Rhodium catalysed hydroformylation of alkenes using highly fluorophilic phosphines
Dave J.
Adams
a,
James A.
Bennett
a,
David J.
Cole-Hamilton
b,
Eric G.
Hope
*a,
Jonathan
Hopewell
a,
Jo
Kight
a,
Peter
Pogorzelec
b and
Alison M.
Stuart
a
aDepartment of Chemistry, University of Leicester, Leicester, UK LE1 7RH. E-mail: egh1@le.ac.uk; Fax: + 44 116 252 3789
bCatalyst Evaluation and Optimisation Service (CATS), School of Chemistry, University of St. Andrews, St. Andrews, UK KY16 9ST
First published on 14th October 2005
Abstract
Highly fluorophilic phosphines incorporating at least one aromatic ring containing two directly attached perfluoroalkyl groups have been synthesised, their partition coefficients (organic phase : fluorous phase) measured and their electronic properties probed using 1JPtP data for their trans-[PtCl2L2] complexes. These phosphines have been used as modifying ligands for the rhodium catalysed hydroformylation of 1-octene in perfluorocarbon solvents. Catalyst activity, regioselectivity and the levels of rhodium leaching to the product phase vary with the substitution patterns of the modifying ligands that do not correlate with the electronic properties or partition coefficients of these ligands, but can be interpreted in terms of differences in the resting states of the catalysts.
Introduction
The formation of long chain linear aldehydes by hydroformylation using cobalt-based catalysts is a very important industrial process for the production of plasticisers, soaps and detergents.1 Although rhodium-based catalysts give higher selectivities and reactivities in the laboratory than those used in the industrial systems, they have not been employed industrially because they decompose as the high boiling products are distilled away from the reaction medium. Consequently, there is considerable interest in developing alternative methodologies for catalyst–product separation for this process. Horváth and Rábai have shown that fluorous biphasic systems can be highly successful in the separation of catalyst from products post-reaction,2 and our work has been developed using triarylphosphines and by working in neat perfluorocarbon solvents.3 We have recently commissioned a reactor in which the hydroformylation of alkenes under fluorous biphase conditions can be carried out over prolonged periods under continuous conditions with full recycling of the fluorous phase.4The critical factor in designing an efficient catalyst for application in fluorous biphasic systems is the high solubility of the catalyst in the perfluorinated solvent.5 In a rhodium-catalysed hydroformylation system, the solubility is conferred on the rhodium by the addition of perfluoroalkyl ‘ponytails’ to the modifying phosphine ligands and, to date, only monodentate phosphines with, at most, three ponytails have been evaluated for hydroformylation in fluorous solvents.2–4,6 However, whilst the incorporation of further ponytails is expected to increase the solubility of the phosphine and, hence, the catalyst in the perfluorinated solvents offering better recovery and recyclability under fluorous biphase conditions, there are few examples of phosphines with more than three ponytails. Those reported in the literature include perfluoroalkylated analogues of dppe,7,8 a ligand scaffold that affords poor regioselectivity in the hydroformylation of alkenes, ligands incorporating a silyl spacer group,8 which allows up to three ponytails per ring and, most recently, Sinou et al. have described a phosphine with two ponytails per ring incorporating OCH2 spacer groups,9 which has also been shown to offer poor regioselectivity in the hydroformylation of alkenes.6 We have been investigating phosphines incorporating perfluoroalkyl groups directly attached to the aromatic ring i.e. with no additional spacer group in order to exploit the high electron-withdrawing ability of the perfluoroalkyl groups which has been shown to be beneficial in hydroformylation reactions.3 Such phosphines also have the advantage that they are highly fluorophilic {e.g. P(C6H4-4-C6F13)3 has a partition coefficient of 85 : 15 {perfluorodimethylcyclohexane(PP3) : toluene} compared to 47 : 53 for P(C6H4-4-CH2CH2C6F13)3}. In an attempt to improve the retention of the fluorous-derivatised rhodium hydroformylation catalysts in the fluorous phase, here we report the preparation of a series of phosphines having two directly attached perfluoroalkyl groups per aromatic ring and their application in the rhodium-catalysed hydroformylation of 1-octene in perfluorocarbon solvents.
Results and discussion
Bromobenzenes such as 1 (Scheme 1) have been reported by a number of authors.10 In general, these have been prepared by the copper-mediated coupling reaction of a perfluoroalkyl iodide with a 1,3-dihalobenzene, followed by bromination using NBS. For the C6F13 analogue 1a, van Koten et al. found that the separation of the bromobenzene and unreacted starting material was very difficult and they were unable to purify this material completely.10a However, Pozzi et al. have described the successful preparation of the C8F17 analogue 1c by the reaction of 1,3,5-tribromobenzene with two equivalents of pentadecafluoro-n-octyl iodide,11 and we have found that this method allows the formation of analytically pure 1a and 1c in moderate yields.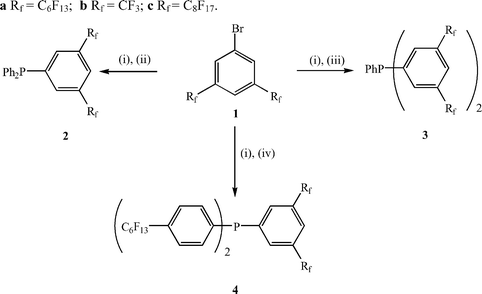 | ||
| Scheme 1 (i) n-BuLi, ether; (ii) Ph2PCl; (iii) PhPCl2; (iv) ClP(C6H4-4-C6F13)2. (a) Rf = C6F13; (b) Rf = CF3; (c) Rf = C8F17. | ||
Formation of a range of perfluoroalkylated phosphines was achieved by lithiation of 1a or 1b {3,5-bis(trifluoromethyl)bromobenzene} at low temperature with n-BuLi followed by quenching with either diphenylchlorophosphine, dichlorophenylphosphine or bis(4-tridecafluoro-n-hexylphenyl)chlorophosphine (Scheme 1).12 In all cases, the reaction was complicated by the low solubility of 1a at these low temperatures, which necessitated the use of a very high dilution. Nonetheless, this methodology has allowed the formation of one phosphine with two perfluoroalkyl groups on the same aromatic ring, 2a, and two phosphines with four perfluoroalkyl groups directly attached to the aromatic rings in two different ways, 3a and 4a. Unfortunately, all attempts at reaction of the aryl lithiate of 1a with PCl3 failed to give the desired tris-derivatised phosphine with six ponytails. In contrast, the straightforward synthesis of P{C6H3-3,5-(CF3)2}3 with six trifluoromethyl groups suggests that this is a solubility or steric issue rather than an electronic one.13 The even lower solubility of 1c at low temperatures actually precluded all of our attempts to prepare phosphine ligands containing C6H3-3,5-(C8F17)2 groups using this methodology. Reaction of 1c with n-BuLi in ether, ether–hexane or ether–PP2 mixtures between −80 °C and room temperature only resulted in the virtual quantitative recovery of the starting aryl bromide. Further experiments indicated that, at low temperatures (−80 to −60 °C), no reaction of 1c with n-BuLi occurs such that, on addition of P–Cl reagents, only butyl-derivatised phosphines are formed. In contrast, at higher temperatures (−60 to 0 °C), lithiation at the 4-position occurs preferentially over lithium-bromine exchange and the resulting aryl lithiate is too sterically crowded to react with any P–Cl reagent. Consequently, on work-up, the starting material is regenerated or, following the addition of D2O, the analogous deuteride can be isolated (Scheme 2). An alternative strategy can be employed for the synthesis of phosphine ligands containing C6H3-3,5-(C8F17)2 groups (Scheme 3) involving the copper-mediated cross coupling reaction of the perfluoroalkyliodide with an appropriately substituted bromoarylphosphine oxide followed by reduction.14 However, our observations on the influence of 3,5-disubstitution on the regioselectivity and metal leaching levels in the rhodium-catalysed hydroformylation of 1-octene in perfluorocarbon solvents (vide infra) indicated that this substitution pattern was unlikely to be beneficial for this catalytic process, and we restricted work on this strategy to proof of principle with the synthesis and characterisation of 2c.
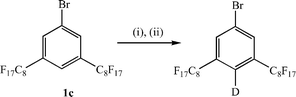 | ||
| Scheme 2 (i) n-BuLi, ether, −60 °C; (ii) D2O. | ||
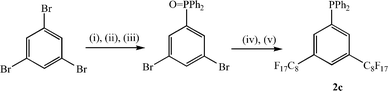 | ||
| Scheme 3 (i) n-BuLi, ether; (ii) Ph2PCl; (iii) H2O2; (iv) C8F17I, Cu, bipy, DMSO, C6H5F, 100 °C; (v) HSiCl3, ether. | ||
The partition coefficients (Table 1) show that these methodologies can provide phosphines with extremely high affinities for the perfluorinated solvents, with 4a having the highest reported partition coefficient for any triarylphosphine, and rivalling those for the trialkylphosphines.5 It is clear, however, that the positioning of the fluorous ponytails on the triarylphosphines is extremely important and that the presence of the underivatised phenyl ring in 3a has a negative impact on its partition coefficient, i.e. although 3a and 4a both have the same percentage fluorine (w/w) and the same number of ponytails, 4a has a much superior partition coefficient than 3a. In addition, the data for 3b and 4b illustrate that, even with up to 59% w/w fluorine, the CF3 groups are not sufficiently fluorophilic to confer high solubility in perfluorocarbon solvents, limiting the applications of complexes incorporating these ligands for catalysis under fluorous biphase conditions (vide infra, Table 2).
| Phosphine | % Fluorine (w/w) | % Tol : % PP3a | 1 J PPt/Hzb |
|---|---|---|---|
| a Determined gravimetrically. b Coupling constant for trans-[PtCl2L2] dissolved in CDCl3 at 25 °C. c Data taken from ref. 20. d Data taken from ref. 15. | |||
| PPh3 | 0 | 100 : 0 | 2635c |
| 2a | 55 | 39 : 61 | 2692 |
| 2c | 59 | 24 : 76 | 2695 |
| P(C6H4-4-C6F13)3 | 61 | 15 : 85 | 2719d |
| 3a | 64 | 10 : 90 | 2741 |
| 4a | 64 | 1 : 99 | 2743 |
| 3b | 43 | 88 : 12 | 2739 |
| 4b | 59 | 48 : 52 | 2741 |
| Phosphine | Conv.b (%) | Octanec (%) | Isomc (%) | Branchedc (%) | n-Nonanalc (%) | l : bd | Rate constante (s−1) | Rh lossf |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: solvent perfluoromethylcyclohexane (PP2; 4 ml); temperature 70 °C; catalyst generated in situ from [Rh(CO)2(acac)] and ligand, Rh : P ratio 1 : 10; time 90 min. b Conversion (100%−% residual 1-octene). c Percentage of product by mole fraction. Isomerised products = sum (2-, 3- and 4-octene), generally > 95% 2-octene; Branched products = sum (2-propylhexanal + 2-ethylheptanal + 2-methyloctanal), generally > 98% 2-methyloctanal. d l : b = ratio of linear to all branched aldehyde products. e All reactions were found to be 1st order in [1-octene] to over 80% 1-octene consumption. f Rhodium loss to the organic product as ppm by weight. nd = not determined; (organic phase deeply coloured due to the presence of rhodium and, consequently, outside the range of the ICP-MS). g Data taken from ref. 3. | ||||||||
| P(C6H4-4-C6F13)3g | 97.8 | 0.4 | 3.7 | 12.8 | 80.9 | 6.3 | 1.9 | 7292 |
| 2a | 95.9 | 0.3 | 2.0 | 17.3 | 76.4 | 4.4 | 1.9 | nd |
| 3a | 98.9 | 1.0 | 3.3 | 20.1 | 74.6 | 3.9 | 1.2 | 225700 |
| 4a | 98.5 | 0.4 | 3.3 | 16.9 | 77.9 | 4.7 | 2.5 | 7486 |
| 3b | 98.3 | 0.3 | 3.3 | 15.4 | 79.3 | 5.1 | 3.3 | nd |
| 4b | 97.3 | 0.4 | 3.7 | 14.5 | 78.7 | 5.4 | 3.7 | nd |
As expected, the perfluoroalkyl groups have a substantial effect on the electronic behaviour of the phosphine centres, and we have evaluated this effect using the magnitude of the 1JPPt coupling constants for trans-[PtCl2L2], (L = phosphine; Table 1), since the corresponding cis complexes could not be formed for 3a or 3b presumably due to steric constraints. This is consistent with our earlier work where we have shown that, although exclusively cis-[PtCl2L2] are obtained from the reactions of the mono- and bis-derivatised para-substituted ligands {PPh2(C6H4-4-C6F13) and PPh(C6H4-4-C6F13)2},15 inseparable mixtures of cis- and trans-isomers were obtained in the same reaction with P(C6H4-4-C6F13)3,15 PPh2(C6H4-3-C6F13) and PPh(C6H4-3-C6F13)216 and exclusively trans-[PtCl2L2] with P(C6H4-3-C6F13)3,16 as a consequence of steric congestion. In all cases, a cumulative increase in the magnitude of the coupling constant, in comparison to those for triphenylphosphine and P(C6H4-4-C6F13)3 was observed as the number of perfluoroalkyl substituents increased. However, as can be seen for the data for 3a and 4a, the electron-withdrawing effect is independent of the position of the perfluoroalkyl groups on the aryl rings. For comparison, we have also prepared analogues of 3a and 4a containing a number of CF3 groups, 3b and 4b respectively. These data imply that, electronically, these phosphines are almost identical to their C6F13 counterparts, confirming that the electronic influence of the perfluoroalkyl substituents is independent of chain length.
These phosphines have been investigated in the hydroformylation of 1-octene, Scheme 4 and Table 2, and the data compared directly with those for P(C6H4-4-C6F13)3.3,4 As can be seen, the bis-meta-perfluoroalkyl substitution results in catalysts which are less selective to the desired linear aldehyde than the para-substituted phosphines. However, it is clear that the selectivity of the catalyst is not simply related to the electronic environment of the phosphine since, despite the almost identical 1JPPt values for 3a and 4a, the rates of reaction in the rhodium catalysed hydroformylation of 1-octene using these ligands differ dramatically and the l : b ratios are also different. It is known that bulky phosphines can give rise to different mechanisms in the hydroformylation of alkenes, resulting in lower selectivity to the linear aldehyde,15 and it seems that 3a is sufficiently bulky for this to occur. At the same time, despite it having a slightly higher partition coefficient than that of P(C6H4-4-C6F13)3, significant leaching of rhodium into the product phase occurs for ligand 3a. Initially this may seem slightly surprising, but it could be accounted for in two ways. We have found that the identity of the perfluorinated solvent has a negligible effect when determining the partition coefficients, but the identity of the organic solvent is critical. Here, following catalysis, the catalyst partitions between perfluoromethylcyclohexane (PP2) and, mainly, nonanals, whilst the partition coefficients were measured in a toluene–PP3 biphase. Since nonanal is more polar than toluene, it might be expected that retention of the catalyst in the fluorous phase would actually be better and, certainly, should affect both phosphines similarily. Therefore, the influence of the ligand on catalyst leaching is more likely to be due to the resting state of the catalyst. Several species are known to exist under reaction conditions including [RhH(CO)(L)3], [RhH(CO)2(L)2] and [RhH(CO)3(L)] (L = phosphines),17 and it is likely that, due to the steric bulk of 3a, the resting state of the catalyst is one which includes a smaller number of phosphines than that derived from P(C6H4-4-C6F13)3. Hence, although 3a contains four perfluoroalkyl groups whilst P(C6H4-4-C6F13)3 has only three, it may well be that there is a significant difference in the number of ponytails present in the catalyst resting states. In addition, the presence of one phenyl ring with no ponytails could also influence the large amount of leaching observed in the case of 3a. For the catalyst generated with 4a, the impact of the increased steric requirement appears to be closely matched by the improved fluorophilicity of the additional perfluoroalkyl group to give rhodium leaching levels comparable to those for P(C6H4-4-C6F13)3. The data for the CF3-substituted ligands (3b and 4b) confirm that these ligands are not suitable for catalysis under fluorous biphase conditions and reinforce the implication that steric congestion is an issue in these reactions. Using 3b, a catalyst is generated with a significantly higher rate constant and greater selectivity to n-nonanal than that formed with 3a, which is mirrored in the catalysts formed with 4a and 4b. Once again, there is little difference in the electronic properties of the phosphines, as measured by the 1JPPt coupling constants, which implies that the differences arise from the larger steric bulk of the C6F13 analogues.
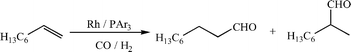 | ||
| Scheme 4 Rhodium-catalysed hydroformylation of 1-octene in perfluorocarbon solvents under 20 bar CO–H2 (1 : 1). | ||
Conclusions
We have successfully prepared highly fluorophilic triaryl phosphines that feature at least one aromatic ring derivatised with two directly attached perfluoroalkyl groups. The effect of the ponytails on the electronic properties of the phosphine has been found to be independent of the structure of the phosphine, but highly dependent on the number of ponytails incorporated. These fluorophilic phosphines have been used successfully for the hydroformylation of 1-octene in fluorocarbon solvents, for which the catalytic data are highly dependent on the structure of the phosphine ligand, in particular the length and substitution patterns of the perfluoroalkyl groups, which can be interpreted in terms of the resting state of the metal catalyst. Further spectroscopic work on these catalytic systems is in progress and may shed further light on this interpretation.Experimental
General remarks
1H, 19F and 31P NMR spectroscopies were carried out on a Bruker ARX250 spectrometer at 250.13, 235.34 and 101.26 MHz or a Bruker DPX300 spectrometer at 300.14, 282.41 and 121.50 MHz respectively and were referenced to external SiMe4 (1H), external CFCl3 (19F) and external H3PO4 (31P) using the high frequency positive convention. Abbreviations for NMR spectral multiplicities are as follows: s = singlet, d = doublet, m = unresolved multiplet. Elemental analyses were performed by the Elemental Analysis Service at the University of North London. Mass spectra were recorded on a Kratos Concept 1H mass spectrometer. (F13C6-4-C6H4)2PCl12 and cis-[PtCl2(MeCN)2]18 were prepared by the literature routes. Gas chromatographic analyses of the catalytic product mixtures were carried out on a Hewlett-Packard 5890 series gas chromatograph equipped with a flame ionisation detector. The gas chromatograph was interfaced with a Hewlett-Packard Chemstation for the determination of peak areas by electronic integration. Concentrations of products were obtained by comparison of peak areas with those obtained from standard solutions plotted on a calibration graph. The method employed a SupelcoTM Meridian MDN-35 low polarity, cross-linked phase comprised of a (35% phenyl)-methylpolysiloxane fused silica capillary column (30 m × 0.25 mm × 0.25 µm). Rhodium analyses of fractions recovered after the catalytic reactions were measured by inductively coupled plasma mass spectrometry (ICP-MS) on an Agilent 7500a instrument. The instrument was modified for direct analyses of the organic fractions by using O2 as a make-up gas to prevent carbon deposition on the sample and skimmer cones. Platinum cones were used together with a self-aspirating nebuliser held at −5 °C. Samples were diluted by 10% in a mixture of xylene and toluene (50 : 50) and ion counts were referenced against calibration curves obtained from standard solutions of [Rh(acac)(CO)2] (acacH is 2,4-pentanedione) in xylene–toluene (50 : 50). Standard solutions were run intermittently between samples to ensure that there were no drifts in the instrument response and that the rate of aspiration was constant.The partition coefficients were determined by dissolving 0.100 g of the required compound in a biphase consisting of toluene (2.00 ml) and PP3 (2.00 ml) at 25 °C. This was stirred for 20 min, allowed to settle for 30 min and a 1.00 ml aliquot removed from each phase. The solvents were removed in vacuo to constant weight.
3,5-Bis(tridecafluorohexyl)bromobenzene, 1a10a
A solution of C6F13I (33.79 g, 0.076 mol) in DMSO (20 ml) was added dropwise over 4 h to a stirred mixture of 1,3,5-tribromobenzene (12.01 g, 0.038 mol) and copper powder (9.64 g, 0.152 mol) in DMSO (60 ml) at 120 °C. The mixture was stirred at 120 °C for 12 h. After cooling to room temperature, water (50 ml) and diethyl ether (50 ml) were added and the mixture filtered. The organic layer was separated, washed with water (2 × 50 ml), dried over MgSO4 and the solvent was removed in vacuo. The off-white solid was distilled in a Kugelrohr oven to yield the product as a colourless oil (bp 90–100 °C, 0.05 mmHg), which formed white crystals upon cooling. These crystals were recrystallized in 80 : 20 ethyl acetate : acetonitrile (9.67 g, 0.012 mol, 32%). Mp = 39–41 °C. Anal. Calc. for C18H3BrF26: C, 27.24; H, 0.38. Found C, 26.94; H, 0.32. m/z (FAB) 792/4 (M+). 1H NMR (CDCl3) 7.86 (2H, s, H-2,6), 7.71 (1H, s, H-4); 19F{1H} NMR (CDCl3) −81.28 (6F, t, 4JFF = 10.0 Hz, CF3), −111.50 (4F, t, 4JFF = 14.2 Hz, α-CF2), −121.81 (4F, m, CF2), −122.11 (4F, m, CF2), −123.20 (4F, m, CF2), −126.57 (4F, m, CF2).[3,5-Bis(tridecafluoro-n-hexyl)phenyl]diphenylphosphine, 2a
mp = 54–56 °C. Anal. Calc. for C30H13F26P: C, 36.81; H, 1.33. Found C, 36.85; H, 1.29. m/z (EI) 898 (M+). 1H NMR (CDCl3) 7.88 (1H, s, H-4), 7.59 (2H, s, H-2,6), 7.25 (6H, m, PhH), 7.18 (4H, m, PhH). 19F{1H} NMR (CDCl3) −81.17 (6F, t, 4JFF = 9.7 Hz, CF3), −111.49 (4F, t, 4JFF = 14.3 Hz, α-CF2), −121.80 (4F, m, CF2), −122.05 (4F, m, CF2), −123.23 (4F, m, CF2), −126.47 (4F, m, CF2). 31P{1H} NMR (C6D6) −4.0 (s).Bis[3,5-bis(tridecafluoro-n-hexyl)phenyl]phenylphosphine, 3a
mp = 56–58 °C. Anal. Calc. for C42H11F52P: C, 32.86; H, 0.72. Found C, 32.72; H, 0.70. m/z (FAB) 1534 (M+). 1H NMR (CDCl3) 7.97 (2H, s, H-4), 7.89 (4H, s, H-2,6), 7.74 (1H, m, PhH), 7.63 (2H, m, PhH), 7.55 (2H, m, PhH). 19F{1H} NMR (CDCl3) −81.26 (12F, t, 4JFF = 10.0 Hz, CF3), −111.57 (8F, t, 4JFF = 13.2 Hz, α-CF2), −121.80 (8F, m, CF2), −122.39 (8F, m, CF2), −123.34 (8F, m, CF2), −126.58 (8F, m, CF2). 31P{1H} NMR (CDCl3) −5.3 (s).Bis(4-tridecafluoro-n-hexylphenyl)-[3,5-bis(tridecafluoro-n-hexyl)phenyl]phosphine, 4a
Oil. Anal. Calc. for C42H11F52P: C, 32.84; H, 0.72. Found C, 33.03; H, 0.57. m/z (FAB) 1534 (M+). 1H NMR (CDCl3) 7.73 (1H, s, H-4), 7.54 (6H, m, PhH), 7.32 (4H, t, 3JHH ≈ 3JHP = 7.8 Hz, H-2,6). 19F{1H} NMR (CDCl3) −81.50 (12F, t, 4JFF = 9.0 Hz, CF3), −111.67 (4F, t, 4JFF = 14.0 Hz, α-CF2), −111.89 (4F, t, 4JFF = 15.0 Hz, α-CF2), −122.02 (8F, m, CF2), −122.52 (4F, m, CF2), −122.67 (4F, m, CF2), −123.43 (8F, m, CF2), −126.82 (8F, m, CF2). 31P{1H} NMR (CDCl3) −6.3 (s).Bis[3,5-bis(trifluoromethyl)phenyl]phenylphosphine, 3b
Oil. Anal. Calc. for C22H11F12P: C, 49.44; H, 2.06. Found C, 49.51; H, 2.00. m/z (FAB) 534 (M+). 1H NMR (CDCl3) 7.82 (2H, s, H-4), 7.64 (4H, d, 3JHP = 6.6 Hz, H-2,6), 7.38 (3H, m, PhH), 7.26 (2H, m, PhH). 19F{1H} NMR (CDCl3) −63.04 (s). 31P{1H} NMR (CDCl3) −3.9 (s).Bis(4-tridecafluoro-n-hexylphenyl)-[3,5-bis(trifluoromethyl)phenyl]phosphine, 4b
mp = 63–64 °C. Anal. Calc. for C32H11F32P: C, 37.14; H, 1.06. Found C, 36.97; H, 1.05. m/z (FAB) 1034 (M+). 1H NMR (CDCl3) 7.83 (2H, s, H-2,6), 7.69 (1H, s, H-4), 7.59 (4H, m, PhH), 7.34 (4H, m, PhH). 19F{1H} NMR (CDCl3) −63.00 (6F, s, CF3), −80.78 (6F, t, 4JFF = 9.3 Hz, CF3), −111.19 (4F, t, 4JFF = 13.6 Hz, α-CF2), −121.35 (4F, m, CF2), −121.97 (4F, m, CF2), −122.78 (4F, m, CF2), −126.21 (4F, m, CF2). 31P{1H} NMR (CDCl3) −5.7 (s).[3,5-Bis(heptadecafluoro-n-octyl)phenyl]diphenylphosphine oxide
A solution of C8F17I (5.46 g, 10 mmol) in benzotrifluoride (10 mL) was added dropwise over 3 h to a stirred mixture of Ph2PO(C6H3-3,5-Br2) (2.18 g, 5.00 mmol),19 activated copper powder (2.25 g, 35.00 mmol) and bipy (0.17 g, 1.04 mmol) in DMSO (5 mL) and benzotrifluoride (40 mL) under nitrogen at 120 °C. The reaction was stirred and heated at 120 °C for 167 h. On cooling to room temperature the mixture was filtered, and the filtrate washed with chloroform (2 × 20 mL), 1 M HCl (2 × 50 mL) and water (50 mL). The organic layer was separated and washed with water (2 × 20 mL). The aqueous layer was washed with ether (2 × 50 mL) and the organic fractions were combined and dried over anhydrous MgSO4. After filtration, the solvent was removed in vacuo and the resulting solid recrystallised from ethanol to give a white solid (1.80 g, 1.62 mmol, 32%). Mp 114–116 °C. Anal. Calc. for C34H13F34OP: C, 36.62; H, 1.17. Found C, 36.73; H, 1.01. m/z (EI) 1115 (MH)+. 1H NMR (CDCl3) 8.05 (2H, d, 3JPH = 11.4 Hz, H-2,6), 7.99 (1H, s, H-4), 7.57 (6H, m, PhH), 7.45 (4H, m, PhH). 19F{1H} NMR (CDCl3) −80.81 (6F, m, CF3), −111.24 (4F, t, 4JFF = 14.0 Hz, α-CF2), −121.10 (4F, m, CF2), −121.89 (12F, m, CF2), −122.69 (4F, m, CF2), −126.20 (4F, m, CF2). 31P{1H} NMR (CDCl3) 27.1 (s).[3,5-Bis(heptadecafluoro-n-octyl)phenyl]diphenylphosphine, 2c
Ph2PO(C6H3-3,5-{C8F17}2) (0.50 g, 0.45 mmol) was dissolved in dry and degassed toluene (20 mL), NEt3 (2.00 g, 2.81 mL, 20 mmol) was added and the stirred solution was heated to 120 °C. HSiCl3 (0.91 g, 0.68 mL, 6.7 mmol) was added dropwise over 2 h and the reaction mixture refluxed for a further 6 h. The solution was then cooled in ice for 30 min and a saturated solution of NaHCO3 (1 mL) was added. The solution was flushed through a short alumina column using hexane (300 mL). On evaporation of the solvent in vacuo a yellow solid was obtained (0.35 g, 0.32 mmol, 71%).Mp = 53–55 °C. Anal. Calc. For C34H13F34P: C, 37.16; H, 1.18. Found C, 37.00; H, 1.17. m/z (FAB) 1098 (M)+. 1H NMR (CDCl3) 7.64 (1H, s, H-4), 7.58 (2H, d, 3JHP = 6.0 Hz, H-2,6), 7.29 (4H, m, PhH) 7.21 (6H, m, PhH). 19F{1H} NMR (CDCl3) −81.02 (6F, t, 4JFF = 10.5 Hz, CF3), −111.28 (4F, t, 4JFF = 14.2 Hz, α-CF2), −121.16 (4F, m, CF2), −121.95 (8F, m, CF2), −122.29 (4F, m, CF2), −122.1 (4F, m, CF2), −126.33 (4F, m, CF2). 31P{1H} NMR (CDCl3) −4.4 (s).
cis- and trans-[PtCl2{Ph2P(C6H3-3,5-[C6F13]2)}2]
Anal. Calc. for C60H26P2Cl2F52Pt: C, 34.93; H, 1.26. Found C, 34.62; H, 1.18. m/z (FAB) 2026 (M–Cl)+. 1H NMR (CDCl3) 7.96 (4H, s, H-2,6), 7.75 (2H, s, H-4), 7.65 (8H, m, PhH), 7.46 (12H, m, PhH). 19F{1H} NMR (CDCl3) −80.82 (12F, t, 4JFF = 9.8 Hz, CF3), −111.35 (8F, t, 4JFF = 13.9 Hz, α-CF2), −121.40 (8F, m, CF2), −122.10 (8F, m, CF2), −122.77 (8F, m, CF2), −126.19 (8F, m, CF2). 31P{1H} NMR (CDCl3) 21.9 (s, 1JPPt = 2692 Hz, trans), 14.5 (s, 1JPPt = 3630, cis).trans-[PtCl2{PhP(C6H3-3,5-[C6F13]2)2}2]
Anal. Calc. for C84H22P2Cl2PtF104: C, 30.24; H, 0.66. Found C, 30.12; H, 0.50. 1H NMR (PP3) 8.00 (8H, s, H-2,6), 7.82 (4H, s, H-4), 7.69 (4H, m, PhH), 7.34 (6H, m, PhH). 19F{1H} NMR (CDCl3) −80.93 (24F, t, 4JFF = 8.5 Hz, CF3), −111.70 (16F, t, 4JFF = 14.2 Hz, α-CF2), −121.38 (16F, m, CF2), −122.06 (16F, m, CF2), −122.77 (16F, m, CF2), −126.20 (16F, m, CF2). 31P{1H} NMR (CDCl3) 22.5 (s, 1JPtP = 2741 Hz).cis- and trans-[PtCl2{P(C6H4-4-C6F13)2(C6H3-3,5-[C6F13]2)}2]
Anal. Calc. for C84H22P2F104Cl2Pt: C, 30.24; H, 0.66. Found C, 30.06. H, 0.60. 1H NMR (CDCl3) 8.02 (4H, s, H-2,6), 7.92 (2H, s, H-4), 7.69 (8H, m, C6H4), 7.18 (8H, m, C6H4). 19F{1H} NMR (CDCl3) −80.96 (24F, m, CF3), −111.21 (8F, t, 4JFF = 14.2 Hz, α-CF2), −111.45 (8F, t, 4JFF = 13.7 Hz, α-CF2), −121.42 (16F, m, CF2), −121.90 (16F, m, CF2), −122.75 (16F, m, CF2), −126.13 (16F, m, CF2). 31P{1H} NMR (CDCl3) 21.7 (s, 1JPPt = 2743 Hz, trans), 13.0 (s, 1JPPt = 3576 Hz, cis).trans-[PtCl2{PPh(C6H3-3,5-[CF3]2)2}2]
Anal. Calc. for C44H22P2F12Cl2Pt: C, 39.61; H, 1.65. Found C, 39.69; H, 1.57. m/z (FAB) 1299 (M–Cl)+. 1H NMR (CDCl3) 7.99 (8H, s, H-2,6), 7.95 (4H, s, H-4), 7.72 (4H, m, PhH), 7.54 (6H, m, PhH). 19F{1H} NMR (CDCl3) −63.12 (s). 31P{1H} NMR (CDCl3) 23.0 (s, 1JPPt = 2739 Hz).cis- and trans-[PtCl2{P(C6H4-4-C6F13)2(C6H3-3,5-[CF3]2)}2]
Anal. Calc. for C64H22P2F64Cl2Pt: C, 32.92; H, 0.94. Found C, 32.81. H, 1.00. 1H NMR (CDCl3) 8.00 (4H, s, H-2,6), 7.95 (2H, s, H-4), 7.69 (8H, m, C6H4), 7.18 (8H, m, C6H4). 19F{1H} NMR (CDCl3) −63.20 (12F, s, CF3), −80.88 (12F, t, 4JFF = 9.9 Hz, CF3), −111.45 (8F, t, 4JFF = 13.9 Hz, α-CF2), −121.40 (8F, m, CF2), −121.93 (8F, m, CF2), −122.84 (8F, m, CF2), −126.06 (8F, m, CF2). 31P{1H} NMR (CDCl3) 21.7 (s, 1JPPt = 2741 Hz, trans), 15.7 (s, 1JPPt = 3599 Hz, cis).Acknowledgements
We thank the Lloyd's Tercentary Foundation (AMS), the Royal Society (EGH, AMS) and the EPSRC (DJA) for financial support.References
- C. D. Frohling and C. W. Kohlpaintner, Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, VCH, 1999, vol. 1, p. 27 Search PubMed.
- (a) I. T. Horváth and J. Rábai, Science, 1994, 266, 72 CrossRef CAS; (b) I. T. Horváth, G. Kiss, R. A. Cook, J. E. Bond, P. A. Stevens, J. Rábai and E. J. Mozeleski, J. Am. Chem. Soc., 1998, 120, 3133 CrossRef CAS.
- (a) D. F. Foster, D. Gudmunsen, D. J. Adams, A. M. Stuart, E. G. Hope, D. J. Cole-Hamilton, G. P. Schwarz and P. Pogorzelec, Tetrahedron, 2002, 58, 3901 CrossRef CAS; (b) D. F. Foster, D. J. Adams, D. Gudmunsen, A. M. Stuart, E. G. Hope and D. J. Cole-Hamilton, Chem. Commun., 2002, 722 RSC.
- E. Perperi, Y. Huang, P. Angeli, G. Manos, C. R. Mathison, D. J. Cole-Hamilton, D. J. Adams and E. G. Hope, Dalton Trans., 2004, 2062 RSC.
- L. P. Barthel-Rosa and J. A. Gladysz, Coord. Chem. Rev., 1999, 190–192, 587 CrossRef CAS.
- A. Aghmiz, C. Claver, A. M. Masdeu-Bultó, D. Maillard and D. Sinou, J. Mol. Catal. A: Chem., 2004, 208, 97 CrossRef CAS.
- E. G. Hope, R. D. W. Kemmitt and A. M. Stuart, J. Chem. Soc., Dalton Trans., 1998, 3765 RSC.
- B. Richter, E. de Wolf, G. van Koten and B.-J. Deelman, J. Org. Chem., 2000, 65, 3885 CrossRef CAS.
- D. Sinou, D. Maillard, A. Aghmiz and A. M. Masdeu I-Bultó, Adv. Synth. Catal., 2003, 345, 603 CrossRef CAS.
- (a) J. van den Broeke, B.-J. Deelman and G. van Koten, Tetrahedron Lett., 2001, 42, 8085 CrossRef CAS; (b) K. Ishihara, S. Kondo and H. Yamamoto, Synlett, 2001, 1371 CrossRef CAS; (c) G. J. Chen and C. Tamborski, J. Fluorine Chem., 1989, 43, 207 CrossRef CAS; (d) J. Duan, L. H. Zhang and W. R. Dolbier, Jr., Synlett, 1999, 1245 CrossRef CAS.
- M. Cavazzini, A. Manfredi, F. Montanari, S. Quici and G. Pozzi, Eur. J. Org. Chem., 2001, 4639 CrossRef CAS.
- B. Croxtall, J. Fawcett, E. G. Hope and A. M. Stuart, J. Chem. Soc., Dalton Trans., 1999, 491 Search PubMed.
- J. A. S. Howell, N. Fey, J. D. Lovatt, P. C. Yates, P. McArdle, D. Cunningham, E. Sadeh, H. E. Gottlieb, Z. Goldschmidt, M. B. Hursthouse and M. E. Light, J. Chem. Soc., Dalton Trans., 1999, 3015 RSC.
- W. Chen and J. Xiao, Tetrahedron Lett., 2000, 41, 3697 CrossRef CAS.
- J. Fawcett, E. G. Hope, R. D. W. Kemmitt, D. R. Paige, D. R. Russell and A. M. Stuart, J. Chem. Soc., Dalton Trans., 1998, 3751 RSC.
- E. G. Hope, R. D. W. Kemmitt, D. R. Paige, A. M. Stuart and D. R. W. Wood, Polyhedron, 1999, 18, 2913 CrossRef CAS.
- See Rhodium catalysed hydroformylation, ed. P. N. W. M. Van Leeuwen and C. Claver, Kluwer, Dordrecht, 2000, and refs. therein Search PubMed.
- F. R. Hartley and C. A. McAuliffe, Inorg. Chem., 1979, 18, 1394 CrossRef CAS.
- E. G. Hope and J. Hopewell, unpublished work.
- J. P. Farr, F. E. Wood and A. L. Balch, Inorg. Chem., 1983, 22, 3387 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |