
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition metal coordination to degradation products in battery electrolytes revealed by NMR and EPR spectroscopy
Jennifer P. Allen ab,
Conrad Szczukacd,
Erlendur Jónssona,
Rüdiger-A. Eichelcd,
Josef Granwehrce and
Clare P. Grey*ab
ab,
Conrad Szczukacd,
Erlendur Jónssona,
Rüdiger-A. Eichelcd,
Josef Granwehrce and
Clare P. Grey*ab
aYusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, Cambridge, UK. E-mail: cpg27@cam.ac.uk
bThe Faraday Institution, Quad One, Harwell Science and Innovation Campus, Didcot OX11 0RA, UK
cInstitute of Energy Technologies (IET-1), Forschungszentrum Jülich GmbH, 52425 Jülich, Germany
dInstitute of Physical Chemistry, RWTH Aachen University, 52056 Aachen, Germany
eInstitute of Technical and Macromolecular Chemistry, RWTH Aachen University, 52056 Aachen, Germany
First published on 29th October 2025
Abstract
The dissolution of transition metals from lithium-ion battery materials contributes to cell failure. Developing a better understanding of transition metal solvation, reactivity, and deposition will help mitigate transition metal dissolution and, thus, facilitate batteries with higher capacity and longer lifetime. In this work, Mn2+ and Ni2+ coordination in degraded LiPF6 carbonate electrolyte solutions is examined via 1H and 19F NMR relaxometry at ambient temperatures and pulsed (double resonance HYSCORE and ENDOR) EPR spectroscopy of the frozen solutions. Critically, the solvation spheres of transition metals in heat-degraded electrolytes are shown to differ from those found in pristine electrolytes, with significant coordination to LiPF6-derived fluorophosphate degradation species—a factor that will impact the mechanisms and the extent of transition metal dissolution. The Mn2+ coordination environment is shown to be affected by adding a variety of species including water, ethylene glycol, and acetylacetonate, increasingly displacing EC and PF6− from inner and outer Mn2+ coordination shells. EPR and NMR studies of electrolytes containing the battery additive and proposed degradation product LiPO2F2 explicitly confirm that the transition metals coordinate to PO2F2− in both the inner and outer sphere. By contrast, in heat-degraded electrolytes, additional protonated (uncharged) fluorophosphate species are also present in the metal solvation shell, as clearly demonstrated via large 1H and 31P hyperfine interactions, seen in the EPR spectra of Mn2+ complexes. To probe transition metal deposition, Mn2+ and Ni2+ were exposed to a wide variety of salts found in the solid electrolyte interphase (SEI), revealing for the first time that the metathesis deposition pathway is viable with many different SEI species.
Broader contextThe dissolution of transition metal ions from essentially all battery cathode active materials is thought to be a significant cause of battery capacity fade. Dissolution can occur as part of cathode degradation processes, and these dissolved transition metals will cross over from cathode to anode, where they may precipitate, potentially contributing to breakdown of the passivating layer that forms on the graphite anode (the solid electrolyte interphase). Surprisingly, little is known about how the transition metals dissolve in battery electrolytes, from the pristine to the more degraded solutions that are representative of practical systems. In order to develop strategies to mitigate dissolution, including the design of new additives, coatings, and electrolytes, a fundamental understanding of how the relevant transition metals are dissolved in both pristine and degraded electrolytes is required. This paper attacks the issue directly by developing new magnetic resonance approaches to study these electrolytes and shows clearly that the coordination of these ions in degraded electrolytes is very different from coordination in the pristine electrolytes, and that these ions are strongly coordinated to a series of anions often found in degraded electrolytes and used as additives. |
Introduction
While the dissolution of transition metals from battery cathode active materials is thought to cause significant battery capacity fade via a myriad of different mechanisms,1–6 comparatively little work has been performed to study the solvation of the transition metals once they are dissolved in the battery electrolyte solution.7–11 Even less attention has been paid to how the solvation may differ between ideal pristine electrolyte solutions and (partly) degraded electrolyte solutions that more accurately represent the use case, particularly in regimes where the rate of transition metal dissolution is enhanced. Although an understanding of the transition metal solvation shell is not generally directly relevant to the bulk properties of the electrolyte solution, transition metal solvation and desolvation are important for transition metal dissolution and deposition behaviour, respectively. For example, a study of metal dissolution in LiNi0.5Mn1.5O4/graphite cells proposed that solvent oxidation at the cathode surface generates β-diketonate species that are adsorbed on the surface, chelating the transition metals and facilitating metal dissolution.12 Another study proposed that the chelation of dissolved Mn2+ by unspecified electrolyte degradation products may create neutral Mn complexes that can be transported through the outer, organic layer of the solid electrolyte interphase (SEI) and deposited at the surface of the inner, inorganic SEI.13 One density functional theory (DFT) study showed that P–F bonds in PF6− were longer in the presence of Mn2+, facilitating the formation of MnF+ and the reactive Lewis acid, PF5.8 In combination with experiments showing severe discolouration of electrolyte solutions stored with Mn2+,8,14 this suggests that dissolved transition metals may actually contribute to bulk electrolyte decomposition reactions even before their deposition at the anode occurs.The process of transition metal dissolution may be driven by or coupled with electrolyte decomposition reactions occurring at the cathode; for instance, acidic oxidation or hydrolysis products may leach transition metals from the cathode.12,15–20 Thus, when transition metal dissolution occurs, it is likely that the electrolyte solution is already degraded to some degree, and it is therefore vital to consider how transition metals are then solvated in the degraded electrolyte solution. However, computational studies of transition metal coordination in battery electrolyte solutions have largely focused on the components of the pristine electrolyte.7,8 One simulation study of Mn2+ dissolution processes incorporated ethylene carbonate (EC) decomposition products and F− on the modelled LiMn2O4 (LMO) surface, but simulated the electrolyte solution as 32 EC molecules.21 Notably, modelling studies that have incorporated coordinating degradation species have shown that they do have an effect: one study incorporated F− into a diethyl carbonate solution and found that F− may facilitate the dissolution of Ni and Mn from LiNi0.5Mn1.5O4 (LNMO);22 another simulation study found that biacetyl complexation at the surface of LiNixMnyCo1−x−yO2 (NMC) increased Ni dissolution, which was then confirmed experimentally.23 This is further supported by an experimental study showing enhanced dissolution from charged LiNi0.5Mn0.3Co0.2O2 (NMC532) cathodes stored with a solution containing the salt lithium acetylacetonate (Li(acac)), as compared to the control electrolyte containing LiPF6.24 Another study found that transition metals dissolved from delithiated cathode materials in the presence of carbonate solvents tended to coordinate to anionic species.10 Operando studies have shown that exposure of cathode materials to water-contaminated electrolyte solutions enhanced transition metal dissolution,25 while exposure to thermally degraded electrolyte solutions did not appear to enhance transition metal dissolution, complicating the picture.26 If transition metals are indeed coordinated to degradation products in solution, whether thermally, chemically, or electrochemically generated, then models of how dissolution (solvation) and deposition (desolvation) occur may benefit from the incorporation of soluble electrolyte decomposition species in the simulated electrolyte solution. Although simulation of additional components is increasingly computationally expensive, targeted experiments may serve as a basis for computational work by identifying the solution species that are most likely to affect transition metal dissolution.
The role of PF6− degradation products in battery chemistry is complex. While alkylfluorophosphates have been proposed to trigger autocatalytic decomposition of the electrolyte solution,27 and while difluorophosphate generated in situ may serve as an indicator of PF6− hydrolysis,28–31 difluorophosphate itself does not appear to be harmful in cells: studies using a small percentage of added LiPO2F2 in the electrolyte solution have shown that it enhances cell performance32–38 by modifying the SEI at the cathode33,34,37,38 and anode34,36,37 surfaces, offering protective benefits, such as minimising impedance growth. With respect to transition metal dissolution, previous work has shown that added LiPO2F2 results in lower levels of transition metals deposited at the negative electrode.32–35 It has been proposed that the PO2F2− additive may decompose into PO43− and PO3F2−, which may induce precipitation of dissolved metals before they migrate to the anode (presumably at the cathode or in the separator).32,35,39–41
Another pathway to prevent transition metal deposition, rather than inducing precipitation of transition metals before they reach the anode, involves sequestering them in solution. Chelating agents have been added to cells to prevent the deposition of dissolved metals by modifying the electrolyte solution, separator, or anode. In the electrolyte solution, transition metals have been sequestered by using crown ether,14 aza-crown ether,42 or 1,2-bis(diphenylphosphino)ethane43 additives, or a specialised chelating polymer gel electrolyte.44 Highly concentrated electrolyte solutions have been suggested to prevent the deposition of dissolved Mn2+ by increasing the formation of contact ion pairs and aggregates, in turn decreasing the favourability of incorporation in the SEI.9 Numerous functionalised separators have been employed to trap dissolved transition metals in cells, with varying degrees of selectivity and efficiency,45,46 and at the negative electrode, an aza-crown ether functionalised binder has been proposed.42 Chelating agents have also been used in the cathode to coordinate the metals and prevent dissolution into the electrolyte solution, for example, by functionalising the cathode binder47–50 or conductive carbon.51 The effects of transition metal chelation in lithium-ion cells are complex, and it may be beneficial or harmful, depending on the context. Storage with 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 ethylene carbonate:ethyl methyl carbonate (EC:EMC, w/w) solution containing lithium acetylacetonate, Li(acac), has been shown to enhance Mn and Co dissolution (not Ni dissolution) from charged NMC532, although dissolution from pristine cathodes was unaffected.24 However, acetylacetone has also been used to chelate Mn and prevent its dissolution from LMO spinel: rinsing electrode materials with an acac solution and then sintering the materials was found to reduce Mn dissolution.52,53 This strategy succeeded because the acac solution leached Mn from the LMO surface, creating a Mn-deficient surface layer that was less prone to dissolution.52,53 These results suggest that an understanding of chelation is important and can directly be used to prevent cell degradation.
:7 ethylene carbonate:ethyl methyl carbonate (EC:EMC, w/w) solution containing lithium acetylacetonate, Li(acac), has been shown to enhance Mn and Co dissolution (not Ni dissolution) from charged NMC532, although dissolution from pristine cathodes was unaffected.24 However, acetylacetone has also been used to chelate Mn and prevent its dissolution from LMO spinel: rinsing electrode materials with an acac solution and then sintering the materials was found to reduce Mn dissolution.52,53 This strategy succeeded because the acac solution leached Mn from the LMO surface, creating a Mn-deficient surface layer that was less prone to dissolution.52,53 These results suggest that an understanding of chelation is important and can directly be used to prevent cell degradation.
Generally, it is not entirely clear what fraction of transition metals remains in the electrolyte as soluble complexes and what fraction precipitates at the cathode or deposits at the anode, and how this is affected by the species present in solution, motivating further studies. In a cell, where many species are present simultaneously, it is difficult to predict how the formation of soluble chelate complexes may impact metal deposition. Both strategies outlined above, of inducing transition metal precipitation at the cathode and of sequestering metal ions in solution, target the prevention of metal deposition at the anode, and both strategies require an understanding of transition metal solvation, complexation, and desolvation behaviours.
In this work, we study the coordination of Mn2+ and Ni2+ in a degraded carbonate-based electrolyte. These species are of interest because Mn2+ is thought to be more harmful to cells than other dissolved metals,3,5,54–56 and Ni2+ is the most abundant metal in nickel-rich lithium-ion battery cathode materials; both ions represent typical divalent5,20,57–60 paramagnetic dissolved transition metal ions. Mn2+ and Ni2+ species are studied separately in this work, such that results may be applicable to any chemistry containing either ion, including NMC, LMO, LNMO, LiNixCoyAl1−x−yO2 (NCA), LiNiO2 (LNO), LiMnxFe1−xPO4 (LMFP), etc. The paramagnetism of these ions can be exploited to examine interactions between electron and nuclear spins, i.e., hyperfine interactions, using the complementary techniques of electron paramagnetic resonance (EPR) and nuclear magnetic resonance (NMR) spectroscopy, as illustrated in our previous publication on pristine electrolytes.11 Pulsed EPR identifies nearby magnetically-active nuclei through their effect on the paramagnetic centre's electronic spin states, whereas paramagnetic NMR probes the nuclear spins’ relaxation driven by nearby paramagnetic centres. Since the nuclear spins relax on a slower timescale than electron spins, the electron–nuclear (hyperfine) interactions can be studied via pulsed EPR experiments. These hyperfine interactions are described by interaction tensors represented through the coupling constants Ax, Ay, and Az. A non-zero isotropic component (Ax + Ay + Az)/3 indicates electron spin density at the position of the nucleus, generally implying that an inner sphere (bonded) complex is formed, and an anisotropic part describes all through-space dipolar contributions from both inner and outer sphere complexes. Cryogenic temperatures are typically required for pulsed EPR experiments, resulting in a loss of dynamic information, but instead allowing the static compositions and conformations of the transition metal complexes to be determined. EPR studies of the Ni2+ ion with its S = 1 electron spin are, however, difficult due to its rapid electronic relaxation.61 Paramagnetic NMR spectroscopy complements the EPR method because it can be carried out at ambient temperatures, encoding information on dynamics, and may be used to indirectly characterise the solvation shell of most paramagnetic transition metals, with direct relevance here for Ni2+.62
The coordination sphere of Mn2+ and Ni2+ in degraded carbonate-based electrolytes is approached in two ways in this work. First, transition metal-containing electrolytes are heated and subsequently examined by pulsed EPR and NMR spectroscopy. Second, transition metal coordination to externally added species is investigated, including difluorophosphate,28–32 water,10,63–67 acac−,12 and ethylene glycol (EG),68–70 because these and related species have been proposed to form, and/or coordinate to transition metals, in degraded battery electrolytes. EG is a hydrolysis product of EC,68,69 and some of the authors have previously proposed the dianion's role in vanadyl coordination in V4+-containing solutions.70 Lithium difluorophosphate is also commonly used as an electrolyte additive or co-salt.37,71 In pristine electrolytes, the transition metal solvation shell is largely comprised of EC;7,8,11 however, we show herein by using a combination of EPR and NMR spectroscopies that the solvation shell differs noticeably in degraded electrolytes. Neutral degradation species such as water and ethylene glycol appear to displace ethylene carbonate, while some PF6− remains, largely in the outer solvation shell, at ambient temperature for charge balance. Anionic species such as PO2F2−, which was observed to form upon heating of electrolyte solutions, and acac− appear to have a strong binding affinity and displace both EC and PF6− from the Mn2+ coordination shell. The effect of dissolved Mn2+ toward enhancing electrolyte decomposition, which has been previously proposed,8,14 is then studied with NMR spectroscopy.
We use heat-treated electrolyte solutions to simulate electrolyte degradation, rather than sourcing cycled electrolytes from lithium-ion cells for the following key reasons. Firstly, using electrolytes from lithium-ion cells would cause various changes to the electrolyte composition that could have significant effects on the measured paramagnetic relaxation enhancement. The most notable of these is, of course, transition metal dissolution, both from the cathode in the lithium-ion cell and the cell casing.72 It would not be possible in this way to obtain an electrolyte that contained only Mn2+ or only Ni2+. Furthermore, electrolyte degradation reactions in the cell would result in changes to the salt concentration, solvent species, and viscosity that would in turn skew the baseline relaxation. The final reason is a practical limitation of the electrolyte volume: NMR relaxometry experiments used 500 μL of undiluted electrolyte (rather than a small volume of electrolyte in a large volume of deuterated solvent), preventing the use of electrolytes extracted from small-scale cells. Had many cells been used to gather a sufficient quantity of degraded electrolyte for all NMR experiments, there would be differences in the electrolyte extracted from each cell, leading to inconsistency between samples. In this case, it was our judgement that adding pristine electrolyte to a sealed container (a closed system) and heating it shows that it is specifically and necessarily the generation of degradation products that is affecting the transition metal solvation, which is later explored through the addition of LiPO2F2. Nevertheless, building on the insights obtained from the results described below, future studies will focus on incorporating electrochemically-degraded electrolyte solutions sourced from full cells.
Lastly, we explore the deposition of transition metals in the SEI by metathesis, a mechanism that has been suggested for dissolved Mn2+,73–76 Ni2+,77,78 and Co2+;79 even Li+ is known to exchange with other Li+ in the SEI.80 Ex situ ion exchange experiments have shown by ion chromatography that Li3PO4 addition39 and Na2PO3F addition40 cause precipitation of Mn2+, Ni2+, and Co2+ from electrolyte solutions. Inductively coupled plasma mass spectrometry (ICP-MS) exchange experiments have shown Mn2+ precipitation from electrolyte solutions upon addition of Li2O, LiF, and Li2CO3;75 Mn2+ exchange with the latter two salts is further supported by XPS analysis.74 Thus, the effect of a large series of lithium salts—some of which have been shown to be present in the SEI—on transition metal solubility was investigated in this work. Specifically, we examine the precipitation rates of Mn2+ and Ni2+ using inductively coupled plasma optical emission spectroscopy (ICP-OES) following exposure to 12 different organic and inorganic lithium salts.
Materials and methods
Preparation of electrolyte solutions
The bis(trifluoromethane)sulfonimide (TFSI) salts Mn(TFSI)2 (Solvionic, 99.5%) and Ni(TFSI)2 (Alfa Aesar, ≥97%) were used to model transition metals dissolved from battery cathodes. EPR samples used concentrations of 8 mM Mn(TFSI)2 and NMR samples used concentrations of 1 mM Mn(TFSI)2 and 1 mM Ni(TFSI)2, except where specified otherwise. The main electrolyte solution used in this work comprised 1 M LiPF6 in 3:7 ethylene carbonate:ethyl methyl carbonate (EC:EMC, v/v). For NMR experiments, this was sourced premixed (soulbrain MI PuriEL R&D 280); for EPR experiments, all solutions were mixed in-house using LiPF6 (Sigma Aldrich, ≥99.99% trace metals basis), EC (Sigma Aldrich, 99%, anhydrous), and EMC (Sigma Aldrich, 99%). Owing to the different electrolyte sources, it is likely that EPR samples contained more water impurities than the NMR samples. EPR experiments also used 1 M LiTFSI (Alfa Aesar, 98+%) in 3:7 EC:EMC, mixed in-house. To mimic electrolyte degradation, samples were heat-treated (at either 35 °C or 45 °C for EPR samples and 60 °C for NMR samples) or underwent addition of LiPO2F2 (American Elements, 99%, recrystallised in-house), lithium acetylacetonate (Li(acac), Sigma Aldrich, 97%), distilled H2O/D2O (Sigma Aldrich, 99.9 atom%), and ethylene glycol (Sigma Aldrich, 99.8%, anhydrous). Additionally, deuterated dimethyl sulfoxide (d6-DMSO, Sigma Aldrich, NMR-grade), methanol (MeOD, Sigma Aldrich, NMR-grade), and acetonitrile (CD3CN, Fluorochem and Sigma Aldrich, NMR-grade), were added to some EPR and NMR samples to probe metal coordination to these species. All solutions were prepared under argon and all salts were dried under dynamic vacuum at 100 or 110 °C.
EPR spectroscopy
Mixing, storage, and heat-treatment of electrolyte solutions was performed in Eppendorf tubes made from polypropylene, which is HF-resistant. Solutions were transferred into 2 mm outer diameter (O.D.) quartz EPR tubes (Wilmad, CFQ) for X-band, and 0.9 mm O.D. Suprasil tubes (Wilmad) for Q-band experiments. Tubes were sealed and transferred from the glovebox into the pre-cooled EPR resonator within 15 min after preparation. A Bruker ElexSys E580 spectrometer equipped with an EN4118X-MD4 resonator for X-band and an EN5107-D2 resonator for Q-band frequencies was used for experiments at a temperature of 20 K. Microwave amplification was performed with a 1 kW travelling-wave tube (TWT) pulse amplifier at X-band and with a 150 W TWT at Q-band; radiofrequency amplification was done with a 150 W amplifier.Field-swept pulsed EPR spectra were obtained using the Hahn echo pulse sequence π/2-τ-π-τ-echo and a two-step phase cycle. For X-band, τ = 200 ns and pulse durations τπ/2 = 12 ns and τπ = 24 ns; for Q-band, τ = 160 ns and pulse durations τπ/2 = 20 ns and τπ = 40 ns.
Hyperfine sublevel correlation81 (HYSCORE) spectra were recorded using the sequence π/2-τ-π/2-t1-π-t2-π/2-τ-echo with τ = 80, 120 ns, τπ/2 = 12 ns, and τπ = 24 ns. Multiple τ-values were used to remove blind spot artifacts and t1, t2 were incremented by 20 ns starting from 80 ns. A four-step phase cycle was applied. Two-dimensional time traces were baseline-corrected with a third-order polynomial, apodised with a Hamming window function, and zero filled to 1024 points in each dimension, followed by 2D Fourier transformation and calculation of the absolute value. Davies-type electron nuclear double resonance (ENDOR) experiments were conducted at Q-band using the pulse sequence π–D–π/2-τ-π-τ-echo with tπ/2 = 100 ns, tπ = 200 ns, and τ = 450 ns. During the delay D, an rf π-pulse lasting 10 μs was applied.
DFT calculations and EPR spectra simulation
Density functional calculations were conducted using ORCA 5.0.1.82 Structures were geometry optimised with the hybrid functional TPSSh,83,84 the zeroth-order regular approximation (ZORA),85,86 and def2-TZVP(-f) basis sets87 in their ZORA-recontracted version.88 Decontracted def2/J auxiliary basis sets were employed for the resolution-of-identity and chain-of-spheres approximation. The convergence criterion was set to tight self-consistent-field. Zero-field splitting and hyperfine coupling tensors were calculated using the hybrid functional PBE089 and ZORA. ZORA-def2-TZVP(-f) basis sets were modified through full s shell decontraction and addition of three steep Gaussians, as explained elsewhere.90,91 Spin–orbit coupling was calculated using the spin–orbit mean-field approximation (SOMF(1X)).92 Integration accuracy was set to defgrid3 and radial accuracy IntAcc to 11 for manganese and 9 for all other atoms.EPR spectra were simulated using EasySpin v6.0.0-dev.4993,94 running in Matlab v2021b (MathWorks). HYSCORE spectra were simulated considering all magnetic nuclei with hyperfine tensors as obtained from DFT calculations using the simulation module saffron. Second-order perturbation theory and the product rule were used. Input parameters were set as used experimentally and electronic relaxation times as T1e = 80 μs and T2e = 0.4 μs, which are in the range of those experimentally determined. Post-processing was equivalent as for experiments. 1H powder Davies ENDOR spectra were simulated at Q-band frequencies assuming one coupled 1H nucleus using the simulation module salt. The obtained spectra were multiplied with the Davies ENDOR detection function.95,96
NMR spectroscopy
1H and 19F NMR relaxation measurements of electrolyte solutions were performed at ∼25 °C on a Bruker Avance III HD 300 MHz spectrometer using a Bruker double-channel MicWB40 probe. NMR spectra were collected on a Bruker Avance III HD 500 MHz spectrometer using a BBO probe, with a sealed capillary of C6D6 added to the NMR tube for field locking. No deuterated solvents were used for measurements at 300 MHz. Spin–lattice T1 measurements were performed using the inversion recovery pulse sequence; spin–spin T2 measurements were performed using the Carr–Purcell–Meiboom–Gill (CPMG) pulse sequence97,98 with echo spacings of τ = 2 ms for most solutions. For 19F measurements of Mn2+-containing solutions, echo spacings of 1 ms were used, due to faster relaxation. NMR tubes were filled in an argon glovebox and sealed with J-Young valves.LiOH (Alfa Aesar, 99.995%), LiF (Sigma Aldrich, 99.995%), Li3PO4 (Sigma Aldrich), Li2CO3 (Alfa Aesar, 99.998%), Li2O (Alfa Aesar, 99.5%), Li2O2 (Acros Organics, 95%), LiOMe (Sigma Aldrich, 98%), LiOEt (Sigma Aldrich, 95%), Li2C2O4 (Alfa Aesar, 99+%), LiOCHO (Alfa Aesar, 98%), LiOAc (Sigma Aldrich, 99.95%), Li(acac) (Sigma Aldrich, 97%). All salts were dried under vacuum at 100 °C before use. In plastic vials, 1 mL of 3:7 EC:EMC (v/v)—diamagnetic or containing 1 mM Mn(TFSI)2 or 1 mM Ni(TFSI)2—was added to 20 mg of each salt (10 mg for LiOMe, LiOEt, and Li2O2; 40 mg for LiF). Solutions were shaken twice per day; after two days, solutions were removed from the argon atmosphere and 150 μL of solution was pipetted off the top and added to 2.5% aqueous nitric acid. Samples were analysed for Li, Mn, and Ni concentration via ICP-OES.ICP-OES analysis
ICP-OES measurements were performed on an iCAP 7400 Duo ICP-OES Analyzer in axial mode (Thermo Fisher Scientific). Calibration points were measured with multi-element standard solutions at concentrations of 0.005–20.0 ppm.Results and discussion
EPR spectroscopy
The following sections contain results from pulsed EPR spectroscopy of frozen electrolytes performed at a temperature of 20 K. Measurements were optimised for detecting ligand hyperfine couplings of Mn(II) complexes, which exhibit sufficiently long electronic relaxation times for this technique.Field-swept EPR spectra
Fig. 1 shows X-band Hahn-echo-detected pulsed EPR spectra of frozen 1 M LiPF6 in 3:7 EC:EMC (LP57) electrolyte containing 8 mM Mn2+. The freshly prepared sample (lower spectrum) exhibits six central transition resonances corresponding to hyperfine coupling to the six nuclear spin states of the I = 5/2 nucleus 55Mn. The additional features associated with each of these peaks arise from a rhombic zero-field splitting interaction, which characterises the symmetry of the complex. This spectrum was assigned in our previous work to a sixfold coordinated Mn(II) complex coordinated by the oxygen atoms of the carbonyl functionalities from mainly EC, and minorly EMC, with PF6− located in the second solvation shell.11 When the identical sample is stored at 35 °C for 24 h, representing a mild heating protocol, the distinct six central transitions are less pronounced and the very broad outer transitions at around 300 and 400 mT are seen more clearly (middle spectrum). An analysis of the degraded electrolyte using 19F solution NMR revealed the presence of PO2F2− and/or HPO2F2 species at a concentration of approximately 9 mM. To explore the effect of the PO2F2− anion further, frozen electrolytes with LiPO2F2 salt added in a Mn2+:PO2F2− ratio of either 1:2 or 1:10 (corresponding to 16 and 80 mM LiPO2F2 solutions) were prepared. These now exhibit almost featureless pulsed EPR spectra, where the sharp central transitions are barely visible. Despite the different Mn2+:PO2F2− ratios, both samples give rise to almost identical spectra.
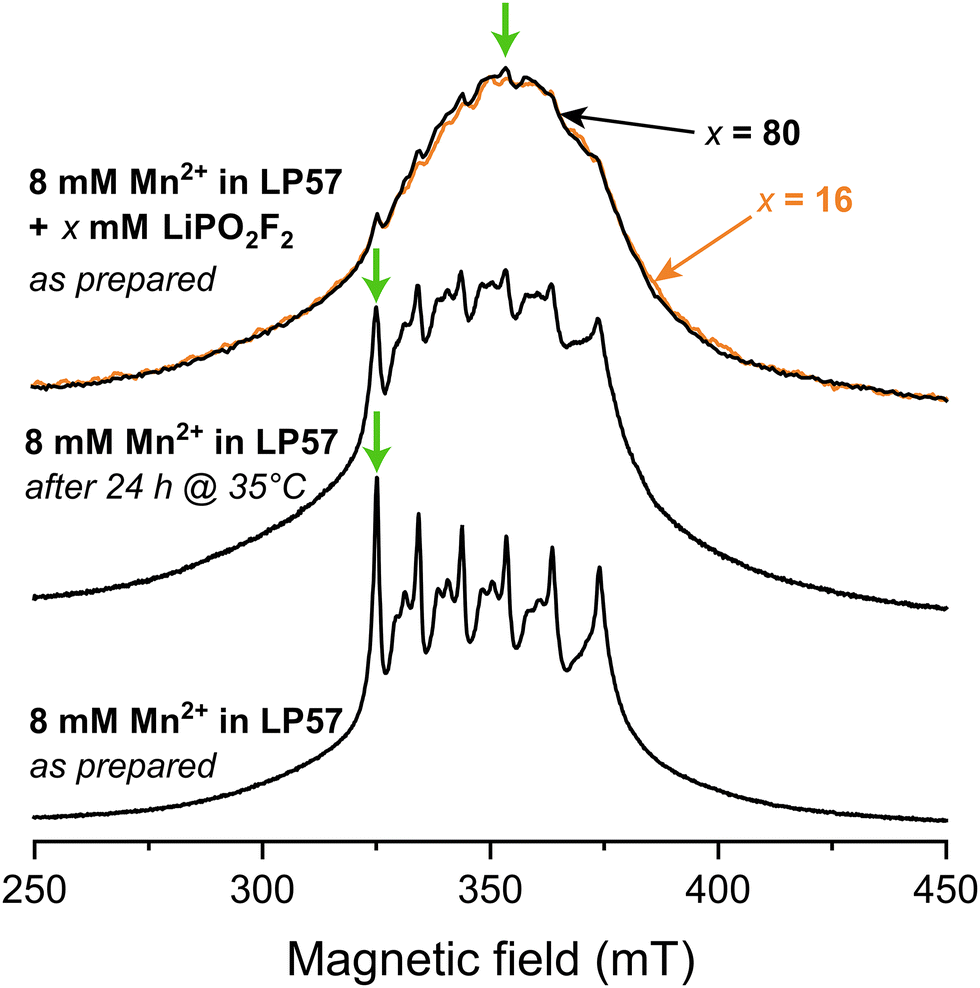 | ||
| Fig. 1 Field-swept echo-detected X-band pulsed EPR spectra, recorded at 20 K. Frozen samples of 8 mM Mn(TFSI)2 dissolved in 1 M LiPF6 in 3:7 EC:EMC (LP57) are investigated freshly prepared (bottom), after heating at 35 °C (middle), and with 16 mM (orange) or 80 mM (black) LiPO2F2 added (top). Orange and black arrows (top) point at the partially overlapping spectra for samples with 16 and 80 mM LiPO2F2 addition, respectively. Green arrows show the magnetic fields used for the hyperfine spectroscopy experiments (Fig. 2). | ||
The increasing intensity away from the sharp central transitions and line broadening observed for the mildly heated and LiPO2F2-containing samples indicate contributions from complexes exhibiting either a large zero-field splitting, a large distribution of zero-field splitting constants, and/or strain effects.99–101 To estimate the relative sizes of these interactions, the zero-field splitting interactions of sixfold coordinated Mn(II) complexes containing carbonate and fluorophosphate ligands in the first coordination shells were investigated by DFT. The zero-field splitting magnitude follows the series: carbonate (6 EC) < 6 HPO2F2/6 PO2F2− ≪ mixed PO2F2−/HPO2F2 ≈ mixed carbonate/PO2F2− ligands (SI, Fig. S1). Sixfold PO2F2− coordination is, however, considered unlikely due to an overall complex charge of −4. HPO2F2 was investigated because a trace amount of water may react with LiPF6 under mildly elevated temperature to form HPO2F2, a species which is only weakly acidic.102 Although the calculated absolute values of zero-field splitting constants are often unreliable,101 the increase in zero field splitting for complexes containing PO2F2−/HPO2F2 in the Mn2+ coordination shell could explain the increase in broadening from the pristine sample to the mild heat-treated and LiPO2F2-additive samples. To further investigate the coordination to Mn2+, hyperfine-targeted pulsed EPR measurements were applied to investigate the hyperfine interactions with the various NMR active nuclei.
X-band hyperfine sublevel correlation (HYSCORE) spectroscopy
The above-introduced samples of Mn(II) in 1 M LiPF6/3:7 EC:EMC (LP57), either freshly prepared, mildly heat-treated, or with added LiPO2F2, were first analysed by HYSCORE spectroscopy (Fig. 2, upper row). This four-pulse, two-dimensional EPR experiment is performed at a constant magnetic field (i.e., at one particular position in the EPR spectrum, as marked in Fig. 1 with green arrows) and detects coupling to magnetic nuclei in the solvation sphere(s) of the Mn(II) ion, with the nuclear transitions να,β (for S = 1/2) being observed, to first order, at frequencies| να,β = νI ± A/2 if |A/2| < νI |
 | ||
| Fig. 2 Experimental (upper row) and calculated (lower row) HYSCORE spectra at 9.8 GHz (X-band; 20 K). Spectra from samples containing 8 mM Mn(TFSI)2 dissolved in 1 M LiPF6 in 3:7 EC:EMC (left), with mild heat-treatment at 35 °C for 24 h (centre), and addition of 80 mM LiPO2F2 (right) are shown. Experiments were conducted at the magnetic field positions indicated by green arrows in Fig. 1. The diagonal is shown with a dashed line. Anti-diagonal (solid) lines are shown at relevant nuclear Larmor frequencies. Calculations are shown for three clusters, [Mn(EC)6]2+, [Mn(PO2F2)3(HPO2F2)3]− and [Mn(PO2F2)2(EC)4]. | ||
After mild heating of the Mn(II)-containing sample, the resonance at ν1H splits, indicating a larger 1H hyperfine interaction, and new resonances centred at ν31P and ν19F appear, indicating a change of the Mn2+ coordination shell. The most intense signals cluster around the 31P anti-diagonal with hyperfine coupling constants of around A = 5.5–10 MHz. While the intensity of HYSCORE signals is determined by the number of coupled nuclei, the coordination number is difficult to directly determine, as the signal intensifies when A/2 approaches νI (ν31P = 5.6 MHz) and is also affected by experimental parameters. The separate ν31P signal on the diagonal indicates additional, more remote 31P-containing species in outer solvation spheres with weaker but non-zero hyperfine coupling constants. Coupling parameters of A(19F) = 1.3 MHz and A(1H) = 0.6 MHz are extracted from the observed maximum antidiagonal intensity at the respective Larmor frequencies. The HYSCORE signals exhibit a specific curvature, which is pronounced for 1H and to some degree for 31P and 19F: generally, the further away resonances are from the diagonal, the more these resonances move away from the anti-diagonal toward higher frequencies, as discussed in more detail in ref. 103. This line shape results from a continuous variation of hyperfine tensor values due to a conformational distribution rather than exactly defined atomic positions in the frozen state,91,103,104 which is also consistent with the observed line broadening in Fig. 1. The shift away from the nuclear Larmor frequency towards higher frequency directly on the diagonal, as best seen for 31P but also to some degree for 1H, is indicative of the dipolar contribution to the hyperfine interaction.105 The signal at low ν1,2 results from imperfect microwave pulses and limited excitation bandwidth.
For the sample with added LiPO2F2, only signals due to coupling to 31P and 1H are resolved. A doublet on the ν31P antidiagonal from strongly coupled 31P spins is detected at identical frequencies as observed for the mildly heated sample; however, it is far less intense. In contrast, the intensity maximum is found on the diagonal, the signal arising from a large number of weakly interacting and thus more remote 31P spins. The 1H signal is also located on the diagonal and is not as clearly split as for the mildly heated sample, again indicating more remote 1H nuclei.
DFT calculations of Ax,y,z and subsequent spectra simulation (Fig. 2, lower row) allow a variety of structural models to be compared to the experimental data. For the pristine sample, a simulated HYSCORE spectrum of a Mn(II) complex sixfold coordinated by EC provides a good match to the experiment. The broadening of the diagonal peak, seen in the spectral simulations and assigned to weak coupling to nearby 1H spins, arises from both the broadening due to T2e relaxation, with the values used in this simulation specified in the Methods section, and the slightly varying hyperfine tensors for all 24 involved weakly coupled 1H nuclei. In the experiment, nuclei from outer solvation shells and conformational differences between complexes also contribute to the broadening, which are both disregarded in all simulations. Mn(II) complexes with sixfold coordination containing either both fluorophosphate and EC ligands, or both HPO2F2 and EC ligands resemble the experimental spectrum of the mildly heat-treated sample, consistent with complexes proposed above. Other structure models, including bidentate oxygen coordination of PO2F2−, or PF6− coordination as a source for 31P and 19F couplings, are precluded, since they lead to hyperfine coupling values or simulated HYSCORE features that are not consistent with the experimental spectra as, for example, the coupling to the 31P spins is too weak (Fig. S2). The underlying ratio of protic HPO2F2 to aprotic PO2F2− is not extractable, because the effect of protonation on the simulated spectrum is minor (Fig. S2). The weaker peak seen on the diagonal near the 31P frequency in the experimental spectrum is assigned to P-containing complexes that are in more distant (outer sphere) coordination shells: all P-containing ligands—even PF6−—in the first coordination shell give rise to pronounced 31P hyperfine coupling.
DFT seems to underestimate hyperfine coupling strengths, particularly for 19F and 1H, as also observed for predicted 55Mn coupling tensors (Table S1). As discussed previously,106,107 d5 systems still prove particularly challenging for DFT due to the large electron–electron exchange and correlation interactions, also manifested in DFT functional sensitivity (Fig. S1 and S2). Apart from DFT errors, the optimised lowest-energy structures also likely deviate significantly from the experimental situation. The proposed Mn(II) fluorophosphate structures, and in particular the lowest-energy location of the proton in the HPO2F2 ligands, depend on the HPO2F2:PO2F2− ratio (Fig. S3). Whereas in [Mn(HPO2F2)6]2+, all –OH groups face away from the center of the complex, with Mn–H distances of 5.0–5.5 Å, inter-ligand hydrogen bonds are formed in [Mn(PO2F2)3(HPO2F2)3]−, resulting in [–Mn–O–P–O–H–O–P–O–] rings with shorter Mn–H distances of ∼3.9 Å. Hydrogen bonding via smaller rings, e.g. [–Mn–O–P–O–H–O–], is also conceivable. Additionally, outer solvation shells might affect bond lengths, angles, and dihedrals, which have a large effect on all hyperfine coupling tensors of the complex,91,103 which might explain deviations between experimental and simulated spectra.
Considering the sample with LiPO2F2, the experimental spectrum is noticeably different from the mildly heated electrolyte sample's HYSCORE spectrum and does not reflect pure inner sphere fluorophosphate coordination. Assuming a lack of protic HPO2F2 in this solution and charge neutrality, the first structural model considered was a [Mn(PO2F2)2(EC)4] complex. The reduced symmetry and therefore larger zero-field splitting (Fig. S1) could explain the broad field-swept EPR spectrum (Fig. 1). Although the intensity contribution of 1H resonances increases relative to complexes with no EC molecules, agreement with the experimental HYSCORE spectrum is still poor. Nonetheless, this and related complexes are still likely responsible for the weak signals along the 31P anti-diagonal, with associated couplings to 19F and 1H too weak to be resolved. The intense 31P resonance near the diagonal indicates the presence of a superimposed second complex type, the stronger 1H diagonal peak than seen for the degraded electrolyte suggesting that more EC ligands are present in the Mn2+ coordination shell. In contrast to the mildly heated sample, fluorophosphate is present as unprotonated PO2F2−. A preference of PO2F2− for Li+ might hinder Mn2+ coordination, placing it in outer solvation shells of Mn2+ resulting in an intense 31P signal on the diagonal, indicating weaker hyperfine interactions. The underlying Mn(II) carbonate complex would account for the 1H diagonal signal. Additional experiments show that the relative ratio of strongly to weakly coupled 31P increases with the LiPO2F2:Mn2+ ratio (Fig. S4, ratio ranging from 2:1 to 10:1). This indicates that PO2F2− is more likely to move from an outer to the inner solvation shell with increasing PO2F2− concentration, following the law of mass action.
Another possible explanation of the HYSCORE spectra involves the formation of Mn(II) complexes containing side products as ligands. A number of (oligo-)organo-(fluoro)phosphates can form through PO2F2− reaction with EC or EMC,108,109 which could bind to Mn2+ via carbonate fragments that are chemically bonded to phosphate(s), accounting for the HYSCORE 31P and 1H signals. However, we consider such degradation products unlikely because significant amounts only form above 40 °C,110 and the 19F NMR spectra of these samples showed that the dominant degradation product is PO2F2−/HPO2F2.
Q-band ENDOR
To support the assumption that trace moisture is responsible for HPO2F2 formation in the mildly heated sample, Davies-type electron nuclear double resonance (ENDOR) spectroscopy at Q-band was performed. In an ENDOR experiment, radiofrequency irradiation νrf drives nuclear transitions and, thus, modifies the evolution of magnetization under the (isotropic and anisotropic) hyperfine interaction, altering the electron spin echo intensity. This experiment often allows the principal components of the hyperfine tensor Ax,y,z to be extracted, unlike in a HYSCORE experiment, where the modulation amplitude tends to vanish for orientations aligned with the principal axes.111 Here, the method is used to compare the 19F–Mn2+ and 1H–Mn2+ hyperfine interactions in multiple electrolytes, including electrolytes containing either LiPF6 or LiTFSI salts, before and after heating and with added water (Fig. 3). Their corresponding 1D field-swept EPR spectra are displayed in Fig. S5. The pristine LiPF6 electrolyte sample exhibits an intense ENDOR signal at around ν1H (50.45 MHz) assigned to carbonates, and a low-intensity signal around ν19F assigned to PF6− in outer solvation shells (Fig. 3, left panel, bottom spectrum).11 After mild heating (24 h at 35 °C), the hyperfine coupling to both 19F and 1H is larger; the signals centred around ν19F become more intense, with the more distinct features indicating better defined complexes. Since fluorophosphates result from LiPF6 decomposition, pristine and heated (24 h at 45 °C) samples were also prepared in a LiTFSI-based, but otherwise identical, electrolyte for comparison. The spectrum of the pristine LiTFSI electrolyte resembles that of the LiPF6 electrolyte. After heating, however, a very different ligand environment is detected, with couplings (predominantly from 1H) that are in very good agreement with the spectral fingerprint of [Mn(OH2)6]2+.112,113 Water has clearly been formed, either as part of an electrolyte degradation reaction or because it has entered into the NMR tube during the heating step. When 1 vol% (0.56 M) H2O was intentionally added to the LiPF6 electrolyte pristine sample, a very similar spectrum to that seen for the LiTFSI heated sample, i.e., [Mn(OH2)6]2+, is observed. However, after heating this sample for 24 h at 45 °C, the spectrum (Fig. 3, top right) is essentially identical to that of the mildly heated sample without water addition, indicating that fluorophosphate has formed, and suggesting that fluorophosphate coordination is preferred over H2O coordination.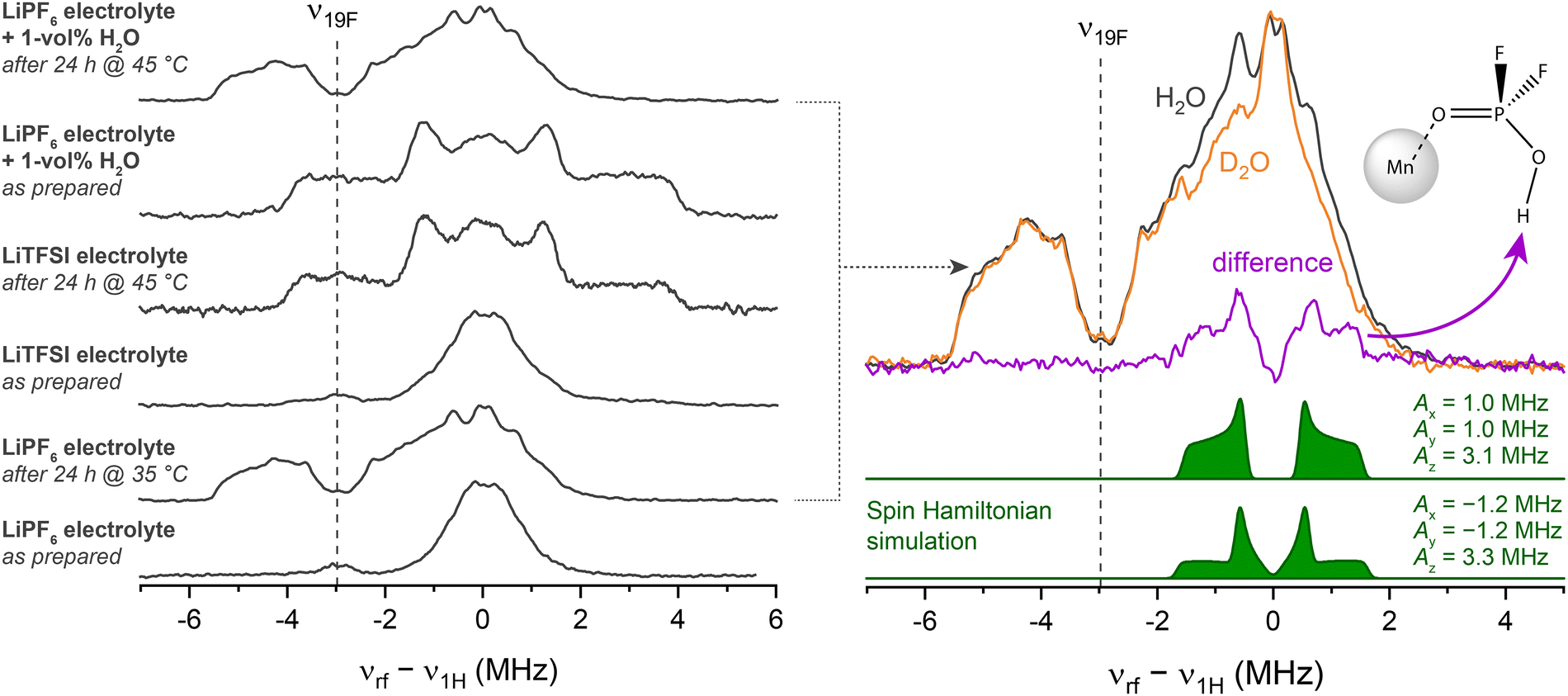 | ||
| Fig. 3 Davies-type electron nuclear double resonance (ENDOR) spectra recorded at 34 GHz (Q-band) of a series of samples containing 8 mM Mn2+ in a 3:7 EC:EMC solvent with 1 M Li salt (LiPF6 and LiTFSI) as indicated (left). The radiofrequency axis is shifted to yield the 1H Larmor frequency at 0 MHz. Right: ENDOR spectra of the LiPF6-containing electrolyte with either 1 vol% H2O (black) or D2O (orange). Spin Hamiltonian fits to these spectra (green) were adjusted to reproduce the difference spectrum (purple). The complex shown, Mn(HPO2F2), was used to simulate the spectra, with simulations for two different axial hyperfine tensors being shown. | ||
The ENDOR spectrum of this mildly heated (wet) LiPF6 electrolyte sample contains multiple overlapping hyperfine coupling patterns, possibly from more than one species. To reduce complexity, D2O instead of H2O was added to the solution so as to form DPO2F2 via
| LiPF6 + 2D2O → DPO2F2 + 3DF + LiF | (1) |
NMR spectroscopy
The paramagnetic relaxation theory underpinning this section can be found in our previous work,11 and in many other reviews;114 a condensed version is also presented in the SI.NMR relaxometry of degraded electrolyte solutions
To understand how coordination may change between pristine and degraded electrolyte solutions, pristine solutions were heated at 60 °C for five days and the spin lattice R1 (= 1/T1) relaxation rates were measured daily. Relaxation rates of 1 M LiPF6 in 3:7 EC:EMC electrolyte solutions containing either 1 mM Mn(TFSI)2 or 1 mM Ni(TFSI)2 are presented in Fig. 4 as paramagnetic relaxation rates (R1p). Values were calculated by subtracting the relaxation rates R1d of pristine diamagnetic electrolyte solutions from the measured relaxation rates, R1p = R1 − R1d,62 where R1d for 1 M LiPF6 in 3:7 EC:EMC v/v is 0.6307 ± 0.0002 s−1 for 1H of EC and 0.352 ± 0.003 s−1 for 19F of PF6−, as determined in our previous work.115 This method assumes that the R1d relaxation rates of pristine solutions remain constant over a period of heating, or with the addition of a small amount of contaminant. We suggest that minor compositional changes to the electrolyte should not significantly affect bulk properties such as viscosity, and that the concentration of degradation species is small relative to the other solution components: 1 M LiPF6, 4.5 M EC, and 6.8 M EMC.
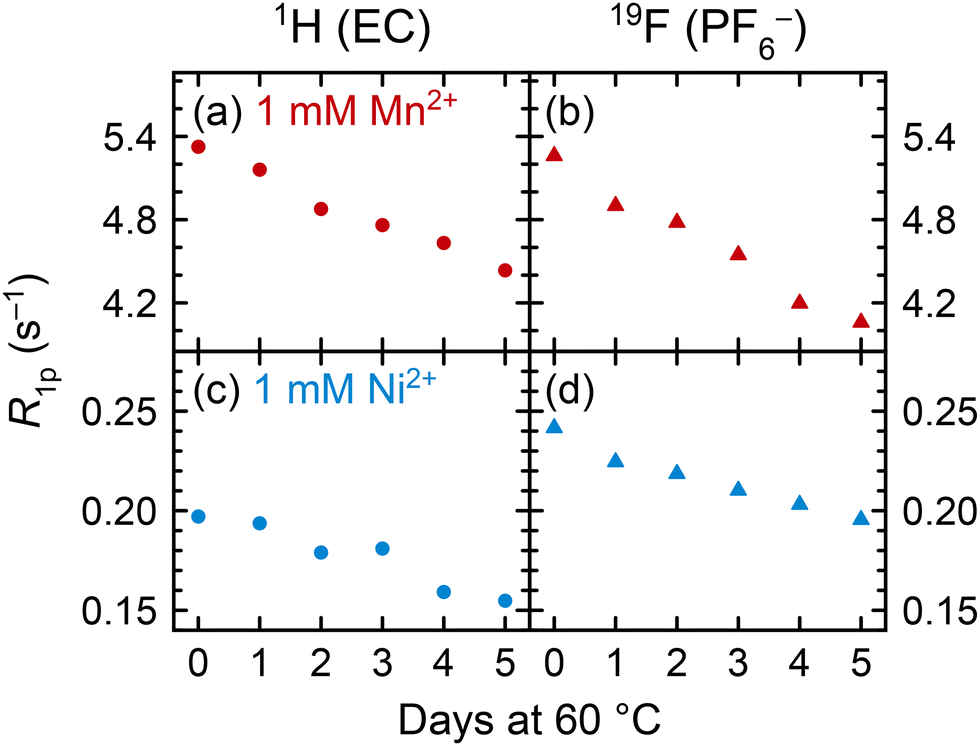 | ||
| Fig. 4 (a) and (c) 1H EC (circles) and (b) and (d) 19F PF6− (triangles) longitudinal paramagnetic relaxation rates (R1p) of Mn2+- or Ni2+-containing electrolyte solutions across five days of heating at 60 °C. Electrolyte solutions comprised 1 M LiPF6 in 3:7 EC:EMC (v/v) with 1 mM Mn(TFSI)2 (red) or Ni(TFSI)2 (blue). Solutions were cooled to ambient temperature (∼25 °C) before relaxation rates were measured. | ||
In both Mn2+- and Ni2+-containing electrolyte solutions, the 1H (EC) and 19F (PF6−) R1p values show a clear decrease across the heating period. With no substantial change in the total concentration of solution components, the decrease in R1p values indicates a decrease in the number of EC or PF6− molecules in the transition metal solvation shells. This suggests that the transition metal ions must be preferentially coordinating to a new component in the degraded electrolyte. (Electrolyte degradation species were not observable in these samples owing to both the paramagnetic ions and the use of a solid-state spectrometer for these measurements; however, later NMR experiments with heated diamagnetic electrolyte showed PO2F2− and HF as the major degradation species (Fig. 7), which is also consistent with analysis of heated EPR samples.) While the lifetimes of the different Mn2+/Ni2+ complexes may change, affecting NMR relaxation times,11 the EPR results in the previous section clearly indicate changes in the nature of the complexes present in the degraded electrolyte, consistent with the NMR results shown here.
Transition metal coordination to PO2F2−
To isolate the effect of PO2F2− on transition metal coordination, LiPO2F2 was directly added to pristine electrolyte solutions. Fig. 5a shows NMR spectra for 1 M LiPF6 in 3:7 EC:EMC (v/v) containing added LiPO2F2, Mn2+, or both; Fig. 5b shows relaxation measurements for solutions containing 1 mM Mn2+ or Ni2+ and 0–50 mM LiPO2F2.
 | ||
| Fig. 5 (a) 19F spectra and (b) 1H EC and 19F PF6− relaxation data of pristine electrolyte solutions to which LiPO2F2 was added. 19F spectra are shown for solutions containing LiPO2F2, Mn2+, or both; inset shows enlargement of the PO2F2− resonance. Longitudinal paramagnetic relaxation rates (R1p) and R2/R1 ratios are shown for the 1H EC (circles) and 19F PF6− resonances (triangles), in solutions containing Mn2+ (red) or Ni2+ (blue). Electrolyte solutions comprised 1 M LiPF6 in 3:7 EC:EMC (v/v). | ||
As expected, the 19F NMR spectrum of the diamagnetic solution containing LiPO2F2 (Fig. 5a, black line) shows narrow doublets for both PF6− and PO2F2−, while the solution that contains Mn2+ but not LiPO2F2 (red line) shows a broadened PF6− doublet only. However, in the solution containing both Mn2+ and LiPO2F2 (purple line), the PF6− doublet is much narrower than its counterpart in the Mn2+-only solution; additionally, the PO2F2− doublet is severely broadened relative to the PO2F2− doublet in the diamagnetic solution, indicating faster relaxation. The same behaviour can be seen in Fig. 5b, panel (ii), where the 19F PF6− R1p values decrease in Mn2+-containing solutions as LiPO2F2 is added.
In Fig. 5b, panels (i–iv), the 1H (EC) and 19F (PF6−) R1p values decrease with increasing LiPO2F2 concentration. 1H EMC relaxation rates also decrease (not shown). The diamagnetic relaxation rates should not be affected in these samples as the solution viscosity increase due to the addition of 0–50 mM LiPO2F2 is likely minor. Regardless, a viscosity increase would lead to an increase rather than a decrease in relaxation rates, as the solutions are in the fast motion regime.11 The decrease in R1p values is consistent with the results in Fig. 4, where EC and PF6− relaxation rates slowed as PO2F2− and other fluorophosphates were generated in situ by heating. The decrease in R1p values in Fig. 5b begins to level off at ∼10–25 mM LiPO2F2 addition, indicating a very strong competition for Ni2+/Mn2+–PO2F2− complexation over Ni2+/Mn2+–PF6− interactions/complexes. The larger decrease in 19F R1p rates over the 1H values suggests that EC is still involved in binding to Ni2+/Mn2+ even in the presence of PO2F2−, which is consistent with the EPR results.
Fig. 5b, panel (v) shows the R2/R1 ratios for the same solutions studied in panels (i)–(iv). The only instance where the R2/R1 ratio exceeds 2 is the 19F PF6− relaxation in Mn2+-containing solutions (red triangles), where the R2/R1 ratio decreases from 9.0 to 5.4 as 50 mM LiPO2F2 is added. This indicates that a different relaxation mechanism is driving the R2 rates in this system. As discussed in our previous paper,11 and explored further in the SI, one potential source of an R2/R1 ratio greater than one is a contribution of the contact (through-bond) relaxation mechanism to R2, while the major source of R1 relaxation originates from a dipolar (though-space) interaction. Thus, an R2/R1 ratio of ≫1 could be ascribed to a short-lived inner sphere complex. Since the hyperfine interaction for a Mn2+–PF6− inner sphere complex should remain the same with added LiPO2F2, the convergence of R2 towards R1 could then be ascribed to a decrease in the relative fraction of inner sphere vs. outer sphere Mn2+–PF6− complexes. However, the HYSCORE EPR spectrum shows no evidence of an inner sphere complex—i.e., no strong 19F/31P–Mn2+ hyperfine interaction is seen, while the ENDOR spectrum of the same sample reveals only a weak interaction likely due to an anisotropic (dipolar, through-space) interaction, indicating the presence of some PF6− anions in the outer coordination shells in the frozen electrolytes. Thus, the time that a PF6− anion spends on average in an inner sphere complex must be extremely short, so that it is not captured in the frozen solution. It is also possible that a second relaxation mechanism involving a change in the dynamics of exchange in and out of the outer sphere coordination shell may contribute to some of the observed changes in the relaxation processes, but to test this proposal is beyond the scope of this work.
While it could be argued that the PO2F2− addition slowed the 1H and 19F relaxation rates by causing Mn2+ precipitation, it has been shown that LiPO2F2 does not cause transition metal precipitation in battery electrolytes.32 Two experiments in this work also suggest that precipitation does not occur: (i) when a solution of 1 mM Mn2+ in 3:7 EC:EMC was saturated with LiPO2F2, ICP-OES measurement revealed that the Mn concentration in solution was unchanged; (ii) the NMR spectra in Fig. 5a show that when 10 mM LiPO2F2 is added to a solution of 1 M LiPF6 in 3:7 EC:EMC + 1 mM Mn2+, the PO2F2− resonance becomes extremely broad, but does not disappear, indicating that it is interacting with Mn2+ but is still present in solution. Notably, the result of transition metal coordination to PO2F2− in solution, shown by both EPR and NMR, may also apply to other cell chemistries, as PF6− salts are used in beyond-lithium systems, including sodium-ion, potassium-ion, and calcium-ion cells.116
Transition metal coordination to other model degradation species
We now explore Mn2+ and Ni2+ coordination to two other degradation species, ethylene glycol (EG) and acetylacetonate (acac−) (Fig. 6). The solubility of Li(acac) in the electrolyte solution was found to be very small, so data are presented here for solutions that are saturated with Li(acac), roughly estimated to occur at ∼5 mM Li(acac). Fig. 6a shows that after ethylene glycol addition to Mn2+- or Ni2+-containing solution, the 1H (EC) R1p values decrease, suggesting that ethylene glycol replaces (at least some) EC in the Mn2+ and Ni2+ solvation shells. Interestingly, instead of the 19F PF6− R1p values decreasing, as occurs upon LiPO2F2 addition, the R1p values increase. This suggests that PF6− is in closer proximity to Mn2+ or Ni2+ when ethylene glycol is present. It is not likely that the fraction of coordinated PF6− increases, because EC coordination is preferred over PF6− coordination and so the displacement of EC by an even more preferred coordination agent should not then facilitate additional PF6− binding. | ||
| Fig. 6 Relaxation behaviour upon either (a) ethylene glycol or (b) Li(acac) addition to paramagnetic electrolyte solutions. R1p values and R2/R1 ratios are shown for 19F PF6− resonances (triangles) and 1H EC resonances (circles) in electrolyte solutions containing 1 mM Mn(TFSI)2 (red) or Ni(TFSI)2 (blue). The electrolyte solution comprises 1 M LiPF6 in 3:7 EC:EMC (v/v). | ||
Concurrently, the 1H (EC) R2/R1 ratio remains constant and the 19F (PF6−) R2/R1 ratio decreases in Mn2+-containing solutions. It is noted here that the absolute value of R2 decreases, not just the R2/R1 ratio—if the R2/R1 decrease were caused by R2 remaining constant as R1 increased, then R2 would only decrease by 11–18%, not 43–63% as is observed. Together, the longitudinal and transverse relaxation behaviour is consistent with the interpretation of an inner solvation shell mostly composed of EC, which is displaced by ethylene glycol: the smaller size of ethylene glycol compared to EC results in a smaller first solvation shell, resulting in a closer approach by PF6− ions in the second solvation shell to Mn2+ or Ni2+, along, potentially, with different dynamics for PF6− exchange, and an increase in R1 and decrease in R2. It is possible that EG binding reduces the albeit small probability that PF6− forms an inner sphere complex, also reducing the R2/R1 ratio.
We have previously suggested coordination to the ethylene glycol dianion for vanadyl ions dissolved in degraded LiPF6 electrolyte solutions, supported by pulsed EPR measurements.70 We note that the experiments in Fig. 6a used neutral ethylene glycol, but it is possible that if it exists in situ as the dianion, then the transition metal charge would be satisfied by the anionic ethylene glycolate, and coordination to PF6− may become significantly less favoured. The acidity of the electrolyte solution likely helps determine the favourability of transition metal coordination by determining whether potential coordinating ligands are neutral or charged.
The addition of Li(acac) to Mn2+-containing electrolyte (Fig. 6b) causes similar results as addition of LiPO2F2 (Fig. 5b), where the 1H (EC) and 19F (PF6−) R1p values both decrease and the 19F (PF6−) R2/R1 ratio also decreases, suggesting both EC and PF6− are displaced from the inner and outer spheres. Notably, the ICP-OES results that will be discussed later show that Li(acac) causes precipitation of ∼9% of 1 mM Mn2+ in 3:7 EC:EMC, which may have contributed to the observed decrease in R1p values. Still, the qualitative interpretation of the NMR data here and the conclusion that acac− affects the Mn2+ solvation shell is reasonable, because the dramatic change in the 19F (PF6−) R2/R1 ratio shows that the results in Fig. 6b are not only due to loss of overall Mn2+ in solution. Had a small amount of Mn2+ simply precipitated, and the overall concentration of Mn2+ were slightly smaller, the 19F (PF6−) R2/R1 ratio would not be expected to change significantly.
Electrolyte degradation induced by dissolved transition metals
Previous work has asserted that dissolved Mn2+ can itself induce PF6− degradation.8,14 This is now explored in work presented in Fig. 7, which shows the 19F (PF6−) and 1H (EC) relaxation rates, as well as the measured concentrations of PO2F2− and HF generated, in solutions of 0.1 mM and 1.0 mM Mn2+. In one group of samples, Mn2+ was added before heating at 60 °C for ten weeks, and in the other group, Mn2+ was added after heating; if the Mn2+ ions were inert, results should be identical. NMR of the heated diamagnetic sample prior to any Mn2+ addition indicated that PO2F2− and HF were the major degradation species. Mn concentrations were confirmed with ICP-OES and are indicated in parentheses in Fig. 7a; we note that duplicate samples were prepared from separate stock solutions of Mn2+, accounting for the small variation in Mn concentrations between some duplicates.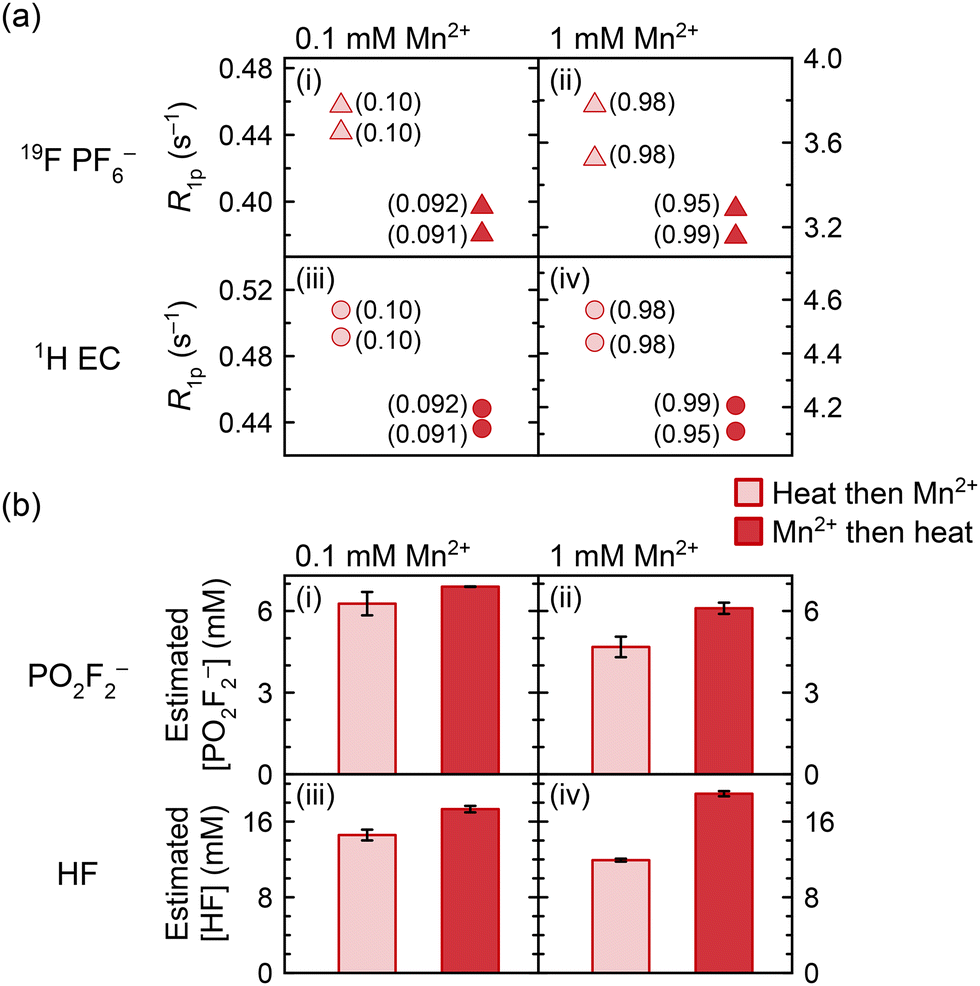 | ||
| Fig. 7 (a) 19F PF6− (triangles) and 1H EC (circles) longitudinal paramagnetic relaxation rates (R1p) and (b) estimated concentrations of PO2F2− and HF in electrolyte solutions to which Mn2+ was added before (red) or after heating (pink) at 60 °C for ten weeks. Values next to the relaxation data indicate the Mn concentration in mM measured by ICP-OES. PO2F2− estimated concentrations were derived from integrating 19F spectra relative to PF6− (assuming 1 M PF6−); HF estimated concentrated were derived from 19F spectra as well as from 1H spectra relative to EC (assuming 4.5 M EC). Electrolyte solutions comprised 1 M LiPF6 in 3:7 EC:EMC (v/v), to which 0.1 or 1.0 mM Mn(TFSI)2 was added. | ||
Since the generation of fluorophosphate degradation species results in reduced PF6− and EC relaxation rates, as the fluorophosphates compete for the Mn2+ ions (Fig. 4 and 5), the relaxation rates may be used as a proxy to monitor the degree of degradation in electrolyte solutions with the same Mn2+ concentration. In Fig. 7a, 1H (EC) and 19F (PF6−) longitudinal paramagnetic relaxation rates are slower in all samples that were heated in the presence of Mn2+, compared to samples where Mn2+ was added after heating. However, ICP-OES also showed a small but significant difference in the concentration of the nominally 0.1 mM samples, where the samples with Mn2+ addition before heating contained 0.1 mM Mn but the samples with Mn2+ addition after heating contained 0.09 mM Mn (attributed to experimental error in solution preparation). This difference in concentration is sufficient to explain most of the relaxation difference between the datasets. While small differences in Mn concentrations were also seen in the samples nominally containing 1.0 mM of Mn2+, the sample containing 0.99 mM Mn added pre-heating produced slower relaxation rates than both samples containing 0.98 mM Mn added post-heating. This suggests that paramagnetic samples underwent more degradation than diamagnetic samples; i.e., the Mn(TFSI)2 addition caused an albeit small amount of additional electrolyte degradation.
The samples were then diluted 10× with d6-DMSO to improve the visibility of the degradation peaks, as has been discussed elsewhere.117 Further NMR analysis concerning the effects of deuterated solvents, including d6-DMSO, CD3CN, and MeOD, on the Mn2+ solvation shell is provided in the SI (Fig. S7); the most significant effects are observed with DMSO, which appears to displace EC from the solvation shell. The resulting 19F spectra were used to estimate the concentrations of PO2F2− and HF (Fig. 7b). These are still only estimates, as the d6-DMSO does not completely prevent the broadening of degradation peaks; hence, comparisons are only drawn between samples containing the same Mn2+ concentrations, and not between the 0.1 and 1.0 mM datasets. In all cases, the PO2F2− and HF concentrations are larger in the samples that were heated in the presence of Mn2+. It is possible that some of the degradation enhancement is due to the Mn(TFSI)2 addition requiring several transfers during the preparation and dilution of stock solutions, which may have caused minor contamination from trace adsorbed water on vial or pipette surfaces, despite work being carried out in an argon glovebox. That said, the degradation enhancement is larger when more Mn2+ is present: there is a ∼10% increase in PO2F2− concentration and ∼19% increase in HF concentration with 0.1 mM Mn2+ added, and a ∼30% increase in PO2F2− concentration and ∼59% increase in HF concentration with 1.0 mM Mn2+ added, even with potential underestimation of degradation species at 1.0 mM due to signal broadening. The apparent dose–response relationship confirms that at least some of the degradation enhancement is specifically due to the Mn(TFSI)2, consistent with the relaxometry results in Fig. 7a.
Previous work on electrolytes heated with 0 versus 100 or 300 mM Mn2+ has shown a dramatic effect of Mn2+ on electrolyte degradation, with paramagnetic solutions turning brown after 8 days at 55 °C.8,14 It was proposed that Mn2+ coordination to PF6− facilitated the formation of reactive PF5.8 (We note that a brown discolouration may also indicate MnO2 formation and possibly oxygen/water contamination.) In comparison, the degradation of samples in our work is relatively minor, particularly considering that samples were heated at 60 °C for ten weeks. No difference in colouration was observed between diamagnetic and paramagnetic samples after heating. The EPR and NMR data (Fig. 2 and 5, respectively) clearly show that Mn2+ preferentially interacts with PO2F2− over PF6− in the inner coordination shell, which is also confirmed by DFT calculations (Table S2). At small Mn2+ concentrations, such as those used in this work, the probability that PF6− enters the Mn2+ inner coordination shell once Mn2+ is surrounded by PO2F2− may be reduced significantly, consistent with the 19F NMR studies. Thus, the PF6− decomposition effect may become self-limiting, especially in cases where LiPO2F2 is used as an electrolyte additive. At the large Mn2+ concentrations used in previous studies, far more PO2F2− would be required to coordinate all Mn2+, and complete coordination to PO2F2− may not be possible, resulting in “free” Mn2+ and far more electrolyte degradation. The dissolved Mn2+ concentration in cells is generally small as Mn2+ migrates to the negative electrode and deposits there; furthermore, when dissolution occurs in situ into an already-degraded electrolyte, PO2F2− may already exist in solution, which is not the case when Mn2+ model compounds are added to pristine electrolyte. Therefore, while the results in Fig. 7 are consistent with previous observations of Mn2+-induced electrolyte degradation, it also appears that this effect is unlikely to be a main contributor to electrolyte degradation in cells, with most of the harm of dissolved transition metals occurring once they are deposited at the anode. These results also show that NMR relaxometry may be used in these systems as a probe for electrolyte degradation, as long as the concentration of paramagnetic ions is constant; this method may also be applied in other systems, if a degradation product coordinates more closely to the transition metal ions than to the pristine electrolyte species.
Fig. 8 shows ICP-OES results for the concentration of Mn2+ or Ni2+ in 3:7 EC:EMC (v/v) after 1 mM solutions were shaken with various lithium salts and stored for two days. All lithium salts studied induced some amount of transition metal precipitation, including the organic lithium salts, suggesting that metals may be able to precipitate directly in both the outer organic SEI and the inner inorganic SEI. The addition of Li2O2 to Mn2+-containing solution resulted in the solution immediately turning brown in colour, likely due to the oxidation of Mn2+ to yield the brown MnO2 precipitate. The largest differences between Ni2+ and Mn2+ precipitation behaviour were observed with Li2O, LiF, and LiOMe, where more Mn2+ precipitation occurred, and with LiOEt and Li(acac), where more Ni2+ precipitation occurred. Generally, this seems to suggest a possible preference for Mn2+ precipitation with harder, more inorganic components, and Ni2+ precipitation with softer, more organic components, which is consistent with the classification of Mn2+ as a hard cation and Ni2+ as borderline.118,119 This may explain why some studies that have examined metals both in the electrolyte solution and on the anode surface have shown that a greater fraction of dissolved Mn2+ is deposited compared to dissolved Ni2+ or Co2+.41,120 Interestingly, with Li(acac), not only did ∼91% of Mn2+ remain in solution, but the lithium concentration in solution increased from 0.474 ± 0.004 mM when Li(acac) was stored in a diamagnetic solution to 3.43 ± 0.02 mM when Li(acac) was stored in a Mn2+-containing solution; i.e., an additional 2.96 mM Li dissolved, or ∼3:1 Li:Mn ratio. This enhanced Li dissolution suggests that Li/Mn exchange occurs, and Mn2+ chelation by acac− is favourable, but the product is soluble. This ICP-OES result also supports the interpretation of the relaxometry results in Fig. 6b.
 | ||
| Fig. 8 (a) Mn2+ or (b) Ni2+ concentrations in 3:7 EC:EMC (v/v) solutions after lithium salts were added to solutions containing ∼1 mM of Mn(TFSI)2 or Ni(TFSI)2. The initial concentration is indicated by the dashed line (1.07 mM for both Mn2+ and Ni2+); bars indicate the new transition metal concentration two days after lithium salt addition. All concentrations were measured by ICP-OES. Error bars show the range obtained for duplicate experiments (i.e., measurements on two different solutions). | ||
While previous studies have exposed transition metal ions to cycled graphite anodes,73,75–79 specific learnings from those experiments are complicated by the fact that the exact SEI composition is unknown, and it is therefore unknown which species the transition metal ions are exchanging with. We also note that the exact nature of an SEI greatly depends on the specific electrolyte composition used and particularly any SEI additives employed. This experiment expands on that previous work, as well as previous work studying ex situ exchange with some inorganic lithium salts,39,74,75 revealing that transition metals can exchange with many different species, including organic salts.
The ICP-OES results show that various SEI salts can induce transition metal precipitation out of EC:EMC solution, lending credence to metathesis deposition mechanisms. (We note that this is not necessarily mutually exclusive with possible reduction mechanisms; the first step for any reduction that may occur is initial incorporation into the SEI, and one such mechanism occurs via ion exchange.) However, when metals are surrounded by suitably coordinating species in a degraded electrolyte solution, such as PO2F2−/HPO2F2, desolvation and precipitation at the anode surface may become less favourable. More exchange-type experiments that are explicitly conducted in the presence of degradation species such as PO2F2− may help determine the realistic likelihood of transition metal metathesis. The combined EPR, NMR, and ICP-OES results in this work present a spectrum of coordination behaviour that influences transition metal deposition. With species like PF6−, Mn2+ coordination is not preferred; with species like PO2F2− or acac−, Mn2+ coordination is preferred but the complexes are soluble; and with species like PO43− or OH−, Mn2+ precipitates almost completely. The location where dissolved transition metals ultimately precipitate will likely depend on which species are nearby: if a favourable species encouraging precipitation is present when Mn2+ dissolves, it will more likely deposit on the cathode or separator; otherwise, Mn2+ may bind to species in solution and either remain in solution or deposit on the anode, depending on the strength and favourability of coordination. A clearer understanding of transition metal complexation behaviours in battery electrolytes may facilitate strategies that prevent transition metal dissolution, encourage precipitation of metal ions at the cathode, and/or enable sequestration of metal ions in solution, ultimately preventing deposition and harmful reactions at the anode.
Conclusions
Paramagnetic solution NMR and pulsed one and two-dimensional EPR and ENDOR were used to probe the coordination of Mn2+ and Ni2+ in degraded electrolytes, probing the electron–nuclear interactions from two perspectives. Heat-degraded electrolyte solutions, or solutions containing simulated degradation species, were examined to unravel transition metal coordination to specific solution components of interest. Although in pristine electrolyte solutions the solvation shell is mainly comprised of the solvent molecule EC, the transition metal solvation shell is considerably changed in degraded electrolyte solutions; these results illustrate the power of this joint approach in determining the structures of metal complexes in pristine and degraded electrolytes.Pulsed EPR experiments of frozen solutions show that in heat- and water-degraded LiPF6 electrolytes, Mn2+ is coordinated to a combination of anionic and protonated fluorophosphate species. This is consistent with measurements of NMR relaxation rates showing that EC and PF6− are increasingly displaced in the Mn2+ and Ni2+ inner and outer coordination spheres when electrolytes are heated. When water or D2O is added to pristine, non-degraded electrolyte solutions containing either LiPF6 or LiTFSI salts, EPR experiments show that it enters the Mn2+ first coordination sphere and displaces EC to form [Mn(OH2)6]2+/[Mn(OD2)6]2+; however, in LiPF6-containing electrolytes, the protonated fluorophosphate species that arise from PF6− hydrolysis, specifically HPO2F2, ultimately displace the water from the first coordination shell. The electron-1H hyperfine coupling tensors obtained by simulations of the ENDOR experiments indicate a Mn–H distance of between 3.7 and 4.8 Å, consistent with an HPO2F2 inner sphere complex. EPR and NMR of solutions containing added LiPO2F2 complement the results from heated electrolyte solutions, indicating that PO2F2− is present in both inner sphere and outer sphere Mn2+ complexes.
While EPR experiments of frozen solutions allow for a more detailed structural analysis of Mn2+ complex formation, the use of NMR relaxometry permits a more rapid screening of the effect of additives and electrolyte degradation on complex formation. For example, NMR experiments showed that the addition of ethylene glycol to the electrolyte causes the displacement of EC in the first solvation shell of Mn2+ and Ni2+ (as seen for PO2F2−), now also reducing the size of the first solvation shell. Addition of the larger acetylacetonate ligand displaced both EC and PF6−. Examination of samples heated with and without Mn2+ indicated that dissolved transition metals likely do not contribute significantly to decomposition of the bulk electrolyte solution in the period after dissolution and before deposition, as has been previously suggested. This is because transition metals are present in small concentrations and coordination to typical electrolyte degradation species is preferred over coordination to, and decomposition of, pristine electrolyte components. Lastly, ICP-OES experiments targeting the strength and extent of transition metal coordination showed that many lithium-containing salts proposed to be present in the SEI appear to induce some amount of transition metal exchange, typically associated with precipitation of the corresponding transition metal salts.
This work demonstrates the importance of considering degradation species in the transition metal solvation shell, which must be considered when designing representative experimental model systems and when performing computational work. This work also supports the metathesis mechanism of transition metal incorporation into the SEI, while identifying differences in how Mn2+ and Ni2+ are desolvated, which may affect the likelihood of metal deposition and the activity of deposited metals towards electrolyte decomposition. The phenomenon of transition metal coordination to solution degradation species that is shown in this work may present an additional, largely overlooked mechanism for dissolution behaviour. New insights about transition metal coordination may be leveraged to reduce transition metal dissolution and deposition, preventing battery degradation and extending lifetime.
Conflicts of interest
There are no conflicts to declare.Data availability
In addition to the NMR, EPR, and DFT figures and tables in the supplementary information (SI), we include ZIP files with further supporting data for EPR and DFT studies of Mn coordination. These include CSV files with hyperfine coupling constants and XYZ files with final structures of all complexes. Supplementary information: Further DFT calculations, experimental and simulated EPR spectra, and NMR relaxometry. See DOI: https://doi.org/10.1039/d5ee01250c.Acknowledgements
JPA thanks the Natural Sciences and Engineering Research Council of Canada for funding; JPA and CPG acknowledge support from the Faraday Institution under grant number FIRG001 and from the Royal Society via a professorship to CPG; CS acknowledges computing resources granted by RWTH Aachen University under project rwth0701; EJ and CPG acknowledge support from the ERC grant BATNMR no 835073.Notes and references
- W. Choi and A. Manthiram, J. Electrochem. Soc., 2006, 153, A1760 CrossRef CAS.
- H. Zheng, Q. Sun, G. Liu, X. Song and V. S. Battaglia, J. Power Sources, 2012, 207, 134–140 CrossRef CAS.
- J. A. Gilbert, I. A. Shkrob and D. P. Abraham, J. Electrochem. Soc., 2017, 164, A389–A399 CrossRef CAS.
- C. Zhan, T. Wu, J. Lu and K. Amine, Energy Environ. Sci., 2018, 11, 243–257 RSC.
- R. Jung, F. Linsenmann, R. Thomas, J. Wandt, S. Solchenbach, F. Maglia, C. Stinner, M. Tromp and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, A378–A389 CrossRef CAS.
- O. C. Harris, S. E. Lee, C. Lees and M. Tang, JPhys Energy, 2020, 2, 032002 CrossRef CAS.
- Y.-K. Han, K. Lee, S. Kang, Y. S. Huh and H. Lee, Comput. Mater. Sci., 2014, 81, 548–550 CrossRef CAS.
- C. Wang, L. Xing, J. Vatamanu, Z. Chen, G. Lan, W. Li and K. Xu, Nat. Commun., 2019, 10, 3423 CrossRef PubMed.
- X. Cui, J. Sun, D. Zhao, J. Zhang, J. Wang, H. Dong, P. Wang, J. Zhang, S. Wu, L. Song, N. Zhang, C. Li and S. Li, J. Energy Chem., 2023, 78, 381–392 CrossRef CAS.
- S.-B. Son, Z. Zhang, J. Gim, C. S. Johnson, Y. Tsai, M. Kalensky, S. Lopykinski, O. Kahvecioglu, Z. Yang, A. T. Montoya and I. Bloom, J. Phys. Chem. C, 2023, 127, 1767–1775 CrossRef CAS.
- J. P. Allen, C. Szczuka, H. E. Smith, E. Jónsson, R.-A. Eichel, J. Granwehr and C. P. Grey, Phys. Chem. Chem. Phys., 2024, 26, 19505–19520 RSC.
- A. Jarry, S. Gottis, Y.-S. Yu, J. Roque-Rosell, C. Kim, J. Cabana, J. Kerr and R. Kostecki, J. Am. Chem. Soc., 2015, 137, 3533–3539 CrossRef CAS PubMed.
- I. A. Shkrob, A. J. Kropf, T. W. Marin, Y. Li, O. G. Poluektov, J. Niklas and D. P. Abraham, J. Phys. Chem. C, 2014, 118, 24335–24348 CrossRef CAS.
- X. Luo, L. Xing, J. Vatamanu, J. Chen, J. Chen, M. Liu, C. Wang, K. Xu and W. Li, J. Energy Chem., 2022, 65, 1–8 CrossRef CAS.
- A. Du Pasquier, A. Blyr, P. Courjal, D. Larcher, G. Amatucci, B. Gérand and J.-M. Tarascon, J. Electrochem. Soc., 1999, 146, 428–436 CrossRef CAS.
- D. R. Gallus, R. Schmitz, R. Wagner, B. Hoffmann, S. Nowak, I. Cekic-Laskovic, R. W. Schmitz and M. Winter, Electrochim. Acta, 2014, 134, 393–398 CrossRef CAS.
- D. Huang, C. Engtrakul, S. Nanayakkara, D. W. Mulder, S.-D. Han, M. Zhou, H. Luo and R. C. Tenent, ACS Appl. Mater. Interfaces, 2021, 13, 11930–11939 CrossRef CAS.
- J. C. Hestenes, J. T. Sadowski, R. May and L. E. Marbella, ACS Mater. Au, 2023, 3, 88–101 CrossRef CAS PubMed.
- Y. Tesfamhret, H. Liu, E. J. Berg and R. Younesi, RSC Adv., 2023, 13, 20520–20529 RSC.
- L. J. Reinschlüssel, L. Reuter, P. Rapp, M. Bock, A. Berger, M. A. Schilling and H. A. Gasteiger, J. Electrochem. Soc., 2025, 172, 060536 CrossRef.
- K. Leung, Chem. Mater., 2017, 29, 2550–2562 CrossRef CAS.
- N. N. Intan, K. Klyukin and V. Alexandrov, ACS Appl. Mater. Interfaces, 2019, 11, 20110–20116 CrossRef CAS PubMed.
- H. R. Morin, D. G. Graczyk, Y. Tsai, S. Lopykinski, H. Iddir, J. C. Garcia, N. Dietz Rago, S. Trask, L. Flores, S.-B. Son, Z. Zhang, N. M. Johnson and I. Bloom, ACS Appl. Energy Mater., 2020, 3, 2565–2575 CrossRef CAS.
- R. Sahore, D. C. O’Hanlon, A. Tornheim, C.-W. Lee, J. C. Garcia, H. Iddir, M. Balasubramanian and I. Bloom, J. Electrochem. Soc., 2020, 167, 020513 CrossRef CAS.
- J. Lüchtefeld, H. Hemmelmann, S. Wachs, K. J. Mayrhofer, M. T. Elm and B. B. Berkes, J. Phys. Chem. C, 2022, 126, 17204–17211 CrossRef.
- J. Lüchtefeld, M.-Y. Lee, H. Hemmelmann, S. Wachs, C. Behling, K. J. J. Mayrhofer, M. T. Elm and B. B. Berkes, ACS Omega, 2023, 8, 32606–32614 CrossRef.
- C. L. Campion, W. Li, W. B. Euler, B. L. Lucht, B. Ravdel, J. F. DiCarlo, R. Gitzendanner and K. M. Abraham, Electrochem. Solid-State Lett., 2004, 7, A194 CrossRef CAS.
- C. L. Campion, W. Li and B. L. Lucht, J. Electrochem. Soc., 2005, 152, A2327–A2334 CrossRef CAS.
- A. V. Plakhotnyk, L. Ernst and R. Schmutzler, J. Fluorine Chem., 2005, 126, 27–31 CrossRef CAS.
- S. Wiemers-Meyer, M. Winter and S. Nowak, Phys. Chem. Chem. Phys., 2016, 18, 26595–26601 RSC.
- K. Nagayama, K. Kamioka, E. Iwata, H. Oka, Y. Tokunaga and T. Okada, Electrochemistry, 2001, 69, 6–9 CrossRef CAS.
- S. Klein, P. Harte, S. van Wickeren, K. Borzutzki, S. Röser, P. Bärmann, S. Nowak, M. Winter, T. Placke and J. Kasnatscheew, Cell Rep. Phys. Sci., 2021, 2, 100521 CrossRef CAS.
- J. Chen, L. Xing, X. Yang, X. Liu, T. Li and W. Li, Electrochim. Acta, 2018, 290, 568–576 CrossRef CAS.
- C. Wang, L. Yu, W. Fan, J. Liu, L. Ouyang, L. Yang and M. Zhu, ACS Appl. Energy Mater., 2018, 1, 2647–2656 CrossRef CAS.
- S. Klein, L. Haneke, P. Harte, L. Stolz, S. van Wickeren, K. Borzutzki, S. Nowak, M. Winter, T. Placke and J. Kasnatscheew, ChemElectroChem, 2022, 9, e202200469 CrossRef CAS.
- B. Yang, H. Zhang, L. Yu, W. Fan and D. Huang, Electrochim. Acta, 2016, 221, 107–114 CrossRef CAS.
- G. Yang, J. Shi, C. Shen, S. Wang, L. Xia, H. Hu, H. Luo, Y. Xia and Z. Liu, RSC Adv., 2017, 7, 26052–26059 RSC.
- W. Zhao, G. Zheng, M. Lin, W. Zhao, D. Li, X. Guan, Y. Ji, G. F. Ortiz and Y. Yang, J. Power Sources, 2018, 380, 149–157 CrossRef CAS.
- S. Klein, P. Harte, J. Henschel, P. Bärmann, K. Borzutzki, T. Beuse, S. van Wickeren, B. Heidrich, J. Kasnatscheew, S. Nowak, M. Winter and T. Placke, Adv. Energy Mater., 2021, 11, 2003756 CrossRef CAS.
- S. Klein, S. van Wickeren, S. Röser, P. Bärmann, K. Borzutzki, B. Heidrich, M. Börner, M. Winter, T. Placke and J. Kasnatscheew, Adv. Energy Mater., 2021, 11, 2003738 CrossRef CAS.
- Y. Tesfamhret, PhD thesis, Uppsala University, 2022.
- H. Kim, J. G. Lee, S.-M. Kim, J. Kim, J. Soon, J. B. Lee, J. Mun, J. H. Ryu and S. M. Oh, Meet. Abstr., 2016, MA2016-03, 1049 CrossRef.
- S. Y. Park, S. Park, H. Y. Lim, M. Yoon, J. Choi, S. K. Kwak, S. Y. Hong and N. Choi, Adv. Sci., 2023, 10, 2205918 CrossRef CAS PubMed.
- Y.-G. Cho, S. H. Jung, J. Jeong, H. Cha, K. Baek, J. Sung, M. Kim, H. T. Lee, H. Kong, J. Cho, S. J. Kang, J. M. Park and H.-K. Song, ACS Appl. Mater. Interfaces, 2021, 13, 9965–9974 CrossRef CAS PubMed.
- A. Banerjee, Y. Shilina, B. Ziv, J. M. Ziegelbauer, S. Luski, D. Aurbach and I. C. Halalay, J. Electrochem. Soc., 2017, 164, A6315–A6323 CrossRef CAS.
- J. Seo, J. Im, M. Kim, D. Song, S. Yoon and K. Y. Cho, Small, 2024, 20, 2312132 CrossRef CAS PubMed.
- M.-H. Ryou, S. Hong, M. Winter, H. Lee and J. W. Choi, J. Mater. Chem. A, 2013, 1, 15224 RSC.
- T. Dong, P. Mu, S. Zhang, H. Zhang, W. Liu and G. Cui, Electrochem. Energ. Rev., 2021, 4, 545–565 CrossRef CAS.
- N.-Y. Kim, J. Moon, M.-H. Ryou, S.-H. Kim, J.-H. Kim, J.-M. Kim, J. Bang and S.-Y. Lee, Adv. Energy Mater., 2022, 12, 2102109 CrossRef CAS.
- C. Zou, Y. Huang, L. Zhao, W. Ren, Z. Zhao, J. Liu, X. Li, M. Wang, B. Guo and Y. Lin, ACS Appl. Mater. Interfaces, 2022, 14, 14226–14234 CrossRef CAS PubMed.
- H. Saneifar, K. Zaghib and D. Bélanger, ACS Appl. Energy Mater., 2020, 3, 647–657 CrossRef CAS.
- G. G. Amatucci, A. Blyr, C. Sigala, P. Alfonse and J. M. Tarascon, Solid State Ionics, 1997, 104, 13–25 CrossRef CAS.
- A. Blyr, A. Du Pasquier, G. Amatucci and J.-M. Tarascon, Ionics, 1997, 3, 321–331 CrossRef CAS.
- S. Solchenbach, G. Hong, A. T. S. Freiberg, R. Jung and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, A3304–A3312 CrossRef CAS.
- O. C. Harris, Y. Lin, Y. Qi, K. Leung and M. H. Tang, J. Electrochem. Soc., 2020, 167, 013502 CrossRef CAS.
- X. Zhang, Z. Cui and A. Manthiram, Adv. Energy Mater., 2022, 2103611 CrossRef CAS.
- Y. Terada, Y. Nishiwaki, I. Nakai and F. Nishikawa, J. Power Sources, 2001, 97–98, 420–422 CrossRef CAS.
- J. Wandt, A. Freiberg, R. Thomas, Y. Gorlin, A. Siebel, R. Jung, H. A. Gasteiger and M. Tromp, J. Mater. Chem. A, 2016, 4, 18300–18305 RSC.
- L. Hanf, J. Henschel, M. Diehl, M. Winter and S. Nowak, Electrophoresis, 2020, 41, 697–704 CrossRef CAS PubMed.
- J. P. Allen and C. P. Grey, Chem. Commun., 2023, 59, 1677–1680 RSC.
- A. Abragam and B. Bleaney, Electron Paramagnetic Resonance of Transition Ions, Clarendon Press, 1970 Search PubMed.
- I. Bertini, C. Luchinat, G. Parigi and E. Ravera, NMR of Paramagnetic Molecules: Applications to Metallobiomolecules and Models, Elsevier, 2016 Search PubMed.
- M. Jiang, B. Key, Y. S. Meng and C. P. Grey, Chem. Mater., 2009, 21, 2733–2745 CrossRef CAS.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A1361–A1377 CrossRef CAS.
- A. T. S. Freiberg, M. K. Roos, J. Wandt, R. de Vivie-Riedle and H. A. Gasteiger, J. Phys. Chem. A, 2018, 122, 8828–8839 CrossRef CAS PubMed.
- X. Ren, J. Wang, Z. Peng and L. Lu, Chem. Sci., 2018, 9, 231–237 RSC.
- Y. Yu, P. Karayaylali, Y. Katayama, L. Giordano, M. Gauthier, F. Maglia, R. Jung, I. Lund and Y. Shao-Horn, J. Phys. Chem. C, 2018, 122, 27368–27382 CrossRef CAS.
- U. Heider, R. Oesten and M. Jungnitz, J. Power Sources, 1999, 81–82, 119–122 CrossRef CAS.
- M. Metzger, B. Strehle, S. Solchenbach and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, A1219 CrossRef CAS.
- C. Szczuka, R.-A. Eichel and J. Granwehr, ACS Appl. Energy Mater., 2022, 5, 449–460 CrossRef CAS.
- L. Liu, S. Gu, S. Wang, X. Zhang and S. Chen, RSC Adv., 2020, 10, 1704–1710 RSC.
- Z. Ruff, C. Xu and C. P. Grey, J. Electrochem. Soc., 2021, 168, 060518 CrossRef CAS.
- C. Zhan, J. Lu, A. J. Kropf, T. Wu, A. N. Jansen, Y.-K. Sun, X. Qiu and K. Amine, Nat. Commun., 2013, 4, 2437 CrossRef PubMed.
- H. Shin, J. Park, A. M. Sastry and W. Lu, J. Power Sources, 2015, 284, 416–427 CrossRef CAS.
- D. R. Vissers, Z. Chen, Y. Shao, M. Engelhard, U. Das, P. Redfern, L. A. Curtiss, B. Pan, J. Liu and K. Amine, ACS Appl. Mater. Interfaces, 2016, 8, 14244–14251 CrossRef CAS PubMed.
- C. Zhan, X. Qiu, J. Lu and K. Amine, Adv. Mater. Interfaces, 2016, 3, 1500856 CrossRef.
- V. Meunier, M. L. D. Souza, M. Morcrette and A. Grimaud, J. Electrochem. Soc., 2022, 169, 070506 CrossRef CAS.
- H. Xu, Z. Li, T. Liu, C. Han, C. Guo, H. Zhao, Q. Li, J. Lu, K. Amine and X. Qiu, Angew. Chem., Int. Ed., 2022, 61, e202202894 CrossRef CAS PubMed.
- Q. Xie, J. Chen, L. Xing, X. Zhou, Z. Ma, B. Wu, Y. Lin, H. Zhou and W. Li, J. Energy Chem., 2022, 69, 389–396 CrossRef CAS.
- P. Lu and S. J. Harris, Electrochem. Commun., 2011, 13, 1035–1037 CrossRef CAS.
- P. Höfer, A. Grupp, H. Nebenführ and M. Mehring, Chem. Phys. Lett., 1986, 132, 279–282 CrossRef.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- E. Van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- R. Van Leeuwen, E. Van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 1272–1281 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. A. Pantazis, X.-Y. Chen, C. R. Landis and F. Neese, J. Chem. Theory Comput., 2008, 4, 908–919 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Neese, Inorg. Chim. Acta, 2002, 337, 181–192 CrossRef.
- N.-A. Stamos, E. Ferentinos, M. Chrysina, C. P. Raptopoulou, V. Psycharis, Y. Sanakis, D. A. Pantazis, P. Kyritsis and G. Mitrikas, Inorg. Chem., 2020, 59, 3666–3676 CrossRef CAS PubMed.
- F. Neese, J. Chem. Phys., 2005, 122, 034107 CrossRef PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- S. Stoll and R. D. Britt, Phys. Chem. Chem. Phys., 2009, 11, 6614–6625 RSC.
- C. Fan, P. E. Doan, C. E. Davoust and B. M. Hoffman, J. Magn. Reson. (1969), 1992, 98, 62–72 CrossRef CAS.
- P. E. Doan, N. S. Lees, M. Shanmugam and B. M. Hoffman, Appl. Magn. Reson., 2010, 37, 763–779 CrossRef PubMed.
- H. Y. Carr and E. M. Purcell, Phys. Rev., 1954, 94, 630–638 CrossRef CAS.
- S. Meiboom and D. Gill, Rev. Sci. Instrum., 1958, 29, 688–691 CrossRef CAS.
- T. A. Stich, S. Lahiri, G. Yeagle, M. Dicus, M. Brynda, A. Gunn, C. Aznar, V. J. DeRose and R. D. Britt, Appl. Magn. Reson., 2007, 31, 321–341 CrossRef CAS PubMed.
- T. M. Atkins, J. H. Walton, M. P. Singh, S. Ganguly, O. Janka, A. Y. Louie and S. M. Kauzlarich, J. Phys. Chem. C, 2017, 121, 1948–1956 CrossRef CAS PubMed.
- C. Duboc, M.-N. Collomb and F. Neese, Appl. Magn. Reson., 2010, 37, 229–245 CrossRef.
- J. W. Larson and B. Su, J. Chem. Eng. Data, 1994, 39, 33–35 CrossRef CAS.
- C. Szczuka, P. Jakes, R.-A. Eichel and J. Granwehr, Adv. Energy Sustainability Res., 2021, 2, 2100121 CrossRef CAS.
- S. A. Dikanov and A. T. Taguchi, Appl. Magn. Reson., 2020, 51, 1177–1200 CrossRef CAS.
- A. Pöppl and L. Kevan, J. Phys. Chem., 1996, 100, 3387–3394 CrossRef.
- F. Neese, J. Chem. Phys., 2003, 118, 3939–3948 CrossRef CAS.
- M. L. Munzarova, P. Kubacek and M. Kaupp, J. Am. Chem. Soc., 2000, 122, 11900–11913 CrossRef CAS.
- M. Kubot, L. Frankenstein, E. Muschiol, S. Klein, M. Esselen, M. Winter, S. Nowak and J. Kasnatscheew, ChemSusChem, 2022, e202202189 Search PubMed.
- G. Gachot, P. Ribière, D. Mathiron, S. Grugeon, M. Armand, J.-B. Leriche, S. Pilard and S. Laruelle, Anal. Chem., 2011, 83, 478–485 CrossRef CAS PubMed.
- V. Kraft, W. Weber, M. Grützke, M. Winter and S. Nowak, RSC Adv., 2015, 5, 80150–80157 RSC.
- S. Van Doorslaer, eMagRes, John Wiley & Sons, Ltd, Chichester, UK, 2017, vol. 6, pp. 51–70 Search PubMed.
- D. van Ormondt, R. de Beer, M. Brouha and F. de Groot, Z. Naturforsch., A, 1969, 24, 1746–1751 CAS.
- S. Un, Inorg. Chem., 2013, 52, 3803–3813 CrossRef CAS PubMed.
- A. J. Pell, G. Pintacuda and C. P. Grey, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 111, 1–271 CrossRef CAS PubMed.
- J. P. Allen, C. A. O’Keefe and C. P. Grey, J. Phys. Chem. C, 2023, 127, 9509–9521 CrossRef CAS PubMed.
- J. Zhang, X. Yao, R. K. Misra, Q. Cai and Y. Zhao, J. Mater. Sci. Technol., 2020, 44, 237–257 CrossRef CAS.
- J. P. Allen and C. P. Grey, J. Phys. Chem. C, 2023, 127, 4425–4438 CrossRef CAS PubMed.
- S. F. A. Kettle, Coordination Compounds, Thomas Nelson and Sons, London, 1969 Search PubMed.
- J. E. Huheey, E. A. Keiter and R. L. Keiter, Inorganic Chemistry: Principles of Structure and Reactivity, HarperCollins College Publishers, New York, 4th edn, 1993 Search PubMed.
- W. M. Dose, I. Temprano, J. P. Allen, E. Björklund, C. A. O’Keefe, W. Li, B. L. Mehdi, R. S. Weatherup, M. F. L. De Volder and C. P. Grey, ACS Appl. Mater. Interfaces, 2022, 14, 13206–13222 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |