
Recent advances in fabricating injectable hydrogels via tunable molecular interactions for bio-applications
Wenshuai
Yang
 ab,
Jingsi
Chen
b,
Ziqian
Zhao
b,
Meng
Wu
b,
Lu
Gong
b,
Yimei
Sun
b,
Charley
Huang
b,
Bin
Yan
c and
Hongbo
Zeng
*b
ab,
Jingsi
Chen
b,
Ziqian
Zhao
b,
Meng
Wu
b,
Lu
Gong
b,
Yimei
Sun
b,
Charley
Huang
b,
Bin
Yan
c and
Hongbo
Zeng
*b
aZhongyuan Critical Metals Laboratory, Zhengzhou University, Zhengzhou 450001, Henan, China
bDepartment of Chemical & Materials Engineering, University of Alberta, Edmonton, Alberta T6G 1H9, Canada. E-mail: hongbo.zeng@ualberta.ca
cNational Engineering Laboratory for Clean Technology of Leather Manufacture, College of Biomass Science and Engineering, Sichuan University, Chengdu 610065, China
First published on 7th November 2023
Abstract
Hydrogels with three-dimensional structures have been widely applied in various applications because of their tunable structures, which can be easily tailored with desired functionalities. However, the application of hydrogel materials in bioengineering is still constrained by their limited dosage flexibility and the requirement of invasive surgical procedures. Compared to traditional hydrogels, injectable hydrogels, with shear-thinning and/or in situ formation properties, simplify the implantation process and reduce tissue invasion, which can be directly delivered to target sites using a syringe injection, offering distinct advantages over traditional hydrogels. These injectable hydrogels incorporate physically non-covalent and/or dynamic covalent bonds, granting them self-healing abilities to recover their structural integrity after injection. This review summarizes our recent progress in preparing injectable hydrogels and discusses their performance in various bioengineering applications. Moreover, the underlying molecular interaction mechanisms that govern the injectable and functional properties of hydrogels were characterized by using nanomechanical techniques such as surface forces apparatus (SFA) and atomic force microscopy (AFM). The remaining challenges and future perspectives on the design and application of injectable hydrogels are also discussed. This work provides useful insights and guides future research directions in the field of injectable hydrogels for bioengineering.
Introduction
Hydrogels with highly porous structures and water retention ability have been widely applied in designing soft electronics,1–3 artificial skins,4–6 sensors,7,8 and actuators9,10 owing to their high electrical conductivity and outstanding stretchability. Moreover, due to their biomimetic structure and desirable biocompatibility,11–13 hydrogels also share many common characteristics with human tissues,14,15 making them a more promising candidate than traditional biomaterials when used in biomedical applications involving controlled drug delivery16,17 and soft tissue engineering.18,19 Despite these advantages, the application of conventional hydrogels in the bio-application field is limited by the inconvenience of the implantation process of subcutaneous hydrogels.20–22 Moreover, soft hydrogel materials with limited mechanical properties are easily deformed when subjected to external forces,15,23,24 which will damage the integrity of their polymer network and eventually lead to the loss of functionality.To address this issue, injectable hydrogels that can be directly applied to the target site through a syringe have been developed.25–27 Compared to conventional pre-formed hydrogels, injectable hydrogels greatly simplify the operation process28 and minimize tissue invasion29 in various biomedical engineering applications, such as targeted drug delivery30–32 and precise wound dressing.33,34 Injectable hydrogels can be designed through either in situ formation and/or shear thinning strategies.35 For the injectable hydrogel with in situ formation property, the hydrogel precursor solution is prepared under mild conditions and can be transported through syringe injection due to its low viscosity.36 Then, the hydrogel solution will convert into a gel state at the target site through a sol–gel transition triggered by the external stimulus, such as a temperature37–39 or pH40–42 change. For the stimuli-responsive in situ formation injectable hydrogel, the aqueous pre-gel solution can be transported to the target tissue site under mild conditions by simple injection. The solution then converts into its gel state due to the change in physiochemical conditions, such as temperature or pH, at the injection site. The stimulus responsive injectable hydrogel can be applied to improve the efficiency of cancer treatment by specifically releasing the drugs at target cancer tissues to reduce the side effects of cancer therapy.43 The in situ formation injectable hydrogel can also be designed to match the mechanical properties of the target tissue to accelerate the tissue regeneration process.44 Recently, shear-thinning hydrogels provided an alternative strategy to construct injectable hydrogels by reducing their viscosity in the injection process with increasing shear rate and restoring to the original gel state upon relaxation.45–50 The viscosity (η) of the shear thinning hydrogel should not be higher than 1 Pa s at a high shear rate (∼100 s−1) in the injection process to avoid causing needle clogging,51 which enables a pre-formed hydrogel with desired properties to be directly delivered to the target site in the in vivo system by syringe injection.52–54 Shear-thinning injectable hydrogels enable the delivery of pre-formed hydrogels with tailored physiochemical properties into in vivo environments via injection. Moreover, the shear-thinning injectable hydrogel can recover its hydrogel network and rapidly restore its modulus without being affected by physiochemical conditions of the bio-environment after the injection process, indicating its great potential as a wound dressing material.55 Injectable hydrogels with in situ formation and/or shear thinning properties can be designed by incorporation of reversible physical bonds, such as hydrogen bonding56–58 and hydrophobic interaction,44,59 or dynamic covalent interactions, including dynamic Schiff base interactions60,61 and disulfide bonds62 into a hydrogel network. For example, temperature-responsive injectable hydrogels can be designed by incorporating a thermally responsive polymer, such as poly(N-isopropyl acrylamide) (PNIPAM), into their a hydrogel network. The PNIPAM polymer remains soluble at temperatures below its lower critical solution temperature (LCST).63 When the temperature increases above the LCST, the PNIPAM polymer chains collapse, resulting in a sol–gel transition process. Alternatively, injectable hydrogels can also be triggered by a change in pH. For instance, dynamic Schiff-base hydrogels remain in a sol state under acidic conditions but rapidly transition to a gel state under basic conditions due to the formation of reversible imine bonds between aldehydes (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and primary amine groups (–NH2). Additionally, injectable hydrogels based on hydrogen bonds utilize their shear-thinning properties. During the injection process, the hydrogen bonds in the hydrogel network are disrupted under high-stress conditions. They then restore themselves upon relaxation after injection due to their reversible nature.64–66 This remarkable self-healing ability is primarily driven by the dynamic movements of polymer chains driven by physical and/or chemical interactions. Specifically, physically crosslinked self-healing hydrogels including hydrophobic interaction-based hydrogels can be fabricated by incorporating polymers with hydrophobic segments, and hydrogen bond-based hydrogels can be crafted using H-bond-rich polymers such as polyvinyl alcohol (PVA)67 or alginate.68 Chemically crosslinked hydrogels can achieve their self-healing properties by incorporating specific dynamic covalent bonds between polymer chains, such as Schiff-base interactions,69 disulfide bonds,70 and more. These chemical bonds help the hydrogel to reform and reestablish its network structure after damage to achieve self-healing, making the injectable hydrogel a promising candidate for most bioengineering applications.71–74
O) and primary amine groups (–NH2). Additionally, injectable hydrogels based on hydrogen bonds utilize their shear-thinning properties. During the injection process, the hydrogen bonds in the hydrogel network are disrupted under high-stress conditions. They then restore themselves upon relaxation after injection due to their reversible nature.64–66 This remarkable self-healing ability is primarily driven by the dynamic movements of polymer chains driven by physical and/or chemical interactions. Specifically, physically crosslinked self-healing hydrogels including hydrophobic interaction-based hydrogels can be fabricated by incorporating polymers with hydrophobic segments, and hydrogen bond-based hydrogels can be crafted using H-bond-rich polymers such as polyvinyl alcohol (PVA)67 or alginate.68 Chemically crosslinked hydrogels can achieve their self-healing properties by incorporating specific dynamic covalent bonds between polymer chains, such as Schiff-base interactions,69 disulfide bonds,70 and more. These chemical bonds help the hydrogel to reform and reestablish its network structure after damage to achieve self-healing, making the injectable hydrogel a promising candidate for most bioengineering applications.71–74
Both in situ formation and shear-thinning-based injectable hydrogels rely on a sol–gel transition to restore the hydrogel network after injection, utilizing reversible and dynamic bonds within the hydrogel network. Therefore, it holds significant importance to investigate the roles of different types of reversible molecular interactions during the hydrogel injection process, which can provide a deeper understanding of the hydrogel's sol–gel transition behavior and offer new insights into the design of novel injectable hydrogels. Force-measuring techniques, such as surface forces apparatus (SFA)75–78 and atomic force microscopy (AFM),79–82 allow for the direct measurement of intermolecular interactions, which enables a quantitative understanding of the self-healing behaviors of injectable hydrogels. Specifically, SFA allows in situ and real-time force measurements between opposing surfaces in a vapor or a fluid with an absolute surface separation precision of less than 0.1 nm with a force sensitivity of <10−8 N.83–89 On the other hand, AFM allows force measurements between a nanoscopic or microscopic probe and a substrate surface with a typical force sensitivity of 10−11 N.90–94 The AFM probe can also be functionalized to perform direct force measurements at the single-molecule level, allowing quantification of molecular interactions among different components in the hydrogel at the molecular level.95–97
Conventional preformed hydrogels are prone to deformation and damage under high shear pressure during the injection process due to their limited mechanical properties, which will damage their structural integrity and eventually lead to the loss of hydrogel functionality. Consequently, the use of traditional preformed hydrogels in bio-engineering fields is still constrained by their restricted dosage flexibility and the necessity for invasive surgical procedures. Compared to traditional hydrogels, injectable hydrogels that can be directly delivered to target sites through facile syringe injection offer distinct advantages by simplifying the implantation process and minimizing tissue invasion in various bio-engineering applications. In this review, we summarize our recent progress in the development of injectable hydrogels based on reversible physical interactions (such as hydrogen bondings and hydrophobic interactions), dynamic covalent interactions (including dynamic Schiff base interactions and disulfide bonds), and mussel-inspired interactions (such as metal–ligand coordination and cation–π interactions). We also explore their applications in bioengineering fields, such as wound dressing, anti-biofouling, sterilization, and stimulus-responsive drug release. The underlying interaction mechanisms involved in the hydrogel system were further elucidated by using SFA and AFM, providing valuable insights into the shear-thinning, stimulus-responsive, and self-healing behavior of the prepared hydrogels. Furthermore, we discuss the remaining challenges and future perspectives for the development of current injectable hydrogels, providing inspiration and strategies for designing advanced multifunctional injectable hydrogels for various biomedical applications.
Reversible physical molecular interactions
Hydrophobic interactions
Hydrophobic interactions present a promising approach for the development of injectable hydrogels through the formation of hydrophobic domains as crosslinks to construct a hydrogel network. For instance, Meng et al. fabricated a silk fibroin-based injectable hydrogel based on hydrophobic interactions using hydrophobic monomer stearyl methacrylate (C18M) and an amphiphilic regenerated silk fibroin (RSF).98 The C18M as a hydrophobic monomer was initially solubilized within amphiphilic RSF solutions to form a hydrogel system consisting of hydrophobic domains (Fig. 1(a)). As shown in Fig. 1(b) and (c), the as-prepared hydrogel showed a shear thinning behavior because the hydrophobic associations between C18M and RSF micelles were disrupted with high shear stress in the injection process, and the hydrogel network structure can be quickly restored upon relaxation due to the reversible nature of the hydrophobic interaction. Besides, the mechanical properties of the as-prepared hydrogel can be modulated by changing the C18M concentration in the hydrogel. With the C18M content increasing from 0 to 30 mM L−1, the Young's modulus of the resulting hydrogel increased from 0.16 to 0.37 kPa, which can be attributed to the formation of denser hydrophobic interactions in the hydrogel network. Moreover, as shown in Fig. 1(d), the as-prepared hydrogel can automatically recovery its network after damage via resemble of C-18 micelle by the hydrophobic interaction.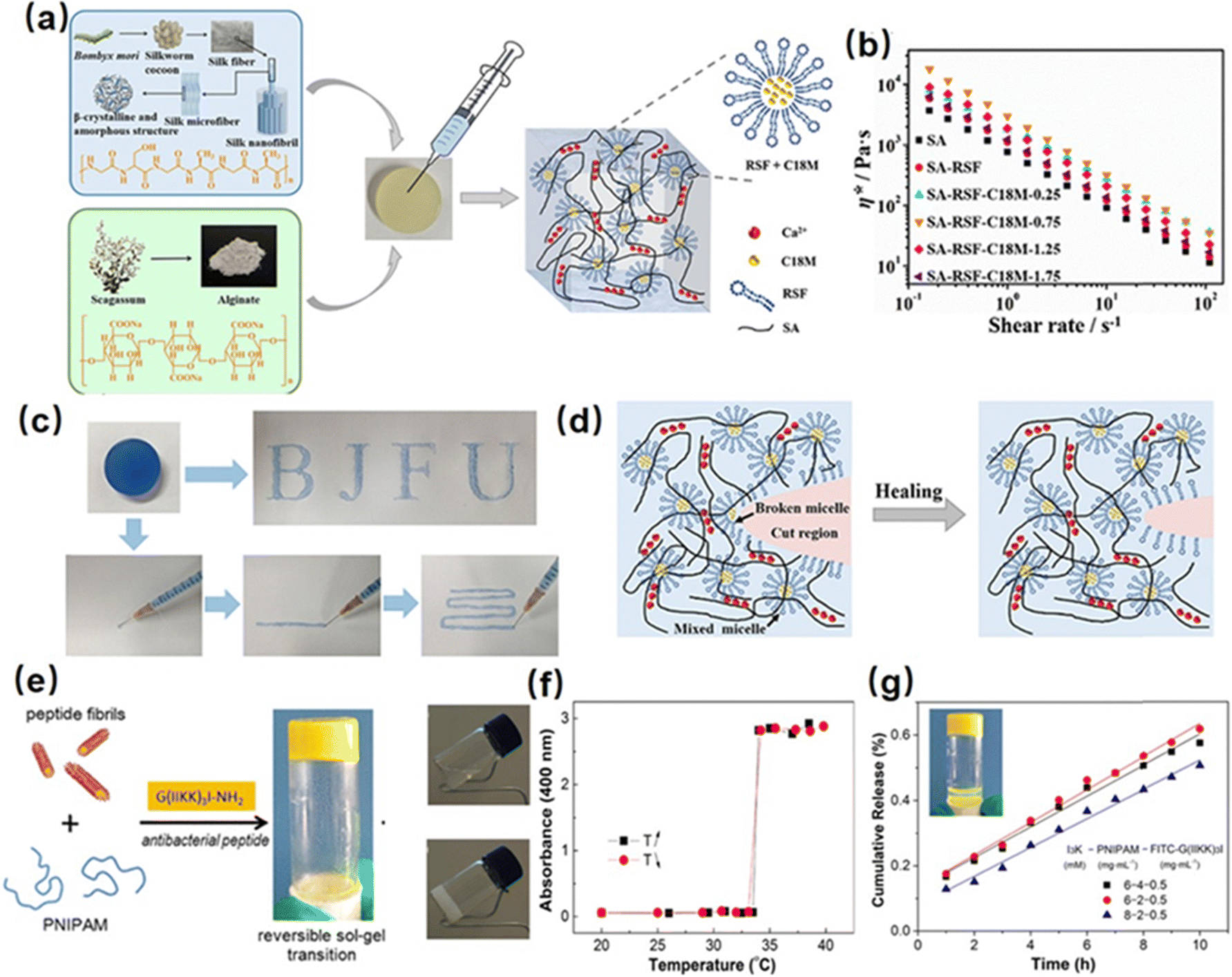 | ||
| Fig. 1 (a) Structure of a silk fibroin-based injectable hydrogel based on hydrophobic interaction using the hydrophobic monomer stearyl methacrylate (C18M) and an amphiphilic regenerated silk fibroin (RSF), (b) and (c) shear thinning and injectability, (d) and self-healing mechanism of the as-prepared hydrogel. (Reproduced with permission from ref. 98. Copyright 2020, American Chemical Society.) (e) Structure of a reversible thermal responsive injectable peptide hydrogel by facilely mixing the peptide nanofibrils with PNIPAAm, (f) variation of turbidities of the hydrogel with temperature changes, (g) drug releasing property of the as-prepared hydrogel. (Reproduced with permission from ref. 63. Copyright 2019, American Chemical Society.) | ||
Recently, poly(N-isopropyl acrylamide) (PNIPAAM) is widely used to construct hydrophobic interaction hydrogels due to its temperature-responsive property. Specifically, PNIPAAM has a lower critical solution temperature (LCST) of around 32–34 °C, at which it undergoes a coil-to-globule conformational transition, thus leading to a sol–gel transition via the formation of hydrophobic domains in the hydrogel network. For instance, Tan et al. synthesized a thermo-sensitive polymer by using alginate as the backbone and PNIPAAm as the pendant group (AAlg-g-PNIPAAm).99 The as-prepared AAlg-g-PNIPAAm co-polymer showed a thermo-sensitive sol–gel property with an LCST of around 35 °C. Below the LCST, the AAlg-g-PNIPAAm co-polymer remains soluble, allowing easy injection through a syringe due to its low viscosity. By increasing the temperature higher than LCST, the extended AAlg-g-PNIPAAm polymer chain would collapse into aggregation due to the dehydration of PNIPAAM, thus enabling hydrogel formation. Moreover, the viscosity of the formed hydrogel can be easily modulated by changing the PNIPAAm content in the AAlg-g-PNIPAAm co-polymer, making the as-prepared injectable hydrogel a great candidate for tissue engineering applications. Besides, Cao et al. fabricated a reversible thermal responsive injectable peptide hydrogel by facilely mixing the peptide nanofibrils with PNIPAAm (Fig. 1(e)).63 At a temperature over LCST (around 32 °C), as shown in Fig. 1(f), the PNIPAAm would collapse into small aggregations due to the hydrophobic interaction and connect with different peptide nanofibrils as a solidified hydrogel 3D network, inducing the sol–gel transition process and increasing solution turbidity. Moreover, as shown in Fig. 1(g), the as-prepared injectable hydrogel can be used as a thermal-sensitive drug-releasing platform by encapsulating the antibacterial peptide into the hydrogel network, which showed a sustained and controlled linear drug-releasing ability with the temperature over LCST.
The hydrophobic interaction between nonpolar polymers plays a crucial role in fabricating hydrophobic interaction-based injectable hydrogels via the formation of small aggregates of hydrophobic moieties by rearranging the structures of surrounding water molecules. However, the direct measurement and quantification of the hydrophobic interaction between two hydrophobic surfaces remain challenging due to the interference of “pseudo-hydrophobic interactions”,100 including the bridging of micro or nanobubbles and/or the electrostatic interactions due to surface charge. Zeng et al. used SFA coupled with a top-view optical microscope to directly measure the hydrophobic interactions between two polystyrene (PS) surfaces and proposed a three-regime interaction model,101 indicating that the long-range hydrophobic interaction (over 20 nm) was mainly attributed to the bridging of micro or sub-micro scope bubbles on PS substrates; an intermediate attractive regime detected around 10 nm could be due to the bridging of nanobubbles or the possibly enhanced proton hopping in water; a short-range interaction detected within several nanometers could be mainly due to the water structuring effects close to the hydrophobic surface. Moreover, the range of detected hydrophobic interaction decreased with increasing NaCl and CaCl2 concentrations, which is related to the formation and stability of air bubbles on hydrophobic surfaces. In order to eliminate the interference of dissolved gases and bubbles on the measured hydrophobic interaction, the following study prepared stable and smooth hydrophobic surfaces by chemically bonding hydrophobic polymers (poly(dimethylsiloxane) (PDMS)) onto the substrates.102 The hydrophobic interactions measured using SFA exhibit an exponential decay with a decay length of ∼0.3 to ∼2 nm, providing a quantitative description of the attraction between two hydrophobic polymer films.
Hydrogen bonding interactions
Hydrogel bonding is one of the most developed strategies for constructing injectable hydrogels owing to its modulated strength, high biocompatibility, stimulus-response, and fast dissociation/association behavior.103–106 For instance, Zhao et al. reported a hydrogen bonding supramolecular injectable hydrogel based on the copolymerization of N-acryloyl glycinamide and N-(2-hydroxypropyl) methacrylamide.107 The as-prepared hydrogel showed shear thinning injectability at 37 °C due to the incorporation of reversible hydrogen bonds into the hydrogel network. Specifically, under high-stress conditions during the injection process, the hydrogen bonding domains in the hydrogel can be disrupted, leading to a decrease in viscosity to 0.1 Pa s. Then, the hydrogel network can restore itself upon relaxation due to the reversible nature of the hydrogen bonds. Moreover, the super hydrophilic hydrogel also showed low organ toxicity and considerable anti-biofouling properties with a protein adsorption amount of only about 4.8 μg cm−2, which can be applied as anti-biofouling implantable material; However, injectable hydrogels only based on hydrogen bondings generally suffered from an intrinsically weak structure with poor stability due to competition of polar water molecules with the formation of intermolecular hydrogen bonds.108,109 Thus, most hydrogen bonding-based hydrogel generally showed limited mechanical properties and may easily dissemble under aqueous conditions. To further improve the stability of hydrogen bonding-based injectable hydrogels, other dynamic reversible bonds such as hydrophobic interactions or electrostatic interactions were introduced to improve their mechanical properties.110–112 For instance, Wang et al. fabricate a guar gum–Cu2+(CG–Cu) hydrogel based on hydrogen bonding by simply dissolving CG into the aqueous solution of CuCl2 at room temperature.113 The incorporation of Cu2+ not only contributed to dissolving the CG polymer as a homogeneous solution, but also enhanced the mechanical strength of CG–Cu hydrogels by the electrostatic interaction between Cu2+ and GC polymers. The as-prepared hydrogel had excellent biocompatibility and could be degraded after 21 days of subcutaneous implantation, which can be designed as a bio-degradable wound dressing material.In our previous work, we reported a supramolecular injectable hydrogel as an internal wound dressing material by the assembly of ABA triblock copolymer (ANGNA) via the formation of hydrogen bondings and hydrophobic interactions between the collapsed P(NIPAM-co-NA6ACA) blocks under gastric conditions (Fig. 2(a)).114 As shown in Fig. 2(b), the as-prepared ANGNA hydrogel exhibited reversible sol–gel transition behavior through multiple cooling–heating cycles with fully restored mechanical properties, and a stable hydrogel network can be instantaneously formed by injecting a 4 °C-preserved co-polymer solution into a PBS buffer solution (pH 3) at 37 °C. The injectability of the as-prepared hydrogel was further demonstrated by “writing” letters of “U of A” via injecting the co-polymer solution onto a platform at 37 °C (Fig. 2(c)). The sol–gel transition behavior at 37 °C can be attributed to the synergistic effect of hydrogen bonds and hydrophobic interactions. Specifically, at a temperature higher than LCST, the NIPAAm parts of the AGNGA co-polymer will dehydrate and result in the formation of hydrophobic domains within the hydrogel network, which further protects the hydrogen bonds formed between NA6ACA, enabling the rapid and reversible sol–gel transition. Additionally, as shown in Fig. 2(d), the ANGNA hydrogels rapidly merged into one single piece after bringing two freshly cut hydrogels together under acidic conditions, and the oscillatory strain sweep measurement also indicated that the hydrogel can fully regain its original mechanical after damage, which can be attributed to the incorporation of reversible hydrophobic interactions and hydrogen bonds into the hydrogel network. Furthermore, the biocompatibility of the ANGNA hydrogel was also evaluated, as shown in Fig. 2(e), the cell viabilities are above 90% for both HeLa and MRC-5 cells, indicating low cytotoxicity of the as-prepared ANGNA copolymer. Moreover, the ANGNA hydrogel dressing showed excellent anti-biofouling properties, which can effectively prevent the accumulation of debris, bacteria, and other microorganisms to cause infections.
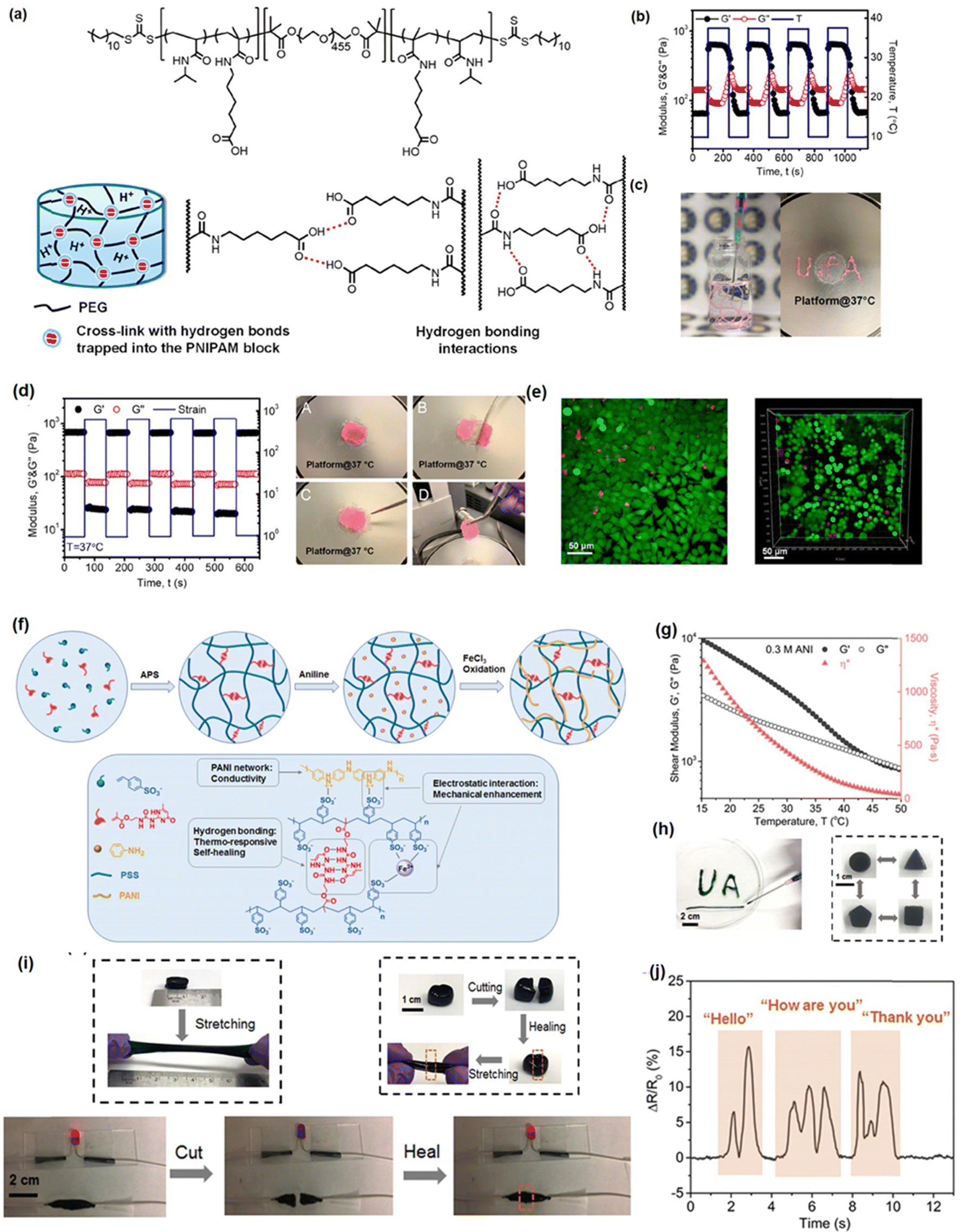 | ||
| Fig. 2 (a) The structure of the injectable ANGNA hydrogel based on the hydrogel bonding via the self-assembly of the ABA triblock copolymer, (b) thermal responsiveness, (c) injectability, (d) self-healing, and (e) biocompatibility properties of the as-prepared ANGNA hydrogel. (Reproduced with permission from ref. 114; Copyright 2021, American Chemical Society); (f) the structure of the Upy-based injectable hydrogel and its (g) shear thinning property, (h) shape-reform and injectable ability, (i) stretchability and self-healing, (j) application as a conductive sensor. (Reproduced with permission from ref. 57; Copyright. 2019, American Chemical Society.) | ||
Recently, by mimicking the DNA double helix structure in biological self-assembly systems, a multiple hydrogen-bonding motif, 2-ureido-4[1H]-pyrimidinone (UPy),115–118 designed by Meijer and co-workers, has been widely used to construct supramolecular network due to its strong dimerization (>106 M−1 in CHCl3) via quadruple hydrogen bonding.119 Taking advantage of the reversible, strong, directional quadruple hydrogen bonding between Upy dimers,120,121 several injectable hydrogels have been designed based on UPy dimers. For example, Zhang et al. prepared self-healing injectable hydrogel based on co-polymerization of UPy and acrylamide monomers.56 The as-prepared hydrogel can be easily injected through an 18-gauge needle due to its shear-thinning property and retains its self-healing properties under physiological pH and temperature conditions due to reversible hydrogen bondings. To further improve the mechanical properties of UPy-based hydrogels, our group recently developed a novel injectable hydrogel with excellent stretchability, conductivity, and self-healing capability by using UPy groups as cross-linkers into polyaniline/poly(4-styrenesulfonate) (PANI/PSS) network (Fig. 2(f)).57 As shown in Fig. 2(g), the as-prepared hydrogel showed a thermo-responsive sol–gel transition property with its shear moduli and viscosity significantly reducing with the temperature increasing from 10 to 50 °C, which can be attributed to the weakened hydrogen-bonding interactions between the UPy dimers at elevated temperatures. Therefore, as shown in Fig. 2(h), the as-prepared hydrogel can be easily shaped and injected into the target site through a syringe after maintaining a temperature higher than the gel–sol transition temperature at 32 °C. Moreover, the addition of Fe3+ ions into the hydrogel network further enhances its mechanical and conductive properties, making the as-prepared hydrogel a stretchable wearable electronics with excellent strain sensing performances. As shown in Fig. 2(i), the as-prepared hydrogel showed excellent stretchability (≈300%) and rapid self-healing ability, which can fully restore its mechanical properties and electrical conductivity within 30 s upon damage by the reversibility of the hydrogen bonds in the hydrogel network. Moreover, taking advantage of its superior conductivity (13 S m−1), the as-prepared hydrogel gave distinct signals in response to different words (Fig. 2(j)), indicating its great potential as an ultra-sensitive sensor.
To better understand the role of hydrogen bonding in the injectable and self-healing processes of as-prepared hydrogels, SFA has been employed for directly measuring the hydrogen bonding interaction between different components involved in the hydrogel network at the nano-scale. The setup of SFA for measuring the normal forces between two surfaces is shown in Fig. 3(a),122 two back-silvered micas with the same thickness (1–5 μm) were first glued onto two cylindrical silica disks (radius R = 2 cm), which were further coated with the polymer solution by dip-coating method. Then, two coated-surfaces were mounted into the SFA chamber in a cross-cylinder configuration with the PBS buffer injected into the confined space between the two surfaces. The interaction forces between surface-coated mica surfaces were measured as a function of the separation distance between two mica surfaces, which was determined by the multiple beam interferometry with optical interference fringes of equal chromatic order (FECO); thus, the surface forces can be determined based on Hooke's law.88,123–126 In our previous work, we quantified the reversible hydrogen-bonding between ANGNA co-polymers by measuring the interaction between ANGNA-coated mica surfaces under neutral and acidic conditions.114 As shown in Fig. 3(b), no adhesion was measured between ANGNA-coated mica surfaces under neutral conditions, which was possibly caused by the electrostatic repulsion between charged polymers at neutral conditions, thus suppressing the formation of intermolecular hydrogen bonds between two ANAGA-coated mica surfaces. In contrast, a significant and reversible adhesion of ∼7.9 mN m−1 (Fig. 3(c)) was detected between ANGNA-coated mica surfaces under acidic conditions. As shown in Fig. 3(d) and (e), such significant and reversible adhesion was likely originated from the formation of intermolecular hydrogen bonds between two mica surfaces because the carboxyl groups of ANGNA are protonated and electroneutral at low pH, leading to the intimate contact between carboxyl groups and/or amide groups on the ANGNA polymer. Overall, the SFA force measurement results demonstrated that the reversible hydrogen bonds formed between ANGNA polymers played critical roles in the sol–gel transition and self-healing property of the as-prepared hydrogel under neutral and acidic conditions.
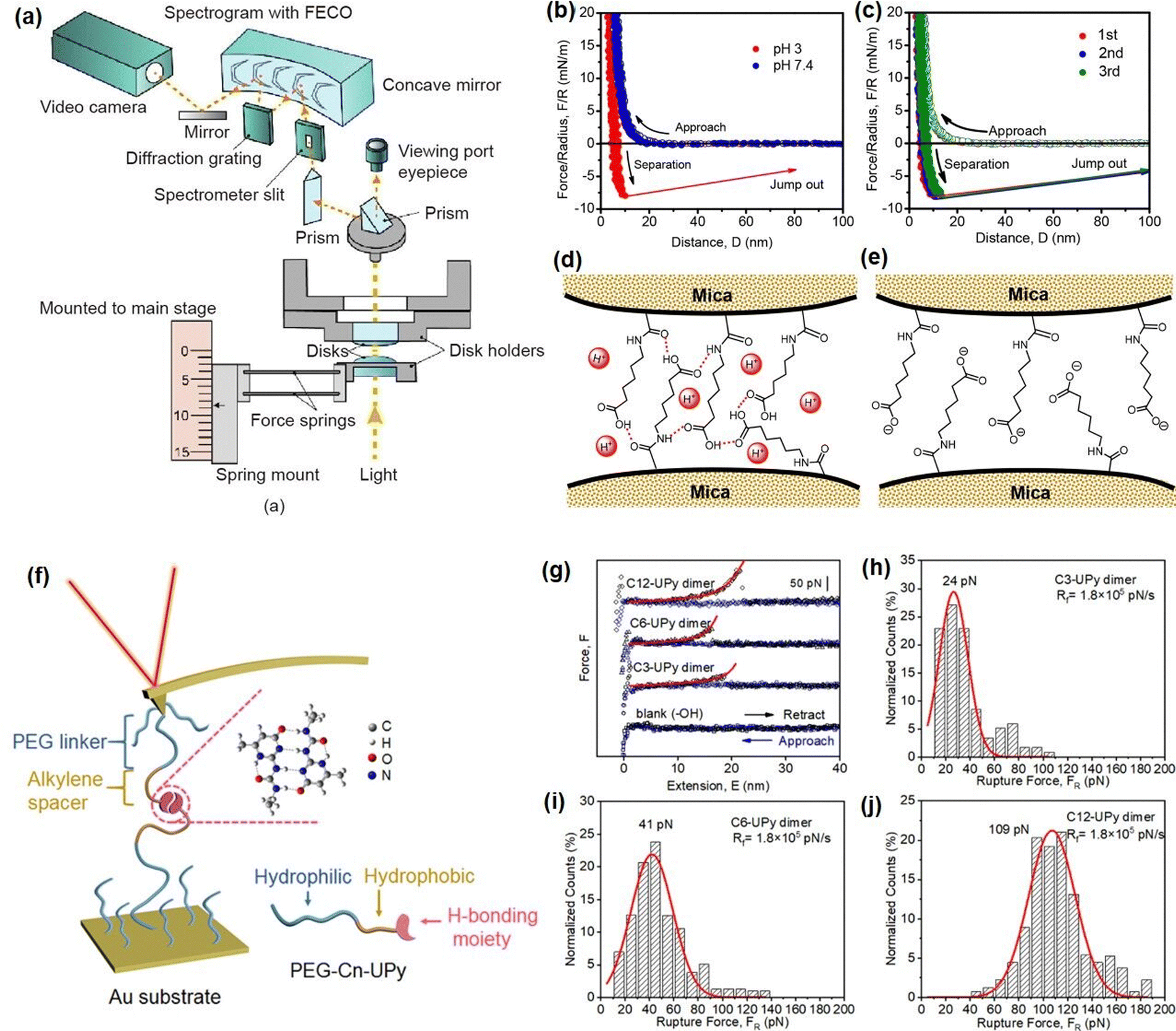 | ||
| Fig. 3 (a) Schematic diagram of the SFA setup for measuring the interaction forces between two surfaces (reproduced with permission from ref. 122; Copyright 2021 Elsevier); force–distance profiles indicated the (b) interaction forces between ANGNA at pH 3 or 7.4 conditions and (c) reversible interaction forces between ANGNA. (d) and (e) Schematics illustration of the interaction mechanism between ANGNA (reproduced with permission from ref. 114; Copyright 2021, American Chemical Society); (f) schematic diagram of the AFM-SMFS setup for measuring the interaction between UPy dimer, (g) –(j) force measurement results of Upy dimers with different lengths of hydrophobic spacer (reproduced with permission from ref. 127; Copyright 2019, American Chemical Society). | ||
AFM-based single-molecule force spectroscopy (AFM-SMFS) has also been widely applied to directly measure the inter/intramolecular interaction forces at the single-molecular level to provide useful insights into the underlying interaction mechanisms involved in the hydrogel assembly process.128–130 In our recent work, we quantitatively characterized the hydrogen-bonding interactions between UPy dimers using AFM-SMFS to investigate the effect of hydrophobic interactions on the intermolecular hydrogen bonding in an aqueous environment.127 The experiment set-up is shown in Fig. 3(f), the hydrogen-bonding UPy moieties were attached to the AFM tip and the Au substrate with the assistance of a PEG linker. The alkylene spacers of varying lengths (carbon chain number = 3, 6, and 12) are incorporated between the UPy groups and PEG linker to investigate the hydrophobic effects on UPy. As shown in Fig. 3(g)–(j), the rupture force between UPy dimers significantly increased from 24 pN to 109 pN when the carbon chain number of alkylene spacers increased from 3 to 12. This synergistic effect between hydrogen bonding and hydrophobic interaction can be attributed to the hydrogen-bonded structures that are stabilized in the hydrophobic region created by the hydrophobic interactions in an aqueous environment, which provides a fundamental understanding for the future designing mechanical robust injectable UPy-based hydrogels.
Dynamic covalent interactions
Dynamic Schiff base
Dynamic Schiff base is formed by the reversible crosslinking of the aldehydes (–CHO) with the primary amine groups (–NH2),131–133 which has been widely applied to construct injectable hydrogels due to its dynamic reversibility and pH-dependent properties.69,134–136 Moreover, the Schiff-base hydrogels are attractive candidates as a bio-composable drug delivery platform because the imine bonds would easily decompose in a mildly acidic environment,137 thus achieving precise drug release based on the pH difference in tumor or inflammation tissues.138,139 Most reported Schiff base injectable hydrogels were generally constructed by using natural polymers, such as chitosan140 or cellulose,141 and aldehyde-modified PEG.142 For instance, Qu et al. reported an injectable hydrogel with self-healing and pH-responsive properties based on the Schiff-base reaction between chitosan and dibenzaldehyde-terminated poly(ethylene glycol) (PEGDA),31 which demonstrated a pH-controlled sol–gel transition property due to reversibility of dynamic Schiff-base under mildly acidic and neutral conditions. The in vitro drug release test of the as-prepared hydrogel demonstrated its pH-controlled drug release behavior, indicating its potential to be used as a smart platform for drug delivery. Besides, Yang et al. fabricated an injectable hydrogel for removing proinflammatory factors by using the glycosaminoglycan oxidized chondroitin sulfate (OCS), the cationic polyethylenimine (PEI), and the antibiotic tobramycin (Tob) via the Schiff base reaction.143 The as-prepared hydrogel showed excellent shear thinning injectability due to the incorporation of dynamic reversible imine bonds in the hydrogel network, which would be disrupted during the injection process and re-formed upon relaxation at the target site. Moreover, the OCS/PEI/Tob hydrogel can effectively scavenge anionic proinflammatory molecules and bacteria due to the antibacterial properties of PEI and Tob, making the as-prepared hydrogel a promising wound dressing material for preventing bacterial infections and excessive inflammation. However, it is still a challenge to design the Schiff base hydrogel with a precisely controlled structure to achieve desired functionalities, such as high stability and sensitivity, as well as injectable and self-healing properties for wound healing, blood capillary formation, and controlled drug release.
Recently, our group developed several novel Schiff-base injectable self-healing hydrogels with precisely designed structures to achieve tunable mechanical properties and ultra-sensitive stimulus-response properties. For example, a novel injectable, self-healing, and pH-responsive hydrogel (PMF-S) drug delivery platform has been successfully prepared based on a Schiff-base reaction between aldehyde-containing copolymers and amine-modified silica nanoparticles by simply mixing copolymer and nanoparticle solutions (Fig. 4(a)).51 Incorporation of amine-modified silica nanoparticles into a hydrogel system not only can improve the stability and pH responsiveness sensitivity of hydrogels but also can be used as drug delivery platforms to anchor drug molecules. As shown in Fig. 4(b), the as-prepared PMF-S hydrogel could be freely extruded through 23-gauge needles and write letters of “UA” without clogging, and the viscosity dropped below 1 Pa s with the shear rate increasing to 50 s−1. This shear-thinning property can be attributed to the dynamic reversible imine bonds in the hydrogel system, as the imine bonds in the hydrogel network will be disrupted and lead to a decrease in its viscosity. Moreover, as shown in Fig. 4(c), the self-healing property of the PMF-S hydrogel was demonstrated by bringing two pieces of cracked hydrogel into contact, and an integrated free-standing hydrogel was formed due to the formation of dynamic Schiff base interaction, which was further confirmed by the oscillatory strain sweep test. The PMF-S hydrogel also showed pH-responsive properties as the rapid and reversible sol–gel transition can be achieved by changing the pH due to the hydrolysis and re-form of the dynamic Schiff base bonds under the acidic and neutral conditions (Fig. 4(d)), which made the PMF-S hydrogel applicable as a pH-responsive drug delivery platform for precise drug release. As shown in Fig. 4(e), the releasing behavior of the model drugs, Congo red and BSA can be well manipulated by slightly changing the pH of 0.2 in the faintly acidic range, indicating its great potential as an in vivo precise drug delivery platform.
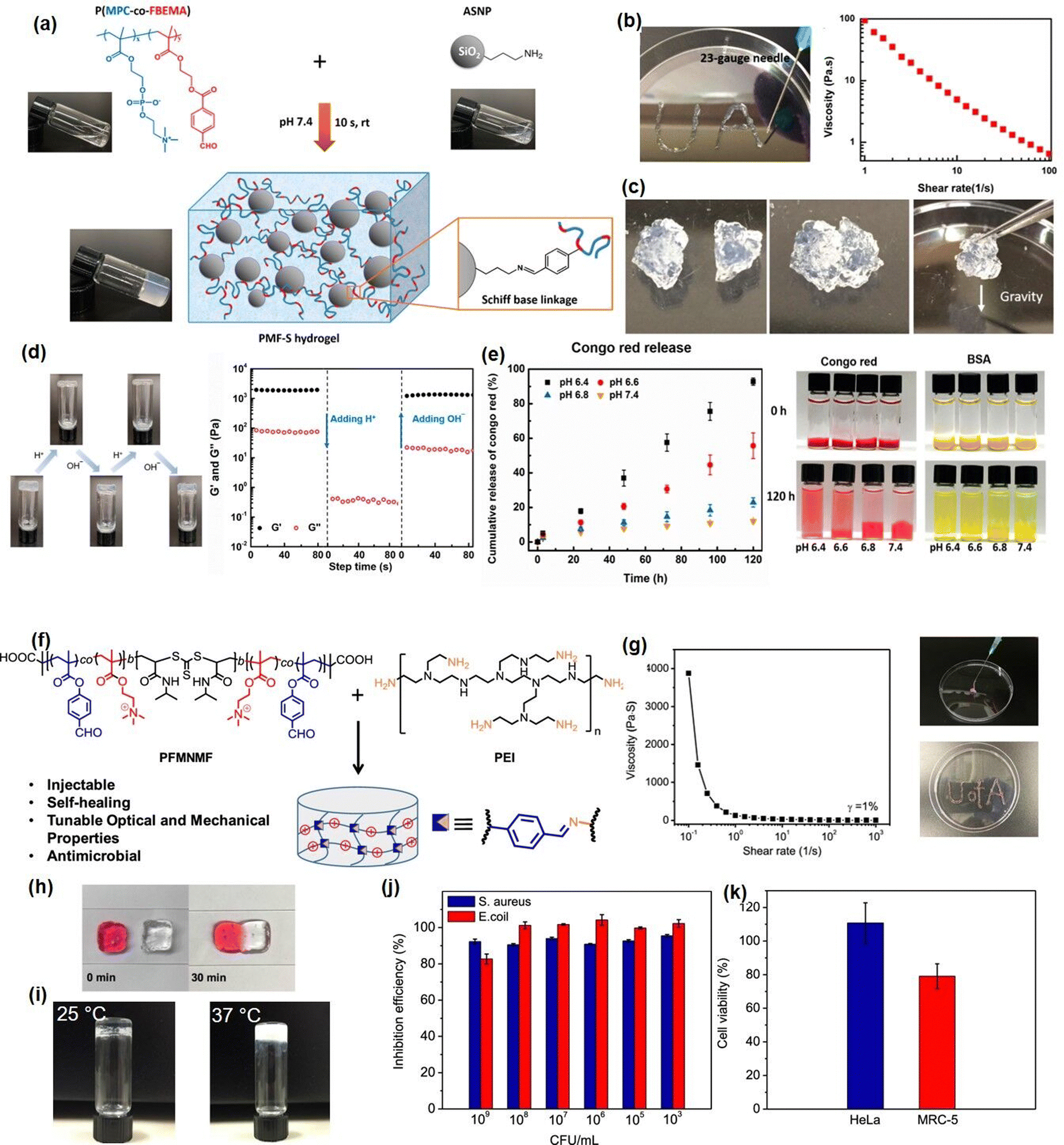 | ||
| Fig. 4 (a) The Schiff base hydrogel fabricated by using amine-modified silica nanoparticles and PMPC copolymers with (b) injectability, (c) self-healing ability, (d) pH-response, and (d)–(e) drug-release properties (reproduced with permission from ref. 51; Copyright 2020, American Chemical Society); (f) the Schiff base hydrogel constructed from PEI and benzaldehyde-functionalized ABA copolymer, demonstrated (g) shear thinning injectability, (h) self-healing, (i) thermal responsiveness, (j) antibacterial activity, and (k) biocompatibility (reproduced with permission from ref. 61; Copyright 2019, American Chemical Society). | ||
To further expand the application of Schiff-base reaction injectable hydrogel in bioengineering fields, we reported a multifunctional Schiff-base injectable hydrogel with tunable mechanical and antimicrobial properties by facile mixing branched polyethylenimine (PEI) with the ABA block co-polymer (PFMNMF).61 As shown in Fig. 4(f), the ABA block copolymer was composed of the thermosensitive PNIPAAM as the central group, and the trimethylammonium cationic monomer and the aldehyde-containing monomer as the terminal group, which can react with PEI through dynamic Schiff base reaction to build a hydrogel network. As shown in Fig. 4(g) and (h), the viscosity of the as-prepared hydrogel dramatically dropped from 4000 Pa s−1 to almost 0 Pa s−1 at a shear rate of 10 s−1, and the hydrogel can be healed at the target site within 30 min, which can be attributed to the incorporation of dynamic reversible imine bonds in the hydrogel. Moreover, as shown in Fig. 3(i), the mechanical property can be modulated by increasing the storage modulus of the hydrogel from ∼1000 to ∼2500 Pa by changing the temperature from 25 to 37 °C, the increase in its mechanical strength can be attributed to the hydrophobic domains formed by NIPAAM within the hydrogel at a temperature higher than LCST, which serve as additional crosslinks and lead to a compact hydrogel network. Notably, the as-prepared hydrogel can effectively kill more than 90% of both S. aureus and E. coli due to the synergistic effect of the quaternary amine groups of the copolymer and the protonated amine groups of the PEI (Fig. 4(j)). Moreover, the biocompatibility of the hydrogel was evaluated by using both cancer cells (HeLa) and fibroblasts (MRC-5) and more than 80% was retained for both cells (Fig. 4(k)), indicating its great potential as wound dressing material.
The dynamic reversibility and pH-responsive property of the Schiff base reaction between PEI and PFMNMF is illustrated at the nano-scale using SFA force measurement with the experiment setup shown in Fig. 5(a).61 As shown in Fig. 5(b), the adhesion between the PEI-coated surface and the PFMNMF-coated mica surfaces was measured to be 6.0 mN m−1 due to the formation of dynamic Schiff base chemistry between the two surfaces. Fig. 5(c) showed that the adhesion between two surfaces increased with increasing contact time, which can be attributed to the increasing number of dynamic imine bonds formed between the two surfaces with longer contact time. Moreover, to further investigate the gel–sol–gel transition behavior of the hydrogel (Fig. 5(d)), the interaction forces between PEI and PFMNMF copolymer were measured at different pH values, as shown in Fig. 5(e) and (f), the adhesion between two surfaces decreased from 6.0 mN m−1 to 2.5 mN m−1 with the pH decreasing from 7.4 to 6.4, and there was no adhesion detected between these two surfaces at pH = 5.4, demonstrating the ultrasensitive pH response property of the hydrogel, which is consistent with the sol–gel transition behavior of the as-prepared hydrogel in acidic and neutral conditions due to the pH sensitivity of the dynamic imine bonds.
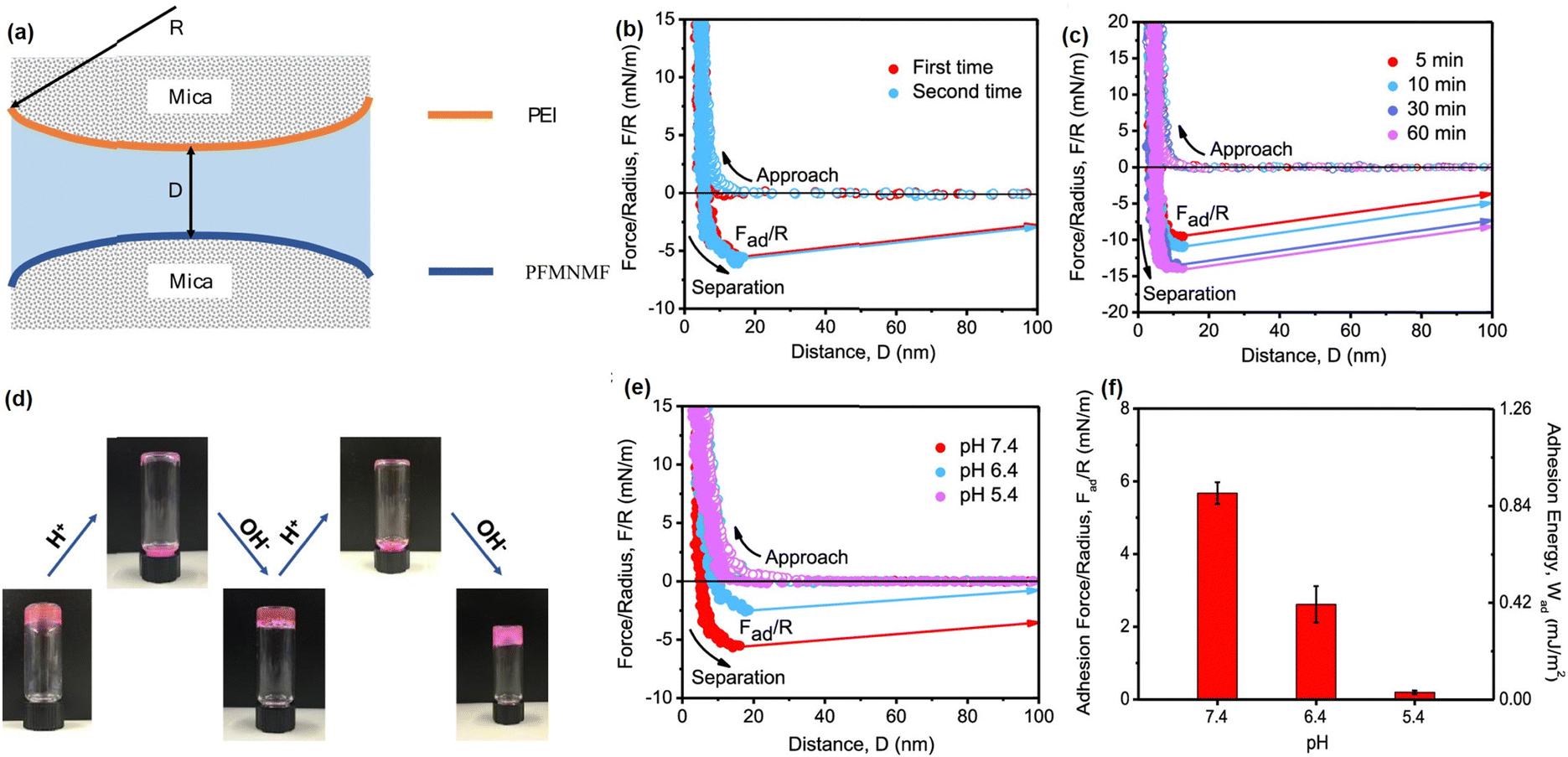 | ||
| Fig. 5 (a) The SFA force measurement setup for measuring the Schiff base reaction. The force–distance profiles illustrated the (b) reversibility of dynamic imine bonds and (c) effect of contact time on the interaction forces between PEI and PFMNMF; (d) reversible gel–sol–gel transitions by changing pH, (e) force–distance profiles and (f) normalized adhesion force and energy between PEI and PFMNMF with pH at 7.4, 6.4, and 5.4 (reproduced with permission from ref. 61; Copyright 2019, American Chemical Society). | ||
Disulfide bond
Disulfide chemistry is particularly attractive as one of the commonly found bonds in biological systems for folding proteins into their hierarchical structure.144 The disulfide bond can be exchanged rapidly with thiols based on the thiol/disulfide exchange reactions that depend on pH, temperature, and redox potential.145,146 In the thiol/disulfide exchange process, a thiolate anion acts as a nucleophile and attacks a sulfur atom in a disulfide bond, which cleaves the existing disulfide bond and re-form a new disulfide bond between the thiolate and the other sulfur atom.147,148 Disulfide bonds have been extensively investigated using single-molecule force spectroscopy. These studies aimed to elucidate the significance of intramolecular disulfide bonds in protein structures.149 Specifically, a protein molecule containing disulfide bonds was attached to the AFM probe and stretched at a constant velocity, nearly reaching its full contour length. During this process, a consistent discrepancy was observed where the protein's contour length was consistently shorter than expected. This can be attributed to the presence of intramolecular disulfide bridges in the protein structure, effectively creating shortcuts and limiting its extensibility. To further investigate the disulfide reduction reaction of disulfate bonds, recent AFM single-molecule force measurements were used to capture the dynamics of the reduction and reformation of individual intramolecular disulfides.150 During the protein stretching process, the protein unfolded and exposed the disulfide bond to an L-cysteine solution, which can attack the disulfide bond through nucleophilic attack. This leads to changes in the protein's extensibility over time. Many injectable hydrogels have been designed based on thiol/disulfide exchange reactions. For example, Barcan et al. fabricated a self-healing injectable hydrogel based on the thiol/disulfide exchange reactions by using an ABA triblock where the terminal A block was pendant 1,2-dithiolanes and the central B block was water-soluble PEG.151 The terminal A block was cross-linked with 1,2-dithiolanes to form a stable hydrogel via reversible disulfide exchange reactions, endowing the as-prepared hydrogel with excellent self-healing behavior that restores its structure even after deformation at 800% strain. Moreover, the as-prepared hydrogel can be easily injected through a syringe based on its pH and/or thermal-induced sol–gel transition property since the disulfide bonds would break at acidic (pH = 1) and high temperature (55 °C) and recover under neutral (pH = 7) and low temperature (25 °C) conditions. Besides, to further enhance the stability of the disulfide bonding hydrogel under acidic conditions, as shown in Fig. 6(a), Yu et al. reported an injectable self-healing hydrogel by simply mixing the thiol functionalized F127 and dithiolane modified PEG based on the reversible thiol/disulfide exchange reaction.70 The hydrogel solution could be injected using a 23 G syringe and then transformed to a stable gel state at body temperature (37 °C) due to the formation of disulfide bonds (Fig. 6(b)). Moreover, as shown in Fig. 6(c) and (d), the as-prepared hydrogel can rapidly self-heal under both neutral or even mildly acidic conditions, and this self-healing behavior was relatively fast and completely reversible based on the repeated dynamic strain step test, which can be attributed to the increased reactivity of the disulfide bonds due to the ring tension of the cyclic disulfide. Moreover, to further investigate the bioengineering application of disulfide bonding hydrogels, Choh et al. reported injectable hyaluronic acid (HA) hydrogels by modifying HA with pyridyl dithiol and then reacting with dithiol crosslinker via a thiol/disulfide exchange reaction (Fig. 6(e)).152 The as-prepared hydrogel can be rapidly formed in minutes after injection under physiological pH conditions due to the incorporation of highly dense disulfide bonds in the hydrogel. Moreover, the as-prepared hydrogel showed high biocompatibility (Fig. 6(f)) and can be used for the encapsulation of different types of cells, such as fibroblasts and adult stem cells, for up to 7 days in cell culture, indicating its great potential in in vivo cell therapy.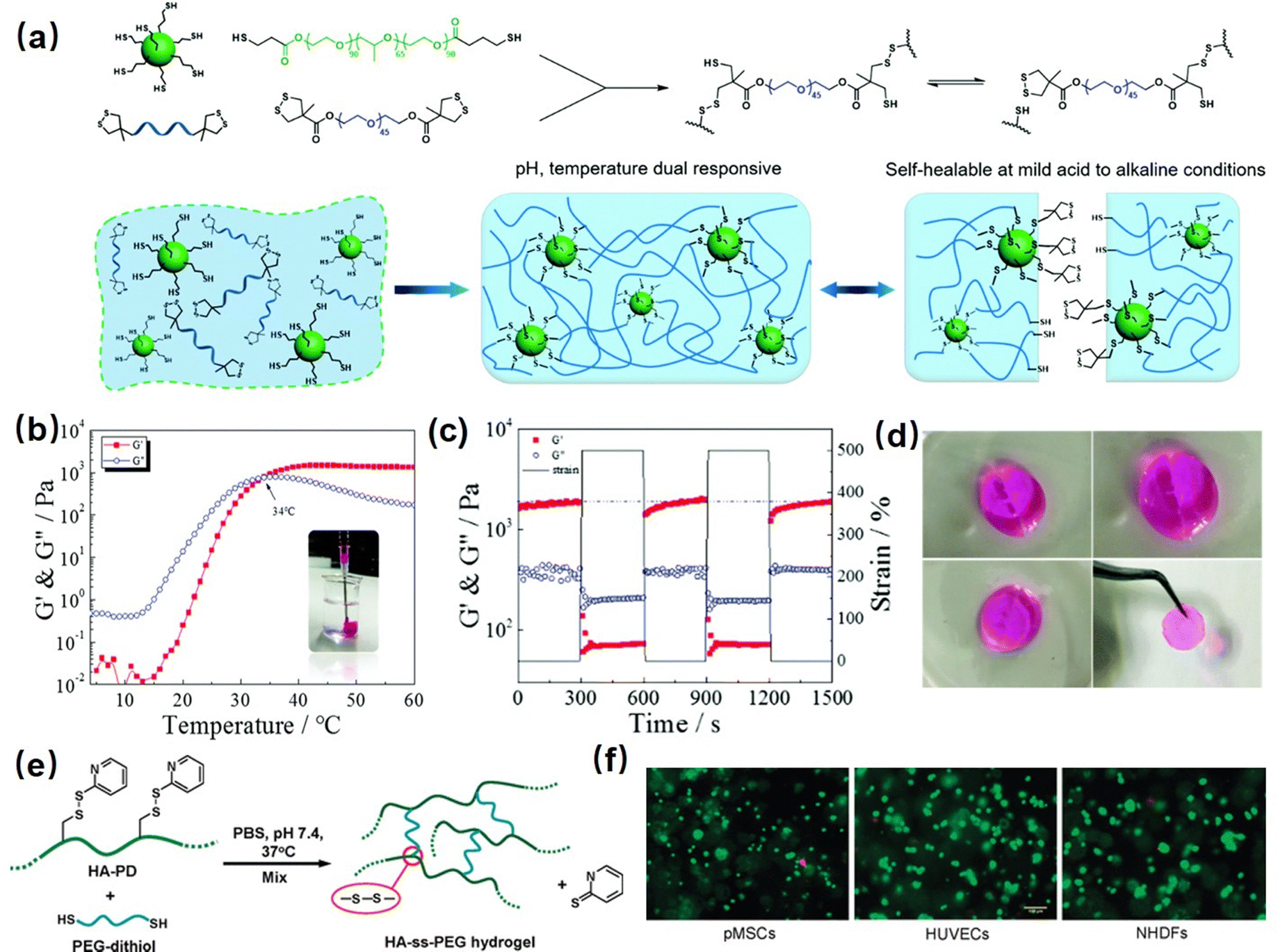 | ||
| Fig. 6 (a) The disulfide-bond based injectable hydrogel constructed by mixing thiol functionalized F127 and dithiolane modified PEG, (b) the temperature-responsive injectable and (c) and (d) the self-healing property of the as-prepared hydrogel (reproduced with permission from ref. 70. Copyright 2017, Royal Society of Chemistry); (e) another disulfide cross-linked HA hydrogel via the thiol-disulfide exchange reaction between HA derived with pyridyl disulfide and PEG-dithiolate, (f) the live/dead cell viability staining of cells encapsulated in disulfide-cross-linked HA hydrogels. (Reproduced with permission from ref. 152. Copyright 2011, American Chemical Society.) | ||
Mussel-inspired injectable hydrogels
Many marine organisms such as mussels and barnacles exhibit intriguing wet-adhesion properties.153–155 For example, robust underwater adhesion of mussels can be achieved via the formation of a bio-adhesive layer between the mussel byssal thread and substrates with the mussel foot proteins (Mfps).156,157 Previous studies have focused on investigating this unique wet adhesion property by elucidating the structure of Mfps, a catecholic amino acid called 3,4-dihydroxyphenyl-L-alanine (DOPA), which was demonstrated to be the major component of Mfps and significantly contribute to the remarkable underwater adhesion via multiple interactions,158 such as metal–ligand coordination,86 hydrogen bonding,95 π–π interaction,159 hydrophobic interaction,160 cation–π interaction,161etc. DOPA can be further oxidized into reactive quinone species and undergo Michael addition and Schiff base reaction to crosslink with other Mfps to achieve rapid solidification on various surfaces under complex seawater conditions.162–164 Inspired by this rapid Mfps solidification behavior, many mussel-inspired injectable hydrogels have been designed by using catechol-based polymers. For example, Xu et al. prepared an injectable hydrogel as a potential drug delivery platform by using oxidized-carboxymethylcellulose and carboxymethylchitosan,165 which showed good injectability attributed to the shear thinning property of the dynamic imine-cross-linked hydrogel. The as-prepared hydrogel also exhibited good bio-compatibility and sustained drug-release properties for up to a week in both in vitro and in vivo tests. Zhang et al. reported an injectable hydrogel by using alginate-dopamine, chondroitin sulfate, and regenerated silk fibroin (AD/CS/RSF)166 and can be readily injected using a double injection syringe through in situ formation within 3 s. The formed AD/CS/RSF hydrogel also showed high adhesive properties to porcine skin tissue with a lap shear strength of 120 kPa due to the formation of reversible hydrogen bonds, π–π interactions, and Schiff base reaction. Recently, by mimicking the metal-induced Mfps complexation process in the mussel adhesion process, injectable hydrogels with tunable properties have been designed by applying the reversible coordinative bonding between metal ions and DOPA. For example, Wang et al reported a mechanically strong or biodegradable injectable hydrogel by crosslinking catechol-modified methacryloyl chitosan with methacryloyl chitosan via catechol-Fe3+ chelation.167 The as-prepared hydrogel showed intrinsic antibacterial capability and can be facilely injected into the target site and achieved robustly adhered onto the bio-tissue with a lap shear strength up to 18.1 kPa, which can be used to promote healing of bacteria-infected wounds. Moreover, Choi et al. prepared bio-inspired tissue-adhesive gelatin hydrogels by coordinating DOPA-modified gelatin with Fe3+ ions.168 Upon adding Fe3+ ions, a sticky hydrogel can be formed within seconds due to the rapid complexation of DOPA and Fe3+. The in vivo test showed that the bleeding site of a live rat was effectively arrested after hydrogel injection due to its appropriate bio-adhesion ability and mechanical properties.In our previous research, as shown in Fig. 7(a), we reported a mussel-inspired injectable hydrogel (DNODN) based on the self-assembly of an ABA triblock polymer composed of catechol-functionalized PNIPAAM and hydrophilic PEG.169 The UV-vis absorption spectra of the DNODN solution are shown in Fig. 7(b); the characteristic peak of catechol groups at 280 nm remained unchanged up to 48 h, demonstrating that the catechol groups in the hydrogel matrix remained unoxidized, which enhanced the hydrogen bonding and π–π interaction between catechol groups in the hydrogel. Moreover, the as-prepared hydrogel showed thermal responsive properties, and the free-standing DNODN hydrogel can be in situ formed by injecting the hydrogel solution preserved at 4 °C into 37 °C DI water (Fig. 7(c)), which can be attributed to the sol–gel transition triggered by the dehydration of the PNIPAAM block at a temperature over 37 °C. Moreover, as shown in Fig. 7(d), the self-healing property of the DNODN hydrogel was demonstrated by observing two hydrogel fragments healing automatically into one integral piece. In contrast, the oxidized DNODN hydrogel lost its self-healing ability due to the oxidation of catechol groups, resulting in the formation of an irreversible quinone crosslinking. Additionally, the antifouling property of the as-prepared hydrogel was confirmed by seeding human intestinal Caco-2 cells onto the hydrogel surface followed by fluorescence imaging characterization. As shown in Fig. 7(e), compared to the uncoated substrate, the DNODN hydrogel-coated substrate effectively prevents the formation of Caco-2 cell biofilms due to surface hydration, where the hydration layer acts as a physical barrier to prevent Caco-2 cell adhesion. The ABA block polymer structure also provides a versatile platform for tailoring different functionalities into injectable hydrogels. For example, in our following work, the central PEG polymer in the ABA block polymer was replaced with a quaternary amine to endow the injectable hydrogel with antibacterial properties,171 making the as-prepared hydrogel a promising wound dressing or biomedical implant material.
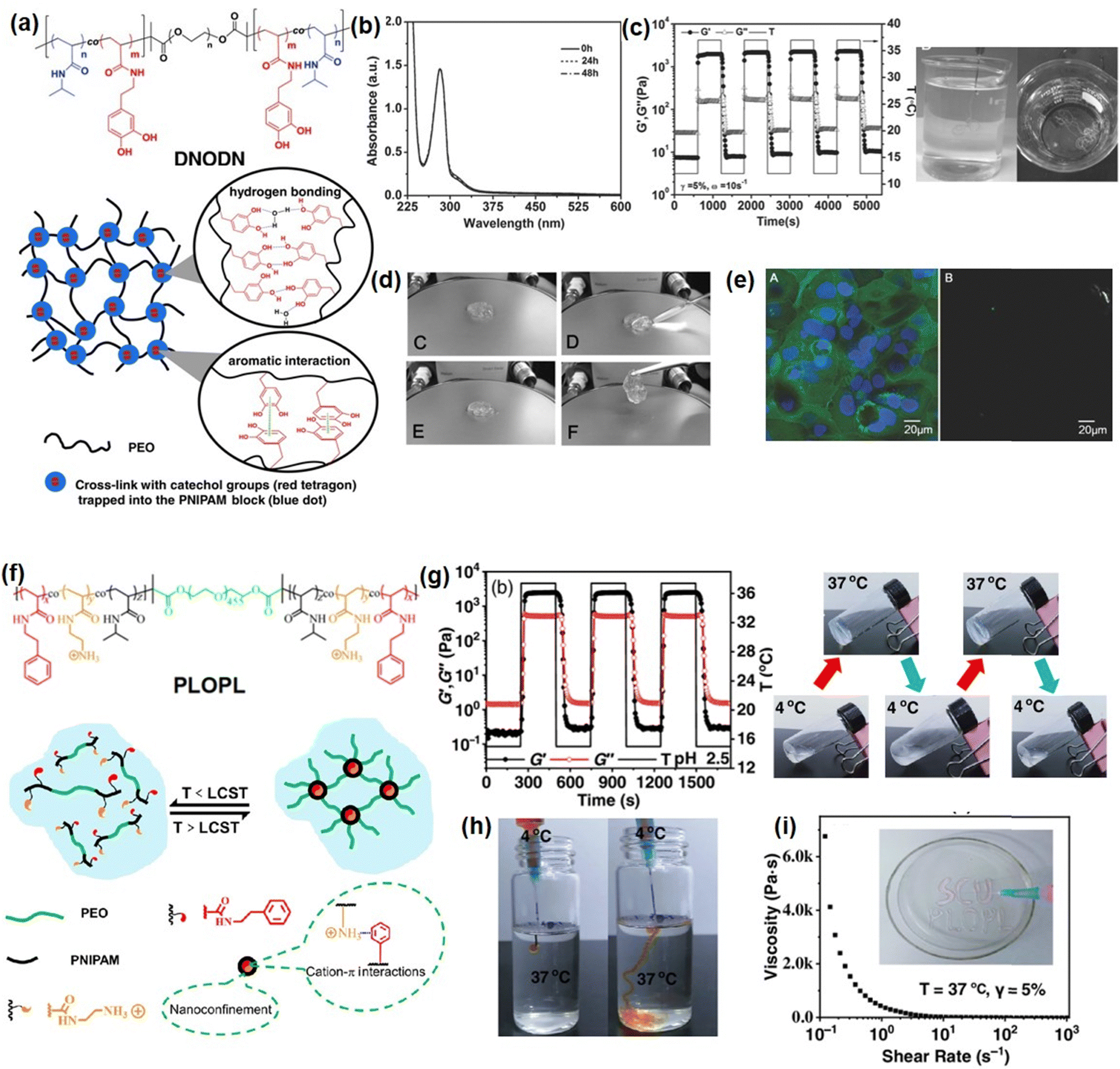 | ||
| Fig. 7 (a) Chemical structure of ABA copolymer DNOND and the proposed structure of as-prepared mussel-inspired hydrogels, (b) UV-vis spectra of a DNODN solution, (c) injectability, (d) self-healing ability, (e) anti-cell fouling property (reproduced with permission from ref. 169; Copyright 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim); (f) chemical structure of PLOPL copolymer and the proposed structure of formed cation–π hydrogel, and its (g) thermal responsive, (h) injectability, (i) shear thinning properties. (Reproduced with permission from ref. 170; Copyright 2022 Chinese Chemical Society.) | ||
Recently, it was reported that robust mussel adhesion in complex seawater also relies on the cation–π interaction between the aromatic cationic moieties in Mfps, such as lysine by displacing surface salt from substrate the with its cationic groups.172 By mimicking this bio-adhesion process, the cation–π based injectable hydrogel can be designed by using the complexation and coacervation between the polyelectrolytes with positively charged ions (cations) and electron-rich π components.173 Recently, we fabricated an injectable hydrogel based on cation–π interactions between cationic amine and aromatic phenyl of ABA block polymer (PLOPL) (Fig. 7(f)),170 which was composed of a modified PNIPAAM incorporated with cationic and aromatic components as A block and a hydrophilic poly(ethylene oxide) (PEO) as B block. As shown in Fig. 7(g), the PLOPL copolymer exhibited rapid and reversible sol–gel transition behavior at a temperature above the LCST, which was triggered by the synergistic effect of the cation–π and hydrophobic interactions between the cationic/aromatic components and NIPAAM in the PLOPL copolymers. Taking advantage of its thermal sensitive property, a 4 °C polymer solution could be injected readily into a 37 °C water bath to form a stable hydrogel immediately (Fig. 7(h)). Moreover, the formed PLOPL hydrogel showed a shear thinning property where the viscosity of the PLOPL hydrogel decreased sharply with a shear rate from 0.1 to 1000 s−1 (Fig. 7(i)) due to the dynamic reversible nature of the cation–π interaction. Moreover, the letters “SCU PLOPL” can be easily written by injecting PLOPL hydrogel via a needle, demonstrating its outstanding injectability behavior at human body temperature.
Although great efforts have been devoted to designing novel injectable hydrogels by mimicking the adhesion/cohesion process of Mfps, only limited reports have focused on studying the intermolecular mechanisms between the different components in the designed mussel-inspired hydrogels. To provide a better understanding of the underlying mechanism of the mussel-inspired adhesion/cohesion process, we investigated the mechanical nature of cation–π interactions in aqueous media directly by using SFA.174 The experiment setup is shown in Fig. 8(a), one mica surface was coated with positively charged poly-L-lysine (PLL) (primary amine, NH3R+) as typical cations and another mica surface was coated with one of three aromatic polymers: poly-L-tryptophan (PTrp), poly-L-tyrosine (PTyr), and polystyrene (PS) with indole, phenol, and benzene structures to provide π systems. As shown in Fig. 8(b), the adhesion strengths between PLL and the three aromatic polymers follow the order PTrp > PS > Pty, and the cation–π interaction strength of PS was over twice than that of PTyr in aqueous solution due to their different polymer backbone structures. Moreover, the adhesion between PTrp and PLL decreased from about 2.0 to about 0.36 mJ m−2 with increasing KNO3 concentration from 0.05 to 0.2 M (Fig. 8(c)). Since the electrostatic interaction energy does not significantly change in the salt concentration range (0.05–0.25 M), the reduced adhesion between PLL and PTrp was mainly attributed to the weakening cation–π interaction due to the competition of K+ ions with the primary amine on PLL. Besides, we also investigated the reversible DOPA–Fe3+ coordination interaction by measuring the interaction between mfp-1 coated mica surfaces in the absence and presence of Fe3+ at two different concentrations (Fig. 8(d)).86 Specifically, no adhesion between two mfp-1-coated mica surfaces was measured when two mica surfaces were brought into contact and separated without Fe3+ (Fig. 8(e)), which was consistent with the previous report. In contrast, as shown in Fig. 8(f), significant and reversible adhesion (∼6 mN m−1) was measured with 10 μM Fe3+ due to the formation of triscatecholato–iron complexes through coordination reaction between DOPA and Fe3+ ions. However, the adhesion between two mfp-1 was abolished, with the Fe3+ concentrating further increasing to 100 μM (Fig. 8(g)), which was due to the triscatecholato–Fe3+ complexes collapsing into nonbridging monocatecholato–iron form under this condition. To further demonstrate the chelation between Fe3+ and DOPA, as shown in Fig. 8(h), only repulsive forces were measured in the presence of 10 μM Fe3+ and EDTA, which was consistent with the greater affinity of EDTA for Fe3+ ions than for DOPA.
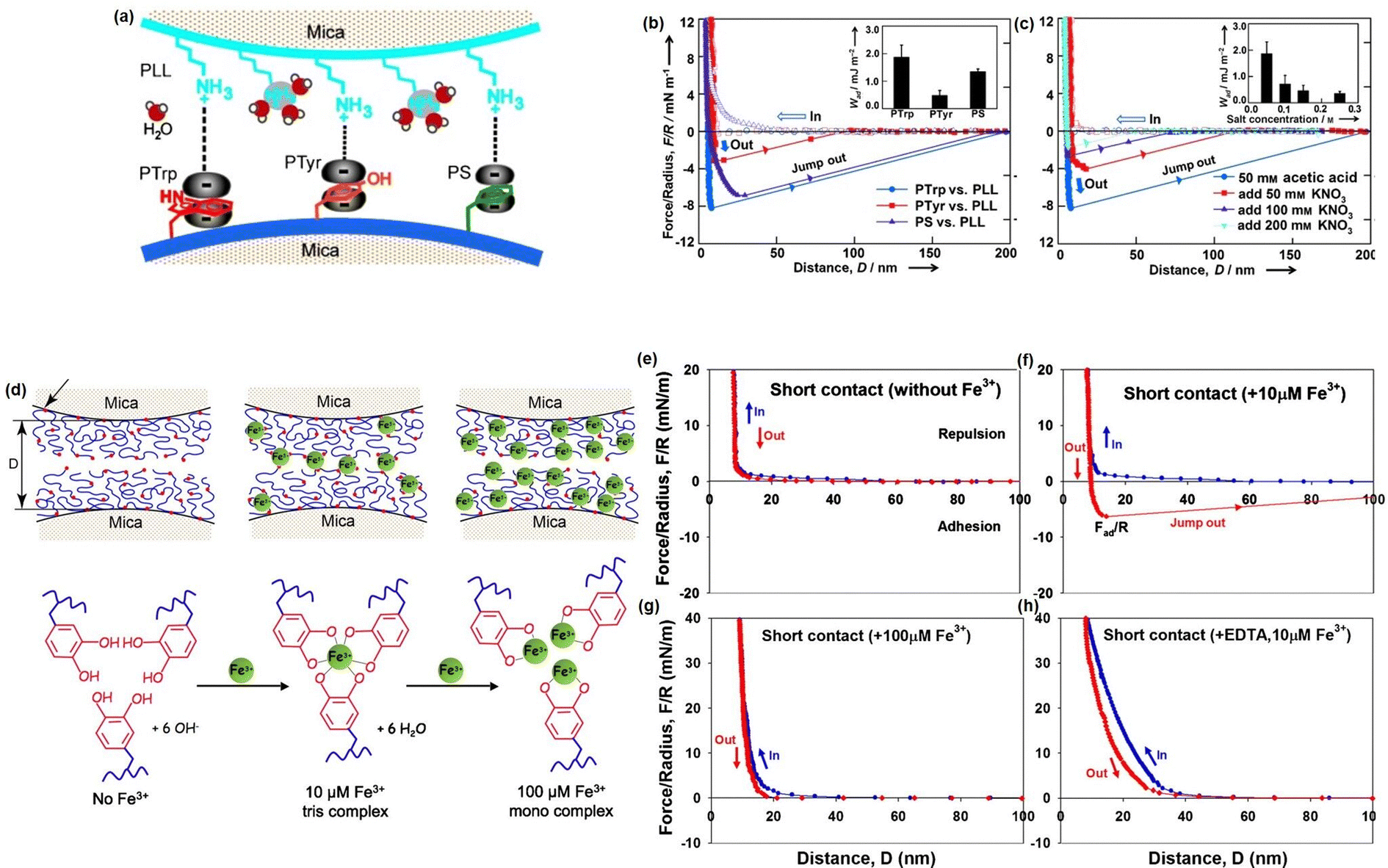 | ||
| Fig. 8 (a) The SFA setup for measuring the cation–π interaction between positively charged side groups of PLL and aromatic side groups of polymers (PTrp, PTyr, or PS). The force–distance profiles that illustrated the effects of (b) aromatic groups and (c) salt concentration on the cation–π interaction (reproduced with permission from ref. 174; Copyright 2013, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim); (d) the SFA setup for measuring the interactions between mfp-1 with increasing Fe3+ concentration, and the force–distance profiles that illustrated the effect of the (e)–(h) Fe3+ concentration and (g) EDTA on the bridging adhesion between mfp-1 (reproduced with permission from ref. 86. Copyright 2010, National Academy of Sciences). | ||
Summary and future perspectives
Hydrogels are widely applied in various bioengineering fields, such as drug delivery, tissue engineering, and wound dressing, due to their biomimetic structure with high biocompatibility and biodegradability. Recently, injectable hydrogels that can be delivered directly to the target site via syringe injection have garnered significant attention because they simplify the implantation process and minimize tissue invasion. Moreover, stimulus-responsive injectable hydrogels can serve as drug delivery platforms with controlled and sustained drug release behavior. Compared to other delivery systems, injectable hydrogels do not cause inflammation due to their biocompatibility and biodegradable properties. In this review, we summarize our recent progress in fabricating self-healing injectable hydrogels based on reversible physical bonding, such as hydrogen bonding and hydrophobic interactions, dynamic covalent bonding, including dynamic Schiff base reactions and disulfide bonding, as well as mussel-inspired interactions. The as-prepared injectable hydrogels can be easily injected into the target site based on the stimulus-responsive in situ formation and/or shear-thinning properties, and they can fully restore their 3D networks and maintain their structural integrity after damage through the self-healing process. Moreover, most injectable hydrogels also exhibit antifouling, antibacterial, pH/thermal responsiveness, and conductive properties, making them promising candidates as implantable materials, smart drug release platforms, and wound dressing materials. In order to gain a fundamental understanding of the reversible molecular interactions involved in the processes of hydrogel injection and self-healing, we employed nanomechanical tools, such as SFA and AFM, to elucidate the molecular interaction mechanisms within the hydrogel systems. The results of force measurements demonstrate the stimulus-responsive behavior and dynamic, reversible nature of interactions, including hydrogen bonding, Schiff base reactions, coordination reactions, and cation–π interactions. These findings correlate the injectable and self-healing behavior of hydrogels with the underlying intermolecular interactions, providing researchers with improved fundamental understanding and inspiration for designing advanced injectable self-healing hydrogels with desired functionalities for various bioengineering applications.Despite the significant efforts devoted to designing novel injectable hydrogels, there are still certain limitations that must be addressed to enhance their application in bioengineering fields. For example, many injectable hydrogels with promising shear-thinning or rapid in situ formation properties often exhibit low mechanical strength due to the incorporation of non-covalent bonds. To meet the demands of most bioengineering applications, which require robust mechanical properties, there is a need for the inclusion of multiple cross-linking bonds or the development of novel cross-linking mechanisms in the future. Moreover, injectable hydrogels with self-healing properties have been widely investigated to enhance the mechanical properties of injectable hydrogels by restoring their structure after damage. However, most investigations into the self-healing property of injectable hydrogels have been conducted in in vitro settings, rather than in vivo conditions. This discrepancy makes it challenging to predict their self-healing performance when used as wound dressings or implantable materials in practical applications. Another aspect of injectable hydrogels that requires further improvement is their in vivo biocompatibility. While many reported injectable hydrogels have demonstrated excellent biocompatibility, there is limited research on the cytotoxicity of degradation products from injectable hydrogels, including monomers, incorporated nanoparticles, or unreacted crosslinkers. This knowledge gap may hinder the application of injectable hydrogels as biodegradable drug delivery platforms.
It is also noted that while most researchers have focused on developing advanced injectable hydrogels with tailored functionalities or modifications by designing new crosslinking systems, the fundamental understanding of the reversible molecular interactions involved in hydrogel systems remains unclear. Direct force measurement between the different components within hydrogel networks, using advanced tools such as AFM and SFA, can provide a molecule-level interpretation of the mechanisms governing the self-healing, injectable, as well as stimulus-responsive properties of hydrogels. This approach provides new insights and shapes the future development of advanced injectable hydrogels with desired functionalities for various bioengineering applications.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canada Research Chairs Program (H. Zeng).References
- X. Liu, J. Liu, S. Lin and X. Zhao, Mater. Today, 2020, 36, 102–124 CrossRef CAS.
- C. Wang, C. Wang, Z. Huang and S. Xu, Adv. Mater., 2018, 30, 1801368 CrossRef.
- Y. Zhou, C. Wan, Y. Yang, H. Yang, S. Wang, Z. Dai, K. Ji, H. Jiang, X. Chen and Y. Long, Adv. Funct. Mater., 2019, 29, 1806220 CrossRef.
- R. Fu, L. Tu, Y. Zhou, L. Fan, F. Zhang, Z. Wang, J. Xing, D. Chen, C. Deng and G. Tan, Chem. Mater., 2019, 31, 9850–9860 CrossRef CAS.
- N. Gogurla, B. Roy and S. Kim, Nano Energy, 2020, 77, 105242 CrossRef CAS.
- B. Xue, H. Sheng, Y. Li, L. Li, W. Di, Z. Xu, L. Ma, X. Wang, H. Jiang and M. Qin, Natl. Sci. Rev., 2022, 9, nwab147 CrossRef CAS.
- Y.-Z. Zhang, K. H. Lee, D. H. Anjum, R. Sougrat, Q. Jiang, H. Kim and H. N. Alshareef, Sci. Adv., 2018, 4, eaat0098 CrossRef PubMed.
- G. Balakrishnan, J. Song, A. S. Khair and C. J. Bettinger, J. Mater. Chem. B, 2023, 11, 5544–5551 RSC.
- F.-m Cheng, H.-x Chen and H.-d Li, J. Mater. Chem. B, 2021, 9, 1762–1780 RSC.
- X. Li, X. Cai, Y. Gao and M. J. Serpe, J. Mater. Chem. B, 2017, 5, 2804–2812 RSC.
- S. Pourshahrestani, E. Zeimaran, N. A. Kadri, N. Mutlu and A. R. Boccaccini, Adv. Healthcare Mater., 2020, 9, 2000905 CrossRef CAS PubMed.
- X. Xue, Y. Hu, Y. Deng and J. Su, Adv. Funct. Mater., 2021, 31, 2009432 CrossRef CAS.
- J. Liu, S. Qu, Z. Suo and W. Yang, Natl. Sci. Rev., 2021, 8, nwaa254 CrossRef CAS PubMed.
- G. Su, J. Cao, X. Zhang, Y. Zhang, S. Yin, L. Jia, Q. Guo, X. Zhang, J. Zhang and T. Zhou, J. Mater. Chem. A, 2020, 8, 2074–2082 RSC.
- A. Vedadghavami, F. Minooei, M. H. Mohammadi, S. Khetani, A. R. Kolahchi, S. Mashayekhan and A. Sanati-Nezhad, Acta Biomater., 2017, 62, 42–63 CrossRef CAS PubMed.
- T. Lai, J. Yu and W. Tsai, J. Mater. Chem. B, 2016, 4, 2304–2313 RSC.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 1–17 Search PubMed.
- Y. Yi, C. Xie, J. Liu, Y. Zheng, J. Wang and X. Lu, J. Mater. Chem. B, 2021, 9, 8739–8767 RSC.
- J. A. Hunt, R. Chen, T. van Veen and N. Bryan, J. Mater. Chem. B, 2014, 2, 5319–5338 RSC.
- L. Zhang, Z. Cao, T. Bai, L. Carr, J.-R. Ella-Menye, C. Irvin, B. D. Ratner and S. Jiang, Nat. Biotechnol., 2013, 31, 553–556 CrossRef CAS.
- A. Al Abboodi, J. Fu, P. M. Doran, T. T. Tan and P. P. Chan, Adv. Healthcare Mater., 2014, 3, 725–736 CrossRef CAS.
- J. Wu, Q. Ding, A. Dutta, Y. Wang, Y.-H. Huang, H. Weng, L. Tang and Y. Hong, Acta Biomater., 2015, 16, 49–59 CrossRef CAS.
- A. B. South and L. A. Lyon, Angew. Chem., 2010, 122, 779–783 CrossRef.
- V. Can, Z. Kochovski, V. Reiter, N. Severin, M. Siebenbürger, B. Kent, J. Just, J. R. P. Rabe, M. Ballauff and O. Okay, Macromolecules, 2016, 49, 2281–2287 CrossRef CAS.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- M. M. Pakulska, B. G. Ballios and M. S. Shoichet, Biomed. Mater., 2012, 7, 024101 CrossRef PubMed.
- Y. Sun, D. Nan, H. Jin and X. Qu, Polym. Test., 2020, 81, 106283 CrossRef CAS.
- Y. Zhao, Z. Cui, B. Liu, J. Xiang, D. Qiu, Y. Tian, X. Qu and Z. Yang, Adv. Healthcare Mater., 2019, 8, 1900709 CrossRef PubMed.
- K. H. Bae, L.-S. Wang and M. Kurisawa, J. Mater. Chem. B, 2013, 1, 5371–5388 RSC.
- M. S. Gil, T. Thambi, V. G. Phan, S. H. Kim and D. S. Lee, J. Mater. Chem. B, 2017, 5, 7140–7152 RSC.
- J. Qu, X. Zhao, P. X. Ma and B. Guo, Acta Biomater., 2017, 58, 168–180 CrossRef CAS PubMed.
- A. P. Mathew, S. Uthaman, K.-H. Cho, C.-S. Cho and I.-K. Park, Int. J. Biol. Macromol., 2018, 110, 17–29 CrossRef CAS PubMed.
- Z. Ma, W. Song, Y. He and H. Li, ACS Appl. Mater. Interfaces, 2020, 12, 29787–29806 CAS.
- Y. Dong, M. Rodrigues, X. Li, S. H. Kwon, N. Kosaric, S. Khong, Y. Gao, W. Wang and G. C. Gurtner, Adv. Funct. Mater., 2017, 27, 1606619 CrossRef.
- F. Rizzo and N. S. Kehr, Adv. Healthcare Mater., 2021, 10, 2001341 CrossRef CAS PubMed.
- X. Ding, Y. Wang, J. Liu, P. Zhang, G. Li, T. Sun and C. Xiao, Chem. Mater., 2021, 33, 5885–5895 CrossRef CAS.
- K. Dutta, R. Das, J. Ling, R. M. Monibas, E. Carballo-Jane, A. Kekec, D. D. Feng, S. Lin, J. Mu and R. Saklatvala, ACS Omega, 2020, 5, 17531–17542 CrossRef CAS.
- M. Zou, R. Jin, Y. Hu, Y. Zhang, H. Wang, G. Liu, Y. Nie and Y. Wang, J. Mater. Chem. B, 2019, 7, 4402–4412 RSC.
- Z. Ma, D. M. Nelson, Y. Hong and W. R. Wagner, Biomacromolecules, 2010, 11, 1873–1881 CrossRef CAS.
- A. Gonsalves, P. Tambe, D. Le, D. Thakore, A. S. Wadajkar, J. Yang, K. T. Nguyen and J. U. Menon, J. Mater. Chem. B, 2021, 9, 9533–9546 RSC.
- H. Ding, B. Li, Z. Liu, G. Liu, S. Pu, Y. Feng, D. Jia and Y. Zhou, Adv. Healthcare Mater., 2020, 9, 2000454 CrossRef CAS.
- L. Zhang, Y. Zhou, D. Su, S. Wu, J. Zhou and J. Chen, J. Mater. Chem. B, 2021, 9, 5887–5897 RSC.
- S. Kumar and A. Bajaj, Biomater. Sci., 2020, 8, 2055–2073 RSC.
- J.-A. Yang, J. Yeom, B. W. Hwang, A. S. Hoffman and S. K. Hahn, Prog. Polym. Sci., 2014, 39, 1973–1986 CrossRef CAS.
- M. Guvendiren, H. D. Lu and J. A. Burdick, Soft Matter, 2012, 8, 260–272 RSC.
- Y. S. Zhang and A. Khademhosseini, Science, 2017, 356, eaaf3627 CrossRef.
- H. J. Lee, P. T. Le, H. J. Kwon and K. D. Park, J. Mater. Chem. B, 2019, 7, 3374–3382 RSC.
- X. Ding, J. Gao, H. Awada and Y. Wang, J. Mater. Chem. B, 2016, 4, 1175–1185 RSC.
- N. Zandi, E. S. Sani, E. Mostafavi, D. M. Ibrahim, B. Saleh, M. A. Shokrgozar, E. Tamjid, P. S. Weiss, A. Simchi and N. Annabi, Biomaterials, 2021, 267, 120476 CrossRef CAS PubMed.
- A. Baidya, R. Haghniaz, G. Tom, M. Edalati, N. Kaneko, P. Alizadeh, M. Tavafoghi, A. Khademhosseini and A. Sheikhi, ACS Appl. Mater. Interfaces, 2022, 14, 42852–42863 CrossRef CAS.
- M. Wu, J. Chen, W. Huang, B. Yan, Q. Peng, J. Liu, L. Chen and H. Zeng, Biomacromolecules, 2020, 21, 2409–2420 CrossRef CAS.
- X. Ding, J. Gao, Z. Wang, H. Awada and Y. Wang, Biomaterials, 2016, 111, 80–89 CrossRef CAS PubMed.
- C. Loebel, C. B. Rodell, M. H. Chen and J. A. Burdick, Nat. Protoc., 2017, 12, 1521–1541 CrossRef CAS PubMed.
- P. T. Smith, A. Basu, A. Saha and A. Nelson, Polymer, 2018, 152, 42–50 CrossRef CAS.
- S. Uman, A. Dhand and J. A. Burdick, J. Appl. Polym. Sci., 2020, 137, 48668 CrossRef CAS.
- G. Zhang, T. Ngai, Y. Deng and C. Wang, Macromol. Chem. Phys., 2016, 217, 2172–2181 CrossRef CAS.
- J. Chen, Q. Peng, T. Thundat and H. Zeng, Chem. Mater., 2019, 31, 4553–4563 CrossRef CAS.
- X. Ding and Y. Wang, J. Mater. Chem. B, 2017, 5, 887–906 RSC.
- X. Du, Y. Liu, X. Wang, H. Yan, L. Wang, L. Qu, D. Kong, M. Qiao and L. Wang, Mater. Sci. Eng., C, 2019, 104, 109930 CrossRef CAS PubMed.
- S. Li, M. Pei, T. Wan, H. Yang, S. Gu, Y. Tao, X. Liu, Y. Zhou, W. Xu and P. Xiao, Carbohydr. Polym., 2020, 250, 116922 CrossRef CAS PubMed.
- W. Wang, L. Xiang, L. Gong, W. Hu, W. Huang, Y. Chen, A. B. Asha, S. Srinivas, L. Chen and R. Narain, Chem. Mater., 2019, 31, 2366–2376 CrossRef CAS.
- S.-W. Wu, X. Liu, A. L. Miller II, Y.-S. Cheng, M.-L. Yeh and L. Lu, Carbohydr. Polym., 2018, 192, 308–316 CrossRef CAS PubMed.
- M. Cao, Y. Wang, X. Hu, H. Gong, R. Li, H. Cox, J. Zhang, T. A. Waigh, H. Xu and J. R. Lu, Biomacromolecules, 2019, 20, 3601–3610 CrossRef CAS PubMed.
- X. Mao, R. Cheng, H. Zhang, J. Bae, L. Cheng, L. Zhang, L. Deng, W. Cui, Y. Zhang and H. A. Santos, Adv. Sci., 2019, 6, 1801555 CrossRef PubMed.
- J. Qu, X. Zhao, Y. Liang, T. Zhang, P. X. Ma and B. Guo, Biomaterials, 2018, 183, 185–199 CrossRef CAS.
- V. Panwar, A. Babu, A. Sharma, J. Thomas, V. Chopra, P. Malik, S. Rajput, M. Mittal, R. Guha and N. Chattopadhyay, J. Mater. Chem. B, 2021, 9, 6260–6270 RSC.
- G. Wu, K. Jin, L. Liu and H. Zhang, Soft Matter, 2020, 16, 3319–3324 RSC.
- D. Zhao, M. Feng, L. Zhang, B. He, X. Chen and J. Sun, Carbohydr. Polym., 2021, 256, 117580 CrossRef CAS PubMed.
- J. Xu, Y. Liu and S.-H. Hsu, Molecules, 2019, 24, 3005 CrossRef CAS PubMed.
- H. Yu, Y. Wang, H. Yang, K. Peng and X. Zhang, J. Mater. Chem. B, 2017, 5, 4121–4127 RSC.
- G. D. Cha, W. H. Lee, S.-H. Sunwoo, D. Kang, T. Kang, K. W. Cho, M. Kim, O. K. Park, D. Jung and J. Lee, ACS Nano, 2022, 16, 554–567 CrossRef CAS.
- X. Jiang, X. Yang, B. Yang, L. Zhang and A. Lu, Carbohydr. Polym., 2021, 273, 118547 CrossRef CAS PubMed.
- Z. Wang, Y. Zhang, Y. Yin, J. Liu, P. Li, Y. Zhao, D. Bai, H. Zhao, X. Han and Q. Chen, Adv. Mater., 2022, 34, 2108300 CrossRef CAS PubMed.
- Y. Gao, Z. Li, J. Huang, M. Zhao and J. Wu, J. Mater. Chem. B, 2020, 8, 8768–8780 RSC.
- L. Xiang, J. Zhang, L. Gong and H. Zeng, Soft Matter, 2020, 16, 6697–6719 RSC.
- L. Xiang, L. Gong, J. Zhang, L. Zhang, W. Hu, W. Wang, Q. Lu and H. Zeng, Phys. Chem. Chem. Phys., 2019, 21, 20571–20581 RSC.
- L. Gong, L. Xiang, J. Zhang, J. Chen and H. Zeng, Langmuir, 2019, 35, 15914–15936 CrossRef CAS PubMed.
- W. Wang, Z. Zeng, L. Xiang, C. Liu, D. Diaz-Dussan, Z. Du, A. B. Asha, W. Yang, Y.-Y. Peng and M. Pan, ACS Nano, 2021, 15, 9913–9923 CrossRef CAS.
- Y. Zhou, Y. Qian, J. Wang, X. Qiu and H. Zeng, Biomacromolecules, 2020, 21, 3231–3241 CrossRef CAS PubMed.
- M. Wu, M. Pan, C. Qiao, Y. Ma, B. Yan, W. Yang, Q. Peng, L. Han and H. Zeng, Chem. Eng. J., 2022, 450, 138212 CrossRef CAS.
- M. Pan, K.-C. T. Nguyen, W. Yang, X. Liu, X.-Z. Chen, P. W. Major, L. H. Le and H. Zeng, Chem. Eng. J., 2022, 434, 134418 CrossRef CAS.
- X. Peng, W. Wang, W. Yang, J. Chen, Q. Peng, T. Wang, D. Yang, J. Wang, H. Zhang and H. Zeng, J. Colloid Interface Sci., 2022, 618, 111–120 CrossRef CAS PubMed.
- J. N. Israelachvili and P. M. McGuiggan, J. Mater. Res., 1990, 5, 2223–2231 CrossRef CAS.
- J. N. Israelachvili and G. Adams, Nature, 1976, 262, 774–776 CrossRef CAS.
- D. S. Hwang, H. Zeng, Q. Lu, J. Israelachvili and J. H. Waite, Soft Matter, 2012, 8, 5640–5648 RSC.
- H. Zeng, D. S. Hwang, J. N. Israelachvili and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12850–12853 CrossRef CAS PubMed.
- C. Zhang, L. Xiang, J. Zhang, C. Liu, Z. Wang, H. Zeng and Z.-K. Xu, Chem. Sci., 2022, 13, 1698–1705 RSC.
- J. Israelachvili, Y. Min, M. Akbulut, A. Alig, G. Carver, W. Greene, K. Kristiansen, E. Meyer, N. Pesika and K. Rosenberg, Rep. Prog. Phys., 2010, 73, 036601 CrossRef.
- A. Faghihnejad, K. E. Feldman, J. Yu, M. V. Tirrell, J. N. Israelachvili, C. J. Hawker, E. J. Kramer and H. Zeng, Adv. Funct. Mater., 2014, 24, 2322–2333 CrossRef CAS.
- H. Zeng, Polymer adhesion, friction, and lubrication, John Wiley & Sons, 2013 Search PubMed.
- C. Shi, D. Y. Chan, Q. Liu and H. Zeng, J. Phys. Chem. C, 2014, 118, 25000–25008 CrossRef CAS.
- C. Shi, B. Yan, L. Xie, L. Zhang, J. Wang, A. Takahara and H. Zeng, Angew. Chem., Int. Ed., 2016, 55, 15017–15021 CrossRef CAS PubMed.
- X. Cui, J. Liu, L. Xie, J. Huang, Q. Liu, J. N. Israelachvili and H. Zeng, Angew. Chem., 2018, 130, 12079–12084 CrossRef.
- J. Chen, Q. Peng, X. Peng, H. Zhang and H. Zeng, Chem. Rev., 2022, 122, 14594–14678 CrossRef CAS PubMed.
- L. Xie, L. Gong, J. Zhang, L. Han, L. Xiang, J. Chen, J. Liu, B. Yan and H. Zeng, J. Mater. Chem. A, 2019, 7, 21944–21952 RSC.
- J. Zhang, X. Lu, C. Shi, B. Yan, L. Gong, J. Chen, L. Xiang, H. Xu, Q. Liu and H. Zeng, Chem. Eng. J., 2019, 358, 842–849 CrossRef CAS.
- J. Chen and H. Zeng, Langmuir, 2022, 38, 12999–13008 CrossRef CAS PubMed.
- L. Meng, C. Shao, C. Cui, F. Xu, J. Lei and J. Yang, ACS Appl. Mater. Interfaces, 2019, 12, 1628–1639 CrossRef.
- R. Tan, Z. She, M. Wang, Z. Fang, Y. Liu and Q. Feng, Carbohydr. Polym., 2012, 87, 1515–1521 CrossRef CAS.
- J. W. Tyrrell and P. Attard, Phys. Rev. Lett., 2001, 87, 176104 CrossRef CAS PubMed.
- A. Faghihnejad and H. Zeng, Soft Matter, 2012, 8, 2746–2759 RSC.
- S. H. Donaldson Jr, A. Røyne, K. Kristiansen, M. V. Rapp, S. Das, M. A. Gebbie, D. W. Lee, P. Stock, M. Valtiner and J. Israelachvili, Langmuir, 2015, 31, 2051–2064 CrossRef.
- I. M. Geisler and J. P. Schneider, Adv. Funct. Mater., 2012, 22, 529–537 CrossRef CAS.
- H. Liu, X. Hu, W. Li, M. Zhu, J. Tian, L. Li, B. Luo, C. Zhou and L. Lu, Chem. Eng. J., 2023, 452, 139368 CrossRef CAS.
- H. Tan, C. Xiao, J. Sun, D. Xiong and X. Hu, Chem. Commun., 2012, 48, 10289–10291 RSC.
- D. Wang, Y. Xia, D. Zhang, X. Sun, X. Chen, S. Oliver, S. Shi and L. Lei, ACS Appl. Polym. Mater., 2020, 2, 1587–1596 CrossRef CAS.
- X. Zhao, J. Yang, Y. Liu, J. Gao, K. Wang and W. Liu, Chem. Eng. J., 2021, 404, 127096 CrossRef CAS.
- J. L. Cook, C. A. Hunter, C. M. Low, A. Perezral Engi and J. G. Vinter, Angew. Chem., Int. Ed., 2007, 46, 3706–3709 CrossRef CAS PubMed.
- M. Yang, X. Fei, J. Tian, L. Xu, Y. Wang and Y. Li, J. Mater. Chem. B, 2022, 10, 6026–6037 RSC.
- C. Shao, H. Chang, M. Wang, F. Xu and J. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 28305–28318 CrossRef CAS PubMed.
- Q. Cheng, S. Ding, Y. Zheng, M. Wu, Y.-Y. Peng, D. Diaz-Dussan, Z. Shi, Y. Liu, H. Zeng and Z. Cui, Biomacromolecules, 2021, 22, 1685–1694 CrossRef CAS PubMed.
- K. Lei, K. Wang, Y. Sun, Z. Zheng and X. Wang, Adv. Funct. Mater., 2021, 31, 2008010 CrossRef CAS.
- Y. Wang, M. Yang and Z. Zhao, Carbohydr. Polym., 2023, 310, 120723 CrossRef CAS PubMed.
- W. Wang, Z. Zeng, L. Xiang, C. Liu, D. Diaz-Dussan, Z. Du, A. B. Asha, W. Yang, Y.-Y. Peng and M. Pan, ACS Nano, 2021, 15, 9913–9923 CrossRef CAS.
- S. H. Söntjens, R. P. Sijbesma, M. H. van Genderen and E. Meijer, J. Am. Chem. Soc., 2000, 122, 7487–7493 CrossRef.
- H. M. Keizer, J. J. González, M. Segura, P. Prados, R. P. Sijbesma, E. Meijer and J. de Mendoza, Chem. – Eur. J., 2005, 11, 4602–4608 CrossRef CAS PubMed.
- S. H. Söntjens, R. P. Sijbesma, M. H. van Genderen and E. Meijer, Macromolecules, 2001, 34, 3815–3818 CrossRef.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. Dankers and E. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS PubMed.
- G. Ligthart, H. Ohkawa, R. P. Sijbesma and E. Meijer, J. Am. Chem. Soc., 2005, 127, 810–811 CrossRef CAS PubMed.
- J. Choi, S. Kim, J. Yoo, S.-H. Choi and K. Char, Macromolecules, 2021, 54, 6389–6399 CrossRef CAS.
- H. Kautz, D. Van Beek, R. P. Sijbesma and E. Meijer, Macromolecules, 2006, 39, 4265–4267 CrossRef CAS.
- J. Zhang and H. Zeng, Engineering, 2021, 7, 63–83 CrossRef CAS.
- H. Zeng, Y. Tian, T. H. Anderson, M. Tirrell and J. N. Israelachvili, Langmuir, 2008, 24, 1173–1182 CrossRef CAS PubMed.
- Q. Lu, J. Wang, A. Faghihnejad, H. Zeng and Y. Liu, Soft Matter, 2011, 7, 9366–9379 RSC.
- L. Xiang, J. Zhang, W. Wang, L. Gong, L. Zhang, B. Yan and H. Zeng, Acta Biomater., 2020, 117, 294–301 CrossRef CAS.
- L. Xiang, J. Zhang, L. Gong, L. Han, C. Zhang, B. Yan, J. Liu and H. Zeng, Langmuir, 2019, 35, 15639–15650 CrossRef CAS PubMed.
- J. Chen, M. Wu, L. Gong, J. Zhang, B. Yan, J. Liu, H. Zhang, T. Thundat and H. Zeng, J. Phys. Chem. C, 2019, 123, 4540–4548 CrossRef CAS.
- J. Hou, N. Li, W. Zhang and W. Zhang, Acta Biomater., 2023, 157, 310–320 CrossRef CAS.
- M. I. Giannotti and G. J. Vancso, ChemPhysChem, 2007, 8, 2290–2307 CrossRef CAS PubMed.
- F. Oesterhelt, M. Rief and H. Gaub, New J. Phys., 1999, 1, 6 CrossRef.
- Y. Zhang, L. Tao, S. Li and Y. Wei, Biomacromolecules, 2011, 12, 2894–2901 CrossRef CAS.
- P. Kovaricek and J.-M. Lehn, J. Am. Chem. Soc., 2012, 134, 9446–9455 CrossRef CAS.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- H. Guo, S. Huang, A. Xu and W. Xue, Chem. Mater., 2022, 34, 2655–2671 CrossRef CAS.
- M. R. Arkenberg, H. D. Nguyen and C.-C. Lin, J. Mater. Chem. B, 2020, 8, 7835–7855 RSC.
- B. Yan, J. Huang, L. Han, L. Gong, L. Li, J. N. Israelachvili and H. Zeng, ACS Nano, 2017, 11, 11074–11081 CrossRef CAS PubMed.
- T. Lei, M. Guan, J. Liu, H.-C. Lin, R. Pfattner, L. Shaw, A. F. McGuire, T.-C. Huang, L. Shao and K.-T. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5107–5112 CrossRef CAS PubMed.
- H. Ding, P. Tan, S. Fu, X. Tian, H. Zhang, X. Ma, Z. Gu and K. Luo, J. Controlled Release, 2022, 348, 206–238 CrossRef CAS.
- N. Xiao, H. Liang and J. Lu, Soft Matter, 2011, 7, 10834–10840 RSC.
- R. Antony, T. Arun and S. T. D. Manickam, Int. J. Biol. Macromol., 2019, 129, 615–633 CrossRef CAS PubMed.
- N. Chen, H. Wang, C. Ling, W. Vermerris, B. Wang and Z. Tong, Carbohydr. Polym., 2019, 225, 115207 CrossRef CAS PubMed.
- C. Mo, L. Xiang and Y. Chen, Macromol. Rapid Commun., 2021, 42, 2100025 CrossRef CAS PubMed.
- C. Yang, J. Dawulieti, K. Zhang, C. Cheng, Y. Zhao, H. Hu, M. Li, M. Zhang, L. Chen and K. W. Leong, Adv. Funct. Mater., 2022, 32, 2111698 CrossRef CAS.
- D. Fass and C. Thorpe, Chem. Rev., 2018, 118, 1169–1198 CrossRef CAS.
- H. F. Gilbert, Methods Enzymol., 1995, 251, 8–28 CAS.
- D. M. Rothwarf and H. A. Scheraga, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 7944–7948 CrossRef CAS.
- P. A. Fernandes and M. J. Ramos, Chem. – Eur. J., 2004, 10, 257–266 CrossRef CAS PubMed.
- C. Jacob, G. I. Giles, N. M. Giles and H. Sies, Angew. Chem., Int. Ed., 2003, 42, 4742–4758 CrossRef CAS PubMed.
- A. P. Wiita, S. R. K. Ainavarapu, H. H. Huang and J. M. Fernandez, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7222–7227 CrossRef CAS PubMed.
- A. E. Beedle, M. Mora, S. Lynham, G. Stirnemann and S. Garcia-Manyes, Nat. Commun., 2017, 8, 15658 CrossRef CAS.
- G. A. Barcan, X. Zhang and R. M. Waymouth, J. Am. Chem. Soc., 2015, 137, 5650–5653 CrossRef CAS PubMed.
- S.-Y. Choh, D. Cross and C. Wang, Biomacromolecules, 2011, 12, 1126–1136 CrossRef CAS PubMed.
- H. G. Silverman and F. F. Roberto, Mar. Biotechnol., 2007, 9, 661–681 CrossRef CAS.
- N. Bandara, H. Zeng and J. Wu, J. Adhes. Sci. Technol., 2013, 27, 2139–2162 CrossRef CAS.
- W. Yang, M. Pan, J. Zhang, L. Zhang, F. Lin, X. Liu, C. Huang, X. Z. Chen, J. Wang and B. Yan, Adv. Funct. Mater., 2022, 32, 2109989 CrossRef CAS.
- D. S. Hwang, H. Zeng, A. Srivastava, D. V. Krogstad, M. Tirrell, J. N. Israelachvili and J. H. Waite, Soft Matter, 2010, 6, 3232–3236 RSC.
- Q. Lu, E. Danner, J. H. Waite, J. N. Israelachvili, H. Zeng and D. S. Hwang, J. R. Soc., Interface, 2013, 10, 20120759 CrossRef.
- L. Li and H. Zeng, Biotribology, 2016, 5, 44–51 CrossRef.
- B. Yang, N. Ayyadurai, H. Yun, Y. S. Choi, B. H. Hwang, J. Huang, Q. Lu, H. Zeng and H. J. Cha, Angew. Chem., 2014, 126, 13578–13582 CrossRef.
- L. Xie, X. Cui, L. Gong, J. Chen and H. Zeng, Langmuir, 2020, 36, 2985–3003 CrossRef CAS PubMed.
- S. Kim, A. Faghihnejad, Y. Lee, Y. Jho, H. Zeng and D. S. Hwang, J. Mater. Chem. B, 2015, 3, 738–743 RSC.
- Q. Wei, K. Achazi, H. Liebe, A. Schulz, P. L. M. Noeske, I. Grunwald and R. Haag, Angew. Chem., Int. Ed., 2014, 53, 11650–11655 CrossRef CAS PubMed.
- W. Yang, Z. Zhao, M. Pan, L. Gong, F. Wu, C. Huang, X. Wang, J. Wang and H. Zeng, J. Colloid Interface Sci., 2022, 625, 628–639 CrossRef CAS PubMed.
- Q. Guo, J. Chen, J. Wang, H. Zeng and J. Yu, Nanoscale, 2020, 12, 1307–1324 RSC.
- L. Xu, S. Tang, H. Yang, M. Liang, P. Ren, D. Wei, J. He, W. Kong, P. Liu and T. Zhang, J. Mater. Chem. B, 2022, 10, 6338–6350 RSC.
- F.-X. Zhang, P. Liu, W. Ding, Q.-B. Meng, D.-H. Su, Q.-C. Zhang, R.-X. Lian, B.-Q. Yu, M.-D. Zhao and J. Dong, Biomaterials, 2021, 278, 121169 CrossRef CAS PubMed.
- L. Wang, X. Zhang, K. Yang, Y. V. Fu, T. Xu, S. Li, D. Zhang, L. N. Wang and C. S. Lee, Adv. Funct. Mater., 2020, 30, 1904156 CrossRef CAS.
- Y. C. Choi, J. S. Choi, Y. J. Jung and Y. W. Cho, J. Mater. Chem. B, 2014, 2, 201–209 RSC.
- L. Li, B. Yan, J. Yang, L. Chen and H. Zeng, Adv. Mater., 2015, 27, 1294–1299 CrossRef CAS.
- B. Yan, C. He, S. Chen, L. Xiang, L. Gong, Y. Gu and H. Zeng, CCS Chem., 2022, 4, 2724–2737 CrossRef CAS.
- L. Li, B. Yan, J. Yang, W. Huang, L. Chen and H. Zeng, ACS Appl. Mater. Interfaces, 2017, 9, 9221–9225 CrossRef CAS.
- B. D. B. Tiu, P. Delparastan, M. R. Ney, M. Gerst and P. B. Messersmith, Angew. Chem., 2020, 132, 16759–16767 CrossRef.
- H. Geng, P. Zhang, Q. Peng, J. Cui, J. Hao and H. Zeng, Acc. Chem. Res., 2022, 55, 1171–1182 CrossRef CAS PubMed.
- Q. Lu, D. X. Oh, Y. Lee, Y. Jho, D. S. Hwang and H. Zeng, Angew. Chem., 2013, 125, 4036–4040 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |