
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA robust heterogeneous chiral phosphoric acid enables multi decagram scale production of optically active N,S-ketals†
Aitor
Maestro
 *ab,
Bhanwar K.
Malviya
bc,
Gerald
Auer
d,
Sándor B.
Ötvös
bc and
C. Oliver
Kappe
*bc
*ab,
Bhanwar K.
Malviya
bc,
Gerald
Auer
d,
Sándor B.
Ötvös
bc and
C. Oliver
Kappe
*bc
aDepartment of Organic Chemistry I, University of the Basque Country, UPV/EHU, Paseo de la Universidad 7, 01006 Vitoria-Gasteiz, Spain. E-mail: aitor.maestro@ehu.eus
bInstitute of Chemistry, University of Graz, NAWI Graz, A-8010 Graz, Austria. E-mail: oliver.kappe@uni-graz.at
cCenter for Continuous Flow Synthesis and Processing (CC FLOW), Research Center Pharmaceutical Engineering GmbH (RCPE), A-8010 Graz, Austria
dDepartment of Earth Sciences, University of Graz, NAWI Graz Geocenter, A-8010 Graz, Austria
First published on 6th March 2024
Abstract
Asymmetric organocatalysis has been recognized as one of the “top 10 emerging technologies” in chemistry by IUPAC in 2019. Its potential to make chemical processes more sustainable is promising, but there are still challenges that need to be addressed. Developing new and reliable enantioselective processes for reproducing batch reactions on a large scale requires a combination of chemical and technical solutions. In this manuscript, we combine a robust immobilized chiral phosphoric acid with a new packed-bed reactor design. This combination allows scaling up of the enantioselective addition of thiols to imines from a few milligrams to a multi-decagram scale in a continuous flow process without physical or chemical degradation of the catalyst.
Introduction
Asymmetric organocatalysis is a highly effective method for producing enantiomerically pure compounds. Since its introduction in the field around 20 years ago,1 it has become one of the primary catalytic strategies for obtaining optically active molecules,2 alongside metal-catalysis3 and biocatalysis.4 Enzymes have a limited range of stability when it comes to temperature and solvents, while metal complexes often require moisture-free or oxygen-free conditions. On the other hand, organocatalysts are more versatile and easily overcome these limitations. The potential of asymmetric organocatalysis was recognized by IUPAC in 2019 as one of the “top ten emerging technologies in chemistry with potential to make our planet more sustainable”5 and was further highlighted by the Nobel Prize awarded to List and MacMillan in 2021. As a result, the scientific community has high expectations for the future of asymmetric organocatalysis.6 However, the required catalyst loading, costly catalysts and limited scalability are challenges that still need to be addressed in order to utilize asymmetric organocatalysis as a general and efficient tool. This is evidenced by the limited applications of organocatalysis in industry and patent application processes in recent years.7In the last decade, several authors have made efforts to develop robust and recyclable organocatalysts that minimize the cost of large-scale organocatalytic processes.8 Moreover, by combining immobilized catalysts with continuous flow technology, it has been possible to intensify those processes, achieving high yield, enantiocontrol and increased catalyst turnover numbers (TON) with respect to the analogous batch processes.9 While most of organocatalytic processes, either in batch or flow, only allow the milligram scale synthesis of optically active molecules, examples reporting multi gram syntheses are scarce.10
Developing highly recyclable catalysts that can be used in continuous processes with similar performance to the analogous batch reactions would increase the productivity, minimizing the effect of the high environmental factor (E-factor) known for the preparation of some organocatalysts.11 Following our previous work on immobilized chiral phosphoric acids,12 we aimed to evaluate the robustness of the immobilized TRIP (PS-TRIP) catalyst (Scheme 1A), developed by Pericàs in 2016.13 To date, PS-TRIP and closely related catalysts have mostly been used in reactions that require several hours to complete and with no or limited background reaction ensuring high chemical selectivity.12–14 Analyzing the performance of PS-TRIP towards a reaction with a strong background effect represents a challenge since a minimal decrease in the catalyst activity with respect to the corresponding homogeneous variant would lead to a drastic decrease in enantiocontrol.
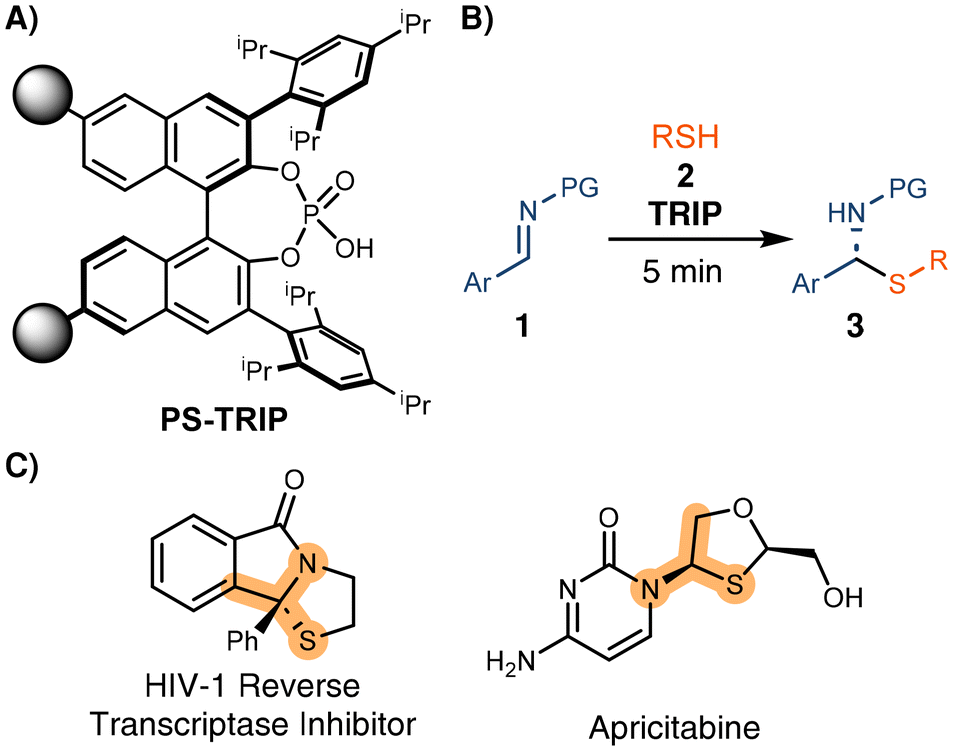 | ||
| Scheme 1 A) Pericàs's PS-TRIP catalyst. (B) Enantioselective synthesis of N,S-ketals. (C) Selected examples of pharmaceuticaly active N,S-ketals. | ||
In 2011, Antilla and coworkers reported the synthesis of N,S-acetals through a highly enantioselective addition of thiols to imines (Scheme 1B).15 This reaction is particularly interesting for our purpose since full conversion to the racemate was reported after just a few minutes of reaction even in the absence of any catalyst. Additionally, similar processes have been used to synthesize active pharmaceutical ingredients (Scheme 1C),16 increasing the interest in merging this asymmetric methodology with continuous flow processing.
Results and discussion
The activity of the PS-TRIP catalyst for the enantioselective thiol addition was explored in a flow set-up consisting of two separate reagent feeds: solutions of imine 2 (1.0 equiv.) and thiol 3a (1.0 equiv.), respectively. The reagent streams were pumped at a flow rate of 100 μL min−1 each and were combined before entering a packed bed reactor containing 0.5 g of the supported catalyst (Table 1). This corresponded to a residence (contact) time on the catalyst bed of approx. 12 min. Although the optimal conditions from preliminary batch tests allowed obtaining up to 91% ee using a toluene/acetone 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mixture as a solvent (see ESI, Table S1†), the chiral HPLC analysis showed no enantiocontrol in the initial flow experiment (Table 1, entry 1). We hypothesized that, due to the high reaction rate, quantitative conversion may be reached before entering the catalyst bed. In order to minimize the residence time of the combined reaction feed in the absence of the PS-TRIP catalyst, the effect of the flow rate was evaluated, obtaining up to 52% ee (Table 1, entries 2–6). However, no further improvement was achieved when higher flow rates were used. As an alternative, a new end-fitting was designed for the packed bed reactor in which both reagents are directly combined, thereby avoiding the pre-mixing of the reaction feeds (Fig. 1A and B). The new reactor design allowed increased enantioselectivities of up to 80%, which gradually decreased with the flow rate due to the reduced residence time (Table 1, entries 7–10). In addition, using the thiol in excess was found beneficial for the enantiocontrol (Table 1, entries 11–14).
:3 mixture as a solvent (see ESI, Table S1†), the chiral HPLC analysis showed no enantiocontrol in the initial flow experiment (Table 1, entry 1). We hypothesized that, due to the high reaction rate, quantitative conversion may be reached before entering the catalyst bed. In order to minimize the residence time of the combined reaction feed in the absence of the PS-TRIP catalyst, the effect of the flow rate was evaluated, obtaining up to 52% ee (Table 1, entries 2–6). However, no further improvement was achieved when higher flow rates were used. As an alternative, a new end-fitting was designed for the packed bed reactor in which both reagents are directly combined, thereby avoiding the pre-mixing of the reaction feeds (Fig. 1A and B). The new reactor design allowed increased enantioselectivities of up to 80%, which gradually decreased with the flow rate due to the reduced residence time (Table 1, entries 7–10). In addition, using the thiol in excess was found beneficial for the enantiocontrol (Table 1, entries 11–14).
 | ||
| Fig. 1 Catalyst bed design. (A) Initial set up. Imine and thiol are premixed before entering the catalyst bed. (B) Second design, no pre-mixing before the packed bed reactor. (C) Final setup with spiral mixing element installed inside the packed-bed reactor. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 2a (equiv.) | Flow rate (mL min−1) | Reactor | Conv.a (%) | eeb (%) | |
| Pump 1 (1) | Pump 2 (2a) | |||||
| a Conv. HPLC area%. b Analyzed by chiral HPLC. | ||||||
| 1 | 1.00 | 0.1 | 0.1 | A | >98 | 0 |
| 2 | 1.00 | 0.25 | 0.25 | A | >98 | 10 |
| 3 | 1.00 | 0.5 | 0.5 | A | >98 | 25 |
| 4 | 1.00 | 0.75 | 0.75 | A | >98 | 43 |
| 5 | 1.00 | 1.0 | 1.0 | A | >98 | 52 |
| 6 | 1.00 | 1.5 | 1.5 | A | >98 | 51 |
| 7 | 1.00 | 0.5 | 0.5 | B | >98 | 79 |
| 8 | 1.00 | 1.0 | 1.0 | B | >98 | 80 |
| 9 | 1.00 | 1.5 | 1.5 | B | >98 | 75 |
| 10 | 1.00 | 2.0 | 2.0 | B | >98 | 70 |
| 11 | 1.11 | 1.9 | 2.1 | B | >98 | 78 |
| 12 | 1.22 | 1.8 | 2.2 | B | >98 | 82 |
| 13 | 1.35 | 1.7 | 2.3 | B | >98 | 90 |
| 14 | 1.50 | 1.6 | 2.4 | B | >98 | 93 |
| 15 | 1.50 | 1.6 | 2.4 | C | >98 | 92 |
| 16 | 1.22 | 1.8 | 2.2 | C | >98 | 92 |
| 17 | 1.11 | 1.9 | 2.1 | C | >98 | 90 |
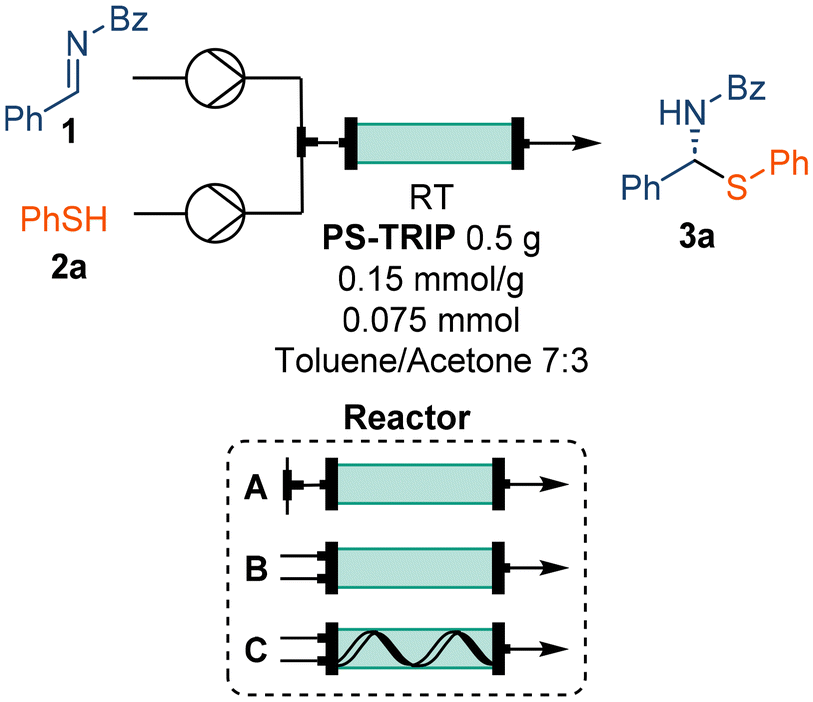
Despite the promising results obtained during the optimization process, this set-up was not suitable for longer continuous runs due to some overpressure generated. The continuous flow of the reagent solutions compressed the catalyst bed leading to a significant pressure drop after 5–10 minutes of processing time. To overcome this limitation, a Teflon spiral inside the catalyst bed was designed to release system pressure and to aid maintaining process stability (Fig. 1C).
As a control experiment, pure solvent (toluene/acetone 7:3) was pumped at 20.0 mL min−1 for 1 h without any pressure increase. Importantly, the spiral not only ensured increased process stability, but also allowed reducing the excess of 3 to only 1.2 equivalents without losing enantioselectivity (Table 1, entries 15–17).
With the optimized set-up in hand, we performed a continuous long run for 6 h under steady state conditions. The reaction mixture was collected directly into a 0.25 M solution of NaOCl to quench the excess of thiol used in the reaction (Scheme 2). The overall process was followed by off-line HPLC with samples taken and analyzed every 30 min. We were pleased to find no decrease in either conversion or enantioselectivity and a constant system pressure of around 1 bar, showcasing the robustness of the process. During the long run, precipitation of a white solid was observed after a few minutes of collection time allowing to isolate the pure product by simple filtration (Scheme 2C). The precipitate was washed with water and cyclohexane to afford 19.44 g of pure product (94% yield, 92% ee).
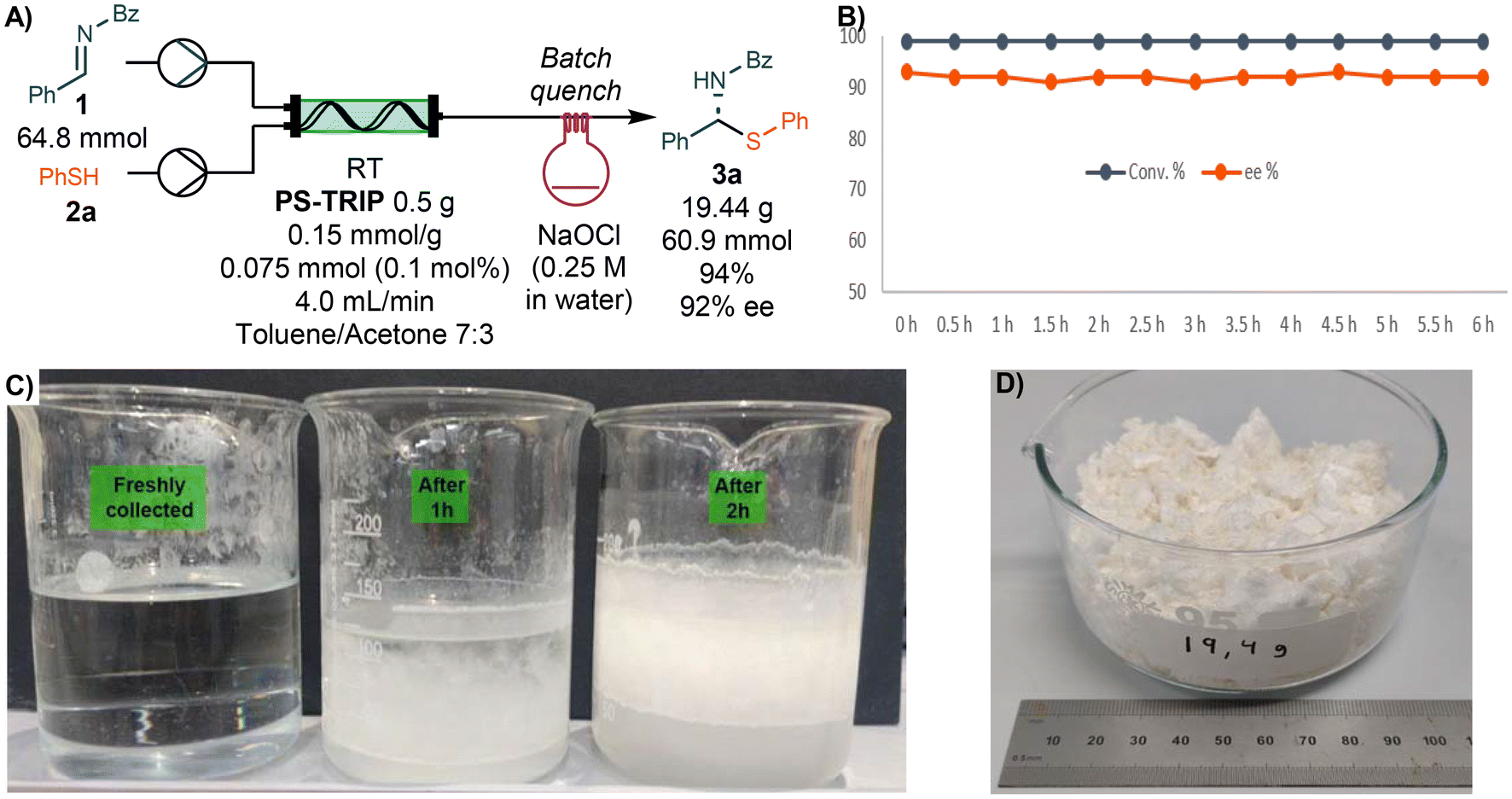 | ||
| Scheme 2 A) Optimized set-up and long run. (B) Conversion and ee of 3a over the time (HPLC area %). (C) Evolution of the product precipitation in the quench flask over 2 h. (D) Isolated product after 6 h run. | ||
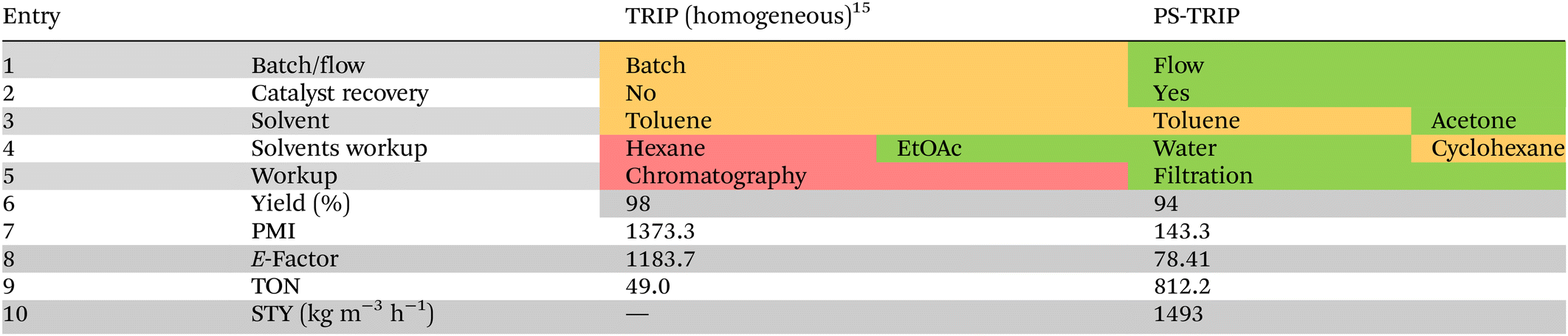
To demonstrate the superior performance of the PS-TRIP catalyst in the 6 h flow process with respect to the previously reported batch methodology,15 both processes were evaluated in detail by means of quantitative green metrics and qualitative sustainability indicators according to the CHEM21 toolkit (Table 2).17 Regarding the solvents used in the reaction, part of the toluene required in batch was replaced for the environmentally more acceptable acetone, and hexane required for the batch purification was replaced for the less problematic cyclohexane (Table 2, entries 4 and 5).18 Overall, both processes showed isolated yields above 90%. However, isolating the product by filtration minimizes the waste generation by reducing the amount of required solvents and completely avoiding the classical chromatography on silica gel (Table 2, entry 5). This highlights the benefit of employing immobilized organocatalysts, not only for the catalyst recovery but also for simplifying the purification step. As a consequence, the process mass intensity (PMI) and environmental factor (E-factor) were reduced around ten times in our process with respect to the literature precedent (Table 2, entries 7 and 8). In particular, the PMI was reduced from 1373 in batch to 143 in flow, while the E-factor was reduced from 1183 to 78. This highlights the benefit of the flow process by minimizing the waste generated, with the consequent economic and environmental effects.
|
Regarding the heterogeneous organocatalyst, a turnover number (TON) of 812 was calculated for the 6 h run (0.1 mol%) with respect to the literature value of 49 for the batch process (Table 2, entry 9). It should also be noted that all the experiments done within this project were carried out using the same sample of PS-TRIP catalyst and no deactivation was observed, indicating a higher accumulative TON. Finally, a space–time yield (STY) of 1493 kg m−3 h−1 was calculated for the flow process (Table 2, entry 10). Therefore, using a continuous flow process combined with an easily recyclable catalyst is clearly advantageous to the analogous homogeneous reactions in batch.
Next, we extended the scope of the reaction to several other thiols (Scheme 3). The reaction worked similarly for several thiophenol derivatives, regardless of the electronic or steric effect of the substituents, with isolated yields above 90% and enantioselectivities above 85% in all cases. However, less reactive benzylic thiols required increased residence times in order to afford high yield and stereocontrol. The functional group tolerance of the reaction was further tested by reacting a free carboxylic acid-containing thiol. Even though the reaction gave good yield, only 54% ee was achieved at 1 mL min−1 flow rate. In contrast, the analogous ester provided a slightly higher yield and up to 89% ee under the same reaction conditions.
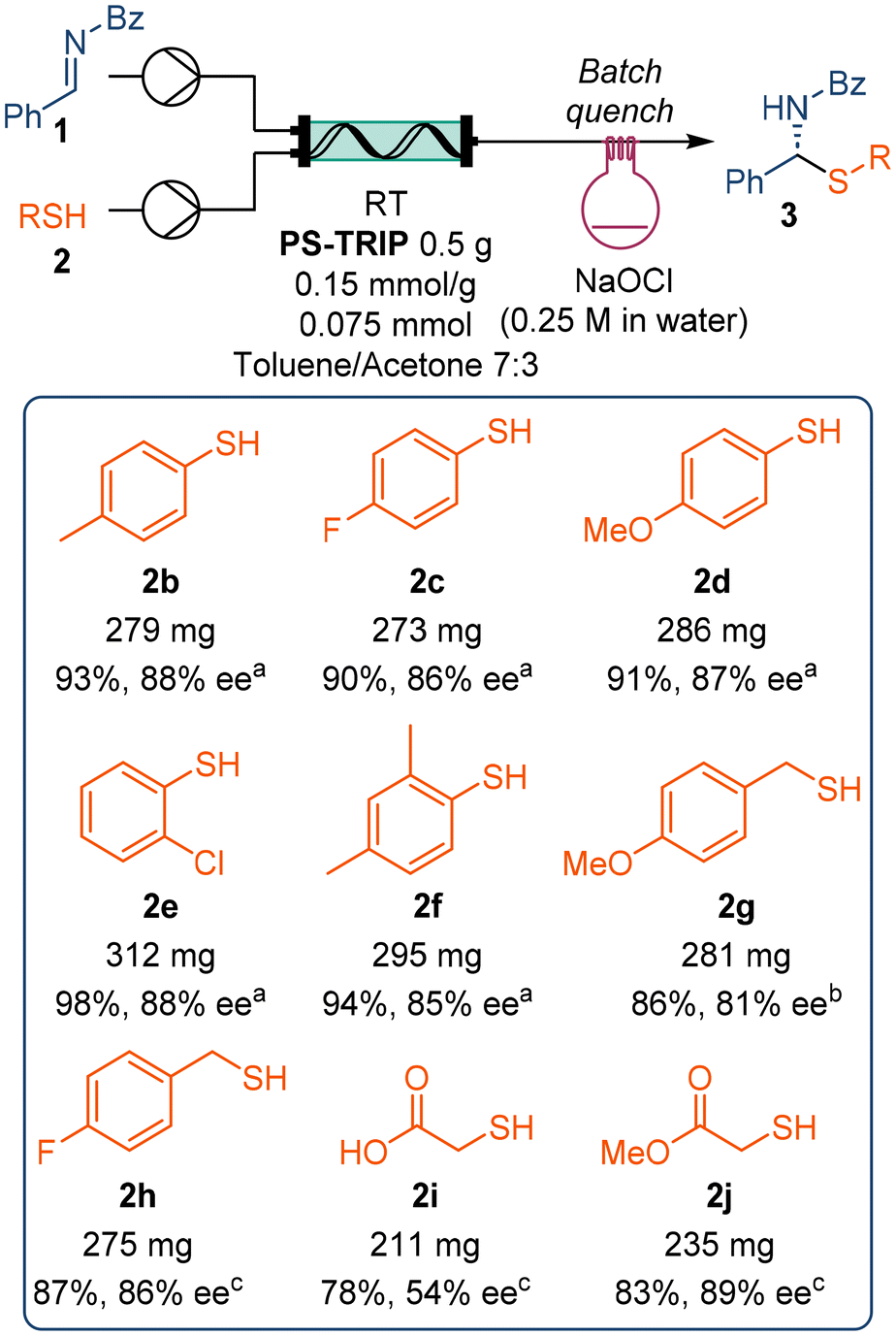 | ||
| Scheme 3 Scope of thiols explored in the flow reaction. a4.0 mL min−1, collected for 5 min. b2.0 mL min−1, collected for 10.0 min. c1.0 mL min−1, collected for 20.0 min. | ||
The as-prepared PS-TRIP catalyst was analyzed by optical and electronic microscopy techniques to shed light into the morphological and chemical structure. As expected in a polystyrene-supported catalyst, characteristic C–H bending peaks are present at 695 and 745 cm−1 on the FTIR analysis. Aromatic C–C stretching bands are also visible at 1450 and 1490 cm−1 (Fig. 2A). From the morphological point of view, 0.3–0.5 mm size pale-yellow particles with needles and sharp edges can be observed by optical microscopy (Fig. 2B). Scanning electron microscopy (SEM) shows smooth surfaces with some holes and cavities in the catalyst particles (Fig. 2C). In order to analyze potential alterations in catalyst structure and thus evaluate catalyst stability and robustness, the same analyses were performed on catalyst samples used during this project. Only a minor difference was observed during analysis of fresh and used catalyst samples; namely, the polishing of the particle surface (Fig. 2E).
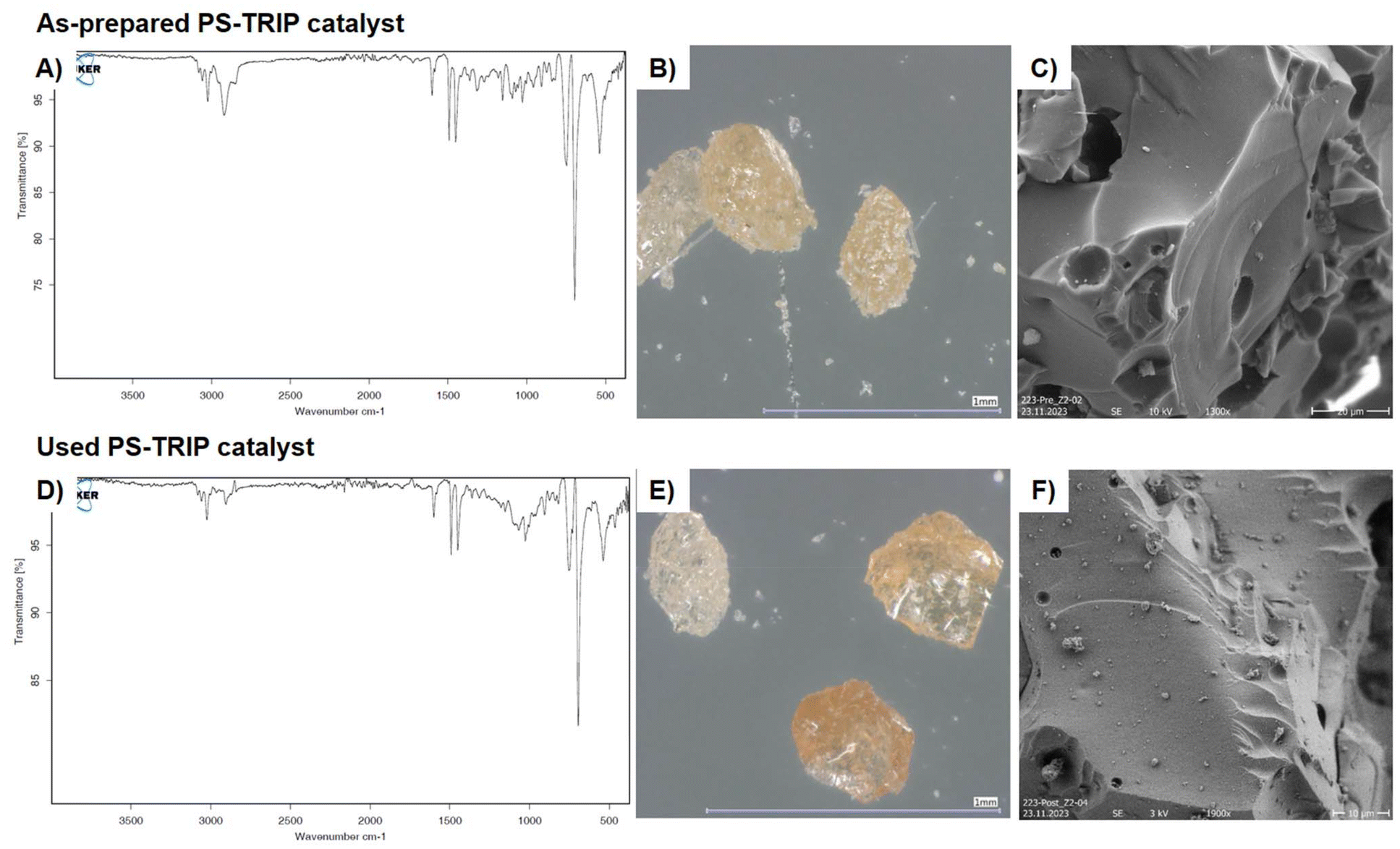 | ||
| Fig. 2 Catalyst characterization: (A) FTIR, as-prepared PS-TRIP. (B) Optical microscopy, as prepared PS-TRIP. (C) SEM, as prepared PS-TRIP. (D) FTIR, used PS-TRIP. (E) Optical microscopy, used PS-TRIP. (F) SEM, used PS-TRIP. | ||
This might be due to the erosion of polymer grains during the flow process. Moreover, when analyzing the catalyst by SEM, some particle deposition was observed (Fig. 2F). After analysis by FTIR and SEM-EDX, the deposited particles were found to contain Si and Al as the main elements, suggesting their identification as glass wool, the material that is used to contain the catalyst bed (see ESI Fig. S5, S9 and 10†). Therefore, no appreciable physical or chemical degradation of the catalyst can be observed after the reaction.
Conclusions
A continuous flow process using a highly recyclable PS-TRIP organocatalyst has been developed, allowing scaling up of the enantioselective addition of thiols to imines from just a few milligrams to multiple decagrams. By using a laboratory scale reactor (2.17 mL), high productivity (6.5 g h−1 gresin−1; STY 1493 kg m−3 h−1) was achieved in an overall greener process with a significant waste reduction. Remarkably, all the experiments carried out in this project, including the reaction optimization, a 6 h continuous flow run and the scope, were performed by using only 0.5 g of the PS-TRIP catalyst without observing any deactivation of its activity (TON >800, <0.1 mol%). This was confirmed by a control test reaction for the synthesis of 3a at the end of the project, as well as by the chemical and physical analysis of the used catalyst. All those tests showed no significant alterations that could affect the performance of the PS-TRIP for future applications.Furthermore, the design of a new packed bed reactor that minimizes the racemic background reaction is also presented, allowing to obtain higher enantiocontrol in challenging reactions. In summary, a heterogeneous PS-TRIP catalyst along with a new reactor resulted in an advantageous combination that will open new frontiers for future applications in the field.
Experimental
Procedure for filling the packed-bed reactor
The packed bed reactor was prepared as shown in Fig. S3.† The adjustable end of the Omnifit® glass column (10 mm ID) was opened and 0.5 g of dry catalyst was loaded (dried overnight at 40 °C in a vacuum oven). The reactor was then filled with toluene to swell the resin, closed and adjusted as required. Both ends were closed with glass wool to prevent the catalyst particles from clogging the system.General procedure for the enantioselective synthesis of N,S-ketals in continuous flow
0.5 g of PS-TRIP catalyst was loaded into a modified adjustable Omnifit® glass column (10 mm ID). Before the reactions, the catalyst bed was swollen by pumping toluene/acetone 7:3 at 4.0 mL min−1 for 5 min. The stock solutions (in toluene/acetone 7:3) of imine 1 (0.10 M, 1.80 mL min−1, 1.0 equiv.) and 2a (0.1 M, 2.20 mL min−1, 1.2 equiv.) were pumped independently (4.0 mL min−1 overall flow rate) and combined in the modified Omnifit column containing the PS-TRIP catalyst by using a Syrris® Asia syringe pump. The pressure of the system generated by the packed bed reactor during the long run was around 1 bar. The reaction outcome was quenched by pumping directly into an aqueous solution of 0.25 M NaClO.
Quenching for 30 min of reaction in continuous flow
For analysis purposes, 30 min fractions were independently collected. The reaction mixture was pumped through a 0.25 M solution of NaClO (30 mL) After 1h precipitation of the product was observed and the crude was allowed to further precipitate for 6 h to ensure complete precipitation of the product.Work up for 6 h reaction
The combined fractions were filtered and washed with water (3 × 200 mL) and cyclohexane (3 × 250 mL). The cake was dried in a vacuum oven at 50 °C at 1 torr for 12 h to give the pure product (19.44 g, 94% yield, 92% ee).Conflicts of interest
There are no conflicts to declare.Acknowledgements
AM acknowledges funding from the Department of Education of the Basque Government (postdoctoral fellowship). SBO thanks the Austrian Science Fund (FWF) for financial support through project P34397-N. Open Access funding provided by University of Basque Country.References
- Pioneering examples in asymmetric organocatalysis:
(a) W. S. Jen, J. J. M. Wiener and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 9874–9875 CrossRef CAS
; (b) K. A. Ahrendt, C. J. Borths and D. W. C. Macmillan, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS
- Recent reviews:
(a) B. Han, X. H. He, Y. Q. Liu, G. He, C. Peng and J. L. Li, Chem. Soc. Rev., 2021, 50, 1522–1586 RSC
- P. S. Steinlandt, L. Zhang and E. Meggers, Chem. Rev., 2023, 123, 4764–4794 CrossRef CAS PubMed
-
(a) E. Cigan, B. Eggbauer, J. H. Schrittwieser and W. Kroutil, RSC Adv., 2021, 11, 28223–28270 RSC
- F. Gomollón-Bel, Chem. Int., 2019, 41, 12–17 Search PubMed
-
(a) M. H. Aukland and B. List, Pure Appl. Chem., 2021, 93, 1371–1381 CrossRef CAS
-
(a) J. Zhang, S.-X. Lin, D.-J. Cheng, X.-Y. Liu and B. Tan, J. Am. Chem. Soc., 2015, 137, 14039–14042 CrossRef CAS PubMed
- For related reviews see:
(a) M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., Int. Ed., 2006, 45, 4732–4762 CrossRef CAS PubMed
-
(a) D. Zhao and K. Ding, ACS Catal., 2013, 3, 928–944 CrossRef CAS
- Examples in batch:
(a) S. Li, J. W. Zhang, X. L. Li, D. J. Cheng and B. Tan, J. Am. Chem. Soc., 2016, 138, 16561–16566 CrossRef CAS PubMed
-
(a) R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC
-
(a) A. Maestro, B. S. Nagy, S. B. Ötvös and C. O. Kappe, J. Org. Chem., 2023, 88, 15523–15529 CrossRef CAS PubMed
- L. Clot-Almenara, C. Rodríguez-Escrich, L. Osorio-Planes and M. A. Pericàs, ACS Catal., 2016, 6, 7647–7651 CrossRef CAS
-
(a) J. Lai, M. Fianchini and M. A. Pericàs, ACS Catal., 2020, 10, 14971–14983 CrossRef CAS
- G. K. Ingle, M. G. Mormino, L. Wojtas and J. C. Antilla, Org. Lett., 2011, 13, 4822–4825 CrossRef CAS PubMed
- Some examples:
(a) R. A. Unhale, N. Molleti, N. K. Rana, S. Dhanasekaran, S. Bhandary and V. K. Singh, Tetrahedron Lett., 2017, 58, 145–151 CrossRef CAS
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC
-
(a) D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization of PS-TRIP, and all the synthesized N,S-ketals, as well as the reactor design. See DOI: https://doi.org/10.1039/d4gc00019f |
| This journal is © The Royal Society of Chemistry 2024 |