
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering strategies in the rational design of Cu-based catalysts for electrochemical CO2 reduction: from doping of elements to defect creation
Sheraz
Yousaf
a,
Iqbal
Ahmad
 *b,
Muhammad
Farooq Warsi
a and
Asad
Ali
*c
*b,
Muhammad
Farooq Warsi
a and
Asad
Ali
*c
aInstitute of Chemistry, The Islamia University of Bahawalpur, Baghdad-ul-Jadeed Campus, Bahawalpur-63100, Pakistan
bDepartment of Chemistry, Allama Iqbal Open University, 44000 Islamabad, Pakistan. E-mail: iqbal.ahmad@aiou.edu.pk; iqbalahmadchem@gmail.com
cEnergy Engineering, Division of Energy Science, Luleå University of Technology, 97187 Luleå, Sweden. E-mail: asad.ali@associated.ltu.se
First published on 26th July 2024
Abstract
Copper (Cu)-based catalyst design for electrochemical CO2 reduction (e-CO2R) has attracted much interest. This is because it could help climate change by converting CO2 into useful chemical products. This study considers a range of techniques used to optimize Cu-based catalysts, from element doping to defect engineering. Each technique has its advantages as well as its own unique problems. Doping Cu with noble metals such as silver (Ag) can result in very high catalytic activity and selectivity, but it also has disadvantages in terms of cost and long-term stability. In contrast, defect engineering which uses Cu as a material is both economically viable and sustainable. Maintaining stability and reliability is a demanding task that requires precise control. In addition, the single-atom approach has been a breakthrough method. It can efficiently and cheaply accommodate multiple carbon materials from CO2, and other than this it is quite stable and steady. It is possible for us to gain control over the active sites at an atomic level, even if we have inefficiency and selectivity problems that remain to be resolved. Due to this method, the chemical toolbox for Cu-based catalyst design has been expanded with many other tricks in addition. As they are flexible and can be tailored to specific applications or requirements, new types of Cu-based catalysts will be able to help in e-CO2R. When technologies mature, their sustainable deployment and global impact will depend on rigorous environmental impact assessments. It is important to emphasize the importance of Cu-based catalysts in the fight against climate change. In this crucial undertaking the paper also highlights the need for further research, innovations, and collaboration between nations.
 Sheraz Yousaf | Dr Sheraz Yousaf completed his doctorate under the supervision of Prof. Dr M. Farooq Warsi at The Islamia University of Bahawalpur, Pakistan. Dr Yousaf is specialized in the field of electrocatalysis, with a focus on the development of various electrocatalysts for energy conversion and storage applications. He has made significant contributions to advancing the understanding and performance of electrocatalysts through experimental approaches. Dr Yousaf's work is widely recognized, and he has published numerous articles in prestigious journals. For more information about his research and publications, please visit his Google Scholar profile (Sheraz Yousaf – Google Scholar), ORCID profile, and Scopus profile. |
 Iqbal Ahmad | Dr Iqbal Ahmad is an Assistant Professor at the Department of Chemistry, Allama Iqbal Open University. He completed his PhD in Chemistry from Quaid-i-Azam University, Islamabad, Pakistan, in 2017. He then secured a postdoctoral fellowship at the School of Chemical Engineering and Technology, Xian Jiaotong University, China, in 2019, and received the prestigious International Postdoctoral Exchange Fellowship from the Chinese government. Dr Ahmad has published over 70 research articles, and his research expertise includes electrocatalysis, particularly oxygen evolution reaction (OER), hydrogen evolution reaction (HER), and CO2 reduction reaction (CO2RR), as well as supercapacitors. |
 Muhammad Farooq Warsi | Dr Muhammad Farooq Warsi, after completing MPhil degree in Physical Chemistry from Quaid-i-Azam University (Islamabad-Pakistan), joined the physical-organic research laboratory of Professor Dr Victor Chechik as a PhD fellow, at the Department of Chemistry, The University of York, United Kingdom. He completed the PhD degree in 2010 in the research area of gold nanoparticle based MRI contrast agents. Immediately after PhD award, in January 2011, he joined as Assistant Professor of Physical Chemistry, at the Islamia University of Bahawalpur-Pakistan. Dr Warsi established the electrochemistry and nanocatalysts research laboratory, which is one of the state-of-the-art research laboratories, at Southern Punjab universities in Pakistan. He has published more than 250 research articles since he joined the Islamia University of Bahawalpur (Pakistan). His research interest includes electrocatalysts, supercapacitors, photocatalysis and electrochemical sensors. Currently, Dr Warsi is working as Professor of Physical Chemistry, in the Institute of Chemistry at The Islamia University of Bahawalpur-Pakistan. |
 Asad Ali | Dr Asad Ali received his PhD degree in chemical engineering and technology from Guangxi University, China, in 2022. He is a postdoctoral researcher in the Division of Energy Engineering at Luleå University of Technology, Sweden. He has authored over 50 high-quality reviews and research articles published in journals such as Electrochemical Energy Reviews, Advanced Energy Materials, Chemical Engineering Journal, Journal of Materials Chemistry A, Nano Research, etc. His research focuses on preparing nanomaterials and their applications for hydrogen production and biomass (lignin) conversion to valuable products. |
1. Introduction
As global carbon dioxide (CO2) levels rise and climate change becomes more entrenched the world faces an ongoing dilemma of how to cope with environmental problems. Very recently, there has been considerable consultation on sustainable technologies.1 Copper plays a crucial role in enabling sustainable technologies and copper-containing catalysts are totally unique as they convert CO2 into feedstocks for chemical production. Here's a new way of thinking: where CO2 is considered as a resource, not a pollutant. This novel approach to carbon economy has the potential of being both sustainable and circular.2 This review focuses on Cu-containing catalysts and their use in e-CO2R. It explores the engineering skills required for the rational design and synthesis of these catalysts. Engineering strategies include techniques such as selectively adding elements through doping, inducing defects within crystal lattices and using the single-atom approach to control overactive sites for innovation in precision. Conversion of CO2 to beneficial compounds through reactions is the focus of e-CO2R. This entire area of research has the urgency to find some solution as the emissions are becoming an issue for global warming today. This is done through capturing CO2 from sources such as power plants, industrial processes and the atmosphere. The captured CO2 is converted into products that have various uses. Different methods could be used to collect CO2 which are post-combustion capture, pre-combustion capture, and direct air capture.3The capture and storage of carbon is an essential step in limiting CO2 emissions. CO2 can be employed in the production of goods in transformation processes. The output depends on which products one wants to create as well as what resources are available. Common types of CO2 conversion include electrochemical, photocatalytic and biological reduction, using different reaction agents, such as metal coatings at the nanoscale.4 Catalysts such as copper and silver metal catalysts for electrochemical reduction are very important.5,6 And semiconductor materials are important in these strategies. It is essential to supply energy to drive these conversion reactions, which can be sourced from diverse renewable sources like geothermal, wind, or residual energy from industrial processes. Energy sources that prioritize both efficiency and sustainability not only promote CO2 emission reduction but also contribute to lowering the overall carbon footprint.
Carbon dioxide reduction can produce various valuable products such as fuels, chemicals, and materials. Typical outputs comprise of methane, methanol, formic acid, carbon monoxide, and diverse hydrocarbons. Product selection is influenced by market demand, feasibility of application, and environmental factors. It has the potential to mitigate climate change by reducing greenhouse gas emissions.7 The environmental impact of CO2 conversion processes must be assessed, taking into account resource availability, energy efficiency, and waste management. This review centers on the investigation of engineering approaches in the logical design of Cu-based catalysts for e-CO2R. This review seeks to clarify the complex world of catalyst optimization with a wide range of techniques covered, from element doping to defect engineering. By exploring the underlying theories and mechanisms of e-CO2R, it establishes the foundation for a sophisticated comprehension of the various approaches used in catalyst design. The aim of this endeavor is to create a comprehensive resource for researchers, engineers, and policymakers to guide future advancements in the field by consolidating and synthesizing the wealth of knowledge accumulated across various research domains.
2. Fundamentals and mechanisms involved in electrochemical CO2 reduction
To assess the performance of a system and to determine the effectiveness of an electrocatalyst, several fundamental parameters could be considered for evaluation of the e-CO2R process. Their assessments are done using some common key parameters employed in evaluating the reaction. Faradaic efficiency gives the ratio of electrons employed in generating the desired CO2R products with respect to the total number of electrons that flow through the system and therefore can be used as an indication of selectivity for specific products.8 The rate at which electrons are transferred by electron transfer processes occurring over a particular unit surface area is reported as current density and is given in mA cm−2. Higher current densities would indicate faster kinetics for CO2R and higher catalytic activity.9,10Further, the energy efficiency assesses the energy consumption of the electrochemical CO2 reduction process. It quantifies the amount of electrical energy required to produce a certain amount of desired products. Higher energy efficiency indicates a more sustainable and economically viable process.11,12 The TOF represents the number of CO2 molecules converted into the desired product per active site on the catalyst per unit time.13 It provides information about the intrinsic catalytic activity of the electrocatalyst, independent of the electrode size or current density.14 Further, its dependence on geometrical activity, specific activity and mass activity is schematically shown in Fig. 1.
 | ||
| Fig. 1 Dependence of TOF on geometrical activity, specific activity and mass activity. | ||
Product selectivity refers to the distribution of different carbonaceous products obtained during CO2 reduction. It evaluates the electrocatalyst's ability to favor specific target products over others. Selectivity can be determined through product analysis using techniques such as gas chromatography, mass spectrometry, or NMR spectroscopy. Fig. 2 shows the schematic representation of production of various organic products during an electrocatalysis based experiment.15,16
 | ||
| Fig. 2 Schematic illustration of production of different gaseous and liquid products over the surface of electrocatalyst's active sites. | ||
The electrocatalyst's stability and durability are important for long-term operation. Evaluating the catalyst's performance over time and under different operating conditions helps to determine its durability and potential for practical use.17 The catalyst loading is the amount of material used on an electrode. Optimizing the catalyst loading improves the electrode's efficiency and the usage of the catalyst. Learning about the kinetics of CO2R can give us important information about the mechanisms and rate-limiting steps.18 We can use cyclic voltammetry and electrochemical impedance spectroscopy to analyse reaction kinetics. A comparative study can help to give an idea of how well an electrocatalyst is performing according to different catalyst materials and electrode configurations as well as working modes. Evaluation procedures vary according to study objectives and desired CO2R products and also the choice of evaluation parameters should suit the study objectives and the products of CO2R that are wanted. The performance, selectivity and potential of e-CO2R technology can be evaluated by analysing these parameters.
A multitude of intermediate products and steps lie in the reduction of CO2 on the electrocatalyst's surface. Catalyst material and experimental conditions affect the catalytic mechanism. Generally, certain key steps are taken to be necessary. First, CO2 molecules come to rest on the electrocatalyst's surface. Adsorption happens due to weak interactions, such as van der Waals forces, or chemisorption, in which CO2 forms chemical bonds on the surface of the catalyst. The adsorbed carbon dioxide is activated upon breaking of its C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, allowing subsequent reduction. To carry out this process, energy must be provided, and both the potential applied and the electronic properties of the catalyst can affect it.19
O bond, allowing subsequent reduction. To carry out this process, energy must be provided, and both the potential applied and the electronic properties of the catalyst can affect it.19
Activated CO2 can undergo intermediate formation steps, resulting in the production of various reaction intermediates. Frequently encountered intermediates comprise carbon monoxide (*CO), formate (*HCOO),20 formyl (*CHO),21 and carboxyl (*COOH) species.5 Electrons from the cathode electrode are transferred to the adsorbed reaction intermediates. The electron transfer is facilitated by the potential difference applied to the electrochemical cell. Protons from aqueous electrolytes can participate in the reaction. Protons react with adsorbed intermediates to produce reduced carbon compounds. The electrochemical reduction of CO2 generates reduced carbon compounds that subsequently desorb from the catalyst surface. This step facilitates the collection of products for subsequent utilization or analysis.
Electrochemical CO2 conversion involves a range of mechanistic pathways leading to the formation of various products. Here are different mechanisms for the electrochemical CO2 conversion to different products. CO2 reduction to CO typically involves a 2-electron transfer process. Mechanistically, CO2 is first adsorbed onto the catalyst surface, followed by the electrochemical reduction of CO2 to the carboxyl intermediate. The carboxyl intermediate further undergoes protonation and electron transfer steps to form CO and water.
This mechanism is preferred at low overpotentials and in the absence of other competing reduction pathways. It occurs most often on Cu-based catalysts.
| CO2 → CO2* |
| CO2 + * → COOH + O |
| CO2 + * → COOH + O |
| CO* → CO |
| CO2 + 2H+ + 2e− → CO + H2O E° = −0.52 V vs. SHE |
| CO2 → CO2* |
| CO2 + * → CO + O |
| *CO + H+ + e− → *HCOO |
| HCOO + H+ + e− → HCOOH + O |
| CO2 + 2H+ + 2e− → HCOOH E° = −0.61 V vs. SHE |
| CO2 → CO2* |
| CO2 → CO2* |
| *CO + H+ + e− → *HCOO |
| *HCOO + H+ + e− → CHO + O |
| *CHO + 3H+ + 3e− → CH3OH + H2O |
| CO2 + 6H+ + 6e− → CH3OH + H2O E° = −0.38 V vs. SHE |
| CO2 → CO2* |
| CO2 + * → CO + O |
| *CO + 2H+ + 2e− → *CH2O + H2O |
| *CH2O + *CH2O → *C2H4O + H2O |
| *C2H4O + 2H+ + 2e− → C2H4 + H2O |
| CO2 + 8H+ + 8e− → C2H4 + 2H2O E° = −0.04 V vs. SHE |
| CO2 → CO2* |
| CO2 + * → CO + O |
| *CO + 4H+ + 4e− → *CH3OH + H2O |
| *CH3OH + H+ + e− → *CH3 + H2O |
| *CH3 + 3H+ + 3e− → CH4 + H2O |
| CO2 + 8H+ + 8e− → CH4 + 2H2O E° = −0.24 V vs. SHE |
 | ||
| Fig. 3 Possible different routes for the production of C1 and C2 products over the surface of polycrystalline copper upon e-CO2R. Reproduced with permission from ref. 11. Copyright 2023, American Chemical Society. | ||
Modern electrocatalysis research aims to enhance comprehension of these mechanisms and create efficient catalysts that can boost CO2 reduction selectivity and activity. This will facilitate the development of sustainable and economically possible carbon capture and utilization technologies.
3. Importance of Cu-based catalysts in e-CO2R
Cu-based catalysts have gained significant importance in electrochemical CO2 reduction for several reasons like their high selectivity for C–C bond formation. Copper-based electrocatalysts show a great deal of selectivity in the formation of C–C bonds during the reduction of CO2.23 Due to this, it is possible to synthesize valuable hydrocarbon compounds such as ethylene (C2H4) and methane (CH4), highly varied in industrial applications as fuels or for use in chemical feedstocks. Natural limit defects of these Cu nanosheets (2–14 nm) were designed to produce ethylene with a high ethylene faradaic efficiency of 83.2% from carbon dioxide. These defects enhance the formation of ethylene by promoting C–C coupling through reaction intermediates and hydroxyl ions.24 In the same way, CuO nanoparticles were made and deposited on conductive carbon materials, resulting in a higher faradaic efficiency (FE) of over 70% for ethylene but only 30% for hydrogen.The catalyst exhibited durability and selectivity for ethylene production, due to its active domains and high local pH.25 The Cu–polyamine hybrid catalyst significantly improved ethylene production selectivity by 87% at −0.47 V. Additionally, the full-cell energetic efficiency reached 50%. Raman measurements indicated that incorporating polyamine in a Cu electrode enhances surface pH, CO content, and intermediate stabilization. This demonstrates the positive effect of polymer incorporation on product selectivity.26 The tw-Cu foil electrocatalyst has dense twin boundaries and shows a faradaic efficiency of 86.1 ± 5.3% for CH4. It also reduces the barrier for reduction and has high selectivity for CH4.27 Moreover, copper-based catalysts have shown favorable kinetics for CO2 reduction reactions.28 They possess lower overpotential, resulting in a reduced energy requirement for driving the desired electrochemical reactions. This characteristic is responsible for an increase in the energy efficiency of the process.
Copper-based catalysts can have different oxidation states and undergo structural changes during electrochemical processes.29,30 The versatility of Cu catalysts makes them able to form and react with a greater variety of intermediates, leading to different products from the same source. Copper, widely available and plentiful, is an inexpensive choice for massive carbon dioxide reduction. Copper catalysts have low cost and can be used widely. Cu-based electrocatalysts are preferred because of their adjustable catalytic properties. It is possible to change the catalytic properties of Cu-based catalysts by controlling particle size, morphology, and surface composition. Researchers can optimize the catalyst performance and customize selectivity for specific CO2 reduction products. Copper catalysts are stable and durable for electrochemical CO2 reduction. They resist deactivation and degradation, allowing them to operate efficiently for a long time. Cu-based catalysts are important in electrochemical CO2 reduction because they can efficiently produce valuable carbon-based compounds with high selectivity. Their abundance, cost-effectiveness, tunable properties, and stability make them ideal for scaling up CO2 reduction technologies and using carbon dioxide sustainably in various industrial processes.31,32
Although Cu-based electrocatalysts show promise for CO2 conversion, there are still some challenges and current issues that need to be addressed. The key issues associated with Cu-based electrocatalysts for CO2 conversion include their low faradaic efficiency. Cu catalysts often exhibit low faradaic efficiency for specific CO2 reduction products like C2 products.33–35 Undesired side reactions, such as hydrogen evolution and competitive reduction pathways, can limit the selectivity towards desired products like ethylene or methane.36 Enhancing the faradaic efficiency and selectivity for these materials remains a major challenge. Cu based electrocatalysts can suffer from stability issues during prolonged CO2 conversion reactions.37,38 A hydrophobic coating of long-chain alkanethiols on Cu dendrites enhances CO2 reduction selectivity by creating a plastron effect. This creates a gas layer on the electrode, which increases the concentration of CO2 in that area. The current electrode has drawbacks. Future work should focus on promoting stable hydrophobicity on high-surface-area microporous electrodes. This supports the idea that gas diffusion electrodes can produce stable C2 products, which may explain the low long-term stability of electrocatalytic surfaces.39 Surface restructuring, agglomeration, or dissolution of Cu nanoparticles may occur, leading to decreased catalytic activity and selectivity over time.40 Improving the stability of Cu-based catalysts under harsh reaction conditions is critical for their practical applications. Additionally, the reduction of CO2 on Cu surfaces often requires a high overpotential, meaning a higher energy input is needed to drive the electrochemical reactions efficiently. Reducing the overpotential would enhance the energy efficiency of the process and make it more economically viable.41 Diffusion of CO2 to the catalyst surface can be a limiting factor, especially in dense electrode structures or in the case of thick catalyst layers. Insufficient CO2 transport to the catalyst surface can lower the reaction rate and efficiency. Efficient CO2 mass transport is important for catalyst designs and electrode architectures.42
Cu-based electrocatalysts are susceptible to poisoning or deactivation by adsorbed species or impurities in the reaction environment.43,44 Oxygen- or sulfur-containing species that are adsorbed can block active sites or alter the electronic properties of the catalyst, leading to reduced activity and selectivity.45 Mitigating catalyst poisoning effects is crucial for ensuring long-term performance. Despite the abundance and cost-effectiveness of copper (Cu), scaling up copper-based catalysts for large-scale CO2 conversion processes is still challenging.
Innovative synthesis approaches are important for addressing challenges in CO2 conversion with Cu-based electrocatalysts. Various synthesis techniques can precisely control catalyst composition, morphology, and structure. These approaches improve the selectivity and activity of Cu-based catalysts by giving them specific properties. Metals can be added through alloying or surface modification. Bimetallic or composite catalysts can exhibit improved catalytic performance and selectivity by leveraging the synergistic effects between Cu and other metals. Bottom-up assembly methods can be used to develop nanostructured Cu catalysts, providing control over particle size, shape, and surface properties. This leads to improvements, in both the efficiency and stability of reactions. Researchers have the opportunity to employ synthesis techniques in order to design electrocatalysts based on copper that exhibit improved performance, selectivity, stability and scalability for efficient conversion of CO2.
4. Mechanistic insights from spectroscopic techniques
Recent discoveries and developments from spectroscopic techniques such as XAS, Raman and IR spectroscopy have provided mechanistic knowledge of CO2 reduction on Cu-based electrode catalysts. XAS, especially XANES and EXAFS, demonstrates the change of the Cu oxidation state and the ability of local geometry around Cu, which represents the transition from Cu(II), Cu(I) to active Cu(0) during the catalytic process. Raman spectroscopy identifies surface species such as CO and CHO which play important roles in the reaction route map while IR spectroscopy is useful in gaining information on the adsorbed species and reaction intermediates. Each of these spectroscopic strategies combined provides a detailed description of the dynamic processes occurring at the catalyst surface and with CO2, which is profitable for the development of enhanced and selective electrocatalysts. EC-RSoXS was employed to study the real-time and real-space particle interactions as well as the chemical surroundings of Cu nanocatalysts in e-CO2R. The changes of Cu nanoparticle properties depending on the particle size and interaction with soft X-ray beams were analyzed. The 7-nanometer nanoparticles were primarily oxidized to CuO, whereas the larger 18-nanometer particles showed more resistance to damage. EC-RSoXS combines soft XAS (X-ray absorption spectroscopy) and SAXS (small-angle X-ray scattering) methods to give a complete picture of how chemical and physical states affect the interparticle dynamics of nanocatalysts.46 Furthermore, another research study has showcased that the utilization of Ce–Cu2O, a catalyst characterized by a unique orbital coupling between high-order Ce4+ 4f and O 2p, brings about significant improvements in e-CO2RR (Fig. 4). Throughout a 7-hour operational run, the as-synthesized CeCu2O sample consistently generated C2H4 and demonstrated a C2H4/CO ratio 1.69 times higher than that of the Cu2O catalyst. The Ce–Cu2O catalyst maintained stable Cu+ species during the entire CO2RR process.47 | ||
| Fig. 4 Molecular orbital representation of Ce4+ 4f O 2p Cu+ 3d coupling (A). Ex situ sXAS measurements of the O-K edge in CeCu2O and Cu2O catalysts (B). Ex situ sXAS at the Ce-M4,5 edge in Ce–Cu2O (C). Ce–Cu2O and Cu2O O2-TPD spectra (D). Formulation Energy requirenments for OVs in Ce–Cu2O and Cu2O, is presented in (E). (F) A schematic showing how the connection between the high-order orbitals 4f and 2p in Ce–Cu2O prevents CuO deactivation and shields the Cu+ site from electron attack during the CO2RR. Reproduced with permission from ref. 47. Copyright 2023, American Chemical Society. | ||
In another facet of this research, pure Cu, a metal known for catalyzing the conversion of CO2 to hydrocarbons with high FE, was utilized for e-CO2RR. Researchers closely monitored the structural dynamics of Cu during the electrochemical process using techniques such as electrochemical scanning tunneling microscopy and Raman spectroscopy. Through control of polarization potential and in situ synthesis time, it was discovered that polycrystalline Cu surfaces could give rise to Cu nano-cuboids. Additionally, a graphene monolayer was grown on the Cu surface to create smaller features with enhanced catalytic activity.48
Furthermore, the study employed in situ HERFD XAS to investigate the potential dependency of a single Cu layer, revealing significant differences compared to bulk Cu electrodes. Unlike thick Cu electrodes, which exhibit two redox transitions, only one redox transition was observed for 1 ML Cu/Au(111) and 1 ML Cu/Pt. This single Cu layer exhibited an anodic shift at the onset of Cu oxidation. The considerable tensile strain experienced by a single Cu monolayer when deposited on Au, Pt, and platinum-group metals was found to influence the redox behavior and interaction with e-CO2R intermediates. This tensile strain allowed for tunability of the ratio of methane to ethylene formed.49 Moreover, the research demonstrated that the incorporation of s-ZnO into s-CuO during the fabrication of a series of sputtering electrocatalysts significantly altered the product profile of the CO2RR. Specifically, the efficiency of CH4 generation increased, while the production of C2+ products decreased under ideal conditions. In situ XAS analysis revealed that the coordination number of Cu sites played a crucial role in CH4 formation, which was influenced by the presence of Zn sites and led to a reversal of Ostwald ripening during the CO2RR.50
Furthermore, in another research study, the scientists employed the carbon nanoparticle moderator method, wherein a copper-complex catalyst was confined in a membrane electrode assembly. Real-time XAS experiments demonstrated that increased carbon nanoparticle loadings reduced the coordination number of metallic copper. The system operated continuously for over 110 hours, achieving a CO2-to-methane selectivity of 62%, a methane partial current density of 136 mA cm−2, and a copper coordination number of 4.2.51 Lei et al. have designed nanocrystals of Cu2(OH)2CO3, Cu(OH)2, and CuO for e-CO2R. In situ XRD and Raman spectroscopy confirmed the complete reduction of precursors to Cu(0), and Cu(0) was identified as the active species responsible for the improved selectivity. Initially, the strong C2+ selectivity was attributed to the small grain size of the resulting Cu. Interestingly, Cu nanocrystals synthesized from Cu(OH)2 and Cu2(OH)2CO3 were found to possess substantial tensile stresses, as indicated by in situ XRD. These stresses play a crucial role in promoting hydrogenation of *CO and C–C coupling, ultimately enhancing the overall e-CO2RR selectivity while reducing HER rates.52
5. Theoretical calculations and computational studies
Theoretical calculations and computational studies, particularly those involving density functional theory (DFT), play a crucial role in the design and optimization of catalysts for CO2 reduction. These approaches allow researchers to model and predict the electronic structure, potential energy surfaces, and reaction pathways of catalyst materials at an atomic level. A DFT study found that Cu–M bimetallic catalysts have notable effects on the reaction barrier of CO2RR and HER and display catalytic activity. The Cu–Pd catalyst is better for CO2RR than for HER. This information can help in designing effective electrocatalysts and syngas production applications.53 By simulating interactions between CO2 and catalyst surfaces, computational studies can identify active sites, determine the most favorable reaction intermediates, and predict product selectivity. This theoretical insight complements experimental findings, guiding the rational design of catalysts with enhanced activity and selectivity. For instance, DFT calculations have helped in optimizing the composition and structure of Cu-based catalysts to improve their performance, ensuring a more efficient conversion of CO2 to valuable chemicals.The self-supporting and through-hole structure of CuSAs/TCNFs facilitated an increase in the number of useful Cu atoms while simultaneously reducing the number of incorporated metal atoms in the material. These individual Cu atoms played a pivotal role in the e-CO2RR. Notably, the process yielded stable C1 products with a current density of −93 mA cm−2, maintaining stability in water for over 50 hours. DFT simulations suggested relatively high binding energy for the *CO intermediate in isolated Cu atoms. These findings suggest the potential for CO conversion into other valuable products, such as methanol, contributing to the reduction of CO levels.54 Besides, ethanol synthesis emerged as one focal point: copper nitrogen-doped carbon materials with porphyrin-like Cu graphene structures played a critical role. The reversible transformation of porphyrin-like Cu sites to metallic copper nanoclusters complicates understanding their specific activities. A computational method was used to model the structure of the Cu–N–C material. Employing DFT calculations, the main focus of the work was on e-CO2R for ethanol and ethylene production. In both CO reduction and ethanol production, Cu nanoclusters are significantly better than single sites. This all-encompassing piece of research helps to further our knowledge of Cu-based catalysts for many different CO2 conversion processes.55
Further expanding the investigation, the research delved into the CO2RR process catalyzed by a material housing a single Cu site within an N-doped carbon surface. Utilizing periodic DFT computations, distinct surface models were developed for the first CuN4 site and a secondary Cu cluster at negative potentials. While the feasibility of the single-site surface was found to be thermodynamically challenging, the mechanistic study highlighted the significant activity of the Cu13N4 nanocluster surface in driving CO2R. Notably, the catalytic activity of the Cu, N-doped carbon with a single site heavily depended on the presence of Cu13N4 nanoclusters on its surface, owing to their unique capability to facilitate the crucial C–C coupling reactions required for C2 product formation.55
DFT simulations were also employed to investigate the binding energies of CO2R intermediates. Surface slab models for Cu(111), Sn(200), and Cu3Sn(002) were constructed based on XRD data. The binding energy of H* on Cu3Sn(002) was similar to that on Cu(111), while CO* binding energy on Cu3Sn(002) was lower. This difference, along with the energetically preferable CO desorption process over CO* reduction, contributed to improved CO selectivity compared to Cu(111). The presence of surface Sn dramatically impacted HCOOH formation selectivity, allowing for control over two-electron reduction product selectivity through Cu–Sn alloying.56 In the same study, DFT calculations were employed to examine the adsorption properties of CO2 on clusters of Cu55, Cu54Ni1, and Cu42Ni13, featuring highly symmetric cub-octahedral, decahedral, and icosahedral structures. The composition of these clusters was found to influence the energy required for CO2 adsorption, with the icosahedral Cu42Ni13 cluster demonstrating the highest CO2 adsorption capacity.57
Further, using DFT calculations, the impact of Zr doping on CO2 hydrogenation to CHO in icosahedral Cu NPs was investigated. The findings indicated that Zr doping of Cu NPs improved CO2 adsorption and hydrogen dissociation. Zr atoms played a crucial role in stabilizing chemical intermediates by lowering energy barriers.58 Additionally, the structure, activity, and selectivity of the B-doped copper (B@Cu(111)) catalyst were probed with DFT simulations. It was revealed that CO adsorption and activation occurred at the B site, with CO favoring coupling to *C2O2. B played a crucial role in the conversion of carbon dioxide to ethanol and in capturing CO. The electronic structure and Bader charge analysis demonstrated that B stabilized C2 intermediates by providing both empty and occupied orbitals for CO adsorption and activation.59 In another study, the DFT simulations were employed to study e-CO2R using Cu–X catalysts. Thermodynamic studies linked the O affinity of dopant atom sites and the Cu site's selectivity towards C2 hydrocarbons and alcohols. Doping Cu–X catalysts with strong O affinity atoms, such as boron, favored the ethylene route. Non-metal doping on Cu surfaces often improved ethane selectivity while suppressing ethanol selectivity. The effect of electrical characteristics on intermediate adsorption and inherent selectivity was also addressed.60
In a related study, Chang and coworkers employed DFT calculations to investigate the highly selective formation of C2 during CO2RR on different copper surfaces, with a particular focus on Cu(711). The Cu(711) surface presents a unique challenge due to the presence of a high dipole–dipole repulsive force, which excludes the formation of an OC–CO intermediate via the CO–CO route. Instead, adsorbed CO undergoes protonation, leading to the creation of CHO or COH intermediates. Lowering the activation energy for C–C bond formation with adsorbed CO is achievable through the production of COH or CHO. In contrast, Cu(100) and Cu(111) surfaces are thermodynamically unfavorable for these reactions. Given the increased likelihood of COH participation in the C–C coupling processes of CO2RR, its stabilization on the Cu(711) surface is more probable.61 Additionally, the benefits of Cu cluster-doped C3N4 as a CO2 reducer were investigated utilizing DFT calculations. Cu clusters were identified as ideal active sites for CO2 adsorption and activation due to their structure and electrical properties, facilitating the separation and transport of photo-generated electrons and holes. The study revealed that C–O carbon atoms in gas-phase molecules could link with C–N4 carbon atoms in the substrate, reducing the adsorption energy. Furthermore, it evaluated how the energy requirements for producing CO through the CO2R process compare to those of the rival HER reaction. The reaction barrier for C2 production on Cu4–C3N4 was found to be larger than the HER reaction barrier due to the dimerization of *CO.62
In addition, Xue et al. developed efficient and selective electrocatalysts for the e-CO2RR. Twenty-one MOF supported flexible, self-adaptive dual-metal-site pairs (DMSPs) were screened using DFT calculations. For e-CO2R to C2H6 and C2H5OH, the computed limiting potential is −0.87 V, while MOF-808-EDTA-FePt shows even better activity at −0.35 V for CO2R to C1 products.63 Furthermore, DFT was employed to investigate the adsorption, structural changes, energy profiles, and reaction processes caused by CO coverage on the active sites of the Cu2O(111) surface. Due to the presence of unsaturated Cu atoms, the coordinatively unsaturated Cu site (CuCUS) on the Cu2O(111) surface is shown to be highly reactive towards CO adsorption and subsequent reactions. C–C coupling is favored because the energy barrier of *CH2 dimerization is lowered at higher CO coverage, specifically between 0.13 and 0.25. The maximum barrier energy occurs in the *CHO route, making the C–C coupling the rate-determining step.64
Moreover, Das et al. modeled the CuAl2O4(111) surface for CO2 conversion to DME using DFT calculations, elucidating the importance of dopants (Ga and Zn) in modifying the catalyst's active center. They also revealed Ga's role in changing the acidic site of the CuAl2O4(111) surface, enabling methanol conversion to DME.65 Furthermore, twenty bimetallic M–Cu–BTCs were subjected to DFT calculations to examine their CO2 adsorption and activation properties. Linear adsorption emerged as a more effective means of interaction between CO2 molecules and most bimetallic MOFs, except for Pt–Cu–BTC and Pd–Cu–BTC. CO2 activation was observed on W–, Mo–, Re–, and Os–Cu–BTC complexes. While CO2 adsorption was stronger on Os–Cu and Re–Cu nodes, W–Cu–BTC and Mo–Cu–BTC showed promise for investigating the underlying mechanisms due to their higher H2 adsorption capacities. The study also explored both the carboxyl (COOH*) and formate (HCOO*) routes of CO2 hydrogenation with H2 molecules on W–Cu and Mo–Cu complexes. While the energy barrier for W–Cu–BTC was 1.27 eV, it was only 1.10 eV for Mo–Cu–BTC.66
6. Engineering strategies in the rational design of Cu-based catalysts for electrochemical CO2 reduction
There are various synthesis techniques utilized to manipulate the composition, structure, and morphology of copper-based catalysts to reduce CO2. The choice of synthesis methods plays a part in determining performance and selectivity. Some strategies for synthesizing copper-based catalysts are introduced here.6.1. Single crystal approach
The single crystal approach includes the use of single-crystal surfaces with well-defined atomic arrangements to design electrocatalysts for e-CO2RR. In this context, the key to developing and making electrocatalysts is creating catalysts with specific surfaces of the single crystal. Adjusting the crystallographic orientation and atomic arrangements can provide more information about the catalytic mechanism and develop highly effective and selective catalysts. A number of different ways can be used with this technology to create materials with the desired properties. Epitaxial growth, surface etching, atomically accurate deposition, selective growth, template-based growth, and tip-based scanning tunnelling microscopy (STM) manipulation are all examples of this kind of technique. That is one important advantage of being able to design active sites consisting of single atoms. In this field, researchers have become interested in making electrocatalysts that can convert CO2 to CH3OH using single-atom Cu. It can be seen from first-principles calculations that the Co@Cu SA catalyst has potential to catalyze CO2 reactions. The d-band of the catalyst becomes narrowed and its center shifts up, making it more stable for CO2. As a result, energy barriers of the reaction are significantly reduced. This phenomenon enables us to selectively and efficiently produce CH3OH, which has a profound effect on the entire procedure. The reaction pathway, CO2 → COOH* → CO* → COH* → CHOH* → CH2OH* → CH3OH, is guided by the modified catalyst. It is significant that the SA has a small lattice mismatch, so that it is stable against surface reconstruction. The catalyst's superiority is further highlighted by the difference in activation energy for the reduction of COOH* compared to Cu. Specifically, the activation energy required is 0.54 eV, which is 0.22 eV lower than the 0.76 eV required on Cu and reaction is exothermic. In another aspect, the narrow cobalt d-band plays a vital role in the performance of this catalyst by preventing the buildup of unwanted byproducts. This feature results in a higher yield of CH3OH, further enhancing the overall sustainability and efficiency of the synthesis process. Collectively, the utilization of single-atom catalysts and the precise manipulation of crystallographic orientations hold promise for advancing the field of electrocatalysis and enabling more sustainable energy production through the selective and efficient conversion of CO2 to valuable chemicals like methanol.67The pursuit of high-rate e-CO2RR activity, selectivity, and stability led Sassenburg et al. to investigate five distinct transition metal catalysts (Ag, Au, Pd, Sn, and Cu). Notably, Au and Pd exhibited CO-limiting current densities of 72 and 50 mA cm−2, respectively, emerging as standout contenders among the 88 diverse catalysts examined. The study showcased the convergence of product selectivity for Cu catalysts across both neutral and alkaline environments. Additionally, the investigation highlighted Sn's susceptibility to instability under highly alkaline conditions (Fig. 5). The research encompassed an assessment of catalytic selectivity at 10 and 200 mA cm−2, as well as a fundamental comparison of reaction rates.68
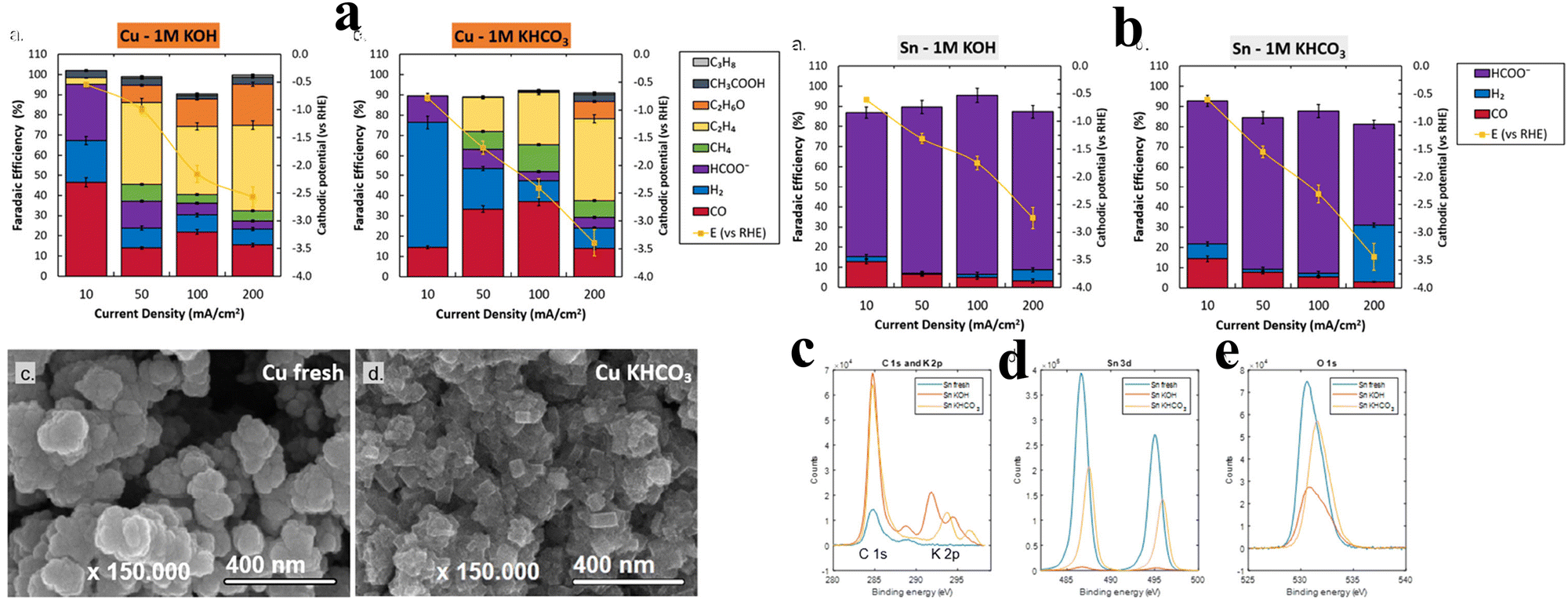 | ||
| Fig. 5 (a) Analysis of electrodes with copper coating: the FE, in relation to activity, is explored across varying cathodic potentials in 1 M KOH and 1 M KHCO3 solutions. Noteworthily, the error bars in both the panels illustrate the findings from distinct experimental runs. Employing HR-SEM, the visual depiction of fresh copper and its state after 1 hour of KHCO3 electrolysis underscores the emergence of the distinct cubic faceting pattern on catalyst's surface. (b) Exploring Sn-coated electrodes: the characterization involves the assessment of faradaic efficiency concerning activity across cathodic potentials in 1 M KOH and 1 M KHCO3 solutions. The error bars in both the panels depict the data variations from distinct experimental iterations. Further insights are gained through X-ray photoelectron spectroscopy (XPS) analyses, specifically examining C 1s and K 2p (c), Sn 3d (d), and O 1s (e) scans before and after subjecting the system to a 200 mA cm−2 experiment in both electrolytes. Reproduced from ref. 68. Open access article, American Chemical Society. | ||
Furthermore, the evolution of a nanotwinned copper electrocatalyst specifically for e-CO2RR aimed at methane production. At a potential of −1.2 ± 0.02 V vs. RHE, the catalyst exhibited impressive selectivity in generating methane, achieving a FE of 86.1 ± 5.3%. The pivotal role of twin boundaries on Cu(111) facets in reducing the barrier for CO hydrogenation underscores their significance in the creation of CH4. Notably, the presence of these twin boundaries on Cu(111) renders the surface more predisposed to CH4 selectivity than a planar copper surface (Fig. 6). This multifaceted investigation and the nuanced understanding of catalyst behavior contribute to the advancement of electrocatalytic strategies for sustainable energy conversion.27
 | ||
| Fig. 6 Structural characterization of twin-bound and polycrystalline copper catalysts: (A) the intricate structure of tw-Cu captured through HRTEM. The cross-sectional image reveals the assembly of twin boundaries (TB). Inset: FFT of the corresponding Cu TEM, which indicates the 〈110〉 axial direction and expression of the {111} planes. (B) Offering a broader perspective, a low-magnification TEM image showcases tw-Cu, where dashed white lines delineate the characteristic twin boundaries. (C) This structural insight is further corroborated by a SEM image of tw-Cu, with white dashed lines again indicating the presence of twin boundaries. (D) A detailed AFM image of tw-Cu discloses a surface roughness of 2.7 nm. (E) The crystallographic texture of the tw-Cu surface is illuminated through EBSD orientation maps. The inset highlights color-coded crystallographic vectors, vividly suggesting a robust (111) texture. (F) XRD patterns of both tw-Cu and pc-Cu reveal the strikingly high orientation toward (111) planes in tw-Cu, in sharp contrast to pc-Cu. This distinction is emphasized through a comparison with the reference sample represented by the black line with PDF number 96-431-3212. e-CO2RR: FEs of (G) tw-Cu and (H) pc-Cu. The data points corresponding to H2, CO, CH4, and C2H4 are color-coded as green, red, orange, and blue, respectively. Visualization of the partial current densities of (I) H2, (J) CO, (K) CH4, and (L) C2H4, where red lines signify tw-Cu and black lines represent pc-Cu. Each error bar emanates from three distinct measurements, while all potentials underwent iR-correction. Reproduced from ref. 27. Open access article, American Chemical Society. | ||
In contrast, the work by Lyu et al. explored the impact of trace copper loading within N-doped graphene quantum dots (NGQDs) on both e-CORR and e-CO2RR. Interestingly, a rise in Cu loading within NGQDs exhibited the capacity to alter the selectivity of catalytic methane oxidation, shifting from primarily C1 products to C2 products. A notable outcome was achieved by introducing copper with an increased atomic size of 2.5 mg cm−2, resulting in an increased selectivity of up to 52% for C2 products, accompanied by a reduced selectivity of 62% for CH4. Elevating the Cu loading further to 3.8 mg cm−2 led to the peak of C2 product production efficiency at 78%. This observation is particularly significant considering the low partial pressure of CO during e-CO2RR. The heightened copper loading becomes imperative to enhance selectivity toward C2 products, thus illustrating the intricate interplay between catalyst composition and product selectivity in the context of sustainable energy conversion strategies.69 With a shift of focus, Qai et al. carried out research experiments to probe the multifaceted role of crystalline structure and valency in oxide-derived copper (OD-Cu) catalysis as well as the mechanism and characteristics of e-CO2R. The experiment also used single crystal Cu(111) foils, enabling regulatory analysis of their intrinsic variation in morphology and crystallography. It was interesting that the overall efficiency of OD-Cu in reducing CO2, and particularly in achieving C2H4 selectivity, was shown to be significantly correlated with the initial oxidation degree of the Cu(111) foil. There is no significant effect on the catalytic activity of catalyst due to grain boundaries and polycrystallinity. These findings highlight a complex web of connections among the structural features, oxidation states, and performance levels of OD-Cu, and therefore make important contributions to the understanding of environmentally friendly energy conversion processes.70
The study examined the catalytic activity of copper in the e-CO2RR and how it varies according to the crystal phase. This was done by depositing ultra-pure Cu, which remained in the 4H and 4H/fcc phases. The study found that non-traditional crystal structures had better overall activity and selectivity in e-CO2RR than the standard fcc-Cu structure towards ethylene. DFT calculations reveal that the 4H phase and 4H/fcc interface of Cu promote the C2H4 formation pathway more efficiently than fcc Cu, resulting in a crystal phase-dependent selectivity towards C2H4. This meant that different crystal phases had different C2H4 selectivity.71 In an effort to research further, a widespread plan was drawn up by researchers. The goal was to effectively use copper-decorated through-hole carbon nanofibers (CuSAs/TCNFs) in e-CO2R for the transformation of CO2 to methanol. The fabricated CuSAs/TCNFs membrane achieved an impressive FE value of 44% for methanol.
6.2. Single atom approach
The single atom approach in the design of electrocatalysts for e-CO2RR involves the dispersion of individual metal atoms on a suitable support material, often carbon-based. This method maximizes the utilization of metal atoms, ensuring that each atom acts as an active site, thereby significantly enhancing catalytic efficiency. By isolating metal atoms, this approach also allows for precise tuning of electronic properties and coordination environments, which can lead to superior selectivity and activity for specific CO2RR pathways. The unique atomic configuration of single atom catalysts (SACs) can facilitate the stabilization of key intermediates and lower energy barriers, contributing to higher overall performance in CO2RR. Chen et al. aimed to increase the efficiency of converting CO2 to methane by designing a Cu-based catalyst that is augmented with a Lewis acid support. For example, metal oxides like Al2O3 and Cr2O3 can do it in this way. They can modulate the electronic structure of Cu atoms according to the requirements of intermediate adsorption. The specific study explored the potential of an ultrathin porous Al2O3 that was highlighted by its numerous Lewis acid sites, to bind Cu atoms at the single atom level. Remarkably, it yielded a notable increase in FE for CH4 synthesis, reaching 62%, operating at a potential of −1.2 V and a current density of 153 mA cm−2. This precise manipulation of individual copper atoms demonstrated a remarkable efficacy in the conversion of carbon dioxide to methane, offering a significant step towards efficient and sustainable energy conversion processes.72 Zhao and his colleagues switched gears in their work, pursuing methanol production through electrolysis of CO2. They developed a method in which copper-monitoring MXene was made with much success. The fourth MAX phases underwent selective etching. Only aluminum and copper from the hybrid A layers were specifically targeted for removal. It makes use of the saturated vapor pressure differences between aluminum and copper products. As a result, installing a Cu single-atom catalyst attains an impressive FE of 59.1% in making CH3OH. The catalyst not only showed promise for electrocatalytic stability but also the potential for e-CO2R because of its unsaturated electronic structure at the copper atom. Because of its unique properties, the e-CO2RR has a low-energy barrier in the rate-determining step. In fact, copper-immobilized MXene plays a key role in promoting the development of sustainable energy solutions.73Researchers considered the e-CO2RR using a carbon substrate featuring coordinated copper atoms within a unique CuN4 structure. This material showed impressive rates of conversion. It converted 55% of ambient CO2 with distilled water as its source into ethanol; more impressively, 80% of dissolved CO was converted to C2-products such as ethanol or ethylene. After all, the high faradaic yield in both cases proved this. Metal copper sites are responsible for catalytic action. Furthermore, the starting material was entirely recoverable.74 Turning attention to another interesting study, Bao et al. illuminated the realm of e-CO2R, employing single copper atoms on MXene nanosheets. The researchers achieved a remarkable selectivity of 98% for multicarbon products and an impressive FE of 71% for ethylene synthesis at a potential of −0.7 V vs. RHE. The process involved the combination of carbon monoxide molecules to form the *CO–CHO species, effectively reducing the free energy barrier of the potential-determining step. This catalyst had something special about it as it continued being active all the way through the 68 hours of the electrolysis process, thereby proving that single copper atoms could produce useful compounds made up of many carbon atoms in a substantial way.75
In a separate project, the effort to achieve e-CO2R led to the fabrication of atomically dispersed Cu catalysts on defective Ag nanowires. This innovative approach hinged upon a site-specific underpotential deposition method and an electrochemical conditioning treatment strategy, resulting in the dispersion of the Cu/Ag(S) catalyst. The integration of Cu single-atom catalysts remarkably elevated the selectivity and activity of the catalyst in CO2 reduction. The alteration of the d-band structure and the facilitated interaction between Cu single-atom catalysts and the conditioned surface's defects played a pivotal role. This augmentation led to an enhanced adsorption of e-CO2RR intermediates, subsequently boosting the efficiency of the process. Impressively, at an e-CO2RR current density of 2.9 mA cm−2, the Cu/Ag(S) catalyst exhibited an outstanding FE of 93.0%, surpassing the performance of the Ag(S) catalyst lacking Cu single-atom catalysts. The presence of Cu SACs in proximity to faulty silver surfaces induced increased microstrain and a downward shift in the center of the d-band, effectively reducing the activation energy required for *CO generation by approximately 0.5 eV. This synergistic interplay resulted in heightened efficiency and specificity in CO2 reduction, particularly in the production of CO and C2 products, underscoring the catalytic competence of the Cu/Ag(S) catalyst.76
In a distinct exploration by Zheng et al., a catalyst consisting of a single lead atom with copper was employed to successfully convert CO2 into formic acid. The catalyst's remarkable 96% FE in formate generation underscores its exceptional performance. The active copper sites regulated the first protonation step extremely accurately so that the reaction went towards formate synthesis without the formation of other products. The authors supported the use of this method for producing valuable liquid fuels with high purity.77 Moreover, Song et al. delved into the catalytic performance of two Cu SACs with asymmetric surfaces in e-CO2RR. The CuN3O/C catalyst exhibited outstanding selectivity for e-CO2RR, boasting a CO FE of over 90% across a potential range of −0.5 to −0.9 V vs. RHE. With a potential of −0.8 V, the selectivity for CO production reached an impressive 96%. In complete contrast, the CuCo3/C catalyst showed terrible CO selectivity even at −0.5 V vs. RHE, only 20.0% CO FE was observed. In particular, the CuN3O/C catalyst displayed a TOF of 2782.6 h−1 at −0.9 V vs. RHE, greatly in excess of the highest TOF recorded for CuCo3, 4.8 h−1.78
Furthermore, the e-CO2R rate can be improved by using Cu single-atom catalysts with adjustable concentrations in addition to nitrogen attachment. In addition to facilitating water dissociation, CH4 synthesis in the presence of nitrogen involves the metal assisting in the binding of hydrogen to make COOH intermediates. At a potential of −1.8 V vs. RHE, CH4 yield for Cu catalysts was as high as 68.2% along with an impressive CH4j of 493.1 mA cm−2.79
So as to convert CO2 into acetate and syngas more efficiently, a new tandem catalyst had to be developed. A cost-effective catalyst has copper nanoparticles that integrate nickel single atoms selectively. These single Ni atoms make CO coverage bigger, and the C–C bond tends to cause an expanded interaction with adjacent Cu nanoparticles to facilitate acetate synthesis. It is worth noting that when flexibility with respect to production of syngas (H2/CO) is desired, the tandem catalyst varies from 0.06![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 19.5:1. The intention of this method is to supply liquid fuels more effectively by following CO2RR for the production of syngas and helping to achieve a higher selectivity. The target is single C2 molecules. This is the first critical step from production in the laboratory to implementing it in industry on a large scale.80Table 1 lists the different kinds of electrocatalysts developed and designed for e-CO2R to different products based on the single crystal and single atom approach.
:1 to 19.5:1. The intention of this method is to supply liquid fuels more effectively by following CO2RR for the production of syngas and helping to achieve a higher selectivity. The target is single C2 molecules. This is the first critical step from production in the laboratory to implementing it in industry on a large scale.80Table 1 lists the different kinds of electrocatalysts developed and designed for e-CO2R to different products based on the single crystal and single atom approach.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | Tw-Cu | 0.1 M KHCO3 | −1.31 | 39.3 | 1.1 | 69.5 | 0.74 | 0.01 | 0.04 | — | — | — | 27 |
| 2 | Pc-Cu | 18.7 | 1.86 | 62.5 | 23.02 | 0.01 | 0.04 | — | — | — | |||
| 3 | Cu@GDE | 1 M KOH | −2.6 | 7 | 18 | 6 | 39 | 4 | — | — | 21 | — | 68 |
| 4 | Cu@GDE | 1 M KHCO3 | −3.4 | 10 | 17 | 3 | 46 | 3 | — | — | 8 | — | |
| 5 | Cu(111)-LO@GDE | 1 M KOH | −1.8 | 40 | 6 | 8 | 8 | — | — | — | — | — | 70 |
| 6 | Cu(111)-MO@GDE | 32 | 4.8 | 12 | 20 | — | — | — | — | — | |||
| 7 | Cu(111)-HO@GDE | 38 | 2.9 | 2.3 | 27.4 | — | — | — | — | — | |||
| 8 | p-Al2O3 | 1 M KOH | −1.4 | 68 | 4 | — | — | 8 | — | — | — | 4 | 72 |
| 9 | Cu/p-Al2O3 SAC | 10 | 10 | 49 | 1 | 9 | — | — | 1 | 9 | |||
| 10 | Poisoned Cu/p-Al2O3 SAC | 19 | 21 | 14 | 2 | — | — | — | — | 9 | |||
| 11 | Cu NPs/p-Al2O3 | 10 | 10 | 39 | 2.5 | 2 | — | — | 12.5 | 13 | |||
| 12 | SA–Cu–MXene | 0.1 M KHCO3 | −1.8 | 75.5 | — | — | — | — | — | 17.5 | 5 | — | 73 |
| 13 | Cu-particles–MXene | — | — | — | — | — | — | 4 | — | — | |||
| 14 | Cu0.5NC | 0.1 M CsHCO3 | −1.3 | 45 | 22 | — | — | — | — | — | 28 | — | 74 |
| 15 | 4H Au@Cu | 0.1 M KHCO3 | −1.13 | 22.5 | 17.5 | 2 | 45 | 8 | — | — | — | — | 71 |
| 16 | 4H/fcc Au@Cu | 29 | 9 | 3 | 39 | 20 | — | — | — | — | |||
| 17 | Cu SAs/TCNFs | 0.1 M KHCO3 | −1.2 | 33 | 45 | — | — | — | — | 25 | — | — | 54 |
| 18 | Cu SAs/CNFs | 45 | 33 | — | — | — | — | 22 | — | — | |||
| 19 | Cu–SA/Ti3C2Tx | 1 M KOH | −0.9 | 18 | — | — | 55 | — | — | — | 25 | — | 75 |
| 20 | Cu-NP/Ti3C2Tx | 80 | — | — | 8 | — | — | — | 10 | 2 | |||
| 21 | BNC–Cu | 0.5 M KHCO3 | −1.46 | 25 | 1 | 72 | 2 | — | — | — | 2 | — | 81 |
| 1 M KOH | |||||||||||||
| 22 | Ag | 0.1 M KHCO3 | −1.4 | 22 | 2 | 76 | |||||||
| 23 | Ag(S) | 25 | 5 | ||||||||||
| 24 | Cu/Ag(S) | — | 52 | 4 | 1 | 6 | — | — | 13 | — | |||
| 25 | Pb1Cu SAAs | 0.5 M KHCO3 | −1.62 (Ag/AgCl) | 12.4 | 0.7 | — | — | 78.4 | — | — | — | — | 77 |
| 26 | CuN3O/C | KHCO3 | −0.9 | — | 96 | — | — | — | — | — | — | — | 78 |
| CuCO3/C | — | 5 | — | — | — | — | — | — | — | ||||
| 27 | Cu SAs-0.1 | KOH | −1.4 | 32 | 12 | 54 | — | — | — | — | — | — | 79 |
| 28 | Ni SACs–Cu NPs | 1 M KOH | −1.0 | 25 | 33 | 5 | 10 | 13 | — | — | 8 | — | 80 |
6.3. Oxidation state engineering
One technique to develop materials for CO2 conversion involves manipulating the oxidation states of elements. Materials scientists can modify the oxidation states of elements to regulate both activity and selectivity. This way, we learn that oxidation states can change material structure, surface chemistry, and reactivity. Through the control of oxidation states, CO2 absorption and charge transfer are made possible, affecting reaction pathways. Numerous methods exist for engineering oxidation states. Through controlled changes made to a material's surface, for example the addition of groups or the creation of defects, the oxidation state of the surface oxide atom can be altered. To adjust oxidation states and improve the catalytic activity of a material, controlled doping involves introducing atoms into the lattice structure. Advanced in situ techniques, as potential modulation tools, allow oxidation states to be manipulated in real time during catalysis and enable dynamic control over reaction pathways. Owing to these techniques, researchers have a wide range of tools that can manipulate oxidation states. They can create materials that efficiently convert CO2 into useful products. According to this idea, one groundbreaking study found that CuxO-decorated graphene oxide (G-CuxO-T) has the potential to be used as an electrocatalyst that is capable of controlling e-Co2R. It was found that the function of the electrocatalyst G-CuxO-2h (x = 1–3) can be controlled by the valency of Cu. Key to this control is the multivalence of Cu. It is Cu(I) above all that has been chosen to improve the adsorption of CO2 and enable the facile desorption of HCOO−, so the catalysis process can be optimized. However, Cu(II) was important in maintaining durability, and thus in enhancing the overall stability.82In a parallel avenue of investigation, another study's focus shifted to the impact of the substrate (Cu2O) on the catalytic selectivity of the deposited Sn catalyst. Notably, the presence of Sn played a significant role in formate formation, with the potential to limit the HER requiring a deposition time of less than 120 seconds. Surface analysis revealed the presence of Cu+ either on or beneath the surface, attributed to kinetic constraints during cathodic Sn deposition and e-CO2R. Positioned between Sn2+ and Sn4+ in terms of electronegativity, Cu+ occupied a midrange position. The Sn-deposited electrode exhibited enhanced catalytic selectivity when the deposition time was under 120 seconds, attributed to the stabilizing influence of residual Cu+ species on the surface of Sn, maintaining it at a positive oxidation state between +2 and +4.83 Concurrently, a separate avenue of inquiry was pursued, leading to the discovery that a Cu/ZnO catalyst could serve as a catalyst for converting CO2 into alcohols, specifically ethanol and methanol. This was achieved through the oxidation of Cu nanoparticles at a low temperature, followed by their amalgamation with ZnO crystalline powder. Through a liquid-phase setup, the study scrutinized ZnO's influence on the restructuring of the Cu-based catalyst and its electrocatalytic efficiency in alcohol production under varying applied potentials. Enhanced carbon–carbon coupling and ethanol generation were attributed to ZnO inducing higher CO productivity on the Cu/ZnO-based electrode compared to the Cu-only counterpart. The production of ethanol within this tandem catalyst, as a precursor to further CO reduction into C2+ alcohols, was directly linked to the abundance of Cu1+ and Cu0 combinations on the CZ catalytic surface.84
Moreover, the research revealed the catalytic role of the original copper electrodes in influencing their future activity and stability. More specifically, it was found that Cu(I) oxide would dissolve because of its low conductivity when dispersing in electrolyte for e-CO2R, but Cu(II) oxide would just as easily be converted into metallic copper. That is to say, when these materials were used as electrodes and maintained at above room temperature they behaved more like liquid crystals than crystalline solids. However, the high activity and selectivity reported in the literature for OD-Cu electrodes could potentially be attributed to the fact that Cu(II) dissolution, which is more prevalent in bulk electrodes like foils or single crystals, exposes the metallic surface beneath the porous Cu(II) oxide structure. The outcomes of this study demonstrated that metallic copper is an excellent electrocatalyst for e-CO2R and that the metal's intrinsic activity can be modified without compromising its conductivity. In order to maintain the stability of the metallic copper matrix phase, moderate doping levels must be used, which might influence activity and selectivity.85 In another study, Cu-MOF/NP catalysts were made from MOF templates. More specifically, the Cu-MOF20/300 nanoparticles proved to be potent electrocatalysts for electrochemical CO generation because of their Cu/Cu2O heterogeneous structure and vast active surface area. Impressively, the Cu-MOF20/300 catalyst demonstrated a high faradaic efficiency (FECO) of 43.8% for CO generation. Furthermore, the electrochemical reaction performance of the flow MEA reactor displayed significant enhancements compared to the conventional H-type setup. The reactor's compact design allowed it to reach current densities in excess of 230 mA cm−2 at a low applied potential, effectively lowering both ohmic and charge transfer resistances. The incorporation of a fillable gas–liquid separation component made it much more sensitive to changes, thereby facilitating detection and estimation.86
To gain deeper insights, the study explored the complex relationship between torsion and porous Cu electrode oxidation state. It turned out that substrates with high tortuosity have lower CO2 content especially in deeper layers. Amazingly high tortuosity in the network resulted in bubbles of CO traveling farther, increasing the chance of CO going back to surface catalytic surfaces. The generation of C2+ products was favored by this phenomenon. The generation of CO, HCOOH, CH4 and C2+ products were elucidated in this study, showing that their production was influenced by CO2 and CO availability and an intricate interplay between them. The Cu oxidation state was used as a means of extracting insight into selectivity from research on yield/efficiency of carbon dioxide reduction. This detailed analysis showed that metallic copper had more favorable C2+ alcohol pathways than copper derived from Cu2+ or Cu+ particles.87 In another facet of their research, scientists also succeeded in regenerating the Cu catalyst in situ for the sustainable reduction of CO2 into various carbon products. Using an AC intermittent working mode, the catalysts went through periodic invigoration by the oxidizing Cu surface. When operated in this effective manner, at a constant current density of 150 mA cm−2 the Cu catalyst remained active for an impressive 120 hours and maintained 40% selectivity throughout.88
The authors demonstrated that, for better efficiency, optimizing the placement of the semiconductor sensitizer and CuOx co-catalyst was better. In addition, the research also explores the role played by thin Cu2O. The findings showed that CO2 reduction was affected by the distribution of the Cu surface's oxidation states. Because of COatop intermediate species and C1 hydrocarbon-product production, electrodeposited Cu electrodes had higher Cu(I) in them. Conversely, the presence of Cu(0) in the initially prepared electrodes hindered hydrocarbon product development. Moreover, the coexistence of Cu(I) and Cu(0) in the CO2-reduction products yields not only CObridge but also COatop, leading to the high selectivity for the C2 product.89 When using HQ-Cu or AN-Cu, both of which are electrodes comprised of mixed oxidation states, a greater selectivity for C2+ products was noted. The distribution and historical development of different Cu species were characterized using FIB-TEM/EELS. Notably, during the steady state of CO2RR, the HQ-Cu electrode underwent complete reduction to metallic Cu, indicating that the observed high C2+ selectivity did not rely on specific Cu oxidation states. To ensure complete reduction to Cu0 prior to CO2RR, the electrodes underwent electro-reduction pretreatment. Further bolstering the pivotal catalytic role of Cu0 and the minimal impact of the initial oxidation state of oxidized Cu, the pretreated electrodes exhibited slightly enhanced selectivity toward C2+ products. Notably, an oxidation–reduction cycle led to crystal fragmentation, observed when oxide crystals in HQ-Cu and AN-Cu were reduced and fragmented into small irregular Cu grains under the negative potential of electro-reduction pretreatment or CO2RR.90
In the pursuit of exceptional CO2RR performance, a 1D composite SnCu-CNS was resourcefully developed, boasting a hierarchical structure of nanotubes and loaded nanorods. This composite showcased a remarkable trait, achieving a partial current density of 205.1 mA cm−2 for formate at 1.0 V vs. RHE cathode potential, and reaching an impressive peak FE of 95.1% within an H-type cell.91 Another study's findings unveiled a synergistic connection between the structural evolution of Cu2O and the surface––interface CO2 reaction. During the cathodic scanning phase of CV, CO* derived from CO2R assumed the role of a stabilizer, effectively preventing the oxidation of metallic Cu. This stabilization process expedited the shift from inert Cu2O to the more dynamic Cu/Cu2O under regular operational conditions. Importantly, the incorporation of Cu/Cu2O led to a substantial increase in FE for ethanol, elevating it from 5.15% to an impressive 56.56%, and significantly fostered the generation of C2 products.92
To ensure the stability of Cu+ sites during the CO2RR, the researchers designed PdCu3N, a catalyst built upon the antiperovskite type Cu3N. Remarkably, the incorporation of negative-valence Pd into the catalyst significantly augmented the synthesis of C2 products, resulting in a substantial increase in the FE of C2 (78.2%) compared to the Cu3N catalyst with only single Cu+ sites (5.6%).93 Concurrently, another study delved into the utilization of Cu nanosheets decorated with small Cu2S nanocrystals as catalysts for the e-CO2R into C2H5OH (Fig. 7). The investigation showcased that in an H-cell containing 0.1 M KHCO3, the total current density reached 45 mA cm−2, accompanied by a substantial JC2H2OH of approximately 20.7 mA cm−2. The advantageous attributes of decorating Cu nanosheets with nanocrystals encompassed a non-flat, stepped surface, a high localized positive charge of Cu+, and well-established interfaces between Cu+ and Cu0. These attributes collectively enhanced CO2 adsorption, facilitated charge transfer for CO2RR kinetics, facilitated the dissociation of the *COOH intermediate into *CO, heightened the affinity of *CO on the catalyst, reduced the energy barrier of CC coupling, created favorable spatial conditions for C2H5OH production, and facilitated the spontaneous hydrogenation of *CH2CHO to *CH3CHO.94
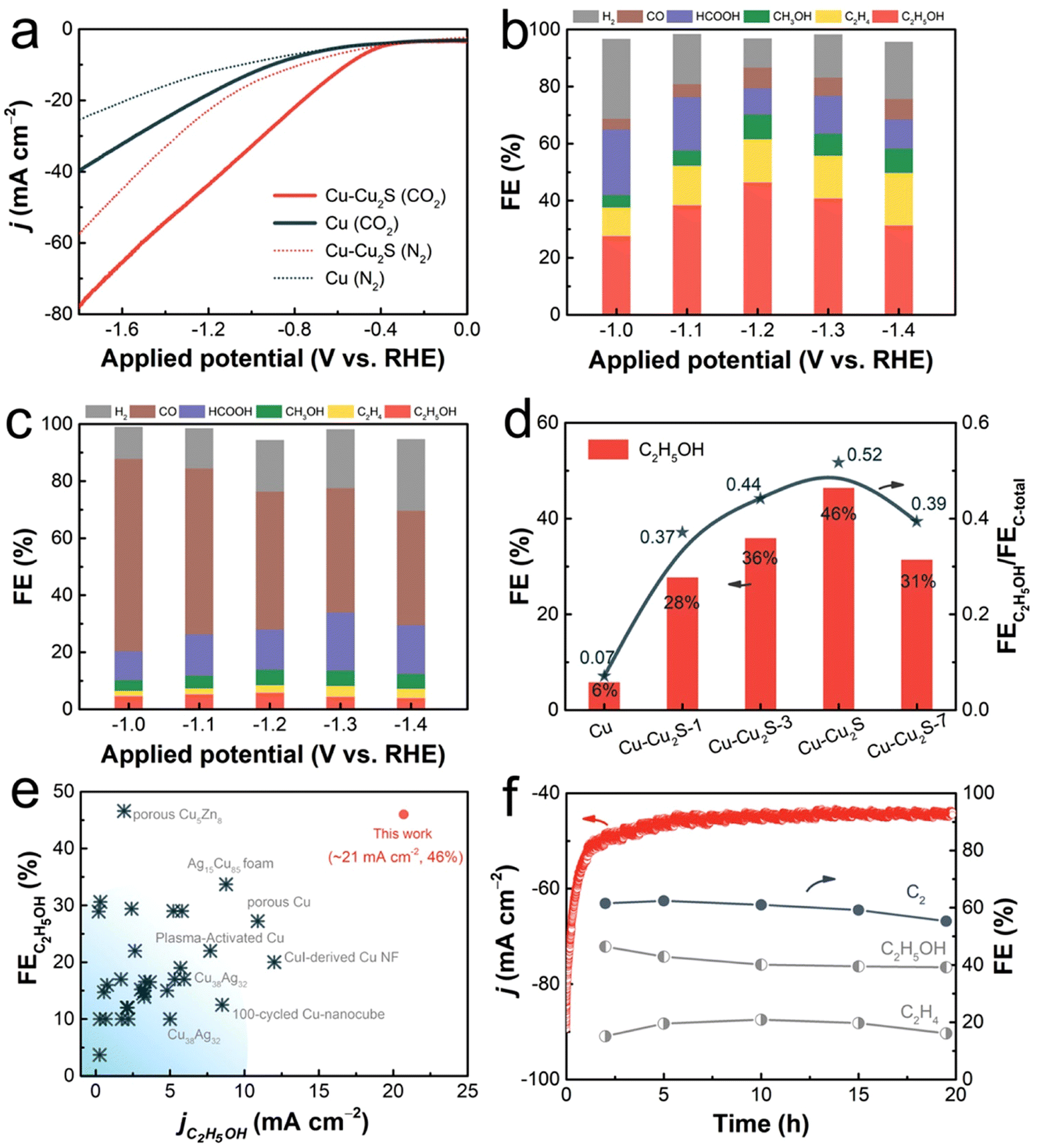 | ||
| Fig. 7 Electrochemical activity of the manufactured catalysts for CO2R. In a N2- and CO2-saturated 0.1 M KHCO3 aqueous electrolyte (a), the LSV curves of pure Cu and CuCu2S are shown. Distributions of the products of the catalyzed CO2RR for (b) CuCu2S nanosheets and (c) pure Cu nanosheets at various applied potentials. At 1.2 V (against RHE), FEC2H5OH and FEC2H4OH/FEC products for CuCu2S, pure Cu, and CuCu2S-1,3,7 in an H-cell containing 0.1 M KHCO3 solution (d). The comparison of partial current density for C2H5OH and the FEC2H5OH of CuCu2S with those of previously reported catalysts. Some examples of these catalysts are copper nanocubes, copper nanofibrils, nanofibrils generated from copper ions, copper alloys, copper activated in plasma, copper foam, and porous copper (e). 20 hours of stability at 1.2 V (against RHE) in terms of geometrical current density and field strength for carbonaceous compounds catalyzed by Cu and Cu2S (f). Reproduced with permission from ref. 94. Copyright 2023, American Chemical Society. | ||
In a related investigation, the selective production of CH4 and C2H4 in the CO2RR process on CuOx catalysts with varying oxidation states and reductant agents was investigated. The study identified the optimal oxidation state of the copper catalyst, leading to the highest selectivity for C2H4 (FE = 53%). The study explored several potential reaction pathways, including CC coupling, hydrogenation of CO to produce CHO, direct dimerization of CO to produce C2H4, and hydrogenation of CO to yield CHO or COH.95Table 2 presents an overview of different electrocatalysts designed by following the oxidation state engineering approach for e-CO2R to a number of fuels.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | G-CuxOT | 0.5 M KHCO3 | −0.8 | 64 | — | — | — | 9 | — | 2 | — | — | 82 |
| 2 | Cu foil | 0.5 M KHCO3 | −1.2 | 25 | 22 | — | — | 20 | — | — | — | — | 83 |
| 3 | Soaked Cu 0 s | 24 | 31 | — | — | 37 | — | — | — | — | |||
| 4 | Sn foi | 30 | 7 | — | — | 68 | — | — | — | — | |||
| 5 | S Sn | 22 | 8 | — | — | 73 | — | — | — | — | |||
| 6 | Cu calc | −1.4 | 68 | 3 | 2 | — | 9 | — | — | 5 | — | 84 | |
| 7 | CZ calc | 78 | 5 | 1 | — | 4 | — | 1 | 7 | — | |||
| 8 | PVD Cu films @ 25 °C | 100 mM KHCO3 | −1.8 Ag/AgCl | 76 | 2 | 9 | 9 | 3 | — | — | 1 | — | 85 |
| 9 | PVD Cu films @ 150 °C | 67 | 1 | 13 | 12 | 1 | — | — | 6 | — | |||
| 10 | Cu/MOF 20/300 | 0.5 M KHCO3 | −0.66 | 62 | 35 | — | — | — | — | — | — | — | 86 |
| 11 | OD-Cu-16 | 0.1 M KHCO3 | −1.0 | — | 22 | 2 | 15 | 23 | — | — | — | — | 87 |
| 12 | OD-Cu-32 | — | 23 | 3 | 21 | 21 | — | — | — | — | |||
| 13 | OD-Cu-45 | — | 5 | 4 | 31 | 14 | — | — | — | — | |||
| 14 | OD-Cu-70 | — | 3 | 6 | 28 | 17 | — | — | — | — | |||
| 15 | Cu/PTFE 200 nm | 1 M KHCO3 | 100 mA cm−2 | 17 | 4 | 16 | 38 | 4 | — | — | 10 | — | 88 |
| 16 | Cu/PTFE 700 nm | 19 | 7 | 1 | 42 | 8 | — | — | 17 | — | |||
| 17 | Cu/PTFE 1000 nm | 18 | 9 | 1 | 42 | 10 | — | — | 37 | — | |||
| 18 | Electrodeposited Cu | 0.1 M KHCO3 | −1.0 | 53 | — | 32 | — | 9 | — | — | — | — | 89 |
| 19 | As-prepared Cu | 49 | 3 | — | — | 14 | — | — | — | — | |||
| 20 | CV-treated Cu | 10 | 5 | 38 | 42 | 8 | — | — | — | — | |||
| 21 | Electropolished Cu | 0.1 M KHCO3 | −1.0 | 70 | 11 | 2 | 15 | — | — | — | — | — | 90 |
| 22 | HQ-Cu | 30 | 2 | 5 | 27 | — | — | — | — | — | |||
| 23 | AN-Cu | 29 | 3 | 3 | 23 | — | — | — | — | — | |||
| 24 | SnCu-CNS | 0.1 M KHCO3 | −1.0 | 9 | 28 | — | — | 63 | — | — | — | — | 91 |
| 25 | o-Cu2O | 0.1 M KHCO3 | −1.06 | 30 | 22 | — | 13 | 12 | — | — | 5 | — | 92 |
| 26 | Cu/Cu2O-CV | 9 | 8 | — | 9 | 14 | — | 5 | 58 | 5 | |||
| 27 | Ce–Cu2O | 0.5 M KHCO3 | −1.1 | — | 8 | — | 11 | — | — | — | — | — | 47 |
| 28 | Cu2O | — | 4 | — | 8 | — | — | — | — | — | |||
| 29 | Cu3N | 1 M KOH | −1.1 | 19 | 43 | — | 2 | 36 | — | — | — | — | 93 |
| 30 | Pdδ−–Cu3N | 9 | 10 | — | 39 | 4 | — | — | 32 | — | |||
| 31 | Pd0.1 | — | 90 | 1 | — | — | — | 9 | — | — | 96 | ||
| 32 | Cu | — | 89 | — | — | — | — | 11 | — | — | |||
| 33 | CuPd0.05 | — | 65 | — | — | — | — | 35 | — | — | |||
| 34 | CuPd0.1 | — | 93 | — | — | — | — | 7 | — | — | |||
| 35 | CuPd0.5 | — | 87 | — | — | — | — | 13 | — | — | |||
| 36 | CuPd2 | — | 89 | 1 | — | — | — | 10 | — | — | |||
| 37 | CuPd5.7 | — | 91 | 1 | — | — | — | 8 | — | — | |||
| 38 | Cu–Cu2S | 0.1 M KHCO3 | −1.1 | 18 | 3 | — | 14 | 19 | — | 6 | 38 | — | 94 |
| 39 | Pristine Cu nanosheets | 15 | 57 | — | 3 | 15 | — | 3 | 5 | — | |||
| 40 | CuOx−1 | 0.1 M KHCO3 | −1.5 | — | — | 40 | 20 | — | — | — | — | — | 95 |
6.4. Alloying approach
The use of alloying is a versatile strategy in designing electrocatalysts for CO2R. By combining metals catalysts with customized properties can be created to enhance efficiency and selectivity in converting CO2 into products. This approach plays a role in tackling challenges related to clean energy conversion. Researchers can carefully mix metals to optimize the binding strength for CO2 and its byproducts, minimize overpotentials and improve catalytic performance. Importantly alloying can result in effects where the resulting alloy's properties surpass those of metals, making it an invaluable tool in the pursuit of sustainable CO2R.There are two primary types of alloying approaches used in electrocatalysis: pure metallic alloys and compound alloys. In pure metallic alloys, metals are blended to create alloys with enhanced catalytic activity and selectivity. Metals are selected based on their characteristics and desired catalytic outcomes. On the other hand, in compound alloys, a metal is combined with a metal element or a compound. This approach introduces active sites and modifies the electronic structure of the alloy leading to improved performance in CO2R.
Gunji et al. have explored the electrocatalytic selectivity of Pd-based bimetallic nanoparticles for CO2 reduction. PdZn NPs/CB and PdCu NPs/CB exhibited selectivity for HCOO− production, while PdSn NPs/CB and PdAg3 NPs/CB favored CO generation. Notably, PdZn NPs/CB displayed a higher selectivity for HCOO−, achieving approximately 100% faradaic efficiency for HCOO− generation at −0.1 V. The presence of PdSn NPs and PdAg3 NPs/CB suppressed HCOO− production, leading to CO selectivity. The underlying mechanism driving this selectivity is currently undergoing further theoretical investigation.97 Additionally, CuZn catalysts with varying Zn contents were developed to enhance CO2R into various products, particularly HCOOH. These catalysts were used as cathodes in PEC reactions and electrodes in electrochemical CO2 reduction. The CuZn-0.5 catalyst exhibited significantly higher current density compared to Cu-foil, with a FE of 60%. The synergistic interaction between Cu and Zn sites played a crucial role in increasing the selectivity for HCOOH during CO2R.98
In comparison to an electrodeposited Cu electrode, the Cu76Sn24 alloy, composed of Cu3Sn, demonstrated reduced H2 and hydrocarbon formation during CO2R. The alloy exhibited selectivity for CO generation across all applied potentials, with an increase in HCOOH production as the potential was increased. Furthermore, another study, by analyzing the structure of the active site, highlights that Cu–Sn binary catalysts have shown improved selectivity. To mitigate hydrogen generation in electrochemical operations, Sn single atoms should replace nearby terrace Cu atoms. The active Cu edge atoms are responsible for this mechanism. These structures maintain thermodynamic stability under reducing conditions. It is advisable to avoid creating competing structures during material preparation. Substituting Sn prevents the HER process by disrupting the local 3-fold symmetry required for efficient H adsorption at Cu edges. By facilitating CO formation from CO2 through charge transfer from the Sn atom to the Cu edge atoms, increased selectivity for CO is achieved.99 Similarly, Yoo et al. have fabricated 3D-h CuSn in order to examine the impact of composition and geometry on CO2R. Remarkably, the FECO was significantly increased to 98.6% at −0.45 V vs. RHE when tiny Sn NPs partially covered the Cu surface. Effective CO production was demonstrated at this low Cu/Sn ratio. This change in the FE landscape for the CO2R is attributed to the hierarchical nanoscale architecture, which generates a strong localized electric field, resulting in K+ accumulation near the catalyst surfaces.100
Using a co-reduction strategy, Tang et al. have developed a Cu-decorated Cu2O catalyst with tunable activity by adjusting the amount of Ag. The modified CuAg-0.75% catalyst displayed enhanced catalytic performance in producing liquid multi-carbon compounds, particularly ethanol. It achieved a maximum JliquidC2+ of 288 mA cm−2 and a jethanol of 214 mA cm−2 at a low overpotential. DFT simulations suggested that the CC coupling process might benefit from the overflow of CO from sites with strong CO adsorption on Cu sites.101 In another experiment, a CuNiZn alloy was used as the electrocatalyst for the e-CO2R and e-COR. The primary hydrocarbons produced were alkenes, while formate, carbon monoxide, and methane were byproducts of the initial CO2R.102 Another study highlighted the ability to manipulate bimetallic Cu–Pd nanoalloys to achieve specific morphologies and compositions. Spherical Cu–Pd nanoalloys with a Cu/Pd molar ratio of 1/0.3 exhibited the highest FE for CO conversion, while dendritic Cu–Pd nanoalloys were efficient for H2 synthesis via HER (65.2% at a polarized potential of −0.87 V).103
In addition, Liu et al. investigated the electrical and nano/mesostructural effects of uniformly produced Au–Cu bimetallic films, with an emphasis on their utilization in e-CO2R. The introduction of Au drastically altered the d-band electronic structure, leading to a reduction in the binding strength of CO. The binding energy in the d-band could be tuned by varying the composition of the alloy. Increasing the Au content was found to enhance e-CO2R activity and selectivity, demonstrating that the electronic effect alone was not sufficient to overcome the linear scaling relationship. Interestingly, copper oxide/hydroxide species on Au–Cu surfaces were less stable compared to those on pure copper, despite the presence of gold. Alloying Cu with Au could prevent the formation of HCOO− by reducing the adsorption energy of *OCHO. The generation of HCOO− was profoundly affected by the electrical effect and d-band structure, although the activity did not scale linearly with changes in composition.104
Again, for e-CO2R, Cu–Co bimetallic nanoparticles were synthesized on porous carbon using a similar process as adapted by Guo et al. These catalysts achieved current densities as high as 62.1 mA cm−2 in an electrolyte containing 0.5 M [Bmim]PF6 in MeCN, resulting in a FECO of 97.4%. Catalytic activity was increased due to the interaction between Cu and Co, which increased the electrochemical surface area and CO2 adsorption capacity.105 However, Velasco-Vélez et al. have discovered that adding Zn heteroatoms to a copper metal lattice did not significantly boost the creation of profitable multi-carbon species during the e-CO2RR. The presence of Cu+ species in the reaction pathway had little effect on these species.106 Further, the impact of metal content and particle size of CuCo nanoparticles generated via inverse micellar encapsulation on e-CO2RR activity was investigated. The results demonstrated that initially both metals were oxidized, and even a small amount of Co significantly enhanced the activity of CuCo nanoparticles. As particle size increased, e-CO2RR outperformed HER.107
Furthermore, pulsed laser ablation in liquids (PLALs) was used to produce noble metal-free multi-principal element alloy nanoparticles (MPEA-NPs) for the e-CO2R. The electrochemical testing confirmed the unique activity of MPEA-NPs stabilized with poly-(diallyldimethylammonium chloride) (PDADMAC) for CO2R. It was discovered that Al8Cr17Co17Cu8Fe17Ni33 MPEA-NPs exhibited highly efficient catalysis for CO2R, potentially replacing costly noble metal electrocatalysts.108 Okatenko et al. had shown that the stability of Cu nanoparticles under CO2RR had been significantly enhanced by the addition of Ga. Ga-Added nanoparticles had remained stable for up to 20 hours, whereas the parent Cu nanoparticles had decayed quickly (Fig. 8). Employing various experimental and theoretical methods based on the known reconstruction mechanism of Cu nanoparticles under CO2RR, the researchers had provided an explanation for this observation. They had hoped that their findings would lay the foundation for the development of alloyed Cu-based nano-catalysts that could withstand CO2RR and other reducing reaction conditions. Similar mechanisms to prevent Cu reconstruction and produce more stable nanoparticles had been found in the study for metals with higher oxophilicity and strong M–Cu bonding energies.109
 | ||
| Fig. 8 CuGa NPs were synthesized and characterized. (A) A streamlined approach to synthesis. Bright field TEM images of Cu, CuGa4, CuGa17, and CuGa37 NPs (B)–(E) and their matching XRD patterns (F). These results point to the synthesis of CuGa NPs with a consistent shape, extremely similar diameters, and varying Ga concentrations. How well the CuGa NPs electrocatalyze CO2RR. (g) Mean FE (left axis) and geometric current density (right axis) for Cu, CuGa4, CuGa17, and CuGa37 NPs at various applied potentials (NP loading was held constant at 15 g on glassy carbon with a geometric area of 1.33 cm2) during 45 min CO2RR in CO2-saturated 0.1 M KHCO3. The advancement in CH4 production using CuGa NPs is indicated by the ratio of the FE for CH4 and C2H4 across the same potential range (h). Reproduced with permission from ref. 109. Copyright 2023, American Chemical Society. | ||
Foucher et al. have presented a strategy for the synthesis of Cu–Pt nanocrystals of varying sizes and crystal shapes. A Cu-rich core had been found to be enclosed in a Pt-rich layer, as determined by a thorough STEM-EDS study. Cu–Pt particles had been stable as catalysts because, after being annealed at 800 °C, they had become intermetallic CuPt particles. Non-intermetallic CuPt had been found to be stable at 400 °C in H2 or O2 using in situ STEM analysis. The production of a Cu oxide layer, which would cover and render Pt inactive, had been prevented by the intermetallic CuPt phase. They had shown that copper alloys didn't always oxidize and decay as quickly as feared, and that nanoscale CuPt structures could help conserve otherwise costly Pt. CuPt particle stability had been of significant interest and may be useful in a number of different chemical processes.110 In a related context, another study delved into bimetallic CuAg electrodes. These electrodes had bulk compositions closely matching their surface compositions, primarily consisting of Cu and Ag crystallites. During CO2RR, the presence of CO adsorbates induced the segregation of initially dissolved Cu in the Ag phase, resulting in Cu surface enrichment on the Ag-rich bimetallic electrodes. CO formation primarily occurred on the surface of the Ag domains, while CO reduction was exclusive to the Cu domains. The distribution of products changes when they are produced on a stable CuAg electrode. Changing from pure Cu electrodes significantly decreases hydrogen generation and favors carbonyl compounds.111
The study proposed an alternate route using the chemical named DAT to co-electrodeposit high-surface-area CuAg alloys. Instead of nucleus formation the way this method worked merely prevents nucleation, producing more evenly distributed samples of Cu and Ag atoms in the CuAg-wire. Using a flow electrolyzer, we found that CuAg-wire samples were more active and coherent in terms of selectivity compared to CuAg-poly and Cu-wire in the e-CO2R. Products such as C2H4 and C2H5OH were detected.112 Seeking a different avenue of study, Pd@Au nanoparticles were developed by impregnating Au nanoparticles with specified amounts of Pd. Intriguingly, it was shown that at varying Pd loadings the catalytic activity for CO2R was non-linear. In addition, the bimetallic Pd–Au surfaces with isolated Pd presented lower energy barriers for CO2 activation and were less prone to poisoning by strongly binding *CO intermediates.113
Moreover, the research delved into the impact of the starting Cu/Zn ratio and the extent of alloying between Cu and Zn components on the product selectivity of CuZn nanoparticles. Under CO2R conditions, CH4 was predominantly generated near the metallic Cu in proximity to ZnO, while the exclusive production of CO and H2 resulted from the reduction of ZnO species and the enhanced Cu–Zn contact.114 Additionally, Li et al. have attained FE exceeding 95% in industrial-scale CO2 reduction to CO, by using a novel antimony–copper single-atom alloy catalyst (Sb1Cu). This catalyst offered improved selectivity and throughput due to the atomic-level SbCu interface, which facilitated CO2 adsorption while reducing CO* binding (Fig. 9).9
 | ||
| Fig. 9 CO2RR performance over Sb1Cu catalysts and Cu. FEs of all CO2RR products at different current densities and the associated j–V curve for Sb1Cu-5 (a). In situ DEMS measurement of the production of (up) CO and (down) C2H4 during CO2RR on Cu and Sb1Cu-5 catalysts (b). Stability test at 100 mA cm−2 current density in MEA for more than 10 hours. GC analysis shows an average FECO of about 95% (c). Different reported electrocatalysts for CO2RR-to-CO (d) Cu-based and (e) non-Cu-based catalysts' performance measures. Product FEs on (f) Cu, (g) Sb, and (h) Sb1Cu-1.5 compared. The error bars show the average difference between three different readings. Reproduced from ref. 9. Open access article, Nature. | ||
In a different facet of the research, bimetallic Au0.5Cu0.5 NPs with a narrow size distribution were synthesized using inverse micelle encapsulation. Smaller NPs exhibited higher current density during e-CO2R due to their larger population of low-coordinated sites, as confirmed by X-ray spectroscopy. While faradaic selectivity showed less size dependence, with H2 being the predominant product at approximately 80% selectivity, larger NPs yielded a 7% increase in CO output and a 7% decrease in H2 production.115Table 3 provides a comprehensive overview of different designed electrocatalysts for e-CO2RR following the pure metallic alloy approach on Cu-based materials.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | Pd NPs/CB | 0.1 M KHCO3 | −1.0 | 63 | 38 | — | — | 3 | — | — | — | — | 97 |
| 2 | Ag NPs/C | 43 | 63 | — | — | 3 | — | — | — | — | |||
| 3 | PdZn NPs/CB | 44 | 17 | — | — | 42 | — | — | — | — | |||
| 4 | PdCu NPs/CB | 53 | 57 | — | — | 1 | — | — | — | — | |||
| 5 | PdSn NPs/CB | 62 | 15 | — | — | 1 | — | — | — | — | |||
| 6 | PdAg3 NPs/CB | 18 | 84 | — | — | 1 | — | — | — | — | |||
| 7 | Cu | 0.1 M KHCO3 | −1.09 | 18 | 5 | — | — | 10 | — | — | — | — | 56 |
| 8 | Cu3Sn (Cu76Sn24) | 5 | 38 | — | — | 43 | — | — | — | — | |||
| 9 | Sn | 5 | 3 | — | — | 89 | — | — | — | — | |||
| 10 | Cu | 0.1 M KHCO3 | −1.1 | 45 | 4 | — | — | 19 | — | — | — | — | 98 |
| 11 | CuZn-0.1 | 40 | 5 | — | — | 33 | — | — | — | — | |||
| 12 | CuZn-0.5 | 26 | 7 | — | — | 60 | — | — | — | — | |||
| 13 | CuZn-1 | 28 | 10 | — | 16 | 31 | — | — | — | — | |||
| 14 | Zn | 20 | 55 | — | — | 15 | — | — | — | — | |||
| 15 | Flat Cu–Sn | 0.1 M KHCO3 | −1.0 | — | 77 | — | — | — | — | — | — | — | 100 |
| 16 | 3D-h-Cu–Sn | −0.4 | — | 91 | — | — | — | — | — | — | — | ||
| 17 | Cu2O | −0.75 | 20 | 22 | 7 | 28 | 4 | — | — | 13 | — | 101 | |
| 18 | CuAg-0.25% | 19 | 18 | 8 | 34 | 1 | — | — | 16 | — | |||
| 19 | CuAg-0.75% | 16 | 19 | 5 | 37 | 1 | — | — | 17 | — | |||
| 20 | CuAg-2% | 14 | 19 | 2 | 45 | 1 | — | — | 15 | — | |||
| 21 | CuNiZn | 0.1 M KHCO3 | −1.2 | 68 | 4 | 2 | 2 | 38 | — | — | — | — | 102 |
| 22 | Cu–Pd-D | 0.5 M KHCO3 | −0.87 | 65 | 30 | — | — | — | — | — | — | — | 103 |
| 23 | Cu–Pd-0.8 | 9 | 86 | — | — | — | — | — | — | — | |||
| 24 | Cu–Pd-C | 71 | 48 | — | — | — | — | — | — | — | |||
| 25 | Cu–Pd-0.3 | 5 | 92 | — | — | — | — | — | — | — | |||
| 26 | Cu–Pd-S | 25 | 55 | — | — | — | — | — | — | — | |||
| 27 | Cu–Pd-0.1 | 40 | 53 | — | — | — | — | — | — | — | |||
| 28 | Pd/C | 68 | 37 | — | — | — | — | — | — | — | |||
| 29 | Au | 0.1 M KHCO3 | −0.8 | 22 | 78 | — | — | 1 | — | — | — | — | 104 |
| 30 | Au75Cu25 | 42 | 66 | — | — | 2 | — | — | — | — | |||
| 31 | Au50Cu50 | 58 | 49 | — | — | 3 | — | — | — | — | |||
| 32 | Au25Cu75 | 63 | 29 | — | — | 4 | — | — | — | — | |||
| 33 | Cu | 59 | 12 | — | — | 13 | — | — | — | — | |||
| 34 | Cu | 0.5 M [Bmim]PF6 | −2.1 | 77 | 23 | — | — | — | — | — | — | — | 105 |
| 35 | CuCo0.4 | 50 | 50 | — | — | — | — | — | — | — | |||
| 36 | CuCo0.6 | 12 | 88 | — | — | — | — | — | — | — | |||
| 37 | CuCo1.0 | 2 | 99 | — | — | — | — | — | — | — | |||
| 38 | CuCo1.9 | 22 | 79 | — | — | — | — | — | — | — | |||
| 39 | Co | 66 | 22 | — | — | — | — | — | — | — | |||
| 40 | Cu:Zn 100:0 |
100 mM KHCO3 | −1.8 Ag/AgCl | 38 | 2 | 3 | 30 | — | — | — | — | — | 106 |
| 41 | Cu:Zn 91:9 |
32 | 5 | 7 | 31 | — | — | — | — | — | |||
| 42 | Cu:Zn 83:17 |
30 | 8 | 5 | 35 | — | — | — | — | — | |||
| 43 | Cu:Zn 50:50 |
23 | 10 | 17 | 28 | — | — | — | — | — | |||
| 44 | Cu:Zn 9:91 |
67 | 20 | 18 | 4 | — | — | — | — | — | |||
| 45 | Cu:Zn 0:100 |
70 | 25 | 28 | 1 | — | — | — | — | — | |||
| 46 | Co | 0.1 M KHCO3 | −1.1 V | 98 | 1 | — | — | 1 | — | — | — | — | 107 |
| 47 | Cu30Co70 | 93 | 2 | — | — | 6 | — | — | — | — | |||
| 48 | Cu50Co50 | 92 | 3 | — | — | 5 | — | — | — | — | |||
| 49 | Cu70Co30 | 92 | 4 | — | — | 5 | — | — | — | — | |||
| 50 | Cu90Co10 | 82 | 5 | — | — | 13 | — | — | — | — | |||
| 51 | Cu | 87 | 8 | — | — | 8 | — | — | — | — | |||
| 52 | Cu | 0.1 M KHCO3 | −1.0 | 67 | 7 | 6 | 7 | 11 | — | — | — | — | 109 |
| 53 | CuGa4 | 58 | 8 | 3 | 9 | 14 | — | — | — | — | |||
| 54 | CuGa17 | 80 | 6 | 2 | 1 | 3 | — | — | — | — | |||
| 55 | CuGa37 | 85 | 6 | 2 | 3 | — | — | — | — | ||||
| 56 | Cu wire | 0.1 M KHCO3 | −0.8 | — | 15 | — | 35 | — | — | — | 28 | — | 112 |
| 57 | Cu–Ag poly | 8 | — | 27 | — | — | — | 16 | — | ||||
| 58 | Cu–Ag wire | 7 | — | 55 | — | — | — | 26 | — | ||||
| 59 | Cu | 0.1 M KHCO3 | −0.8 | 58 | — | — | — | — | — | — | — | 113 | |
| 60 | Pd2@Cu98 | 48 | — | — | — | — | — | — | — | ||||
| 61 | Pd5@Cu95 | 30 | 58 | — | — | 7 | — | — | — | — | |||
| 62 | Pd10@Cu90 | 41 | — | — | 10 | — | — | — | — | ||||
| 63 | Pd20@Cu80 | 40 | — | — | 22 | — | — | — | — | ||||
| 64 | Pd | 9 | — | — | 38 | — | — | — | — | ||||
| 65 | Cu | 0.1 M KHCO3 | −1.35 | 52 | 1 | 40 | 3 | 2 | — | — | — | — | 114 |
| 66 | Cu90Zn10 | 32 | 2 | 62 | 1 | 2 | — | — | — | — | |||
| 67 | Cu70Zn30 | 25 | 4 | 70 | 2 | 3 | — | — | — | — | |||
| 68 | Cu50Zn50 | 50 | 5 | 68 | 2 | 4 | — | — | — | — | |||
| 69 | Cu30Zn70 | 51 | 38 | 9 | 1 | 5 | — | — | — | — | |||
| 70 | Cu10Zn90 | 52 | 42 | 2 | 1 | 1 | — | — | — | — | |||
| 71 | Zn10 | 53 | 43 | 1 | 1 | 1 | — | — | — | — | |||
| 72 | Ni–N–C | 0.5 M KHCO3 | −1.0 | — | 70 | — | — | — | — | — | — | — | 116 |
| 73 | Cu–Ni–C | — | 12 | — | — | — | — | — | — | — | |||
| 74 | Ni/Cu–N–C | — | 92 | — | — | — | — | — | — | — | |||
| 75 | Cu | 0.5 M KHCO3 | −100 | 11 | 68 | — | 7 | 9 | — | — | 3 | — | 9 |
| 76 | Sn | 31 | 34 | — | 30 | — | — | — | — | ||||
| 77 | Sb1–Cu-1.5 | 3 | 80 | — | 3 | 2 | — | — | 4 | — | |||
| 78 | Sb1–Cu-5 | 2 | 96 | — | — | 1 | — | — | — | — | |||
| 79 | AuCu-5 nm | 0.1 M KHCO3 | −1.2 | 83 | 13 | 3 | — | — | — | — | — | — | 115 |
| 80 | Cu100Bi | 50 mA cm−2 | 50 | 8 | — | — | — | — | — | — | — | 117 | |
| 81 | Cu50Bi | 70 | 14 | 3 | 2 | — | — | — | — | — | |||
| 82 | Cu10Bi | 20 | 44 | — | — | — | — | — | — | — | |||
| 83 | Cu5Bi | 19 | 31 | — | — | 42 | — | — | — | — | |||
Lai et al. have reported that the inclusion of the organic additive 1-Br2 can enhance CO2R activity and selectivity in Cu alloys containing Co, In, Mn, and Zn. This covered the way for the development of hybrid electrocatalysts with improved performance in CO2R to higher-order products. It was noteworthy that these compounds displayed strong correlations with CH4 and C2+, underscoring a fundamental limitation in selectivity tuning through alloying. The hybrid organic–inorganic catalysts demonstrated the potential to exert control over branching ratios within the CO2R reaction network.118 Further, a novel activation approach by Nafion on Cu(OH)2-derived Cu was applied for transforming this material into an effective e-CO2RR catalyst for flow cell applications (Fig. 10). Notably, under optimized conditions, a FEC2H4 of 44% and a total CO2 reduction FE of 83% were achieved at 300 mA cm−2, showcasing exceptional performance. This activation strategy showcased its efficacy by promoting C2H4 production and simultaneously mitigating the occurrence of hydrogen evolution reactions.119
 | ||
| Fig. 10 ANCu@Nafion catalysts with varying Nafion percentages were fabricated at 0.1 V, and their Nyquist plots from 0.01 Hz to 100 kHz (A) and their product distribution FEs at 300 mA cm−2 (B) were plotted. The CO2 reduction kinetics of ANCu@Nafion-28.4 wt% in a 1 M KOH electrolyte (C) along with the corresponding current density versus product distribution kinetics (D), voltage profile and C2H4 kinetics (E), and product distribution kinetics (F) over the course of an 8000-second test. Reproduced from ref. 119. Open access article, Wiley. | ||
Further, Chang et al. investigated the structural changes, CO2RR activity, and selectivity of the Ag@Cu core–shell system. Employing a straightforward polyol reduction strategy, researchers manipulated the compositions and structures of nanoparticles (NPs). The study underscored the significance of synergistic interactions between Ag and Cu, which led to elevated CO or hydrocarbon production within specific NP configurations, such as Ag@Cu-5 or Ag@Cu-20.120 Additionally, the synthesis of granular particles featuring a Cu-based nanostructure and approximately 10 nm nanopores was undertaken using a Na-melt technique (Fig. 11). The process involved the electrochemical extraction of calcium from Cu5Ca, facilitating the further evolution of these nanostructures. The application of a large negative voltage to the Cu5Ca particles resulted in the generation of platelet Cu particles with comparable nanopores.121
 | ||
| Fig. 11 Schematic representation of the synthesis of Cu5Ca and its derivatives (A). The P-XRD patterns for Cu5Ca (PDF 04-007-1585) and Cu (PDF 00-004-0836) synthesized at 400, 600, and 750 °C from a Na melt-synthesized sample with a nominal molar ratio of Cu/Ca = 3/1 (B). SEM micrographs of raw Cu powder (a), 400 (b), 600 (c), and 750 °C (d) Na-melt-synthesized samples (C). Current efficiencies for CO2R products at different potentials for (a) Cu5Ca and (b) Cu samples. Each applied potential lasted for 1 hour (D). Reproduced with permission from ref. 121. Copyright 2022, American Chemical Society. | ||
Remarkable findings emerged from the study of bimetallic Cu–Sn catalysts, unveiling spontaneous surface transformations driven by local galvanic corrosion during CO2RR. The distinct oxidation potential gap between Cu and Sn induced accelerated corrosion of the thin Sn layer on Cu foil, prompting the emergence of finer Sn patterns and outward Sn migration. This intriguing redox interplay fostered thinner Sn patterns and showcased the novel Cu/p-Sn 3-nm catalysts, boasting an exceptional catalytic selectivity of 58.1% at −1.0 VRHE.122 Further, in the pursuit of effective ethylene synthesis via CO2RR, a groundbreaking CeO2/CuO electrocatalyst was synthesized. This catalyst, with innovatively integrated CeO2 quantum dots, harnessed heterointerfaces to enhance CO2 and CO adsorption, while stabilizing Cu+ for CO2RR, remarkably surpassing both CuO and CuO–CeO2 hybrid counterparts.123
Moreover, the electrochemical process unraveled the potential of CuWO4 to shine as an exceptional electrocatalyst for CO2R, surpassing conventional CuO.124 Further, the synthesis of a new composite by combining Cu–BTC with GrO yielded a transformative breakthrough, showcasing augmented adsorption capabilities. With the infusion of GrO, the composite's BET surface area and pore volume were elevated compared to its Cu–BTC precursor. This composite displayed a significantly elevated capacity for CO2 adsorption, while the CH4 adsorption remained essentially unaltered.125 Embracing innovative methodologies, Huang et al. harnessed a unique seeded-growth colloidal technique to synthesize AgCu NDs, unraveling intricate interplays between varying Cu domain sizes. This approach, indicates dimer systems such as CuRu, CuRh, and CuIr, presented a gateway to synthetic control over Cu and Ag nanoparticle counterparts. Remarkably, Ag1Cu1.1 NDs exhibited a transformative 3.4-fold surge in FE toward C2H4, along with a twofold increase in total CO2RR activity compared to similar-sized Cu NPs.126 A meticulous approach involving the electrodeposition of Sn onto porous Cu hollow fibers illuminated pathways to enhancing current density and reaction selectivity in electrochemical CO2R. Skilled optimization of the Sn2+/Sn4+ ratio on the HFGDE surface yielded substantial enhancements in the FE of formate.127 Unveiling a pathway to dual-loop CO2 capture during gasification, Rahman et al. showcased the potential of pellets merging CuO and limestone. Intriguingly, core-in-shell Ca50Cu40Cem10 pellets exhibited rapid CuO reduction in the presence of CO and H2.128
Continuing this scientific journey, elevated-temperature pyrolysis in a reducing atmosphere was harnessed to successfully produce monometallic Cu/C, Ni/C, and intermetallic Cu0.85Ni0.15/C nanomaterials from phase-pure MOFs Cu–BTC, Ni–BTC, and Cu0.85Ni0.15–BTC. Structural analyses unveiled the creation of a substitutional solid solution, the Cu0.85Ni0.15/C nanoalloy, within these MOF-derived nanomaterials, showcasing a significant dispersion of active metal sites on the carbonaceous substrate. With an impressive selectivity for CH3OH and a high FE reaching up to 60% at 0.40 V, the intermetallic nanoalloy outperformed conventional monometallic catalysts in enhancing the efficiency of CO2RR.129 In a parallel exploration, Wannakao et al. expressed in the field of e-CO2R on metal/WC surfaces, employing density functional theory simulations to unravel activity and selectivity trends involving 13 different metal species. The findings revealed that metals such as Zn/WC, Ag/WC, and Au/WC, known for their limited interactions with reaction intermediates, produced CO as a significant product with a limiting potential similar to their pristine metal surfaces. These core–shell M/WC systems showcased potential to reduce the demand for costly metals in contemporary catalytic processes. Conversely, Cu/WC and Pd/WC surfaces exhibited a comparatively low capability for reducing CO2 to CO.130 In another facet of investigations, Zhang et al. explored the functions of Cu and Sn in selective e-CO2R by designing a series of Cu–Sn-based composites as model catalysts. By varying the ratio of copper to silver, e-CO2RR's products can be adjusted to produce either formate or carbon monoxide. Remarkably, both Cu1Sn1 and Cu20Sn1 emerged as excellent catalysts for formate and CO oxidation, exhibiting high selectivity and partial current density. While Cu20Sn1 featured a structure consisting of a single Sn atom doped into a Cu atom, characterization of Cu1Sn1 unveiled a CuSn alloy core with a minimal quantity of a Cu-doped SnO shell.131Table 4 provides a complete summary of different designed electrocatalysts for e-CO2RR following the compound alloying approach on Cu-based materials.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | Cu-Raw | 0.1 M KHCO3 | −1.0 | 48 | 7 | 12 | — | — | — | — | — | — | 132 |
| 2 | Cu-50 | 38 | 11 | 11 | — | — | — | — | — | — | |||
| 3 | Cu-100 | 33 | 10 | 8 | — | — | — | — | — | — | |||
| 4 | Cu-200 | 37 | 7 | 6 | — | — | — | — | — | — | |||
| 5 | Flat-Cu–Sn | 0.1 M KHCO3 | −1.0 | 75 | — | — | — | — | — | — | — | 100 | |
| 6 | Cu–Sn rod | 51 | — | — | — | — | — | — | — | ||||
| 7 | 3D-h Cu–Sn | 68 | — | — | — | — | — | — | — | ||||
| 8 | AN-Cu | 1 M KOH | −0.1 | 41 | 9 | 6 | 24 | 3 | — | — | 10 | 2 | 119 |
| 9 | AN-Cu@Nafion (0.07) | 35 | 8 | 1 | 26 | 2 | — | — | 17 | 4 | |||
| 10 | Ah NPs | 0.1 M KHCO3 | −1.16 | 29 | 63 | — | — | — | — | — | — | — | 120 |
| 11 | Ag@Cu-5 | 29 | 68 | — | — | — | — | — | — | — | |||
| 12 | Ag@Cu-7 | 29 | 70 | 2 | — | — | — | — | — | — | |||
| 13 | Ag@Cu-10 | 53 | 21 | 13 | 4 | — | — | — | — | — | |||
| 14 | Ag@Cu-15 | 53 | 10 | 20 | 13 | — | — | — | — | — | |||
| 15 | Ag@Cu-20 | 45 | 5 | 23 | 16 | — | — | — | — | — | |||
| 16 | Ag@Cu-25 | 44 | 5 | 21 | 17 | — | — | — | — | — | |||
| 17 | Cu NPs | 58 | 7 | 17 | 19 | — | — | — | — | — | |||
| 18 | Cu5Ca | 0.5 M KHCO3 | −2.5 Ag/AgCl | — | 3 | 1 | 24 | — | — | — | 11 | — | 121 |
| 19 | Cu samples | — | 3 | 11 | 8 | — | — | — | 3 | — | |||
| 20 | Cu | 0.1 M KHCO3 | −1.2 | — | 2 | — | — | 8 | — | — | — | — | 122 |
| 21 | Cu/p-Sn 3 nm | — | 52 | — | — | 12 | — | — | — | — | |||
| 22 | Cu/p-Sn 20 nm | — | 36 | — | — | 62 | — | — | — | — | |||
| 23 | Cu/p-Sn 30 nm | — | 3 | — | — | 98 | — | — | — | — | |||
| 24 | CeO2/CuO-50% | 1 M KOH | 250 mA cm−2 | — | — | — | — | 42 | — | — | — | — | 123 |
| 25 | CuO | — | — | — | — | 2 | — | — | — | — | |||
| 26 | CeO2/CuO-50%-PM | — | — | — | — | 1 | — | — | — | — | |||
| 27 | CeO2/CuO-50%-bulk | — | — | — | — | 14 | — | — | — | — | |||
| 28 | CeO2/CuO-20% | — | — | — | — | 32 | — | — | — | — | |||
| 29 | CeO2/CuO-37.5% | — | — | — | — | 38 | — | — | — | — | |||
| 30 | CeO2/CuO-50% | — | — | — | — | 42 | — | — | — | — | |||
| 31 | CeO2/CuO-70% | — | — | — | — | 28 | — | — | — | — | |||
| 32 | Ag NPs | 0.1 M KHCO3 | −1.1 | 45 | 68 | 1 | — | — | — | — | — | 126 | |
| 33 | Cu NPs | 52 | 13 | 8 | 12 | 12 | — | — | — | — | |||
| 34 | Ag1–Cu1.1 NDs | 34 | 18 | 19 | 41 | 19 | — | — | — | — | |||
| 35 | CuSn (50 s) HFGDEs | 0.5 M KHCO3 | −1.4 | — | — | — | — | 50 | — | — | — | — | 127 |
| 36 | CeO2 | 0.1 M KHCO3 | −1.4 | 78 | 3 | — | — | 2 | — | — | 2 | — | 133 |
| 37 | 5-CuO/CeO2 | 57 | 17 | 2 | 3 | 1 | — | 2 | 9 | — | |||
| 38 | 60-CuO/CeO2 | 45 | 2 | 27 | 55 | 1 | — | 2 | 3 | — | |||
| 39 | CuO | 39 | 2 | 10 | 21 | 5 | — | — | 16 | — | |||
| 40 | Cu/C | −1.0 | — | 10 | 10 | — | — | — | 13 | — | — | 129 | |
| 41 | Cu0.85Ni0.15/C | — | 3 | 4 | — | — | — | 2 | — | — | |||
| 42 | Ni/C | — | 16 | — | — | — | 2 | — | — | ||||
| 43 | Pure Sn | 0.5 M KHCO3 | −1.1 | 9 | 15 | — | — | 70 | — | — | — | — | 131 |
| 44 | Pure Cu | 41 | 41 | — | — | 16 | — | — | — | — | |||
| 45 | Cu1Sn1 | 4 | 9 | — | — | 87 | — | — | — | — | |||
| 46 | Cu20Sn1 | 5 | 78 | — | — | 25 | — | — | — | — | |||
6.5. Composite engineering approach
Engineering strategies enable designing electrocatalysts for e-CO2R providing adaptable solutions to improve catalytic performance. The composite engineering approach involves integrating materials or phases with functionalities into one catalyst system. By selecting and combining these components researchers can optimize the activity, selectivity and stability of catalysis effectively addressing the challenges associated with CO2R. The significance of composite engineering lies in its ability to harness the strength of various materials to create synergistic effects. For instance, a composite catalyst may include supports, active catalytic sites and promoters to enhance CO2 adsorption, electron transfer and intermediate binding. This tailored approach enables adjustment of catalyst properties to meet requirements, for reducing CO2R. Different methods such as co-precipitation, sol–gel synthesis and chemical vapor deposition can be employed to produce composite catalysts with controlled structures. Overall composite engineering plays a role in advancing electrocatalysis by providing pathways for developing sustainable technologies for converting CO2 that are essential, in combating climate change and transitioning towards a greener energy future.The research group of Cao and co have utilized microporous nitrogen-rich MOFs known as Cu-BTT to serve as precursors for the synthesis of Fe–Cu-BTT. Through a meticulously controlled pyrolysis process, this endeavor yielded FexCu–N–C compounds featuring a diverse array of Fe–Nx sites. The introduction of Fe induced notable structural alterations, including an increase in the BET surface area and total pore volume, a reduction in Cu nanoparticle density, and an augmentation of Fe–Nx sites within the carbon matrix. This structural evolution resulted in an enhanced CO generation, with Fe0.07Cu–N–C800 exhibiting the highest CO2 transfer efficiency at 48.5%.134 In the context of e-CO2RR conducted in neutral aqueous solutions, a novel organic–inorganic composite was crafted by Li et al. via a hydrothermal synthesis method. Remarkably, this composite demonstrated sustained performance without degradation even after 24 hours of continuous e-CO2R. It comprised a 3D g-C3N4 nanosheet-CNT interconnected framework adorned with a polymeric CoPc layer. This reported innovative method offered several advantages, including an increased electrochemically active surface area with numerous active sites, improved homogeneous immobilization at high catalyst loading, and reinforced contacts between the molecular catalyst and the conductive support.135 Furthermore, CoPc-based and O-linked MOFs were predicted to exhibit reduced activation energies in the synthesis of the carboxyl intermediate, as evidenced by density functional theory calculations. This reduction in activation energy translated into improved catalytic activity and selectivity. These findings underscored the potential for fine-tuning MOFs' activity and selectivity in catalysis through the meticulous design and selection of functional ligands, offering a significant degree of modular tunability. Notably, for the e-CO2R to CO using reticular framework materials, it was demonstrated that these materials served as exceptional cathode materials, boasting high current density and excellent selectivity toward the CO product at a relatively low overpotential of 0.63 V.136
To optimize support connections and catalytic efficiency, He et al. developed a dimer catalyst, a minimal bimetallic catalyst structure. This innovation opened up the possibility of breaking scaling relationships by expanding the parameter space for optimizing attributes while retaining non-monotonic property correlations. The dimers, formed from Group 10 (Ni, Pd, Pt) and Group 11 (Cu, Ag, Au) elements, were supported at single vacancy sites of graphene. These dimers featured an anchoring atom that transferred a large number of electrons to the graphene and an antenna atom that was much less positively charged and protruded from the graphene surface. All clusters exhibited strong binding energies to the defective graphene, indicating their stability. Notably, the stability of the alloy dimers, such as Cu2, Ag2, and Au2, was significantly improved compared to their monometallic forms. These dimers were selected for further investigation as potential candidates for the e-CO2R to CH4 using the first-principles approach and the computational hydrogen electrode model. Among the options studied, Pt2, AgNi, Pd2, and AgPt showed the lowest overpotential values when compared to standard Cu electrodes. While the oxygen binding strength of the catalytic dimer provided a semi-quantitative description of the reactivity of the supported dimers, the free energies of OCHO* and OH* correlated well with O*, whereas the free energies of CHO* and HCOOH* exhibited significant variations in their reactivity.137
Further, for the e-CO2R to carbon, an inexpensive and robust Cu-based hybrid catalyst was created. The catalyst, a polymer of Cu phthalocyanine coated on carbon nanotubes, showed 80% faradaic yield for CO synthesis at low overpotentials and was selective for CO production. Since Cu phthalocyanine polymerization was selective for proton reduction under the same conditions, this resulted in a marked increase in reaction selectivity. Although the material initially displayed isolated Cu sites in phthalocyanine-like CuN4 coordination, the Cu atoms were transformed to Cu nanoparticles under operational conditions.138 In addition, the research findings showed that CoP2O6 played a crucial role in enhancing the efficiency of the Cu/C catalyst for formate production through e-CO2R (Fig. 12). Utilizing ZIF-67 as the carbon precursor, the resulting CoP2O6/HCS–Cu catalyst not only increased current density but also reduced overpotential. Importantly, the catalyst's efficiency in e-CO2R was found to be modulated by the Cu/Co ratio. Impressively, this catalyst exhibited the most negative overpotential (−0.4 mV) and achieved the highest current density (34.5 mA cm−2) for formate production during e-CO2R. Furthermore, at −0.66 V vs. RHE, formate was generated with an exceptional FE of 89.9%.139
 | ||
| Fig. 12 The LSV curves were generated through experiments conducted in a CO2-saturated 0.5 M KHCO3 electrolyte using a scan rate of 10 mV s−1 (a). The formate, CO, and H2 FEs were determined at a specific potential for the CoP2O6/HCS and CoP2O6/HCS–Cu catalysts (b)–(e). The current density (f), Tafel slopes (g), and electrochemically active surface area were determined for the CoP2O6/HCS and CoP2O6/HCS–Cu catalysts. The detection of products resulting from e-CO2R on the CoP2O6/HCS and CoP2O6/HCS–Cu20 catalysts was carried out in situ (i). This was achieved by utilizing a Pt UME tip CV response, with a scan rate of 10 mV s−1, at a substrate potential of −1.1 V vs. Ag/AgCl. The electrolyte employed was a 0.5 M solution of KHCO3 that was saturated with CO2 under atmospheric pressure. The experiments were carried out at a temperature of 293 K. Reproduced with permission from ref. 139. Copyright 2023, American Chemical Society. | ||
In a separate study, solvothermal processing was employed to vulcanize copper hydroxide nanowire arrays (S–CNWs) with a specific amount of cuprous sulfide. These S–CNWs were subsequently investigated for their e-CO2R under neutral conditions. The research revealed a strong correlation between the degree of vulcanization of the catalyst and formate selectivity. When vulcanized to an optimal extent, the FE could exceed 60%, with almost no generation of additional e-CO2RR products. Further, the introduction of a small quantity of sulfur was found to facilitate the formation of formate by lowering the energy barrier associated with generating the OCHO intermediate.140 In a related endeavor, hydrothermal processing followed by pyrolysis was employed to synthesize various Cu–Ni alloys tailored for e-CO2R. In a 0.5 M KHCO3 electrolyte, the optimized CuNi/NCNT catalyst exhibited a remarkable FECO of 94.8% for CO2R and a substantial current density of 26.6 mA cm−2. The addition of nickel to copper facilitated the formation of a crucial intermediate, COOH*, which played a pivotal role in boosting e-CO2RR activity. Promisingly, the current density further increased to 124.6 mA cm−2 with an FECO of 92.8% at 1.1 V versus RHE.141
Additionally, Daiyan et al. created metal–carbon composites with dual active sites to produce H2 and CO during e-CO2RR. Co@CoNC-900 composites exhibited a wide potential window within which they could produce a tunable syngas ratio of 1:1. By adjusting the HER active sites, this syngas ratio could be shifted from 1:1 to 3:2, unlocking new possibilities for the production of a diverse range of fuels and chemicals using the well-established FT process in the industry.142 In a separate research effort, the impact of Nafion/solvent formulation on CO2 electrolysis employing CuO as a model catalyst was examined. The composition of Nafion/solvent exerted a significant influence on the activity, selectivity, and stability of e-CO2RR. The effects of Nafion/solvent formulation on CO2 electrolysis were attributed to three primary factors: the evolution of the catalyst structure, alteration of the local microenvironment, and accumulation of surface adsorbates. Nafion played a stabilizing role in preserving product distributions by preventing morphological rearrangement and delamination of weakly bound catalysts. The surface coverage of CO and local concentrations of CO2 and H2O in the reaction microenvironment were influenced by adjusting the thickness of Nafion coatings and the connectivity of their inner networks via modifications to the catalyst ink formulations.143 Furthermore, researchers produced catalysts derived from CuO for e-CO2RR, which exhibited resistance to a single thermal treatment but could be removed in two steps. g-C3N4-supported CuO catalysts favored the production of CH4, whereas CuO displayed the highest selectivity toward C2H4. Various factors, including particle growth, the ensemble effect due to varying catalyst loading densities, and hybridization and stabilization of the catalyst–support relationship, contributed to this disparity. Scientists aimed to design catalyst systems that would result in a more tightly regulated product distribution of hydrocarbons over H2 or CO by carefully controlling experimental conditions.144
Another research study reports on the reaction of the metal precursor with the nitrogen-doped carbon support to generate (sub)nanosized metal oxide clusters under ex situ conditions. The study encompassed a variety of M/NC catalysts. The results indicated that the catalytic activity for CO formation increased while using transition metal based electrocatalysts. Co/NC exhibited the highest activity at 0.5 V vs. RHE, while Ni/NC demonstrated good activity and selectivity. Fe/NC, Co/NC, and Ni/NC outperformed other metal hydroxides due to phase contraction and HCO3 insertion into the layered structure of metal hydroxides.145 Furthermore, nanophase-separated Cu#ZrO2 was successfully produced through the internal oxidation of the Cu51Zr14 precursor composite alloy (Fig. 13). The material featured nanoscale lamellae of Cu metal and tetragonal ZrO2, creating a stable Cu–ZrO2 interface. Its uniformly dispersed catalytic sites and abundant metal–oxide interfaces contributed to increased selectivity for e-CO2RR-to-FA conversion. CO2 adsorption led to protonation over the nearby Cu sites, generating FA through the formation of O(CH)O.146
 | ||
| Fig. 13 The p-XRD patterns of the Cu51Zr14 composite alloy (A) and Cu#ZrO2 (B) were analyzed. The HAXPES profiles of the Cu51Zr14 precursor alloy (black) and Cu#ZrO2 (red) were analyzed in the Zr 3d region (C) and the Cu 2p region (D). The product distribution of e-CO2RR over Cu#ZrO2 was quantified after 3 hours of electrolysis at a potential of −0.9 V vs. RHE (E). The e-CO2RR performance was evaluated for different catalysts, including a mixture of Cu and YSZ (F), Cu#ZrO2 (G), commercial Cu (H), and Cu2O (I). Reproduced from ref. 146. Open access article, American Chemical Society. | ||
Furthermore, another research study explored the use of a triphenylene-core porous structure as a means to modify the performance of Cu atomic catalysts in e-CO2RR. The triangular pores within this structure served as anchoring sites for Cu atoms, forming complexes that facilitated the hydrogenation of CO2, a rate-limiting step in e-CO2RR. Functionalization of these structures with oxygen and nitrogen resulted in reduced overpotentials for e-CO2RR on Cu@TPGDY, which decreased from 0.26 V to 0.16 V and 0.08 V, respectively.147 In another facet of e-CO2RR research, scientists devised a novel catalyst model using a Cu nanosheet coated with a thin layer of carbon. This carbon coating acted as a barrier, inhibiting surface reconstruction at high reducing potentials and preventing the surface oxidation of metallic Cu. Operating at 1.0 V and a current density of 20.3 mA cm−2, the C/Cu/C nanosheet exhibited remarkable stability and a high FE of 47.8% for the production of CH4. The carbon layers not only enhanced the catalyst's electrical conductivity but also facilitated streamlined electron transport.148 Furthermore, another research study showcased the synthesis and application of ultrasmall Cu nanocrystals uniformly distributed in nitrogen-doped carbon as catalysts for e-CO2RR to produce ethanol and formate. These compounds' selective production necessitated specific potentials. At a low potential, the FE for ethanol reached 43.7%, while that for formate was 63.5%. The electronic environment of Cu was influenced by the nitrogen-doped carbon, facilitating proton–electron transport.149 The overview of the composite engineering strategy utilized to create a number of electrocatalysts for e-CO2RR to different sustainable products is given in Table 5.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | Cu–N–C800 | 0.1 M KHCO3 | −1.2 Ag/AgCl | 58 | 11 | — | — | 22 | — | — | — | — | 134 |
| 2 | Fe0.01Cu–N–C800 | 49 | 27 | — | — | 20 | — | — | — | — | |||
| 3 | Fe0.07Cu–N–C800 | 44 | 48 | — | — | 4 | — | — | — | — | |||
| 4 | Fe0.13Cu–N–C800 | 41 | 46 | — | — | 5 | — | — | — | — | |||
| 5 | CoPPc@g-C3N4–CNTs | 0.5 M KHCO3 | −0.55 | 35 | 43 | — | — | — | — | — | — | — | 135 |
| 6 | CoPc–Cu–NH | KHCO3 | −0.95 | 41 | 59 | — | — | — | — | — | — | — | 136 |
| 7 | CoPc–Cu–O | 42 | 59 | — | — | — | — | — | — | — | |||
| 8 | NiPc–Cu–NH | 62 | 37 | — | — | — | — | — | — | — | |||
| 9 | NiPc–Cu–O | 50 | 48 | — | — | — | — | — | — | — | |||
| 10 | CuPolyPc@CNT | 0.1 M CsHCO3 | −1.1 | 42 | 53 | — | — | — | — | — | — | — | 138 |
| 11 | CoP2O6/HCS | 0.5 M KHCO3 | −0.76 | 68 | 32 | — | — | — | — | — | — | — | 139 |
| 12 | CoP2O6/HCS–Cu10 | 41 | 28 | — | — | 31 | — | — | — | — | |||
| 13 | CoP2O6/HCS–Cu20 | 25 | 9 | — | — | 70 | — | — | — | — | |||
| 14 | CoP2O6/HCS–Cu40 | 11 | 18 | — | — | 73 | — | — | — | — | |||
| 15 | CF | KHCO3 | −1.1 | 62 | 1 | — | — | 3 | — | — | 2 | 140 | |
| 16 | CNWs | 62 | — | 1 | 22 | — | — | 3 | |||||
| 17 | S–CNWs-2 | 23 | 1 | — | — | 60 | — | — | — | — | |||
| 18 | Cu2.4Ni/NCNT | 0.5 M KHCO3 | −0.9 V | 8 | 92 | — | — | — | — | — | — | — | 141 |
| 19 | CuNi/NCNT | 5 | 95 | — | — | — | — | — | — | — | |||
| 20 | Cu0.36Ni/NCNT | 25 | 75 | — | — | — | — | — | — | — | |||
| 21 | Cu/NCNT | 16 | 84 | — | — | — | — | — | — | — | |||
| 22 | Ni/NCNT | 16 | 84 | — | — | — | — | — | — | — | |||
| 23 | NCNT | 6 | 92 | — | — | — | — | — | — | — | |||
| 24 | Co–N–C | 0.1 M KHCO3 | −0.8 | 76 | 19 | — | — | — | — | — | — | — | 142 |
| 25 | Co@ CoNC-800 | 61 | 38 | — | — | — | — | — | — | — | |||
| 26 | Co@ CoNC-900 | 48 | 43 | — | — | — | — | — | — | — | |||
| 27 | Co@ CoNC-1000 | 38 | 59 | — | — | — | — | — | — | — | |||
| 28 | CuO/Nafion/solvent formulations (0.5:75 vol) |
0.1 M K2SO4 | −50 mA cm−2 | 28 | 10 | 1 | 39 | 7 | — | — | 6 | — | 143 |
| 29 | g-C3N4 | −1.2 | 92 | — | — | — | — | — | — | — | — | 144 | |
| 30 | as-syn CuO | 65 | 4 | 6 | 11 | — | — | — | — | — | |||
| 31 | as-syn CuO/C3N4 | 78 | 4 | 6 | 2 | — | — | — | — | — | |||
| 32 | as-syn Cu/C3N | 77 | 5 | 11 | 2 | — | — | — | — | — | |||
| 33 | Mn/N–C | 1 M KHCO3 | 20 mA cm−2 | 82 | 18 | — | — | — | — | — | — | — | 145 |
| 34 | Fe/N–C | 79 | 19 | — | — | — | — | — | — | — | |||
| 35 | Co/N–C | 70 | 25 | — | — | — | — | — | — | — | |||
| 36 | Ni/N–C | 90 | 10 | — | — | — | — | — | — | — | |||
| 37 | Cu/N–C | 79 | 26 | — | — | — | — | — | — | — | |||
| 38 | Zn/N–C | 71 | 19 | — | — | — | — | — | — | — | |||
| 39 | Cu + YSZ | 0.1 M KHCO3 | −0.9 Ag/AgCl | 21 | 2 | — | — | 42 | — | — | — | — | 146 |
| 40 | Cu#ZrO2 | 19 | 8 | — | — | 82 | — | — | — | — | |||
| 41 | Commercial Cu | 55 | 5 | — | — | — | — | — | — | — | |||
| 42 | Cu2O | 43 | 1 | — | — | 8 | — | — | — | — | |||
| 43 | CuNCs@p-Cu/g-Cu | 0.1 M KHCO3 | −1.15 | 45 | 1 | 9 | 41 | 5 | — | — | — | — | 48 |
| 44 | Cu nanosheet | 0.5 M KHCO3 | −1.0 | — | — | 27 | 24 | — | — | — | — | — | 148 |
| 45 | C/Cu/C nanosheet | — | — | 48 | 10 | — | — | — | — | — | |||
| 46 | Cu/NC–NSs | KOH | −0.37 | — | — | — | — | 3 | — | — | 45 | — | 149 |
6.6. Surface engineering approach
Surface engineering is a multifaceted strategy employed in the design of electrocatalysts for e-CO2R. Its importance stems from the critical role of catalyst surfaces in dictating reaction pathways and kinetics during electrochemical CO2 conversion. Surface engineering encompasses various techniques aimed at tailoring the catalyst's surface properties to enhance its activity, selectivity, and stability. Among these techniques are surface anchoring, molecular surface functionalization, interfacial engineering and speciation of Cu surface, which involve the precise attachment of catalytic sites, often metal nanoparticles, onto a support material.A comparison of CO2RR performance of blank Cu, Cu modified with PPT (Cu-PPT), and Cu modified with QAPPT (Cu-QAPPT) revealed that attaching Pip+ cations onto the Cu electrode reduced competing HER and enhanced CO2RR selectivity. This enhancement was attributed to reduced interfacial water concentration, an increase in “ice-like” water, and the presence of *COatop, *CObridge, and CO–CO dimer species at the Cu/QAPPT interface.150 Additionally, Ye et al. successfully engineered a bi-phasic p-Cu@Cu2O nanosphere, comprising a Cu nanocrystal core and a Cu2O nanoprism shell. These nanospheres exhibited exceptional e-CO2R, with a maximum FEC2H4 of 79% at −1.18 V vs. RHE. Phase engineering facilitated synergistic control of the CO2 reactant and *CO intermediate adsorption behaviors, resulting in improved CO–CO coupling and increased production of C2+ products. During e-CO2R, CO2 molecules were absorbed by the faceted Cu2O shell, where they underwent reduction to *CO intermediates capable of escaping.151
In another study, Cu dendrites were coated with PTFE at varying concentrations to create a hydrophobic structure with a stable WCC wetting state. This hydrophobic coating enabled the catalyst surface to efficiently take in gas, increasing the number of triphase contacts for gas diffusion. Consequently, the reaction of *CO was enhanced, with an increase in CO2 and intermediate *CO on the catalyst surface. Hydrophobic Cu dendrites exhibited a twofold increase in the intrinsic electrochemical activity of their active sites compared to untreated Cu dendrites, leading to more efficient conversion of CO2 into liquid fuel products such as CH3OH and C2H5OH.152 Xie et al. also unveiled that increased activity and selectivity for C2H4 generation were achieved by creating C–Cu nanosheets through the in situ reduction of C–CuO nanosheets during e-CO2RR (Fig. 14). In a gas diffusion flow cell, FEC2H4 reached 56.2%, and the current density reached 171.0 mA cm−2. Both electrochemical measurements and XAS spectra confirmed the presence of uncoordinated edge sites in the nanosheets. C–Cu nanosheets displayed exceptional activity and selectivity for e-CO2RR to C2H4. They disclosed that Cu atoms with a coordination number of around 5 played a crucial role in assisting the development of a *CHO intermediate, thereby enhancing the coupling reaction of *CO and *CHO intermediates.153
 | ||
| Fig. 14 Gas diffusion flow cell with a 1.0 M KOH electrolyte with FEs of (a) C2H4 (a) and (b) H2, CO, C6H5OH, and formate on C–Cu nanosheets. In a gas diffusion flow cell with an Ar-saturated 1.0 M KOH electrolyte, the corresponding total and ethylene currents (Jtotal and Jethylene) are measured at varying potentials and the differences between them are iR compensated (c). When utilizing a gas diffusion flow cell and an electrolyte of 1.0 M KOH, a summary of the performance of Cu-based electrocatalysts for the formation of C2H4 is provided (d). ATR-FTIR spectra of C–Cu nanosheets (e) and Cu powder (f) taken in situ during e-CO2RR at varying potentials in a CO2-saturated 0.1 M KHCO3 electrolyte. Reproduced with permission from ref. 153. Copyright 2023, Elsevier. | ||
In a separate development, researchers devised a method to modify Cu2O nanocubes to encapsulate carbon dioxide and reduce its concentration. This molecular-scale wrapping was made possible by the CuAAC layer development, transitioning from surface-confined to random as a function of temperature. As a result of this modification, the increased hydrophobicity of the nanocubes dampened hydrogen evolution, making self-controlled modification the optimal condition for CO2 reduction electrocatalysis.154 In a different study, Cu nanocubes had been synthesized with rGO modifications through a straightforward electrochemical method. This modification had shifted the post-C–C coupling selectivity from ethylene to ethanol, enhancing local CO concentration on Cu sites. The tandem electrocatalysis approach had led to reduced formate synthesis, increased selectivity, and intrinsic activity towards C2+ products during e-CO2R. Notably, at 0.9 V, the Cu NCbs rGO-60 catalyst had exhibited the highest C–C bond coupling efficiency, favoring ethanol with a 76.84% FE. NCbs rGO-120 and NCbs rGO-60 catalysts had achieved selectivity ratios of approximately 107 and 85.34 for C2+ oxygenates/C2+ hydrocarbon (ethylene) products, respectively.155
In an overarching overview, the utilization of organic chemicals to modify the Cu surface for e-CO2 conversion had been explored. Catalytic activity had been found to be heavily influenced by the local microenvironment of the electrode surface, encompassing factors like hydrophobicity, electric field, pH, and intermediate coverage. The tunability and designability of organic compounds had offered numerous avenues for enhancing the catalytic activity of Cu-based catalysts. However, challenges remained, such as improving the conductivity and stability of organic compounds. The enhancement of catalytic activity of organic/inorganic hybrid catalysts would benefit from the design and synthesis of well-defined organic molecules or polymers with excellent conductivity and durability. Machine learning with artificial intelligence has the potential to expedite catalyst manufacturing by predicting crucial reaction parameters, ultimately leading to Cu-based catalysts with high activity, selectivity, and stability for CO2RR.156 In the meantime, the discussion also underscored the need to move beyond single-crystal studies and design Cu nanocrystals (NCs) in order to better understand the structural dependence of CO2RR. The view of NCs is a necessary tool, elucidating the reaction pathway and identifying size-dependent selectivity trends for C2 products. Most studies have focused on shapes exposing high-index facets. Therefore, it is extremely important to enrich the family of Cu NC shapes. The steps for rebuilding the catalyst, primarily driven by a dissolution–redeposition cycle, can be better understood by using NCs to identify the transient species involved.157
In another study, rough-edged Ag NPs/CuO MNSs were synthesized on a Cu foil for e-CO2R to CO. The heterostructure exhibited high electrical conductivity, abundant active sites, and a superior electronic structure, leading to enhanced CO2 adsorption and activation. The critical COOH intermediate in the pathway from e-CO2R to CO was stabilized at the Ag–CuO contact. The best Ag1.01%/CuO catalyst achieved a maximum FECO of 91.2% at a partial current density of 10.5 mA cm−2 and 0.7 V in 0.1 M KHCO3. The catalyst demonstrated good catalytic stability, maintaining a FECO of over 80% in the potential region.158 Additionally, a novel bimetallic InSn catalyst was developed using a potential-driven volume diffusion approach at room temperature (Fig. 15). This catalyst effectively increased current density by selectively reducing CO2 to formate. The catalyst featured an InSn hybrid core, a nanoshell structure with multiple interfaces, a multicomponent core–shell heterostructure, and a novel configuration that potentially accelerated CO2 and HCO3 equilibrium on the InSn interface.159
 | ||
| Fig. 15 CO2RR effectiveness. Current density (top) and FE of CO, H2, and formate on In40Sn40 at different applied potentials (a). Formate production FE values and partial current densities for In, Sn, and InSn catalysts vs. various In concentrations at 0.97 V vs. RHE (b). Each catalyst's FEformate at varying potentials (c). Partial current densities of each catalyst, expressed in a formate formation (d). EIS plots of all the produced samples were analyzed (e). Electrolysis of In40Sn40 for 100 hours at 0.97 V vs. RHE (f). Comparison with literature in terms of formate current density and FEformate with 90% iR-correction (g). Standard deviations from at least three separate measurements of the sample are used to create the error bars. Reproduced with permission from ref. 159. Copyright 2023, American Chemical Society. | ||
In addition, the effectiveness of GDEs loaded with copper nanocubes for e-CO2R in a zero-gap, MEA-type electrolyzer was investigated. These GDEs produced ethylene (jC2H4 up to about 80 mA cm−2) and propylene, a valuable industrial product, at higher rates compared to nanocube and sputtered Cu catalysts. Although flooding occurred in nanocube GDEs at current densities exceeding 150 mA cm−2, they maintained high ethylene production rates even during flooded operation. Researchers are currently focusing on optimizing mass loading and developing mitigation methods to prevent the loss of ethylene selectivity in Cu nanocube-loaded electrodes due to flooding.160 In a separate study, electrocatalytic TDPA-capped CuAg NCs were synthesized using a novel approach that involved investigating near-stoichiometric ligand-to-copper (P/Cu) ratios and utilizing Cu(II) acetate monohydrate. Depending on the diameter of the Cu NC precursor, one or two galvanic replacement steps were employed to form sub-10 nm Cu@Ag core–shells and Cu/Ag nanodimers, as confirmed through HRTEM and spectroscopy. The relationship between composition and selectivity was explored in the range of 14 to 41% Ag by spreading the nanocrystals on carbon black at high packing densities. The CO2RR selectivity transitioned from ethylene to ethanol production between CuNCs/C and CuAg/C-based electrodes, resulting in a 2- to 3-fold increase in the oxygenate-to-hydrocarbon ratio at moderate Ag atom %age levels. Cu/Ag0.14/C nanodimers and Cu@Ag0.17/C core–shells achieved a total C2 and C3 selectivity of 50% at neutral bulk pH and 0.1 A cm−2, suggesting a substantial impact from local CO concentration and CO-spillover on catalytic patterns at high CO2RR rates.161
Moreover, another research endeavor proposed that the e-CO2R to ethylene can be significantly improved by utilizing an ionic liquid (IL) in combination with a Cu electrocatalyst. The IL was demonstrated to alter the electronic structure of the Cu catalyst, facilitating *CO dimerization and enhancing ethylene generation. This method achieved a remarkably high 77.3% FEethylene in a water-based electrolyte and a standard H-type cell.162 Additionally, researchers have coated a layer of PTFE that conforms itself to a Cu nanoneedle (NN). By increasing the amount of PTFE at the end of a nanoneedle, the electric and thermal field strength rapidly varies. This in turn accelerates the C–C coupling of C2 and CO intermediate production. This method also enabled C2 generation at a TOF of 11.5 ± 0.3 s−1 Cu_site−1 and a FEC2 of 85.4 ± 1.5% under partial currents exceeding 250 mA cm−2.163 The effect of plasma pretreatment of the Cu surface on the overall surface was unusual. This, in turn, caused huge differences in the way the CO2RR products distributed. The observed differences in CO2RR activity seen across the series are related to changes in the electrochemically active surface area. The roughened surface has a stronger inclination to CO formed through e-CO2R than does the electrochemically polished surface. This phenomenon implies that as the size of the surface increased, the percentage of the adsorbed H to CO showed a decreasing trend. As surface roughness increased, the fractions of products containing two or more carbons also changes. The ratio of current densities for C2+ to C1 products displayed an increasing trend with increasing surface roughness, mainly due to the production of CH4 increasing on substrates with roughness levels above 3.164
Meanwhile, during the course of e-CO2R, the selectivity to C2H4 was greatly improved through the formation of the Cu nanoparticle substructure from spheres to cubes. The development was initiated by a weak adhesion of Cu NPs to the graphene oxide substrate. However, with time these Cu nanocubes turned into smaller, less organized nanoparticles, thus reducing the tendency to select for C2H4. To meet this challenge, an innovative approach appeared in which 2D- or “flat”-graphene with nitrogen doping served as an anchoring framework for Cu nanocubes. This relied on the strong interactions between Cu NPs and the pyridinic nitrogen atoms that covered the surface of 2D graphene.165 Simultaneously, Cu2O nanostructures with precisely controlled crystal facets were synthesized through a hydrothermal methodology. The catalytic ability and alcohol selectivity of these catalysts were responsible to the exposed crystal facets, ultimately dictating their performance in e-CO2R. The electrochemical evaluation of the Cu2O-o catalyst, decorated with (111) crystal facets, revealed an impressive high partial current density and 35.4% selectivity for alcohol generation at 0.3 V vs. RHE.166 Transitioning to the TB catalyst, the most elongated among the examined 1D active sites, it was synthesized with density adjustments ranging from 0 to 105 cm−1. Research findings illuminated TB's remarkable electrocatalytic ability, characterized by an exceedingly high local jCH4 of 1294 mA cm−2, accompanied by inherent CH4 selectivity fixed at 92%. Computational simulations, in alignment with DFT, affirmed that TB atoms outpaced flat atoms by a staggering factor of 104 in protonating CO*.167
In a comprehensive comparative analysis deploying gas-diffusion electrodes and flowing alkaline catholytes, researchers meticulously scrutinized the electrocatalytic performance of Cu nanocubes and nanospheres in CO2R. While an uptick in catalytic efficiency ensued with the use of a more concentrated alkaline catholyte, it became evidently clear that Cu nanocubes surpassed their nanospheric counterparts when catalyzing CO2R toward ethylene.168 Furthermore, the research unveiled that liquid C2+ products could be produced at a significantly accelerated rate due to the synergistic interplay of Cu and Pb within Cu/Pb core/shell nanocrystals. The partial current density for CO2RR in a flow cell system over CuPb-0.7/C reached an impressive 294.4 mA cm−2, and the total free energy associated with C2+ products stood at 73.5 kJ mol−1. Notably, in terms of generating C2+ liquid products, the total free energy achieved an outstanding 49.5%, accompanied by a partial current density of 196.8 mA cm−2. These results surpassed the performance of most reported Cu-based catalysts for CO2RR. When CuPb-0.7/C was used as a catalyst for CO2 reduction, the cooperation between Cu and Pb made it easier to pass through the intermediate product of COOH and then into C2+ products through the production of *OCCOH.169
In the search for affordable and environmentally friendly e-CO2R catalysts, research teams have developed a new bimetallic alloy/oxide nanowire catalyst with a core–shell structure. The core of this catalyst boasts high electrical conductivity, while the catalytic activity and selectivity are guaranteed by the amorphous Cu-doped SnO2 shell. Computational analyses underscored the pivotal role of the Cu-doped SnO2 layer in curbing the electrocatalytic generation of formate and hydrogen. Remarkably, when operating at 0.9 V versus RHE, this electrocatalyst achieved an impressive FE of 78% for formate production at a partial current density of 30 mA cm−2.170 Another noteworthy development in the field of CO2R was the monodisperse core/shell Cu/In2O3 nanoparticles, resulting in the effective production of syngas. The H2/CO ratios in syngas could be fine-tuned over a broad range, with a FE for syngas generation exceeding 90% through adjustments in the thickness of the In2O3 shell and applied voltage. Compared to other reported electrocatalysts for syngas production, the C–Cu/In2O3-0.4 catalyst emerged as the top performer, delivering high current densities at remarkably low overpotentials. The incorporation of compression strain along with Cu doping in the In2O3 shell was identified as a key factor responsible for elevating CO production activity and enhancing the H2/CO ratio.171Table 6 presents the properties of different electrocatalysts for e-CO2R to sustainable products designed by following the surface anchoring strategy.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | Blank Cu | 0.1 M KHCO3 | −1.0 | 1 | 83 | — | — | — | — | — | — | — | 150 |
| 2 | Cu-PPT | 1 | 87 | — | — | — | — | — | — | — | |||
| 3 | Cu-QAPPT | 42 | 16 | — | — | 20 | — | — | — | — | |||
| 4 | p-Cu@Cu2O | 0.1 M KHCO3 | −0.98 | 18 | 18 | 1 | 35 | 3 | — | — | 13 | 151 | |
| 5 | i-Cu@Cu2O | — | — | — | 4 | — | — | — | — | — | |||
| 6 | r-Cu@Cu2O | — | — | — | 2 | — | — | — | — | — | |||
| 7 | Untreated Cu dendrite | 0.1 M KHCO3 | −1.0 | — | — | — | — | 3.5 | — | 3 | 2 | — | 152 |
| 8 | Hydrophobic Cu dendrite | — | — | — | — | — | — | 4.5 | 2.8 | — | |||
| 9 | C–Cu nanosheets | 0.1 M KHCO3 | −1.0 | 25 | 22 | 1 | 31 | — | — | — | — | — | 153 |
| 10 | Cu powder | 62 | 2 | 1 | 10 | — | — | — | — | — | |||
| 11 | @3AZ rt-1h | 0.1 M KHCO3 | −1.4 | 65 | 1 | 28 | 2 | — | — | — | — | — | 154 |
| 12 | @3AZ rt-20h | 37 | 1 | 43 | 2 | — | — | — | — | — | |||
| 13 | @3AZ 80 °C 1 h | 37 | 1 | 40 | 4 | — | — | — | — | — | |||
| 14 | @3AZ 80 °C 20 h | 33 | 1 | 43 | 1 | — | — | — | — | — | |||
| 15 | @2AZ r-t 20 h | 41 | 1 | 35 | 2 | — | — | — | — | — | |||
| 16 | Cu NCbs | 0.1 M KHCO3 | −1.1 | — | — | 20 | 33 | — | — | 13 | 34 | — | 155 |
| 17 | Cu NCbs-rGO/60 | — | — | 10 | 32 | 4 | — | 2 | 35 | — | |||
| 18 | Cu NCbs-rGO/120 | — | — | 18 | 55 | 1 | — | — | 45 | — | |||
| 19 | Ag1.55%/CuO | 0.1 M KHCO3 | −0.4 | 59 | 40 | — | — | — | — | — | — | — | 158 |
| 20 | Ago.68%/CuO | 32 | 63 | — | — | — | — | — | — | — | |||
| 21 | Ag1.01%/CuO | 41 | 56 | — | — | — | — | — | — | — | |||
| 22 | CuO | 40 | 53 | — | — | — | — | — | — | — | |||
| 23 | Sn | 0.5 M KHCO3 | −1.1 | — | — | — | — | 65 | — | — | — | — | 159 |
| 24 | In | — | — | — | — | 44 | — | — | — | — | |||
| 25 | In10Sn40 | — | — | — | — | 72 | — | — | — | — | |||
| 26 | In20Sn40 | — | — | — | — | 73 | — | — | — | — | |||
| 27 | In40Sn40 | 5 | 3 | — | — | 86 | — | — | — | — | |||
| 28 | In60Sn40 | — | — | — | — | 80 | — | — | — | — | |||
| 29 | Cu-Cubes GDE (loading 440 μg cm−2) | 0.1 M KHCO3 | 100 mA cm−2 | — | 62 | — | 10 | — | — | — | — | — | 160 |
| 30 | Cu/Ag0.14 | 0.1 M CsHCO3 | 0.1 A cm−2 | 11 | 50 | 6 | 18 | 2 | — | — | 15 | — | 161 |
| 31 | Cu/Ag0.17 | 8 | 83 | 1 | 1 | 2 | — | — | 3 | — | |||
| 32 | Cu/Ag0.41 | 13 | 84 | — | — | 2 | — | — | — | — | |||
| 33 | IL@Cu | 0.1 M KHCO3 | −1.19 | 49 | 10 | 1 | 40 | — | — | — | — | — | 162 |
| 34 | Cu | 62 | 8 | 2 | 22 | — | — | — | — | — | |||
| 35 | Cu-NP | 0.1 M KHCO3 | −1.5 | 43 | 2 | 3 | 16 | 20 | — | — | 6 | — | 163 |
| 36 | Cu-NN | 43 | 4 | 4 | 24 | 4 | — | — | 21 | — | |||
| 37 | Cu-PTFE NNs (70%) | 25 | 2 | 3 | 37 | 2 | — | — | 33 | — | |||
| 38 | Cu-PTFE NNs (90%) | 18 | 2 | 2 | 34 | 2 | — | — | 43 | — | |||
| 39 | Cu-PTFE NNs (99%) | 10 | 1 | 1 | 55 | 1 | — | — | 41 | — | |||
| 40 | EC polished Cu | 0.1 M CsHCO3 | −1.0 | 34 | 17 | 4 | 25 | 7 | — | — | 5 | — | 164 |
| 41 | After N2 plasma | 30 | 8 | 4 | 34 | 6 | — | — | 5 | — | |||
| 42 | After Ar plasma | 28 | 3 | 2 | 43 | 4 | — | — | 10 | — | |||
| 43 | After O2 plasma | 33 | 3 | 3 | 42 | 3 | — | — | 11 | — | |||
| 44 | 20 wt% Cu/GO | 66 | 3 | 2 | 10 | 10 | — | — | 2 | — | 165 | ||
| 45 | 40 wt% Cu/GO | 53 | 4 | 4 | 8 | 20 | — | — | — | — | |||
| 46 | 60 wt% Cu/GO | 45 | 12 | 9 | 22 | 5 | — | — | — | — | |||
| 47 | 80 wt% Cu/GO | 51 | 10 | 8 | 25 | — | — | — | — | ||||
| 48 | Cu2O-c | 0.5 M KHCO3 | −0.3 | 2 | 21 | 28 | — | 2 | — | 5 | — | — | 166 |
| 49 | Cu2O-o | 11 | 40 | 5 | — | 5 | — | 6 | — | — | |||
| 50 | Cu2O-t | 5 | 37 | 1 | — | 4 | — | 11 | — | — | |||
| 51 | Cu2O-u | 4 | 10 | 16 | — | — | — | 2 | — | — | |||
| 52 | Cu | 1 | 25 | — | — | — | — | 20 | — | — | |||
| 53 | CuO | 1 | — | — | — | — | — | 4 | — | — | |||
| 54 | Nt-16 | 0.2 M KHCO3 | −1.6 | 20 | 1.9 | 59 | 3.6 | 19 | — | — | — | — | 167 |
| 55 | Nt-43 | 26 | 1.6 | 49 | 1.4 | 16 | — | — | — | — | |||
| 56 | Nt-77 | 40 | 1.5 | 38 | 1.2 | 15 | — | — | — | — | |||
| 57 | Nt-265 | 42 | 0.8 | 32 | 1.8 | 18 | — | — | — | — | |||
| 58 | Ut-Cu | 47 | 0.38 | 28 | 1.5 | 20 | — | — | — | — | |||
| 59 | Cu nanocubes | 0.1 M KHCO3 | −0.8 | 5 | 24 | 51 | 5 | — | — | — | — | 168 | |
| Cu nanospheres | 5 | 34 | 1 | 20 | 22 | — | — | — | — | ||||
| 60 | CuPd0.5/C | 1 M KOH | 100 mA cm−2 | 10 | 20 | — | 34 | — | — | — | 18 | — | 169 |
| 61 | Cu3Sn/CF | 0.5 M KHCO3 | −0.9 | 64 | 4 | — | — | 37 | — | — | — | — | 170 |
| 62 | C–Cu | 0.5 M KHCO3 | −0.9 | 64 | 4 | — | — | 32 | — | — | — | — | 171 |
| 63 | C–Cu/In2O3-0.4 | 38 | 52 | — | — | 10 | — | — | — | — | |||
| 64 | C–Cu/In2O3-0.8 | 42 | 48 | — | — | 10 | — | — | — | — | |||
| 65 | C–Cu/In2O3-1.5 | 34 | 21 | — | — | 40 | — | — | — | — | |||
| 66 | Acid-treated In foil | 88 | — | — | — | 17 | — | — | — | — | |||
The e-CO2R into enantiomeric amino acids had represented one of the earliest instances of producing complex C3+ products with C–N bonds. This achievement was made possible by reducing reaction barriers in the synthesis of C3+ compounds through the utilization of chiral twist sites on catalyst surfaces. This discovery underscored the viability of a universal strategy aimed at enhancing the intrinsic catalytic activities of materials for the production of C3+ species, while also showcasing the potential of chiral twist sites for generating specific biomolecules starting from CO2 and ammonium bicarbonate.172 In a significant development, inorganic CuNCs underwent functionalization with an imidazolium ligand, resulting in the creation of hybrid CO2RR catalysts that were subject to comprehensive investigation. The findings revealed that, at higher applied potentials, the ImPF6 ligand exhibited a preference for CO2RR over HER and formates over ethylene across the studied potential range. The structural stability of cubic CuNCs and their strong interactions with the ligands facilitated surface modification experiments. However, it was noteworthy that the promoter effect of the imidazolium ligand dissipated during the reconstruction of spherical CuNCs. These insights highlighted the pressing need for hybrid catalysts characterized by either ligands impeding the rebuilding processes or stable NC morphologies.173
In a stunning revelation of scientific achievement, previous researches unveiled the remarkable enhancement of copper catalytic activity through the application of various imidazolium salts. These chemical modifications yielded impressive improvements in ethylene selectivity, all while effectively suppressing the production of H2, CH4, and ethanol by skillfully manipulating substituents in organic modifiers. Amazingly, the modified electrodes exhibited an ethylene selectivity of up to 73%, with C2 product selectivity rising as high as 82%. Even after a demanding 19 hours of continuous use, the total current densities remained virtually indistinguishable between the modified electrodes and their bare copper counterparts. The nuanced steric and electronic characteristics of substituents on imidazolium nitrogen atoms took center stage, dictating product distribution with the finesse to fine-tune selectivity through adjustments in film deposition potential and alkyl chain length.174
To explore this further, researchers entered the e-CO2R process excitedly in a novel way using Cu/SnOx heterostructured nanoparticles combined with CNTs. As the ratio of Cu/SnOx was carefully adjusted, the potential for modulating the primary product was revealed – intermediate hydrocarbons, CO2, formaldehyde or formic acid.175 Further, the addition of CTAB to an electrolyte was found to significantly change the selectivity of unmodified Cu for formate production during e-CO2R. In near-neutral aqueous electrolytes, this produced a remarkable 82% selectivity and a 56-fold increase in partial current density.176
Moreover, an innovative technique was described for manufacturing a catalyst containing Ni/Cu dual sites on MOF-templated porous carbon. This approach facilitated uniform dispersion of dual sites with adjacent NiN4 and CuN4 moieties. The presence of Cu sites was found to lower the formation barrier of *COOH on surrounding Ni sites, making the rate-limiting step more accessible. As a result, the Ni/CuNC catalyst with N4Ni/CuN4 moieties exhibited an exceptionally high intrinsic activity and achieved impressive FECO values (99.2% at 0.79 V versus RHE and greater than 95% from 0.39 to 1.09 V vs. RHE).177 In addition to these findings, shed light on the role of metal–supramolecular couples constructed via stacking interactions in facilitating a pivotal phase of C2+ product production, specifically C–C coupling (Fig. 16). The selectivity towards C2+ products was attributed to the paired structure of metal sites and the van der Waals interaction between the secondary coordination sphere and the adsorbed intermediates. Remarkably, they marked the first detailed exploration of engineering metal–supramolecular couples with catalytic activity for multicarbon-selective electrochemical reduction.178
 | ||
| Fig. 16 The P-XRD patterns of the GDE without any modification and the GDEs with the Cu(salophen)–carbon composite catalyst were analyzed before and after electrolysis (a). The Cu 2p XPS spectrum was examined, and the Cu LMM Auger spectrum was included as an inset (b). A TEM image was taken to visualize the structure of the catalyst (c). The Cu K-edge EXAFS spectrum was analyzed, and the fit in R-space was determined (d). The Cauchy wavelet transform of the Cu K-edge X-ray absorption spectrum was studied for the post-electrolysis Cu(salophen)–carbon composite catalyst (e). A structural model was used for EXAFS fitting (f). Reproduced with permission from ref. 178. Copyright 2023, American Chemical Society. | ||
Moreover, the research proposed an approach aimed at enhancing the selectivity of e-CO2R, with a specific focus on C2 products, notably ethanol. Impressively, CuBr treated with DDT molecules yielded remarkable results, a high ethanol-to-ethylene ratio and an impressive FE of 72%. This achievement stemmed from the inhibition of Br migration and the complete reduction of CuBr through DDT adsorption onto CuBr, leading to the formation of a novel and highly stable Br-doped Cu–thiol interface during the catalytic process. Additionally, the incorporation of Br species into Cu was hypothesized to stabilize high-valence Cu species and direct selectivity towards C2 products, particularly ethanol, as evidenced in the study.179 In parallel, another research study's findings unveiled a significant breakthrough in the realm of e-CO2R. The incorporation of ZrO2 and the meticulous tuning of nanocavities within CuO were identified as critical factors that facilitated the dimerization and protonation of CO, ultimately yielding C2H4. Notably, this approach also acted as an impediment to the antagonistic HER. The capacity for rational manipulation of e-CO2R performance was established through the modulation of ZrO2 incorporation levels and the control of flaws in CuO. Notably, over CuO@ZrO2 in an H-cell configuration, the FE and cathodic energy efficiency (EE) for C2H4 generation witnessed substantial enhancements compared to bare CuO, with FE increasing from 19.7% to 47.6% and cathodic EE from 11% to 26.8%. Furthermore, with a flow cell architecture operating at a high current density of 250 mA cm−2, the overall FE and EE for C2H4 were significantly boosted to 84.3% and 54.7%, respectively.180
In a related domain, the e-CO2R to CO in an aqueous environment, utilizing metal nanoparticle catalysts such as Au nanoparticles, witnessed significant advancement through the implementation of a molecular surface functionalization strategy.181 Concurrently, the research delved into the utilization of a one-step solvothermal methodology for the synthesis of copper hydroxyphosphate catalysts tailored for deployment in e-CO2RR. Electrochemical investigations brought to light the remarkable component-specific efficiency of these catalysts. The FE exhibited by polyhydroxyl Cu5(OH)4(PO4)2 reached 37.4% at −1.6 V, surpassing that of Cu2(OH)PO4 by over 100% in terms of current density.182
In a parallel research avenue, an innovative strategy revolved around surface molecule functionalization, presenting a potential breakthrough for catalytic applications. Specifically, the 4H/fcc Au-MMT configuration outperformed its counterpart, 4H/fcc Au-OAm, by a significant margin in terms of e-CO2RR performance within H-type cells. This surface functionalization strategy also exhibited the capacity to enhance the selectivity of the flow cell for CO. DFT simulations conducted on 4H/fcc Au nanorods functionalized with OAm and MMT ligands revealed opposing modulation effects. In both the fcc and 4H phases, surface MMT activation of Au was observed, resulting in a e-CO2RR process characterized by minimal reaction barriers. In contrast, OAm exerted effects such as reduced electroactivity and the introduction of spatial barriers for intermediate adsorption on the surface.183 An overview of electrocatalysts designed by following the molecular surface functionalization approach is tabulated in Table 7.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | L-CCFs | 0.25 M NH3·H2O | — | — | — | — | 1 | — | — | 10 | — | 172 | |
| 2 | ACFs | — | — | — | — | 1 | — | — | 4 | — | |||
| 3 | Cucub | 0.1 M KHCO3 | −1.05 | 65 | 7 | 2 | 20 | 3 | — | — | 1 | — | 173 |
| 4 | Cucub–ImPF6 | 67 | 7 | 1 | 14 | 10 | — | — | — | ||||
| 5 | Modified polycrystalline copper (12) | 0.1 M KHCO3 | −1.14 | 39 | 5 | 10 | 20 | 21 | — | — | 3 | — | 174 |
| 6 | CuOy-CNT-#1 | 0.1 M KHCO3 | −1.09 | 46 | 4 | 35 | 10 | 6 | — | — | — | — | 175 |
| 7 | CuOy/SnOx-CNT-#2 | 50 | 5 | 25 | 10 | 10 | — | — | — | — | |||
| 8 | CuOy/SnOx-CNT-#7 | 12 | 76 | — | — | 12 | — | — | — | — | |||
| 9 | CuOy/SnOx-CNT-#12 | 14 | 13 | — | — | 80 | — | — | — | — | |||
| 10 | SnOx-CNT-#13 | 23 | 25 | — | — | 52 | — | — | — | — | |||
| 11 | Oxide-derived Cu | 0.5 M KHCO3 | −0.9 | 45 | 2 | 2 | 1 | 50 | — | — | — | — | 176 |
| 12 | Cu-CTAB | 48 | 1 | — | — | 54 | — | — | — | — | |||
| 13 | N/C | — | 25 | — | — | — | — | — | — | — | 177 | ||
| 14 | Cu–N–C | — | 50 | — | — | — | — | — | — | — | |||
| 15 | Ni–N–C | — | 51 | — | — | — | — | — | — | — | |||
| 16 | Ni/Cu–N–C | — | 85 | — | — | — | — | — | — | — | |||
| 17 | Cu(salophen)-coated GDE | 1 M KOH | −1.0 | 30 | 20 | 3 | 38 | 6 | — | — | 3 | — | 178 |
| 18 | CuBr | — | −1.10 | 24 | 4 | 25 | — | — | — | — | — | 179 | |
| 19 | CuBr-DDT | 18 | 20 | 18 | — | — | — | — | — | ||||
| 20 | CuO@ZrO2 | 0.1 M KHCO3 | −1.1 | — | 9 | 7 | 48 | 7 | — | — | — | — | 180 |
| 21 | CuO | — | 2 | 2 | 25 | 9 | — | — | — | — | |||
| 22 | ZrO2 | — | — | — | — | 7 | — | — | — | — | |||
| 23 | Cu2(OH)PO4 | 0.1 M KHCO3 | −1.0 | 38 | 8 | — | — | — | — | — | — | — | 182 |
| 24 | Cu5(OH)4(PO4)2 | 46 | 8 | — | 9 | — | — | — | — | — | |||
| 25 | 4H/fcc Au-MMT | 1.0 M KHCO3 | −1.0 | 70 | — | — | — | — | — | — | — | 183 | |
| 26 | 4H/fcc Au-OAm | 53 | — | — | — | — | — | — | — | ||||
Zhang et al. designed a Cu–Zn alloy/Cu–Zn aluminate oxide composite system for the e-CO2R to valuable C2+ products. Decomposition of the Cu–Zn–Al-LDH precursor signals the beginning of this novel composite system. Following this, the precursory catalyst was transformed into a composite catalyst consisting of the Cu–Zn alloy and Cu–Zn aluminate oxide. These two components were replicated in a lattice that results in interface after interface crisscrossing. An optimized Cu9Zn1/Cu0.8Zn0.2Al2O4 catalyst displayed massive current and atom-light effects (though only for C2+ products): 88.5% FE at 400 mA cm−2 when operating at 1.15 V vs. RHE.184 Simultaneously, another study delved into the e-CO2R into methane, leveraging a Cu foil modified with ZnOx nanoparticles. Notably, the peak FE for methane production was achieved at 1.1 V, reaching approximately 36%, and it exhibited no significant deterioration over time. This catalytic activity was attributed to the induction of active sites for methane production, identified as cupric ions, at the Cu/ZnOx interface. The interaction between Cu and ZnOx provided stability to these active species, prolonging their catalytic lifespan. Importantly, the synthesis process for this system was characterized by its swiftness, simplicity, cost-effectiveness, and straightforwardness. Furthermore, the scalability of methane production was achieved by expanding the surface area of the Cu foil.185
Furthermore, in the pursuit of achieving exceptional selectivity and stability in the e-CO2R to C2+ products, Liu et al. developed a heterostructured catalyst comprising 2D Cu MOFs and Cu2O nanoparticles (Cu2O@Cu-BDC). This catalyst consistently generated stable C2+ products with remarkable FE of 72.1% in an H-type cell and 58.2% in a flow cell setup. The key to its success lay in the stabilization of Cu+ active sites at the interfaces formed between Cu2O and Cu-BDC within the 2D Cu MOFs. This stabilization, in turn, enhanced C–C coupling reactions and improved the adsorption of critical intermediates involved in the production of C2+ products.186 Additionally, a one-step reduction technique had been employed to create a bimetallic Cu and SnOx nanocomposite on an amorphous rGO support, aiming to enhance formate synthesis through the interplay between the materials, heterointerfaces, high oxophilicity, and electrochemical surface area. The production of SnOx nanoparticles had hindered C–C coupling and had reduced H2 and CO generation, ultimately leading to enhanced formate selectivity and endurance. Remarkably, even after 6 hours of electrolysis, the nanocomposite had maintained 85% of its original current density.187 In the quest to improve C2+ selectivity in e-CO2R, researchers had devised a method for in situ modification of the Cu+/Cu0 interface within oxide-derived Cu catalysts. Tests of the R-Cu with Br catalyst's performance in e-CO2R had unveiled outstanding stability, complemented by its excellent activity and selectivity in producing C2+ products. The cooperative Cu+/Cu0 interface, which played a pivotal role in conferring remarkable selectivity for C2+ products, had substantially lowered the energy barrier for CO–CO coupling and had repressed the HER, thereby enhancing the efficiency of CO–CO coupling.188
In a related context, the interactions between Cu and the electrolyte were found to be of paramount importance in the context of CO2R in aqueous solutions. When CuO electrodes were modified with various polymers such as PVDF, PE, PVA, and PVP, the FE for C2H4 generation doubled, and the partial current density nearly tripled. This advantageous effect was primarily governed by the hydrophobic environments created by PVDF and PE polymers on the Cu surface. Hydrophobic polymer coatings resulted in a higher surface pH, inhibiting HER and promoting the reduction of CO2 to C2H4.189 To illustrate uniform Cu dispersion onto the CeO2 support and the fine-tuning of interfacial characteristics, Yin et al. had demonstrated the fabrication of heterostructured Cuy/CeO2 nanorods through chemical prelithiation and galvanic displacement. The Cu decoration had facilitated the activation of CO2 and its subsequent conversion to the *CO intermediate on the surrounding Cu surface, generating a new strong-binding site at the Cu and CeO2 interface. The trends in the production of C1 and C2+ products in e-CO2R had exhibited a decreasing and volcano-shaped pattern as the extent of Cu decoration had increased. Through in situ ATR-SEIRAS and Raman measurements, it had been found that the Cu surface had generated two distinct types of *COLFB and *COHFB intermediates, with a high coverage of *COHFB promoting the creation of C1 products and a high coverage of *COLFB contributing to the formation of C2+ products.190 Moreover, it was found that during e-CO2RR the S–CuSn catalyst goes through dynamic changes. There are five stages in this complicated process: CuS reduction; S leaching from Cu2SnS3; Cu24Sn20 alloy generation; Sn metal formation; and the constitution of a stable structure in the form of co-doped Cu24Sn20 alloys with S/Sn. This reconstructed S–CuSn catalyst gave a maximum CO2 FE of 91.5% and formate FE of 96.4%, owing to its exemplary activity in the e-CO2R toward formate.191
Furthermore, metallurgy-dealloying techniques were employed to craft a novel CeO2Cu amorphous heterostructure. This catalyst, through its interfacial sites, increased the adsorption strength of CO and stabilized CH2CHO, guiding the reaction toward C2+ alcohol production. The catalyst significantly enhanced the inherent activity and selectivity of Cu-based catalysts in the synthesis of C2+ alcohols, achieving a remarkable FE of 32.9% at a low potential.192 Besides, Rudd et al. investigated the creation of foams and their morphology, crystal structure, and catalytic efficiency under varying conditions. They also studied how foams reacted to CO2 electrolysis and examined the potential impact of urea as an additive. Both s unmodified and urea-treated foam electrodes exhibited a variety of cub-octahedra and dendrites. However, changes in the shape, crystallinity, and surface content of copper foams after 35 minutes of electrolysis resulted in their deactivation as catalytic materials.193
Additionally, the study examined two electrode families comprised of Cu NPs loaded at varying concentrations (ranging from 0.25 to 2.0 mg cm−2) as e-CO2RR catalysts (Fig. 17). At low loadings, the predominant reduction product at the electrodes was CH4, while at higher loadings, CO became the dominant product. Interestingly, at moderate concentrations (0.5 to 1.0 mg cm−2), C2H4 selectivity was maximized. This behavior was attributed to the presence of low-density NPs, which reduced the likelihood of CO dimerization, ultimately leading to increased CH4 generation. Bulk CO generation increased with higher loading, increasing the probability of dimerization and thus favoring C2H4 synthesis. Additionally, capacitance was found to slow the outward flow of charges from the electrode as NP loading increased further.194
 | ||
| Fig. 17 Comparison of electrodes consisting of 25 nm nanoparticles (NPs) with varying loadings: at 200 mA cm−2, (a) assessing selectivity, (c) evaluating equivalent bulk CO generation and dimerization rates and (e) normalizing equivalent CO generation and dimerization rates to the electrode's electrochemically active surface area (ECSA); at 267 mA cm−2, (b) analyzing selectivity, (d) calculating equivalent bulk CO generation and dimerization rates, and (f) ECSA-normalized equivalent CO generation and dimerization rates. Each electrode underwent testing on multiple occasions. The bulk and ECSA-normalized equivalent CO generation and dimerization rates in (c)–(f) were computed based on the average selectivity values from at least two sets of experiments conducted at different catalyst loadings and current densities. Reproduced with permission from ref. 194. Copyright 2023, Elsevier. | ||
Likewise, Tafazoli et al. highlighted a substantial difference in the FEs of C2 products between low and high current density electrodeposited Cu oxides. In situ SERS was employed to monitor changes in surface chemistry under CO2RR conditions, revealing rapid shifts in Cu oxidation states on porous high current density electrodes but not on low current density electrodes. The formation of Cu2(CO3)(OH)2 was linked to Cu oxidation states, surface intermediates, and product analyses, affecting adsorbed intermediates and C2 products based on local HCO3− concentration and pH during Cu2(CO3)(OH)2 production. The high current density surface was easily terminated by Cu2(CO3)(OH)2, inhibiting further CO2RR. As a result, HER was enhanced at the expense of C2 products due to decreased CO2 concentration near the surface, driven by HCO3− consumption and resulting in higher alkalinity over the electrode. The strong adsorption of CO molecules over the high current density electrode terminated by Cu2(CO3)(OH)2 might account for the removal of the C2 pathway.195
In addition, Yang et al. demonstrated increased selective adsorption of Cl at dual-phase interfaces using a Cu-based catalyst with abundant Cu+ species. This facilitated CO2-to-C2+ conversion in neutral fluids by providing local *CO coverage, which favored energetically advantageous CO dimerization. The employment of the dual-phase method to enhance *CO intermediates' coverage has broader implications for improving CO2R catalysis in various electrolytes and other electrochemical processes.196 Also, single-atom-thick Cu–Sn surface alloys (Cu97Sn3 and Cu99Sn1) were created for effective e-CO2R. These alloys exhibited different catalytic selectivity compared to pure Cu100 and Cu70Sn30 bulk alloys due to their distinct geometric and electronic structures. The Cu97Sn3 catalyst achieved 98% FECO at low overpotential, showcasing the influence of the local coordinating environment in isolated Cu–Sn bonding, as demonstrated by density functional theory simulations.197
Additionally, a practical method for stabilizing copper with silica and producing CuSiOx amorphous nanotube catalysts that are resistant to rebuilding was introduced by Tan et al. The Cu–O–Si interfacial sites are ultra-stable in the CO2RR without evident reconstruction due to the strong interfacial connection between Cu and silica, resulting in high CO2-to-CH4 selectivity (72.5% after 12 hours of testing) and stability (FECH4 remains over 60% after 12 hours of testing) (Fig. 18). In a flow cell device, an impressive CO2-to-CH4 conversion rate of 0.22 mol cm−2 s−1 was also attained, suggesting a viable path toward the development of highly active and stable Cu-based CO2R catalysts.198Table 8 presents a comparative overview of different electrocatalysts synthesized by the interfacial engineering approach for e-CO2R to sustainable products.
 | ||
| Fig. 18 e-CO2R performance: LSV curves of CuSiOx, CuGeO3, and CuO under CO2 or Ar purging conditions (a). FEs of CO2R products at varying potentials (b) and (c). FE for CH4 production by CuSiOx compared with other catalysts from previous studies after iR corrections (d). Time-dependent total current density and FECH4 generated by CuSiOx measured at −1.6 V vs. RHE over a 12-hour period (e). A comparative analysis of this research with prior investigations regarding electrocatalytic CO2-to-CH4 conversion, conducted in a flow cell device (f). Reproduced with permission from ref. 198. Copyright 2023, American Chemical Society. | ||
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | CuZn/CuZnAl2O4 | 2 M KOH | −1.1 | 4 | 4 | — | 38 | 2 | — | — | 34 | — | 184 |
| 2 | Cu/CuAl2O4 | 18 | 4 | — | 35 | 4 | — | — | 25 | — | |||
| 3 | CuZn | 17 | 18 | 2 | 17 | 2 | — | — | 21 | — | |||
| 4 | 0D-CuZn | 0.1 M KHCO3 | −1.1 | — | — | 35 | 18 | — | — | — | — | — | 185 |
| 5 | 0D-Cu | — | — | 8 | 30 | — | — | — | — | — | |||
| 6 | Cu | — | — | 6 | 2 | — | — | — | — | — | |||
| 7 | Cu2O@Cu-BDC | 0.1 M KBr | −1.1 | 17 | 21 | 2 | 25 | 6 | — | — | — | — | 186 |
| 8 | Cu-BDC | 38 | 6 | 5 | 14 | 2 | — | — | — | — | |||
| 9 | Cu2O | 40 | 5 | 8 | 26 | 5 | — | — | — | — | |||
| 10 | Ni–N–C@900 °C | 0.1 M KHCO3 | −1.1 | 93 | 12 | — | — | — | — | — | — | — | 199 |
| 11 | Ni–N–C@700 °C | 77 | 8 | — | — | — | — | — | — | — | |||
| 12 | NiNPs–N–C | 94 | 7 | — | — | — | — | — | — | — | |||
| 13 | N–C | 85 | 6 | — | — | — | — | — | — | — | |||
| 14 | CuSnOx/rGO | 0.5 M KHCO3 | −0.99 | 2 | 21 | — | — | 22 | — | — | — | — | 187 |
| 15 | Cu3SnOx/rGO | 3 | 28 | — | — | 28 | — | — | — | — | |||
| 16 | Cu0.33SnOx/rGO | 3 | 27 | — | — | 69 | — | — | — | — | |||
| 17 | R–Cu | 0.1 M KHCO3 | 1.0 | 18 | 1 | 1 | 52 | 2 | — | — | 12 | — | 188 |
| 18 | R–Cu–Br | 38 | 1 | 8 | 38 | 1 | — | — | 5 | — | |||
| 19 | CuO | 0.5 M KHCO3 | −1.02 | 51 | 12 | 2 | 18 | — | — | — | — | — | 189 |
| 20 | CuO-PVDF | 40 | 10 | 2 | 25 | — | — | — | — | — | |||
| 21 | Cu0.04/CeO2 | 0.1 M KHCO3 | −1.0 | 23 | 32 | — | 2 | 33 | — | — | — | — | 190 |
| 22 | Cu0.12/CeO2 | 31 | 17 | — | 24 | 22 | — | — | — | — | |||
| 23 | Cu0.32/CeO2 | 25 | 20 | — | 20 | 23 | — | — | 7 | — | |||
| 24 | Cu0.4/CeO2 | 24 | 10 | 4 | 23 | 24 | — | — | 7 | — | |||
| 25 | Cu0.6/CeO2 | 29 | 18 | — | 13 | 38 | — | — | — | — | |||
| 26 | S–CuSn | 0.5 M KHCO3 | −0.8 | 8 | 10 | — | — | 50 | — | — | — | — | 191 |
| 27 | S–Cu | −0.65 | 66 | — | — | — | 3 | — | — | — | — | ||
| 28 | S–Sn | −0.99 | 9 | 9 | — | — | 41 | — | — | — | — | ||
| 29 | CeO2–Cu | 1.0 M KOH | −0.6 | — | — | — | — | — | — | — | 30 | — | 192 |
| 30 | CuZn | −0.7 | — | — | — | — | — | — | — | 38 | — | ||
| 31 | Grain boundary rich Cu | −0.8 | — | — | — | — | — | — | — | 17 | — | ||
| 32 | CuAg | −0.8 | — | — | — | — | — | — | — | 19 | — | ||
| 33 | Cu2S–Cu–V | −1.0 | — | — | — | — | — | — | — | 19 | — | ||
| 34 | Nanoporous Cu | −0.7 | — | — | — | — | — | — | — | 10 | — | ||
| 35 | CuAg wires | −0.7 | — | — | — | — | — | — | — | 10 | — | ||
| 36 | Cu nanoparticles | −0.8 | — | — | — | — | — | — | — | 15 | — | ||
| 37 | Porous Cu | −0.8 | — | — | — | — | — | — | — | 15 | — | ||
| 38 | CuAu | −1.0 | — | — | — | — | — | — | — | 25 | — | ||
| 39 | CF-18H | 0.1 M KHCO3 | −1.05 | 74.3 | 1.7 | 0.3 | 11.6 | 2.6 | — | — | 2.5 | — | 193 |
| 40 | CF-18H-100U | 90.9 | 0.8 | 0.7 | 8 | 2.4 | — | — | 1.5 | — | |||
| 41 | CuZn/CuZnAl2O | 2 M KOH | −1.1 | 4 | 6 | — | 38 | 2 | — | — | 34 | — | 184 |
| 42 | Cu/CuAl2O4 | 26 | 4 | — | 33 | 4 | — | — | 27 | — | |||
| 43 | CuZn | 17 | 20 | 4 | 16 | — | — | — | 21 | — | |||
| 44 | Cu NPs (3 mg cm−2) | 1.0 M KOH | 200 mA cm−2 | 22 | 44 | — | 16 | 10 | — | — | — | — | 194 |
| 45 | S–ZnO | 1 M KOH | −1.06 | 38 | 30 | — | — | 8 | — | — | — | — | 50 |
| 46 | S–CuO | 21 | 5 | 3 | 40 | — | — | — | 12 | — | |||
| 47 | S–Cu5Zn1Ox | 48 | 12 | 2 | 18 | — | — | — | 6 | — | |||
| 48 | S–Cu1Zn3Ox | 28 | 14 | 3 | 16 | — | — | — | 7 | — | |||
| 49 | CuSiOx | 1 M KOH | −1.3 | 19 | 4 | 43 | 16 | — | — | — | — | — | 198 |
| 50 | CuO | 20 | 18 | 10 | 18 | — | — | — | — | — | |||
| 51 | Cu100 | 0.5 M KHCO3 | −1.0 | 22 | 18 | — | 28 | 28 | — | — | — | — | 197 |
| 52 | Cu97Sn3 | 5 | 84 | — | — | 9 | — | — | — | — | |||
| 53 | Cu70Sn30 | 2 | 5 | — | — | 90 | — | — | — | — | |||
Continuing with this line of investigation, tin doped CuO NSs were prepared and their catalytic activity was found to be higher than that of pure CuO catalysts. The excellent performance of the NSs was mainly attributed to the increased CO2˙ adsorption. The synergistic relationships between SnO2 and CuO were found to be beneficial for e-CO2R.200 In another facet of research, La2CuO4 perovskites with engineered grain boundaries (GBs) were developed as effective electrocatalysts for e-CO2RR. These tailored nanoparticles exhibited a strong FE toward C2H4, a desirable product of CO2R. The enhanced selectivity for the C2 product was attributed to their high adsorption affinity for CO intermediates. The introduction of strain generated by the twin boundaries (TBs) in La2CuO4 nanoparticles led to increased CuO and CuLa bond lengths, further improving FEC2H4.201 Advancing this research, a microfluidic flow cell was utilized to reduce Cu2O precursors under CO2RR conditions, resulting in the formation of catalysts with a porous Cu cavity (Fig. 19). These catalysts exhibited a Cu(111) facetted cavity with dimensions of approximately 108 ± 23 nm. Remarkably, these structures led to an increase in both the FE for C2+ products and the partial current density, all achieved at a low applied voltage. This was attributed to the entrapment of numerous *CO intermediates within the cavities, preventing their easy migration between cavities. DFT calculations and electrochemical Raman spectra suggested that C–C coupling primarily occurred via the CO–CHO coupling mechanism.202
 | ||
| Fig. 19 The study focuses on characterizing the synthesized Cu2O pre-catalyst and the Cu cavity catalysts. The XRD pattern (a), Raman spectra (b), and XPS of Cu 2P (c) and Cu LMM (d) were obtained for the synthesized Cu2O precatalyst and the Cu cavity catalysts. “GDL” refers to the commercial gas diffusion layer. The performance of e-CO2RR was tested in a flow cell reactor. The major products of the e-CO2RR were studied on both cavity Cu (e) and solid Cu (f) surfaces at various applied potentials in a 1 M KOH solution. Reproduced with permission from ref. 202. Copyright 2023, American Chemical Society. | ||
In the realm of catalytic advancements, a groundbreaking achievement was made by immobilizing OHad-introduced noncovalent interactions (NCI) in the reduction of CO2 on SnO nanosheet catalysts. This innovative approach yielded remarkable results, including a high FE of 90.5% and a substantial partial current density of 61.8 mA cm−2. The catalytic mechanism revolved around the rapid protonation of CO2 into formate on the SnO(001) surface through activation. The introduction of NCI by OHad, coupled with its role in enhancing hydrogen adsorption, facilitated efficient regeneration, enabling continuous operation. Notably, OHad-induced NCIs outperformed their H2-annealed counterparts in promoting formate reduction across various catalysts, including CuO, Bi2O3, and TiO2.203 Another notable advancement in catalytic science involved the creation of a Cu2O(CO) nanocubic catalyst through controlled thermal topological transformation of Cu2O nanocubes under a CO atmosphere at 275 °C. This catalyst exhibited a unique structural configuration characterized by high-density nanograin boundaries, which exposed Cu(100) facets, Cu[n(100) × (110)] step sites, and Cu0/Cu+ interfaces. During the CO2RR process, it demonstrated exceptional performance, achieving a FEC2+ of 77.4%, and showcased remarkable stability even under high polarization and current densities. The abundance of Cu0/Cu+ interfaces played a pivotal role in increasing the adsorption density of *CO intermediates, thereby contributing to the high selectivity for C2+ products. Furthermore, the presence of rich nanograin boundaries significantly improved the catalytic stability, underscoring the critical importance of surface structures in Cu-based catalysts for achieving high C2+ selectivity during CO2RR, especially under industrially relevant current densities.204
Synthesis of methanol using Cu/ZnO/Al2O3 catalysts at an industrial level has long been a mystery, and it is mainly because we are not certain of where the active sites are located. A large-scale machine learning atomic simulation was harnessed to investigate thousands of reaction paths connected to both CO2 and CO hydrogenations on Cu–Zn surfaces predicted by thermodynamic calculations to be stable. This extensive study revealed several key insights. Firstly, under reaction conditions, Zn decorates at the step-edge of Cu(211) up to 0.22 monolayers (ML), indicating a dynamic surface modification process. Additionally, the investigation highlighted that CO2 hydrogenation overwhelmingly governs the methanol synthesis process. Moreover, the formation of [–Zn–OH–Zn–] chains on Cu(111) surfaces was observed under reaction conditions, suggesting the pivotal role of CO in the mixed gas environment.205 Transitioning to the selectivity of the carbon reduction reaction (CRR) aimed at producing C2+ products on oxidized Cu-based catalysts, it was discovered that the oxidation state of the surface Cu active site plays a pivotal role. At lower oxidation states, weak *CHO adsorption is observed at the Cu site, rendering the transition from *CO to *CHO unfavorable. As the oxidation state of Cu increases, the C–C coupling step shifts toward *CHO dimerization, and the *CO to *CHO transition becomes more favorable. Remarkably, the optimal oxidation state for CC coupling was identified to be around +0.5. Importantly, the selectivity for C2+ products can be tailored through doping with transition metals, which have the capacity to alter the Cu oxidation state. The presence of these metal dopants can be discerned through the utilization of artificial intelligence clustering based on physical characteristics.206 Notably, one promising avenue for enhancing the electrocatalytic performance of copper involves surface promotion of copper with alumina (AlOx) clusters. This strategy has led to the development of highly selective and stable catalysts capable of catalyzing the transformation of CO2 into ethylene and other multicarbon compounds. The catalyst, formed from Cu–aluminum (Cu–Al) layered double hydroxide nanosheets and promoted with AlOx, has demonstrated remarkable performance in this regard, offering potential solutions for sustainable carbon utilization and emission reduction.207Table 9 presents an overview of different materials designed by following the Cu surface engineering strategy for e-CO2R to different valuable products.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | La2CuO4 NBs | 0.1 M KHCO3 | −1.1 | 38 | 10 | 4 | 43 | — | — | — | — | — | 201 |
| 2 | La2CuO4 NRs | 55 | 7 | 8 | 25 | — | — | — | — | — | |||
| 3 | Bulk La2CuO4 | 18 | 84 | 2 | — | — | — | — | — | — | |||
| 4 | Bulk CuO | 0.5 M KHCO3 | −1.0 | — | — | 8 | — | — | — | — | — | — | 200 |
| 5 | CuO NS | — | — | 12 | 4 | — | — | — | — | — | |||
| 6 | 1% SnO2–CuO NS | — | — | 12 | 21 | — | — | — | — | — | |||
| 7 | 3% SnO2–CuO NS | — | — | 15 | 24 | — | — | — | — | — | |||
| 8 | 5% SnO2–CuO NS | — | — | 13 | 13 | — | — | — | — | — | |||
| 9 | 10% SnO2–CuO NS | — | — | 11 | 8 | — | — | — | — | — | |||
| 10 | Cavity Cu | 1 M KOH | −0.7 | 11 | 9 | 8 | 23 | 4 | — | — | 6 | — | 202 |
| 11 | Solid Cu | 31 | 7 | 19 | 15 | 3 | — | — | 2 | — | |||
| 12 | Sn | 0.5 M KHCO3 | −0.95 | — | — | — | — | 37 | — | — | — | — | 203 |
| 13 | H–SnO | — | — | — | — | 40 | — | — | — | — | |||
| 14 | SnO | 10 | 20 | — | — | 41 | — | — | — | — | |||
| 15 | SnO2 | — | — | — | — | 41 | — | — | — | — | |||
| 16 | Ag-as prepared | 0.1 M KHCO3 | −1.1 | 63 | 2 | 8 | — | 18 | — | — | 6 | — | 208 |
| 17 | Ag-5 mint O2 plasma | 75 | 1 | 2 | — | 13 | — | — | 16 | — | |||
| 18 | Pt-as prepared | 83 | 1 | 2 | — | 10 | — | — | 3 | — | |||
| 19 | Pt-5 mint O2 plasma | 91 | 1 | 1 | — | 8 | — | — | 2 | — | |||
| 20 | Cu2O | 0.1 M KHCO3 | −1.0 | — | — | — | 45 | — | — | — | 17 | — | 204 |
| 21 | Cu2O(CO) | — | — | — | 55 | — | — | — | 13 | — | |||
| 22 | Cu2O(H2) | — | — | — | 38 | — | — | — | 17 | — | |||
| 23 | Cu after 1 hour | 0.1 M KHCO3 | −1.0 | — | 1.6 | — | 0.25 | — | — | — | — | — | 209 |
| 24 | Cu2O after 1 hour | — | 8 | — | 1.5 | — | — | — | — | — | |||
| 25 | NH2–Cu after 1 hour | — | 22 | — | 12 | — | — | — | — | — | |||
| 26 | NH2–Cu after 12 hours | — | 15 | — | 14 | — | — | — | — | — | |||
| 27 | 4H Au@Cu | 0.1 M KHCO3 | −1.06 | 18 | 52 | — | 14 | 9 | — | — | — | — | 71 |
| 28 | 4H/fcc Au@Cu | 22 | 23 | 3 | 43 | 7 | — | — | — | — | |||
| 29 | Od-Cu | 0.1 M KHCO3 | −1.05 | — | — | — | 38 | — | — | — | — | — | 207 |
| 30 | AP-Cu | — | — | — | 32 | — | — | — | — | — | |||
| 31 | Hd-Cu | — | — | — | 50 | — | — | — | — | — | |||
6.7. Doping strategy
Doping plays a role in developing e-CO2R. It involves intentionally introducing foreign atoms, such as metals or non-metals, into the catalyst structure to modify its electronic and chemical properties. This technique is extremely significant in the field of electrocatalysis because it allows for adjustment of the catalyst's ability to react with and select CO2 conversion products.Xu et al. have investigated the e-CO2R efficiency of Cu, Ni, and Mn doped CNTs. The overpotential values of Cu, Ni, and Mn doped CNT(8,0) and CNT(6,6) catalysts were found to be lower than those obtained with individual metal catalysts.210 Additionally, the Ce–Cu nanoparticles (NPs) exhibited exceptional performance in e-CO2RR, particularly in the production of the C2H4 product, with Ce–Cu-2 NPs achieving an impressive 53% FE for C2H4 at 150 mA cm−2. The addition of Ce to Cu resulted in an increase in the number of sites for catalysis and a reduction in particle size. This played a role in enhancing the activity of Ce–Cu NPs. Experimental evidence, including XANES and XPS analyses, indicated that the presence of Ce altered the oxidation state of Cu and caused the formation of OVs. Furthermore in situ SERS experiments supported the idea that Ce doping helped to stabilize CuOx species enabling adsorption of CO and subsequent interactions between carbon atoms.211
In order to explore how various compositions and structures of Cu–ZrO2 catalysts affect the conversion of CO2 to CH3OH through hydrogenation, a group of researchers led by Mao synthesized a range of catalysts. The catalytic performance of these catalysts was found to be influenced by factors like OVs, low valence copper species, the surfaces of ZrO2 and the size of pores. Methanol selectivity was particularly impacted by the interface between low-valence Cu species and ZrO2, where OVs were abundant.212 In another study, the research focused on the synthesis of Pd and Cu doped g-C3N4 nanotubes with a porous structure. These nanotubes exhibited exceptional gas-phase CO oxidation activity and durability, along with enhanced e-CO2R activity attributed to their high surface area.213 In a different study, a hydrothermal synthesis method was used to prepare a Cu-doped BiVO4 catalyst for e-CO2R (Fig. 20). This catalyst had excellent electrocatalytic performance, with the highest FEHCOO− of 87.15% at −1.0 V versus RHE, and a maximum current density of −20.68 mA cm−2 at …1.3 V versus RHE. The introduction of Cu was conducive to changing the electronic structure of BiVO4 and produced more active adsorption sites. Also, in a photo-electrocatalytic system, a WO3 nanotube array was made into a photoanode, which further improved the catalyst's performance. In the PEC system, formate yield was 3.3-fold higher at −0.9 V versus RHE than in the electrochemical system. Reflecting this fact is that there is a synergistic effect between photocatalysis and electrocatalysis.214
 | ||
| Fig. 20 The provided images (a)–(f) display SEM images of BiVO4 samples doped with varying concentrations of copper (Cu), including pristine (a) and 3% (b), 5% (c), 8% (d), 10% (e), and 15% (f) Cu-doped BiVO4. TEM images of undoped BiVO4 (g) and BiVO4 doped with 8% Cu (h). EDS of 8% Cu-doped BiVO4 (i). The study also examines the XRD pattern of pristine BiVO4 and Cu-doped BiVO4 (j and k). Reproduced with permission from ref. 214. Copyright 2023, Elsevier. | ||
A remarkable C2+ FE of 76.4% was found for Cu–Al MONFs at a high current density of 600 mA cm−2, demonstrating their superior ability in the electrocatalytic reduction of e-CO2R to C2+ products. This marked a significant difference compared to CuO nanofibers, which exhibited weak C2+ products and excessive hydrogen evolution. The introduction of aluminum through doping modified the electronic structure of Cu, optimizing intermediate binding and CC coupling, while also creating a mesoporous structure during the etching process.215 The study explored the in situ activation of Sn-doped Cu bimetallic electrocatalysts using pristine SnO2-decorated CuO nanoparticles. These catalysts demonstrated near-unity selectivity for CO2 to CO conversion and maintained steady performance for up to 15 hours. Ex situ electron microscopy, X-ray spectroscopy, and diffraction experiments shed light on the native shape and structure of these electrocatalysts. The in situ activated Sn-doped Cu electrocatalysts significantly enhanced the electrochemical conversion of CO2 to CO, achieving a record faradaic efficiency of 98% for CuO-0.4% Sn at 0.75 V applied potential versus RHE. In situ Raman spectroscopy and XRD measurements revealed catalyst activation and adsorbed species, both dependent on time and potential. The findings indicated that clean surface CuO was rapidly reduced within seconds, paving the way for the development of high-performance, low-cost electrocatalysts by doping Cu-based electrocatalysts with post-transition metals.216 Another study introduced Cu-doped CeO2 nanocrystals coated with carbon, synthesized through a one-step pyrolysis of MOFs (Fig. 21). These nanocrystals exhibited a significant CH4 partial current density of 138.6 mA cm−2, coupled with the highest FECH4 of 80.3%. The outstanding activity and selectivity of these nanocrystals were attributed to the synergistic effect of carbon encapsulation and the Cu/CeO2 active component.217
 | ||
| Fig. 21 The performance of e-CO2RR was evaluated for various samples in the flow cell. The FEs of all the products for the Cu/CeO2@C catalyst were measured at different applied potentials (a) and (b). The comparison of CH4 selectivity and CH4 partial current density among various samples (c). The CH4 selectivity and partial current density of Cu/CeO2@C were compared to those of previously reported electrocatalysts (d). A long-term stability test was conducted on Cu/CeO2@C at a potential of −1.5 V for a duration of 9 hours (e). Reproduced with permission from ref. 217. Copyright 2023, American Chemical Society. | ||
Kim et al. demonstrated that the production rate of CO over a CoCu SAA catalyst with trace doping levels of Co atoms in Cu could be doubled compared to that based on bare Cu. The material also displayed a high jC2H4 of 282 mA cm−2 at 1.01 V vs. RHE in a neutral electrolyte. This alloy improved the coverage of *CO intermediates necessary for the synthesis of multicarbon products and accelerated the conversion of CO2 to CO. The selectivity of CoCu SAA was enhanced toward ethylene over ethanol due to its active sites, which promoted the deoxygenation of *HOCCH, resulting in a yield of 15.6% ethylene.218 A high-performance Vo-CuO(Sn) catalyst containing OVs for e-CO2RR-to-CO conversion was produced using one-pot synthesis. The electrode's FECO was greater than 95% across a broad range of operating potentials, and its partial current density for CO was 35.22 mA cm−2. The catalyst's high activity was further demonstrated by its ability to achieve an optimal 99.9% FE at a low potential.219 Through in situ structural rebuilding of a CuS–Bi2S3 heterojunction precursor, a CDB electrocatalyst with an ampere-level current density and remarkable durability for e-CO2RR to formate was created. The catalyst's high conversion rate can be attributed to the electron-rich surface, a critical point both for its stability and activity, and it could effectively lower the kinetic barrier in the reaction.220
In another investigation, researchers explored the Cu/Sn ratio for e-CO2RR to CO. In undertaking experiments to use Sn-decorated Cu electrodes, they found that the catalyst is believed to be a Cu–SnO overlayer. These studies showed that for particle shaped electrodes, the CO partial current density becomes saturated at high overpotentials and then decreases. However, this phenomenon was not seen in dendritic shaped electrodes. Dendrite morphology increased the concentration of CO2 at the electrode surface, thus maintaining relatively constant FECO over a wide potential range.221 Another study explored electrochemically produced C2H6 from CO2 using a Cu2O-derived Cu working electrode and PdCl2 in a 0.1 M KHCO3 catholyte. A FE of up to 30.1% at a potential of 1.0 V was achieved. The process involved the initial reduction of CO2 at Cu sites, followed by hydrogenation using PdClx to produce C2H6. This approach required the presence of both copper and PdClx sites for effective reduction, and the conversion efficiency of other palladium-based particles was found to be comparatively lower.222 In another investigation, the study had revealed extremely porous Fe-CNPs through a process of amorphous SiO2 coating, carbonizing, and acid leaching. The presence of these nanoparticles had greatly aided the e-CO2RR to CO process, with their FE in a 1.0 M KHCO3 electrolyte reaching 98.8%. The research had also shown that the selectivity for e-CO2R had changed with porosity. Additional investigations on ZIF-derived carbon catalysts with Ni- and Co-doping had indicated that mesopores and macropores might enhance CO selectivity.223
Furthermore, Li et al. had demonstrated that doping Cu with p-block metal atoms could enhance e-CO2R to C2+ products at high current density by inducing p-d orbital hybridization. At a potential of −1.07 V vs. RHE, the CuGa catalyst had achieved a remarkable C2+ FE of 81.5% at a current density of 0.9 A cm−2. The catalyst had maintained its high C2+ productivity at 1.1 A cm−2 and a FE of 76.9%. P-d hybridization between Cu and Ga had increased C2+ selectivity, strengthened binding to the *CO intermediate, reduced the reaction energy barrier for CC coupling, and guaranteed an abundance of catalytic reactive sites.224 In a different study, Ni-substituted CuO and CuO catalysts had been synthesized using solution combustion synthesis. The research had demonstrated the significance of ionic nickel substitution through electrochemical analyses of Cu0.9Ni0.1O and Cu0.95Ni0.05O, both of which had exhibited exceptional redox-active behavior and higher electrocatalytic performance compared to CuO. Ionic connection between Ni2+ and Cu2+ had been crucial, as evidenced by the enhanced electrocatalytic activities of Cu0.9Ni0.1O and Cu0.95Ni0.05O in comparison to Ni metal and NiO-supported CuO. Steady-state chronoamperometry at a fixed potential of 0.2 V vs. RHE in a CO2-saturated electrolyte had also revealed the high activity and stability of nickel-doped catalysts. On Cu0.9Ni0.1O, the FE% values of CH4 and C2H4 had been roughly 3 and 4 times higher than those on CuO, significantly increasing the hydrocarbon selectivity.225 In another investigation, Al-PILC doped with Cu2+ ions and mixed metal pillars had both increased basal spacing, indicating effective dispersion of aluminum and copper. This increased surface area and volume had facilitated the adsorption and reduction of CO2. It is noteworthy that the CO2R reaction activity has been found to increase with the proportion of copper in the composite electrode.226 Information on a number of representative catalysts prepared through metal doping to convert CO2 into organic fuel is given in Table 10.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | Ce–Cu-3 NPs | 1 M KOH | 100 mA cm−2 | — | 35 | — | 47 | — | — | — | — | — | 211 |
| 2 | Ce–Cu-2 NPs | — | 38 | — | 47 | — | — | — | — | — | |||
| 3 | Ce–Cu-1 NPs | — | 42 | — | 38 | — | — | — | — | — | |||
| 4 | Cu NPs | — | 69 | — | 12 | — | — | — | — | — | |||
| 5 | Cu-doped BiVO4 | 0.5 mol L−1 NaHCO3 | −1.0 | 7 | — | — | — | 16 | — | — | — | — | 214 |
| 6 | Cu-doped BiVO4-3% | 3 | — | — | — | 48 | — | — | — | — | |||
| 7 | Cu-doped BiVO4-5% | 4 | — | — | — | 67 | — | — | — | — | |||
| 8 | Cu-doped BiVO4-8% | 1 | — | — | — | 87 | — | — | — | — | |||
| 9 | Cu-doped BiVO4-10% | 5 | — | — | — | 60 | — | — | — | — | |||
| 10 | Cu-doped BiVO4-15% | 16 | — | — | — | 45 | — | — | — | — | |||
| 11 | Cu3Al MONFs | 1 M KOH | 100 mA cm−2 | 12 | 51 | 18 | 9 | — | — | 8 | 1 | 215 | |
| 12 | Cu ONFs | 30 | 13 | 1 | 19 | 15 | — | — | 13 | 2 | |||
| 13 | Cu foil | 0.1 M KHCO3 | −0.75 | 57 | 19 | — | 9 | 13 | — | — | — | — | 216 |
| 14 | CuO | 41 | 40 | — | 3 | 14 | — | — | — | — | |||
| 15 | CuO-0.4% Sn | 3 | 98 | — | — | — | — | — | — | — | |||
| 16 | CuO-0.6% Sn | 17 | 77 | — | — | — | — | — | — | — | |||
| 17 | CuO-0.8% Sn | 27 | 68 | — | — | — | — | — | — | — | |||
| 18 | CeO@C | 0.1 M KOH | −1.1 | — | — | 5 | — | — | — | — | — | — | 217 |
| 19 | Cu/CeO@C | 13 | 12 | 60 | 9 | — | — | — | 1 | 1 | |||
| 20 | Cu/CeO | — | — | 45 | — | — | — | — | — | — | |||
| 21 | Bare CuO | 1 M KHCO3 | 100 mA cm−2 | 40 | 32 | 8 | 13 | — | 0.2 | 4 | — | 218 | |
| 22 | 0.2% Co-doped CuO | 27 | 41 | 0.4 | 8 | 19 | — | 0.2 | 3 | — | |||
| 23 | 1.0% Co-doped CuO | 27 | 32 | — | 9 | 26 | — | 0.2 | 3 | — | |||
| 24 | 3.0% Co-doped CuO | 30 | 26 | 0.3 | 5 | 35 | — | 0.1 | 2 | — | |||
| 25 | 5.0% Co-doped CuO | 50 | 6 | 0.3 | 7 | 27 | — | 0.5 | 5 | — | |||
| 26 | Vo–CuO(Sn) | 0.1 M KHCO3 | −1.03 | 9 | 91 | — | — | — | — | — | — | — | 219 |
| 27 | Vo–CuO | 15 | 75 | — | 2 | 8 | — | — | — | — | |||
| 28 | CuO | 31 | 19 | — | 5 | 45 | — | — | — | — | 220 | ||
| 29 | Cu-doped bismuth | 1 M KOH | −1.05 | — | — | — | — | 92 | — | — | — | — | |
| 30 | Cu–Sn dendrite | 0.1 M KHCO3 | −1.1 | 18 | 75 | — | 2 | 4 | — | — | 2 | — | 221 |
| 31 | Cu–Sn particle | 62 | 2 | — | 19 | 10 | — | — | 8 | — | |||
| 32 | Electropolished Cu | 24 | 1 | 6 | 20 | 3 | — | — | 10 | ||||
| 33 | Cu2O-derived Cu | 85 | 1 | 1 | 10 | 1 | — | — | 8 | ||||
| 34 | Cu2O-derived Cu with PdCl2 | 26 | 1 | 1 | 4 | 2 | — | — | 10 | ||||
| 35 | Fe-CNPs-w/o | 1.0 M KHCO3 | −0.88 | 48 | — | — | — | — | — | — | — | 223 | |
| 36 | Fe-CNPs | — | 59 | — | — | — | — | — | — | — | |||
| 37 | Ni-CNPs | — | 60 | — | — | — | — | — | — | — | |||
| 38 | Ni-CNPs-w/o | — | 45 | — | — | — | — | — | — | — | |||
| 39 | Co-CNPs | — | 38 | — | — | — | — | — | — | — | |||
| 40 | Co-CNPs-w/o | — | 15 | — | — | — | — | — | — | — | |||
| 41 | Cu | 1 M KOH | 1.1 | 38 | 10 | — | 29 | 2 | — | — | 19 | — | 224 |
| 42 | CuGa-II | 20 | 2 | — | 46 | 2 | — | — | 22 | — | |||
| 43 | Cu0.9Ni0.1O | 0.5 M NaHCO3 | −0.2 | 25 | 2 | 29 | 13 | — | — | — | — | — | 225 |
| 44 | Cu0.95Ni0.05O | 28 | 3 | 19 | 8 | — | — | — | — | — | |||
| 45 | CuO | 35 | 3 | 9 | 5 | — | — | — | — | — | |||
In a study, Yuan et al. explored novel materials for electrochemical reduction; they investigated Cu2S–X materials with varying starting morphologies (Fig. 22). These materials were successfully transformed into S–Cu2O–X through electrochemical reduction. By strategically optimizing the electronic structure and increasing the surface area via microstructure rebuilding, the efficiency of the catalyst in e-CO2R reduction and formate selectivity was significantly enhanced. Notably, the team demonstrated the ability of S–Cu2O-14 catalysts in formate synthesis, achieving an impressive 18% increase in current and a remarkable selectivity of 66.1%.227
 | ||
| Fig. 22 The FEHCOO− and partial current density were measured for Cu(OH)2, S-Cu2O-2, S-Cu2O-6, S-Cu2O-10, and S-Cu2O-14 electrodes at various applied potentials ranging from −0.6 V to −1.2 V vs. RHE. Reproduced with permission from ref. 227. Copyright 2023, Elsevier. | ||
In a parallel effort, using a co-doping approach Dai et al. converted CO2 to CH4 using copper-based catalysts. The Cu–N2B2 planar unit within their B-doped Cu–Nx set-up was the main focus of concern. Incorporation of boron into the coordination layer facilitated methane synthesis by increasing the strength of binding towards both CO* and CHO* intermediates. This strategic approach has been very successful, with the maximum FE for methane reaching as high as 73% and a peak methane partial current density of −462 mA cm−2 at −1.94 V vs. RHE. This demonstrates the excellent methane yield of the B-doped Cu–Nx material. The underlying reaction mechanism of the Cu–N2B2 coordination complex was exactly analyzed, employing a comprehensive two-dimensional reaction phase diagram. This multifaceted investigation contributes valuable insights into the design and performance of copper-based catalysts for efficient and sustainable CO2 conversion processes.81 Another research study led to alternative breakthrough as scientists ingeniously dispersed Cu and Zn onto microporous N-doped carbon, resulting in a remarkable catalyst tailored for the e-CO2R to produce CH4. The catalyst's performance metrics surpassed expectations, showcasing above-average efficiency, production rate, partial current density, and an exceptional 45-hour lifespan. The success was attributed to the unique combination of microporosity, excellent electrical conductivity, and the synergistic effect of Cu and Zn surface areas, which played pivotal roles in elevating its overall effectiveness.228 Simultaneously, Zhang et al. delved into the electrocatalytic landscape, scrutinizing the potential of various SACs and DACs for CO2RR. The results unveiled the secure attachment of Cu and Er atoms to the graphene framework, with ErNC emerging as the prime choice for facilitating the conversion of CO2 to CO. Among these options, CuErNC-I exhibited remarkable catalytic ability, favoring the production of CO molecules with minimal energy barriers.229 In a parallel endeavor, scientists harnessed the power of a Cu electrocatalyst supported by Ni–Ni–C, facilitating the transformation of carbon dioxide into hydrocarbons. Through ammonia-driven deposition precipitation, they effectively deposited copper nanoparticles onto carbon black and nickel–nitrogen-doped carbon substrates. The results, presented at potentials of −1.0 V vs. RHE, unveiled the impressive activity of these particles in driving the synthesis of methane and ethylene. Particularly notable, Cu nanoparticles supported by Ni–N–AC demonstrated a reduced C1/C2 product ratio, leading to a tenfold increase in CO generation and a twofold increase in partial ethylene current density.230
In a separate investigation, Patra et al. observed a phenomenon of high selectivity for C2+ hydrocarbons in boron-doped CuO, achieved along with a simultaneous reduction in overpotential requirements. The operational spectra of ATR-SEIRAS unveiled distinct CO intermediate species present on BCuO, attributed to the active sites of Cu+ and Cu0. This nuanced insight was linked to the availability of multiple CO adsorption sites and heightened CO binding energy within BCuO.231 In a separate advancement, Yang et al. introduced an innovative approach to CO2R through the utilization of copper selenide nanocatalysts. This introduction marked a novel path to methanol synthesis, boasting exceptional FE and current density at remarkably low overpotentials.232 Another study offered a method to fabricate Br-doped CuO multilamellar mesoporous nanosheets on Cu foam for e-CO2R. These nanosheets showcased a prolonged retention time for carbonaceous intermediates and provided numerous active sites for CO2 adsorption. Through the incorporation of Br ions, the electronic structure of CuO was transformed, inducing the formation of OVs and thereby amplifying CO2 adsorption capacity. With an admirable FE of 53.3%, the optimized Br1.95%–CuO emerged as a catalyst of choice for driving the e-CO2R to C2H5OH.233 Saxena et al. demonstrated that CuCo2Se4 was a highly selective catalyst for converting carbon dioxide into carbon-rich, high-value compounds. It generated acetic acid and ethanol at low potentials and formic acid in trace amounts at high potentials.234
Another study highlighted a high-performance e-CO2RR electrocatalyst (ONCuO) sustaining good selectivity for the C2+ product over time. Utilizing oxide-derived CuO and heteroatom doping, oxygen-assisted nitrogen doping on CuO resulted in defects such as OVs and grain boundaries. The selectivity for the C2+ product was 77%, and ONCuO remained stable for 22 hours.235 Ma et al. revealed that sulfur-modified Cu2O electrocatalysts could provide more targeted product dispersion (Fig. 23). The selectivity of these electrocatalysts for formate was extremely sensitive to the orientation of the Cu2O crystal faces. The improved S3-Cu2O-70 electrocatalyst displayed a formate efficiency of 90% and an extended lifetime.236
 | ||
| Fig. 23 The synthesis and subsequent characterization of a particular structure. Schematic representation of the process for preparing electrocatalysts (a). The SEM images of S0-Cu2O-70 (b) and S3-Cu2O-70 (c) are shown. TEM images of S0-Cu2O-70 and S3-Cu2O-70, along with their corresponding SAED patterns as insets, are presented. The HRTEM images of S0-Cu2O-70 (f) and S3-Cu2O-70 (g) are shown. The elemental mappings for S0-Cu2O-70 (h) and S3-Cu2O-70 (i) are also shown. Reproduced with permission from ref. 236. Copyright 2023, Wiley. | ||
The mechanism of the e-CO2RR for the formation of highly selective formic acid was studied using sulfur-modified Cu electrodes. It was discovered that most sulfur existed in unstable forms, with the stability of individual sulfur atoms dependent on their immediate surroundings and symmetry. The study observed that CO* coverage was up to 3.9 times higher than on clean Cu surfaces due to the significant adsorption of CO* by sulfur atoms.237 The selective production of formate using a sulfide-derived copper (SD-Cu) catalyst was attributed to stronger CO intermediate binding from subsurface sulfur atoms.238Table 11 provides an overview for evaluating the electrochemical performance of different electrocatalysts designed by following the non-metal doping strategy for conversion of CO2 to sustainable fuel.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | Cu(OH)2 | 0.1 M KHCO3 | −1.2 | 37 | — | — | — | 61 | — | — | — | — | 227 |
| 2 | S-Cu2O-2 | 54 | — | — | — | 43 | — | — | — | — | |||
| 3 | S-Cu2O-6 | 35 | — | — | — | 63 | — | — | — | — | |||
| 4 | S-Cu2O-10 | 35 | — | — | — | 63 | — | — | — | — | |||
| 5 | S-Cu2O-14 | 33 | — | — | — | 65 | — | — | — | — | |||
| 6 | Cu–SAs/NC | KHCO3 | −1.2 | — | — | 11 | — | — | — | — | — | — | 228 |
| 7 | Zn-SAs/NC | — | — | 12 | — | — | — | — | — | — | |||
| 8 | CuZn-SAs/NC | — | — | 70 | — | — | — | — | — | — | |||
| 9 | Ni–N-AC | 0.1 M KHCO3 | −1.07 | 48 | 52 | — | — | — | — | — | — | — | 230 |
| 10 | Ni–N-EC | 46 | 46 | — | — | — | — | — | — | — | |||
| 11 | Cu-VC | — | 3 | 12 | 14 | 12 | — | — | — | — | |||
| 12 | Cu-EC | — | 3 | 22 | 15 | 8 | — | — | — | — | |||
| 13 | Cu–Ni–N-AC | — | 10 | 15 | 30 | — | — | — | — | — | |||
| 14 | CuO | 1 M KOH | −0.6 | 20 | 31 | 2 | 23 | 15 | — | — | 3 | 1 | 231 |
| 15 | B–CuO | 13 | 35 | 2 | 32 | 7 | — | — | 4 | 1 | |||
| 16 | Cu2O | 0.5 M H2SO4 [Bmim]PF6–CH3CN–H2O | −2.1. Ag/Ag+ | 78 | 5 | — | — | 11 | — | 6 | — | — | 232 |
| 17 | Cu2S | 43 | 20 | — | — | 17 | — | 19 | — | — | |||
| 18 | Cu2Se | 33 | 12 | — | — | 31 | — | 23 | — | — | |||
| 19 | Cu1.63Se(1/3) | 2 | — | — | 21 | — | 78 | — | — | ||||
| 20 | CuSe | 35 | 28 | — | — | 22 | — | 19 | — | — | |||
| 21 | CuS | 63 | 15 | — | — | 10 | — | 12 | — | — | |||
| 22 | CuO | 42 | 23 | — | — | 22 | — | 5 | — | — | |||
| 23 | Cu | 62 | 20 | — | — | 10 | — | 3 | — | — | |||
| 24 | CuO | 0.1 M KHCO3 | −1.2 | 62 | 32 | — | — | — | — | — | — | — | 233 |
| 25 | Br1.95%–CuO | — | 18 | 8 | 37 | — | — | 31 | — | ||||
| 26 | CuCo2Se4 | 0.3 M NaHCO3 | −0.9 | 2 | — | — | — | 26 | — | 17 | 24 | 26 | 234 |
| 27 | CuO | 0.1 M KHCO3 | −1.0 | 26 | 6 | — | 43 | 5 | — | — | 10 | — | 235 |
| 28 | 5-N–CuO | 58 | 8 | — | 24 | 24 | — | — | 3 | — | |||
| 29 | 15-N–CuO | 45 | 8 | — | 35 | 35 | — | — | 6 | — | |||
| 30 | 25-N–CuO | 38 | 6 | — | 38 | 38 | — | — | 7 | — | |||
| 31 | 5-ON–CuO | 23 | 8 | — | 49 | 49 | — | — | 9 | — | |||
| 32 | 15-ON–CuO | 24 | 5 | — | 54 | 54 | — | — | 9 | — | |||
| 33 | 25-ON–CuO | 21 | 5 | — | 52 | 52 | — | — | 10 | — | |||
| 34 | c-Cu2O | 0.1 M KHCO3 | −1.1 | 20 | 21 | — | 25 | 20 | — | — | — | — | 236 |
| 35 | t-Cu2O | 23 | 18 | — | 32 | 18 | — | — | — | — | |||
| 36 | o-Cu2O | 33 | 16 | — | 19 | 22 | — | — | — | — | |||
| 37 | S3-c-Cu2O | 23 | 1 | — | 1 | 77 | — | — | — | — | |||
| 38 | S3-t-Cu2O | 24 | 3 | — | — | 74 | — | — | — | — | |||
| 39 | S3-o-Cu2O | 24 | 1 | — | — | 73 | — | — | — | — | |||
| 40 | N–Cu | KOH | −1100 mA cm−2 | 18 | 11 | — | 48 | 1 | — | — | 19 | — | 239 |
| 41 | P–Cu | 80 | 1 | 3 | 4 | 1 | — | — | 5 | — | |||
| 42 | S–Cu | 43 | 3 | 2 | 28 | 2 | — | — | 21 | — | |||
| 43 | O–Cu | 43 | 2 | 28 | 15 | 1 | — | — | 9 | — | |||
| 44 | Cu | 53 | 3 | 1 | 23 | 3 | — | — | 11 | — | |||
| 45 | Cu-Foil | 0.1 M KHCO3 | −0.9 | 38 | 11 | — | — | 14 | — | — | — | — | 238 |
| 46 | SD-Cu1 | 38 | 45 | — | — | — | — | — | — | — | |||
| 46 | S-DCu2 | 39 | 37 | — | — | — | — | — | — | — | |||
6.8. Facet engineering approach
The facet engineering is particularly important in catalyst design for e-CO2R because it can control the catalytic properties of designed materials. In this technique, the faces of catalyst materials are responsible for the reactivity towards desired reaction products. In the facet engineering approach, the basic idea is to pick crystalline faces for making catalysts that facilitate pathways while minimizing undesired side reactions. On a larger scale, this approach also optimizes the efficiency and cost-effectiveness of CO2 capture and conversion systems. There are various methods available for incorporating the facet engineering strategy into CO2RR catalyst designs. A typical way is to synthesize catalyst materials under controlled growth conditions so as to have specific crystal planes exposed, or by using templates to do it. Another approach would be to selectively etch facets already present in the catalysts, thereby bringing out the reactive site(s) of interest instead. Additionally surface modification techniques like depositing layers of metals or oxides onto facets can be utilized to engineer the surface properties of the catalysts. Researchers can tap into the power of facet engineering to fully utilize catalyst materials. This opens up opportunities for developing selective catalysts for CO2RR, which play a vital role in reducing greenhouse gas emissions and advancing sustainable energy conversion technologies.Gao et al. have introduced a novel approach to immobilize ionic liquids on Cu, forming a collaborative “Cu-ionic liquid heterointerface” for e-CO2RR. The process involves dissolving Cu2+ and AFIL in a H2SO4 electrolyte, directing their positive ions toward the GDL cathode, and embedding the deposited Cu. This embedding effect enhances Cu(100) production and incorporates more AFIL due to AFIL's capping influence. Consequently, Cu-IL/GDL exhibits smaller Cu particles with embedded AFIL, resulting in a lamellar particle structure. Operating under a total current density of 73.59 mA cm−2 at 1.17 V vs. RHE, this synergistic interaction between Cu and AFIL yields high selectivity for C2 products, notably C2H4 (40.67%). Especially, Cu-IL/GDL maintains relatively stable performance, with only a 3.8% decrease in current density observed after 5 hours of operation.240 Yu et al. have developed copper nanosheet arrays on copper foils, which promote the formation of multi-carbon products, resulting in a remarkable multi-carbon to single-carbon ratio of 7.2 (Fig. 24). Notably, potassium ions exhibit a fivefold higher adsorption density on the surfaces of copper nanosheets compared to pure copper foils.241
 | ||
| Fig. 24 Illustration of the electrochemical characteristics and efficacy of Cu foil and CuNS-0.8 (A). The CV curves of CuNS were obtained after reducing the CuONS at a potential of −0.8 V vs. RHE (B). The OH− adsorption profiles of CuNS-0.8 and Cu foil are shown in the left and right panels, respectively (C). The plot shows the differences in charging current density between CuNS-0.8 and Cu foil as a function of scan rates (D). The LSV curves of CuNS-0.8 and Cu foil were obtained in a CO2-saturated 0.1 M KHCO3 electrolyte using a scan rate of 10 mV s−1 (E). This study examines the product distributions and current densities of Cu foil and CuNS-0.8 at various potentials. The error bars indicate the standard deviation of three measurements (F). The partial current densities of C2H4 and C2+ products were measured on CuNS-0.8 and Cu foil at various potentials (G). The study also investigates the variation in the C2H4 to CH4 ratio on CuNS-0.8 and Cu foil under different potentials (H). The study investigates the variation in the ratio of C2+ products to C1 products on CuNS-0.8 and Cu foil under different potentials (I). Reproduced with permission from ref. 241. Copyright 2023, Elsevier. | ||
Additionally, researchers have harnessed colloidal techniques to craft Cu-based nanocrystals with tailored dimensions, shapes, compositions, and interior structures. Importantly, their exceptional activity and selectivity for C2+ during CO2R make them promising candidates for electrocatalysis.242 Shifting the focus to a different investigation, Mangione et al. investigated the geometry of copper electrodes in the context of e-CO2R, particularly over copper nanocubes. These cube-shaped copper catalysts, characterized by 100-degree terraces, 110-degree edges, and 111-degree corners, exhibit enhanced activity and selectivity in the production of ethylene. Nevertheless, the molecular mechanisms underlying this behavior have remained elusive. They also uncovered a dual facet mechanism occurring at the interface of the 100 terraces and 110 edges, highlighting that the reactivity of shape-controlled nano-catalysts extends beyond facet selectivity observed in single crystals.243 In a separate study examining the effectiveness of Cu(111) ML as a catalyst in the CO2 hydrogenation process, it is revealed that the Cu(111) ML surface exhibits higher adsorption energies for intermediates compared to the bulk Cu(111) surface. This preference for the creation of the *CO intermediate over the *COH intermediate leads to selective hydrogenation to CH3OH. The study identifies two rate-limiting processes in CO2 hydrogenation: the hydrogenation of CO2 to COOH and the hydrogenation of CO to CHO. Notably, when compared to bulk Cu and Cu nanocrystal-based catalysts, the predicted working potential for selective CO2 hydrogenation to CH3OH is lower on the Cu(111) ML surface.244
In another study, Cu nano-catalysts were tested in a gas-fed flow cell, showcasing their facet-dependent selectivity. When compared to a standard H-cell setup, this configuration reduced hydrogen generation while increasing FE towards CO2 reduction products. The interplay between the system and catalyst effects yielded cutting-edge product selectivity even at low applied voltage and high current densities. In 1 M KOH, Cu cubes exhibited a mass activity of 700 mA mg−1 and a selectivity of up to 57% for ethylene, while Cu octahedra achieved a selectivity of up to 51% for methane.245 In a related effort, Kim et al. employed a modified CVD graphene growth technique to create a Cu catalyst featuring a high density of step-sites and a high-facet atomic arrangement. This catalyst stood out for its remarkable ethanol productivity, achieving 40% FE at 0.9 V vs. RHE. This represents one of the highest values reported for copper-catalyzed CO2 conversion to ethanol. Furthermore, the fabrication process for this catalyst is simple and scalable.246
In another investigation, the researchers explored the selectivity of Cu catalysts for CO2RR by employing Cuoh and Cucub in a tandem arrangement with CO-producing Agsph. Both Cu–Ag catalyst configurations exhibited greater selectivity and activity toward C2H5OH and C2H4 compared to CH4 and H2. Cuoh–Ag catalysts displayed higher selectivity for C2H5OH than Cucub–Ag, with the C2H5OH to C2H4 ratio reaching 2.4. DFT calculations and a simple model revealed that active sites on the Cucub–Ag catalyst preferred *CHx and *CO coupling, followed by subsequent reduction, while more extended facets continued to promote ethylene. Cuoh–Ag catalysts exhibited higher selectivity toward ethanol.247 Furthermore, DFT simulations shed light on the facet-dependent nature of Cu electrode reactions, electrochemical potential, and product selectivity in CO2R. Cu(111) facilitates the synthesis of both methane and ethylene from a single CH2 species by promoting the formation of a COH* intermediate. In contrast, Cu(100) exhibits a preference for generating the CHO* intermediate at lower potentials, leading to ethylene production through C–C coupling and the reduction of C2 species. The competition between COH* and CHO* intermediates depends on the applied potential, impacting reaction rates and potential requirements, particularly involving the breaking of the C–OH bond. The partial reconstruction of the Cu(100) surface is proposed as a factor contributing to the observed differences between density functional theory predictions and electrokinetic studies.248
In another research endeavor, Cu2O NPs with tunable morphology were synthesized for efficient electrocatalysis in the conversion of CO2 to C2+ products. Among various Cu2O catalyst forms, the o-Cu2O catalyst exhibited superior efficiency and current density for C2+ generation during CO2RR. Impressively, it demonstrated stability during electrolysis at −1.1 V vs. RHE for 12 hours without destabilization. The abundance of active sites and the preservation of high-index faces in o-Cu2O led to increased C–C coupling during CO2RR.249 Additionally, the synthesis of three distinct Ag–Cu JNS-100s was achieved through confined growth of Cu on one of the six symmetrical faces of Ag NCs. Among these, the Ag65–Cu35 JNS-100 demonstrated the highest FEC2H4 (54%) and FEC2+ product (72%), showcasing its superior performance in CO2R towards C2+ products. The observed shifts in C2H4 production and the C2+/C1 product ratio hold promise for improved CO2RR outcomes.250
In a separate study, two nanoscale forms of copper molybdate (CuMoO4 and Cu3Mo2O9) were synthesized, with their crystal structures significantly influencing their electrochemical properties. In a 0.1 M alkaline solution, Cu3Mo2O9 exhibited improved catalytic performance for both HER and OER, indicating its potential as a bifunctional water splitting catalyst in alkaline environments. The enhanced activity was attributed to the presence of more active sites, potentially arising from OVs or the unique structure of Cu3Mo2O9 crystals.251 In another investigation, a new Cu2O NP film was produced using square-wave electrochemical redox cycling of high-purity Cu foils. This cathode achieved up to 98% FE for e-CO2R to practically pure formate under 45 atm of CO2 in bicarbonate catholytes. A two-electrode high-pressure electrolysis cell, combined with a recently designed NiFe hydroxide–carbonate anode in a KOH/borate anolyte, achieved a high energy conversion efficiency of up to 55.8% for continuous formate synthesis. Importantly, it was discovered that a Cu2O(111) oriented film was crucial for reducing CO2 efficiently when producing formate.2
Using density functional theory, Zheng et al. conducted an analysis of the e-CO2R on four distinct Pd3Au alloy bimetal catalyst surfaces. The findings demonstrated that the catalytic activity and selectivity were influenced by the catalysts' d-band populations, charge distributions, and morphologies. The Pd3Au(100) surface exhibited strong HER selectivity at low applied potentials, while the Pd3Au(110) surface was more active in formic acid production. The (211) surface exhibited high selectivity for methanol synthesis during e-CO2RR.252 In another study, a dynamic deposition-etch-bombardment approach was employed to regulate Cu(100) facets in e-CO2R, achieving a FE of 86.5% and a full-cell electricity conversion efficiency of 36.5% towards C2+ products in a flow cell setup. The single-pass yield for C2+ products from the electrode assembly system reached 13.2% at 12 A.253 Additionally, in another investigation, the impact of high-index facets on e-CO2R was explored. Through SWP treatment, copper NW catalysts with high selectivity for C2+ products and excellent suppression of side reactions were synthesized (Fig. 25). The FE of SW-Cu NWs at −1.1 V was found to be 57%, surpassing the FE of the original Cu NWs.254Table 12 offers a broad overview of different designed electrocatalysts for e-CO2RR following the facet engineering approach on Cu-based materials.
 | ||
| Fig. 25 LSV curves were obtained for Cu NWs and single-walled copper NWs (SW-Cu NWs) in N2- and CO2-saturated aqueous 1 mol L−1 KCl electrolytes. The scanning rate used in this study was 5 mV s−1 (A). The FE of each product was measured on both Cu NWs and SW-Cu NWs electrodes at various applied potentials (B). The distribution of Cu NWs and SW-Cu NWs in terms of product distribution is examined. The gray C1 compound consists of CO and HCOOH, while the red C2+ compound is composed of C2H4, C2H5OH, and n-propanol. The blue compound is H2 (C). The stability of SW-Cu NWs was tested at a potential of −1.1 V vs. RHE (D). Reproduced with permission from ref. 254. Copyright 2022, Wiley. | ||
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | GDL | KCl KHCO3 | — | 90.5 | — | — | 2.5 | — | — | — | — | — | 255 |
| 2 | IL/GDL | −1.18 | 82.9 | — | — | 2.6 | — | — | — | — | — | ||
| 3 | Cu/GDL | −1.16 | 31.2 | — | — | 22.8 | — | — | — | — | — | ||
| 4 | Cu-IL/GDL | −1.17 | 15.7 | — | — | 40.6 | — | — | — | — | — | ||
| 5 | Cu foil | 0.1 M KHCO3 | −1.0 | 29 | 16 | 16 | 19 | 23 | — | 1 | 1 | — | 256 |
| 6 | CuNS-0.8 | 32 | 2 | 3 | 37 | 7 | — | — | 9 | — | |||
| 7 | A-CuNWs | 0.5 M KHCO3 | −1.02 | 7 | 1 | 3 | 56 | — | — | — | — | — | 242 |
| 8 | H-CuNWs | 12 | 4 | 3 | 50 | — | — | — | — | — | |||
| 9 | Cusph | 0.1 M KHCO3 | −0.79 | 47 | 4 | 6 | 24 | 3 | — | — | 3 | — | 245 |
| 10 | Cucub | −0.75 | 12 | 10 | 3 | 60 | 2 | — | — | 3 | — | ||
| 11 | Cuoh | −0.96 | 40 | 10 | 40 | 5 | 2 | — | — | — | |||
| 12 | Cu film | 0.1 M KHCO3 | −1.2 | 20 | 2 | 55 | 10 | 5 | — | — | 2 | — | 246 |
| 13 | Wrinkled Cu | 30 | 2 | 32 | 10 | 5 | — | — | 5 | — | |||
| 14 | Cu film | 0.1 M KCl | 10 | 2 | 32 | 24 | 3 | — | — | 30 | — | ||
| 15 | Wrinkled Cu | 15 | 2 | 32 | 23 | 8 | — | — | 30 | — | |||
| 16 | Cuoh | 0.1 M KHCO3 | −1.1 | 39 | 3 | 15 | 10 | 8 | — | — | 7 | — | 247 |
| 17 | Cuoh–Ag | 32 | 50 | — | 2 | 2 | — | — | — | — | |||
| 18 | Ag | 38 | 52 | — | — | — | — | — | — | — | |||
| 19 | Cucu | 22 | 3 | 13 | 32 | 13 | — | — | 5 | — | |||
| 20 | Cucu–Ag | 20 | 55 | 1 | 8 | — | — | — | — | — | |||
| 21 | Ag | 38 | 52 | — | — | — | — | — | — | — | |||
| 22 | o-Cu2O | 1.0 M KCl | −1.1 | 25 | 7 | 34 | 13 | — | — | 15 | — | 249 | |
| 23 | d-Cu2O | 27 | 5 | 3 | 19 | 29 | — | — | 9 | — | |||
| 24 | c-Cu2O | 28 | 8 | 2 | 15 | 32 | — | — | 9 | — | |||
| 25 | Ag65–Cu35 JNS-100 | 0.1 M KHCO3 | −1.0 | 18 | 22 | 4 | 52 | 14 | — | — | — | — | 257 |
| 26 | Ag50–Cu50 JNS-100 | 16 | 23 | 1 | 54 | 16 | — | — | — | — | |||
| 27 | Ag25–Cu75 JNS-100 | 25 | 21 | 1 | 34 | 22 | — | — | — | — | |||
| 28 | 0.5h-SW-Cu2O/Cu @ 1 atm CO2 | 0.5 M KHCO3 | −1.04 | 68.9 | 1 | 8.9 | — | 16.1 | — | — | 4.9 | — | 2 |
| 29 | 0.5h-SW-Cu2O/Cu @ 45 atm CO2 | 8.4 | 1 | — | — | 79.4 | — | — | 3.7 | — | |||
| 30 | 0.5h-SW-Cu2O/Cu @ 60 atm CO2 | 16 | 1 | — | — | 81.2 | — | — | 1 | — | |||
| 31 | HRS-Cu | 0.1 M KHCO3 | −0.95 | 10 | 1 | 2 | 53 | — | — | — | 22 | — | 253 |
| 32 | Cu NWs | 1 M KCl | −1.1 | 78 | 2 | — | 8 | 1 | — | — | 2 | — | 258 |
| 33 | SW-Cu NW | 28 | 10 | — | 32 | 4 | — | — | 10 | — | |||
6.9. Coordination engineering approach
In the design of e-CO2RR catalysts, coordination engineering has taken on great importance as a means of achieving control over the behavior of the materials involved. This approach works through influencing the coordination environments surrounding metal atoms in catalyst structures. Coordination environment manipulation, however, can have a great effect on reaction rate and selectivity. Accordingly, the importance of coordination engineering lies in its potential to fine-tune the binding energies of CO2 and reaction intermediates so as to steer the pathways of reactions. These coordination sites designed by scientists are able to be observed as selective for specific e-CO2R products. However, to implement the coordination engineering strategy for designing electrocatalysts, there are several methods that can be employed. One used approach involves designing coordination polymers where the coordination environment around metal centers might be set up via ligand choices and metal ions in the medium. Another technique involves modifying surfaces using techniques like layer deposition, which allows for controlled deposition of catalytically active metal atoms onto support materials enabling the creation of well-defined sites for coordination reactions. Furthermore, when two distinct metals are deliberately combined the creation of catalysts can result in improved performance by leveraging synergistic effects. By employing coordination engineering techniques, we can effectively optimize CO2R catalysts thereby promoting the advancement of sustainable carbon capture and utilization technologies and facilitating the transition towards a low carbon future.In a comprehensive exploration of e-CO2R, multiple Cu-doped ZIF-8 samples were synthesized. These Cu-doped ZIF-8 catalysts exhibited excellent selectivity for methane and carbon monoxide, with the highest FECH4 achieved by Cu30%ZIF-8 at −1.6 V vs. Ag/AgCl. Furthermore, Cu30%ZIF-8 demonstrated a peak current density of −40 mA cm−2 at −2.1 V vs. Ag/AgCl, indicating high catalytic efficiency.259 Furthermore, the copper(II) bis-triazine bipyridine complex supported on carbon black was designed by Garcia et al. for e-CO2R. The electrocatalytic performance of the catalyst was monitored using differential mass spectrometry. They exposed the simultaneous production of methanol, formic acid, formaldehyde, carbon monoxide, and methane, indicating that the catalyst lacked product selectivity. Optimal results, with a FE of around 22%, were achieved with both 2.5% and 5% composition of the Cu complex on carbon black, making them suitable for polymeric electrolytic reactor fuel cell applications.260 Furthermore, a separate investigation showcased the potential of porphyrin-based catalysts for CO2 conversion. For this, a molecular Cu–porphyrin built porous framework demonstrated an excellent FE of 73.6%, indicating the promise of porphyrin-based catalysts in CO2 conversion (Fig. 26). Interestingly, this system produced only carbon monoxide as a hydrocarbon on the Cu–porphyrin support. Abundant copper-active sites efficiently absorbed CO2, and finite-element modeling highlighted the importance of isolating CO intermediates for hydrocarbon production.261
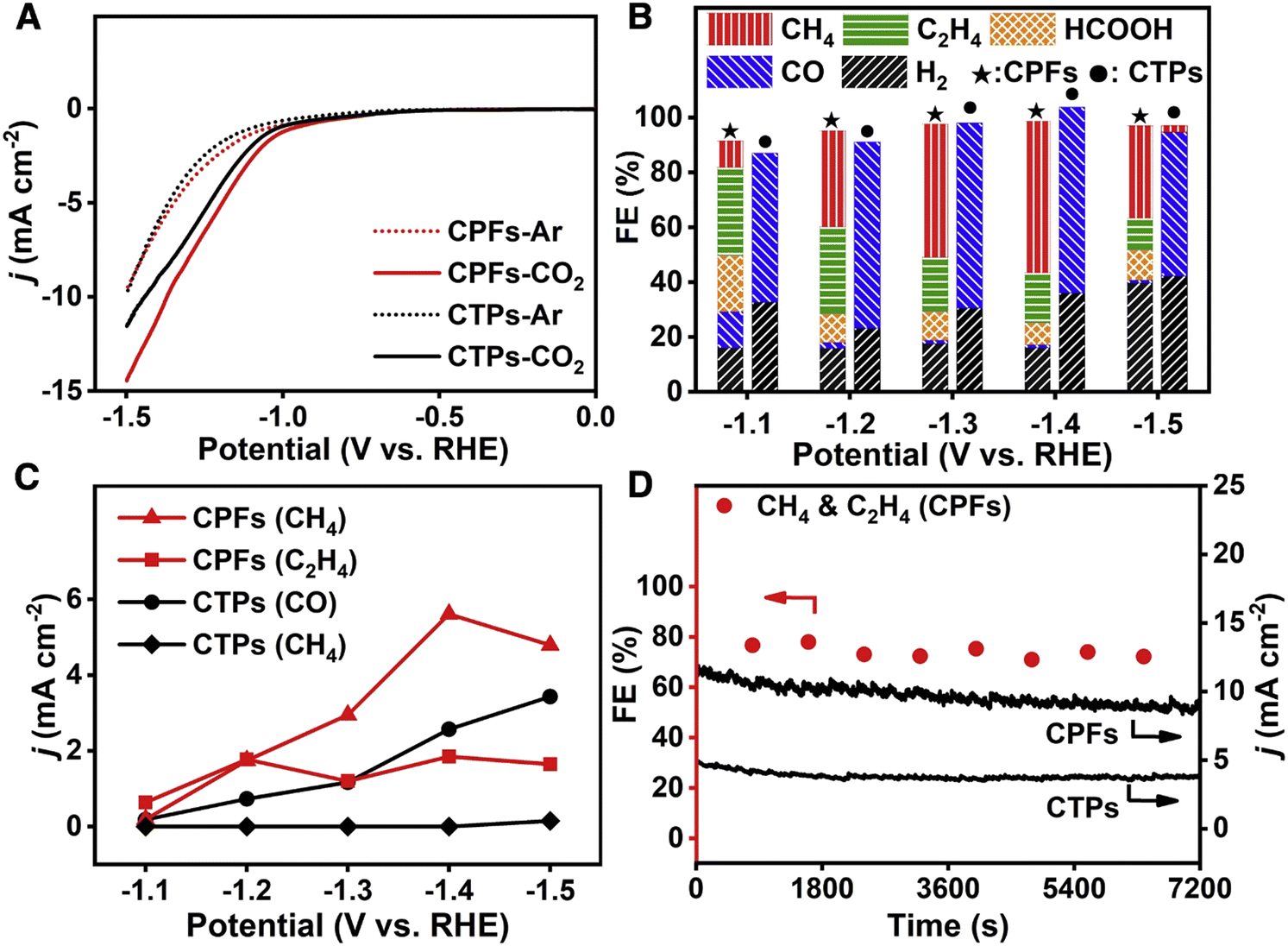 | ||
| Fig. 26 e-CO2R performance of carbon-based transition metal catalysts (CTPs) and carbon-based precious metal-free catalysts (CPFs) was evaluated by LSV curves at a scan rate of 10 mV s−1 in both Ar- and CO2-saturated solution (A). The FEs of various products (B). The partial current densities of various products (C). The FE and current density of CPFs and CTPs were measured at a potential of 1.4 V versus RHE (D). Reproduced from ref. 261. Open access article, Elsevier. | ||
Furthermore, the research proposed the utilization of the generalized coordination number (GCN) as an activity descriptor for e-CO2RR on Cu surfaces. This approach established linear scaling relationships between CO2RR intermediates and GCN at various surface sites, including cavities, step edges, twists, and adatoms. The activity-GCN volcano plot was employed to calculate the theoretical overpotential limit on Cu surfaces, revealing that overpotentials exceeding 0.12 V could not be reduced by surface engineering alone. It was suggested that dimerized surfaces might minimize the overpotential for converting CO2 to methane. Adatom surface engineering showed potential for reducing CO2RR overpotentials while inhibiting competitive HER.262 In a complementary effort, Zhou et al. have proposed the optimization of cobalt–nitrogen functionalized materials for CO2R processes through coordination engineering. This approach involved constructing a volcano diagram to assess activity levels in relation to CO adsorption energies. The study revealed that the absence of bonding in Co–O bonds, in contrast to Co–C or Co–N bonds in cobalt-centered motifs, contributed to increased catalytic activity. Moreover, the variation in vacancy formation energy of the cobalt atom emerged as a predictive factor for catalytic activity.263
Besides, another experiment showed that porous hollow copper microspheres (H-Cu MPs) were produced by combining the properties of EDTA-2Na to chelate ions and their natural ability to self-assemble. The adsorption of EDTA anions induced modifications in the geometrical structure and electronic distribution of H-Cu MPs, facilitating the selective production of ethene.264 In a related development, the Cu2O@Cu-MOF system was created through in situ etching, dissolving, and restructuring, resulting in a hybrid catalyst for the e-CO2R. The system exhibited an impressive overall FE of 79.4%, particularly excelling in forming hydrocarbon products, notably CH4. The remarkable FE ratio of 3.89 for CH4 to C2H2 underscores the excellent selectivity of this catalyst.265 Moreover, the results of another investigation unveiled the potential of Cu catalysts with varying Sn molar ratios for CO2RR. Cu/Sn catalysts containing less than 1.5% Sn exhibited higher FE in CO production, while maintaining nearly identical FEs in hydrocarbon production. Notably, the Cu/Sn-1.5 sample achieved a substantial CH4 partial current density of −27.5 mA cm−2 at −1.25 V vs. RHE.266 Furthermore, the research demonstrated the remarkable e-CO2R to C2H4 facilitated by a crystalline CuPc/C catalyst, which exhibited both high selectivity and reactivity. The crystallinity of CuPc was identified as a critical factor for efficient and selective C2H4 synthesis.267
In a related study, crystalline single-chain models (Cu-PzH, Cu-PzCl, Cu-PzBr, and CuPzI) were constructed to investigate product selectivity during e-CO2 in a flow cell. Predictions indicated different selectivity for CH4 and C2H4 due to variations in the lengths and dihedral angles between bi-copper atoms, which were contingent on the coordination microenvironment. Cu-PzH, at 1.0 V and a high current density, exhibited the highest FEC2H4 (60%) among all materials, while Cu-PzI, under the same conditions, showed the highest FECH4 (52%). These selectivity differences were found to be due to the synergistic effects between the surrounding catalytic active sites and the adsorption ability of the catalytically active center on critical reaction intermediates.268 Furthermore, excellent performance in selective CO2R into C2 compounds has been demonstrated when using a metal-azolate framework with a tetranuclear copper(I) cluster as a catalyst for e-CO2R. The multinuclear copper cluster in CuBPZ, which may encourage C–C coupling to create C2 chemicals, is believed to be responsible for the observed performance.269 In a complementary effort to enhance the selectivity of e-CO2R to CH4, Zhang et al. developed a single crystal of a copper(II) complex containing hydroxy groups, known as 5,10,15,20-tetrakis(3,4-dihydroxyphenyl)porphyrin copper(II) (Cu-PorOH). Cu-PorOH was found to serve as a highly efficient heterogeneous electrocatalyst for catalyzing the e-CO2R to methane. The hydroxy groups within Cu-PorOH construct solid three-dimensional frameworks and stabilize intermediate species, resulting in a remarkable FE of 51.3% for CH4 and a substantial partial current density of 23.2 mA cm−2 at 1.5 V vs. RHE in an H-cell.270
Additionally, the research highlights that ligand-protected Cu-hydride nanoclusters, such as Cu32H20L12, exhibit exceptional selectivity for e-CO2R at modest overpotentials. When copper clusters contain negatively charged hydrides, HCOOH becomes the preferred reduction product due to its lower overpotential compared to CO. However, at low overpotentials, hydrogen evolution competes more favorably than the lattice hydride process. Electrochemical studies of the Cu32H20L12 cluster support these predictions, indicating that HCOOH is the dominant product at low overpotential, while hydrogen production becomes more prominent at higher overpotentials.271 In a related effort to enhance the activity and stability of GDE for CO2 gas diffusion electrolysis, a hydrophobic molecule was incorporated into the Cu catalyst layer. This modification enabled strong-current electrolysis without flooding, due to the 1-octadecanethiol-modified Cu catalyst layer on the GDE. The modified GDE exhibited robust C2+ FEs of over 70.0% in the current density range of 100 to 800 mA cm−2, with a peak FEC2+ of 85.2% at 800 mA cm−2.272 In another study aimed at enhancing e-CO2R to acetic acid, Xiao et al. developed helical porphyrinic MOF CuTCPP on the surface of Cu(OH)2 nanoarrays. Electrodeposition was employed to create the nanoarrays on Cu foil, followed by immersion in a TCPP solution to assemble the chiral HCuTCPP@Cu(OH)2 nanoarrays. The electrocatalytic results demonstrated a high FEaceticacid of 26.1% at a potential of −1.6 V vs. Ag/Ag+, surpassing the performance of nHCuTCPP@Cu(OH)2 (19.8%).273
In a related research effort, the feasibility of e-CO2R into valuable alcohols was investigated. The study employed an elongated and fluorinated porphyrin structure to single out three model bicentric copper complexes, elucidating the relationship between their structure, properties, and performance. It was found that complexes with strong intramolecular tension and coordination asymmetry exhibited more active Cu centers. The hybrid structure of a Cu cluster and a partly reduced O-containing hexaphyrin ligand enabled the production of up to 32.5% ethanol and 18.3% n-propanol from CO2.274
In another research endeavor, a method was used to generate ultra-stable copper dendrites (Cu CF) with a high FEC2. The Cu CF was found to have a FEC2 of 90.6% and the partial current density was 453.3 mA cm−2. A 400 h stable electrolysis at 800 mA and even a ground-breaking stable operation at a large industrial current of 10 A were achieved in the membrane electrode assembly (MEA) form. The formation of a coordination compound was credited with having solved the problem. It generates a Cu(II) carboxylate on the surface of Cu CF, ensuring the stability of the Cu+ state and the material's hydrophobicity.275 Furthermore, the electrocatalyst is critical to obtaining and managing product selectivity in e-CO2R. In their study, two types of 2D MOFs were investigated, copper naphthalenedicarboxylate (Cu-UNDC) and copper benzenedicarboxylate (Cu-UBDC), to see how their ligand environment affected their selective properties and themselves. When ligand modifications were made, the MOF structure was altered, as well as the electrical environment around the copper center. This resulted in changes to product selectivity. The research team found that ligand modifications changed the structure of the MOF and thereby also its electrical environment around the copper center – which affected product selectivity. Under light exposure, Cu-UNDC had a FE of 24.3% for C2 products, and Cu-UBDC 26.2%.276 In addition, Han et al. combined the traditional HKUST-1 layout using the atomic layer infiltration (ALI) method to control the determining conditions at the Cu metal location. When Zn–O–Zn sites were added uniformly to HKUST-1, its FECO increased significantly from 20–30% to 70–80%. This change was made by reinforcing contact bonds and thus increasing the enthalpy of CO2 adsorption, as evidenced by density functional theory simulations.277
In another study related to the e-CO2R to high-energy-density C2+ products, the creation of carbon–carbon bonds was explored, particularly the OC–COH coupling pathway over the CO dimerization pathway. Density functional theory simulations in this study demonstrated that the adsorption of *CO species coupled with their hydrogenation species, *COH, took precedence over CO dimerization on low-coordinated copper sites. Remarkably, at 300 mA cm−2, the researchers achieved a FEC2 of 77.8% using a fragmented Cu catalyst with an abundance of low-coordinated sites.278 In a separate study, Cu2SnS3 nanoplates were synthesized and shown to be formate-selective over a wide potential and current range. When tested in a flow cell with a gas-diffusion electrode, the thiocyanate-capped Cu2SnS3 nanoplates exhibited a maximum formate FE of 92% and partial current densities of up to 181 mA cm−2. The high formate selectivity was attributed to favorable adsorption of HCOO* intermediates on cationic Sn sites, which were electrically regulated by thiocyanates coupled to neighboring Cu sites.279
Furthermore, the research evaluated the performance of MOFs for the e-CO2RR, contrasting those based on square–pyramidal CuO5 nodes with those based on square-planar CuO4 nodes. With a FE of 56% and a current density of 11.4 mA cm−2 at 1.4 V vs. RHE, the MOF (Cu-DBC) built from CuO5 nodes and catechol-derived ligands demonstrated excellent performance in the e-CO2R to CH4. On the other hand, Cu-HHTP and Cu-THQ were two additional MOFs that only produced CO as a reduced product. The study also showed that the CuO5 active sites can effectively hydrogenate CO into CH4 because the energy levels of metal d-orbitals in the square–pyramidal CuO5 site are higher than those in the square-planar CuO4 site, leading to the formation of CH4 rather than CO.280 Moreover, oxide-derived Cu (OD-Cu)-type catalysts were designed by Lu et al. which showed improved C2+ production, maybe due to Cu sites that are undercoordinated. Undercoordinated Cu sites have not been investigated for their stability in alkaline e-CO2R environments. Different crystalline phases and Sr/Cu ratios in strontium copper oxide catalysts were prepared. The C2+ FE of a SrCuO2 tetragonal phase catalyst is 53%, demonstrating its remarkable selectivity for C2+ products. SrCuO2 catalysts can retain or recover oxidized Cu species after being subjected to reductive e-CO2R conditions, as shown by ex situ X-ray absorption spectroscopy.281 The different electrocatalysts designed by following the coordination engineering approach are summarized in accordance with their CO2 conversion to different valuable products in Table 13.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | Cu30%ZIF-8 | 0.1 M KHCO3 | −1.2 Ag/AgCl | 84 | 7 | 1 | — | — | — | — | — | — | 259 |
| 2 | Cu20%ZIF-8 | 80 | 8 | 2 | — | — | — | — | — | — | |||
| 3 | Cu10%ZIF-8 | 82 | 8 | — | — | — | — | — | — | — | |||
| 4 | 1% Cu complex | — | −1.2 | — | — | — | — | — | — | 9 | — | — | 282 |
| 5 | 2.5% Cu complex | — | — | — | — | — | — | 21 | — | — | |||
| 6 | 5% Cu complex | — | — | — | — | — | — | 13 | — | — | |||
| 7 | 10% Cu complex | — | — | — | — | — | — | 20 | — | — | |||
| 8 | 20% Cu complex | — | — | — | — | — | — | 19 | — | — | |||
| 9 | CPFs | 0.1 KHCO3 | −1.1 | 18 | 12 | 31 | 20 | — | 9 | — | — | 283 | |
| 10 | CTPs | 33 | 55 | — | — | — | — | — | — | — | |||
| 11 | H-Cu MPs | 0.1 M KHCO3 | −0.91 | 22 | 4 | 17 | 50 | 4 | — | — | — | — | 264 |
| 12 | Cu2O@Cu-MOF | 0.1 M KHCO3 | −1.71 | — | — | 62 | 17 | — | — | — | — | — | 284 |
| 13 | Cu-MOF | — | — | 35 | 11 | — | — | — | — | — | |||
| 14 | Cu2O | — | — | 3 | 42 | — | — | — | — | — | |||
| 15 | Cu | −1.25 | 22 | 16 | 12 | 12 | — | — | — | — | — | 266 | |
| 16 | Cu/Sn-0.5 | 9 | 31 | 13 | 3 | — | — | — | — | — | |||
| 17 | CuSn-1.5 | 7 | 43 | 15 | 3 | — | — | — | — | — | |||
| 18 | Crystalline CuPC | KHCO3 | −1.6 Ag/AgCl | — | 5 | 5 | 26 | — | — | — | — | — | 285 |
| 19 | Non-crystalline CuPC | — | 28 | 2 | 6 | — | — | — | — | — | |||
| 20 | Crystalline CuPC (restored) | — | 5 | 5 | 35 | — | — | — | — | — | |||
| 21 | I-Cu-PzH | 1 M KOH | −1.1 | 10 | 23 | 9 | 48 | — | — | — | — | — | 286 |
| 22 | II-Cu-PzCl | 22 | 20 | 18 | 30 | — | — | — | — | — | |||
| 23 | III-Cu-PzBr | 20 | 18 | 20 | 30 | — | — | — | — | — | |||
| 24 | IV-Cu-PzI | 22 | 14 | 39 | 20 | — | — | — | — | — | |||
| 25 | CuBPZ | 0.1 M KHCO3 | −1.1 | 20 | 28 | 2 | — | 2 | — | — | 45 | — | 287 |
| 26 | Cu-PorOH | KHCO3 | −1.2 | — | 8 | 7 | 8 | — | — | — | — | 288 | |
| 27 | H-GDE | 1.0 M KOH | 100 mA cm−2 | 13 | 10 | 1 | 26 | 2 | — | — | 38 | — | 289 |
| 28 | Cu(OH)2 | — | −1.4 | — | — | — | — | 19 | — | — | — | — | 273 |
| 29 | nH-CuTCPP@Cu(OH)2 | — | — | — | — | 14 | — | — | — | — | |||
| 30 | H-CuTCPP@Cu(OH)2 | — | — | — | — | 6 | — | — | — | — | |||
| 31 | Hex-2Cu–O | 0.1 M KHCO3 | −1.0 | 13 | 28 | — | 4 | 27 | — | — | 7 | — | 274 |
| 32 | Hex-2Cu–2O | 45 | — | — | 5 | 33 | — | — | — | ||||
| 33 | Oct-2Cu | 60 | 38 | — | — | — | — | — | — | ||||
6.10. Site engineering approach
Site engineering can be used to design e-CO2R catalysts by precisely controlling the catalytic sites on the surface of a material. In this method, the sites such as metal atoms and defects are selectively used. By doing that, the reactivity and selectivity of the catalyst in reducing CO2 into different products can be optimized. Researchers can enhance the ability of these catalysts to facilitate desired chemical transformations while minimizing side reactions by designing and engineering these sites. Such a level of control is needed for shaping catalysts to produce CO2RR products and, therefore, site engineering is a powerful tool for developing sustainable energy conversion and carbon capture technologies. Many methods can be used to put the site engineering tactics into practice for designing CO2RR catalysts. The favored approach is to deposit metal nanoparticles or single metal atoms onto a support material to gain control over the sites. On the catalyst surface, as pointed out earlier, defect engineering can also be considered as a form of site engineering. This strategy involves developing catalysts within a molecular framework that show promise for a defined and specific active site. Researchers can improve the efficiency of catalysts by tuning the arrangement and properties of these sites. As a matter of fact, it facilitates the design of catalysts, which produce CO2RR products that are indispensable in the energy-efficient use of captured carbon dioxide and in response to the global demand to address the problem of climate change.To enhance CO2RR performance under a range of calcination conditions, MOF Cu(BTC) was designed with tunable valence states of Cu(II), Cu(I), and Cu(0) sites. Catalysts for the reactions of air, hydrogen, and nitrogen were all created using variations of the Cu/C-T-gas system. C2H4 was generated by Cu/C-T-air catalysts, which may have been due to the presence of both Cu(I) and Cu(II) and an abundance of VO. After 12 hours, the Cu/C-450-air demonstrated remarkable CO2RR characteristics, with a FEC2H4 of 34.8% at 1.1 V vs. RHE. Smaller grain size and a thinner carbon layer may have led to greater exposure to active sites and faster electron transport in the Cu/C-T-H2 system, leading to the production of C2H4, while in the Cu/C-T-N2 system, CH4 was generated.290
In parallel work, Liu et al. have provided a framework for exploring intrinsic activity differences between e-ND-Fe(II) and c-ND-Fe(II) in the context of CO2-to-CO conversion. Atomic Fe catalysts with changeable porosity architecture were synthesized via the pyrolysis of N-rich bimetallic Fe/Zn 2D ZIF, shedding light on the impact of ND configurations on the CO2RR activity of Fe–N4 active sites. Electrochemical studies revealed that the concentration of e-ND-Fe(II) in A-Fe@NG-LixKy catalysts was positively correlated with CO2RR activity. DFT calculations and in situ ATR-IR spectroscopy studies demonstrated higher electron density surrounding e-ND-Fe(II), which reduced CO bonding on metal sites, favoring CO2RR. The optimized sample A-Fe@NG-Li1K3 showed excellent performance, with a maximum FECO of 95%.291 Furthermore, Zhu et al. have optimized electrosynthesis pathways for selective fuel production by studying aqueous e-CO2R on Cu. Electrocatalysts with voltage- and facet-dependent CO2R selectivity were devised and produced from nine large-area single-crystal Cu foils with different surface orientations.292 Furthermore, the 2Bn-Cu@UiO-67 catalyst, doped with N-heterocyclic carbene (NHC), was designed and it achieved the e-CO2R to CH4 at a current density of 420 mA cm−2 with an outstanding FE of 81%. The catalyst maintained a FECH4 of greater than 70%, and it achieved a record-breaking turnover frequency of 16.3 s−1.293
Moreover, Cheng et al. investigated the active sites of OD-Cu catalysts for e-CO2R to multi-carbon compounds. Utilizing molecular dynamics simulation and density functional theory computations, they described realistic models of the OD-Cu surface and explored more than 150 surface locations. Ethylene formation was observed at planar-square sites and convex-square sites, while alcohol formation was favored at step-square sites due to the geometric influence of stabilizing acetaldehyde intermediates and destabilizing Cu–O interactions.294 Furthermore, lamellae of copper phyllosilicate were created in this research with CuO particles that were uniformly scattered and hence readily reducible to Cu+ and Cu0. Synthesizing a series of CuO/CuSiO3 with varying Cu loadings yielded Cu+–Cu0 sites. On 20% Cu/CuSiO3, the e-CO2R activity was greatly improved, with a FEC2H4 of 51.8% and a FEC2+C2 of 82% being attained. Activation of CO2, adsorption of *CO, and C–C coupling were all shown to involve Cu0 and Cu+, as calculated by in situ ATR-IR and DFT.295
Furthermore, Guan et al. showed that a nitrogen-coordination technique can be used to evenly distribute single-atom Cu catalysts over nitrogen-doped carbon. Nitrogen in nitrogen-doped carbon frameworks with Cu–Nx topologies promotes efficient dispersion and attachment of atomic Cu species. The pyrolysis temperature allows for fine-tuning of the doping concentrations and Cu–Nx combinations. For C–C coupling to occur and C2H4 to be produced, a very high concentration of Cu (4.9% mol) is required. The production of CH4 as C1 products is favored at low Cu concentrations due to the considerable distance between Cu and Nx species.296 Moreover, another research study investigated physical vapor deposition as a viable method for growing large-format Cu thin films intended for e-CO2R. These films epitaxially grew on Al2O3(0001), Si(100), and Si(111) substrates, each exhibiting a distinct out-of-plane orientation. X-ray pole figures revealed that Cu can be oriented in both low and high Miller index directions depending on the single-crystal substrate's orientation. In situ structural investigations identified three distinct Cu surface structures. Electrochemical tests demonstrated that thin-film orientations with more undercoordinated sites exhibit higher activity and selectivity for C–C coupling. Cu(751), characterized by fewer nearest neighbors on the Cu(S) − [n(110) (100)] surface, achieved the highest oxygenate/hydrocarbon ratio at 0.89 V vs. RHE.297
Additionally, the creation of a conductive di-nuclear cuprous complex with a short CuCu contact, formed by combining 1H-[1,10]phenanthrolin-2-one with Cu(I) ions, resulted in Cuophen. This complex demonstrated robust activity for the e-CO2R to C2H4, achieving a FE of 55.11% and a current density of 580 mA cm−2. Notably, a *CO species bridged two copper ions, forming a stable intermediate transition state and facilitating C–C interaction due to the reduced CuCu distance.298 In addition to this, a novel single-site copper coordination polymer known as Cu(OH)BTA was formed from e-CO2R using renewable electricity (Fig. 27). This polymer demonstrated a C2H4 selectivity 1.5 times higher than its metallic analogue and maintained a stable structure during the reaction. The presence of adjacent Cu in the polymer allows for the formation of a *OCCHO intermediate after CO hydrogenation, acting as a dual site. These polymers enable full-device CO2 electrolysis to run for 67 hours at an applicable current within a membrane electrode assembly.299
 | ||
| Fig. 27 The study investigated the FEs of various products at different applied potentials using Cu(OH)BTA (a) and Cu(OH)BTA-derived Cu (b) in a 1 M KOH electrolyte. The study also investigated the formation rates of C2+ products and C2H4 under various applied potentials using Cu(OH)BTA and Cu(OH)BTA-derived Cu in a 1 M KOH electrolyte (c). The study also measured the FEs of various gas products within the voltage range of 3.0–4.0 V using a membrane-electrode assembly (MEA) device (d). The electrosynthesis of C2H4 was performed in a MEA with a geometric electrode area of 4 cm−2. The catalyst maintained its overall current and ethylene FE for a duration of 67 hours (e). The error bars for the uncertainty in FE represent one standard deviation, which is calculated using three independent runs. Reproduced from ref. 299. Open access article, Nature. | ||
Moreover, a technique has been established to achieve the site-selective generation of AgPd nanodendrites on Au nanoparticles by using H2PdCl4 to galvanically replace the Ag layers on Au@Ag nanoprisms and a coreduction process involving silver and palladium ions. The selectivity of AgPd nanodendrite deposition on Au nanoprisms is determined by the concentration of H2PdCl4. These nanoprisms exhibit a strong electrochemical activity for CO2R, particularly at the most favorable reduction potential for CO2 at 0.18 V vs. RHE.300 Furthermore, a novel technique has been developed to create atomically scattered Fe–N5 sites on defect-rich porous carbon nanofibers through electrospinning and annealing. The Fe–N5/DPCF electrocatalyst exhibits exceptional e-CO2RR, with a substantial jCO of 9.4 mA cm−2, high FECO (>90%), and sustained stability even after 25 hours of electrolysis. This catalyst operates reliably for an extended period and provides a high power density of 1.3 mW cm−2.301 Additionally, Varandili et al. have introduced Cu/CeO2−x heterodimers (HDs) to illustrate the synergistic effects at metal/metal oxide interfaces that enhance selectivity in the e-CO2RR. The Cu/CeO2−x HDs achieve up to 80% selectivity against CO2RR and 54% FE for methane at −1.2 V vs. RHE, which is five times higher than that in the case of physically mixed Cu and CeO2−x nanocrystals. In situ and in operando X-ray absorption spectroscopy studies reveal the partial conversion of Ce4+ to Ce3+ in the HDs during CO2RR.302 Furthermore, to construct catalysts with high selectivity towards C2 products in e-CO2R, a Cu foil kinetic model featuring numerous nanocavities and a larger reaction rate constant k is presented by Han et al. This model promotes the production of C2H4 by increasing the concentration of adsorbed CO and decreasing the C–C coupling barrier for CO intermediates. The C2H4/CH4 ratio on commercial Cu foil treated with cyclic voltammetry is 4.11, aligning with this concept and representing an 18-fold increase compared to untreated Cu foil.303
Further, a Cu-based MOF as a catalyst was used by Zhu et al. with a FE of 92%/88%, and the partial current density was 9.8/18.3 mA cm−2 at a potential of 1.2/−1.3 V, making it the most active system for producing methane. The trigonal pyramidal Cu(I)N3 produced on-site acts as the electrochemical active site, stabilizing critical intermediates and suppressing HER.304 Furthermore, three Cu-MOFs with varying copper(II) site distributions were reported. The FE of Cu1 was four times higher than that of FEH2, while the FE of Cu2 was twice as high.305 The different electrocatalysts and their e-CO2R products developed by the site engineering approach are tabulated in Table 14.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | Cu/C-550-air | 0.1 M KHCO3 | −1.1 | — | — | — | 27 | — | — | — | — | — | 306 |
| 2 | Cu/C-450-air | — | — | — | 35 | — | — | — | — | — | |||
| 3 | Cu/C-350-air | — | — | — | 26 | — | — | — | — | — | |||
| 4 | Cu/C-550-N2 | — | — | 20 | 6 | — | — | — | — | — | |||
| 5 | Cu/C-450-N2 | — | — | 35 | 11 | — | — | — | — | — | |||
| 6 | Cu/C-350-N2 | — | — | 12 | 25 | — | — | — | — | — | |||
| 7 | Cu/C-550-H2 | — | — | — | 28 | — | — | — | — | — | |||
| 8 | Cu/C-450-H2 | — | — | — | 33 | — | — | — | — | — | |||
| 9 | Cu/C-350-H2 | — | — | — | 16 | — | — | — | — | — | |||
| 10 | Cu2S/CM | 0.1 M KHCO3 | −1.1 | 50 | — | — | — | — | — | — | — | — | 307 |
| 11 | A-Fe@NG-Li1K3 | 0.5 M KHCO3 | −0.75 | 22 | — | — | — | — | — | — | — | 291 | |
| 12 | Fe/Mn–N–C | 0.1 M KHCO3 | −1.0 | 33 | 65 | — | — | — | — | — | — | — | 308 |
| 13 | Fe–N–C | 75 | 30 | — | — | — | — | — | — | — | |||
| 14 | Mn–N–C | 85 | 12 | — | — | — | — | — | — | — | |||
| 15 | N–C | 68 | 10 | — | — | — | — | — | — | — | |||
| 16 | 20% Cu/CuSiO3 | 0.1 M KHCO3 | −1.1 | 16 | 14 | 4 | 53 | — | — | — | 8 | — | 309 |
| 17 | Cu–N–C-800 | 0.1 M KHCO3 | −1.0 | — | — | 3 | 9 | — | — | — | — | — | 296 |
| 18 | Cu–N–C-900 | — | — | 7 | 1 | — | — | — | — | — | |||
| 19 | Cuophen | 0.1 M KHCO3 | −1.0 | 69 | 2 | 6 | 10 | — | — | — | — | — | 310 |
| 20 | Cu(OH)BTA | 0.1 M KHCO3 | −0.9 | 35 | 10 | 1 | 42 | 2 | — | — | 8 | — | 311 |
| 21 | Cu(OH)BTA-derived Cu | 10 | 43 | — | 30 | 5 | — | — | 2 | — | |||
| 22 | DPCF | 0.5 M KHCO3 | −0.7 | — | 82 | — | — | — | — | — | — | — | 312 |
| 23 | Fe–Nx–/PCF | — | 50 | — | — | — | — | — | — | — | |||
| 24 | Fe–N5–/DPCF | — | 63 | — | — | — | — | — | — | — | |||
| 25 | Cu (15 nm)/CeO2 HDs | 0.1 M KHCO3 | −1.1 | 40 | 1 | 9 | 25 | 15 | — | — | 1 | 302 | |
| 26 | Cu (24 nm)/CeO2 HDs | 25 | 16 | 12 | 31 | 4 | — | — | — | — | |||
| 27 | Cu (36 nm)/CeO2 HDs | 20 | 5 | 40 | 18 | 1 | — | — | — | — | |||
| 28 | Cu (54 nm)/CeO2 HDs | 35 | 14 | 23 | 8 | 13 | — | — | — | — | |||
| 29 | Fe–N–C | — | −0.7 | 65 | 43 | — | — | — | — | — | — | — | 313 |
| 30 | Ni–N–C | 10 | 80 | — | — | — | — | — | — | — | |||
| 31 | Mn–N–C | 72 | 30 | — | — | — | — | — | — | — | |||
| 32 | Co–N–C | 92 | 10 | — | — | — | — | — | — | — | |||
| 33 | Cu–N–C | 62 | 28 | — | — | — | — | — | — | — | |||
6.11. Strain engineering approach
The approach of strain engineering has become a strategy for designing catalysts for e-CO2R. Its significance lies in the ability to finely adjust the properties of materials, especially copper and other metal catalysts, to enhance CO2RR performance. By incorporating controlled strains into the crystal structure of catalyst materials, researchers can manipulate the arrangement of atoms on the surface. This has an effect on the selectivity and activity of the catalyst allowing for the production of carbon-based products like ethylene or ethanol while minimizing unwanted byproducts. The goal of strain engineering is to improve the efficiency of CO2R by developing new catalyst formulations. Thus, it holds promise as a sustainable path toward energy solutions based on chemistry and also possible new carbon capture technologies. There is a way to practice strain engineering on CO2RR catalyst design. One common technique is nano-structuring through methods such as electrodeposition and chemical vapor deposition, which create nanomaterials with specific shapes and sizes. Another effective method is surface doping or alloying, which involves introducing atoms or molecules onto the catalyst surface to induce strain and alter its properties. Furthermore, subjecting the catalyst material to stress or pressure can induce strain, which has demonstrated potential in improving the performance of CO2RR.Kim et al. have shown that CO2 distribution on model-strained Cu(001) surfaces can be controlled using strain engineering (Fig. 28). The kinetics of CO2R to single-carbon products are influenced by tensile strain, which moves the center of the d-band closer to the Fermi level as the film thickness decreases. While C2+ product activity remains stable, CO2R reduction kinetics slow down, leading to fewer single-carbon products. Increases in adsorption energy and surface coverage of CO and H are suggested by the greater d-band center, which improves selectivity for C2+ products relative to CO and CH4. Hydrogen adsorption becomes more attractive as tensile strain rises, surpassing the faradaic current for CO2R.314
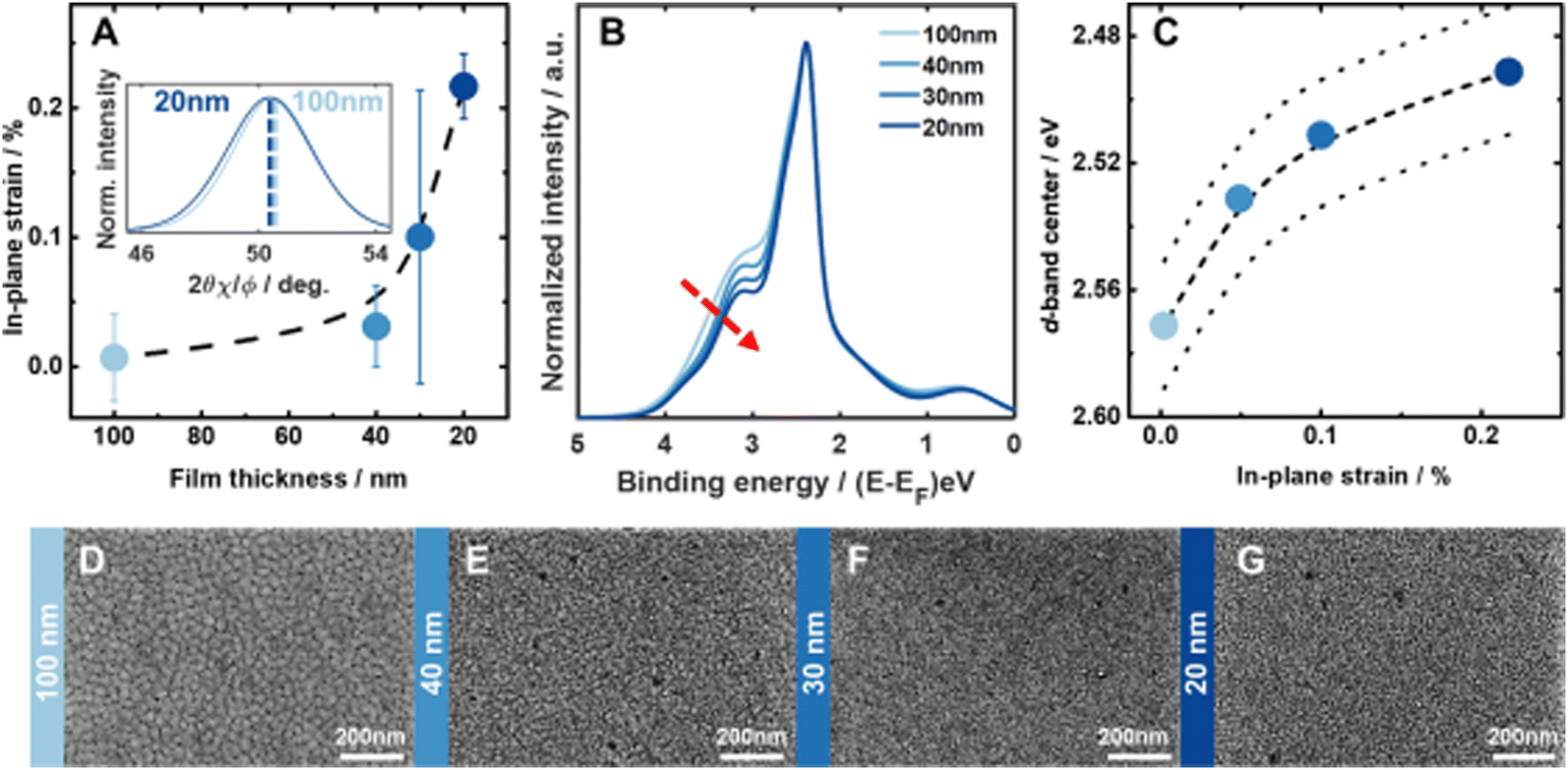 | ||
| Fig. 28 The study examines the structural properties of epitaxial Cu(001) on single-crystal Si(001). The in-plane strain at the surface was measured as a function of film thickness using grazing-incidence XRD. The inset illustrates the decrease in the angle of the Bragg peak as the film thickness decreases (A). The obtained He–I ultraviolet photoelectron spectroscopic valence band spectra is represented in (B). The d-band center from the ultraviolet photoelectron spectroscopy is examined as a function of in-plane strain (C). The 99% confidence interval is represented by dotted lines. SEM images of Cu(001) films with varying thicknesses were obtained. The thicknesses of the films are as follows: (D) 100 nm, (E) 40 nm, (F) 30 nm, and (G) 20 nm. Reproduced with permission from ref. 314. Copyright 2021, American Chemical Society. | ||
Additionally, Kang et al. have demonstrated the effectiveness of self-supported PS–Cu catalysts with tensile strain and a porous microstructure. These catalysts outperform Pt foil at high current densities and exhibit superior pH-universal catalytic activity for the HER compared to Cu NPs and Cu foam. Importantly, PS–Cu displays resistance to corrosion and remains stable over time.315 Moreover, an analysis of the impact of water solvation and uniaxial lattice strain on CO2RR across Cu(211) and Cu(100) surfaces has been conducted by Du and An. The results reveal that tensile strain enhances the binding strength of COOH and CHO, while diminishing the binding strength of CO and COH. This phenomenon arises due to the distinct strain effects induced at the surface site by their respective binding arrangements. Notably, the study highlights the variability between implicit solvation corrections and explicit water solvation, which is crucial for accurately estimating the solvation effect on CO2RR.316
By analyzing the pure ligand effect on Cu and the combined influence of ligand and strain effects on Cu, Maark et al. have investigated two types of bimetallic hetero-surfaces: Cui/Mj/Cuk sandwiched surfaces and Cu/Mj overlayers. To mitigate the ligand impact independently, several strategies were employed, including moving the M layer deeper into the Cu lattice, placing two MLs of M directly below the top Cu layer, and combining these two strategies in the sandwiched surfaces. By doubling the number of Cu MLs in the overlayers, the ligand impact was effectively reduced. The research findings indicate that the ligand effect strengthens the binding energies of all adsorbates in Cu1/M1/Cu4 and Cu1/M5, leading to an increase in overpotential for Rh and a slight decrease for Ni.317 Furthermore, another study delves into the investigation of adsorption energies for critical intermediates in CO2R processes over core–shell type heterostructure catalysts. It explores the impacts of strain and ligand effects on these adsorption energies. Notably, the research observes that the adsorption of *CO and *COH is less sensitive to strain compared to the adsorption of *COOH and *CHO on the Cu(111) surface, revealing that the adsorption energy variations are dependent on the specific adsorbate. The findings highlight certain catalyst models among Cu–M hetero-layered slab structures that exhibit promise for reducing overpotential during e-CO2R due to the interplay of strain and ligand effects. Additionally, the research emphasizes that strain-insensitive adsorbates like *CO and *COH tend to benefit more from the ligand effect.318
In a separate study, an initial metastable Au overlayer was synthesized using a two-step solution approach. The success of this approach is attributed to the significant interface strain within the AuCu3@Au core–shell structure, which provided support for the overlayer. The metastable Au exhibited increased FE and enhanced specific activity for CO2R to CO. This increase in the reaction rate can be attributed to the downshift of the energy barrier for the production of the intermediate COOH* due to the upshift in the d-band center.319 Another line of research focuses on the e-CO2RR facilitated by heteroatom-doped Cu-based catalysts, known for their high activity and selectivity. However, the complexity of Cu-based materials often makes it challenging to pinpoint the specific factors responsible for performance improvements. This study highlights that lattice strain adjustment in Cu-based catalysts can be used to control the activity and selectivity of e-CO2RR. Specifically, tensile-strained Sn/Cu alloy catalysts, by suppressing the dimerization process and promoting the formation of HCOOH and H2, enhance CO production due to the predominant adsorption of CO and lower adsorption free energies of COOH.320 Furthermore, in a separate study, a synthetic method is presented to maintain tensile strain in a copper/ceria heterostructure, thereby increasing selectivity for CO2R. This approach employs metal/metal oxides. The research demonstrates that carbonaceous intermediates are better adsorbed due to the tensile strain in the copper domain, and it facilitates *CO dimerization by creating an inadequate electron environment around interfacial Cu sites. Consequently, this method leads to a maximum FE of 76.4% and increases the efficiency of converting half-cell power to 49.1%.321
In a separate investigation, Feng et al. explore the influence of atomic dopants on the activity of copper-based electrocatalysts during CO2R processes. Notably, doping Cu2O with atomic Gd is found to significantly enhance the catalyst's performance. To achieve this, Gd1/CuOx catalysts were developed, taking advantage of the unique electrical structure and large ion radii of Gd. Gd doping induces tensile strain during the reaction, stabilizing the Cu+ species and resulting in superior catalytic performance. These catalysts achieved a partial current density of 444.3 mA cm−2 at 0.8 V against a hydrogen electrode, with a remarkable FE of 81.4% for C2+ products.322 In another study, bimetallic alloy structures with engineered strain played a crucial role. A remarkable FE of 96.6% for CO2-to-CO conversion was achieved by strategically managing lattice stresses in bimetal MNi alloys using a strain relaxation technique. Molecular dynamics simulations were employed to reveal that the relaxation of strained PdNi alloys can be controlled by increasing the synthesis temperature. DFT simulations further demonstrated that strain relaxation, by maximizing the energies of intermediate production, enhances CO2RR activity and selectivity.323 Furthermore, a novel class of Pd/Cu core/shell icosahedra with a tensile-strained Cu shell has enabled the efficient production of syngas with controllable compositions via the CO2RR. By adjusting the Pd/Cu composition, the molar ratio of H2/CO in syngas can be tuned from 1/1 to 2/1 and 3/1.324Table 15 shows the effect of strain on different electrocatalysts synthesized by the strain engineering approach upon CO2 conversion products.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | fcc Au | 0.1 M KHCO3 | −1.0 | — | 60 | — | — | — | — | — | — | — | 319 |
| 2 | o-AuCu3@fct Au | — | 88 | — | — | — | — | — | — | — | |||
| 3 | Cu2O/CeO2-0.18 | KOH | −0.75 | 12 | 16 | 2 | 38 | 2 | — | — | 15 | — | 321 |
| 4 | Pd ico/C | 0.1 M KHCO3 | −1.0 | 78 | 17 | — | — | — | — | — | — | — | 324 |
| 5 | Pd2.2Cu ico/C | 48 | 37 | — | — | — | — | — | — | — | |||
| 6 | Pd6.1Cu ico/C | 51 | 17 | — | — | — | — | — | — | — | |||
| 7 | Pd4.3Cu ico/C | 63 | 21 | — | — | — | — | — | — | — | |||
| 8 | CuOx | 2 M KOH | −1.0 | 25 | 9 | — | 40 | 2 | — | — | 20 | — | 322 |
| 9 | 6.5% GD1/CuOx | 18 | 8 | 1 | 40 | 1 | — | — | 30 | — | |||
| 10 | s-PdNi/CNFs-1000 | 0.1 M KHCO3 | −1.18 | — | 52 | — | — | — | — | — | — | — | 323 |
| 11 | s-PdNi/CNFs-800 | — | 64 | — | — | — | — | — | — | — | |||
| 12 | PdNi/C | — | 83 | — | — | — | — | — | — | — | |||
| 13 | s-AuNi/CNFs | — | 90 | — | — | — | — | — | — | — | |||
| 14 | s-AgNi/CNFs | — | 68 | — | — | — | — | — | — | — | |||
| 15 | CuO | 1 M KOH | −1.0 | 26 | 19 | — | 28 | 22 | — | — | 5 | — | 320 |
| 16 | Sn–CuO-7.5 | 13 | 75 | — | 15 | — | — | — | |||||
6.12. Precursor derived approach
In the development of catalysts for e-CO2R, the approach that relies on precursor materials is becoming increasingly important. It makes possible the composition and structure of catalysts to be controlled. That involves the creation of catalyst materials by synthesizing them from selected precursor compounds, from which these compounds can be separated once they have been prepared. The main benefit of this method is that it enables the creation of designated catalysts, with chemical compositions, atomic structures and surface properties displayed in an orderly array. And by selecting precursor compounds, and optimizing synthesis conditions, researchers are free to tailor the properties of catalysts to achieve high selectivity and activity in converting CO2 to valuable products such as hydrocarbons or alcohols. There are also scalability and affordability benefits to the precursor-based approach, which therefore provides an encouraging possibility for solving the problems associated with carbon capture and utilization. The precursor-based approach also has a number of techniques for designing CO2 reduction catalysts. In one method, inorganic precursor materials are decomposed to carbon-based catalysts having defined structures and active sites by controlled heating at a temperature or by pyrolysis. Another process is called sol–gel, in which precursor compounds are mixed in a solution and then, by controlled drying and controlled thermal processing, transformed into catalytic materials. Chemical vapor deposition (CVD) is a method that allows the placement of catalysts derived from precursors onto substrates. This enables integration of these catalysts into setups promoting their usefulness in achieving a sustainable and low carbon future. Researchers can effectively utilize the potential of the precursor derived approach, by selecting precursor compounds and optimizing synthesis parameters resulting in the development of highly efficient and selective CO2RR catalysts.In a specific study, using ultrasonic irradiation, Guzmán et al. have developed novel catalysts for e-CO2R, achieving increased selectivity towards H2 and C1 products (CO and formate) in aqueous media (Fig. 29). These catalysts harnessed the synergistic interaction between ZnO and CuO metal oxides, resulting in over a 1.4-fold increase in syngas productivity for the ultrasound-prepared CuZ catalysts. Furthermore, adjusting ultrasonic amplitudes increased the BET surface area by 100%, leading to a more than 14% improvement in FE towards alcohols in the RDE system at a catalyst loading of 0.2 mg cm−2. They have also discussed the electroactivity in terms of precursor concentration, duration of ultrasound-assisted precipitation, and calcination temperature of the synthesized powders, allowing fine-tuning of the physical and chemical properties of the nanoparticles. To optimize the synthesis of *CO intermediates and facilitate their dimerization into multi-carbon products, mesoporous materials with a mean pore size of approximately 20 were engineered to create ideal conditions.325
 | ||
| Fig. 29 The LSV responses, evolution time of total current density for the e-CO2R, FE, and productivity of gas and liquid products were measured for Cu-06 and Cu-06-%30-A (0.6 mg cm−2) in a 1 M KHCO3 aqueous electrolyte at a potential of −0.99 V vs. RHE (a–d). The chronoamperometry responses (e) and FEs were measured for a Cu-06-%30-A catalyst with varying catalyst loading. The tests were conducted in a 1 M KHCO3 aqueous electrolyte at a potential of −0.99 V vs. RHE (f). The FE of the US-prepared CuZ-06-035 (g) and US-prepared Cu-06 (h) catalysts, with a loading of 0.2 mg cm−2, was evaluated in a 1 M KHCO3 aqueous electrolyte at a potential of −0.99 V vs. RHE. Reproduced with permission from ref. 325. Copyright 2023, Elsevier. | ||
In the quest for phase-pure solid Cu3P NPs, Downes et al. have developed a molecular precursor method. This approach involves using a mixture of hydrocarbons and amines as a solvent for the high-temperature decomposition of [Cu(H)(PPh3)]6 to produce Cu3P NPs. The electrocatalytic efficiency of Cu3P NPs for CO2R in CO2-saturated KHCO3 aqueous solutions was thoroughly examined. Cu3P NPs demonstrated exceptional dispersibility, allowing them to be easily deposited onto carbon paper electrodes. However, it was observed that Cu3P NPs exhibited instability under electrocatalytic conditions. Further investigations revealed that increasing the phosphorus content improved electrochemical stability, while decreasing the Cu3P content enhanced stability.326 In another study, nanocomposites of ZIF-L (zeolitic imidazolate framework with large cavities) and GO (graphene oxide) were developed, showcasing excellent electrical conductivity, a substantial active surface area, and robust electrocatalytic performance. Particularly, Cu GNC-VL (Cu zeolitic imidazolate framework-derived nitrogen-doped porous carbon) exhibited a remarkably high current density of 10.4 mA cm−2 at −0.87 V vs. RHE and an impressive FE for ethanol production of 70.52%.327
In a related study, Dou et al. have developed a method for creating CuS nanosheet arrays on a brass mesh through a single-step chemical bath deposition process. When subjected to e-CO2R at the CuS/BM electrode, increased production of HCOO− was observed. This electrochemical process induced reconstruction, resulting in a nanowire network with multiple active centers. The reconstituted Cu(111)/CuS(102) structure was identified as a key contributor to the electrode's strong selectivity towards HCOO− generation. Moreover, the presence of sulfur (S) beneath the Cu0 layer led to reduced binding energies of HCOO* and *COOH compared to the Cu(111) plane, facilitating the production of HCOOH or HCOO−.328 In another innovative approach, synthetic control over Cu NPs and Ag NPs has been achieved through a newly developed seeded-growth colloidal method, resulting in the production of AgCu nanodimers (NDs) with varying Cu domain sizes. This method also applies to other dimer systems like Cu–Ru, Cu–Rh, and Cu–Ir, where two metals are traditionally unmixable in the bulk phase. To examine the compositional and structural sensitivity of e-CO2R in this bimetallic system, model catalysts were made from Ag/Cu nanocrystals. In particular, the Ag1Cu1.1 NDs displayed a 3.4-fold increase in FE for C2H4 and a 2.0-fold improvement in the combined e-CO2RR activity over Cu NPs of comparable size and shape.126
In another research study, Chen et al. controlled the aspect ratio and composition of alloyed nanorods made of Cu through a co-reduction synthesis process using seeds. They demonstrated different catalytic activities for p-nitrophenol reduction by the Cu3Au nanorods tuning across the visible to infrared spectrum.329 Zheng et al. have made significant progress in synthesizing e-CO2RR catalyst materials and created highly efficient Au–Cu knot structures, referred to as NSs in this study. NSs of all types were formed by Cu growth on the concave surface of a Au seed. The final products are obtained by using o-phenylenediamine (OPDA) as a capping agent and Cu(acac)2 as a precursor. A growth mode and high exposure to Au are typical of the anisotropic nature of the products. The Au–Cu NSs exhibited remarkable selectivity for C2+ product generation, with an optimum FE2+ of 67% and a C2+ partial current density of 0.29 A cm−2 at 0.75 V vs. RHE.330 In a separate study, Wang et al. have developed effective catalysts for e-CO2R by generating dense OD-Cu NPs using a three-step process (Fig. 30). This process involved the thermal oxidation of CuOx on a Cu mesh, followed by reduction through annealing, and finally the conversion to OD-Cu by applying a cathodic potential to the partially reduced CuOx. These OD-Cu NPs maintained their shape and exhibited the capability to produce various types of syngas at high rates with modest overpotentials.331
 | ||
| Fig. 30 The FEs of H2 and CO production on OD-Cu NPs were measured at different applied potentials: (a) −0.45 V, (b) −0.50 V, and (c) −0.55 V. The controllable composition of syngas varies depending on the applied potentials (d). Reproduced with permission from ref. 331. Copyright 2020, American Chemical Society. | ||
Additionally, Wang et al. have grown scalable arrays of metallic 2D Cu2Te nanosheets on copper foils. During the growth process, chemical etching and vapor deposition, exposure to potential active sites at the edges of these nanosheets facilitated CO2R to CH4 at lower overpotentials.332 Moreover, Han et al. have employed the atomic layer infiltration method using the HKUST-1 template. By uniformly adding Zn–O–Zn sites throughout HKUST-1, the FE for CO increased from 20–30% to 70–80%. Computational density-functional-theory studies revealed that this modification lowered the activation energy barrier for the reaction and accelerated CO production by increasing the enthalpy of CO2 adsorption.277 Furthermore, a potential electrocatalyst for CO2R has been introduced by Sirisomboonchai et al. This electrocatalyst, consisting of In and Zn deposited over Cu foam, exhibited improved CO2R to CO with a remarkable FE of 93.7% and a 100-hour operational lifetime.333 In a related study, the impact of various sulfur precursors on the structure–property–activity relationship of copper-based catalysts for e-CO2 conversion was investigated by Gao et al. The CuS-TU catalyst, synthesized using thiourea as the sulfur precursor, exhibited a distinctive flower-like structure and the highest sulfur vacancy concentration. This unique structure facilitated interfacial mass transfer during CO2R. The abundance of sulfur vacancies on the catalyst's surface played a crucial role in increasing CO generation through the *COOH route by enhancing the adsorption of COOH intermediates. Subsequently, the CuS-TU catalyst achieved a CO selectivity of 72.67% at −0.51 V vs. RHE.334Table 16 shows different materials designed by the precursor derived approach for e-CO2R.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | CuZ-065-035 | 1 M KHCO3 | −0.99 | 52 | 22 | — | — | 5 | — | — | 10 | — | 325 |
| 2 | CuZ-065-035-%30-A | 80 | 22 | — | — | 2 | — | — | 2 | — | |||
| 3 | Cu GNC-VL | 0.5 M KHCO3 | −1.07 | 40 | — | — | — | 5 | — | 13 | 42 | — | 327 |
| 4 | Cu ZIFL@GO | 60 | 5 | — | — | 9 | — | 14 | 12 | — | |||
| 5 | CuS/BM | 0.5 M KHCO3 | −1.0 | 60 | — | — | — | 48 | — | — | — | — | 328 |
| 6 | Ag NPs | 0.1 M KHCO3 | −1.4 | 92 | 10 | — | — | — | — | — | — | — | 126 |
| 7 | Cu NPs | 34 | 1 | 32 | 8 | 1 | — | — | — | — | |||
| 8 | Ag1–Cu1.1-NDs | 50 | 2 | 30 | 12 | 2 | — | — | — | — | |||
| 9 | Au–Cu Janus | 3 M KOH | −0.75 | — | — | — | 37 | — | — | — | 20 | — | 330 |
| 10 | Au@Cu core–shell | −0.95 | — | — | — | 20 | — | — | — | 10 | — | ||
| 11 | Cu NPs | −0.81 | — | — | — | 37 | — | — | — | 17 | — | ||
| 12 | OD-Cu NA | 0.1 M KHCO3 | −0.55 | 60 | 19 | — | — | — | — | — | — | — | 331 |
6.13. Polymer modification approach
The utilization of polymer modification is an approach in the development of e-CO2R catalysts. It plays a role in enhancing the selectivity, activity and stability of these catalysts. When researchers incorporate polymers into the structure or surface of the catalyst, they achieve some advantages. Polymers serve as stabilizers too, preventing the aggregation or dissolution of species during the CO2RR process. This makes the catalyst more durable. Polymers also are effective as templates for controlling the nanostructure and catalyst material morphology, influencing its performance. Furthermore, the modification of polymers promotes the formation of active sites essential for efficient conversion of CO2 into more valuable products like hydrocarbons or alcohols. The reason this is important is that making CO2RR catalysts better and more efficient by means of such engineering will foster sustainable carbon capture and utilization technologies. Polymer-modified catalysts with catalytic metal cores can be synthesized by multiple methods for designing CO2RR catalysts. One of these is electrochemical deposition, in which polymer films grow onto a catalyst surface allowing precise control of thickness and composition. In another method, polymers can be incorporated into the catalyst matrix when synthesized to produce the material combining the characteristics of metals together with the stabilizing and templating effects caused by polymers. Additionally, researchers may go with a couple of possible synthesis techniques, such as chemical or physical adsorption, to introduce polymer modifiers on the catalyst surface.Poly(ionic liquid) (PIL) NPs with multilamellar chain stacking were prepared by free radical homopolymerization. By manipulating the carbon used in the polymerization process, PIL NPs of various sizes and shell thicknesses could be synthesized. Molecular dynamics simulations corroborated the mechanism of PIL NV formation, emphasizing the role of the polymerization degree of PIL chains in determining assembly behavior. These well-defined PIL NVs with hollow interiors allowed for high loading amounts of ultra-small Cu NPs for e-CO2R. The strong electronic interactions between PIL units and Cu atoms on the surface of Cu NPs resulted in a high FE toward C1 products, primarily CH4.335 In another study, the impact of a poly(4-vinylpyridine) (P4VP) layer on the e-CO2RR on both poly Cu and Au electrodes was investigated by Ye et al. The research revealed that the P4VP layer inhibits the HER and enhances CO2RR, particularly the production of HCOOH. In in situ ATR-SEIRAS tests, fewer CO adsorption bands were observed on P4VP-modified Cu electrodes, while higher ICO2/IHCO3 ratios were detected on P4VP-modified Au electrodes. These observations suggest that the mass transfer of H2O and HCO3 from the bulk to active sites is impeded by the P4VP modification. Importantly, the hydrophilicity of the electrode surface is altered by the presence of P4VP, leading to increased HCOOH production at low overpotentials.336 Furthermore, polymer coatings can modify local pH levels, hydrophilicity/hydrophobicity, and reactant concentrations. Jun et al. have demonstrated that polymer coatings can enhance the catalytic selectivity of Cu-based catalysts in CO2RR while also improving electrocatalytic stability.337
Additionally, the impact of three different ionomers on Cu-catalyzed CO2RR under high-current conditions was investigated. In this investigation, it was observed that the anionic ionomer Nafion had a negligible effect on CO2RR, likely due to the negatively charged nature of the electrode/catalyst interface. However, the cationic ionomers XA-9 and PTP had significant effects on the selectivity of CO2RR with Cu catalysts. Specifically, PTP led to increased HER and formate synthesis, likely due to decreased mass transport. In contrast, XA-9 favored the production of CO at the expense of C2+ products.338 Moreover, another study highlighted the influence of different polymeric binders used during electrode fabrication on the selectivity of CO2RR on Cu electrodes (Fig. 31). The research findings demonstrate that the choice of binder material can significantly modify the selectivity of CO2RR products. Specifically, it was observed that Cu electrodes with polyvinylidene fluoride as the binder favored the production of CH4, while Cu electrodes with polyacrylic acid as the binder exhibited greater selectivity for HCOOH generation.339
 | ||
| Fig. 31 The CO2RR performance of various Cu/C catalysts with different binders was evaluated. The FE of HCOOH, CO, CH4, C2H4, and H2 was evaluated at two different potentials: −0.6 V and −1.4 V (A and B). The partial current density profiles of HCOOH and CH4 are presented (C and D). Reproduced from ref. 339. Open access article, American Chemical Society. | ||
Liang et al. have developed a novel single-site copper coordination polymer known as Cu(OH)BTA. Notably, this polymer exhibits a 1.5 times higher selectivity for producing C2H4 compared to its metallic counterpart (Fig. 32). An interesting feature of this catalyst is that it maintains its structural integrity throughout the reaction. This advantageous characteristic allows for the formation of an energetically favorable *OCCHO intermediate during CO hydrogenation, facilitated by suitably distanced dual sites provided by adjacent copper atoms in the polymer. The study also demonstrates the practical applicability of this polymers by running full-device CO2 electrolysis for 67 hours at an appropriate current in a membrane electrode assembly.311
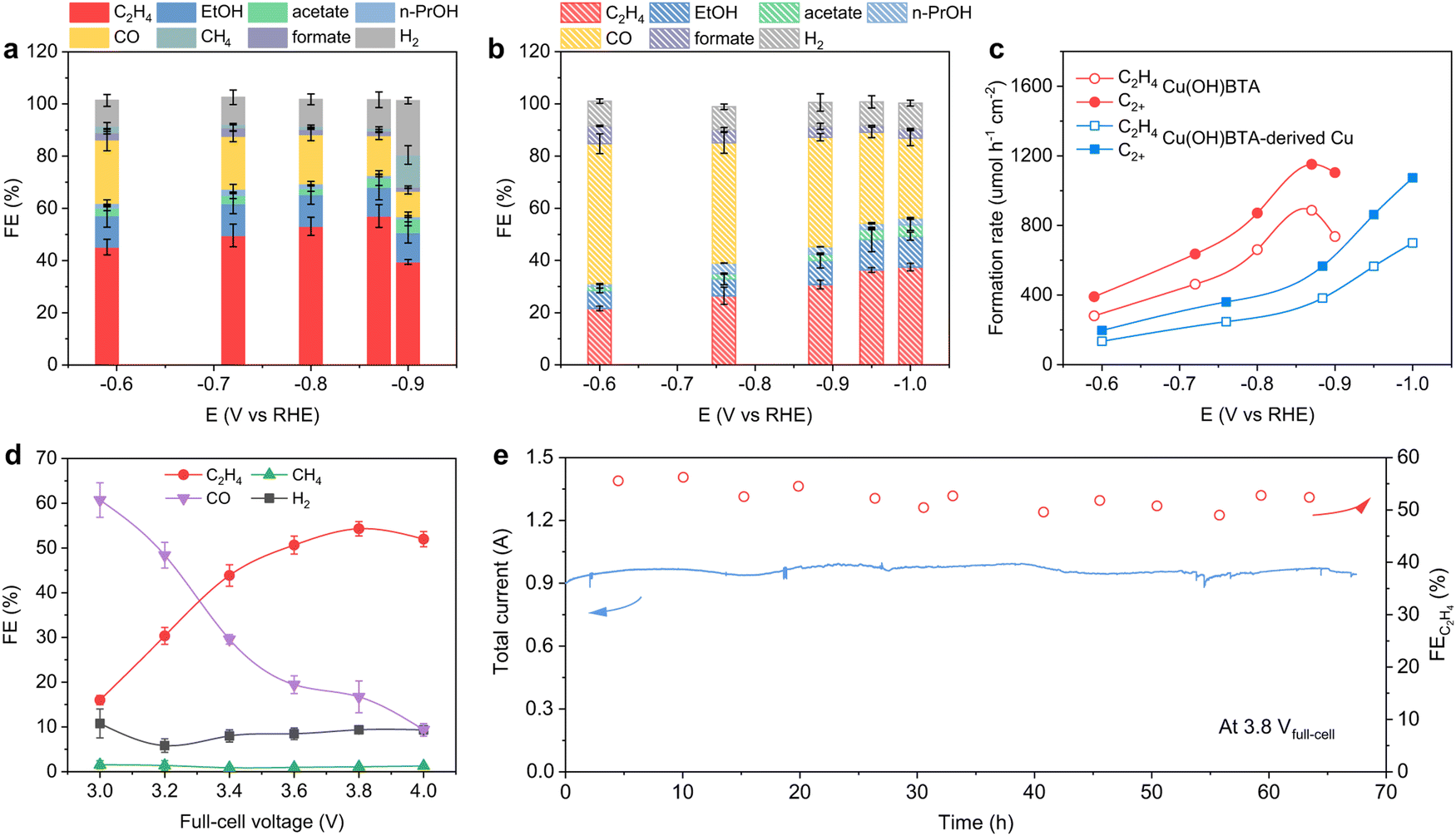 | ||
| Fig. 32 The study investigated the FEs of various products at different applied potentials using Cu(OH)BTA (a) and Cu(OH)BTA-derived Cu (b) in a 1 M KOH electrolyte. The study also investigated the formation rates of C2+ products and C2H4 under varying applied potentials using Cu(OH)BTA and Cu(OH)BTA-derived Cu in a 1 M KOH electrolyte (c). The study also measured the FEs of various gas products within the voltage range of 3.0–4.0 V using a membrane-electrode assembly device (d). The electrosynthesis of C2H4 was conducted in a membrane-electrode assembly with a geometric electrode area of 4 cm−2. The catalyst maintained its overall current and ethylene FE for a duration of 67 hours (e). The error bars for the uncertainty of the FE analysis represent one standard deviation, which is calculated based on three independent samples. Reproduced from ref. 311. Open access article, Nature. | ||
Additionally, Wang et al. have explored a unique approach for enhancing the selectivity of heterogeneous Cu catalysts for CO2RR employing random copolymers as surface modifiers. Among these copolymers, a tricomponent variant demonstrated superior performance in maximizing the FE for ethylene and C2+ products during CO2RR. Surface characterization conducted under CO2RR conditions highlighted the copolymer's resilience, attributed to the presence of a phenylpyridinium component. Advanced techniques, including AFM, SEM, and XPS, were utilized to counter the presence of nanostructuring and electrical effects. Molecular dynamics simulations illustrated that the optimal polymer modifier led to localized increases in CO2 content. Moreover, compared to block copolymers, random copolymers exhibited greater porosity, facilitating improved access and mass transport of reactants.340 Furthermore, Wei et al. demonstrated that coating Cu surfaces with a PANI thin film can significantly alter the catalytic activity and selectivity for CO2RR. The NH group on PANI interacts with CO2 molecules, enriching the presence of CO2 and inhibiting the HER on Cu surfaces. Consequently, the CO2RR current density at the Cu/PANI interface experiences a substantial increase. This transformation leads to a shift from 15% C2+ hydrocarbon production on pure Cu to over 60% on Cu-PANI, making C2+ hydrocarbons the dominant products.341
On another front, researchers have synthesized and evaluated two stable Cu(I)-based coordination polymer electrocatalysts, NNU-32 and NNU-33(H), designed for e-CO2RR within a flow-cell electrolyzer. These electrocatalysts exhibit exceptional selectivity for converting CO2 into CH4, achieving a FE of 82% at −0.9 V vs. RHE. The outstanding selectivity is attributed to the presence of cuprophilic interactions within the catalysts. Furthermore, a dynamic in situ crystal structure transformation from NNU-33(S) to NNU-33(H) enhances cuprophilic interactions and CO2 adsorption. This structural change, involving the substitution of hydroxyl radicals for sulfate radicals, significantly contributes to the superior performance of these electrocatalysts. It is worth noting that cuprophilic contacts play a pivotal role in CH4 production, and the selectivity for CH4 varies with the number and proximity of interacting cuprophiles.342 Furthermore, Li et al. have found that by stabilizing a metal-bound CO intermediate, Nafion-modified electrodes can significantly enhance the production of CH4. To precisely regulate the transfer of protons to the metal–CO intermediate, Cu electrodes were constructed using a polymer blend of Nafion and polyvinylidene fluoride. This precise control mechanism leads to increased rates of production for C2+ products such as ethylene, ethanol, and 1-propanol. Importantly, under these controlled conditions, hydrogen evolution does not occur, as almost all carbon-containing products can be extracted from Cu electrodes equipped with a 15 m Nafion overlayer.343 Furthermore, the e-CO2R to high-energy-density C2+ products necessitates carbon–carbon interactions. In this context, Zhang et al. have explored the use of capping agents and electrochemical treatment to fabricate polycrystalline Cu catalysts. These catalysts exhibit a 34% FE for the production of C2H4 at −1.5 V vs. RHE. Importantly, a critical *COOH intermediate was observed on the Cu surfaces, as confirmed by quasi-in situ total reflection FTIR analysis.344 Different polymers were used for modification of e-CO2R performance of different materials, which is summarized in Table 17.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | NWs/Cu | 0.1 M KHCO3 | −1.0 | 27 | 34 | 7 | 14 | — | — | — | — | — | 335 |
| 2 | Pristine CuNPs | 30 | 15 | 3 | 22 | — | — | — | — | — | |||
| 3 | Poly Cu | 0.1 M KHCO3 | −0.9 | 57 | 12 | 4 | 2 | 27 | — | — | — | — | 336 |
| 4 | P4VP-modified Cu | 95 | 5 | 3 | 2 | 32 | — | — | — | — | |||
| 5 | Cu-P1 | 1 M KOH | −0.97 | 1 | 5 | — | 73 | 1 | — | — | 1 | — | 337 |
| 6 | Cu-P4 | 30 | 3 | 2 | 35 | 15 | — | — | 1 | — | |||
| 7 | Cu | 0.5 M KHCO3 | −0.86 | — | 6 | — | 7 | 15 | — | — | 3 | — | 338 |
| 8 | Cu/Nafion | — | 5 | — | 7 | 6 | — | — | 1 | — | |||
| 9 | Cu/XA-9 | — | 12 | — | 8 | 20 | — | — | 2 | — | |||
| 10 | Cu/PTP | — | 2 | — | — | 10 | — | — | — | — | |||
| 11 | Cu-Nafion | 0.1 M KHCO3 | −1.4 | 28 | 20 | 12 | 10 | 18 | — | — | — | — | 339 |
| 12 | Cu-PAA | 28 | 18 | 6 | 4 | 34 | — | — | — | — | |||
| 13 | Cu-PVDF | 28 | 20 | 12 | 4 | 18 | — | — | — | — | |||
| 14 | Cu(OH)BTA | 0.1 M KHCO3 | −0.6 | 10 | 23 | 2 | 45 | 2 | — | — | 13 | — | 311 |
| 15 | Cu(OH)BTA-derived Cu | 10 | 55 | — | 20 | 7 | — | — | 8 | — | |||
| 16 | Cu | 0.1 M KHCO3 | −1.04 | 39.7 | 0.5 | 25.6 | 22.3 | 1.8 | — | — | 5.9 | — | 340 |
| 17 | Cu-1 | 94.6 | — | 1.5 | — | 0.7 | — | — | — | — | |||
| 18 | Cu-2 | 69.3 | 1 | 14.1 | 5.3 | 6.6 | — | — | — | — | |||
| 19 | Cu-3 | 37.8 | 2.8 | 14 | 21.5 | 11.1 | — | — | 3.4 | — | |||
| 20 | Cu-4 | 34.1 | 0.4 | 33.1 | 19 | 3.8 | — | — | 3.9 | — | |||
| 21 | Cu-5 | 33.6 | 0.5 | 28.1 | 25.4 | 1.1 | — | — | 6 | — | |||
| 22 | Cu-6 | 32.5 | 1.1 | 22.3 | 30.4 | 1 | — | — | 9.3 | — | |||
| 23 | Cu-7 | 16.5 | 1.2 | 4.7 | 49.5 | 2.2 | — | — | 16 | — | |||
| 24 | Cu-8 | 11.9 | 0.7 | 1.9 | 55.6 | 1.9 | — | — | 17.4 | — | |||
| 25 | Cu-9 | 19.2 | 0.4 | 2.8 | 53.8 | 0.9 | — | — | 15.5 | — | |||
| 26 | Cu-10 | 35 | 1 | 11.5 | 36.8 | 1.8 | — | — | 9.5 | — | |||
| 27 | Cu-11 | 36.2 | 2.3 | 8.7 | 31.7 | 9 | — | — | 4.3 | — | |||
| 28 | Cu-12 | 44.8 | 0.5 | 25 | 20.4 | 1.4 | — | — | 5.2 | — | |||
| 29 | Cu-13 | 21.9 | 0.6 | 5.7 | 59.8 | 1.3 | — | — | 11.8 | — | |||
| 30 | Cu-block-8 | 30.5 | 0.6 | 14.1 | 34.5 | 2.4 | — | — | 11 | — | |||
| 31 | Cu | 0.1 M KHCO3 | −1.1 | 45 | 3 | 5 | 5 | 27 | — | — | 10 | — | 341 |
| 32 | Cu-PANI | 17 | 2 | 11 | 30 | 10 | — | — | 13 | — | |||
| 33 | NNU-33(H) | 1 M KOH | −1.0 | 13 | 12 | 63 | 13 | — | — | — | — | — | 342 |
6.14. Support engineering approach
Support engineering plays a role in the design of e-CO2R catalysts as it focuses on selecting and modifying support materials to improve catalytic performance. This approach enables researchers to optimize the sites of the catalyst by providing a conductive platform for reactions. By choosing support materials and manipulating their properties scientists can enhance the efficiency, durability and economic viability of CO2RR catalysts for sustainable carbon capture and utilization technologies. In the strategy of support engineering for CO2RR catalyst design, there are methods that can be employed. One common approach is depositing nanoparticles or single atoms onto support materials like graphene, carbon nanotubes or metal oxides such as TiO2 and ZnO. Another method involves modifying support materials through surface functionalization or doping to influence the binding energies of CO2 and reaction intermediates, thereby customizing the selectivity of the catalyst. Additionally creating materials by combining components with suitable supports offers a versatile solution that combines advantages from both components. In the quest for effective e-CO2R to value-added syngas, a technique has been devised utilizing anion-regulated self-supported Cu3Se2 nanosheet-assembled fibers (Fig. 33). These fibers exhibit an excellent TOF of 1303 h−1 over a wide potential window and accommodate a broad range of H2-to-CO ratios from 0.8 to 6.0. Remarkably, they can sustain bulk electrolysis for 10 hours, owing to their high current density and minimal attenuation.345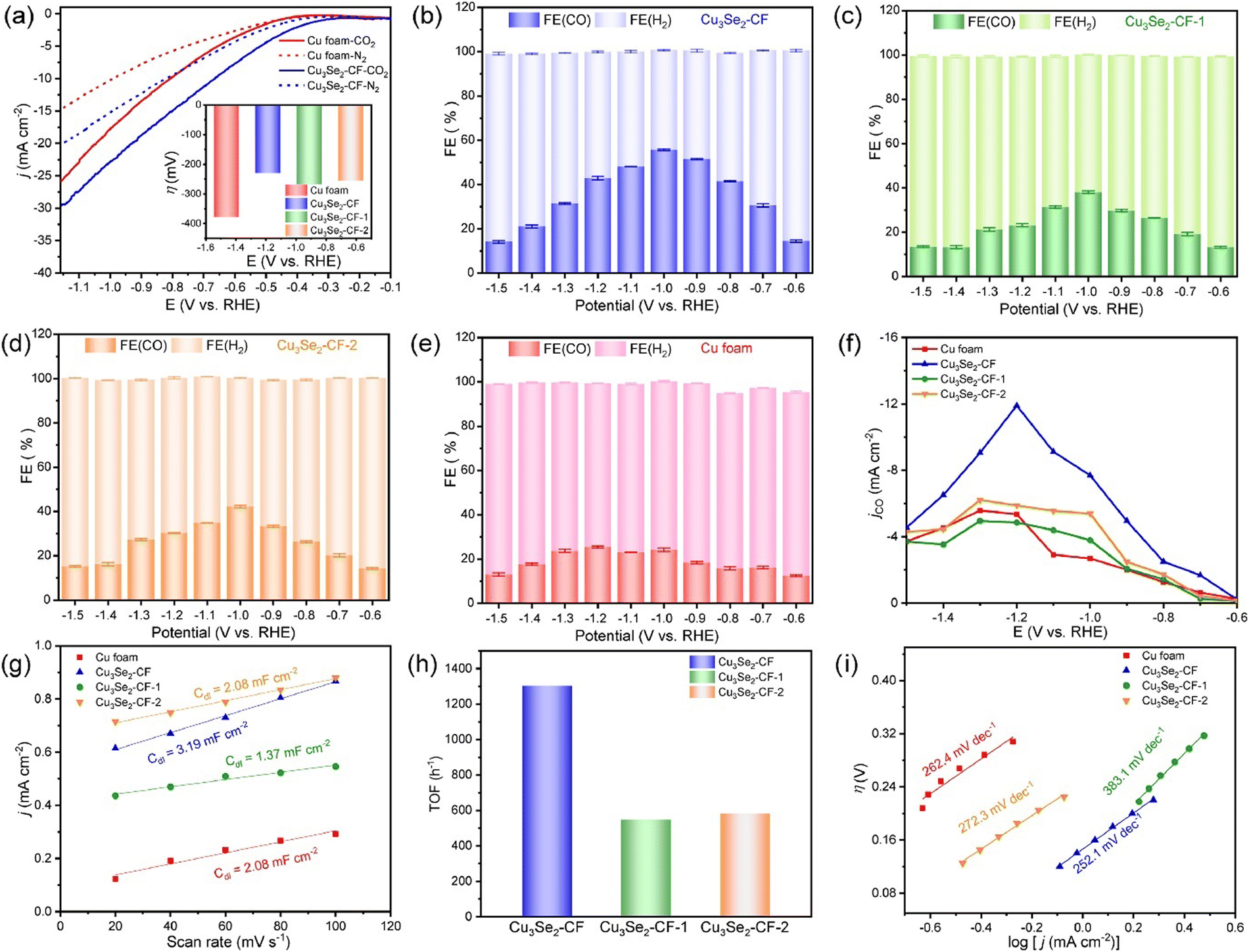 | ||
| Fig. 33 The CO2R of Cu foam and Cu3Se2-CF nanocatalysts. LSV traces were obtained in a CO2-saturated or N2-saturated electrolyte using a scan rate of 5 mV s−1 (a). The inset in (a) shows the onset potential. The total FE was determined for gaseous products only, specifically H2 and CO (b)–(e). The jCO was measured at different applied potentials (f). The double-layer capacitance was measured (g). TOFs were calculated (h). Tafel slopes were determined for Cu foam, Cu3Se2-CF, Cu3Se2-CF-1, and Cu3Se2-CF-2 (i). The data were collected under standard ambient temperature and pressure conditions, using a CO2 stream with a flow rate of 20 sccm. Error bars represent the standard deviation of a minimum of three independent measurements. Reproduced with permission from ref. 345. Copyright 2023, Elsevier. | ||
Furthermore, an oxide-derived composite material supported on N-doped graphene was designed by Dongare et al. It was found that by adjusting the Cu to Zn ratio during the coprecipitation process, one can customize the electrocatalyst's composition to meet specific requirements. Notably, an electrode containing 20 wt% Zn in conjunction with copper exhibits the highest selectivity for ethanol production (FE = 34.25%), while the CuO/NGN electrode demonstrates lower selectivity for ethanol and other C2 compounds.346 Furthermore, pyrolyzing mixed precursors of Cu(NO3)2 and polyvinyl pyrrolidone produces Cu/NC composite electrocatalysts for catalyzing CO2 reduction. Among these, Cu-20/NC stands out due to its substantial Cu nanoparticle size and extensive coverage of the carbon support surface, resulting in highly competitive HER activity. Conversely, Cu-5/NC, with its nanoparticle morphology, favors poor C–C coupling activity and CH4 formation. Meanwhile, Cu-10/NC exhibits remarkable selectivity for CO and hydrocarbons, leading to an impressive 37% FEC2H4 at −1.2 V and enhanced e-CO2R to C2H4 conversion activity, selectivity, and durability.347 Moreover, Gu et al. have demonstrated that the incorporation of Cu onto PCC supports yields intriguing e-CO2R outcomes, particularly with improved selectivity for C2 products like ethanol and ethylene. Among Cu5-PCC, Cu10-PCC, and Cu20-PCC, Cu15-PCC outperforms in various applied potentials, offering a more cost-effective approach to e-CO2R by leveraging affordable and abundant minerals as the foundation for suitable electrocatalysts.348
In another study, Lim et al. have developed a CuSx catalyst for e-CO2R by dipping Cu foil into an industrial CO2-purged electrolyte containing H2S gas. Increasing the H2S concentration significantly improved the maximum FEHCOOH from 22.7% to 71.8%, attributed to the reaction between Cu foil and sulfur species. Notably, these catalysts demonstrated excellent performance in both pure CO2-purged electrolytes and industrial CO2-purged electrolytes, maintaining stability for 12 hours.349 Furthermore, Kim et al. have explored the e-CO2R on copper through pulsed electrolysis. The cathode's potential was cyclically shifted between −0.8 V vs. RHE and −1.15 V versus RHE over durations ranging from 5 seconds to 25 seconds. Notably, static electrolysis at −1.15 V vs. RHE resulted in a decreased FEHER while simultaneously increasing the FE for the CO2RR. However, when pulsed electrolysis was employed, the FEHER decreased, whereas the FE for CO2RR increased. The optimal duration for pulsed electrolysis was found to be 10 s at each potential, achieving the lowest FE for the HER and the highest FE for the CO2RR. Additionally, pulsed electrolysis introduced changes in the ratio of oxygenated to hydrocarbon products and significantly boosted the FE for C2+ products.350
Moreover, by enhancing the geometric partial current density of hydrocarbons through surface modification of copper foam with poly(acrylamide), the faradaic output of ethylene can be doubled. While poly(allylamine) completely impedes CO2R, poly(acrylic acid) exerts only a transient impact. Through mechanisms such as CO activation for dimerization, stabilization of CO dimers via hydrogen bond interactions, and facilitation of CO adsorption in proximity to the polymer, poly(acrylamide) adheres to copper through oxygen on carbonyl groups, thereby increasing the efficiency of ethylene formation. Achieving high CO2 mass transfer rates, utilizing modified poly(acrylamide), and employing porous copper catalysts all contribute to improved ethylene production.351 In another study, the e-CO2R was demonstrated using Bi-doped SnO nanosheets on a Cu foam substrate. Remarkably, the Bi–SnO/Cu foam electrode achieved the highest FE (93%) and HCOOH selectivity (99%), with exceptional long-term stability over 30 hours of operation. The Bi doping played a pivotal role in stabilizing divalent tin on the electrocatalyst, making its reduction to metallic tin more challenging. Additionally, the presence of Cu foam influenced the catalysis by promoting electron transfer, thereby maintaining tin in a positively oxidized state favorable for the adsorption of *OOCH intermediates.352
In a separate investigation, various synthetic approaches were employed to produce Cu nanoparticles with sizes ranging from 10 to 30 nm, supported by VC, KB, and SWNTs. Importantly, these carbon-supported Cu nanocatalysts demonstrated higher selectivity for C2H4 production compared to electrodeposited Cu. The onset potentials for C2H4 generation exhibited significant shifts for well-dispersed 40 wt% Cu/VC and 20 wt% Cu/SWNT catalysts in comparison to electrodeposited Cu.353 In another work, a porous Cu-NC composite catalyst for e-CO2R was synthesized using a scalable glucose blowing process. The catalyst exhibited remarkable efficiency (69% at 590 mV) in converting CO2 to C1 products, underscoring its strong catalytic activity.354 Furthermore, the one-step pyrolysis synthesis of Cu Ps/BCF from butterfly wing carbon fibers and copper salt resulted in a material with unusual geometric features, including low-coordinated active spots at stepped surfaces. Increased conductivity from nitrogen dopants allows CO2 to be reduced to C2H4 at a low applied voltage while maintaining excellent selectivity and long lifetime.355
Additionally, Kordus et al. have analyzed the activity and selectivity of ZnO-supported Cu nanoparticles in methanol synthesis, revealing that the shape of the pre-catalyst (cubic vs. spherical) played a significant role. Cubic Cu particles exhibited higher activity for methanol synthesis but significantly reduced selectivity compared to spherical particles, underscoring the substantial changes in the morphology and composition of the shaped pre-catalysts under reaction conditions.356 Also, another research study has demonstrated that Cu1.8Se nanowires on Cu foam serve as effective electrocatalysts for the selective CO2R to higher-value C2 products, such as C2H4 and EtOH, with promising applications in the chemical and fuel industries. These nanowires exhibit impressive FE of 55% and 24% at −1.1 V and maintain exceptional stability over extended periods. The presence of significant concentrations of CO2 and Se species was likely responsible for the observed high selectivity toward the C2/C1 product.357
In a related study, a series of Cu catalysts with adjustable particle sizes were constructed within a Zn, N-doped carbon matrix generated from ZIF-8. Crystal diameters ranging from 8.9 to 27.3 nm were observed in the enclosed Cu metal catalysts. Notably, catalysts with a grain size of 15.7 nm exhibited the highest FECO (approximately 80%), directly correlating with their performance in the CO2RR.358 In a separate investigation, Lou et al. have conducted experiments in an H-type reaction cell and a continuous-flow membrane electrode assembly (MEA) reactor to evaluate the performance of CuBi bimetallic catalysts in selectively reducing CO2 to formate. The study revealed that co-electrodeposition potentials of 0.6 V yielded the best performance, with the catalyst achieving a remarkable FEformate (98.3%) at a current density of 56.6 mA cm−2 in the continuous-flow MEA reactor, outperforming the H-type reaction cell. Additionally, the study identified the optimal elemental ratio of Cu to Bi at 0.6 V to maximize the catalyst's activity.359
In another breakthrough, a novel fast-reduction approach was employed to create a Cu-decorated porous Bi/Bi2O3 nanofoam, denoted as P-Cu-BiNF. The nanoparticles forming the 3D porous network architecture of P-Cu-BiNF are consistently sized at 15–20 nm. The inclusion of Cu into P-Cu-BiNF enhances CO2R performance by controlling the morphology and local electronic state of Bi/Bi2O3. Remarkably, P-Cu-BiNF exhibits a CO2-to-formate FE exceeding 90% across a wide potential range of 0.78 to 1.08 V, providing a high formate partial current density of 62.7 mA cm−2 at 1.18 V while maintaining exceptional stability over time.360 Additionally, another research study investigated nanosized Cu2O on three different carbon-based substrates, namely BG (positively charged), NG (negatively charged), and rGO (weakly negatively charged), for e-CO2R with promising results. The use of these substrates led to an increase in the FE of C2 products, with the FEC2/FEC1 ratio ranging from 0.2 to 7.1, where rGO/Cu > BG/Cu > pure Cu > NG/Cu. Notably, under CO2R conditions, the negatively charged NG stabilized Cu+ species, thereby improving CO* adsorption and enhancing C–C coupling for C2 products. Consequently, at high current densities, the FEC2+ was significantly increased to around 68%.361 Moreover, it is proposed that introducing different functional groups to Cu based electrocatalysts (N-, G-, COOH, NH2, and OH) can modify their selectivity and, in turn, their ability to adsorb crucial intermediates. Catalysts containing N and OH functional groups exhibit selectivity for C1 products close to 90% during e-CO2R catalysis.362 Conclusively, the support engineering approach is considered as an efficient approach for designing e-CO2R catalysts. An overview of different materials designed by following this approach is given in Table 18.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | Cu foam | 0.1 M KHCO3 | −1.1 | 76 | 24 | — | — | — | — | — | — | — | 345 |
| 2 | Cu3Se2-CF | 52 | 48 | — | — | — | — | — | — | — | |||
| 3 | Cu3Se2-CF-1 | 33 | 67 | — | — | — | — | — | — | — | |||
| 4 | Cu3Se2-CF-2 | 65 | 35 | — | — | — | — | — | — | — | |||
| 5 | CuO-NGN | 0.1 M KHCO3 | −1.0 | — | — | — | — | — | — | — | 3 | — | 346 |
| 6 | CuZn10-NGN | — | — | — | — | — | — | — | 7 | — | |||
| 7 | CuZn20-NGN | — | — | — | — | — | — | — | 13 | — | |||
| 8 | CuZn30-NGN | — | — | — | — | — | — | — | 3 | — | |||
| 9 | CuZn40-NGN | — | — | — | — | — | — | — | 3 | — | |||
| 10 | Cu-0/NC | 0.1 M KHCO3 | −1.0 | 65 | 3 | — | 14 | — | — | — | — | — | 347 |
| 11 | Cu-5/NC | 22 | 23 | 5 | 19 | — | — | — | — | — | |||
| 12 | Cu-10/NC | 16 | 18 | 3 | 8 | — | — | — | — | — | |||
| 13 | Cu-15/NC | 38 | 20 | 1 | 7 | — | — | — | — | — | |||
| 14 | Cu-20/NC | 62 | 6 | 1 | 3 | — | — | — | — | — | |||
| 15 | Pure Cu samples | 25 | 7 | 1 | 17 | — | — | — | — | — | |||
| 16 | Cu5-PCC | 0.1 M KHCO3 | −1.0 | 18 | 2 | 27 | — | — | 6 | — | 348 | ||
| 17 | Cu10-PCC | 10 | 5 | 23 | — | — | 6 | — | |||||
| 18 | Cu15-PCC | 22 | 2 | 12 | 26 | — | — | 16 | — | ||||
| 19 | Cu20-PCC | 15 | 8 | 20 | — | — | 5 | — | |||||
| 20 | CuSx | 0.1 M KHCO3 | −1.0 | — | 32 | — | — | 15 | — | — | — | — | 363 |
| 21 | Cu foam | 0.1 M NaHCO3 | −0.96 | 15 | 1 | 4 | 13 | — | — | — | — | — | 351 |
| 22 | Cu-P-acrylamide | 15 | 2 | 2 | 27 | — | — | — | — | — | |||
| 23 | Cu-P-acrylic acid | 16 | 1 | 3 | 17 | — | — | — | — | — | |||
| 24 | Cu-P-allylamine | 30 | — | 0.5 | — | — | — | — | — | — | |||
| 25 | Bi-SnO/Cu foam | 0.1 M KHCO3 | −1.6 Ag/AgCl | 10 | 2 | — | — | 87 | — | — | — | — | 352 |
| 26 | SnO/Cu foam | 17 | 3 | — | — | 77 | — | — | — | — | |||
| 27 | Cu foam | 58 | 4 | — | — | 15 | — | — | — | — | |||
| 28 | Electrodeposited Cu | 0.1 M KHCO3 | −1.6 | 38 | 37 | 1 | 2 | — | — | — | — | — | 364 |
| 29 | 20 wt% Cu/VC | 50 | 10 | 1 | 2 | — | — | — | — | — | |||
| 30 | 40 wt% Cu/VC | 30 | 20 | 1 | 11 | — | — | — | — | — | |||
| 31 | 20 wt% Cu/SWNT | 40 | 18 | 1 | 17 | — | — | — | — | — | |||
| 32 | 50 wt% Cu/KB | 41 | 17 | 5 | 10 | — | — | — | — | — | |||
| 33 | Cu-NC 1:1 |
0.1 M KHCO3 | −1.0 | — | 10 | — | — | 43 | — | — | — | — | 365 |
| 34 | Cu-NC 2:1 |
— | 13 | — | — | 35 | — | — | — | — | |||
| 35 | Cu-NC 1:2 |
— | 13 | — | — | 42 | — | — | — | — | |||
| 36 | NC | — | 12 | — | — | — | — | — | — | — | |||
| 37 | Cu@Vul | 0.5 M KHCO3 | −1.0 | 52 | 28 | 8 | 3 | — | — | — | — | 366 | |
| 38 | Cu@KB | 43 | 49 | 2 | 5 | — | — | — | — | ||||
| 39 | Cu@MGS | 42 | 48 | 5 | 1 | — | — | — | — | ||||
| 40 | Cu Ps/BCF | 0.1 M KHCO3 | −1.0 | 35 | 2 | 1 | 54 | 8 | — | — | — | — | 355 |
| 41 | Cu1.8Se | 0.1 M KHCO3 | −1.0 | — | 2 | — | 30 | 3 | — | — | 20 | — | 367 |
| 42 | ZNC | 0.1 M KHCO3 | −0.7 | 80 | 20 | — | — | — | — | — | — | — | 358 |
| 43 | Cu@ZNC-1 | 55 | 42 | — | — | — | — | — | — | — | |||
| 44 | Cu@ZNC-2 | 38 | 65 | — | — | — | — | — | — | — | |||
| 45 | Cu@ZNC-3 | 23 | 80 | — | — | — | — | — | — | — | |||
| 46 | Cu@ZNC-4 | 43 | 60 | — | — | — | — | — | — | — | |||
| 47 | CuBi | 0.5 M KHCO3 | −1.07 | — | — | — | — | 98.3 | — | — | — | — | 359 |
| 48 | Bulk BI | 0.5 M KHCO3 | −1.0 | — | — | — | — | 91 | — | — | — | — | 368 |
| 49 | P-Bi | — | — | — | — | 85 | — | — | — | — | |||
| 50 | P-Cu-BiNF | — | — | — | — | 90 | — | — | — | — | |||
6.15. Defect engineering approach
Defect engineering plays a role in designing e-CO2R catalysts as it greatly influences their reactivity, selectivity and overall performance. Defects like vacancies, dislocations and grain boundaries serve as sites for reactions affecting the binding energies of reactants and intermediates. The significance of this approach lies in its ability to customize the surface chemistry of catalyst materials promoting reaction pathways and desired products while minimizing side reactions. By introducing and controlling defects, researchers can enhance the efficiency of CO2 conversion into chemicals and fuels contributing to sustainable energy conversion and carbon capture technologies. To incorporate engineering into CO2RR catalyst design, several methods can be employed. One approach involves synthesizing nanostructured materials with defects like metal nanoparticles with lattice imperfections. Another approach includes treating catalysts after synthesis through annealing in atmospheres to induce formation. Surface modification techniques such as chemical functionalization or atomic layer deposition can also be utilized to introduce controlled defects onto the catalyst surface. Furthermore, combining strain engineering with defect engineering can lead to CO2RR performance improvement.In a related investigation, the study focused on faulty graphene-supported Cu4S2 clusters and isolated Cu4Xn clusters for e-CO2R to C1 products. DFT simulations reveal that among the C1 products on Cu4X2 clusters, CH3OH exhibits the most favorable limiting potential, spanning from Cu4S2 to Cu4Se. When it comes to selectivity and catalytic activity (−0.48 V), the Cu4S2 cluster stands out in the CO2 conversion process. The introduction of defect-engineered graphene increases the robust interaction between the cluster and the substrate, leading to enhanced catalytic performance. Crucially, the Cu4S2/SV catalyst excels in suppressing competing reactions such as HER, CO, and HCOOH generation, thereby exhibiting superior activity in producing CH3OH at low potential.369 In a related study, Zhang et al. investigated e-CO2R, with a specific focus on the use of nano-defective Cu nanosheets as catalysts, aiming to enhance the production of ethylene efficiently. Remarkably, the results demonstrate that at a current density of approximately 60 mA cm−2, the ethylene FE can be significantly increased to an impressive 83.2% when employing these nano-defective Cu nanosheets as catalysts. The key role played by these nano-defects involves providing highly concentrated atomic defects that enrich reaction intermediates while limiting OH− availability. This dual effect facilitates C–C coupling reactions and ultimately enhances ethylene production.24
Simultaneously, another study has successfully synthesized and dispersed CeO2 nanotubes with diameters ranging from 100 to 300 nm and 30 to 50 nm. Importantly, these nanotubes maintain their morphology even after being loaded with Cu and Ni. The interaction between these metals leads to enhanced dispersion and reducibility within the bimetallic Cu–Ni system, effectively reducing the activation barrier for CO adsorption and promoting hydrogenation to CH3OH. Notably, robust Cu–Ni alloy–CeO2 interactions further increase the reducibility of surface CeO2, facilitating the transition from Ce4+ to Ce3+.370 In another significant development, Zhou et al. have conducted hydrothermal synthesis to create catalysts based on MoS2 with varying levels of Cu doping. The incorporation of metallic Cu into the catalyst resulted in improved crystallinity, increased layer thickness, and an increased number of sulfur vacancies. Notably, the 5% Cu-MoS2 catalyst exhibited CH3OH production that was 2.27 times higher than that of the pure catalyst, indicating its superior efficiency in converting CO2 to methanol.371 Furthermore, a La2CuO4 perovskite catalyst was also designed for e-CO2R, leading to the generation of CH4. At −1.4 V vs. RHE, this catalyst achieved a partial current density of 117 mA cm−2 for CH4 production, with a high FE of 56.3%. Interestingly, during cathodic CO2 methanation, the perovskite structure evolves simultaneously with the formation of a Cu/La2CuO4 heterostructure.372
Additionally, Deng et al. have explored the e-CO2R to formate using a sulfur-doped copper catalyst. Sulfur dopants were found to play a crucial role in enhancing the AC–CuSx catalyst's ability to selectively produce formate. The presence of sulfur on the copper surface altered the adsorption strength of adsorbed HCOO*, resulting in increased formate synthesis and reduced *COOH generation, which is an intermediate in the pathway to CO production.373 In another significant development, Wang et al. have successfully created a Cu-doped CeO2 electrocatalyst for the selective CO2R to CH4. The strong interaction between CeO2 and Cu results in the formation of single-atomic-distributed Cu species, which in turn generate numerous OVs in close proximity to one another (Fig. 34). This atomic dispersion of electrocatalytic Cu sites, coupled with the surrounding OVs and the cooperative effect from the CeO2 framework, accounts for the exceptional efficiency of this catalytic site in converting CO2 molecules into CH4.374
 | ||
| Fig. 34 Performances of e-CO2R. CV curves for Cu–CeO2, CeO2, and Cu (a). FE (bars, left y-axis) and jdrp (red curves, right y-axis) of Cu–CeO2-4% (b), pure Cu (c), and undoped CeO2 (d) at different overpotentials. The first five products in the legends at the bottom, which are marked with a red line, are the deep reduction products. A comparison of the FE of samples with different amounts of Cu doping (e). On the left side of the y-axis, the squares show how stable are FECH4 (blue) and FEH2 (black) (f). Total current density (jtotal) of Cu–CeO2-4% at 1.8 V is shown by the red arcs on the right side of the y-axis. Reproduced with permission from ref. 374. Copyright 2018, American Chemical Society. | ||
In the quest for more efficient catalysts for CO2R, Yang et al. have explored the synthesis of a (Cu, N) co-doped SnO2 material through hydrothermal synthesis and calcination (Fig. 35). This doping strategy significantly improved the catalyst's activity, selectivity, and long-term stability. Notably, the catalyst achieved its highest catalytic activity, reaching 54.97%, when co-doped with 7% Cu and N. Furthermore, this doping strategy reduced the material's band gap from 1.058 eV (pure SnO2) to 0.518 eV (7% doped SnOx), enhancing its ability to absorb visible light.375
 | ||
| Fig. 35 The FE of CO2 reduction to formate was investigated using a carbon paper electrode loaded with 7%-(Cu, N)–SnOx at various potentials (a). The faradaic efficiency of CO2R to formate was studied using different electrodes with varying amounts of (Cu, N) doping at a potential of −1.4 V, with an electrolysis time of 1 hour (b). Reproduced with permission from ref. 375. Copyright 2020, Elsevier. | ||
Zhang et al. have developed copper nanowires with a high density of flaws for e-CO2R to C2H4. These nanowires exhibit a broad potential window for C2H4 synthesis and a high selectivity for C2 products.376 In a related study, Wu et al. have inserted point defects and planar defects into the Cu substrate. These defects are then used to successfully construct a highly diluted SnCu polycrystal. The electrical effect is optimized through the combined use of Sn doping and grain boundaries, which prevent H2 evolution and encourage CO2 hydrogenation.377 In another research endeavor, density functional theory calculations were employed to examine CO2R over three defective facets in Cu2O. OVs were found to significantly reduce the activation barrier for C–O bond breakage, from 3.2 to approximately 1 eV. Furthermore, when Cu2O is combined with plasmonic metals, the remaining barrier is further reduced or even eliminated under plasmon-excited states.378 Furthermore, Wang et al. revealed a method for producing binder-free, OV-rich Cu/CuOx in-plane heterostructured nanosheet arrays (Cu/CuOx/CF) using Cu foam as a support. The nanosheet morphology plays a crucial role during electrocatalysis by promoting efficient mass and charge transfer through the exposure of active sites. The electronic structure of these active sites is modified through the synergistic effects of in-plane heterojunctions and OVs, influencing adsorption characteristics and preventing the formation of byproducts.379
Moreover, Zhang et al. have developed a CuPc/DG composite for the e-CO2R into formic acid. The presence of defective graphene in the composite plays a significant role in altering the electronic structure of Cu active centers through π-stacking interactions. The composite exhibits a partial current density of 5.28 mA cm−2 for HCOOH production, a remarkable FE of 44.6% at 0.78 V vs. RHE for CO2RR to HCOOH, and exceptional stability, maintaining its performance for at least 20 hours of continuous reaction.380 Furthermore, in the pursuit of methane generation at single-atomic Cu active sites, Patra et al. have successfully developed a Cu/ceria catalyst with excellent selectivity. It was found that high selectivity during CO2RR is regulated by the calcination temperature due to the increased concentration of OVs on the catalyst surface. Additionally, it was observed that a higher alkaline pH creates more active sites for hydrogen evolution, supporting the hypothesis that pH plays a crucial role in regulating surface termination.381 In a related study, the importance of the Zr–O–Cu interface for CO2R was highlighted. This study utilized Cu nanoparticles for CO2R reduction on a UiO-66 MOF with missing-linker defects. The research revealed that the presence and quantity of linker defects significantly influence the kinetics, efficiency, and side-product production in CO2R. An optimal range of five to seven missing linkers per unit cell was identified, striking a balance between steric effects and binding at the Zr node.382 The crystalline or morphological defect engineering strategy has been adopted as described earlier and a number of designed electrocatalysts are summarized in Table 19. Defect engineering can focus on both crystalline and morphological aspects. The strategies are stated before. Table 19 contains several designed electrocatalysts.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | n-CuNS | 0.1 M KHCO3 | −1.0 | 27 | — | — | 73 | — | — | — | — | — | 24 |
| 2 | OD-Cu | 0.1 M KHCO3 | −0.95 V | 37.4 | 1.17 | 1.3 | 30.5 | 6.4 | — | — | 12.5 | — | 383 |
| 3 | La2CuO4 | 0.1 M KHCO3 | −1.4 | — | — | 50 | — | — | — | — | — | — | 372 |
| 4 | Cu bare | 0.1 M KHCO3 | −2.54 mA cm−2 | 70.1 | — | 7.1 | 4.4 | — | — | — | — | — | 373 |
| 5 | AC–CuSx | −7.49 mA cm−2 | 88.4 | — | 1.6 | 1.6 | 0.2 | — | — | — | — | ||
| 6 | DS-CuSx | −10.45 | 90.9 | — | 0.4 | 2.5 | — | — | — | — | — | ||
| 7 | 0%-(Cu, N)–SnOx | 0.1 M KHCO3 | −1.4 vs. Ag/AgCl | — | — | — | — | 30.21 | — | — | — | — | 375 |
| 8 | 2%-(Cu, N)–SnOx | — | — | — | — | 35.34 | — | — | — | — | |||
| 9 | 5%-(Cu, N)–SnOx | — | — | — | — | 49.21 | — | — | — | — | |||
| 10 | 7%-(Cu, N)–SnOx | — | — | — | — | 54.97 | — | — | — | — | |||
| 11 | 10%-(Cu, N)–SnOx | — | — | — | — | 38.68 | — | — | — | — | |||
| 12 | 15%-(Cu, N)–SnOx | — | — | — | — | 32.22 | — | — | — | — | |||
| 13 | 10h-CuNWs | 1 M KOH | −1.0 | — | — | — | 55 | — | — | — | — | — | 376 |
| 14 | 15h-CuNWs | — | — | — | 58 | — | — | — | — | — | |||
| 15 | 20h-CuNWs | — | — | — | 38 | — | — | — | — | — | |||
| 16 | 25h-CuNWs | — | — | — | 47 | — | — | — | — | — | |||
| 17 | Cu | 2.5 M KOH | −0.9 | 78 | 23 | — | — | — | — | — | — | — | 377 |
| 18 | SnCu | 63 | 37 | 5 | — | — | — | — | — | ||||
| 19 | pc-SnCu | 20 | 30 | 11 | 45 | — | — | — | — | — | |||
| 20 | 0%-Cu/CeO2 | 1 M KOH | −0.89 | 85 | 2 | — | — | — | — | — | — | — | 381 |
| 21 | 0.5%-Cu/CeO2 | 48 | 4 | 32 | 3 | — | — | — | — | — | |||
| 22 | 1%-Cu/CeO2 | 43 | 2 | 42 | 2 | — | — | — | — | — | |||
| 23 | 1.6%-Cu/CeO2 | 50 | 5 | 25 | 4 | — | — | — | — | — | |||
| 24 | 2.3%-Cu/CeO2 | 65 | 3 | 8 | 10 | — | — | — | — | — | |||
6.16. Metal organic framework (MOF) derived approach
The MOF based method to design e-CO2R catalysts is important because researchers can use it to design more environmentally friendly products. What's interesting about MOFs is that they provide a platform that can be easily adjusted to meet the demands of catalyst development. There are lots of MOFs which consist of metal ions bound to ligands, differing in qualitative properties from each other because of their unique structures. By choosing appropriate metal ions and ligands researchers can prepare MOFs that can be used as catalytic materials. The method which is advocated here has control over the composition, structure and surface properties of the catalyst. The MOF derived approach can be used in many ways in the design of CO2RR catalysts. One method widely used is that in which MOFs are thermochemically decomposed by calcination in a controlled atmosphere, hence forming carbon-related materials which have a definite structure and active sites. In situations MOFs can serve as templates, for depositing metal nanoparticles or individual atoms. This enables the development of catalysts that exhibit improved properties. The approach derived from MOFs offers a foundation for designing CO2RR catalysts. By leveraging the characteristics of these materials, we can tackle the urgent issues related to carbon capture and conversion while fostering a sustainable and environmentally friendly future, with reduced carbon emissions.Zhao et al. offered a strategy by using a Cu-based MOF (HKUST-1) and created oxide-derived Cu/carbon (OD Cu/C) catalysts. At −0.1 to −0.7 V vs. RHE, the resultant materials exhibited a total FE of 45.2–71.2%, demonstrating highly selective CO2R to alcohol molecules. Ethanol and methanol yields were optimized to 3.7–13.4 mg L−1 h−1 and 5.1–12.4 mg L−1 h−1, respectively. Near −0.1 V, or 190 mV overpotential, C2H5OH production began to occur.384 In another work, Cu3(BTC)2 (Cu-MOF) was incorporated into carbon paper-based GDEs as a CO2 capture agent. This incorporation resulted in higher FEs of CH4 on GDEs with Cu-MOF weight ratios, greatly improving the conversion efficiency and product selectivity of CO2 electroreduction.385 In a related field, Ma et al. designed CTF-B constrained Cu catalysts with an impressive maximum FE of 81.3% for CO2R. Their investigations revealed the dynamic development of Cu-ACs, active sites for CO2RR, in response to potential, as demonstrated through in vivo XAFS investigations.386 In a complementary study, Kim et al. successfully controlled the shape of Cu-MOFs into nanoparticles with diameters of 80 nm using a seed-mediated solvothermal polyol method. During more than 10 hours of e-CO2RR under neutral conditions, the MOF-based copper catalysts exhibited maximum selectivity towards ethylene and C2+ products. By employing a GDE technique, they maintained high FE and generated a high partial geometric current density toward C2+ products in alkaline environments.387
In a related investigation, a thin film of organic additives like N-toylpyridinium could effectively restrict H+ mass passage to the electrode without impeding CO2 transit. This strategic modification leads to improved production of C2+ products and a reduction in the competitive HER. Modified copper electrodes enhance selective CO2R to C2+ products, even in very acidic electrolytes and at low alkali cation concentrations and operating current densities.388 In a separate investigation, another study explored the effectiveness of electrolysis at a constant potential in six different electrolytes (KHCO3/H2O, TBAB/DMF, KBr/CH3OH, CH3COOK/CH3OH, TBAB/CH3OH, and TBAP/CH3OH) for the CO2R. Notably, KHCO3/H2O and TBAB/CH3OH showed a lower onset potential. Across all electrolytes, the primary product generated was HCOOH, although with varying maximum formic acid concentrations. Interestingly, when using a 0.1 M TBAB/DMF electrolyte, the synthesized MOF material exhibited a high efficiency of 58% in forming both HCOOH and CH3COOH products.389 Furthermore, through a novel approach of carbonizing Cu–BTC at steady temperatures, a new catalyst (Cu2O/Cu@NC-800) was developed. Carbonizing the catalyst at 800 °C significantly increased its activity and selectivity toward formate production. The formation of low-index facets and the uniform distribution of metallic nanoparticles within N-doped carbon frameworks played a key role in enhancing the catalyst's activity. The increased nitrogen content in the Cu2O lattice lowered the binding energy of *OCHO, facilitating easier formate synthesis via the *OCHO pathway.390
In a related context, another research study focused on regulating the structural evolution of porous materials during e-CO2RR to develop new electrocatalysts. To promote the coupling of C radical intermediates for the creation of C2+ products, TCNQ was introduced as a second electron acceptor to expedite the reduction of Cu(II) sites to Cu(I) species. The resulting TCNQ@CuMOP-p/CP catalyst exhibited a high selectivity of 68% for C2+ products at a total current density of 268 mA cm−2 and 2.27 V vs. RHE, making it competitive with the best Cu(II)-MOF-derived catalysts for CO2RR.391 Furthermore, Cheng et al. revealed that the e-CO2RR performance of Cu catalysts derived from MOFs is greatly influenced by the amount and types of nitrogen present in the carbon shell. The remarkable selectivity for the production of ethylene and ethanol can be attributed to the high pyrrolic-N and Cu–N doping in the carbon support, which provides numerous CO2 adsorption sites and facilitates carbon–carbon coupling processes. However, an abundance of graphitic-N and oxidized-N atoms can lead to severe competition from the HER. Therefore, enhancing the synergy between Cu and the nitrogen-doped carbon support requires modifications to the N structure.392
In related research, the synthesis of a single-atom copper catalyst based on MOFs via plasma activation was described by Wei et al. The plasma bombardment increases low-coordinated copper sites and creates OVs, while also forming a hierarchically porous structure that effectively adsorbs reactant molecules. This unique combination of features greatly enhances e-CO2R activity, resulting in a maximum FE of 75.3%. Products with carbon content achieve a total FE of 96.5%.393 Furthermore, the research reveals that homemade e-CO2R reactors can be constructed using CuIn-MOF series catalysts coated on carbon paper. These catalysts have low over-potentials, large current densities, and selective activity toward CO and formic acid. The stability can be enhanced by adjusting the relative molar ratio of Cu/In, which results in a current density between 20.1 and 88.4 mA cm−2. Cu1In1-MOF-SO4 exhibits the highest e-CO2R activity, while Cu1In3-MOF-NO3 and Cu1In3-MOF-SO4 have the highest FECO and FEHCOO− values.394 In another study, MOF-derived In–Cu bimetallic oxides were used in a 0.5 M KHCO3 aqueous solution for the e-CO2R to CO. With a total current density of 11.2 mA cm−2 and a FE of 92.1%, InCuO-0.92 was found to be a very effective and stable catalyst. The improved performance was traced back to the synergistic impact between In and Cu oxides, the increased electrochemical surface area, the greater CO2 adsorption, the lower charge transfer resistance, and the porous mass diffusion structure.395 Furthermore, through in situ modification of MWCNTs, a novel BiCu bimetallic organic framework composite catalyst, SU-101-Cu@2.5C, was effectively developed and synthesized (Fig. 36). The catalyst's efficiency in converting e-CO2R to formate was investigated. The combination of BMOFs and MWCNTs created a synergistic effect that facilitated electron transport, ultimately enabling efficient e-CO2R to formate.396
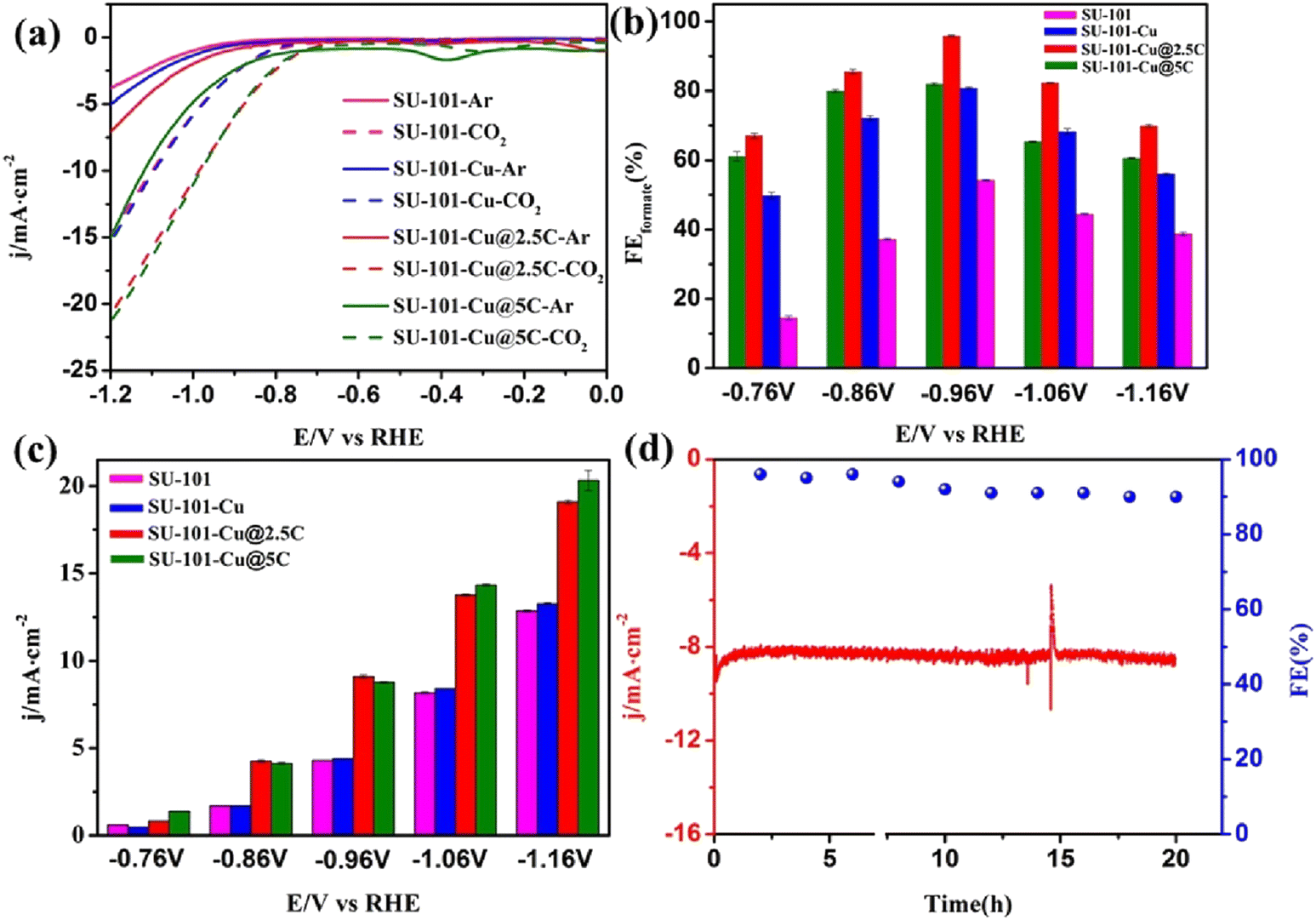 | ||
| Fig. 36 The electrocatalytic performance of SU-101, SU-101-Cu, and SU-101-Cu@xC was evaluated through LSV measurements in CO2 and Ar atmospheres (a). The measurements included different applied potentials, FEformate (b), and current density (c). SU-101-Cu@2.5C was tested for long-term durability at a voltage of −0.96 V vs. RHE for a duration of 20 hours (d). Reproduced with permission from ref. 396. Copyright 2023, Elsevier. | ||
Additionally, Cu-MOF808 was pyrolyzed to produce Cu@ZrO2 with a hollow structure. This transformation allowed for the control of the porosity structure and metal–support interaction through varying pyrolysis temperatures. Notably, the numerous Cu–ZrO2 interfaces created during proper calcination were instrumental in facilitating the hydrogenation of CO2 to methanol. CO2 primarily found absorption and activation sites at the fundamental locations of Cu–ZrO2 interfaces, underscoring the significance of the Cu+/Cu0 ratio. Among these catalysts, CM-300 at 220 °C demonstrated the most remarkable efficacy for converting CO2 into methanol, boasting a 5% conversion rate and an 85% selectivity for methanol.397 Furthermore, electrocatalytic stability and performance in hydrocarbon synthesis can be substantially enhanced by post-synthetic alterations of copper-based MOFs. This improvement involves converting free COOH/–OH groups into amide/amine groups. The modified MOFs exhibit high electrocatalytic performance (total FE = 81%, with FEC1 = 62% and FEC2 = 19%) and remarkable stability.398
Moreover, Liu et al. have developed Cu–N coordinated MOFs for selective C2H4 production, and in situ electrochemical reconstruction was employed to construct highly active Cu/Cu2O nanoclusters. The FE for C2H4 production, measured by partial current density at 1.03 V vs. RHE in the CO2RR, reached 70.2 ± 1.7%.399 In addition, Parambath et al. have utilized a Cu-based MOF, namely HKUST-1. Remarkably, these materials exhibited high selectivity in CO2R to alcohol molecules, with total FE ranging from 45.2% to 71.2% at potentials between −0.1 and −0.7 V vs. RHE. Ethanol and methanol yields were optimized to 3.7–13.4 mg L−1 h−1 and 5.1–12.4 mg L−1 h−1, respectively. The overpotential for CO2 reduction, as indicated by the onset potential for C2H5OH production, was modest and close to −0.1 V.400Table 20 showcases different MOFs designed for e-CO2R to yield valuable products.
| No. | Material | Electrolyte | V vs. RHE | ≈FE% | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | C2H4 | HCOO− | C2H5+ | CH3OH | C2H5OH | C2H3O2− | |||||
| 1 | OD Cu/C-1000 | 0.1 M KHCO3 | −0.7 | — | — | — | — | 9 | — | 15 | 33 | — | 384 |
| 2 | Cu/C1000 | — | — | — | — | 16 | — | 4 | 12 | — | |||
| 3 | Cu-MOF-10% | 0.5 M NaHCO3 | −1.8 | 47 | — | 11 | 16 | — | — | — | — | — | 385 |
| 4 | NNU-50 | — | −1.1 | — | — | 66 | — | — | — | — | — | — | 401 |
| 5 | Cu6MePz | — | — | 22 | — | — | — | — | — | — | |||
| 6 | n-MDC-225 | 1.0 M KOH | −1.01 | 45.9 | 7.3 | 9.8 | 29.6 | 5.8 | — | — | 5.2 | — | 402 |
| 7 | n-MDC-250 | 13.4 | 2.3 | 1.9 | 63.1 | 3.2 | — | — | 12.5 | 1.2 | |||
| 8 | n-MDC-300 | 22.2 | 5.0 | 0.5 | 52.6 | 9.6 | — | — | 2.0 | 7.7 | |||
| 9 | m-MDC-250 | 36.0 | 10.4 | 3.3 | 34.7 | 11.7 | — | — | 5.6 | — | |||
| 10 | m-MDC-265 | 27.7 | 2.7 | 1.4 | 48.3 | 5.9 | — | — | 11.6 | 0.1 | |||
| 11 | m-MDC-300 | 27.8 | 7.7 | 0.7 | 25.1 | 24.6 | — | — | 7.9 | 6.0 | |||
| 12 | Cu | H3PO4/KH2PO4, [K+] = 0.1 M | −1.29 | 95 | 0.14 | — | — | 1 | — | — | — | — | 403 |
| 13 | Cu/10 mM tolyl-pyr | −1.43 | 23.7 | 0.7 | 1.9 | 24.3 | 3.4 | — | — | 23.8 | — | ||
| 14 | Cu2O/Cu@NC-700 | 0.1 M KHCO3 | −0.88 Ag/AgCl | — | — | — | — | 28 | — | — | — | — | 390 |
| 15 | Cu2O/Cu@NC-800 | — | — | — | — | 42 | — | — | — | — | |||
| 16 | Cu2O/Cu@NC-900 | — | — | — | — | 20 | — | — | — | — | |||
| 17 | TCNQ@CuMOP-p/CP | 1.0 M KOH | −1.07 | — | 18 | 1 | 15 | 10 | — | — | 6 | — | 404 |
| 18 | HKUST-1-p/CP | — | 20 | 10 | 5 | — | — | 7 | — | ||||
| 19 | TCNQ@HKUST-1-p/CP | — | 25 | 15 | 8 | — | — | 6 | — | ||||
| 20 | CuMOP-p/CP | — | 18 | 1 | 12 | 8 | — | — | 5 | — | |||
| 21 | PA–CuDBC-1 | 0.5 M KHCO3 | −1.0 | 15 | 15 | 60 | 10 | — | — | — | — | — | 405 |
| 22 | CuDBC | 43 | 42 | — | 1 | — | — | — | — | — | |||
| 23 | PA–CuDBC-2 | 30 | 30 | 30 | 10 | — | — | — | — | — | |||
| 24 | CuIn–MOF–SO4 | 0.5 M KHCO3 | 1.06 | — | 50 | — | — | — | — | — | — | — | 406 |
| 25 | CuIn–MOF–NO3 (Cu/In molar ratio ¼ 1:3) |
— | 38 | — | — | — | — | — | — | — | |||
| 26 | In2O2 | 0.5 M KHCO3 | −1.0 | — | 10 | — | — | — | — | — | — | — | 407 |
| 27 | InCuO-0.15 | — | 30 | — | — | — | — | — | — | — | |||
| 28 | InCuO-0.37 | — | 50 | — | — | — | — | — | — | — | |||
| 29 | InCuO-0.55 | — | 70 | — | — | — | — | — | — | — | |||
| 30 | InCuO-0.72 | — | 83 | — | — | — | — | — | — | — | |||
| 31 | InCuO-0.92 | — | 91 | — | — | — | — | — | — | — | |||
| 32 | SU-101-Cu@5C | 0.5 M KHCO3 | −1.06 | 28 | 2 | — | — | 65 | — | — | — | — | 396 |
| 33 | SU-101-Cu@2.5C | 5 | 2 | — | — | 82 | — | — | — | — | |||
| 34 | SU-101-Cu | 15 | 10 | — | — | 70 | — | — | — | — | |||
| 35 | SU-101 | 43 | 7 | — | — | 45 | — | — | — | — | |||
6.17 Alkaline metal cations and halide anions on Cu electrocatalysts
The modification of Cu electrocatalysts through the adsorption of alkaline metal cations and halide anions has shown significant impact on the e-CO2RR. This section explores the mechanisms and outcomes of such modifications. Alkaline metal cations, such as Li+, Na+, K+, and Cs+, play a crucial role in CO2RR. These cations interact with the electric double layer at the catalyst surface, affecting the local electric field and the binding strength of reaction intermediates. The size and hydration energy of these cations influence their specific adsorption properties, which in turn modify the catalytic activity and selectivity.408 Smaller cations like Li+ have higher charge densities and stronger hydration energies, leading to different adsorption behaviors compared to larger cations like Cs+.409 These differences impact the stabilization of key intermediates such as CO2˙− and *CO. Adsorbed cations can alter the local electric field at the catalyst surface, influencing the electron transfer kinetics. For example, larger cations may create a more diffuse electric field, which could enhance the formation of multi-carbon products by stabilizing the required intermediates.410Halide ions on copper surfaces play a crucial role in e-CO2RR by weakening the C–O bonds of CO2, lowering hydrogenation potential barriers. Understanding halide modified copper electrocatalysts (Cu–X) can help design efficient Cu–X electrocatalysts for e-CO2RR. Factors influencing catalytic performance include morphology, oxidation state, and electrolyte pH/composition.411 Halide anions, including Cl−, Br−, and I−, adsorb on the Cu surface and modify the catalytic properties through electronic and geometric effects. Yuan et al. have presented a method to immobilize Cu-based catalysts during the e-CO2RR, revealing the fundamental mechanism of specific adsorption of halide ions. The method involves pre-reduction in aqueous KX electrolytes and e-CO2RR using non-buffered K2SO4. In situ spectroscopy shows that specific adsorption enhances the adsorption of *CO intermediates, achieving a high selectivity of 84.5% for C2+ products.412 Zhang and co have investigated the impact of alkali metal cations and anions on the current density and product selectivity of e-CO2RR into HCOOH on a SnO2/carbon paper electrode. The results showed that cations promote current density and faradaic efficiencies, while anions decrease current density and FEs for HCOOH formation.413 The current density and FEHCOOH were found to be in the order of Li+ < Na+ < K+ < Cs+ < Rb+. The cation species' effect is attributed to the change in surface charge density influenced by the radius of hydrated cations and the pH values determined by the small number of bare cations. The anions' promotion of current density was found to be in the order of NaHCO3 < NaCl < NaBr < NaI and KHCO3 < KCl ≈ KI < KBr, and the effect on FEHCOOH was in the order of HCO3− < Cl− < Br− < I−. The influence of anions on ERCO2 to HCOOH is mainly due to changes in CO2, H+, and OH− transport, as well as anions' adsorption (Fig. 37).
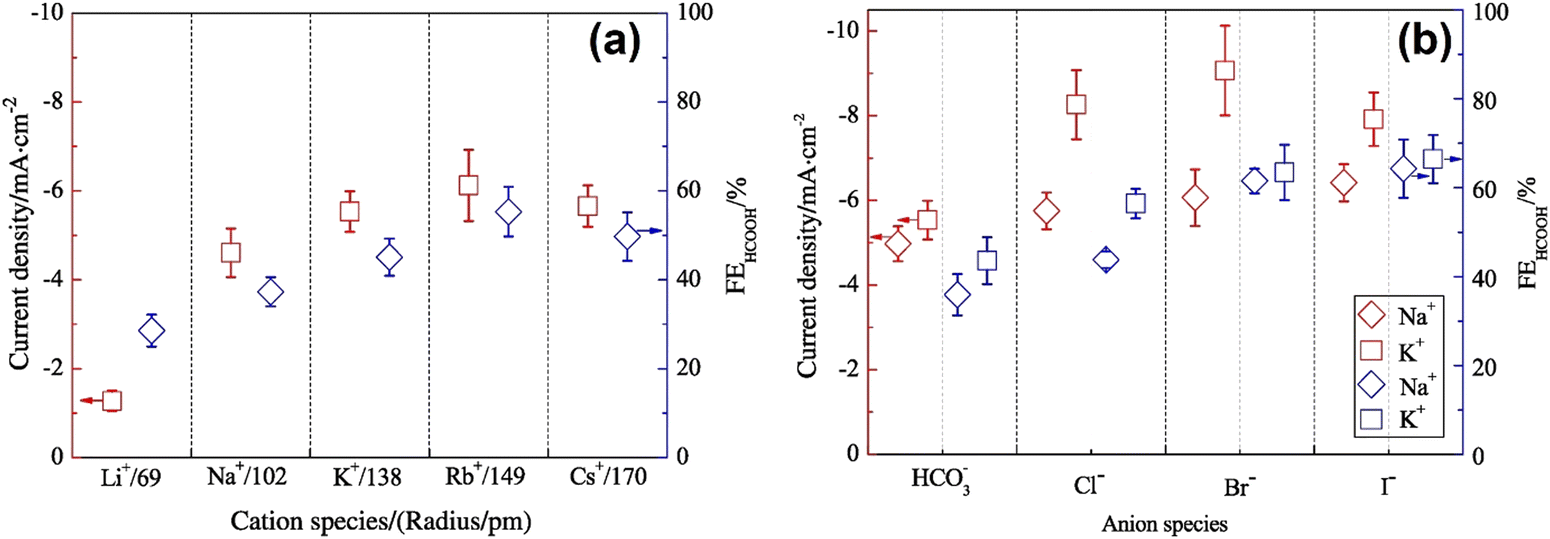 | ||
| Fig. 37 Current density and FEHCOOH as functions of electrolyte type and the cation size, measured using an SnO2/C electrode at −1.4 V in 0.1 mol L−1 MHCO3 (M = Li+, Na+, K+, Rb+, Cs+) and NaX and KX (X = HCO3−, Cl−, Br− and I−), respectively (a and b). Reproduced with permission from ref. 413. Copyright 2020, Elsevier. | ||
7. Summary
In the process of developing technology to fight climate change, e-CO2R copper-based catalysts can be rationally designed, offering hope for the future of this method. Among engineering strategies, the single crystal and atom approach is a standout technique. This innovative technique involves dispersing individual copper atoms onto a support material resulting in efficiency and selectivity for CO2R. By distributing metal atoms scientists can minimize the use of scarce metals while maximizing catalytic activity. Control over sites and this approach are appropriate to address challenges, and offer opportunities for efficiency and selectivity.Another important strategy in designing copper-based catalysts is oxidation state engineering. In designing copper-based catalysts, another important possibility is oxidation state manipulation. The structure of the catalyst allows for optimization of e-CO2R, through tailoring. Balance between activity and selectivity is achieved by this approach. Utilizing alloys is also a robust strategy for designing copper-based catalysts. Copper and metals are strategically combined here. With customized properties, these alloys have been adapted. Compound alloys further take this concept by introducing elements into the alloys, which not only increases their strength but also makes them more accurate. In catalyst design, these strategies allow for fine-tuning of activity and selectivity.
The methods of composite engineering involve the addition of copper-based catalysts to other functional materials in order to increase their stability and activity. Typically, these composites are made up of copper nanoparticles incorporated in a supportive matrix that gives them improved performance. Another way is to add other metals to the structure of copper-based catalysts, to produce changes in their properties and catalytic behavior. This approach may result in high activities and selectivity but sustainability as well as cost are among its drawbacks. On the other hand, nonmetal doping offers an approach to modify the properties of Cu catalysts by introducing nonmetal elements into their structure. This allows researchers to tailor their properties and reactivity for optimized CO2R efficiency and selectivity. Surface anchoring focuses on attaching metal nanoparticles to support materials preventing agglomeration and enhancing stability.
Molecular surface functionalization involves modifying the surface of the catalyst with molecules to enhance CO2 adsorption and reactivity. This strategy improves activity and selectivity by customizing the surface properties of the catalyst. Interfacial engineering aims at optimizing interactions between the catalyst and reactants thereby improving CO2 reduction efficiency. Additionally controlling the chemical species on the surface of Cu through speciation influences its behavior. Facet engineering focuses on manipulating the crystal surfaces of Cu based catalysts to optimize the adsorption and conversion of CO2. The coordination engineering approach involves adjusting the arrangement of Cu atoms in the catalyst, which affects their reactivity. The site engineering approach aims to control the sites on the catalyst surface to optimize CO2 adsorption and conversion. Strain engineering employs chemical methods to introduce strain into the catalyst structure, which impacts its properties and catalytic activity. The catalyst pre-oxidation approach involves controlled oxidation to enhance its performance.
The precursor derived methods use precursor materials to fabricate Cu-based catalysts with the properties desired. As an alternative, polymer modification procedures introduce polymers into the catalyst structure and increase stability and reactivity. This research aims to improve catalyst performance by optimizing the support material from an engineering perspective. The defect engineering method involves carefully forming defects within catalysts as sites for the adsorption and reduction of CO2. By reducing costs, and increasing both activity and selectivity, this process also turns Cu into a primary catalyst material.
8. Conclusions
To sum up, by producing copper-based catalysts for e-CO2R, one can hit three birds with one stone by fighting global warming, alleviating energy tension and facilitating sustainable technologies. Various engineering techniques, from doping of atoms to changing defects, have been studied to improve these catalysts' performance. The single atom approach controls sites effectively and attains outstanding efficiency and selectivity in CO2 reduction. Therefore, this approach will prove an asset in future catalyst modification. Moreover, strategies involving oxidation state manipulation, alloy combinations, and composite designs offer flexibility in catalyst development, providing customized properties that match specific needs. Doping with both metal and non-metal additives, as well as surface functionalization techniques, are ways to modify catalysts for greater reactivity. Interfacial, facet, coordination, site, strain, and speciation engineering approaches allow fine-tuning of catalyst behavior. Catalyst preoxidation, precursor-derived approaches, polymer modification, support engineering, and defect engineering help to further improve the optimization of catalysts. Cost effectiveness plays a vital part in making CO2 utilization a commercial reality. While the costs of such techniques may be raised by approaches like doping with elements, defect engineering with low-cost Cu as the main material offers an economically viable option. For practical applications, stability is essential, and much attention is paid to these problems. Combined with defect engineering, single-atom methods have shown excellent stability, while metal doping with noble metals also helps protect against corrosion and agglomeration. Long-term catalyst reliability can also be achieved through proper defect engineering, strict control of sites for activities, and strain engineering.Based on the comprehensive analysis presented in this review, it is evident that no single approach stands out as the universally best strategy for designing Cu-based electrocatalysts for e-CO2R. However, the combination of several engineering strategies appears to be the most promising pathway. Specifically, integrating doping techniques with surface and defect engineering can synergistically enhance the catalytic performance by improving both the activity and selectivity of the catalysts. This multi-faceted approach leverages the strength of each individual strategy, offering a balanced solution that addresses the various challenges associated with CO2R. Moreover, the incorporation of advanced in situ spectroscopic and computational studies provides critical insights into the reaction mechanisms and intermediate species, guiding the rational design of these hybrid catalysts. This combination not only allows for real-time monitoring and optimization of catalytic processes but also facilitates the prediction and screening of potential modifications at an atomic level. Therefore, a holistic strategy that combines experimental advancements with theoretical insights is recommended as the best approach for the development of high-performance Cu-based electrocatalysts for CO2 electroreduction.
9. Future outlook
Cu-based e-CO2R catalysts project a bright future for further innovation and development. There are several main directions and problems we would like to discuss. In addition, given advances in materials science, future catalysts may be designed to carry out various reactions simultaneously, as applications progress in this area. The realizability of Cu-based catalysts is necessary for real-world usage. Researchers must work out high-capacity synthesis methods and steps. Under industrial conditions, maintaining long-term durability and reliability of Cu-based catalysts is all-important. Taking steps to lessen catalyst deterioration ought to be a target of future research. The integration of catalysts into CO2 capture and utilization systems will facilitate the realization of efficient CO2R processes. Integrating e-CO2R with solar and wind power, among other renewable sources, can make the technology more ecological and manageable. We need to tailor Cu-based catalysts so that they can selectively make ethylene or ethanol or other multi-carbon products in order to satisfy various industrial demands. To ensure these techniques’ overall sustainability, rigorous assessments of the environmental and energy impacts of Cu-based CO2 reduction technology are necessary. Regulations and policies should be introduced as Cu-based catalysts and CO2 utilization technologies mature to support their deployment and promote environmentally sound practices. To accelerate the use of Cu-based catalysts and development of new candidates, a collaborative approach in cooperation with researchers, industry participants, and governments from around the world is required.Data availability
All data and information related to this review article are presented in the main text and supporting references. No new data were generated or analyzed for this review. All sources are properly cited and listed in the reference section. Therefore, no additional data are available for this article.Conflicts of interest
There are no conflicts to declare.References
- L. J. R. Nunes, The Rising Threat of Atmospheric CO2: A Review on the Causes, Impacts, and Mitigation Strategies, Environments, 2023, 10(4), 66 CrossRef.
- J. Li, et al., Electroreduction of CO2 to Formate on a Copper-Based Electrocatalyst at High Pressures with High Energy Conversion Efficiency, J. Am. Chem. Soc., 2020, 142(16), 7276–7282 CrossRef CAS PubMed.
- F. Raganati, F. Miccio and P. Ammendola, Adsorption of Carbon Dioxide for Post-combustion Capture: A Review, Energy Fuels, 2021, 35(16), 12845–12868 CrossRef CAS.
- J. Wu, et al., CO2 Reduction: From the Electrochemical to Photochemical Approach, Adv. Sci., 2017, 4(11), 1700194 CrossRef.
- Y. Wang, J. Liu and G. Zheng, Designing Copper-Based Catalysts for Efficient Carbon Dioxide Electroreduction, Adv. Mater., 2021, 33(46), 2005798 CrossRef CAS.
- K. K. Maniam, et al., Progress in Electrodeposited Copper Catalysts for CO2 Conversion to Valuable Products, Processes, 2023, 11(4), 1148 CrossRef CAS.
- A. Álvarez, et al., Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes, Chem. Rev., 2017, 117(14), 9804–9838 CrossRef.
- P. A. Kempler and A. C. Nielander, Reliable reporting of faradaic efficiencies for electrocatalysis research, Nat. Commun., 2023, 14(1), 1158 CrossRef CAS PubMed.
- J. Li, et al., Selective CO2 electrolysis to CO using isolated antimony alloyed copper, Nat. Commun., 2023, 14(1), 340 CrossRef CAS PubMed.
- J. Medina-Ramos, J. L. DiMeglio and J. Rosenthal, Efficient Reduction of CO2 to CO with High Current Density Using in Situ or ex Situ Prepared Bi-Based Materials, J. Am. Chem. Soc., 2014, 136(23), 8361–8367 CrossRef CAS PubMed.
- S. Nitopi, et al., Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte, Chem. Rev., 2019, 119(12), 7610–7672 CrossRef CAS.
- H. Yang, et al., Performance and long-term stability of CO2 conversion to formic acid using a three-compartment electrolyzer design, J. CO2 Util., 2020, 42, 101349 CrossRef CAS.
- S. Kozuch and J. M. L. Martin, “Turning Over” Definitions in Catalytic Cycles, ACS Catal., 2012, 2(12), 2787–2794 CrossRef CAS.
- S. Anantharaj, P. E. Karthik and S. Noda, The Significance of Properly Reporting Turnover Frequency in Electrocatalysis Research, Angew. Chem., Int. Ed., 2021, 60(43), 23051–23067 CrossRef CAS.
- E. Bertheussen, et al., Quantification of liquid products from the electroreduction of CO2 and CO using static headspace-gas chromatography and nuclear magnetic resonance spectroscopy, Catal. Today, 2017, 288, 54–62 CrossRef CAS.
- G. Zhang, Y. Cui and A. Kucernak, Real-Time In Situ Monitoring of CO2 Electroreduction in the Liquid and Gas Phases by Coupled Mass Spectrometry and Localized Electrochemistry, ACS Catal., 2022, 12(10), 6180–6190 CrossRef CAS PubMed.
- A. Marinoiu, et al., Nitrogen-Doped Graphene Oxide as Efficient Metal-Free Electrocatalyst in PEM Fuel Cells, Nanomaterials, 2023, 13(7), 1233 CrossRef PubMed.
- S.-T. Gao, et al., Theoretical understanding of the electrochemical reaction barrier: a kinetic study of CO2 reduction reaction on copper electrodes, Phys. Chem. Chem. Phys., 2020, 22(17), 9607–9615 RSC.
- J. Bian, et al., Strategies and reaction systems for solar-driven CO2 reduction by water, Carbon Neutrality, 2022, 1(1), 5 CrossRef.
- T. Cheng, H. Xiao and W. A. Goddard, III, Reaction Mechanisms for the Electrochemical Reduction of CO2 to CO and Formate on the Cu(100) Surface at 298 K from Quantum Mechanics Free Energy Calculations with Explicit Water, J. Am. Chem. Soc., 2016, 138(42), 13802–13805 CrossRef.
- X. Cai, et al., Tuning Selectivity in Catalytic Conversion of CO2 by One-Atom-Switching of Au9 and Au8Pd1 Catalysts, CCS Chem., 2021, 3(12), 408–420 CrossRef.
- Y. Pei, H. Zhong and F. Jin, A brief review of electrocatalytic reduction of CO2—Materials, reaction conditions, and devices, Energy Sci. Eng., 2021, 9(7), 1012–1032 CrossRef.
- Y. Zhou and B. S. Yeo, Formation of C–C bonds during electrocatalytic CO2 reduction on non-copper electrodes, J. Mater. Chem. A, 2020, 8(44), 23162–23186 RSC.
- B. Zhang, et al., Highly Electrocatalytic Ethylene Production from CO2 on Nanodefective Cu Nanosheets, J. Am. Chem. Soc., 2020, 142(31), 13606–13613 CrossRef PubMed.
- J. Kim, et al., Branched Copper Oxide Nanoparticles Induce Highly Selective Ethylene Production by Electrochemical Carbon Dioxide Reduction, J. Am. Chem. Soc., 2019, 141(17), 6986–6994 CrossRef.
- X. Chen, et al., Electrochemical CO2-to-ethylene conversion on polyamine-incorporated Cu electrodes, Nat. Catal., 2021, 4(1), 20–27 CrossRef.
- J. Cai, et al., Highly Selective Electrochemical Reduction of CO2 into Methane on Nanotwinned Cu, J. Am. Chem. Soc., 2023, 145(16), 9136–9143 CrossRef PubMed.
- X.-Q. Wang, et al., Cu-based bimetallic catalysts for CO2 reduction reaction. Advanced Sensor and Energy, Materials, 2022, 1(3), 100023 Search PubMed.
- Y. Sun, et al., Importance of the Initial Oxidation State of Copper for the Catalytic Hydrogenation of Dimethyl Oxalate to Ethylene Glycol, ChemistryOpen, 2018, 7(12), 969–976 CrossRef PubMed.
- S. D. McCann and S. S. Stahl, Copper-Catalyzed Aerobic Oxidations of Organic Molecules: Pathways for Two-Electron Oxidation with a Four-Electron Oxidant and a One-Electron Redox-Active Catalyst, Acc. Chem. Res., 2015, 48(6), 1756–1766 CrossRef PubMed.
- W. Hong, et al., Exploring the Parameters Controlling Product Selectivity in Electrochemical CO2 Reduction in Competition with Hydrogen Evolution Employing Manganese Bipyridine Complexes, ACS Catal., 2023, 13(5), 3109–3119 CrossRef PubMed.
- Y. Lei, et al., Recent advances on electrocatalytic CO2 reduction to resources: target products, reaction pathways and typical catalysts, Chem. Eng. J., 2023, 453, 139663 CrossRef.
- J. Zeng, et al., Electrochemical Reduction of CO2 With Good Efficiency on a Nanostructured Cu–Al Catalyst, Front. Chem., 2022, 10, 931767 CrossRef PubMed.
- D. Xu, et al., Electrocatalytic CO2 reduction towards industrial applications, Carbon Energy, 2023, 5(1), e230 CrossRef.
- M. Li, et al., Discovery of single-atom alloy catalysts for CO2-to-methanol reaction by density functional theory calculations, Catal. Today, 2022, 388–389, 403–409 CrossRef.
- T. Ahmad, et al., Electrochemical CO2 reduction to C2+ products using Cu-based electrocatalysts: a review, Nano Res. Energy, 2022, 1, e9120021 CrossRef.
- Y. Jia, et al., Cu-based bimetallic electrocatalysts for CO2 reduction, Adv. Powder Mater., 2022, 1(1), 100012 CrossRef.
- D. S. Ripatti, T. R. Veltman and M. W. Kanan, Carbon Monoxide Gas Diffusion Electrolysis that Produces Concentrated C2 Products with High Single-Pass Conversion, Joule, 2019, 3(1), 240–256 CrossRef.
- D. Wakerley, et al., Bio-inspired hydrophobicity promotes CO2 reduction on a Cu surface, Nat. Mater., 2019, 18(11), 1222–1227 CrossRef.
- C. W. Li, J. Ciston and M. W. Kanan, Electroreduction of carbon monoxide to liquid fuel on oxide-derived nanocrystalline copper, Nature, 2014, 508(7497), 504–507 CrossRef.
- P. Wilde, et al., Is Cu instability during the CO2 reduction reaction governed by the applied potential or the local CO concentration?, Chem. Sci., 2021, 12(11), 4028–4033 RSC.
- T. Burdyny and W. A. Smith, CO2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions, Energy Environ. Sci., 2019, 12(5), 1442–1453 RSC.
- A. Prašnikar, et al., Mechanisms of Copper-Based Catalyst Deactivation during CO2 Reduction to Methanol, Ind. Eng. Chem. Res., 2019, 58(29), 13021–13029 CrossRef.
- J. Zhao, et al., An overview of Cu-based heterogeneous electrocatalysts for CO2 reduction, J. Mater. Chem. A, 2020, 8(9), 4700–4734 RSC.
- P. Forzatti and L. Lietti, Catalyst deactivation, Catal. Today, 1999, 52(2), 165–181 Search PubMed.
- Y. Yang, et al., Operando Resonant Soft X-ray Scattering Studies of Chemical Environment and Interparticle Dynamics of Cu Nanocatalysts for CO2 Electroreduction, J. Am. Chem. Soc., 2022, 144(20), 8927–8931 CrossRef PubMed.
- Y. Sun, et al., Boosting CO2 Electroreduction to C2H4via Unconventional Hybridization: High-Order Ce4+ 4f and O 2p Interaction in Ce–Cu2O for Stabilizing Cu+, ACS Nano, 2023, 17(14), 13974–13984 CrossRef.
- T. H. Phan, et al., Emergence of Potential-Controlled Cu-Nanocuboids and Graphene-Covered Cu-Nanocuboids under Operando CO2 Electroreduction, Nano Lett., 2021, 21(5), 2059–2065 CrossRef PubMed.
- D. Friebel, et al., Structure, Redox Chemistry, and Interfacial Alloy Formation in Monolayer and Multilayer Cu/Au(111) Model Catalysts for CO2 Electroreduction, J. Phys. Chem. C, 2014, 118(15), 7954–7961 CrossRef.
- Y.-P. Huang, et al., In situ probing the dynamic reconstruction of copper–zinc electrocatalysts for CO2 reduction, Nanoscale, 2022, 14(25), 8944–8950 RSC.
- Y. Xu, et al., Low coordination number copper catalysts for electrochemical CO2 methanation in a membrane electrode assembly, Nat. Commun., 2021, 12(1), 2932 CrossRef.
- Q. Lei, et al., Structural evolution and strain generation of derived-Cu catalysts during CO2 electroreduction, Nat. Commun., 2022, 13(1), 4857 CrossRef PubMed.
- B. Xiong, et al., Electrochemical conversion of CO2 to syngas over Cu–M (M = Cd, Zn, Ni, Ag, and Pd) bimetal catalysts, Fuel, 2021, 304, 121341 CrossRef.
- H. Yang, et al., Scalable Production of Efficient Single-Atom Copper Decorated Carbon Membranes for CO2 Electroreduction to Methanol, J. Am. Chem. Soc., 2019, 141(32), 12717–12723 CrossRef PubMed.
- J. G. Rivera de la Cruz and M. Fontecave, Electrochemical CO2 reduction on Cu single atom catalyst and Cu nanoclusters: an ab initio approach, Phys. Chem. Chem. Phys., 2022, 24(26), 15767–15775 RSC.
- M. Morimoto, et al., Experimental and Theoretical Elucidation of Electrochemical CO2 Reduction on an Electrodeposited Cu3Sn Alloy, J. Phys. Chem. C, 2019, 123(5), 3004–3010 CrossRef.
- Y. Yang and D. Cheng, Role of Composition and Geometric Relaxation in CO2 Binding to Cu–Ni Bimetallic Clusters, J. Phys. Chem. C, 2014, 118(1), 250–258 CrossRef.
- H. Li, et al., Insight into the mechanisms of CO2 reduction to CHO over Zr-doped Cu nanoparticle, Chem. Phys., 2021, 540, 111012 CrossRef.
- J.-S. Wang, et al., Strong Boron–Carbon Bonding Interaction Drives CO2 Reduction to Ethanol over the Boron-Doped Cu(111) Surface: An Insight from the First-Principles Calculations, J. Phys. Chem. C, 2021, 125(1), 572–582 CrossRef.
- X. Zhi, et al., Directing the selectivity of CO2 electroreduction to target C2 products via non-metal doping on Cu surfaces, J. Mater. Chem. A, 2021, 9(10), 6345–6351 RSC.
- C.-C. Chang and M.-S. Ku, Role of high-index facet Cu(711) surface in controlling the C2 selectivity for CO2 reduction reaction—a DFT study, J. Phys. Chem. C, 2021, 125(20), 10919–10925 CrossRef CAS.
- F. Zhou, et al., Synergetic effects of Cu cluster-doped g-C3N4 with multiple active sites for CO2 reduction to C2 products: a DFT study, Fuel, 2023, 353, 129202 CrossRef CAS.
- W. Xue, et al., Theoretical Screening of CO2 Electroreduction over MOF-808-Supported Self-Adaptive Dual-Metal-Site Pairs, Inorg. Chem., 2023, 62(2), 930–941 CrossRef CAS.
- R. Duan, et al., Effects of *CO Coverage on Selective Electrocatalytic Reduction of CO2 to Ethylene over Cu2O with Undercoordinated Cu Sites, J. Phys. Chem. C, 2022, 126(49), 20878–20885 CrossRef CAS.
- A. Das, et al., Ga and Zn Atom-Doped CuAl2O4(111) Surface-Catalyzed CO2 Conversion to Dimethyl Ether: Importance of Acidic Sites, J. Phys. Chem. C, 2022, 126(51), 21628–21637 CrossRef CAS.
- X. Dong, et al., Screening of bimetallic M–Cu–BTC MOFs for CO2 activation and mechanistic study of CO2 hydrogenation to formic acid: a DFT study, J. CO2 Util., 2018, 24, 64–72 CrossRef CAS.
- Z. Zhao and G. Lu, Cu-Based Single-Atom Catalysts Boost Electroreduction of CO2 to CH3OH: First-Principles Predictions, J. Phys. Chem. C, 2019, 123(7), 4380–4387 CrossRef.
- M. Sassenburg, et al., Characterizing CO2 Reduction Catalysts on Gas Diffusion Electrodes: Comparing Activity, Selectivity, and Stability of Transition Metal Catalysts, ACS Appl. Energy Mater., 2022, 5(5), 5983–5994 CrossRef.
- X. Lyu, et al., Trace level of atomic copper in N-doped graphene quantum dots switching the selectivity from C1 to C2 products in CO electroreduction, Mater. Today Chem., 2023, 29, 101398 CrossRef.
- P. Qi, et al., Revisiting the Grain and Valence Effect of Oxide-Derived Copper on Electrocatalytic CO2 Reduction Using Single Crystal Cu(111) Foils, J. Phys. Chem. Lett., 2021, 12(16), 3941–3950 CrossRef.
- Y. Chen, et al., Ethylene Selectivity in Electrocatalytic CO2 Reduction on Cu Nanomaterials: A Crystal Phase-Dependent Study, J. Am. Chem. Soc., 2020, 142(29), 12760–12766 CrossRef.
- S. Chen, et al., Lewis Acid Site-Promoted Single-Atomic Cu Catalyzes Electrochemical CO2 Methanation, Nano Lett., 2021, 21(17), 7325–7331 CrossRef PubMed.
- Q. Zhao, et al., Selective Etching Quaternary MAX Phase toward Single Atom Copper Immobilized MXene (Ti3C2Clx) for Efficient CO2 Electroreduction to Methanol, ACS Nano, 2021, 15(3), 4927–4936 CrossRef PubMed.
- D. Karapinar, et al., Electroreduction of CO2 on Single-Site Copper-Nitrogen-Doped Carbon Material: Selective Formation of Ethanol and Reversible Restructuration of the Metal Sites, Angew. Chem., Int. Ed., 2019, 58(42), 15098–15103 CrossRef PubMed.
- H. Bao, et al., Isolated copper single sites for high-performance electroreduction of carbon monoxide to multicarbon products, Nat. Commun., 2021, 12(1), 238 CrossRef PubMed.
- Z. Ma, et al., Atomically Dispersed Cu Catalysts on Sulfide-Derived Defective Ag Nanowires for Electrochemical CO2 Reduction, ACS Nano, 2023, 17(3), 2387–2398 CrossRef PubMed.
- T. Zheng, et al., Copper-catalysed exclusive CO2 to pure formic acid conversion via single-atom alloying, Nat. Nanotechnol., 2021, 16(12), 1386–1393 CrossRef PubMed.
- P. Song, et al., Modulating the Asymmetric Atomic Interface of Copper Single Atoms for Efficient CO2 Electroreduction, ACS Nano, 2023, 17(5), 4619–4628 CrossRef CAS PubMed.
- A. Guan, et al., Nitrogen-Adsorbed Hydrogen Species Promote CO2 Methanation on Cu Single-Atom Electrocatalyst, ACS Mater. Lett., 2023, 5(1), 19–26 CrossRef CAS.
- X. Hu, et al., Tandem Electroreduction of CO2 to Programmable Acetate and Syngas via Single-Nickel-Atom-Encapsulated Copper Nanocatalysts, ACS Mater. Lett., 2023, 5(1), 85–94 CrossRef CAS.
- Y. Dai, et al., Manipulating local coordination of copper single atom catalyst enables efficient CO2-to-CH4 conversion, Nat. Commun., 2023, 14(1), 3382 CrossRef CAS PubMed.
- W. Ni, et al., Efficient electrocatalytic reduction of CO2 on CuxO decorated graphene oxides: an insight into the role of multivalent Cu in selectivity and durability, Appl. Catal., B, 2019, 259, 118044 CrossRef CAS.
- M. Li, et al., Modulated Sn Oxidation States over a Cu2O-Derived Substrate for Selective Electrochemical CO2 Reduction, ACS Appl. Mater. Interfaces, 2020, 12(20), 22760–22770 CrossRef CAS.
- H. Guzmán, et al., CO2 Conversion to Alcohols over Cu/ZnO Catalysts: Prospective Synergies between Electrocatalytic and Thermocatalytic Routes, ACS Appl. Mater. Interfaces, 2022, 14(1), 517–530 CrossRef.
- J.-J. Velasco-Vélez, et al., On the Activity/Selectivity and Phase Stability of Thermally Grown Copper Oxides during the Electrocatalytic Reduction of CO2, ACS Catal., 2020, 10(19), 11510–11518 CrossRef.
- J. Liu, et al., Metal–Organic-Frameworks-Derived Cu/Cu2O Catalyst with Ultrahigh Current Density for Continuous-Flow CO2 Electroreduction, ACS Sustainable Chem. Eng., 2019, 7(18), 15739–15746 CrossRef CAS.
- G. Liu, et al., Elucidating Reaction Pathways of the CO2 Electroreduction via Tailorable Tortuosities and Oxidation States of Cu Nanostructures, Adv. Funct. Mater., 2022, 32(36), 2204993 CrossRef CAS.
- C. A. Obasanjo, et al., In situ regeneration of copper catalysts for long-term electrochemical CO2 reduction to multiple carbon products, J. Mater. Chem. A, 2022, 10(37), 20059–20070 RSC.
- T.-C. Chou, et al., Controlling the Oxidation State of the Cu Electrode and Reaction Intermediates for Electrochemical CO2 Reduction to Ethylene, J. Am. Chem. Soc., 2020, 142(6), 2857–2867 CrossRef CAS.
- Q. Lei, et al., Investigating the Origin of Enhanced C2+ Selectivity in Oxide-/Hydroxide-Derived Copper Electrodes during CO2 Electroreduction, J. Am. Chem. Soc., 2020, 142(9), 4213–4222 CrossRef CAS PubMed.
- X. Jiang, et al., Oxidation State Modulation of Bimetallic Tin-Copper Oxide Nanotubes for Selective CO2 Electroreduction to Formate, Small, 2022, 18(47), 2204148 CrossRef.
- Y. Yang, et al., Operando Constructing Cu/Cu2O Electrocatalysts for Efficient CO2 Electroreduction to Ethanol: CO2-Assisted Structural Evolution of Octahedral Cu2O by Operando CV Activation, ACS Catal., 2022, 12(20), 12942–12953 CrossRef.
- Z. Zhang, et al., Charge-Separated Pdδ−–Cuδ+ Atom Pairs Promote CO2 Reduction to C2, Nano Lett., 2023, 23(6), 2312–2320 CrossRef PubMed.
- Y. Li, et al., Insight into the Electrochemical CO2-to-Ethanol Conversion Catalyzed by Cu2S Nanocrystal-Decorated Cu Nanosheets, ACS Appl. Mater. Interfaces, 2023, 15(15), 18857–18866 CrossRef.
- F. Yang, et al., Optimizing Copper Oxidation State to Promote Ethylene Generation in Efficient Carbon Dioxide Conversion, ACS Sustainable Chem. Eng., 2022, 10(14), 4677–4682 CrossRef.
- H. Pan, et al., Highly Efficient CuPd0.1/γ-Al2O3 Catalyst with Isolated Pd Species for CO2 Hydrogenation to Methanol, ACS Sustainable Chem. Eng., 2023, 11(19), 7489–7499 CrossRef.
- T. Gunji, et al., Preparation of Various Pd-Based Alloys for Electrocatalytic CO2 Reduction Reaction—Selectivity Depending on Secondary Elements, Chem. Mater., 2020, 32(16), 6855–6863 CrossRef CAS.
- S. Ajmal, et al., Zinc-Modified Copper Catalyst for Efficient (Photo-)Electrochemical CO2 Reduction with High Selectivity of HCOOH Production, J. Phys. Chem. C, 2019, 123(18), 11555–11563 CrossRef CAS.
- Y. Ding, Y. Xu and L. Zhang, Tin Alloying Enhances Catalytic Selectivity of Copper Surface: A Mechanistic Study Based on First-Principles Calculations, J. Phys. Chem. Lett., 2021, 12(12), 3031–3037 CrossRef CAS.
- C. J. Yoo, et al., Compositional and Geometrical Effects of Bimetallic Cu–Sn Catalysts on Selective Electrochemical CO2 Reduction to CO, ACS Appl. Energy Mater., 2020, 3(5), 4466–4473 CrossRef CAS.
- H. Tang, et al., Boosting the Electroreduction of CO2 to Ethanol via the Synergistic Effect of Cu–Ag Bimetallic Catalysts, ACS Appl. Energy Mater., 2022, 5(11), 14045–14052 CrossRef.
- G. E. Park, et al., Interfacial Electronic Structures and the Fischer–Tropsch Synthesis Path by Electrochemical CO2/CO Reduction for Ternary CuNiZn Alloys, ACS Appl. Energy Mater., 2023, 6(13), 7258–7273 CrossRef.
- D. Chen, et al., Tailoring the Selectivity of Bimetallic Copper–Palladium Nanoalloys for Electrocatalytic Reduction of CO2 to CO, ACS Appl. Energy Mater., 2018, 1(2), 883–890 CrossRef.
- K. Liu, et al., Electronic Effects Determine the Selectivity of Planar Au–Cu Bimetallic Thin Films for Electrochemical CO2 Reduction, ACS Appl. Mater. Interfaces, 2019, 11(18), 16546–16555 CrossRef.
- W. Guo, et al., Highly Selective CO2 Electroreduction to CO on Cu–Co Bimetallic Catalysts, ACS Sustainable Chem. Eng., 2020, 8(33), 12561–12567 CrossRef.
- J.-J. Velasco-Vélez, et al., Cationic Copper Species Stabilized by Zinc during the Electrocatalytic Reduction of CO2 Revealed by In Situ X-Ray Spectroscopy, Adv. Sustainable Syst., 2023, 7(5), 2200453 CrossRef.
- M. Bernal, et al., CO2 electroreduction on copper-cobalt nanoparticles: size and composition effect, Nano Energy, 2018, 53, 27–36 Search PubMed.
- H. Pérez Blanes, et al., High CO2 reduction activity on AlCrCoCuFeNi multi-principal element alloy nanoparticle electrocatalysts prepared by means of pulsed laser ablation, J. Mater. Res. Technol., 2023, 24, 9434–9440 CrossRef.
- V. Okatenko, et al., Alloying as a Strategy to Boost the Stability of Copper Nanocatalysts during the Electrochemical CO2 Reduction Reaction, J. Am. Chem. Soc., 2023, 145(9), 5370–5383 CrossRef CAS PubMed.
- A. C. Foucher, et al., Synthesis and Characterization of Stable Cu–Pt Nanoparticles under Reductive and Oxidative Conditions, J. Am. Chem. Soc., 2023, 145(9), 5410–5421 Search PubMed.
- E. L. Clark, et al., Electrochemical CO2 Reduction over Compressively Strained CuAg Surface Alloys with Enhanced Multi-Carbon Oxygenate Selectivity, J. Am. Chem. Soc., 2017, 139(44), 15848–15857 CrossRef CAS.
- T. T. H. Hoang, et al., Nanoporous Copper–Silver Alloys by Additive-Controlled Electrodeposition for the Selective Electroreduction of CO2 to Ethylene and Ethanol, J. Am. Chem. Soc., 2018, 140(17), 5791–5797 Search PubMed.
- Y. Wang, et al., Ensemble Effect in Bimetallic Electrocatalysts for CO2 Reduction, J. Am. Chem. Soc., 2019, 141(42), 16635–16642 Search PubMed.
- H. S. Jeon, et al., Operando Insight into the Correlation between the Structure and Composition of CuZn Nanoparticles and Their Selectivity for the Electrochemical CO2 Reduction, J. Am. Chem. Soc., 2019, 141(50), 19879–19887 Search PubMed.
- H. Mistry, et al., Size-dependent reactivity of gold-copper bimetallic nanoparticles during CO2 electroreduction, Catal. Today, 2017, 288, 30–36 Search PubMed.
- J. Zhu, et al., Quasi-Covalently Coupled Ni–Cu Atomic Pair for Synergistic Electroreduction of CO2, J. Am. Chem. Soc., 2022, 144(22), 9661–9671 Search PubMed.
- Y. Wang, et al., Tunable selectivity on copper–bismuth bimetallic aerogels for electrochemical CO2 reduction, Appl. Catal., B, 2022, 317, 121650 CrossRef CAS.
- Y. Lai, et al., Breaking Scaling Relationships in CO2 Reduction on Copper Alloys with Organic Additives, ACS Cent. Sci., 2021, 7(10), 1756–1762 Search PubMed.
- L. Zhou, et al., Synergistic regulation of hydrophobicity and basicity for copper hydroxide-derived copper to promote the CO2 electroreduction reaction, Carbon Energy, 2023, 5(6), e328 CrossRef CAS.
- Z. Chang, et al., The Tunable and Highly Selective Reduction Products on Ag@Cu Bimetallic Catalysts Toward CO2 Electrochemical Reduction Reaction, J. Phys. Chem. C, 2017, 121(21), 11368–11379 CrossRef CAS.
- H. Itahara, et al., Dealloyed Intermetallic Cu5Ca Fine Powders as Nanoporous Electrocatalysts for CO2 Reduction, ACS Appl. Nano Mater., 2022, 5(8), 11991–11996 Search PubMed.
- W. J. Dong, et al., Evidence of Local Corrosion of Bimetallic Cu–Sn Catalysts and Its Effects on the Selectivity of Electrochemical CO2 Reduction, ACS Appl. Energy Mater., 2020, 3(11), 10568–10577 CrossRef CAS.
- S. Wang, et al., Efficiently Electroreducing CO2 to Ethylene on Heterostructured CeO2/CuO, Ind. Eng. Chem. Res., 2022, 61(44), 16445–16452 CrossRef CAS.
- H. Farsi, et al., Nanostructured Tungstate-Derived Copper for Hydrogen Evolution Reaction and Electroreduction of CO2 in Sodium Hydroxide Solutions, J. Phys. Chem. C, 2019, 123(42), 25941–25948 Search PubMed.
- W. Huang, et al., Preparation and Adsorption Performance of GrO@Cu–BTC for Separation of CO2/CH4, Ind. Eng. Chem. Res., 2014, 53(27), 11176–11184 CrossRef CAS.
- J. Huang, et al., Structural Sensitivities in Bimetallic Catalysts for Electrochemical CO2 Reduction Revealed by Ag–Cu Nanodimers, J. Am. Chem. Soc., 2019, 141(6), 2490–2499 CrossRef CAS PubMed.
- H. Rabiee, et al., Tuning the Product Selectivity of the Cu Hollow Fiber Gas Diffusion Electrode for Efficient CO2 Reduction to Formate by Controlled Surface Sn Electrodeposition, ACS Appl. Mater. Interfaces, 2020, 12(19), 21670–21681 CrossRef CAS.
- R. A. Rahman, et al., Investigating the Use of CaO/CuO Sorbents for in Situ CO2 Capture in a Biomass Gasifier, Energy Fuels, 2015, 29(6), 3808–3819 CrossRef CAS.
- S. Payra, et al., Unprecedented Electroreduction of CO2 over Metal Organic Framework-Derived Intermetallic Nano-Alloy Cu0.85Ni0.15/C, ACS Appl. Energy Mater., 2022, 5(4), 4945–4955 CrossRef CAS.
- S. Wannakao, et al., Catalytic Activity and Product Selectivity Trends for Carbon Dioxide Electroreduction on Transition Metal-Coated Tungsten Carbides, J. Phys. Chem. C, 2017, 121(37), 20306–20314 Search PubMed.
- M. Zhang, et al., Tunable Selectivity for Electrochemical CO2 Reduction by Bimetallic Cu–Sn Catalysts: Elucidating the Roles of Cu and Sn, ACS Catal., 2021, 11(17), 11103–11108 CrossRef CAS.
- J. Wen, et al., Restructuring of copper catalysts by potential cycling and enhanced two-carbon production for electroreduction of carbon dioxide, J. CO2 Util., 2022, 56, 101846 Search PubMed.
- H.-D. Cai, et al., Tuning the Interactions in CuO Nanosheet-Decorated CeO2 Nanorods for Controlling the Electrochemical Reduction of CO2 to Methane or Ethylene, ACS Appl. Nano Mater., 2022, 5(5), 7259–7267 CrossRef CAS.
- S.-M. Cao, et al., Iron-doping on Cu–N–C composite with enhanced CO Faraday efficiency for the electrochemical reduction of CO2, J. CO2 Util., 2021, 44, 101418 Search PubMed.
- T.-T. Li, et al., Highly Selective and Active Electrochemical Reduction of CO2 to CO on a Polymeric Co(II) Phthalocyanine@Graphitic Carbon Nitride Nanosheet–Carbon Nanotube Composite, Inorg. Chem., 2020, 59(19), 14184–14192 Search PubMed.
- Z. Meng, et al., Hierarchical Tuning of the Performance of Electrochemical Carbon Dioxide Reduction Using Conductive Two-Dimensional Metallophthalocyanine Based Metal–Organic Frameworks, J. Am. Chem. Soc., 2020, 142(52), 21656–21669 CrossRef CAS.
- H. He, et al., Graphene-Supported Monometallic and Bimetallic Dimers for Electrochemical CO2 Reduction, J. Phys. Chem. C, 2018, 122(50), 28629–28636 CrossRef CAS.
- D. Karapinar, et al., Carbon-Nanotube-Supported Copper Polyphthalocyanine for Efficient and Selective Electrocatalytic CO2 Reduction to CO, ChemSusChem, 2020, 13(1), 173–179 Search PubMed.
- J. Du, et al., CoP2O6-Assisted Copper/Carbon Catalyst for Electrocatalytic Reduction of CO2 to Formate, ACS Nano, 2023, 17(11), 10055–10064 Search PubMed.
- E. Cui, et al., Cu2S Nanoparticle-Modified Copper Hydroxide Nanowire Arrays for Optimized Electrochemical CO2 Reduction Selectivity, ACS Appl. Nano Mater., 2023, 6(11), 9361–9368 CrossRef.
- Y. Ren, et al., Cu–Ni Alloy Nanoparticles Anchored on Nitrogen-Doped Carbon Nanotubes for Efficient CO2 Electroreduction to CO, Energy Fuels, 2023, 37(13), 9289–9296 CrossRef.
- R. Daiyan, et al., Tunable Syngas Production through CO2 Electroreduction on Cobalt–Carbon Composite Electrocatalyst, ACS Appl. Mater. Interfaces, 2020, 12(8), 9307–9315 CrossRef PubMed.
- P. Ding, et al., Elucidating the Roles of Nafion/Solvent Formulations in Copper-Catalyzed CO2 Electrolysis, ACS Catal., 2023, 13(8), 5336–5347 CrossRef.
- C.-L. Sung, et al., g-C3N4 Nanosheet Supported CuO Nanocomposites for the Electrochemical Carbon Dioxide Reduction Reaction, ACS Omega, 2023, 8(8), 7368–7377 CrossRef.
- J. Li, et al., Metal Oxide Clusters on Nitrogen-Doped Carbon are Highly Selective for CO2 Electroreduction to CO, ACS Catal., 2021, 11(15), 10028–10042 CrossRef.
- A. Strijevskaya, et al., Nanophase-Separated Copper–Zirconia Composites for Bifunctional Electrochemical CO2 Conversion to Formic Acid, ACS Appl. Mater. Interfaces, 2023, 15(19), 23299–23305 CrossRef PubMed.
- H. Shen and Q. Sun, Tuning CO2 Electroreduction of Cu Atoms on Triphenylene-Cored Graphdiyne, J. Phys. Chem. C, 2019, 123(49), 29776–29782 CrossRef.
- W. Ni, et al., Surface Reconstruction with a Sandwich-like C/Cu/C Catalyst for Selective and Stable CO2 Electroreduction, ACS Appl. Mater. Interfaces, 2022, 14(11), 13261–13270 CrossRef.
- X. Meng, et al., Ultrasmall Cu Nanocrystals Dispersed in Nitrogen-Doped Carbon as Highly Efficient Catalysts for CO2 Electroreduction, ACS Appl. Mater. Interfaces, 2022, 14(15), 17240–17248 CrossRef.
- Z. Gao, et al., Promoting electrocatalytic CO2 reduction via anchoring quaternary piperidinium cations onto copper electrode, Electrochim. Acta, 2023, 458, 142509 CrossRef.
- M. Ye, et al., Phase engineering of Cu@Cu2O core–shell nanospheres for boosting tandem electrochemical CO2 reduction to C2+ products, Appl. Surf. Sci., 2023, 622, 156981 CrossRef.
- L. Zhao, et al., Regulation of three-dimensional hydrophobic state of copper dendrite adjusts the distribution of liquid products from electrochemical reduction of CO2, Appl. Surf. Sci., 2023, 628, 157369 CrossRef.
- H. Xie, et al., Achieving highly selective electrochemical CO2 reduction to C2H4 on Cu nanosheets, J. Energy Chem., 2023, 79, 312–320 CrossRef.
- T. Umeda, et al., Uniform wrapping of copper(I) oxide nanocubes by self-controlled copper-catalyzed azide–alkyne cycloaddition toward selective carbon dioxide electrocatalysis, Chem. Commun., 2022, 58(58), 8053–8056 RSC.
- E. Mardones-Herrera, et al., Reduced Graphene Oxide Overlayer on Copper Nanocube Electrodes Steers the Selectivity Towards Ethanol in Electrochemical Reduction of Carbon Dioxide, ChemElectroChem, 2022, 9(10), e202200259 CrossRef.
- C. Lu, et al., Organic-moiety-engineering on copper surface for carbon dioxide reduction, Chem. Commun., 2023, 59(45), 6827–6836 RSC.
- K. Rossi and R. Buonsanti, Shaping Copper Nanocatalysts to Steer Selectivity in the Electrochemical CO2 Reduction Reaction, Acc. Chem. Res., 2022, 55(5), 629–637 CrossRef.
- W. Zhang, et al., Efficacious CO2 Adsorption and Activation on Ag Nanoparticles/CuO Mesoporous Nanosheets Heterostructure for CO2 Electroreduction to CO, Inorg. Chem., 2021, 60(24), 19356–19364 CrossRef PubMed.
- S. Chang, et al., Bimetallic In–Sn Core–Shell Catalyst Enabling Highly Efficient Electrocatalytic CO2 Reduction to Formate, ACS Appl. Eng. Mater., 2023, 1(6), 1514–1523 CrossRef.
- G. O. Larrazábal, et al., Investigation of Ethylene and Propylene Production from CO2 Reduction over Copper Nanocubes in an MEA-Type Electrolyzer, ACS Appl. Mater. Interfaces, 2022, 14(6), 7779–7787 CrossRef PubMed.
- D. Choukroun, et al., Mapping Composition–Selectivity Relationships of Supported Sub-10 nm Cu–Ag Nanocrystals for High-Rate CO2 Electroreduction, ACS Nano, 2021, 15(9), 14858–14872 CrossRef PubMed.
- Y. Sha, et al., Anchoring Ionic Liquid in Copper Electrocatalyst for Improving CO2 Conversion to Ethylene, Angew. Chem., Int. Ed., 2022, 61(13), e202200039 CrossRef PubMed.
- B. Yang, et al., Accelerating CO2 Electroreduction to Multicarbon Products via Synergistic Electric–Thermal Field on Copper Nanoneedles, J. Am. Chem. Soc., 2022, 144(7), 3039–3049 CrossRef PubMed.
- K. Jiang, et al., Effects of Surface Roughness on the Electrochemical Reduction of CO2 over Cu, ACS Energy Lett., 2020, 5(4), 1206–1214 CrossRef.
- S. Kuang, et al., Stable Surface-Anchored Cu Nanocubes for CO2 Electroreduction to Ethylene, ACS Appl. Nano Mater., 2020, 3(8), 8328–8334 CrossRef.
- B. Liu, et al., Synthesis of Cu2O Nanostructures with Tunable Crystal Facets for Electrochemical CO2 Reduction to Alcohols, ACS Appl. Mater. Interfaces, 2021, 13(33), 39165–39177 CrossRef PubMed.
- C. Tang, et al., CO2 Reduction on Copper's Twin Boundary, ACS Catal., 2020, 10(3), 2026–2032 CrossRef.
- Y. Wang, et al., Copper Nanocubes for CO2 Reduction in Gas Diffusion Electrodes, Nano Lett., 2019, 19(12), 8461–8468 CrossRef.
- P. Wang, et al., Synergized Cu/Pb Core/Shell Electrocatalyst for High-Efficiency CO2 Reduction to C2+ Liquids, ACS Nano, 2021, 15(1), 1039–1047 CrossRef.
- Y. Wu, et al., Engineering Bimetallic Copper-Tin Based Core–Shell Alloy@Oxide Nanowire as Efficient Catalyst for Electrochemical CO2 Reduction, ChemElectroChem, 2021, 8(14), 2701–2707 CrossRef.
- H. Xie, et al., Boosting Tunable Syngas Formation via Electrochemical CO2 Reduction on Cu/In2O3 Core/Shell Nanoparticles, ACS Appl. Mater. Interfaces, 2018, 10(43), 36996–37004 CrossRef PubMed.
- Y. Fang, et al., Synthesis of amino acids by electrocatalytic reduction of CO2 on chiral Cu surfaces, Chem, 2023, 9(2), 460–471 Search PubMed.
- J. R. Pankhurst, et al., Copper Nanocrystal Morphology Determines the Viability of Molecular Surface Functionalization in Tuning Electrocatalytic Behavior in CO2 Reduction, Inorg. Chem., 2021, 60(10), 6939–6945 CrossRef CAS.
- B. Cheng, et al., Selective CO2 Reduction to Ethylene Using Imidazolium-Functionalized Copper, ACS Appl. Mater. Interfaces, 2022, 14(24), 27823–27832 CrossRef CAS.
- S. Huo, et al., Coupled Metal/Oxide Catalysts with Tunable Product Selectivity for Electrocatalytic CO2 Reduction, ACS Appl. Mater. Interfaces, 2017, 9(34), 28519–28526 CrossRef CAS PubMed.
- Z. Tao, et al., Activating Copper for Electrocatalytic CO2 Reduction to Formate via Molecular Interactions, ACS Catal., 2020, 10(16), 9271–9275 CrossRef CAS.
- H. Cheng, et al., Atomically Dispersed Ni/Cu Dual Sites for Boosting the CO2 Reduction Reaction, ACS Catal., 2021, 11(20), 12673–12681 CrossRef CAS.
- L.-J. Zhu, et al., Copper–Supramolecular Pair Catalyst Promoting C2+ Product Formation in Electrochemical CO2 Reduction, ACS Catal., 2023, 13(8), 5114–5121 CrossRef CAS.
- J. Wang, et al., Fastening Br–Ions at Copper–Molecule Interface Enables Highly Efficient Electroreduction of CO2 to Ethanol, ACS Energy Lett., 2021, 6(2), 437–444 CrossRef CAS.
- X. Li, et al., Selective Electroreduction of CO2 and CO to C2H4 by Synergistically Tuning Nanocavities and the Surface Charge of Copper Oxide, ACS Sustainable Chem. Eng., 2022, 10(19), 6466–6475 CrossRef CAS.
- Z. Cao, et al., A Molecular Surface Functionalization Approach to Tuning Nanoparticle Electrocatalysts for Carbon Dioxide Reduction, J. Am. Chem. Soc., 2016, 138(26), 8120–8125 CrossRef CAS PubMed.
- Z. Wang, et al., Toward highly active electrochemical CO2 reduction to C2H4 by copper hydroxyphosphate, J. Solid State Electrochem., 2023, 27(5), 1279–1287 CrossRef CAS.
- J. Wang, et al., Surface Molecular Functionalization of Unusual Phase Metal Nanomaterials for Highly Efficient Electrochemical Carbon Dioxide Reduction under Industry-Relevant Current Density, Small, 2022, 18(11), 2106766 CrossRef CAS.
- Z.-Y. Zhang, et al., Cu–Zn-based alloy/oxide interfaces for enhanced electroreduction of CO2 to C2+ products, J. Energy Chem., 2023, 83, 90–97 CrossRef CAS.
- R. Wang, et al., Engineering a Cu/ZnOx Interface for High Methane Selectivity in CO2 Electrochemical Reduction, Ind. Eng. Chem. Res., 2021, 60(1), 273–280 CrossRef CAS.
- C. Liu, et al., Highly Selective CO2 Electroreduction to C2+ Products over Cu2O-Decorated 2D Metal–Organic Frameworks with Rich Heterogeneous Interfaces, Nano Lett., 2023, 23(4), 1474–1480 CrossRef CAS.
- M. Kempasiddaiah, et al., Interface-Rich Highly Oxophilic Copper/Tin–Oxide Nanocomposite on Reduced Graphene Oxide for Efficient Electroreduction of CO2 to Formate, ACS Appl. Energy Mater., 2023, 6(5), 3020–3031 CrossRef CAS.
- R. Du, et al., In Situ Engineering of the Cu+/Cu0 Interface to Boost C2+ Selectivity in CO2 Electroreduction, ACS Appl. Mater. Interfaces, 2022, 14(32), 36527–36535 CrossRef CAS.
- H.-Q. Liang, et al., Hydrophobic Copper Interfaces Boost Electroreduction of Carbon Dioxide to Ethylene in Water, ACS Catal., 2021, 11(2), 958–966 CrossRef CAS.
- J. Yin, et al., Customizable CO2 Electroreduction to C1 or C2+ Products through Cuy/CeO2 Interface Engineering, ACS Catal., 2022, 12(2), 1004–1011 CrossRef CAS.
- K. Li, et al., In Situ Dynamic Construction of a Copper Tin Sulfide Catalyst for High-Performance Electrochemical CO2 Conversion to Formate, ACS Catal., 2022, 12(16), 9922–9932 CrossRef CAS.
- T. Kou, et al., Amorphous CeO2–Cu Heterostructure Enhances CO2 Electroreduction to Multicarbon Alcohols, ACS Mater. Lett., 2022, 4(10), 1999–2008 CrossRef.
- J. A. Rudd, et al., Investigation into the Re-Arrangement of Copper Foams Pre- and Post-CO2, Electrocatalysis, 2021, 3(3), 687–703 Search PubMed.
- J. Zeng, et al., Engineering copper nanoparticle electrodes for tunable electrochemical reduction of carbon dioxide, Electrochim. Acta, 2023, 464, 142862 CrossRef.
- S. Tafazoli, et al., In situ surface enhanced Raman spectroscopy investigations on surface transformations of oxide derived copper electrodes during CO2RR, J. Catal., 2023, 423, 118–128 CrossRef.
- P.-P. Yang, et al., Highly Enhanced Chloride Adsorption Mediates Efficient Neutral CO2 Electroreduction over a Dual-Phase Copper Catalyst, J. Am. Chem. Soc., 2023, 145(15), 8714–8725 CrossRef PubMed.
- W. Ren, et al., Isolated copper–tin atomic interfaces tuning electrocatalytic CO2 conversion, Nat. Commun., 2021, 12(1), 1449 CrossRef PubMed.
- X. Tan, et al., Stabilizing Copper by a Reconstruction-Resistant Atomic Cu–O–Si Interface for Electrochemical CO2 Reduction, J. Am. Chem. Soc., 2023, 145(15), 8656–8664 CrossRef CAS.
- F. Ismail, et al., Atomically Isolated Nickel–Nitrogen–Carbon Electrocatalysts Derived by the Utilization of Mg2+ ions as Spacers in Bimetallic Ni/Mg–Metal–Organic Framework Precursors for Boosting the Electroreduction of CO2, ACS Appl. Energy Mater., 2022, 5(8), 9408–9417 CrossRef CAS.
- Y. Lan, et al., SnO2-Modified Two-Dimensional CuO for Enhanced Electrochemical Reduction of CO2 to C2H4, ACS Appl. Mater. Interfaces, 2020, 12(32), 36128–36136 CrossRef CAS PubMed.
- J. Wang, et al., Grain-Boundary-Engineered La2CuO4 Perovskite Nanobamboos for Efficient CO2 Reduction Reaction, Nano Lett., 2021, 21(2), 980–987 CrossRef CAS PubMed.
- L.-X. Liu, et al., Enriching the Local Concentration of CO Intermediates on Cu Cavities for the Electrocatalytic Reduction of CO2 to C2+ Products, ACS Appl. Mater. Interfaces, 2023, 15(13), 16673–16679 CrossRef CAS.
- X.-C. Liu, et al., Tailoring the Electrochemical Protonation Behavior of CO2 by Tuning Surface Noncovalent Interactions, ACS Catal., 2021, 11(24), 14986–14994 CrossRef CAS.
- Q. Wu, et al., Nanograin-Boundary-Abundant Cu2O–Cu Nanocubes with High C2+ Selectivity and Good Stability during Electrochemical CO2 Reduction at a Current Density of 500 mA cm−2, ACS Nano, 2023, 17(13), 12884–12894 CrossRef CAS PubMed.
- Y.-F. Shi, et al., Methanol Synthesis from CO2/CO Mixture on Cu–Zn Catalysts from Microkinetics-Guided Machine Learning Pathway Search, J. Am. Chem. Soc., 2022, 144(29), 13401–13414 CrossRef.
- H. Li, et al., C2+ Selectivity for CO2 Electroreduction on Oxidized Cu-Based Catalysts, J. Am. Chem. Soc., 2023, 145(26), 14335–14344 CrossRef.
- J. Zhang, et al., Surface promotion of copper nanoparticles with alumina clusters derived from layered double hydroxide accelerates CO2 reduction to ethylene in membrane electrode assemblies, Nano Res., 2023, 16(4), 4685–4690 CrossRef.
- F. Scholten, et al., Plasma-Modified Dendritic Cu Catalyst for CO2 Electroreduction, ACS Catal., 2019, 9(6), 5496–5502 CrossRef PubMed.
- H. Deng, et al., Amino Assisted Protonation for Carbon−Carbon Coupling During Electroreduction of Carbon Dioxide to Ethylene on Copper(I) Oxide, ChemCatChem, 2021, 13(20), 4325–4333 CrossRef.
- X. Xu, et al., Density functional theory study of CO2 reduction to CH3OH on the surfaces of metal (Cu, Ni, and Mn)-doped carbon nanotubes, J. Phys. Chem. Solids, 2022, 162, 110537 CrossRef CAS.
- J. Shan, et al., Effective CO2 electroreduction toward C2H4 boosted by Ce-doped Cu nanoparticles, Chem. Eng. J., 2022, 433, 133769 Search PubMed.
- D. Mao, et al., The influence of the compositions and structures of Cu-ZrO2 catalysts on the catalytic performance of CO2 hydrogenation to CH3OH, Chem. Eng. J., 2023, 471, 144605 CrossRef CAS.
- K. Eid, et al., Precise fabrication of porous one-dimensional gC3N4 nanotubes doped with Pd and Cu atoms for efficient CO oxidation and CO2 reduction, Inorg. Chem. Commun., 2019, 107, 107460 Search PubMed.
- F. Gao, et al., Efficient CO2 reduction to formate using a Cu-doped BiVO4 electrocathode in a WO3 photoanode-assisted photoelectrocatalytic system, J. Electroanal. Chem., 2023, 930, 117146 Search PubMed.
- M. Fang, et al., Aluminum-Doped Mesoporous Copper Oxide Nanofibers Enabling High-Efficiency CO2 Electroreduction to Multicarbon Products, Chem. Mater., 2022, 34(20), 9023–9030 Search PubMed.
- S. Yang, et al., Near-Unity Electrochemical CO2 to CO Conversion over Sn-Doped Copper Oxide Nanoparticles, ACS Catal., 2022, 12(24), 15146–15156 Search PubMed.
- Y. Zhang, X.-Y. Zhang and W.-Y. Sun, In Situ Carbon-Encapsulated Copper-Doped Cerium Oxide Derived from MOFs for Boosting CO2-to-CH4 Electro-Conversion, ACS Catal., 2023, 13(2), 1545–1553 Search PubMed.
- B. Kim, et al., Trace-Level Cobalt Dopants Enhance CO2 Electroreduction and Ethylene Formation on Copper, ACS Energy Lett., 2023, 8(8), 3356–3364 Search PubMed.
- X. Zhong, et al., Sn Dopants with Synergistic Oxygen Vacancies Boost CO2 Electroreduction on CuO Nanosheets to CO at Low Overpotential, ACS Nano, 2022, 16(11), 19210–19219 CrossRef CAS.
- H. Shen, et al., In Situ Constructuring of Copper-Doped Bismuth Catalyst for Highly Efficient CO2 Electrolysis to Formate in Ampere-Level, Adv. Energy Mater., 2023, 13(1), 2202818 Search PubMed.
- W. Ju, et al., Sn-Decorated Cu for Selective Electrochemical CO2 to CO Conversion: Precision Architecture beyond Composition Design, ACS Appl. Energy Mater., 2019, 2(1), 867–872 CrossRef CAS.
- C. S. Chen, J. H. Wan and B. S. Yeo, Electrochemical Reduction of Carbon Dioxide to Ethane Using Nanostructured Cu2O-Derived Copper Catalyst and Palladium(II) Chloride, J. Phys. Chem. C, 2015, 119(48), 26875–26882 CrossRef CAS.
- C. Hu, et al., Porosity-Induced High Selectivity for CO2 Electroreduction to CO on Fe-Doped ZIF-Derived Carbon Catalysts, ACS Catal., 2019, 9(12), 11579–11588 Search PubMed.
- P. Li, et al., p–d Orbital Hybridization Induced by p-Block Metal-Doped Cu Promotes the Formation of C2+ Products in Ampere-Level CO2 Electroreduction, J. Am. Chem. Soc., 2023, 145(8), 4675–4682 Search PubMed.
- S. Nellaiappan, et al., Electroreduction of Carbon Dioxide into Selective Hydrocarbons at Low Overpotential Using Isomorphic Atomic Substitution in Copper Oxide, ACS Sustainable Chem. Eng., 2020, 8(1), 179–189 CrossRef CAS.
- M. H. Ravari, A. Sarrafi and M. Tahmooresi, Synthesizing and characterizing the mixed Al,Cu-pillared and copper doped Al-pillared bentonite for electrocatalytic reduction of CO2, S. Afr. J. Chem. Eng., 2020, 31, 1–6 Search PubMed.
- H. Yuan, et al., Dynamic re-construction of sulfur tailored Cu2O for efficient electrochemical CO2 reduction to formate over a wide potential window, Appl. Surf. Sci., 2023, 613, 156130 CrossRef CAS.
- C. An, et al., Cooperated catalytic mechanism of atomically dispersed binary Cu–N4 and Zn–N4 in N-doped carbon materials for promoting electrocatalytic CO2 reduction to CH4, Chem. Eng. J., 2023, 471, 144618 Search PubMed.
- Y. Zhang, et al., CO2 reduction performance of Cu/Er supported on N-doped graphene: a first principles study, Mol. Catal., 2023, 547, 113335 CrossRef CAS.
- D. Choukroun, et al., Bifunctional Nickel–Nitrogen-Doped-Carbon-Supported Copper Electrocatalyst for CO2 Reduction, J. Phys. Chem. C, 2020, 124(2), 1369–1381 CrossRef CAS.
- K. K. Patra, et al., Operando Spectroscopic Investigation of a Boron-Doped CuO Catalyst and Its Role in Selective Electrochemical C–C Coupling, ACS Appl. Energy Mater., 2020, 3(11), 11343–11349 Search PubMed.
- D. Yang, et al., Selective electroreduction of carbon dioxide to methanol on copper selenide nanocatalysts, Nat. Commun., 2019, 10(1), 677 CrossRef CAS PubMed.
- W. Zhang, et al., Br-Doped CuO Multilamellar Mesoporous Nanosheets with Oxygen Vacancies and Cetyltrimethyl Ammonium Cations Adsorption for Optimizing Intermediate Species and Their Adsorption Behaviors toward CO2 Electroreduction to Ethanol with a High faradaic Efficiency, Inorg. Chem., 2021, 60(18), 14371–14381 Search PubMed.
- A. Saxena, et al., Copper Cobalt Selenide as a Bifunctional Electrocatalyst for the Selective Reduction of CO2 to Carbon-Rich Products and Alcohol Oxidation, ACS Appl. Mater. Interfaces, 2023, 15(11), 14433–14446 Search PubMed.
- D. G. Park, et al., Increasing CO Binding Energy and Defects by Preserving Cu Oxidation State via O2-Plasma-Assisted N Doping on CuO Enables High C2+ Selectivity and Long-Term Stability in Electrochemical CO2 Reduction, ACS Catal., 2023, 13(13), 9222–9233 Search PubMed.
- X. Ma, et al., Facet Dopant Regulation of Cu2O Boosts Electrocatalytic CO2 Reduction to Formate, Adv. Funct. Mater., 2023, 33(16), 2213145 CrossRef CAS.
- D. Liu, Y. Liu and M. Li, Understanding How Atomic Sulfur Controls the Selectivity of the Electroreduction of CO2 to Formic Acid on Metallic Cu Surfaces, J. Phys. Chem. C, 2020, 124(11), 6145–6153 Search PubMed.
- K. R. Phillips, et al., Sulfide-Derived Copper for Electrochemical Conversion of CO2 to Formic Acid, J. Phys. Chem. Lett., 2018, 9(15), 4407–4412 CrossRef CAS.
- M. Zheng, et al., Electrocatalytic CO2-to-C2+ with Ampere-Level Current on Heteroatom-Engineered Copper via Tuning *CO Intermediate Coverage, J. Am. Chem. Soc., 2022, 144(32), 14936–14944 Search PubMed.
- L. Gao, et al., Fabrication of Cu(1 0 0) facet-enhanced ionic liquid/copper hybrid catalysis via one-step electro-codeposition for CO2ER toward C2, Fuel, 2022, 322, 124103 Search PubMed.
- J. Yu, et al., Enhancing the electrochemical reduction of carbon dioxide to multi-carbon products on copper nanosheet arrays via cation-catalyst interaction, Cell Rep. Phys. Sci., 2023, 4(4), 101366 Search PubMed.
- Z. Lyu, Y. Shang and Y. Xia, Shape-Controlled Synthesis of Copper Nanocrystals for Plasmonic, Biomedical, and Electrocatalytic Applications, Acc. Mater. Res., 2022, 3(11), 1137–1148 Search PubMed.
- G. Mangione, et al., Dual-facet mechanism in copper nanocubes for electrochemical CO2 reduction into ethylene, J. Phys. Chem. Lett., 2019, 10(15), 4259–4265 CrossRef PubMed.
- S. C. Mandal, et al., Hexagonal Cu(111) Monolayers for Selective CO2 Hydrogenation to CH3OH: Insights from Density Functional Theory, ACS Appl. Nano Mater., 2019, 2(12), 7686–7695 CrossRef.
- G. L. De Gregorio, et al., Facet-dependent selectivity of Cu catalysts in electrochemical CO2 reduction at commercially viable current densities, ACS Catal., 2020, 10(9), 4854–4862 CrossRef.
- J. Y. Kim, et al., High Facets on Nanowrinkled Cu via Chemical Vapor Deposition Graphene Growth for Efficient CO2 Reduction into Ethanol, ACS Catal., 2021, 11(9), 5658–5665 CrossRef.
- P. Iyengar, et al., Elucidating the facet-dependent selectivity for CO2 electroreduction to ethanol of Cu–Ag tandem catalysts, ACS Catal., 2021, 11(8), 4456–4463 CrossRef.
- W. Luo, et al., Facet dependence of CO2 reduction paths on Cu electrodes, ACS Catal., 2016, 6(1), 219–229 CrossRef.
- W. Fu, et al., Promoting C2+ Production from Electrochemical CO2 Reduction on Shape-Controlled Cuprous Oxide Nanocrystals with High-Index Facets, ACS Sustainable Chem. Eng., 2020, 8(40), 15223–15229 CrossRef.
- Y. Ma, et al., Confined growth of silver–copper janus nanostructures with {100} facets for highly selective tandem electrocatalytic carbon dioxide reduction, Adv. Mater., 2022, 34(19), 2110607 CrossRef.
- A. Rahmani and H. Farsi, Nanostructured copper molybdates as promising bifunctional electrocatalysts for overall water splitting and CO2 reduction, RSC Adv., 2020, 10(64), 39037–39048 RSC.
- M. Zheng, et al., The Facet Dependence of CO2 Electroreduction Selectivity on a Pd3Au Bimetallic Catalyst: A DFT Study, Molecules, 2023, 28(7), 3169 CrossRef PubMed.
- G. Zhang, et al., Efficient CO2 electroreduction on facet-selective copper films with high conversion rate, Nat. Commun., 2021, 12(1), 5745 CrossRef PubMed.
- L. Han, et al., Copper nanowire with enriched high-index facets for highly selective CO2 reduction, SmartMat, 2022, 3(1), 142–150 CrossRef CAS.
- L. Gao, et al., Fabrication of Cu(100) facet-enhanced ionic liquid/copper hybrid catalysis via one-step electro-codeposition for CO2ER toward C2, Fuel, 2022, 322, 124103 CrossRef CAS.
- J. Yu, et al., Enhancing the electrochemical reduction of carbon dioxide to multi-carbon products on copper nanosheet arrays via cation-catalyst interaction, Cell Rep. Phys. Sci., 2023, 4(4), 101366 CrossRef CAS.
- Y. Ma, et al., Confined Growth of Silver–Copper Janus Nanostructures with {100} Facets for Highly Selective Tandem Electrocatalytic Carbon Dioxide Reduction, Adv. Mater., 2022, 34(19), 2110607 CrossRef CAS.
- L. Han, et al., Copper nanowire with enriched high-index facets for highly selective CO2 reduction, SmartMat, 2022, 3(1), 142–150 CrossRef CAS.
- A. Ahmad, et al., Cu-doped zeolite imidazole framework (ZIF-8) for effective electrocatalytic CO2 reduction, J. CO2 Util., 2021, 48, 101523 CrossRef CAS.
- L. Garcia, et al., Methanol electrosynthesis from CO2 reduction reaction in polymer electrolyte reactors–fuel cell type using [6,6′-(2,2′-bipyridine-6,6′-diyl)bis(1,3,5-triazine-2,4-diamine)](dinitrate-O) copper(II) complex, Mater. Today Sustainability, 2022, 19, 100177 CrossRef.
- Y. Zhou, et al., Spatial confinement in copper-porphyrin frameworks enhances carbon dioxide reduction to hydrocarbons, Cell Rep. Phys. Sci., 2020, 1(9), 100182 CrossRef.
- Z. Zhao, et al., Generalized surface coordination number as an activity descriptor for CO2 reduction on Cu surfaces, J. Phys. Chem. C, 2016, 120(49), 28125–28130 CrossRef CAS.
- H. Zhou, et al., Coordination Engineering in Cobalt–Nitrogen-Functionalized Materials for CO2 Reduction, J. Phys. Chem. Lett., 2019, 10(21), 6551–6557 CrossRef CAS.
- J. Liu, et al., Controlled Synthesis of EDTA-Modified Porous Hollow Copper Microspheres for High-Efficiency Conversion of CO2 to Multicarbon Products, Nano Lett., 2020, 20(7), 4823–4828 CrossRef CAS.
- X. Tan, et al., Restructuring of Cu2O to Cu2O@ Cu–metal–organic frameworks for selective electrochemical reduction of CO2, ACS Appl. Mater. Interfaces, 2019, 11(10), 9904–9910 CrossRef CAS.
- Y. Wu, et al., Sn Atoms on Cu Nanoparticles for Suppressing Competitive H2 Evolution in CO2 Electrolysis, ACS Appl. Nano Mater., 2021, 4(5), 4994–5003 CrossRef CAS.
- S. Kusama, et al., Crystalline copper(II) phthalocyanine catalysts for electrochemical reduction of carbon dioxide in aqueous media, ACS Catal., 2017, 7(12), 8382–8385 CrossRef CAS.
- R. Wang, et al., Partial Coordination-Perturbed Bi-Copper Sites for Selective Electroreduction of CO2 to Hydrocarbons, Angew. Chem., Int. Ed., 2021, 60(36), 19829–19835 CrossRef CAS PubMed.
- Y. Y. Liu, et al., A Stable Metal-azolate Framework with Cyclic Tetracopper(I) Clusters for Highly Selective Electroreduction of CO2 to C2 Products, Chem. – Asian J., 2022, 17(21), e202200764 CrossRef.
- Y. Zhang, et al., Hydroxy-Group-Functionalized Single Crystal of Copper(II)-Porphyrin Complex for Electroreduction CO2 to CH4, ChemSusChem, 2022, 15(4), e202102528 CrossRef.
- Q. Tang, et al., Lattice-hydride mechanism in electrocatalytic CO2 reduction by structurally precise copper-hydride nanoclusters, J. Am. Chem. Soc., 2017, 139(28), 9728–9736 CrossRef PubMed.
- L. Xue, et al., Hydrophobic 1-octadecanethiol functionalized copper catalyst promotes robust high-current CO2 gas-diffusion electrolysis, Nano Res., 2022, 15, 1393–1398 CrossRef.
- Y.-H. Xiao, et al., Helical copper-porphyrinic framework nanoarrays for highly efficient CO2 electroreduction, Sci. China: Mater., 2022, 65(5), 1269–1275 Search PubMed.
- B. Yang, et al., Electrocatalytic CO2 reduction to alcohols by modulating the molecular geometry and Cu coordination in bicentric copper complexes, Nat. Commun., 2022, 13(1), 5122 CrossRef PubMed.
- M. Fang, et al., Hydrophobic, Ultrastable Cuδ+ for Robust CO2 Electroreduction to C2 Products at Ampere-Current Levels, J. Am. Chem. Soc., 2023, 145(20), 11323–11332 CrossRef PubMed.
- A. K. Singh, et al., Exploring Ligand-Controlled C2 Product Selectivity in Carbon Dioxide Reduction with Copper Metal–Organic Framework Nanosheets, Inorg. Chem., 2023, 62(23), 8803–8811 CrossRef PubMed.
- X. Han, et al., Atomic Layer Infiltration Enabled Cu Coordination Environment Construction for Enhanced Electrochemical CO2 Reduction Selectivity: Case Study of a Cu Metal–Organic Framework, Chem. Mater., 2022, 34(15), 6713–6722 CrossRef.
- K. Yao, et al., Mechanistic Insights into OC–COH Coupling in CO2 Electroreduction on Fragmented Copper, J. Am. Chem. Soc., 2022, 144(31), 14005–14011 CrossRef PubMed.
- Y. Liu, et al., Ligand-Controlled Electroreduction of CO2 to Formate over Facet-Defined Bimetallic Sulfide Nanoplates, Nano Lett., 2023, 23(13), 5911–5918 CrossRef PubMed.
- Y.-Y. Liu, et al., Insight into the Effect of the d-Orbital Energy of Copper Ions in Metal–Organic Frameworks on the Selectivity of Electroreduction of CO2 to CH4, ACS Catal., 2022, 12(5), 2749–2755 CrossRef CAS.
- X. K. Lu, et al., Stabilization of Undercoordinated Cu Sites in Strontium Copper Oxides for Enhanced Formation of C2+ Products in Electrochemical CO2 Reduction, ACS Catal., 2022, 12(11), 6663–6671 CrossRef CAS.
- L. M. S. Garcia, et al., Methanol electrosynthesis from CO2 reduction reaction in polymer electrolyte reactors – fuel cell type using [6,6′-(2,2′-bipyridine-6,6′-diyl)bis(1,3,5-triazine-2,4-diamine)] (dinitrate-O) copper(II) complex, Mater. Today Sustainability, 2022, 19, 100177 CrossRef.
- Y. Zhou, et al., Spatial Confinement in Copper-Porphyrin Frameworks Enhances Carbon Dioxide Reduction to Hydrocarbons, Cell Rep. Phys. Sci., 2020, 1(9), 100182 CrossRef.
- X. Tan, et al., Restructuring of Cu2O to Cu2O@Cu-Metal–Organic Frameworks for Selective Electrochemical Reduction of CO2, ACS Appl. Mater. Interfaces, 2019, 11(10), 9904–9910 CrossRef CAS.
- S. Kusama, et al., Crystalline Copper(II) Phthalocyanine Catalysts for Electrochemical Reduction of Carbon Dioxide in Aqueous Media, ACS Catal., 2017, 7(12), 8382–8385 CrossRef CAS.
- R. Wang, et al., Partial Coordination-Perturbed Bi-Copper Sites for Selective Electroreduction of CO2 to Hydrocarbons, Angew. Chem., Int. Ed., 2021, 60(36), 19829–19835 CrossRef CAS.
- Y.-Y. Liu, et al., A Stable Metal-azolate Framework with Cyclic Tetracopper(I) Clusters for Highly Selective Electroreduction of CO2 to C2 Products, Chem. - Asian J., 2022, 17(21), e202200764 CrossRef CAS PubMed.
- Y. Zhang, et al., Hydroxy-Group-Functionalized Single Crystal of Copper(II)-Porphyrin Complex for Electroreduction CO2 to CH4, ChemSusChem, 2022, 15(4), e202102528 CrossRef CAS PubMed.
- L. Xue, et al., Hydrophobic 1-octadecanethiol functionalized copper catalyst promotes robust high-current CO2 gas-diffusion electrolysis, Nano Res., 2022, 15(2), 1393–1398 CrossRef CAS.
- Z. Yang, et al., Modulable Cu(0)/Cu(I)/Cu(II) sites of Cu/C catalysts derived from MOF for highly selective CO2 electroreduction to hydrocarbons, Vacuum, 2023, 112231 Search PubMed.
- W. Liu, et al., Edge-located Fe–N4 sites on porous Graphene-like nanosheets for boosting CO2 electroreduction, Chem. Eng. J., 2022, 431, 134269 CrossRef CAS.
- C. Zhu, et al., Product-specific active site motifs of Cu for electrochemical CO2 reduction, Chem, 2021, 7(2), 406–420 CAS.
- S. Chen, et al., MOF encapsulating N-heterocyclic carbene-ligated copper single-atom site catalyst towards efficient methane electrosynthesis, Angew. Chem., 2022, 134(4), e202114450 CrossRef.
- D. Cheng, et al., The nature of active sites for carbon dioxide electroreduction over oxide-derived copper catalysts, Nat. Commun., 2021, 12(1), 395 CrossRef CAS.
- X. Yuan, et al., Controllable Cu0–Cu+ sites for electrocatalytic reduction of carbon dioxide, Angew. Chem., 2021, 133(28), 15472–15475 CrossRef.
- A. Guan, et al., Boosting CO2 Electroreduction to CH4via Tuning Neighboring Single-Copper Sites, ACS Energy Lett., 2020, 5(4), 1044–1053 CrossRef CAS.
- C. Hahn, et al., Engineering Cu surfaces for the electrocatalytic conversion of CO2: controlling selectivity toward oxygenates and hydrocarbons, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(23), 5918–5923 CrossRef CAS PubMed.
- J.-M. Heng, et al., A Conductive Dinuclear Cuprous Complex Mimicking the Active Edge Site of the Copper(100)/(111) Plane for Selective Electroreduction of CO2 to C2H4 at Industrial Current Density, Research, 2022, 2022, 0008 CrossRef CAS PubMed.
- Y. Liang, et al., Stabilizing copper sites in coordination polymers toward efficient electrochemical CC coupling, Nat. Commun., 2023, 14(1), 474 CrossRef.
- C. Shan, et al., Site-selective growth of AgPd nanodendrite-modified Au nanoprisms: high electrocatalytic performance for CO2 reduction, Chem. Mater., 2017, 29(14), 6030–6043 CrossRef.
- K. Zhang, et al., Status and perspectives of key materials for PEM electrolyzer, Nano Res. Energy, 2022, 1, e9120032 CrossRef.
- S. B. Varandili, et al., Synthesis of Cu/CeO2−x Nanocrystalline Heterodimers with Interfacial Active Sites To Promote CO2 Electroreduction, ACS Catal., 2019, 9(6), 5035–5046 CrossRef.
- H. Han, et al., Lattice-disorder layer generation from liquid processing at room temperature with boosted nanointerface exposure toward water splitting, Sustainable Energy Fuels, 2022, 6(12), 3008–3013 RSC.
- H.-L. Zhu, et al., Highly Efficient Electroconversion of CO2 into CH4 by a Metal–Organic Framework with Trigonal Pyramidal Cu(I)N3 Active Sites, ACS Catal., 2021, 11(18), 11786–11792 CrossRef CAS.
- T. Yan, et al., Copper(II) Frameworks with Varied Active Site Distribution for Modulating Selectivity of Carbon Dioxide Electroreduction, ACS Appl. Mater. Interfaces, 2022, 14(11), 13645–13652 CrossRef CAS PubMed.
- Z. Yang, et al., Modulable Cu(0)/Cu(I)/Cu(II) sites of Cu/C catalysts derived from MOF for highly selective CO2 electroreduction to hydrocarbons, Vacuum, 2023, 215, 112231 CrossRef CAS.
- T. Dou, et al., Sulfurization-derived Cu0–Cu+ sites for electrochemical CO2 reduction to ethanol, J. Power Sources, 2022, 533, 231393 CrossRef CAS.
- C. Ma, et al., Bimetallic atomic Fe–Mn metal–nitrogen active sites for synergistic enhancement of CO2 electroreduction efficiency, Environ. Funct. Mater., 2022, 1(3), 284–297 Search PubMed.
- X. Yuan, et al., Controllable Cu0–Cu+ Sites for Electrocatalytic Reduction of Carbon Dioxide, Angew. Chem., Int. Ed., 2021, 60(28), 15344–15347 CrossRef CAS PubMed.
- J.-M. Heng, et al., A Conductive Dinuclear Cuprous Complex Mimicking the Active Edge Site of the Copper(100)/(111) Plane for Selective Electroreduction of CO2 to C2H4 at Industrial Current Density, Research, 2022, 2022, 0008 CrossRef CAS PubMed.
- Y. Liang, et al., Stabilizing copper sites in coordination polymers toward efficient electrochemical C–C coupling, Nat. Commun., 2023, 14(1), 474 CrossRef CAS PubMed.
- Z. Li, et al., Coupling Atomically Dispersed Fe–N5 Sites with Defective N-Doped Carbon Boosts CO2 Electroreduction, Small, 2022, 18(38), 2203495 CrossRef CAS.
- X. Wang, et al., Microenvironment and Nanoreactor Engineering of Single-Site Metal Catalysts for Electrochemical CO2 Reduction, Energy Fuels, 2021, 35(12), 9795–9808 CrossRef CAS.
- T. Kim, et al., How Strain Alters CO2 Electroreduction on Model Cu(001) Surfaces, ACS Catal., 2021, 11(11), 6662–6671 CrossRef CAS.
- W.-J. Kang, et al., Strain-Activated Copper Catalyst for pH-Universal Hydrogen Evolution Reaction, Adv. Funct. Mater., 2022, 32(18), 2112367 CrossRef CAS.
- Y. Du and W. An, Effects of Uniaxial Lattice Strain and Explicit Water Solvation on CO2 Electroreduction over a Cu Electrode: A Density Functional Theory Perspective, J. Phys. Chem. C, 2021, 125(17), 9138–9149 CrossRef CAS.
- T. Adit Maark and B. R. K. Nanda, Enhancing CO2 Electroreduction by Tailoring Strain and Ligand Effects in Bimetallic Copper–Rhodium and Copper–Nickel Heterostructures, J. Phys. Chem. C, 2017, 121(8), 4496–4504 CrossRef CAS.
- F. Liu, C. Wu and S. Yang, Strain and Ligand Effects on CO2 Reduction Reactions over Cu–Metal Heterostructure Catalysts, J. Phys. Chem. C, 2017, 121(40), 22139–22146 CrossRef CAS.
- D. Yu, et al., Strain-Stabilized Metastable Face-Centered Tetragonal Gold Overlayer for Efficient CO2 Electroreduction, Nano Lett., 2021, 21(2), 1003–1010 CrossRef CAS.
- Z. Wei, et al., Enhancing Selective Electrochemical CO2 Reduction by In Situ Constructing Tensile-Strained Cu Catalysts, ACS Catal., 2023, 13(7), 4711–4718 CrossRef CAS.
- H. Wang, et al., Strain in Copper/Ceria Heterostructure Promotes Electrosynthesis of Multicarbon Products, ACS Nano, 2023, 17(1), 346–354 CrossRef.
- J. Feng, et al., Improving CO2-to-C2+ Product Electroreduction Efficiency via Atomic Lanthanide Dopant-Induced Tensile-Strained CuOx Catalysts, J. Am. Chem. Soc., 2023, 145(17), 9857–9866 CrossRef.
- J. Hao, et al., Strain Relaxation in Metal Alloy Catalysts Steers the Product Selectivity of Electrocatalytic CO2 Reduction, ACS Nano, 2022, 16(2), 3251–3263 CrossRef.
- H. Li, et al., Strain-Regulated Pd/Cu Core/Shell Icosahedra for Tunable Syngas Electrosynthesis from CO2, Chem. Mater., 2022, 34(17), 7995–8003 CrossRef.
- H. Guzmán, et al., Ultrasound-assisted synthesis of copper-based catalysts for the electrocatalytic CO2 reduction: effect of ultrasound irradiation, precursor concentration and calcination temperature, Sustainable Mater. Technol., 2023, 35, e00557 CrossRef.
- C. A. Downes, et al., Electrocatalytic CO2 Reduction over Cu3P Nanoparticles Generated via a Molecular Precursor Route, ACS Appl. Energy Mater., 2020, 3(11), 10435–10446 CrossRef CAS PubMed.
- Y. Zhang, et al., Cu/Cu2O Nanoparticles Supported on Vertically ZIF-L-Coated Nitrogen-Doped Graphene Nanosheets for Electroreduction of CO2 to Ethanol, ACS Appl. Nano Mater., 2020, 3(1), 257–263 CrossRef CAS.
- T. Dou, et al., CuS Nanosheet Arrays for Electrochemical CO2 Reduction with Surface Reconstruction and the Effect on Selective Formation of Formate, ACS Appl. Energy Mater., 2021, 4(5), 4376–4384 CrossRef CAS.
- S. Chen, et al., Anisotropic Seeded Growth of Cu–M (M = Au, Pt, or Pd) Bimetallic Nanorods with Tunable Optical and Catalytic Properties, J. Phys. Chem. C, 2013, 117(17), 8924–8932 CrossRef CAS.
- Y. Zheng, et al., Seeded Growth of Gold–Copper Janus Nanostructures as a Tandem Catalyst for Efficient Electroreduction of CO2 to C2+ Products, Small, 2022, 18(19), 2201695 CrossRef CAS.
- Y. Wang, et al., Tunable Syngas Formation from Electrochemical CO2 Reduction on Copper Nanowire Arrays, ACS Appl. Energy Mater., 2020, 3(10), 9841–9847 CrossRef CAS.
- H. Wang, et al., Scalable Edge-Oriented Metallic Two-Dimensional Layered Cu2Te Arrays for Electrocatalytic CO2 Methanation, ACS Nano, 2023, 17(5), 4790–4799 CrossRef CAS.
- S. Sirisomboonchai, et al., Efficient CO2 Electrochemical Reduction by a Robust Electrocatalyst Fabricated by Electrodeposition of Indium and Zinc over Copper Foam, ACS Appl. Energy Mater., 2022, 5(8), 9846–9857 CrossRef CAS.
- Y. Gao, et al., Hydrothermal Synthesis of CuS Catalysts for Electrochemical CO2 Reduction: Unraveling the Effect of the Sulfur Precursor, ACS Appl. Energy Mater., 2023, 6(3), 1340–1354 CrossRef CAS.
- X. Pan, et al., Poly(ionic liquid) nanovesicles via polymerization induced self-assembly and their stabilization of Cu nanoparticles for tailored CO2 electroreduction, J. Colloid Interface Sci., 2023, 637, 408–420 CrossRef CAS.
- C. Ye, et al., Enhanced Electrochemical CO2 Reduction to Formate on Poly(4-vinylpyridine)-Modified Copper and Gold Electrodes, ACS Appl. Mater. Interfaces, 2022, 14(40), 45263–45271 CrossRef CAS.
- M. Jun, et al., Polymer-Covered Copper Catalysts Alter the Reaction Pathway of the Electrochemical CO2 Reduction Reaction, ACS Omega, 2022, 7(47), 42655–42663 CrossRef CAS.
- M. Chang, et al., Ionomers Modify the Selectivity of Cu-Catalyzed Electrochemical CO2 Reduction, ChemSusChem, 2023, 16(5), e202201687 CrossRef CAS.
- Q. Chang, et al., Electrochemical CO2 Reduction Reaction over Cu Nanoparticles with Tunable Activity and Selectivity Mediated by Functional Groups in Polymeric Binder, JACS Au, 2022, 2(1), 214–222 CrossRef CAS.
- J. Wang, et al., Selective CO2 Electrochemical Reduction Enabled by a Tricomponent Copolymer Modifier on a Copper Surface, J. Am. Chem. Soc., 2021, 143(7), 2857–2865 CrossRef CAS.
- X. Wei, et al., Highly Selective Reduction of CO2 to C2+ Hydrocarbons at Copper/Polyaniline Interfaces, ACS Catal., 2020, 10(7), 4103–4111 CrossRef CAS.
- L. Zhang, et al., Enhanced Cuprophilic Interactions in Crystalline Catalysts Facilitate the Highly Selective Electroreduction of CO2 to CH4, J. Am. Chem. Soc., 2021, 143(10), 3808–3816 CrossRef.
- J. Li, et al., Polyquinone Modification Promotes CO2 Activation and Conversion to C2+ Products over Copper Electrode, ACS Energy Lett., 2022, 7(11), 4045–4051 CrossRef.
- H. Zhang, et al., Creating Polycrystalline Cu Catalysts via Capping Agents and Electrochemical Treatment for CO2 Reduction to C2H4, Energy Fuels, 2023, 37(1), 529–538 CrossRef.
- Y. Hao, et al., Self-supported copper selenide nanosheets for electrochemical carbon dioxide conversion to syngas with a broad H2-to-CO ratio, Electrochim. Acta, 2023, 449, 142213 CrossRef.
- S. Dongare, N. Singh and H. Bhunia, Oxide-derived Cu–Zn nanoparticles supported on N-doped graphene for electrochemical reduction of CO2 to ethanol, Appl. Surf. Sci., 2021, 556, 149790 CrossRef.
- J. Han, et al., Insight into the effect of surface coverage of carbon support on selective CO2 electroreduction to C2H4 over copper-based catalyst, Appl. Surf. Sci., 2023, 609, 155394 CrossRef CAS.
- L. Gu, J. Ftouni and A. D. Chowdhury, Evaluating carbon dioxide reduction over copper supported on precipitated calcium carbonate via electrochemical route, Mater. Today Chem., 2023, 30, 101539 CrossRef CAS.
- J. W. Lim, et al., Spontaneously Formed CuSx Catalysts for Selective and Stable Electrochemical Reduction of Industrial CO2 Gas to Formate, ACS Appl. Mater. Interfaces, 2020, 12(20), 22891–22900 CrossRef CAS PubMed.
- C. Kim, L.-C. Weng and A. T. Bell, Impact of pulsed electrochemical reduction of CO2 on the formation of C2+ products over Cu, ACS Catal., 2020, 10(21), 12403–12413 CrossRef CAS.
- S. Ahn, et al., Poly-amide modified copper foam electrodes for enhanced electrochemical reduction of carbon dioxide, ACS Catal., 2018, 8(5), 4132–4142 CrossRef CAS.
- X. An, et al., Bi-Doped SnO Nanosheets Supported on Cu Foam for Electrochemical Reduction of CO2 to HCOOH, ACS Appl. Mater. Interfaces, 2019, 11(45), 42114–42122 CrossRef CAS.
- O. A. Baturina, et al., CO2 electroreduction to hydrocarbons on carbon-supported Cu nanoparticles, ACS Catal., 2014, 4(10), 3682–3695 CrossRef CAS.
- A. Vijayakumar, et al., A Nitrogen-Doped Porous Carbon Supported Copper Catalyst from a Scalable One-Step Method for Efficient Carbon Dioxide Electroreduction, ChemElectroChem, 2023, 10(2), e202200817 CrossRef CAS.
- Y. Huo, et al., High Selectivity Toward C2H4 Production over Cu Particles Supported by Butterfly-Wing-Derived Carbon Frameworks, ACS Appl. Mater. Interfaces, 2018, 10(15), 12618–12625 CrossRef CAS.
- D. Kordus, et al., Shape-Dependent CO2 Hydrogenation to Methanol over Cu2O Nanocubes Supported on ZnO, J. Am. Chem. Soc., 2023, 145(5), 3016–3030 CrossRef CAS.
- Y. Mi, et al., Selective formation of C2 products from electrochemical CO2 reduction over Cu1.8Se nanowires, ACS Appl. Energy Mater., 2018, 1(10), 5119–5123 CAS.
- H. Wang, et al., Size Control of Zn, N-doped Carbon Supported Copper Nanoparticles for Effective and Selective CO2 Electroreduction, Catal. Lett., 2023, 153(7), 2115–2124 CrossRef CAS.
- W. Lou, et al., CuBi electrocatalysts modulated to grow on derived copper foam for efficient CO2-to-formate conversion, J. Colloid Interface Sci., 2022, 606, 994–1003 CrossRef CAS PubMed.
- Y. Zhang, et al., Three-dimensional porous copper-decorated bismuth-based nanofoam for boosting the electrochemical reduction of CO2 to formate, Inorg. Chem. Front., 2021, 8(10), 2461–2467 RSC.
- Y. Wang, et al., Effect of Charge on Carbon Support on the Catalytic Activity of Cu2O toward CO2 Reduction to C2 Products, ACS Appl. Mater. Interfaces, 2023, 15(19), 23306–23315 CrossRef CAS PubMed.
- M. Wang, et al., Electrochemical Reduction of CO2 on Copper-Based Electrocatalyst Supported on MWCNTs with Different Functional Groups, Energy Fuels, 2022, 36(11), 5833–5842 CrossRef CAS.
- J. W. Lim, et al., Spontaneously Formed CuSx Catalysts for Selective and Stable Electrochemical Reduction of Industrial CO2 Gas to Formate, ACS Appl. Mater. Interfaces, 2020, 12(20), 22891–22900 CrossRef CAS PubMed.
- O. A. Baturina, et al., CO2 Electroreduction to Hydrocarbons on Carbon-Supported Cu Nanoparticles, ACS Catal., 2014, 4(10), 3682–3695 CrossRef CAS.
- A. Vijayakumar, et al., A Nitrogen-Doped Porous Carbon Supported Copper Catalyst from a Scalable One-Step Method for Efficient Carbon Dioxide Electroreduction, ChemElectroChem, 2023, 10(2), e202200817 CrossRef CAS.
- E. S. Koh, et al., Influence of Support Material on the Structural Evolution of Copper during Electrochemical CO2 Reduction, ChemElectroChem, 2023, 10(5), e202200924 CrossRef.
- Y. Mi, et al., Selective Formation of C2 Products from Electrochemical CO2 Reduction over Cu1.8Se Nanowires, ACS Appl. Energy Mater., 2018, 1(10), 5119–5123 Search PubMed.
- Y. Zhang, et al., Three-dimensional porous copper-decorated bismuth-based nanofoam for boosting the electrochemical reduction of CO2 to formate, Inorg. Chem. Front., 2021, 8(10), 2461–2467 RSC.
- Q. Zhang, et al., High-Performance of Electrocatalytic CO2 Reduction on Defective Graphene-Supported Cu4S2 Cluster, Catalysts, 2022, 12(5), 454 CrossRef.
- Q. Tan, Z. Shi and D. Wu, CO2 Hydrogenation to Methanol over a Highly Active Cu–Ni/CeO2–Nanotube Catalyst, Ind. Eng. Chem. Res., 2018, 57(31), 10148–10158 CrossRef.
- Y. Zhou, et al., Tuning the content of S vacancies in MoS2 by Cu doping for enhancing catalytic hydrogenation of CO2 to methanol, Mol. Catal., 2023, 547, 113288 CrossRef CAS.
- S. Chen, et al., Highly Selective Carbon Dioxide Electroreduction on Structure-Evolved Copper Perovskite Oxide toward Methane Production, ACS Catal., 2020, 10(8), 4640–4646 CrossRef CAS.
- Y. Deng, et al., On the Role of Sulfur for the Selective Electrochemical Reduction of CO2 to Formate on CuSx Catalysts, ACS Appl. Mater. Interfaces, 2018, 10(34), 28572–28581 CrossRef CAS.
- Y. Wang, et al., Single-Atomic Cu with Multiple Oxygen Vacancies on Ceria for Electrocatalytic CO2 Reduction to CH4, ACS Catal., 2018, 8(8), 7113–7119 CrossRef CAS.
- H. Yang, et al., Defect-engineering of tin oxide via (Cu, N) co-doping for electrocatalytic and photocatalytic CO2 reduction into formate, Chem. Eng. Sci., 2020, 227, 115947 CrossRef CAS.
- Y. Zhang, et al., Selective CO2 Reduction to Ethylene Over a Wide Potential Window by Copper Nanowires with High Density of Defects, Inorg. Chem., 2022, 61(50), 20666–20673 CrossRef CAS.
- Q. Wu, et al., Defect-Engineered Cu-Based Nanomaterials for Efficient CO2 Reduction over Ultrawide Potential Window, ACS Nano, 2023, 17(1), 402–410 CrossRef CAS PubMed.
- T. Le, T. Salavati-fard and B. Wang, Plasmonic Energetic Electrons Drive CO2 Reduction on Defective Cu2O, ACS Catal., 2023, 13(9), 6328–6337 CrossRef CAS.
- H. Wang, et al., Cu/CuOx In-Plane Heterostructured Nanosheet Arrays with Rich Oxygen Vacancies Enhance Nitrate Electroreduction to Ammonia, ACS Appl. Mater. Interfaces, 2022, 14(30), 34761–34769 CrossRef CAS PubMed.
- H. Zhang, et al., Selective CO2-to-formic acid electrochemical conversion by modulating electronic environment of copper phthalocyanine with defective graphene, Chin. J. Struct. Chem., 2023, 100089 CrossRef.
- K. K. Patra, et al., Boosting Electrochemical CO2 Reduction to Methane via Tuning Oxygen Vacancy Concentration and Surface Termination on a Copper/Ceria Catalyst, ACS Catal., 2022, 12(17), 10973–10983 CrossRef CAS.
- J. Ye, M. Neurock and D. G. Truhlar, Effect of Missing-Linker Defects on CO2 Hydrogenation to Methanol by Cu Nanoparticles in UiO-66, J. Phys. Chem. C, 2022, 126(31), 13157–13167 CrossRef CAS.
- S. Pablo-García, et al., Mechanistic routes toward C3 products in copper-catalysed CO2 electroreduction, Catal. Sci. Technol., 2022, 12(2), 409–417 RSC.
- K. Zhao, et al., CO2 Electroreduction at Low Overpotential on Oxide-Derived Cu/Carbons Fabricated from Metal Organic Framework, ACS Appl. Mater. Interfaces, 2017, 9(6), 5302–5311 CrossRef CAS.
- Y.-L. Qiu, et al., Selective Electrochemical Reduction of Carbon Dioxide Using Cu Based Metal Organic Framework for CO2 Capture, ACS Appl. Mater. Interfaces, 2018, 10(3), 2480–2489 CrossRef CAS.
- L. Ma, et al., Covalent triazine framework confined copper catalysts for selective electrochemical CO2 reduction: operando diagnosis of active sites, ACS Catal., 2020, 10(8), 4534–4542 CrossRef.
- S. Kim, et al., Grain Boundary-Rich Copper Nanocatalysts Generated from Metal–Organic Framework Nanoparticles for CO2-to-C2+ Electroconversion, Adv. Sci., 2023, 10(9), 2207187 CrossRef.
- W. Nie, et al., Organic Additive-derived Films on Cu Electrodes Promote Electrochemical CO2 Reduction to C2+ Products Under Strongly Acidic Conditions, Angew. Chem., 2023, 135(12), e202216102 CrossRef.
- S.-M. Hwang, et al., Investigation on electroreduction of CO2 to formic acid using Cu3(BTC)2 metal–organic framework (cu-mof) and graphene oxide, ACS Omega, 2020, 5(37), 23919–23930 CrossRef PubMed.
- D. Li, et al., MOF-Derived Cu2O/Cu Nanospheres Anchored in Nitrogen-Doped Hollow Porous Carbon Framework for Increasing the Selectivity and Activity of Electrochemical CO2-to-Formate Conversion, ACS Appl. Mater. Interfaces, 2020, 12(6), 7030–7037 CrossRef.
- L. Li, et al., Control of evolution of porous copper-based metal–organic materials for electroreduction of CO2 to multi-carbon products, Mater. Adv., 2023, 4(8), 1941–1948 RSC.
- Y.-S. Cheng, et al., An MOF-derived copper@ nitrogen-doped carbon composite: the synergistic effects of N-types and copper on selective CO2 electroreduction, Catal. Sci. Technol., 2019, 9(20), 5668–5675 RSC.
- S. Wei, et al., Construction of single-atom copper sites with low coordination number for efficient CO2 electroreduction to CH4, J. Mater. Chem. A, 2022, 10(11), 6187–6192 RSC.
- B. Xu, et al., Anion-regulation engineering toward Cu/In/MOF bimetallic electrocatalysts for selective electrochemical reduction of CO2 to CO/formate, Mater. Rep.: Energy, 2022, 2(3), 100139 Search PubMed.
- W. Guo, et al., Metal–organic framework-derived indium–copper bimetallic oxide catalysts for selective aqueous electroreduction of CO2, Green Chem., 2019, 21(3), 503–508 RSC.
- Y.-H. Zou, et al., Implanting MWCNTs in BiCu-MOFs to enhance electrocatalytic CO2 reduction to formate, Sep. Purif. Technol., 2023, 317, 123806 CrossRef.
- X. Han, et al., Hollow structured Cu@ ZrO2 derived from Zr-MOF for selective hydrogenation of CO2 to methanol, J. Energy Chem., 2022, 71, 277–287 CrossRef.
- N. Rashid, et al., Tailoring Motif and Channel Terminating Groups of Conventional Copper MOFs for Their Enhanced Activity, Selectivity, and Stability toward the Electroreduction of CO2 to Hydrocarbons, ACS Appl. Energy Mater., 2023, 6(3), 1378–1388 CrossRef.
- C. Liu, et al., In Situ Reconstruction of Cu–N Coordinated MOFs to Generate Dispersive Cu/Cu2O Nanoclusters for Selective Electroreduction of CO2 to C2H4, ACS Catal., 2022, 12(24), 15230–15240 CrossRef.
- J. B. Parambath, et al., Copper-Based Metal-Organic Frameworks (MOFs) for Electroreduction of CO2, Catal. Sci. Technol., 2023, 13, 3740–3761 RSC.
- L.-Z. Dong, et al., Porous copper cluster-based MOF with strong cuprophilic interactions for highly selective electrocatalytic reduction of CO2 to CH4, Nano Res., 2022, 15(12), 10185–10193 CrossRef.
- S. Kim, et al., Grain Boundary-Rich Copper Nanocatalysts Generated from Metal-Organic Framework Nanoparticles for CO2-to-C2+ Electroconversion, Adv. Sci., 2023, 10(9), 2207187 CrossRef PubMed.
- W. Nie, et al., Organic Additive-derived Films on Cu Electrodes Promote Electrochemical CO2 Reduction to C2+ Products Under Strongly Acidic Conditions, Angew. Chem., Int. Ed., 2023, 62(12), e202216102 CrossRef.
- L. Li, et al., Control of evolution of porous copper-based metal–organic materials for electroreduction of CO2 to multi-carbon products, Mater. Adv., 2023, 4(8), 1941–1948 RSC.
- S. Wei, et al., Construction of single-atom copper sites with low coordination number for efficient CO2 electroreduction to CH4, J. Mater. Chem. A, 2022, 10(11), 6187–6192 RSC.
- B. Xu, et al., Anion-regulation engineering toward Cu/In/MOF bimetallic electrocatalysts for selective electrochemical reduction of CO2 to CO/formate, Mater. Rep.: Energy, 2022, 2(3), 100139 Search PubMed.
- W. Guo, et al., Metal–organic framework-derived indium–copper bimetallic oxide catalysts for selective aqueous electroreduction of CO2, Green Chem., 2019, 21(3), 503–508 RSC.
- L. D. Chen, Cations play an essential role in CO2 reduction, Nat. Catal., 2021, 4(8), 641–642 CrossRef CAS.
- J. Fernández-Vidal, et al., Effect of alkali-metal cation on oxygen adsorption at Pt single-crystal electrodes in non-aqueous electrolytes, Faraday Discuss., 2024, 248(0), 102–118 RSC.
- B. Pan, Y. Wang and Y. Li, Understanding and leveraging the effect of cations in the electrical double layer for electrochemical CO2 reduction, Chem. Catal., 2022, 2(6), 1267–1276 CrossRef CAS.
- J. J. Masana, et al., Influence of halide ions on the electrochemical reduction of carbon dioxide over a copper surface, J. Mater. Chem. A, 2022, 10(3), 1086–1104 RSC.
- T. Yuan, et al., The effect of specific adsorption of halide ions on electrochemical CO(2) reduction, Chem. Sci., 2022, 13(27), 8117–8123 RSC.
- Q. Zhang, et al., An experimental study of electroreduction of CO2 to HCOOH on SnO2/C in presence of alkali metal cations (Li+, Na+, K+, Rb+ and Cs+) and anions (HCO3−, Cl−, Br− and I−), Chin. J. Chem. Eng., 2020, 28(10), 2549–2554 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |