of encapsulation/sorting techniques.17,20–22 A flexible and efficient in vivo screening platform for directed evolution would allow the growth of cells within droplets and then the addition of the substrate to initiate the enzymatic reaction at a controlled time prior to sorting.
CotA laccase is an oxidoreductase (EC 1.10.3.2) that catalyzes the one-electron oxidation of phenols, polyphenols and anilines with the concomitant four-electron reduction of O2 to water.23 Due to their broad substrate specificity and few requirements, laccases such as CotA have high interest for green industrial applications, such as delignification of wood, biosensors or biofuel cells.24 We designed a flexible droplet-based microfluidic platform that can be used for high-throughput screening of E. coli cells for the CotA enzymatic activity (Fig. 1). This platform involves the following steps: (i) encapsulation of cells in aqueous droplets; (ii) incubation for cell growth and protein expression in droplets; (iii) picoinjection of the fluorogenic substrate; (iv) incubation for enzymatic reactions; (v) sorting of droplets based on the enzymatic activity according to the fluorescence intensity; (vi) recovery of cells from the sorted droplets. We demonstrated that this high-throughput screening platform allows enrichment of specific cells according to the CotA enzymatic activity and should be a powerful tool to screen large libraries for CotA directed evolution experiments. Furthermore, the droplet-based microfluidic platform provides also a powerful high-throughput cell analysis tool to quickly quantify the distribution of the enzymatic activity within a library. We created a library of CotA variants by error-prone PCR and analyzed the library of cells obtained by transformation of the library. In general, such screening tools could be extended to many other laccases or oxidoreductases, enzymes of high industrial interest.
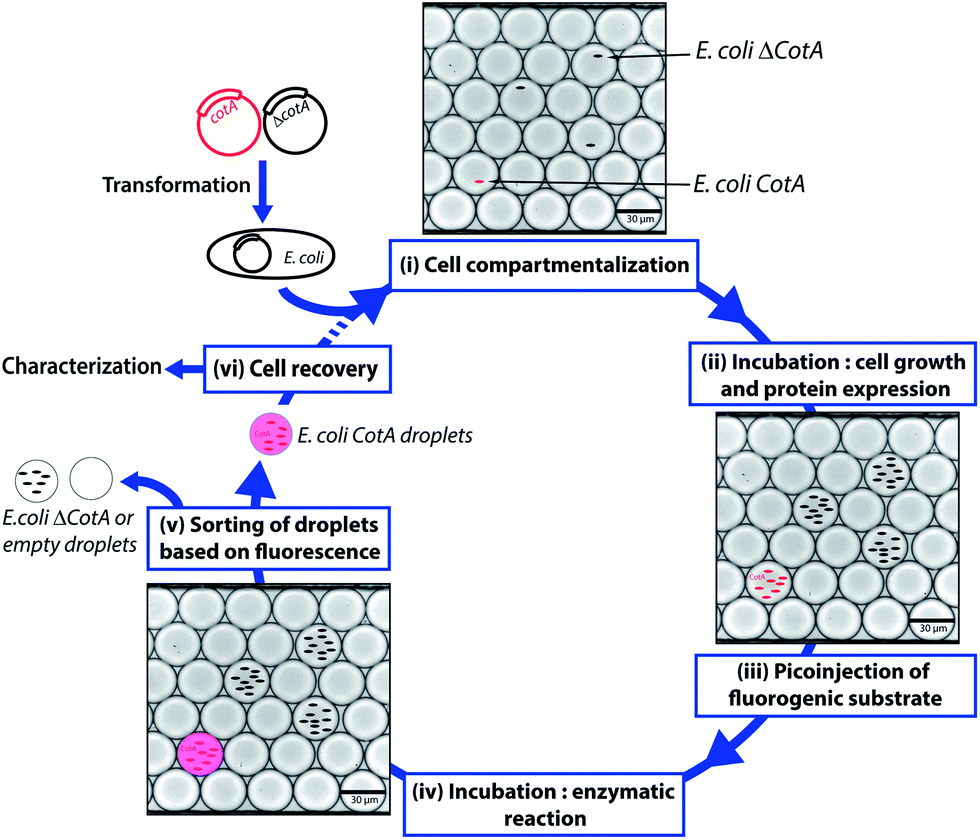 | ||
| Fig. 1 Schematic of the droplet-based microfluidic model screening platform. Wild type CotA gene and a frameshifted inactive variant ΔCotA gene were transformed into E. coli cells to give E. coli CotA and E. coli ΔCotA strains. (i) A mixture of E. coli CotA/E. coli ΔCotA cell suspensions was encapsulated in droplets containing induction medium. (ii) The emulsion was incubated off-chip for cell growth and CotA or ΔCotA enzyme expression within the droplets. (iii) Fluorogenic substrate was picoinjected in every droplet. (iv) The emulsion was incubated off-chip for the enzymatic reaction. (v) The droplets were sorted based on the enzymatic activity according to the fluorescence intensity. (vi) Cells were recovered from the sorted droplets and could be either characterized to determine enrichment in E. coli CotA cells or engaged in another round of selection. | ||
2. Experimental
2.1 Reagents
2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS), ampicillin, D-glucose, ethylenediaminetetraacetic acid (EDTA), and copper(II) chloride were purchased from Sigma-Aldrich. Isopropyl β-D-1-thiogalactopyranoside (IPTG), LB medium and LB agar were obtained from Roth. Dextran Texas Red (10000 MW), dextran fluorescein (10000 MW) and fluorescein were purchased from Life Technologies.
2.2 Fabrication of microfluidic devices
Microfluidic devices were fabricated from poly(dimethylsiloxane) (PDMS) using standard soft-lithography methods.25 A mold of SU8-2015 negative photoresist (MicroChem Corp.) was prepared on a silicon wafer (Siltronix) by UV exposure (MJB3 contact mask aligner; SUSS MicroTec) through a photolithography mask (Selba SA) and subsequent development (SU-8 developer; MicroChem Corp.). A curing agent was added to the PDMS base (Sylgard 184 elastomer kit; Dow Corning Corp.) to a final concentration of 10% (w/w), mixed, and poured onto the mold. After degassing under vaccum to remove air bubbles, the mold was incubated for several hours at 65 °C for PDMS cross-linking. PDMS was then peeled off and inputs and outputs were punched with a 0.75 mm diameter biopsy punch. The structured side of the PDMS slab was bound to a 75 × 50 × 1 mm glass microscope slide (Corning) by exposing both parts to an oxygen plasma (Plasma system FEMTO; Diener Electronic). The device was incubated for several minutes at 65 °C. Finally, an additional hydrophobic surface coating was applied to the microfluidic channel walls by injecting the channels with 1% (w/w) 1H,1H,2H,2H-perfluorodecyltrichlorosilane (97%; ABCR) in HFE7100 fluorinated oil (3 M).Electrodes were included in the microfluidic device as additional channels which were filled with metal: the device was heated at 110 °C and a 51In/32.5Bi/16.5Sn low-temperature solder (Indium Corp.) was melted inside the electrode channels.26 Electrical connections with the solder electrodes were made with short pieces of electrical wire (Radiospares) and strengthened with Loctite 3526 UV glue (Epotecny).
2.3 Optical setup, data acquisition and control system
The optical setup consisted of an Axiovert 200 inverted microscope (Carl Zeiss SAS) mounted on a vibration-dampening platform (Thorlabs GmbH). A 20 mW, 488 nm solid-state laser (LAS; Newporte-Spectraphysics) and a 50 mW, 532 nm solid-state laser (CrystaLaser) were focused through the objective lens (40×/0.6; Carl Zeiss SAS) across a channel within the microfluidic device. Light emitted from fluorescing droplets was captured by the objective, channeled back along the path of the lasers. The emitted light was separated from the laser beam and split between two photomultiplier tubes (PMT, H5784-20; Hamamatsu), which captured the light through a 510 nm bandpass filter (510/20-25; Semrock Inc.) for the green PMT and through a 617 nm bandpass filter (617/73-25, Semrock Inc.) for the orange PMT. The signal output from the PMT was analyzed using a PCI-7831R Multifunction Intelligent Data Acquisition (DAQ) card (National Instruments Corporation) executing a program written in LabView 8.2 (FPGA module, National Instruments Corporation, Fig. S1, ESI†), which allowed the identification of droplets by peaks in fluorescence, as well as the width of each droplet. The data acquisition rate for the system was 100 kHz. A Phantom v4.2 high speed digital camera (Vision Research) was mounted on the top camera port of the microscope to capture digital images during droplet manipulation.Liquids were pumped into the microfluidic devices using standard-pressure infusion-only PHD 22/2000 syringe pumps (Harvard Apparatus Inc.). Syringes (Omnifix-F®; BBRAUN) were connected to the microfluidic devices using 0.6 × 25 mm needle (Terumo) and PTFE tubing with an internal diameter of 0.56 mm and an external diameter of 1.07 mm (Fisher Scientific).
2.4 Description of microfluidic devices
We used aqueous droplets in HFE7500 fluorinated oil (3 M) stabilized against coalescence by block-polymer homemade surfactant, named KryJeffa900 in this report. KryJeffa900 surfactant was prepared from the commercially available carboxylic acid Krytox157-FSH (Dupont) and Jeffamine® polyetheramines (ED 900, Huntsmann) following a synthesis route based on the one described by Holtze et al.27All the microfluidic devices were fabricated with a channel depth of 25 μm. Four different modules were used to compose the platform.
First, a dropmaker module (Fig. S2, ESI†) was used to generate 14 pL droplets (coefficient of variance (CV) = 3%) at 1600 Hz by flow-focusing of the aqueous stream (80 μL h−1) with two streams of HFE7500 fluorinated oil (3 M) (190 μL h−1) containing 1% (w/w) of KryJeffa900 surfactant. A dual-dropmaker module (Fig. S3, ESI†) was used to generate two 14 pL droplets of emulsions simultaneously (the same conditions of operation). The generated emulsions flowed off-chip through PTEF tubing to a syringe.
The second module was a picoinjection device (Fig. S4 and ESI movie “picoinjection.avi”, ESI†) in which droplets were reloaded (70–160 μL h−1) and spaced-out at a flow-focusing junction (Fig. 3b) with HFE7500 fluorinated oil (3 M) (200–600 μL h−1) containing 1% (w/w) of KryJeffa900 surfactant. The picoinjected phase was loaded into a 250 μL gas-tight syringe (Hamilton Company) and pumped into the device (Fig. 3b). A continuous AC field (20 kHz; 200 V) was applied across the electrodes and more than 99.5% of the droplets were picoinjected (∼1 pL; estimated by video analysis) at 1500–3000 Hz (working range of the module) to give droplets of ∼15 pL (CV = 4.5%). The generated emulsions flowed off-chip through PTEF tubing to a glass vial immerged in ice to slow down the enzymatic reaction. The off-chip incubation time at room temperature (25 °C) was determined by the length of the PTFE tubing connected from the chip to the glass vial: droplets were incubated at 25 °C while they were flowing in to the vial (collection) and from the vial (reinjection).
Under screening mode operation, the last module was fluorescence-activated droplet sorting (FADS)16 (Fig. S5, ESI†) in which droplets were reloaded (20–70 μL h−1) and spaced-out at a flow-focusing junction (Fig. 3c) with HFE7500 fluorinated oil (3 M) (500–1000 μL h−1). The droplets were analyzed by the optical setup and fluorescent droplets were sorted at 400 Hz by applying an AC field pulse (30 kHz; 1000–1400 V; 0.5 ms). The sorted droplets were collected in a 1.5 mL microcentrifuge tube (Axygen Inc.).
Under analysis mode operation, the last module was a reloading module (Fig. S6, ESI†) in which droplets were reloaded (50–150 μL h−1) and spaced-out at a flow-focusing junction with HFE7500 fluorinated oil (3 M) (200–400 μL h−1). The droplets were detected by the optical setup and analyzed at ∼1000 Hz.
2.5 Plasmid construction
The plasmid pLOM10 coding for the wild type CotA laccase was kindly provided by Prof. L. O. Martins (Universidade Nova de Lisboa). The plasmid pLOMP3 coding for the inactive frameshifted variant ΔCotA was constructed starting from pLOM10. A mixture of 44 μL of pLOM10 (35 ng μL−1), 5 μL of NEB4 buffer 10× (New England Biolabs) and 1 μL of AatII enzyme (20 U μL−1; New England Biolabs) was incubated for 15 h at 37 °C. T4 DNA Polymerase (3U; New England Biolabs) and 200 μM of dNTP (New England Biolabs) were added and the mixture was incubated for 15 min at 12 °C. The reaction was quenched by adding 10 mM of EDTA and the mixture was incubated for 20 min at 75 °C. After purification using the PCR purification kit (Qiagen), 10 μL of DNA (18 ng μL−1) was mixed with 2 μL of T4 DNA ligase buffer 10× (New England Biolabs), 1 μL of T4 DNA ligase (400 U μL−1; New England Biolabs) and 7 μL of ultrapure water. The mixture was incubated for 3 h at 25 °C. The plasmid was then transformed into Xl10 gold E. coli competent cells (Stratagene). 12 colonies were isolated and sequenced.2.6 Preparation of cell suspensions for encapsulation
Plasmids pLOM10 and pLOMP3 were transformed into E. coli C41(DE3)28 using standard molecular biology protocols to give an active strain (E. coli CotA) and an inactive strain (E. coli ΔCotA) in which the CotA or ΔCotA protein could be produced under the control of the T7lac promoter. 2 mL of LB medium containing ampicillin (0.1 mg mL−1) was inoculated with a single colony of the desired strain. This aliquot was grown for 14 h at 37 °C (230 rpm). The following day, 50 mL of fresh LB medium containing ampicillin (0.1 mg mL−1) and CuCl2 (0.25 mM) was inoculated with 500 μL of preculture. The culture was grown under the same conditions to reach an OD600 of 0.6. 5 mL of culture was centrifuged (10 min; 2000g; 4 °C) and the pellet was resuspended in 5 mL of fresh LB medium containing ampicillin (0.1 mg mL−1), CuCl2 (0.25 mM) and IPTG (1 mM) for induction of protein expression. OD600 was measured and the culture was diluted to have the desired number of cells per droplet volume (OD600 of 1 corresponded to 5 × 108 cells per mL).2.7 CotA and ΔCotA enzymatic activity in droplets
The dual-dropmaker module (Fig. S2, ESI†) was used to produce two emulsions simultaneously. E. coli CotA cell suspension was encapsulated in 14 pL droplets containing 1 μM of dextran fluorescein while E. coli ΔCotA cell suspension was encapsulated in 14 pL droplets containing 5 μM of dextran fluorescein. The binary emulsion was incubated for 15 h at 30 °C and reloaded into the picoinjection module (Fig. S3, ESI†). LB medium containing Amplex®UltraRed (0.5 μM; Life Technologies) was used as the picoinjected phase. After 10 min of incubation at room temperature (25 °C), the emulsion was then reloaded into the reloading module (Fig. S5, ESI†) for analysis.2.8 Screening mode: model selection
E. coli CotA and E. coli ΔCotA cell suspensions were mixed together in the desired ratio prior to encapsulation using the dropmaker module (Fig. S1, ESI†). The emulsion was incubated for 15 h at 30 °C and reloaded into the picoinjection module (Fig. S3, ESI†). LB medium containing Amplex®UltraRed (0.5 μM; Life Technologies) was used as the picoinjected phase. After 10 min of incubation at room temperature (25 °C), the emulsion was then reloaded into the FADS module (Fig. S4, ESI†) for sorting. The sorted droplets were recovered by cutting the collection PTFE tubing and draining last droplets into the collection microcentrifuge tube. To dislodge any droplets remaining in the tubing, it was flushed with 200 μL of fresh LB medium. All the contents of the collection microcentrifuge tube were added to 10 mL of fresh LB medium containing ampicillin (0.1 mg mL−1) and grown for 15 h at 37 °C. As a reference, 250 μL of the initial emulsion was grown under the same conditions.Cells before and after sorting were then identified using two different techniques. On one hand, the sorted cells were plated on LB agar containing ampicillin (0.1 mg mL−1), CuCl2 (0.25 mM), IPTG (1 mM) and ABTS (5 mM), and incubated for 15 h at 30 °C. Plates were then kept at 4 °C for 48 h for coloration to occur. E. coli CotA colonies appeared dark brown while E. coli ΔCotA colonies remained colourless (Fig. 3d2).
On the other hand, sorted cells were plated on LB agar containing ampicillin (0.1 mg mL−1) and incubated for 15 h at 37 °C. 87 colonies were transplanted in a 96-deepwells microtitre plate (Nunc) in 1 mL of fresh LB medium containing ampicillin (0.1 mg mL−1), CuCl2 (0.25 mM) and D-glucose (20 mM) with 3 positive controls (E. coli CotA), 3 negative controls (E. coli ΔCotA) and 3 blanks (growth medium). The microtitre plate was covered by a gas-permeable membrane (Thermofisher) and incubated for 15 h at 37 °C (250 rpm). The microtitre plate was then centrifuged (10 min; 1000g) and the medium was replaced with fresh LB medium containing ampicillin (0.1 mg mL−1), CuCl2 (0.25 mM), and IPTG (1 mM). The microtitre plate was covered by a gas-permeable membrane (Thermofisher) and incubated for 15 h at 30 °C (250 rpm). The CotA enzymatic activity corresponding to each well was tested in a standard 96-well microtitre plate by mixing 50 μL of cells with 50 μL of acetate buffer (50 mM; pH 4.5) containing ABTS (5 mM). The CotA enzymatic activity resulted in green coloration of the solution (Fig. 3d1).
2.9 Library construction
Primers CotAprom (5′-TGTGACTCAGATCTCGATCCCGCGAAATTAATAC-3′) and CotAterm (5′-GGTTATGCTAGTTATTGCTCAGCGG-3′), biotinylated at 5′ ends, were used to PCR-amplify a 1808-bp fragment containing the CotA gene under mutagenesis conditions using the GeneMorph® II Random mutagenesis kit (Stratagene). A mixture of 1 μL of plasmid pLOM10 (100 ng μL−1), 2.5 μL of CotAprom (10 μM), 2.5 μL of CotAterm (10 μM), 1 μL of dNTPs (5 mM each, New England Biolabs), 1 μL of Mutazyme® II (2.5 U μL−1), 5 μL of buffer (10×) and 37.5 μL of RNase/DNase free water (Roth) was subjected to the PCR reaction in a MJ Research thermocycler. The thermal cycle included 240 s initial denaturation at 94 °C followed by 30 cycles of thermal cycling and 420 s of final extension at 72 °C. The thermal cycling protocol consisted of 30 s at 94 °C, 30 s at 55 °C, and 120 s at 72 °C. The PCR products were captured using streptavidin coated magnetic beads (Dynabeads® M-280, kilobaseBinder™ kit, Invitrogen) and released from the magnetic beads using the two cloning sites, BglII and NotI.An “easy-purification” acceptor vector pET-TB was constructed by modifying pET-21a(+) vector (Novagen): pET-21a(+) was digested at BglII/NotI cloning sites and a small oligonucleotide cassette was inserted using K71 (5′-GGCCGCAAGCTTCA-3′) and K72 (5′-GATCTGAAGCTTGC-3′) oligonucleotides phosphorylated at the 5′ end. This plasmid pET-TB was easily purified prior to the cloning step as its digestion at BglII and NotI cloning sites generates a small 14-bp fragment, easily eliminated using a PCR purification kit (Qiagen).
The purified mutagenesis PCR product was inserted into the acceptor vector pET-TB between BglII and NotI cloning sites to give the library plasmid pLOM10mut.
2.10 Analysis mode: library analysis
The library cell suspension was produced as follows: 4 μL of plasmid pLOM10mut was transformed by electroporation into 480 μL of E. coli C41(DE3) competent cells (∼2 × 106 cells per μg of DNA) using standard molecular biology protocols. Cells were then grown for 15 h at 37 °C (230 rpm) in 500 mL of fresh LB medium containing ampicillin (0.1 mg mL−1) and D-glucose (20 mM). 500 mL of fresh LB medium containing ampicillin (0.1 mg mL−1) and CuCl2 (0.25 mM) was inoculated with 500 μL of preculture. The culture was grown at 37 °C (230 rpm) to reach an OD600 of 0.6. 5 mL of culture was centrifuged (10 min; 2000g; 4 °C) and the pellet was resuspended in 5 mL of fresh LB medium containing ampicillin (0.1 mg mL−1), CuCl2 (0.25 mM) and IPTG (1 mM) for induction of protein expression. OD600 was measured and the culture was diluted to 0.2 cells per 14 pL.The dual-dropmaker module (Fig. S2, ESI†) was used to produce simultaneously two emulsions. E. coli CotA cell suspension was encapsulated in 14 pL droplets containing 10 μM of dextran fluorescein while the library cell suspension was encapsulated in 14 pL droplets without any marker. The binary emulsion was incubated for 15 h at 30 °C and reloaded into the picoinjection module (Fig. S3, ESI†). LB medium containing Amplex®UltraRed (0.5 μM; Life Technologies) was used as the picoinjected phase. After 10 min of incubation at room temperature (25 °C), the emulsion was then reloaded into the reloading module (Fig. S5, ESI†) for analysis.
3. Results and discussion
3.1 Screening mode
This work reports a microfluidic screening platform to evolve recombinant CotA laccase expressed in E. coli. We used the following fluorescent assay based on the laccase enzymatic activity: CotA laccase converted the fluorogenic substrate Amplex®UltraRed (AUR) into a fluorescent dye analogous to resorufin (λmax excitation: 568 nm/λmax emission: 581 nm). In order to validate the droplet-based microfluidic screening platform, we have constructed a frameshifted inactive variant ΔCotA.
E. coli CotA and E. coli ΔCotA strains were simultaneously encapsulated using a dual-dropmaker module and a green fluorescent barcode to tag the two populations (Fig. 2a, inset). The binary emulsion was incubated for 15 h at 30 °C and then reloaded into the picoinjection device for fluorogenic substrate injection. The fluorescent signal was measured before injection (Fig. 2a) and after injection and incubation for 10 min at 25 °C by reloading the emulsion in a reloading module (Fig. 2b). E. coli CotA strain showed laccase activity with apparition of orange fluorescent active droplet population (Fig. 2b, 3 [0.6%]; theoretically 1%) while the fluorescent signal of E. coli ΔCotA droplets remained unchanged. It can also be noticed that the droplets are efficiently stabilized by surfactant, as only 0.6% of emulsion is coalesced (Fig. 2b, population 4). This is mainly due to the fact that our dropmaker devices ensured sufficient stabilization time after droplet formation and before the droplets first come into contact.36
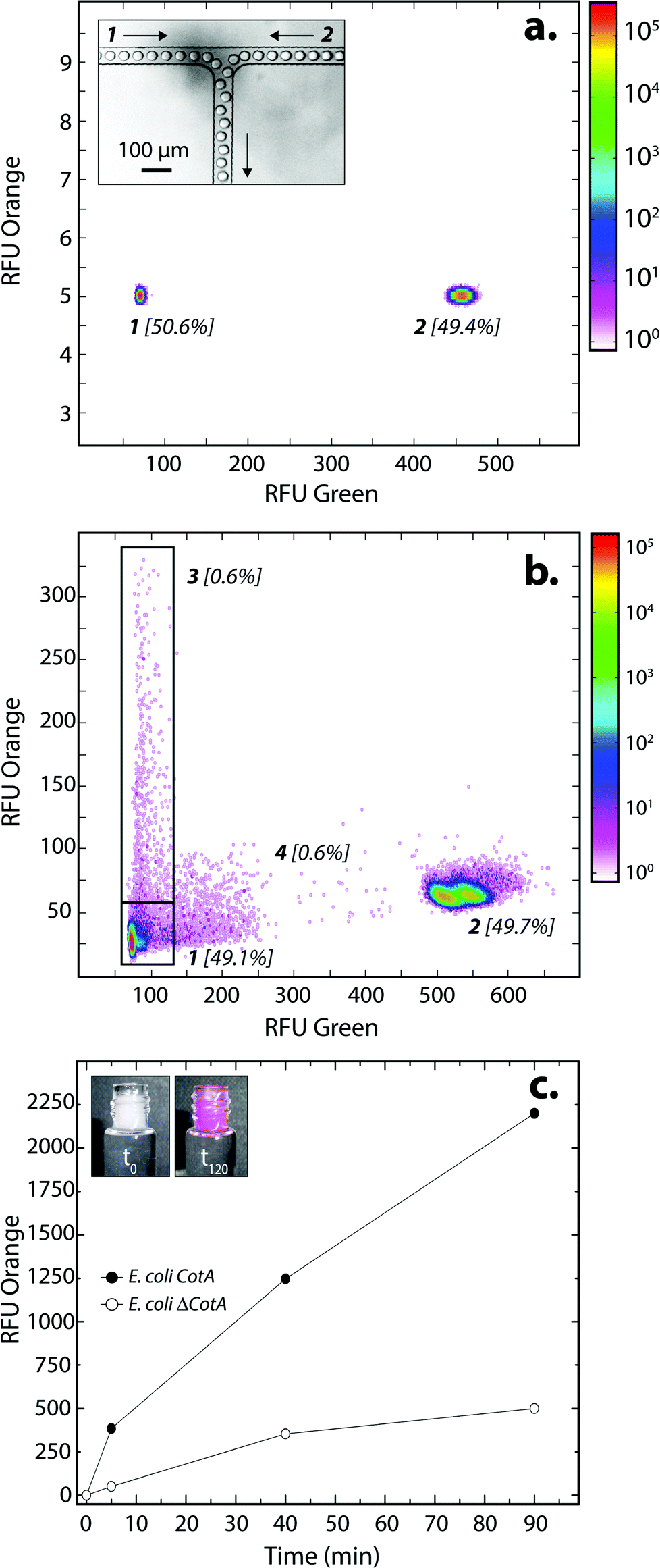 | ||
| Fig. 2 E. coli CotA vs. E. coli ΔCotA laccase activity in droplets. (a) 2D contour plot representing the green fluorescence (corresponding to barcode) and the orange fluorescence (corresponding to the CotA laccase activity) of two populations of 14 pL droplets containing LB inductive medium (LB medium + 0.1 mg mL−1 ampicillin + 1 mM IPTG + 0.25 mM CuCl2) after incubation for 15 h at 30 °C. Population 1 contained E. coli CotA and 1 μM of dextran-fluorescein and population 2 contained E. coli ΔCotA and 5 μM of dextran-fluorescein. The inset shows an image of the dual-dropmaker during encapsulation, focalizing on the area where the two emulsions combined. (b) 2D contour plot representing the green fluorescence and the orange fluorescence of the droplets after picoinjection of the AUR substrate and 10 min of incubation at 25 °C. Two new populations were detected: population 3 showing CotA laccase activity, and a small population 4 due to droplet coalescence. (c) Evolution of the average fluorescent signal of E. coli CotA (dark) and E. coli ΔCotA (white) droplets as a function of incubation time after picoinjection. The inset shows the emulsion in the collection vial after picoinjection of the AUR substrate and 0 (t0) or 120 min (t120) of incubation at 25 °C. | ||
The evolution of the fluorescent signal of the two populations over incubation time was evaluated by reloading the emulsion after 0, 5, 40 and 90 min incubation after picoinjection (Fig. 2c). E. coli CotA active droplets showed a significant increase of the fluorescent signal over time while E. coli ΔCotA droplets showed no activity with a slight increase of the fluorescent signal. This slight increase of the fluorescent signal from the inactive ΔCotA droplets would be due to the leakage of the enzymatic reaction product, a resorufin analogue, from the active droplets mediated by the surfactant.37 The activity ratio E. Coli CotA:E. Coli ΔCotA increased to reach 8 at 10 min and then decreased to remain constant (∼3) while equilibrium between enzymatic kinetics and leakage kinetics would be reached. According to those results, the model selection was validated using an incubation time of 10 min prior to sorting.
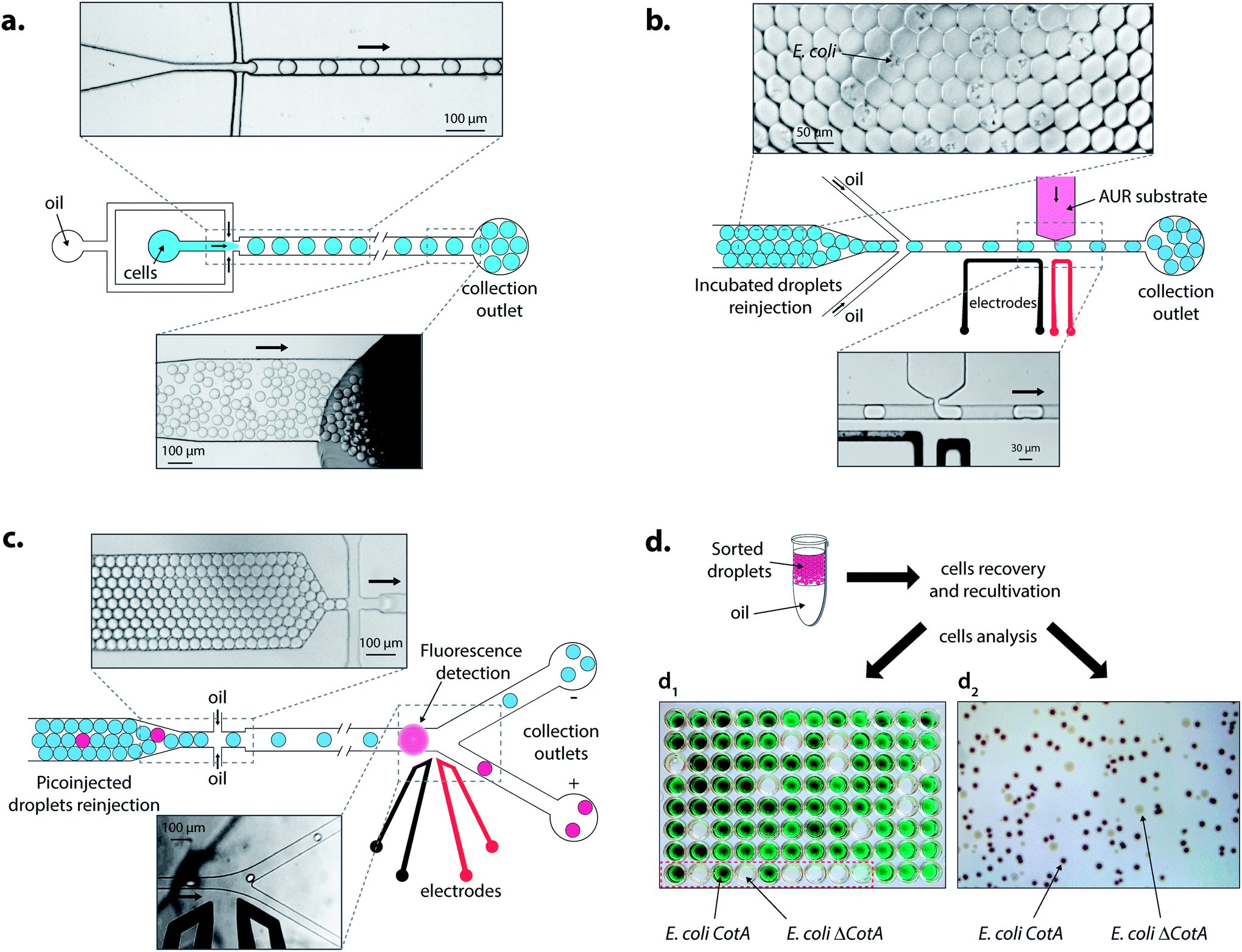 | ||
| Fig. 3 Description of the droplet-based microfluidic screening platform. (a) Cell encapsulation: a mixture of E. coli CotA–E. coli ΔCotA cells in LBind medium was compartmentalized in 14 pL droplets at 0.1 cell per droplet using HFE-7500 fluorinated oil containing 1% (w/w) KryJeffa900 surfactant. Once collected, the emulsion was incubated for 15 h at 30 °C for cell growth and protein expression. (b) Picoinjection of the substrate: incubated droplets were reloaded and spaced using HFE-7500 fluorinated oil containing 1% (w/w) KryJeffa900 surfactant. AUR substrate in LB medium was injected each time a droplet passed in front of the picoinjection head by application of an AC field (20 kHz, 200 V). The emulsion was collected in ice. (c) Fluorescence-activated droplet sorting: after 10 min of incubation at 25 °C, the picoinjected droplets were reloaded and spaced using HFE-7500 fluorinated oil. Fluorescent-activated AC field pulses (30 kHz, 1400 V, 0.5 ms) were used to deflect fluorescent droplets in the positive thinner collection outlet. (d) Cell recovery and characterization: sorted droplets were placed in 10 mL of LB medium containing 0.1 mg mL−1 of ampicillin and incubated for 15 h at 37 °C. Two techniques were then used: (d1) cells were plated on LB agar medium plate. After growth, colonies were then cultivated separately in a microtitre plate. Protein expression was induced and the ABTS substrate was added. Wells showing CotA laccase activity became green while others stayed colourless. Wells in the red square are controls with 3 E. coli CotA (green) and 3 E. coli ΔCotA (colourless) and then 3 without inoculation (colourless), and (d2) cells were directly plated on a LB agar medium plate containing IPTG and ABTS substrates. After growth and coloration, colonies showing CotA laccase activity became dark while others stayed colourless. | ||
Droplets were reloaded in the picoinjection module (Fig. 3b) where the AUR fluorogenic substrate was picoinjected in every droplet (∼1 pL). After 10 min of incubation at 25 °C, the droplets were reloaded in a FADS module for sorting based on the enzymatic activity according to the fluorescence intensity (Fig. 3c). The active droplet population, defined with a fluorescence sorting threshold, was sorted at a rate of 400 Hz and recovered. The sorted cells were grown and characterized to determine sorting efficiencies (Fig. 3d). Two colorimetric assays were developed to identify the sorted cells. In both cases, cells were first grown in LB medium. Then, cells were either recultivated separately in 96-deep well microtitre plates where the colorimetric assay was then performed (Fig. 3d1) or directly plated on colorimetric inductive agar medium (Fig. 3d2). The second identification method was more suitable for analyzing a large number of colonies than the first one, which was limited to 87 colonies and 9 controls per microtitre plate.
In a previous study, the fluorescence-activated droplet sorting module (FADS) has been reported to perform with a low error rate within a specific operating range.17 In that case, the main factor limiting the sorting enrichment is the co-encapsulation events of E. coli ΔCotA cells with E. coli CotA cells. A model for theoretical enrichment was previously described based on the Poisson distribution during encapsulation.17 The theoretical enrichment (ηt) is then given by the equation:
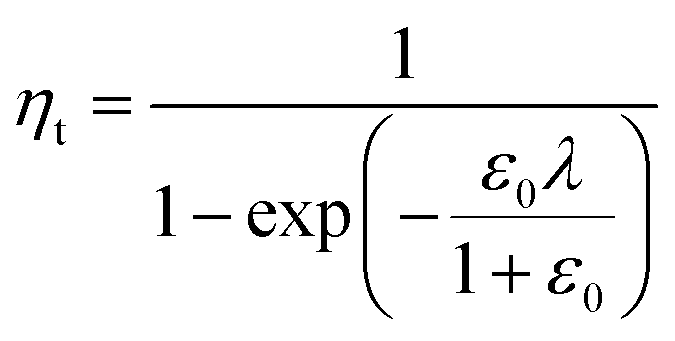
| λ = −ln(1 − k) |
where k ≤ 1 is the fraction of droplets occupied by colonies. A series of sorting was performed at a rate of 400 Hz using different ε0 and λ values to demonstrate that our screening platform followed this model. The results are shown in the table of Fig. 4. For each emulsion, the CotA active droplet population, defined by the red box sorting threshold, was sorted (Fig. 4a1). This sorting threshold was defined with reference to the average fluorescent signal of the inactive droplet population: in the case of a sorting threshold of 8, droplets showing a fluorescence signal 8 times higher than the average fluorescent signal of inactive droplet population were sorted. ε before sorting (ε0) and after sorting (ε1) were determined using the colorimetric assays we developed based on the CotA laccase activity (Fig. 4b). Experimental enrichments ηexp were calculated and plotted against the theoretical model (Fig. 4a2).
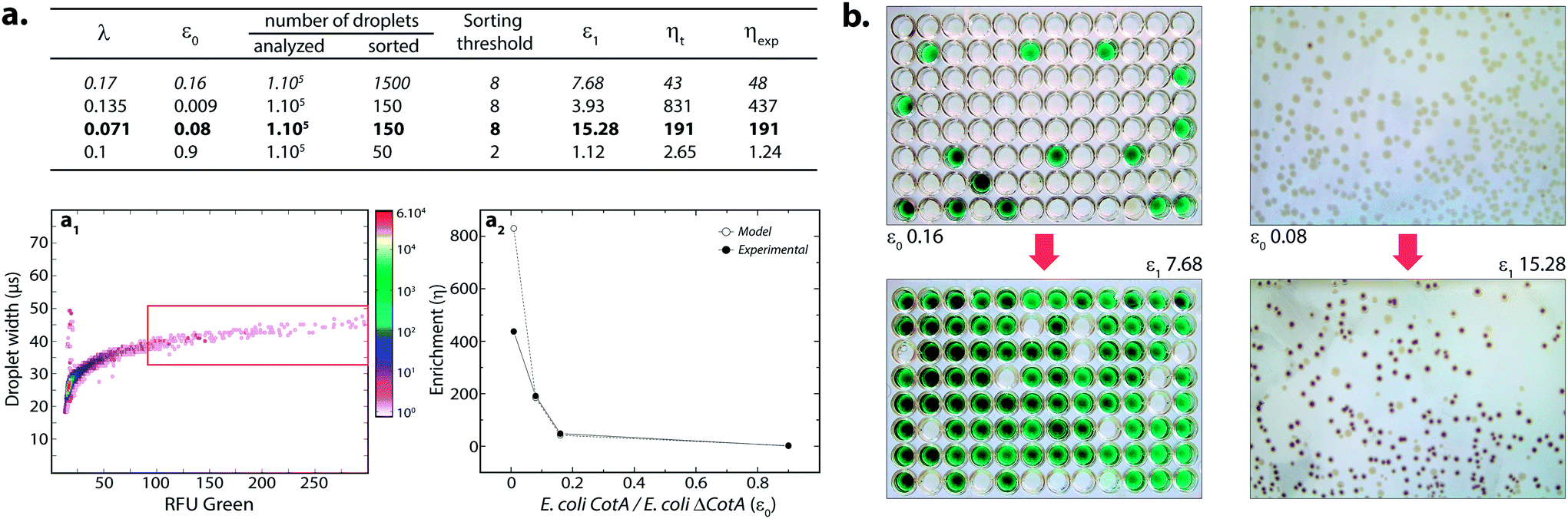 | ||
| Fig. 4 Model selection of the CotA laccase activity. (a) Table summarizing the results for different sorts: λ, ε0, ε1, theoretical (ηt) and experimental (ηexp) enrichments. (a1) 2D contour plot representing the width of the droplets (transit time of the droplet passing the detector; μs) and the orange fluorescence (corresponding to the CotA laccase activity) of 14 pL LBind droplets after compartmentalization of a mixture of E. coli CotA/E. coli ΔCotA, incubation for 15 h at 30 °C, picoinjection of the AUR fluorogenic substrate and incubation for 10 min at 25 °C. Population defined in the red box sorting threshold was sorted. Cell mixtures before and after sorting were analyzed to compare the initial (ε0) and final (ε1) ratios of E. coli CotA to E. coli ΔCotA cells. (a2) Evolution of sorting enrichment as a function of ε0 for the theoretical model (white dots) and experimental data (black dots). (b) Picture of the colorimetric assays performed to determine ε ratios before (ε0) and after sorting (ε1). Microtitre plates on the left refer to data in italic in the table while Petri dishes on the right refer to data in bold in the table. | ||
As expected, experimental data were closely fitting the theoretical model: ηexp was observed to increase decreasing ε or decreasing λ. Sorting at very low ε (0.009) leads to high enrichment. ηexp is only half ηt in that case, which can be explained by the lower sorting threshold (2) implying that more false positive droplets were caught. Those results highlighted the fact that this screening technique offers two operation modes. Working at high λ implies more co-encapsulation events (26% for λ = 1) but higher throughput (250 cells per s for λ = 1) and is suitable for fast enrichment of a library before further fine selection steps. Working with low λ (λ < 0.2) implies less co-encapsulation events (1.8%) and lower throughput (70 cells per s) and is suitable for sorting cells to high purities for fine selection steps.
The major advantage of this droplet-based microfluidic platform relies on the flexibility brought by the injection of the substrate at a defined time. This allows us to adapt the incubation time prior to sorting to the kinetics of the enzymatic reaction and therefore makes the selection occur in the linear range of the kinetics.
3.2 Analysis mode
The droplet-based platform can also operate in analysis mode in which the droplet fluorescence is measured in a reloading module without any sorting. In that case, as FADS is the limiting module in terms of throughput, the analysis of droplets can be performed at very high rates up to 2000 Hz. We constructed a library of 106 variants using error-prone PCR. Amplified fragment was purified using biotin–streptavidin binding and magnetic beads and then cloned into an acceptor vector constructed from pET-21a(+). The library characteristics were evaluated by sequencing twenty variants. It showed, on average, 9–10 mutations per gene that is roughly 2–4 amino acid mutations per variant.The enzymatic activity distribution of this library was analyzed using CotA wild type as an internal reference: E. coli CotA and the library cell suspensions were separately and simultaneously encapsulated (λ = 0.2) using the dual-dropmaker module. The binary emulsion was incubated for 15 h at 30 °C and then reloaded into the picoinjection device for fluorogenic substrate injection. After 10 min of incubation at 25 °C, the enzymatic activity was measured at 1000 droplets per s in a reloading module (Fig. 5). A green fluorescent barcode was used to tag the two populations: the CotA internal reference (1 [52%]) and the library (2 [45%]). The encapsulation rate was measured to be 15% of occupied droplets and 85% of empty one. For each of the sub-population, the active population was defined using a 250 RFU threshold (red line in Fig. 5): the wild type reference with an average fluorescence signal of 625 RFU (11%) and the library with an average fluorescence signal of 425 RFU (0.25%). This high-throughput analysis (75 variants per s) showed that the library contained approximately 2.5% of active mutants or unmutated CotA. As the mutation rate is rather high in this library, it is worth considering that the proportion of unmodified wild type is quite low. Those 2.5% active variants were on average 1.5 times less active than the wild type CotA. This activity distribution within the library is not surprising as the mutation rate is rather high. This platform allows analysis of libraries using high throughputs. In this example, 150 occupied droplets are analyzed in one second, half of them referred to the wild type and the other half to the library. In that case, 75 variants are analyzed per second, 106 in less than 4 hours. It is possible to work at higher λ values that will increase the throughput: λ = 0.5 allows analysis of 106 cells in 1.5 hours with only 9% of co-encapsulation events. Moreover, the internal wild type reference could be used in a smaller ratio than 1:1 with the library, which would increase the throughput of variant analysis. It is then also possible to increase the analysis rate up to 2000 Hz. In that case, with an internal reference–library 1:9 ratio, 106 cells are analyzed in less than 1 h (λ = 0.2) or in 20 min (λ = 0.5). Additional gain in analytical throughput might be obtained by parallelization.38 The efficiency and the throughput could also be theoretically improved by organizing the cells during encapsulation to avoid the Poisson distribution and screen the fully loaded emulsion with single cells per droplet.39
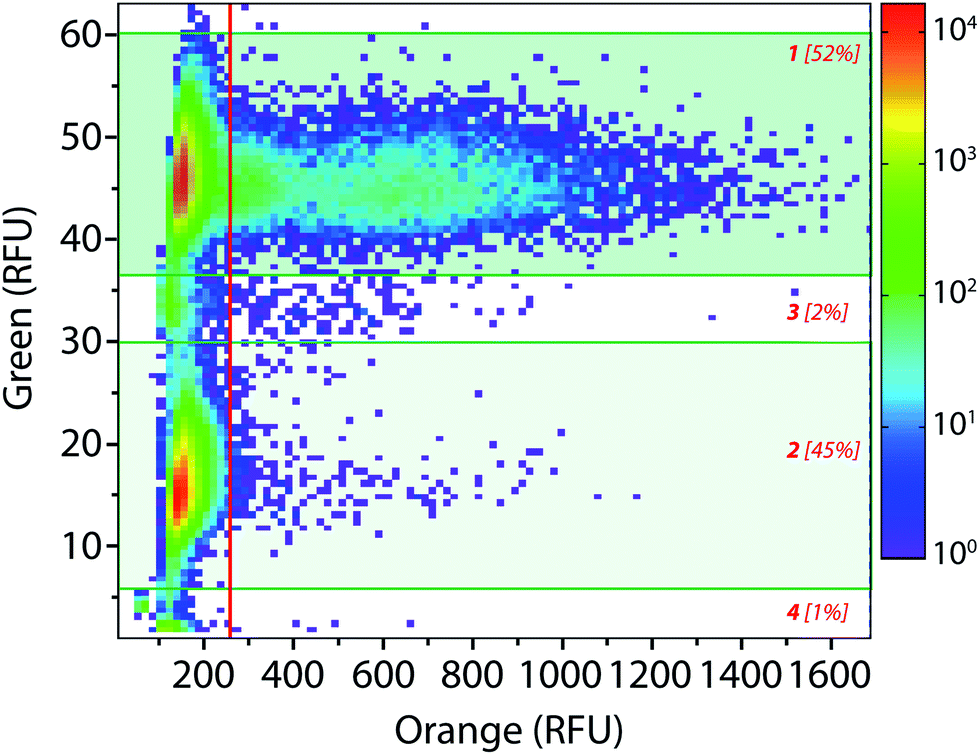 | ||
| Fig. 5 High-throughput library analysis: 2D contour plot representing the green fluorescence (barcode) and the orange fluorescence (enzymatic activity) of a wild type vs. library binary emulsion after picoinjection of the AUR substrate and 10 min of incubation at 25 °C. Population [1] refers to wild type CotA internal reference droplets (10 μM dextran-fluorescein barcode) and population [2] refers to the library droplets (2 μM dextran-fluorescein barcode). Populations [3] and [4] correspond respectively to coalesced droplets and unpicoinjected droplets. | ||
This high-throughput analysis droplet-based microfluidic platform offers a precious tool for directed evolution experiments based on the enzymatic activity. It allows the possibility to rapidly study the activity distribution of large libraries with a tremendous saving of time and reagents. Using this new technology, it is possible to follow the distribution of the activity after each round of mutagenesis or selection. That could be very helpful for choosing mutagenesis methods and directed evolution strategies to follow a desired evolution path and avoid dead ends.
4. Conclusions
We have demonstrated a flexible and efficient droplet-based screening technique able to analyze and sort cells based on a recombinant enzymatic activity. The main merit of this screening platform is the injection of the fluorogenic substrate at a defined time, which offers the possibility to tune the incubation time prior to selection depending on the kinetics of the enzymatic reaction. This high-throughput droplet-based microfluidic platform for directed evolution experiments is used to: (i) evaluate the distribution of the enzymatic activity within a large library at high-throughput (analysis mode; up to 106 variants in 1 h), (ii) rapidly enrich a library in active variants in a high-throughput rate (enrichment mode; up to 106 variants in 1 h), and (iii) select finely variants depending on their enzymatic activity (selection mode; up to 106 variants in 4 h).The screening platform is flexible and could be applied to many kinds of recombinant systems implying ideally the secretion of the enzyme or the display of the enzyme at the cell surface, but also, as in the case of CotA laccase, periplasmic or cytoplasmic enzymes (the lysis of part of the cells within droplets would be required for the substrate to access the enzyme). Moreover, the incubation time prior to sorting could be adapted according to the enzyme kinetics. This platform also gives access to a wide range of controlled selection pressures (pH, temperature, …) as the growth of cells and the enzymatic assay are two distinct steps.
It is worth noting that incubating droplets off-chip for such an enzymatic reaction time scale (0–60 min) introduces a small heterogeneity within the incubation time of each droplet. We are developing an integrated device that can perform picoinjection, incubation on-chip and sorting to attempt higher degrees of control of the time and temperature of incubation. For the longer enzymatic reaction time scale (>60 min), incubation off-chip is necessary and implies that the product of the enzymatic reaction is not leaking out of droplets (which could be overcome by chemical modification of the substrate to make the product more hydrophilic40). Such developments should lead to very robust and universal high-throughput screening techniques for directed evolution.
Acknowledgements
T.B. thanks the ADEME (Agence de Développement de l'Environnement et la Maîtrise de l'Energie) and the Region Alsace for financial support. This work was also supported by the Ministère de l'Enseignement Supérieur et de la Recherche and the Centre National de la Recherche Scientifique (CNRS). F.C. was supported by FP-7 FET OPEN Project E-Flux. The authors would like to thank Jean-François Bartolo for surfactant synthesis, Chaouki Ben Salem for technical assistance and Antoine Drevelle for useful scientific discussions.Notes and references
- S. Bershtein and D. S. Tawfik, Curr. Opin. Chem. Biol., 2008, 12, 151 CrossRef CAS PubMed.
- J. D. Keasling, ACS Chem. Biol., 2008, 3, 64 CrossRef CAS PubMed.
- S. G. Peisajovich and D. S. Tawfik, Nat. Methods, 2007, 4, 991 CrossRef CAS PubMed.
- L. M. Mayr and P. Fuerst, J. Biomol. Screening, 2008, 13, 443 CrossRef CAS PubMed.
- T. Thorsen, S. J. Maerkl and S. R. Quake, Science, 2002, 298, 580 CrossRef CAS PubMed.
- J. Lay, R. Liyanage, S. Borgmann and C. L. Wilkins, TrAC, Trends Anal. Chem., 2006, 25, 1046 CrossRef CAS PubMed.
- D. S. Tawfik and A. D. Griffiths, Nat. Biotechnol., 1998, 16, 652 CrossRef CAS PubMed.
- A. D. Griffiths and D. S. Tawfik, Trends Biotechnol., 2006, 24, 395 CrossRef CAS PubMed.
- R. Seemann, M. Brinkmann, T. Pfohl and S. Herminghaus, Rep. Prog. Phys., 2012, 75, 016601 CrossRef PubMed.
- L. Mazutis, J.-C. Baret and A. D. Griffiths, Lab Chip, 2009, 9, 2665 RSC.
- K. Ahn, J. Agresti, H. Chong, M. Marquez and D. Weitz, Appl. Phys. Lett., 2006, 88, 264105 CrossRef PubMed.
- L. Frenz, A. El Harrak, M. Pauly, S. Bégin-Colin, A. D. Griffiths and J.-C. Baret, Angew. Chem., Int. Ed., 2008, 47, 6817 CrossRef CAS PubMed.
- A. R. Abate, T. Hung, P. Mary, J. J. Agresti and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19163 CrossRef CAS PubMed.
- S. L. Sjostrom, H. N. Joensson and H. Andersson Svahn, Lab Chip, 2013, 13, 1754 RSC.
- L. Frenz, K. Blank, E. Brouzes and A. D. Griffiths, Lab Chip, 2009, 9, 1344 RSC.
- D. Pekin, Y. Skhiri, J.-C. Baret, D. Le Corre, L. Mazutis, C. Ben Salem, F. Millot, A. El Harrak, J. B. Hutchison, J. W. Larson, D. R. Link, P. Laurent-Puig, A. D. Griffiths and V. Taly, Lab Chip, 2011, 11, 2156 RSC.
- J.-C. Baret, O. J. Miller, V. Taly, M. Ryckelynck, A. El Harrak, L. Frenz, C. Rick, M. L. Samuels, J. B. Hutchison, J. J. Agresti, D. R. Link, D. A. Weitz and A. D. Griffiths, Lab Chip, 2009, 9, 1850 RSC.
- M. T. Guo, A. Rotem, J. A. Heyman and D. A. Weitz, Lab Chip, 2012, 12, 2146 RSC.
- A. Fallah-Araghi, J.-C. Baret, M. Ryckelynck and A. D. Griffiths, Lab Chip, 2012, 12, 882 RSC.
- S. L. Sjostrom, Y. Bai, M. Huang, Z. Liu, J. Nielsen, H. N. Joensson and H. Andersson Svahn, Lab Chip, 2014, 14, 806 RSC.
- J. J. Agresti, E. Antipov, A. R. Abate, K. Ahn, A. C. Rowat, J.-C. Baret, M. Marquez, A. M. Klibanov, A. D. Griffiths and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4004 CrossRef CAS PubMed.
- B. Kintses, C. Hein, M. F. Mohamed, M. Fischlechner, F. Courtois, C. Lainé and F. Hollfelder, Chem. Biol., 2012, 19, 1001 CrossRef CAS PubMed.
- F. J. Enguita, L. O. Martins, A. O. Henriques and M. A. Carrondo, J. Biol. Chem., 2003, 278, 19416 CrossRef CAS PubMed.
- M. Fernández-Fernández, M. Ángeles Sanromán and D. Moldes, Biotechnol. Adv., 2013, 31, 1808 CrossRef PubMed.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974 CrossRef CAS PubMed.
- A. C. Siegel, D. A. Bruzewicz, D. B. Weibel and G. M. Whitesides, Adv. Mater., 2007, 19, 727 CrossRef CAS.
- C. Holtze, A. C. Rowat, J. J. Agresti, J. B. Hutchison, F. E. Angile, C. H. J. Schmitz, S. Koester, H. Duan, K. J. Humphry, R. A. Scanga, J. S. Johnson, D. Pisignano and D. A. Weitz, Lab Chip, 2008, 8, 1632 RSC.
- B. Miroux and J. E. Walker, J. Mol. Biol., 1996, 260, 289 CrossRef CAS PubMed.
- M. F. Hullo, I. Moszer, A. Danchin and I. Martin-Verstraete, J. Bacteriol., 2001, 183, 5426 CrossRef CAS.
- L. O. Martins, C. M. Soares, M. M. Pereira, M. Teixera, T. Costa, G. H. Jones and A. O. Henriques, J. Biol. Chem., 2002, 277, 18859 CrossRef PubMed.
- A. Zebda, C. Gondran, A. Le Goff, M. Holzinger, P. Cinquin and S. Cosnier, Nat. Commun., 2011, 2, 370 CrossRef PubMed.
- F. Durand, C. Stimes-Chaumiel, V. Flexer, I. André and N. Mano, Biochem. Biophys. Res. Commun., 2010, 402, 750 CrossRef CAS PubMed.
- T. Beneyton, A. El Harrak, A. D. Griffiths, P. Hellwig and V. Taly, Electrochem. Commun., 2011, 13, 24 CrossRef CAS PubMed.
- T. Beneyton, I. Putu Mahendra Wijaya, C. Ben Salem, A. D. Griffiths and V. Taly, Chem. Commun., 2013, 49, 1094 RSC.
- P. Durao, Z. Chen, C. S. Silva, C. M. Soares, M. M. Pereira, S. Todorovic, P. Hildebrandt, I. Bento, P. F. Lindley and L. O. Martins, Biochem. J., 2008, 412, 339 CrossRef CAS PubMed.
- J.-C. Baret, F. Kleinschmidt, A. El Harrak and A. D. Griffiths, Langmuir, 2009, 25, 6088 CrossRef CAS PubMed.
- T. Chen, A. Wijaya Gani and S. K. Y. Tang, Lab Chip, 2012, 12, 5093 RSC.
- J. Lim, J. Vrignon, P. Gruner, C. S. Karamitros, M. Konrad and J.-C. Baret, Appl. Phys. Lett., 2013, 203704 CrossRef PubMed.
- J. F. Edd, D. Di Carlo, K. J. Humphry, S. Köster, D. Irimia, D. A. Weitz and M. Toner, Lab Chip, 2008, 8, 1262 RSC.
- M. Najah, E. Mayot, I. Putu Mahendra Wijaya, A. D. Griffiths, S. Ladame and A. Drevelle, Anal. Chem., 2013, 85, 9807 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: One picoinjection movie, scheme of the LabView program and a detailed description of microfluidic modules. See DOI: 10.1039/c4an00228h |
| This journal is © The Royal Society of Chemistry 2014 |