
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriborane (B3H7)-mediated regioselective substitution reactions of pyridine derivatives†
Zi-Heng
Fan
a,
Jia-Xin
Kang
a,
Sihan
Jia
a,
Qiao-Jing
Pan
a,
Lei
Cao
a,
Xi-Meng
Chen
a,
Cong-Qiao
Xu
 b,
Yan-Na
Ma
*c and
Xuenian
Chen
*ac
b,
Yan-Na
Ma
*c and
Xuenian
Chen
*ac
aSchool of Chemistry and Chemical Engineering, Henan Key Laboratory of Boron Chemistry and Advanced Materials, Henan Normal University, Xinxiang, Henan 453007, China
bDepartment of Chemistry, Southern University of Science and Technology, Shenzhen 518055, China
cCollege of Chemistry, Zhengzhou University, Zhengzhou, Henan 450001, China. E-mail: mayanna@zzu.edu.cn; xuenian_chen@zzu.edu.cn
First published on 26th July 2024
Abstract
There exists an interplay between borane and a Lewis base in their adducts. However, studies on these adducts so far have mainly focused on the different reactions of B–H bonds with limited attention given to the influence of borane on the chemistry of the Lewis base, except for BF3 and BAr3. Herein, we have synthesized novel borane adducts with pyridine derivatives, Py·B3H7, in which the coordination of B3H7 efficiently achieved the intra-molecular charge transfer. The strong B–N bond in these adducts resulted in the formation of stable dearomatic intermediates of pyridine derivatives, confirmed by 1H and 11B NMR spectroscopy, from which different reactions have transpired to realize C(sp3)–H and C(sp2)–H functionalization under mild conditions. The B3H7 pyridine derivatives are stable and do not dissociate or decompose during the reaction process. The high stability of the B–N bond makes this method a good option for boron-containing drugs with potential for use in boron neutron capture therapy (BNCT).
Introduction
Simple boranes, such as BH3 and BF3, denoted as B, cannot exist stably due to the electron deficiency of the boron atom. Usually they form a Lewis acid–base adduct L·B with a Lewis base (L = THF, Me2S, NH3, Me3N, etc.) through a dative bond. There is no doubt that the interplay between the Lewis acid and base exists in these adducts (Scheme 1a). However, while the influence of L on the properties and reactivity of B has been extensively explored (Scheme 1aI)1 and the steric interrelationship results in frustrated Lewis pair (FLP) complexes that are also well-known (Scheme 1aII),2 the influence of boranes (B) on a Lewis base (L) has been relatively underexplored (Scheme 1aIII).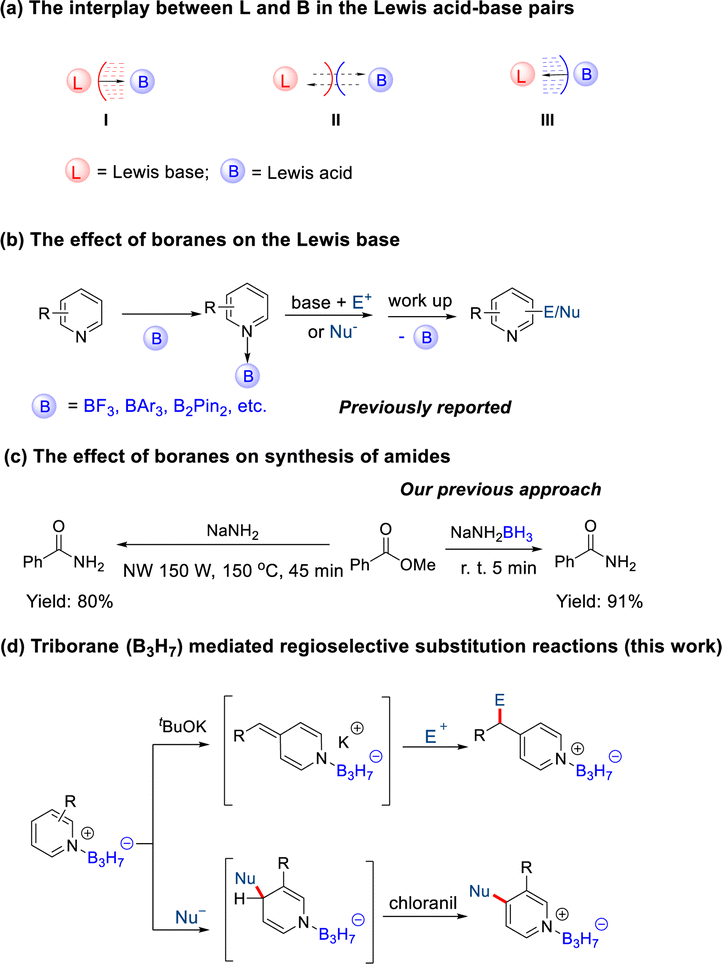 | ||
| Scheme 1 Properties and reactions of borane Lewis base adducts. | ||
Most studies on the influence of boranes (B) on a Lewis base (L) have mainly focused on the impacts of BF3, BAr3, and B2pin2 on pyridine derivatives. BF3 mediated C-2 or C-4 selective functionalization,3 C-2 methyl group deprotonation,4 and asymmetric synthesis of pyridine derivatives5 have been extensively studied. BAr3 and B2pin2 mediated C-3 alkylation,6 trifluoromethylthiolation,7 allylation,8 and cyanation9 of pyridine have become hot research fields recently (Scheme 1b). In contrast, the effects of the boron hydrogen complexes, such as BH3, on the Lewis base have rarely been reported because of their strong reducing ability10 and weak Lewis acidity. Recent studies from our group have found that NaNH2BH3 could directly react with esters to form amides,11 which did not work for NaNH2 (Scheme 1c).12 This result suggested that BH3 could change the reactivity of the Lewis base of adducts.
Multinuclear boranes are usually used as reducing agents in organic synthesis.13 Recently, multinuclear boranes, such as carborane, [B10H10]2−, [B6H6]2−, and [HCB11Cl11]− have also been used as borylation reagents14 or catalysts.15 However, there has been hardly any research into the influence of the Lewis acid properties of multinuclear boranes on reactions.
We have systematically studied the syntheses and properties of octahydridotriborate (B3H8−) and its neutral derivatives (L·B3H7), indicating that B3H7 as a Lewis acid possesses remarkable features.16 Compared with BH3, B3H7 exhibits greater stability, weaker reducing ability, and a higher ability to coordinate with metals.17 Compared with BF3, B3H7 has relatively low acidity. In addition, B3H7 has a steric effect and σ aromaticity. All these properties may enable the B3H7 group to mediate different organic reactions (Scheme 1d).
Results and discussion
Synthesis and properties of Py·B3H7
To explore the effects of B3H7 on a Lewis base, we selected pyridine, which is widely used in pharmaceutical chemistry, functional materials, and natural products as a Lewis base.18 We synthesized Py·B3H7 through a reaction of pyridine with THF·B3H7, formed in situ by reacting KB3H8 with HCl·Et2O. The Py·B3H7 adduct 1a was obtained as a white solid in 91% yield (Scheme 2). This novel compound showed excellent stability towards both acid and base in aqueous solutions at room temperature (see ESI, Fig. S1†), offering an opportunity to examine their reactivity. The results indicated that a rapid dearomatic reaction of pyridine derivatives occurred, which then further reacted with electrophiles or oxidants to provide the rearomatic products (Scheme 1d).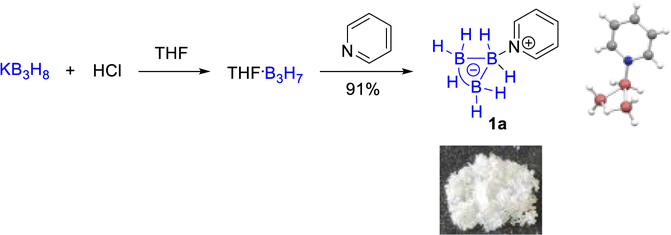 | ||
| Scheme 2 Synthesis of the pyridine B3H7 adduct. | ||
B3H7 mediated dearomatic reaction of Py derivatives
A direct dearomatic reaction of pyridine is impossible under mild conditions. The dearomatic intermediates of BF3 coordinated pyridine derivatives are potential products in most situations.19 We found that the B3H7 mediated dearomatic reaction of 4-methylpyridine rapidly proceeded by reacting with KOtBu to form 4-methylene dihydropyridine at low temperatures. This intermediate was identified spectrally (Fig. 1e) but not isolated, which indicated that B3H7 could stabilize the dihydropyridine derivatives. To explore the universality of such an effect, we conducted a reaction of the B3H7 mediated addition of Grignard reagents to pyridines bearing an electron-withdrawing substituent. Similarly, the dihydropyridine intermediates were stable under these conditions, which could also be characterized by 11B, 1H, and 1H{11B} NMR spectra (ESI, Fig. S7–S9†).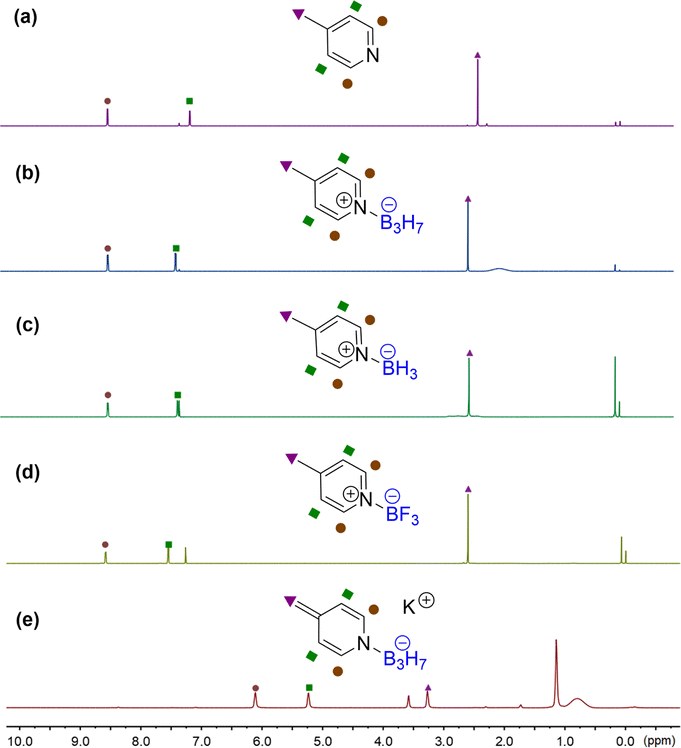 | ||
| Fig. 1 1H NMR in CDCl3 of (a) 4-methylpyridine, (b) 1b, (c) 1c, (d) 1d, and (e) 4-methylene dihydropyridine 3a in THF-d8 at −78 °C. | ||
These observations differ from the BF3 mediated similar reactions of pyridine derivatives under the same conditions where no dihydropyridine derivative intermediates were detected. We further set up control experiments to explore the different influences of BH3, BF3, and B3H7 on pyridine in their adducts. The 1H NMR spectra of 4-methylpyridine of the corresponding B3H7 adduct 1b, BH3 adduct 1c, and BF3 adduct 1d showed that the chemical shifts of the protons at the C3 and C5 position and the methyl downfield-shifted in the adducts in comparison with 4-methylpyridine.
The impact of the BF3 group was the most significant and those of the BH3 and B3H7 groups were almost the same, but the B3H7 group was slightly larger than that of BH3 (Fig. 1a–d). Therefore, we speculated that the impacts of these three Lewis acids on their coordinated Lewis base should be BF3 > B3H7 > BH3.
Surprisingly, the 11B{1H} NMR signals of the B3H7 coordinated 4-methylene dihydropyridine anion 3a and ethyl 4-isopropyl 3-carboxylate dihydropyridine complex 3b changed significantly in comparison with those of their parent complexes. The top boron atom connected with the nitrogen atom shifted to a lower field and the two base boron atoms to a higher field (Fig. 2), in contrast to the general B-signals of the B3H7 adducts.13a Furthermore, the signal shifts in 3a and 3b are different (Fig. 2b and d). The exploration of the reason for such a difference is underway in our laboratory.
 | ||
| Fig. 2 (a) 11B{1H} NMR of 1b in CDCl3. (b) 11B{1H} NMR of 4-methylene dihydropyridine 3a in THF-d8 at −78 °C. (c) 11B{1H} NMR of ethyl nicotinate·B3H76a in CDCl3. (d) 11B{1H} NMR of ethyl 4-isopropyl 3-carboxylate dihydropyridine complex 3b in THF-d8 at room temperature. | ||
B3H7 mediated the nucleophilic substitution reaction of 4-alkylpyridine
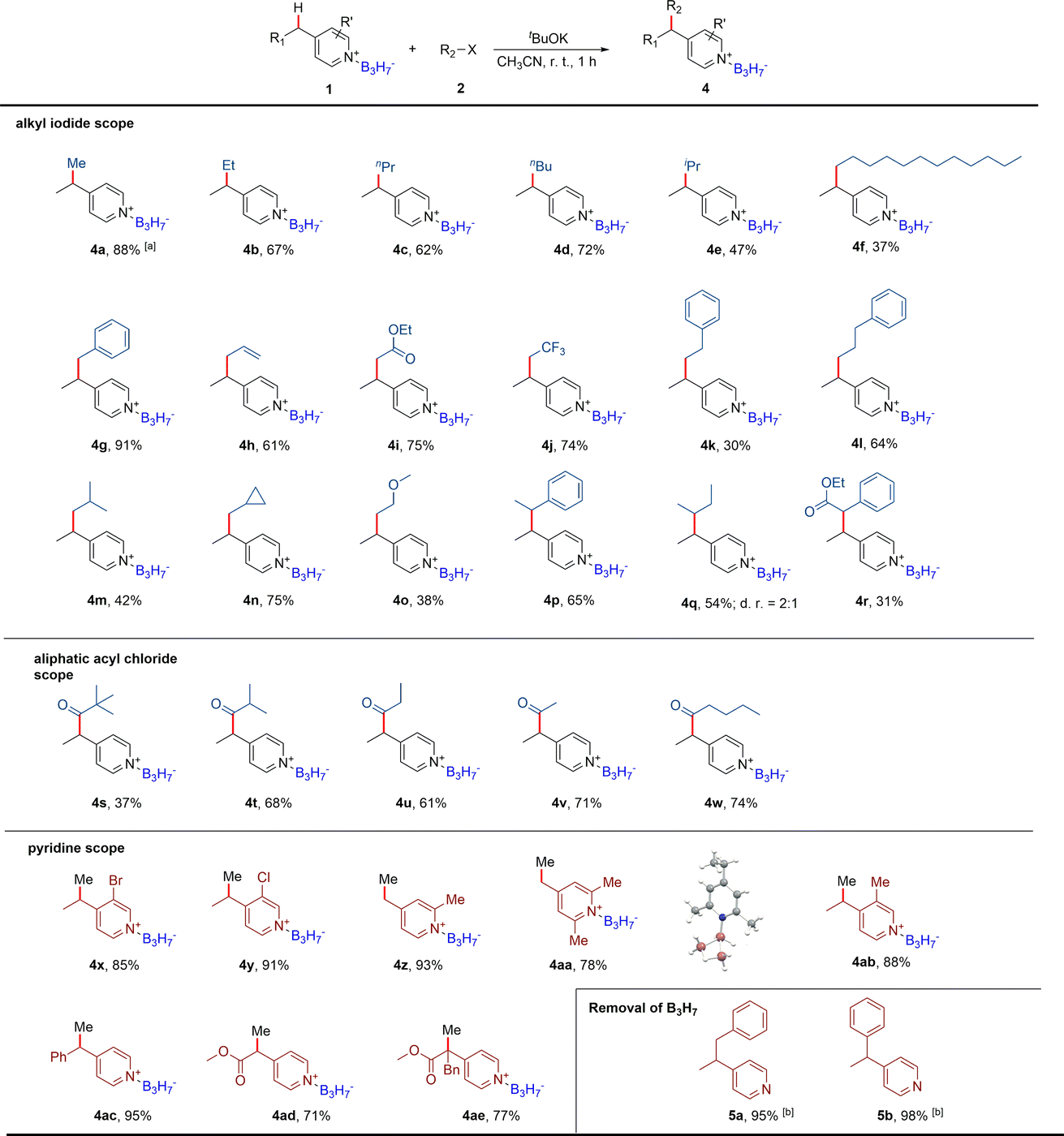
Based on the formation of the dearomatic intermediates, we further explored the B3H7 mediated nucleophilic substitution reactions of pyridine derivatives by selecting the 4-ethylpyridine B3H7 adduct 1e as the model substrate, reacting with methyl iodide (Table S4†). After systematically screening reactions with different bases in various solvents, we found that the methylation product 4a was obtained at an 88% yield in CH3CN when a mild base KOtBu was used (entry 1 in Table S4†). In contrast, no reaction occurred when the BH3 adduct 1f was used (entry 18 in Table S4†) and the product decomposed when the BF3 adduct 1g was used (entry 19 in Table S4†). Only a trace amount of the product was detected. The reaction with 4-ethyl pyridine, without B3H7 coordinated, was also conducted at similar conditions and no reaction occurred (entry 20 in Table S4†). As expected, the starting material 4-ethyl pyridine was collected. These results suggested that the Py·B3H7 adducts have a unique stability and reactivity.Considering the excellent stability of the Py·B3H7 adducts and the importance of boron-containing compounds in BNCT, we further examined the scope of the electrophiles (Table 1). Firstly, different alkyl iodides were detected. As shown in Table 1, regardless of whether the alkyl iodides were primary or secondary, products (4b–4r) were obtained in moderate to good yields. Additionally, this transformation shows a high functional group tolerance, such as allyl, ester, trifluoromethyl, cyclopropyl, and alkoxy were all compatible. We also found that the yields of the reactions using the alkyl bromides were significantly low compared to the alkyl iodines. Encouraged by these results, we further screened other electrophilic reagents and found that acyl chlorides could also be used in this reaction to produce stable Py·B3H7 derivatives (4s–4w). The scope for the pyridine core was assessed next (4x–4ae). Gratifyingly, our method showed a good C4 regioselectivity for the pyridines that contained multiple methyl groups at C2, C3, or C6 (4z–4ab). We thought this was because the high steric hindrance of B3H7 decreased the reactivity of the C2 and C6 positions, and the acidity of the methyl group at the C3 position is relatively weak. It was worth noting that the reaction of 4-methyl Py·B3H7 with methyl iodide gave a mixture of 4-ethyl Py·B3H7, 4-isopropyl Py·B3H7, and 4-tert-butyl Py·B3H7. Introducing methyl groups to the C2 and C6 positions could weaken the acidity of the C4 methyl to give the mono-methylation products (4z, 4aa) in high yields. The structure of 4aa was determined by single-crystal X-ray diffraction. Regardless of whether the substituent group at the C4 position was the primary, secondary, or tertiary alkyl group, the reactions transpired smoothly in high yields. In addition, the halogen atom (4x, 4y) and ester group (4ad, 4ae) were compatible in this transformation.
a Conditions: 1 (0.5 mmol, 1.0 eq.), 2 (1.25 mmol, 2.5 eq.), and base (1.25 mmol, 2.5 eq.) in CH3CN (15 mL) at room temperature for 1 hour. Isolated yield. The d.r. value was determined by 1H NMR analysis of the crude reaction mixture.
b Conditions: 4 (0.5 mmol, 1.0 eq.), MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (5:0.5 mL), 60 °C, 12 h. :H2O (5:0.5 mL), 60 °C, 12 h.
|
|---|
|
To remove B3H7 from pyridine derivatives has also been explored to expand the application of this protocol. After a series of explorations for the reaction conditions, the B3H7 unit was successfully removed with MeOH and H2O at 60 °C, and the corresponding pyridine compounds were obtained in almost quantitative yields (5a, 5b).20
The B3H7 mediated nucleophilic addition of Grignard reagents to electron deficient pyridines and their subsequent oxidation
The BF3 promoted C4 direct alkylation of functionalized pyridines has been reported, in which BF3-mediated 4-isopropyl-dihydropyridine was considered a potential intermediate.4a We have obtained the dearomatic dihydropyridine intermediate by the nucleophilic addition of iPrMgCl·LiCl to ethyl nicotinate B3H7 adduct (Fig. 2d), further oxidation aromatization mediated by chloranil provided the 4-alkylated product 8a in 71% yield (Table 2). Based on this, we examined the scope of Grignard reagents and pyridine derivatives and successfully introduced different alkyl and aryl groups into the C4 position of electron-deficient pyridines (Table 2). In these reactions, moderate yields of products could be obtained (8b–8g). The low yields of 8h and 8i are probably due to using strong bases such as organic lithium reagents. The ethyl nicotinate B3H7 adduct would be destroyed by strong bases to produce the pyridine BH3 adduct, decreasing the yield. In addition, the electronic effects also played an important role in this reaction. The strength and stability of the electron-withdrawing group at the C3 position would affect the yields of products (8j, 8k, 8m, 8o, and 8s). When the pyridine had electron-donating groups, the yield would decrease due to the increase of the electron density at the C4 position (8l, 8n, 8p–8r). For all the low-yield reactions, the starting materials could be detected and recovered, such as the starting material for 8l could be recovered in 72%.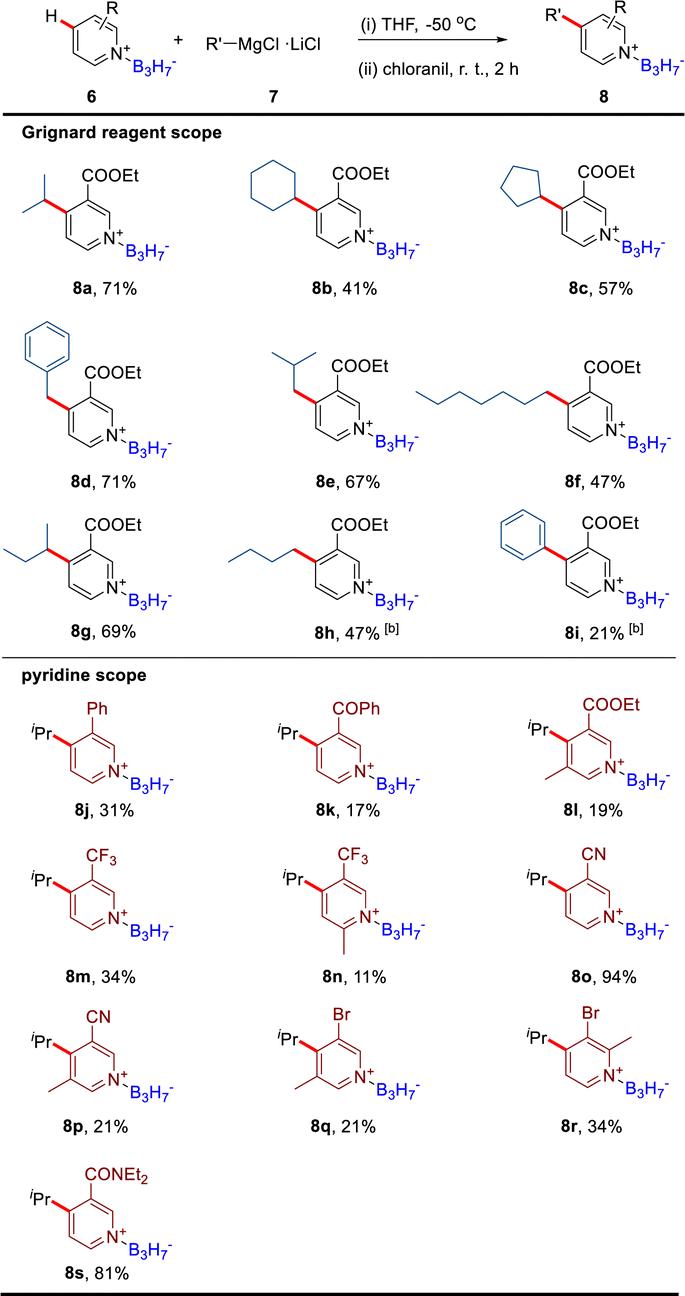
Mechanistic studies
To gain insight into the reaction mechanism, quantum chemical studies using density functional theory (DFT) have been performed. From the theoretical data, the reaction mechanism is proposed, as shown in Fig. 3.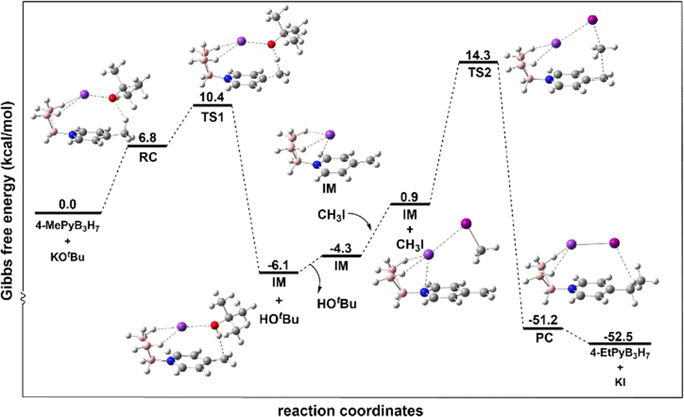 | ||
| Fig. 3 The energy profile of the reaction calculated at M06-2X-D3-SMD/def2-QZVP//B3LYP-D3(BJ)/6-311G(d,p)/SDD level of theory. | ||
First, the reactants 4-methyl Py·B3H7 and potassium tert-butoxide form the reaction complex RC. The relative free energy of RC is 6.8 kcal mol−1. In RC, the K+ cation interacted with the H of the B3H7 fragment, the K–H bond is 2.80 and 2.84 Å, and a hydrogen bond exists between the O of tert-butoxy and the H of the methyl group with a bond length of 2.10 Å. Subsequently, RC traverses transition state TS1 to yield a complex of intermediate (IM) and a tert-butanol with a relative free energy of −6.1 kcal mol−1. From RC to TS1, the K–H bond shortened to 2.77 Å and the O–H bond shortened from 2.10 Å to 1.37 Å. At the same time, the C–H bond enlarged from 1.11 Å to 1.29 Å. The relative free energy of TS1 is 10.4 kcal mol−1, indicating a modest energy barrier of 3.6 kcal mol−1 for this step. Then tert-butanol dissociates, leaving behind IM. Following this, a new complex, IM with CH3I, formed with a relative energy of 0.9 kcal mol−1 with the I–C bond of 2.23 Å. The subsequent reaction of IM and CH3I via transition state TS2 results in the final product. During this step, the I–C bond enlarged to 2.53 Å, and the C–C bond formed with a bond length of 2.47 Å. The relative energy of TS2 is 14.3 kcal mol−1, implying a barrier of 13.4 kcal mol−1 for the second step. With the Walden inversion of CH3, the final product formed and exhibits a relative free energy of −52.5 kcal mol−1. The negative ΔG of the whole reaction and the low energy barrier indicate that this reaction will easily proceed spontaneously. The charge distribution of CH3Py, CH3PyB3H7, KCH3PyB3H7+, and CH2PyB3H7− was further analysed by using Natural Population Analysis (NPA). As delineated in Table S5,† the formation of the N–B dative bond and the preferential association of the K ions near the B3H7 fragment caused the electron concentration to be towards the B3H7 fragment. Such intramolecular charge transfer facilitates the deprotonation.
The proposed mechanism is supported by the experimental results of the capture of the reaction intermediate (Scheme 3a–c). The 1H NMR spectroscopy showed that the B3H7 coordinated 4-methylene dihydropyridine anion intermediate 3a (IM) was formed in an almost quantitative yield by the deprotonation of 4-methyl Py·B3H71b with KOtBu in THF-d8 at −78 °C (Fig. 1e).21 The intermediate of 4-alkalene dihydropyridine was frequently discussed in the literature.22 However, the BH3 and BF3 coordinated 4-methylene dihydropyridine anion intermediates 3c and 3d could not be formed in similar reactions (Scheme 3b and c).
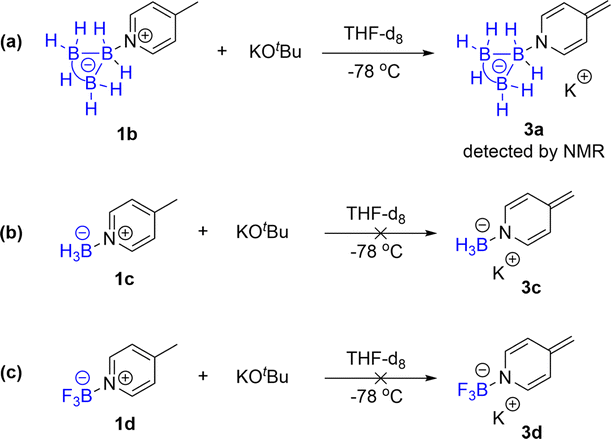 | ||
| Scheme 3 A mechanism study. | ||
These results were consistent with the theoretical prediction that the interaction between the alkali metal cation and the NB3H7 moiety induced the additional intramolecular charge transfer from CH3/CH2 to the B3H7 end to facilitate the reaction and also stabilized the formed 4-methylene dihydropyridine intermediate (Table S5†). This result was further confirmed by the fact that the addition of 18–C-6 to the reaction mixture would decrease the yields from 88% to 54% (Fig. S2†) because the formation of the K-18–C-6 complex weakened the interaction between the K cation and the B3H7 moiety.
10B-labeled 10B3H7 reactions
To evaluate the potential of this protocol in BNCT,23 a 10B-labeled 10B3H7 was introduced to the pyridine core (Scheme 4). Firstly, the 10B-labeled Na10B3H8 was synthesized by the reaction of Na10BH4 with I2. The reaction of 4-ethyl pyridine with the THF·10B3H7 formed the in situ produced 4-ethyl Py·10B3H79 in 60% yield.24 The 10B-labeled 4-ethyl Py·10B3H7 could also react with alkyl iodide under KOtBu to afford the alkylated product 10 in a good yield.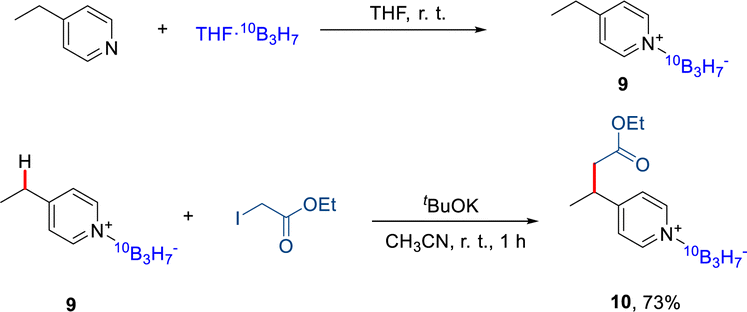 | ||
| Scheme 4 10B-labeled experiments. | ||
Conclusions
In summary, we have synthesized a novel class of Py·B3H7 compounds and studied the reactions of 4-alkyl Py·B3H7 with different electrophiles and pyridine bearing an electron-withdrawing group with nucleophiles. The relatively high stability of the Py·B3H7 adducts results in the formation of stable dearomatic intermediates, dihydropyridine, under mild conditions, from which the rearomatization has further transpired to form different alkylation and acylation products. The mechanistic study reveals that the coordination of the B3H7 unit led to the intra-molecular charge transfer from the pyridine ring to borane, realizing the alkylation and acylation reactions of the C(sp3)–H bond of the C-4 position with excellent regioselectivity and high yields under mild reaction conditions. The B3H7 coordinated dihydropyridine intermediate was obtained in solution and characterized by 1H and 11B NMR. Such an intra-molecular charge transfer also promoted C-4 direct alkylation of the pyridines bearing an electron-withdrawing group to provide the 4-alkylated products. Compared with BH3 and BF3, B3H7 possesses remarkable features, such as relatively higher stability, a weaker reducing ability, and a higher ability to coordinate with metals, as well as steric effects and σ aromaticity. All these properties will enable the B3H7 group to mediate different organic reactions. In addition, stable Py·B3H7 will play a significant role in boron-containing drugs and organic synthesis. Further research into the effect of B3H7 on other series of Lewis bases and the reaction mechanisms is underway in our laboratory.Data availability
Data for all compounds in this manuscript are available in the ESI,† which includes experimental details, characterisation data, computational data and copies of 1H, 13C, 19F, and 11B NMR spectra. The data supporting this article have been included as part of the ESI.† Crystallographic data have been deposited with the Cambridge Crystallographic Data Center as supplementary publication no. CCDC-2308054 (1a) and CCDC-2308050 (4aa).Author contributions
Y.-N. M. and X. C. conceived and designed the study; Z.-H. F., Q.-J. P., L. C., S. J., and X.-M. C. performed the experiments. J.-X. K. and C.-Q. X. performed the mechanistic and DFT studies. Y.-N. M. and X. C. prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 22171246 and 22271256).Notes and references
- (a) B. T. Cho, Chem. Soc. Rev., 2009, 38, 443–452 RSC; (b) J. Yang, Z. Li and S. Zhu, Chin. J. Org. Chem., 2017, 37, 2481–2497 Search PubMed; (c) Y.-J. Yu, F.-L. Zhang, T.-Y. Peng, C.-L. Wang, J. Cheng, C. Chen, K. N. Houk and Y.-F. Wang, Science, 2021, 371, 1232–1240 CrossRef PubMed; (d) J. H. Kim, T. Constantin, M. Simonetti, J. Llaveria, N. S. Sheikh and D. Leonori, Nature, 2021, 595, 677–684 CrossRef PubMed; (e) Z. Chen, Q. Fan, B. Yin, Q. Li and H. Wang, Chin. J. Org. Chem., 2023, 43, 1706–1712 CrossRef; (f) T.-Y. Peng, F.-L. Zhang and Y.-F. Wang, Acc. Chem. Res., 2023, 56, 169–186 CrossRef PubMed; (g) Y.-Q. Miao, X.-Y. Li, Q.-J. Pan, Y. Ma, J.-X. Kang, Y.-N. Ma, Z. Liu and X. Chen, Green Chem., 2022, 24, 7113–7121 RSC; (h) M. Yamashita and K. Nozaki, Bull. Chem. Soc. Jpn., 2008, 81, 1377–1392 CrossRef; (i) R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610–613 CrossRef PubMed.
- (a) G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef PubMed; (b) H. C. Brown, H. I. Schlesinger and S. Z. Cardon, J. Am. Chem. Soc., 1942, 64, 325–329 CrossRef; (c) G. Erős, H. Mehdi, I. Pápai, T. A. Rokob, P. Király, G. Tárkányi and T. Soős, Angew. Chem., Int. Ed., 2010, 49, 6559–6563 CrossRef PubMed; (d) T. Wang, G. Kehr, L. Liu, S. Grimme, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2016, 138, 4302–4305 CrossRef PubMed.
- (a) S. V. Kessar, P. Singh, K. N. Singh and M. Dutt, J. Chem. Soc. Chem. Commun., 1991, 570–571 RSC; (b) S. V. Kessar, P. Singh, R. Vohra, N. Kaur and K. Singh, J. Chem. Soc. Chem. Commun., 1991, 568–570 RSC; (c) M. Jaric, B. A. Haag, A. Unsinn, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2010, 49, 5451–5455 CrossRef PubMed.
- (a) Q. Chen, X. M. du Jourdin and P. Knochel, J. Am. Chem. Soc., 2013, 135, 4958–4961 CrossRef PubMed; (b) M. Jaric, B. A. Haag, S. M. Manolikakes and P. Knochel, Org. Lett., 2011, 13, 2306–2309 CrossRef PubMed; (c) S. M. Manolikakes, M. Jaric, K. Karaghiosoff and P. Knochel, Chem. Commun., 2013, 49, 2124–2126 RSC.
- (a) B. M. Trost and D. A. Thaisrivongs, J. Am. Chem. Soc., 2008, 130, 14092–14093 CrossRef PubMed; (b) B. M. Trost and D. A. Thaisrivongs, J. Am. Chem. Soc., 2009, 131, 12056–12057 CrossRef PubMed; (c) Z.-J. Cai, S.-Y. Wang and S.-J. Ji, Adv. Synth. Catal., 2013, 355, 2686–2692 CrossRef; (d) S. Dutta, J. H. Kim, K. Bhatt, D. R. L. Rickertsen, K. A. Abboud, I. Ghiviriga and D. Seidel, Angew. Chem., Int. Ed., 2023, e202313247 Search PubMed.
- (a) Z. Liu, J.-H. He, M. Zhang, Z.-J. Shi, H. Tang, X.-Y. Zhou, J.-J. Tian and X.-C. Wang, J. Am. Chem. Soc., 2022, 144, 4810–4818 CrossRef PubMed; (b) M. Xu, Z. Wang, Z. Sun, Y. Ouyang, Z. Ding, T. Yu, L. Xu and P. Li, Angew. Chem., Int. Ed., 2022, e202214507 Search PubMed; (c) S. Chakraborty and A. T. Biju, Angew. Chem., Int. Ed., 2022, 135, e202300049 CrossRef.
- (a) X.-Y. Zhou, M. Zhang, Z. Liu, J.-H. He and X.-C. Wang, J. Am. Chem. Soc., 2023, 144, 14463–14470 CrossRef; (b) R. Muta, T. Torigoe and Y. Kuninobu, Org. Lett., 2022, 24, 8218–8222 CrossRef; (c) C. M. Josephitis, H. M. H. Nguyen and A. McNally, Chem. Rev., 2023, 123, 7655–7691 CrossRef; (d) J. Wang, X.-X. Lu, R.-P. Yang, B.-B. Zhang, Z.-H. Xiang, J.-C. Li, L. Liu, S. Chao and X. Shang, Org. Lett., 2023, 25, 8489–8494 CrossRef.
- (a) J.-J. Tian, R.-R. Li, G.-X. Tian and X.-C. Wang, Angew. Chem., Int. Ed., 2023, 62, e202307697 CrossRef; (b) Z. Liu, Z.-J. Shi, L. Liu, M. Zhang, M.-C. Zhang, H.-Y. Guo and X.-C. Wang, J. Am. Chem. Soc., 2023, 145, 11789–11797 CrossRef PubMed; (c) F.-Y. Zhou and L. Jiao, Angew. Chem., Int. Ed., 2022, 61, e202201102Z CrossRef.
- (a) M. Zhang, Q. Zhou, H. Luo, Z.-L. Tang, X. Xu and X.-C. Wang, Angew. Chem., Int. Ed., 2023, 62, e202216894 CrossRef; (b) H. Cao, Q. Cheng and A. Studer, Angew. Chem., Int. Ed., 2023, 62, e202302941 CrossRef PubMed.
- (a) E. Marcantoni, S. Alessandrini, M. Malavolta, G. Bartoli, M. C. Bellucci, L. Sambri and R. Dalpozzo, J. Org. Chem., 1999, 64, 1986–1992 CrossRef PubMed; (b) M. Morimoto, W. Cao, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2021, 143, 2108–2114 CrossRef CAS; (c) M. Ding, J. Chang, J.-X. Mao, J. Zhang and X. Chen, J. Org. Chem., 2022, 87, 16230–16235 CrossRef CAS; (d) J. Wang, M.-Y. Ju, X. Wang, Y.-N. Ma, D. Wei and X. Chen, J. Org. Chem., 2021, 86, 5305–5316 CrossRef CAS PubMed.
- (a) Y. Guo, R.-Y. Wang, J.-X. Kang, Y.-N. Ma, C.-Q. Xu, J. Li and X. Chen, Nat. Commun., 2021, 12, 5964 CrossRef CAS PubMed; (b) Y.-Q. Miao, J.-X. Kang, Y.-N. Ma and X. Chen, Green Chem., 2021, 23, 3595–3599 RSC.
- A. Theppawong, P. Ploypradith, P. Chuawong, S. Ruchirawat and C. Montakarn, Chem.–Asian J., 2015, 10, 2631–2650 CrossRef CAS PubMed.
- (a) J. W. Bae, S. H. Lee, Y. J. Jung, C. O. M. Yoon and C. M. Yoon, Tetrahedron Lett., 2001, 42, 2137–2139 CrossRef CAS; (b) Y. J. Jung, Y. M. Chang, J. H. Lee and C. M. Yoon, Tetrahedron Lett., 2002, 43, 8735–8739 CrossRef CAS; (c) X. Li, T. Mi, W. Guo, Z. Ruan, Y. Guo, Y.-N. Ma and X. Chen, Chem. Commun., 2021, 57, 12776–12779 RSC.
- (a) S. Kim, J. W. Treacy, Y. A. Nelson, J. A. M. Gonzalez, M. Gembicky, K. N. Houk and A. M. Spokoyny, Nat. Commun., 2023, 14, 1671 CrossRef CAS PubMed; (b) X. Mu, J. C. Axtell, N. A. Bernier, K. O. Kirlikovali, D. Jung, A. Umanzor, K. Qian, X. Chen, K. L. Bay, M. Kirollos, A. L. Rheingold, K. N. Houk and A. M. Spokoyny, Chem, 2019, 5, 2461–2469 CrossRef CAS PubMed.
- (a) B. Shao, A. L. Bagdasarian, S. Popov and H. M. Nelson, Science, 2017, 355, 1403–1407 CrossRef CAS; (b) C. N. Kona, R. Oku, S. Nakamura, M. Miura, K. Hirano and Y. Nishii, Chem, 2024, 10, 402–413 CrossRef CAS; (c) M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS.
- (a) X. Chen, Y. Jing, J.-X. Kang, Y. Wang, Y. Guo and X. Chen, Inorg. Chem., 2021, 60, 18466–18472 CrossRef CAS PubMed; (b) M.-Y. Ju, Z.-H. Fan, Y. Ma, Y. Jing, X.-M. Chen and X. Chen, Inorg. Chem., 2023, 62, 8700–8709 CrossRef CAS.
- (a) C. W. Yoon, P. J. Carroll and L. G. Sneddon, J. Am. Chem. Soc., 2009, 131, 855–864 CrossRef CAS; (b) C. W. Yoon and L. G. Sneddon, J. Am. Chem. Soc., 2006, 128, 13992–13993 CrossRef CAS PubMed; (c) A. B. Baylis, G. A. Pressley, E. J. Sinke and F. E. Stafford, J. Am. Chem. Soc., 1964, 86, 5358–5359 CrossRef CAS; (d) B. J. Duke, J. W. Gauld and H. F. Schaefer III, J. Am. Chem. Soc., 1995, 117, 7753–7755 CrossRef CAS.
- (a) Y. Ling, Z. Y. Hao, D. Liang, C. L. Zhang, Y. F. Liu and Y. Wang, Drug Des., Dev. Ther., 2021, 15, 4289–4338 CrossRef CAS PubMed; (b) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (c) C. Campagnuolo, C. Fattorusso, E. Fattorusso, A. Ianaro and B. Pisano, Org. Lett., 2003, 5, 673–676 CrossRef CAS; (d) K. Murakami, S. Yamada, T. Kaneda and K. Itami, Chem. Rev., 2017, 117, 9302–9332 CrossRef CAS.
- (a) Q. Chen, T. León and P. Knochel, Angew. Chem., Int. Ed., 2014, 53, 8746–8750 CrossRef CAS PubMed; (b) Y. Cai, Y. Li, M. Zhang, J. Fu and Z. Miao, RSC Adv., 2016, 6, 69352–69356 RSC; (c) M. Balkenhohl, B. Salgues, T. Hirai, K. Karaghiosoff and P. Knochel, Org. Lett., 2018, 20, 3114–3118 CrossRef CAS PubMed; (d) Q. Zhong, S. Qin, Y. Yin, J. Hu and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 14891–14895 CrossRef CAS.
- (a) H. Dong and H. Berke, J. Organomet. Chem., 2011, 696, 1803 CrossRef CAS; (b) S. Lau, D. Gasperini and R. L. Webster, Angew. Chem., Int. Ed., 2021, 60, 14272–14294 CrossRef CAS PubMed.
- (a) K. Konishi, H. Matsumoto, K. Saito and K. Takahashi, Bull. Chem. Soc. Jpn., 1985, 58, 2294–2297 CrossRef CAS; (b) M. Meanwell, M. B. Nodwell, R. E. Martin and R. Britton, Angew. Chem., Int. Ed., 2016, 55, 1–6 CrossRef; (c) A. I. Lansakara, S. V. S. Mariappan and F. C. Pigge, J. Org. Chem., 2016, 81, 10266–10278 CrossRef CAS; (d) N. Wasfy, F. Rasheed, R. Robidas, I. Hunter, J. Shi, B. Doan, C. Y. Legault, D. Fishlock and A. Orellana, Chem. Sci., 2021, 12, 1503–1512 RSC.
- (a) B. Qian, S. Guo, J. Shao, Q. Zhu, L. Yang, C. Xia and H. Huang, J. Am. Chem. Soc., 2010, 132, 3650–3651 CrossRef CAS PubMed; (b) B. M. Trost and D. A. Thaisrivongs, J. Am. Chem. Soc., 2008, 130, 14092–14093 CrossRef CAS; (c) S. C. Sha, J. D. Zhang, P. J. Carroll and P. J. Walsh, J. Am. Chem. Soc., 2013, 135, 17602–17609 CrossRef CAS; (d) F. Rasheed, J. Q. Shi, T. Zeng, Y. K. Krishna, D. Fishlock and A. Orellana, Org. Lett., 2023, 25, 8628–8633 CrossRef CAS; (e) M. Meanwell, M. B. Nodwell, R. E. Martin and R. A Britton, Angew. Chem., Int. Ed., 2016, 55, 13244–13248 CrossRef CAS PubMed.
- (a) D. Chen, L. Xu, Z. Wang and C. Liu, Chem, 2023, 9, 3212–3223 CrossRef CAS; (b) J. Li, Q. Sun, C. Lu, H. Xiao, Z. Guo, D. Duan, Z. Zhang, T. Liu and Z. Liu, Nat. Commun., 2022, 13, 2143 CrossRef CAS PubMed.
- (a) L. Adams, S. N. Hosmane, J. E. Eklund, J. Wang and N. S. Hosmane, J. Am. Chem. Soc., 2002, 124, 7292–7293 CrossRef CAS; (b) X.-M. Chen, N. Ma, Q.-F. Zhang, J. Wang, X. Feng, C. Wei, L.-S. Wang, J. Zhang and X. Chen, J. Am. Chem. Soc., 2018, 140, 6178–6726 Search PubMed; (c) S. Liang, Y. Ma and X. Chen, Chin. J. Org. Chem., 2023, 43, 1772–1776 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2308054 (1a) and 2308050 (4aa). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03109a |
| This journal is © The Royal Society of Chemistry 2024 |