
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-dimensional nanotube of a metal–organic framework boosts charge separation and photocatalytic hydrogen evolution from water: synthesis and underlying understanding†
Lifang
Liu‡
ab,
Yejun
Xiao‡
c,
Xiangyang
Guo
 a,
Wenjun
Fan
a,
Nengcong
Yang
ab,
Chunmei
Jia
a,
Shengye
Jin
c and
Fuxiang
Zhang
*a
a,
Wenjun
Fan
a,
Nengcong
Yang
ab,
Chunmei
Jia
a,
Shengye
Jin
c and
Fuxiang
Zhang
*a
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian National Laboratory for Clean Energy, Dalian 116023, China. E-mail: fxzhang@dicp.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cState Key Laboratory of Molecular Reaction Dynamics and Dynamics Research Center for Energy and Environmental Materials, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
First published on 31st January 2024
Abstract
One-dimensional (1D) nanostructured inorganic semiconductors have been extensively investigated for efficiently promoting their photocatalytic performances, but it still remains unclear for metal–organic framework (MOF)-based photocatalysis. Herein we present the synthesis 1D Mn-TBAPy MOF nanotubes (denoted as Mn-TBAPy-NT) and give the first demonstration of the marked ability of the 1D nanotube structure to promote charge separation of MOFs relative to that in the Mn-TBAPy single crystal (denoted as Mn-TBAPy-SC), a feature proposed to result from the effect of strain on the nanotubes. As specifically determined using transient absorption (TA) spectroscopy, Mn-TBAPy-NT exhibits a long-lived internal charge-separated (ICS) state (255.6 ns), longer than that for Mn-TBAPy-SC (4.6 ns) and a feature apparently responsible for its over 30-fold promoted hydrogen evolution with a rate of 203.5 μmol h−1 (ca. 10.2 mmol h−1 gcat−1) under visible light and a benchmark apparent quantum efficiency (AQE), of 11.7% at 420 ± 10 nm, among MOF-type photocatalysts. Our results open a new avenue for developing highly efficient MOF-based photocatalysts.
Broader contextMetal–organic framework (MOF) materials have been popularly investigated for their promising solar-to-chemical conversions, but their photocatalytic applications have been impeded by their poor chemical stability in water and sluggish charge separation as well as low efficiency. In this work, we report a water-stable phase of a Mn-TBAPy MOF with a special one-dimensional (1D) nanotube structure prepared by carrying out a simple solvothermal reaction with an emphasis on the solvent effect. This MOF was found to exhibit significantly better charge separation efficiency and photocatalytic performance than its corresponding single crystals. Compared to the layered single crystals, the 1D nanotube was demonstrated to exhibit a strain effect and shortened carrier transfer distance, leading to favorable charge separation and photocatalytic water splitting performance. Upon carrying out an optimization, an apparent quantum efficiency of 11.7% at 420 nm for photocatalytic water splitting to produce hydrogen was achieved—with this value being, according to our literature search, the highest among water-stable MOF-type photocatalysts. |
Introduction
Solar-driven water splitting to produce green hydrogen has been considered a promising approach to providing renewable energy production and addressing increasing energy and environmental issues.1–12 However, it is typically challenged by poor charge separation efficiency that has become a bottleneck severely delaying the development of photocatalysis. Various strategies have been developed to improve the charge separation efficiency of inorganic semiconductor photocatalysts, with examples of these strategies including surface/interface modification,13,14 crystal facet engineering,15–17 heterojunction construction6,18 or Z-scheme,19–21 nanostructures morphology control,8,22,23 piezoelectricity,24etc. Meanwhile, preparation of materials based on one-dimensional (1D) nanotube structures has garnered relatively extensive attention due to their unique electronic properties, enhanced light absorption, high electron mobility, enhanced surface area and catalytic properties.25–29Besides the inorganic semiconductor photocatalysts, metal–organic frameworks (MOFs) have been recently developed to convert solar energy into fuel production in consideration of their advantages including extraordinarily large surface areas, diverse compositions, ordered pores or channels, and designable structures.1,2,30–34 Although a variety of MOF-based photocatalysts have been constructed for splitting water into hydrogen under solar irradiation,3,35–45 they exist predominantly in the form of single crystals or solid blocks, which inevitably hinder charge separation.4 It has been shown that controllable synthesis of nanoscale or low-dimensional MOF photocatalysts can reduce their size or thickness, thereby improving charge separation and performance.46,47 Unfortunately, 1D MOF nanotubes for photocatalysis have been rarely researched, due to the synthesis of MOF nanotubes facing great challenges as a result of the directionality and fragility of their coordination bonds.
We previously developed a water-stable Mn-TBAPy MOF able to absorb a wide range of wavelengths of visible light for promising photocatalytic water splitting, with its structure containing the Mn ion and 1,3,6,8-tetrakis(p-benzoic acid) pyrene (H4TBAPy) ligand.48 Here we introduce the first synthesis of 1D Mn-TBAPy MOF nanotubes (Mn-TBAPy-NT), accomplished by carrying out a simple solvothermal reaction, and describe in detail our examination, based on 1D Mn-TBAPy-NT, of the properties of the special 1D nanostructure and its influence on charge separation as well as water splitting performance. Meanwhile, we also compare these results with those for the Mn-TBAPy single crystal (Mn-TBAPy-SC), synthesized as described in our previous report.48 Strikingly, the special nanotube structure of Mn-TBAPy-NT exhibits a remarkably prolonged lifetime of photogenerated carriers relative to Mn-TBAPy-SC, and is the predominant contributor to its excellent photocatalytic water splitting performance. The hydrogen evolution rate on the Mn-TBAPy-NT nanotube photocatalyst modified with a platinum cocatalyst is about 36 times higher than that of Mn-TBAPy-SC modified in a similar way. Its hydrogen evolution rate reaches ca. 10.2 mmol h−1 gcat−1 in the presence of ascorbic acid (AA) as a sacrificial agent under visible-light irradiation, with a corresponding AQE at 420 ± 10 nm measured to be 11.7%. To our knowledge, this AQE is higher than that of any other water-stable MOF-based photocatalyst reported so far.
Results and discussion
Fig. 1a shows the typical nanotube morphology of Mn-TBAPy-NT, with the uniform 1D tubular structure displaying a length of ca. 4–5 μm and diameter of ca. 40–60 nm. Inspection of a transmission electron microscopy (TEM) image (Fig. 1b) further confirms the nanotube morphology, where an apparent hollow nanotube with a thin wall is observed. The formation of these nanotubes is expected to benefit from its 2D layered structure (Fig. S1, ESI†) with large surface tension, as nanotubes tend to become frizzled during their formation process for most 1D inorganic nanomaterials.49–51 X-ray diffraction (XRD) patterns of the Mn-TBAPy-NT samples resemble those of the Mn-TBAPy-SC single crystal (Fig. 1c and Fig. S2, ESI†), and the effective coordination of the Mn–O bond confirmed in the FT-IR spectra of Mn-TBAPy-SC is similarly observed for the Mn-TBAPy-NT nanotubes (Fig. S3, ESI†), revealing its successful adoption of the crystalline phase. Interestingly, Mn-TBAPy-NT can bear aqueous solutions with a wide range of pH levels (Fig. 1c) with good water stability and absorbs a wide range of wavelengths of visible light with an edge at ca. 510 nm (Fig. 1d), similar to that for Mn-TBAPy-SC (Fig. S4, ESI†).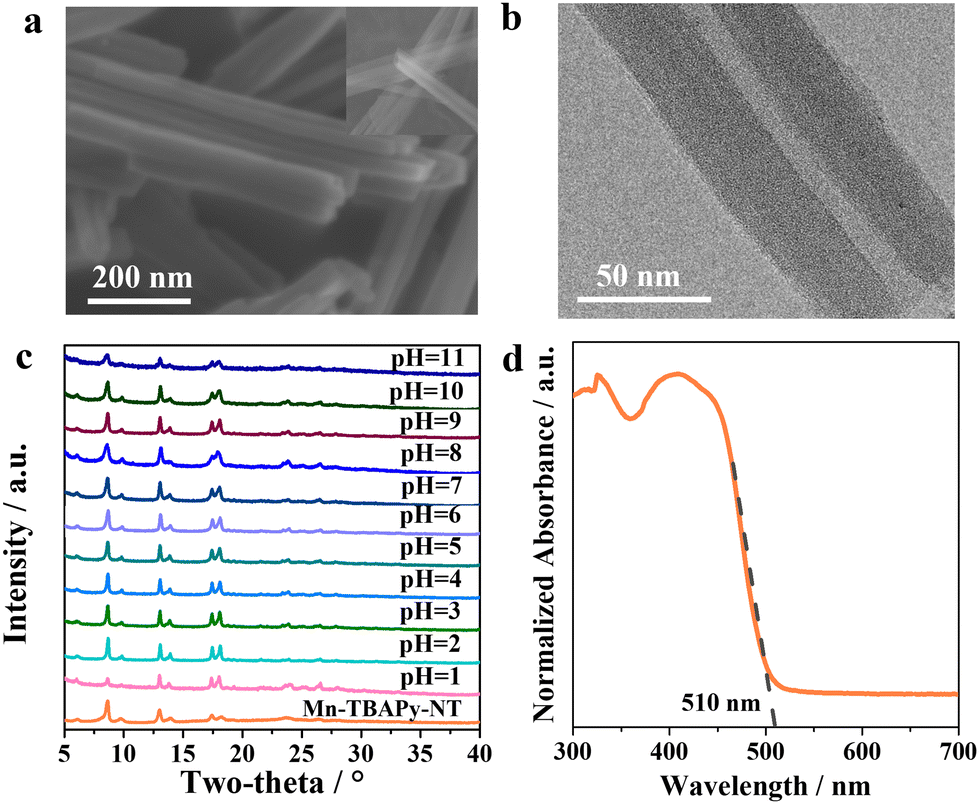 | ||
| Fig. 1 Structural characterizations of 1D Mn-TBAPy-NT nanotubes. (a) SEM and (b) TEM images of the nanotubes. (c) XRD patterns of various sample of the nanotubes soaked in aqueous solutions with different pH values each for 24 h. (d) UV-Vis diffuse reflectance spectrum of the nanotubes. | ||
In our experiments, we first investigated the activities of the MOF photocatalysts with regards to the photocatalytic water splitting to produce hydrogen.
Nanotubes of Mn-TBAPy-NT modified with a platinum cocatalyst (denoted as Pt/Mn-TBAPy-NT) exhibit significantly promoted photocatalytic water splitting to produce hydrogen from aqueous solutions containing ascorbic acid (AA) as a sacrificial agent under visible-light irradiation (Fig. 2a). Specifically, the average H2 evolution rate of Pt/Mn-TBAPy-NT (203.5 μmol h−1) is around 36 times higher than that of Pt/Mn-TBAPy-SC (5.6 μmol h−1), and subjecting Pt/Mn-TBAPy-NT to multiple time-course cycles demonstrate its good photochemical stability (Fig. 2b). The good photocatalytic stability is further confirmed by the lack of any obvious difference between the structures of the Pt/Mn-TBAPy-NT before and after the photocatalytic reaction (Fig. S5–S7, ESI†). Moreover, the measured apparent quantum efficiency (AQE) values of water splitting to produce hydrogen are strongly dependent on the irradiation wavelength, indicating that the water reduction reaction is driven by visible light (Fig. 2c). It is worth noting that the optimized AQE value for the Pt/Mn-TBAPy-NT reaches about 11.7% at 420 ± 10 nm, which is far higher than those of previous typical water-stable MOF-based photocatalysts (Fig. 2d and Table S1, ESI†).
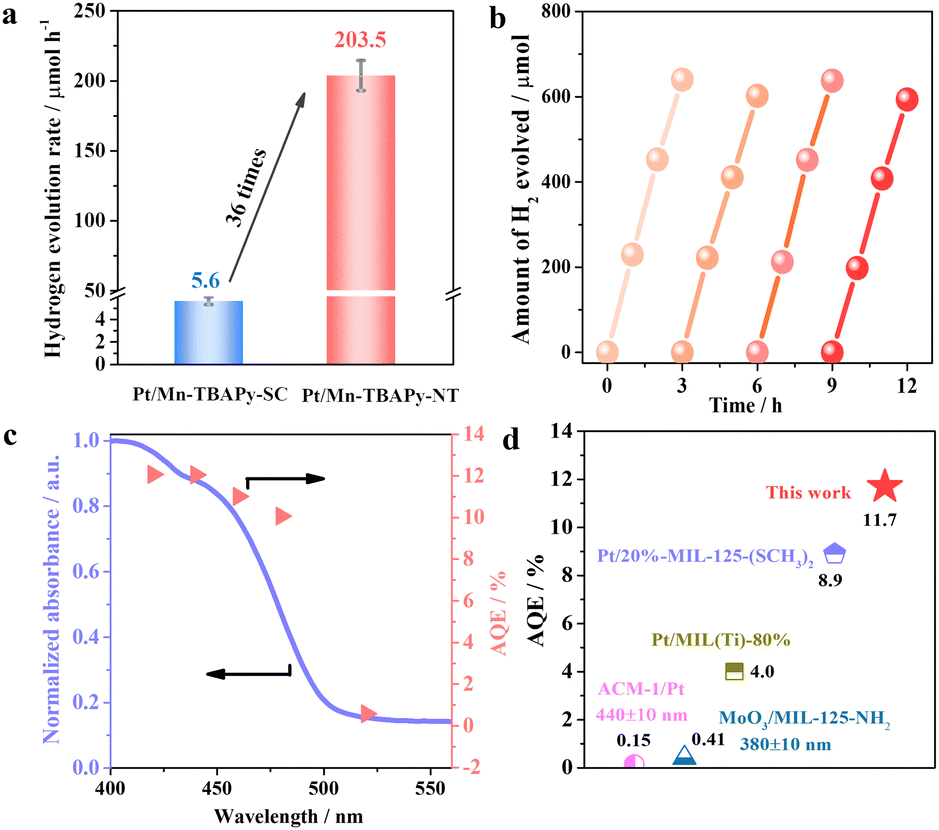 | ||
| Fig. 2 Photocatalytic hydrogen evolution performance from water over a Pt/Mn-TBAPy-NT sample under visible light irradiation. (a) Comparison of H2 evolution rates of Pt/Mn-TBAPy-NT and Pt/Mn-TBAPy-SC. (b) Curves of multiple cycles of activity as a function of reaction time. (c) Apparent quantum efficiencies (AQEs) under different irradiation wavelengths. (d) Comparison of AQE values for typical water-stable MOF-type photocatalysts under irradiation of light with a wavelength of 420 ± 10 nm unless indicated otherwise. | ||
It is generally known that photocatalytic activity is integrally determined by the efficiency of three basic processes, namely light absorption, charge separation and surface catalytic reactions.3,11 In order to gain insight into the extraordinary photocatalytic water splitting activity of Mn-TBAPy-NT nanotubes, the above basic processes are thus analyzed and discussed. First of all, as mentioned before, similar UV-Vis diffuse reflectance spectroscopy curves are observed for Mn-TBAPy-NT and Mn-TBAPy-SC (Fig. 1d and Fig. S4, ESI†). According to the Tauc plot (Fig. S8, ESI†) and Mott-Schottky (Fig. S9, ESI†) plot, both of them exhibit similar band gaps and conduction band (CB) values, demonstrating their similar light absorption efficiencies. Secondly, due to the similar amounts of Pt cocatalyst loaded on Mn-TBAPy-NT and Mn-TBAPy-SC (Fig. S10 and Table S2, ESI†), their surface catalytic efficiencies are expected to be similar, despite their distinct surface areas (Fig. S11, ESI†). However, it should be pointed out that even the H2 evolution rate normalized by the Pt content on Pt/Mn-TBAPy-NT is still about 35 times higher than that of Pt/Mn-TBAPy-SC (Fig. S12, ESI†). On the basis of the above results, we can reasonably deduce that the markedly promoted activity of water splitting to produce hydrogen on the Pt/Mn-TBAPy-NT nanotubes predominantly results from the 1D nanotube structure promoting charge separation.
To gain insight into the charge transfer processes in Mn-TBAPy-NT and Mn-TBAPy-SC photocatalysts, we describe the results first of femtosecond transient absorption (fs-TA) measured to track the photo-induced carrier dynamics. As given in Fig. S13 (ESI†), both samples show a negative bleach peak at 450 nm, assigned as the ground-state bleach (GSB), which is consistent with the UV-Vis absorption spectrum, further indicating that the 1D morphology does not obviously affect the light absorption (Fig. 1d and Fig. S4, ESI†). Besides the bleach peak, their fs-TA spectra similarly exhibit two broad photoinduced absorption (PIA) peaks, located at 609 nm and 690 nm for the Mn-TBAPy-NT sample or 630 nm and 730 nm for the Mn-TBAPy-SC sample, and which can be assigned to the internal charge-separated (ICS) state and the excitonic-state absorption (ESA) of excitons,52 respectively. The population of the ICS (signal amplitude) state is significantly larger in Mn-TBAPy-NT than in Mn-TBAPy-SC, which may be due to the differences between the microenvironments of their coordination bonds caused by the 1D nanotube structure. The result of fitting their kinetics processes by multiexponential functions are shown in Fig. 3a, b, and Table S3 (ESI†). It is worth noting that the internal charge-separated (ICS) state lifetime on the Mn-TBAPy-NT sample is obviously prolonged compared to Mn-TBAPy-SC. To accurately determine the carrier lifetime, nanosecond transient absorption (ns-TA) measurements on the nanosecond to microsecond time scale were also taken, and TA kinetics covering the entire fs-μs time window were obtained after combining these results with the results of fs-TA. The fitting results suggest the average lifetime of ICS state in Mn-TBAPy-NT to be 255.6 ns (Fig. 3c, d and Fig. S14, Table S4, ESI†), more than 50 times longer than that of Mn-TBAPy-SC (4.6 ns). The long-lived (255.6 ns) ICS state contributes to a markedly promoted photogenerated charge separation efficiency of the nanotube structure. A similar trend can be further confirmed by observation of obviously prolonged photoluminescence (PL) lifetimes on the Mn-TBAPy-NT nanotubes (Fig. 3e and Table S5, ESI†), which may be related to the enhanced delocalization of excitons in the ICS state.52
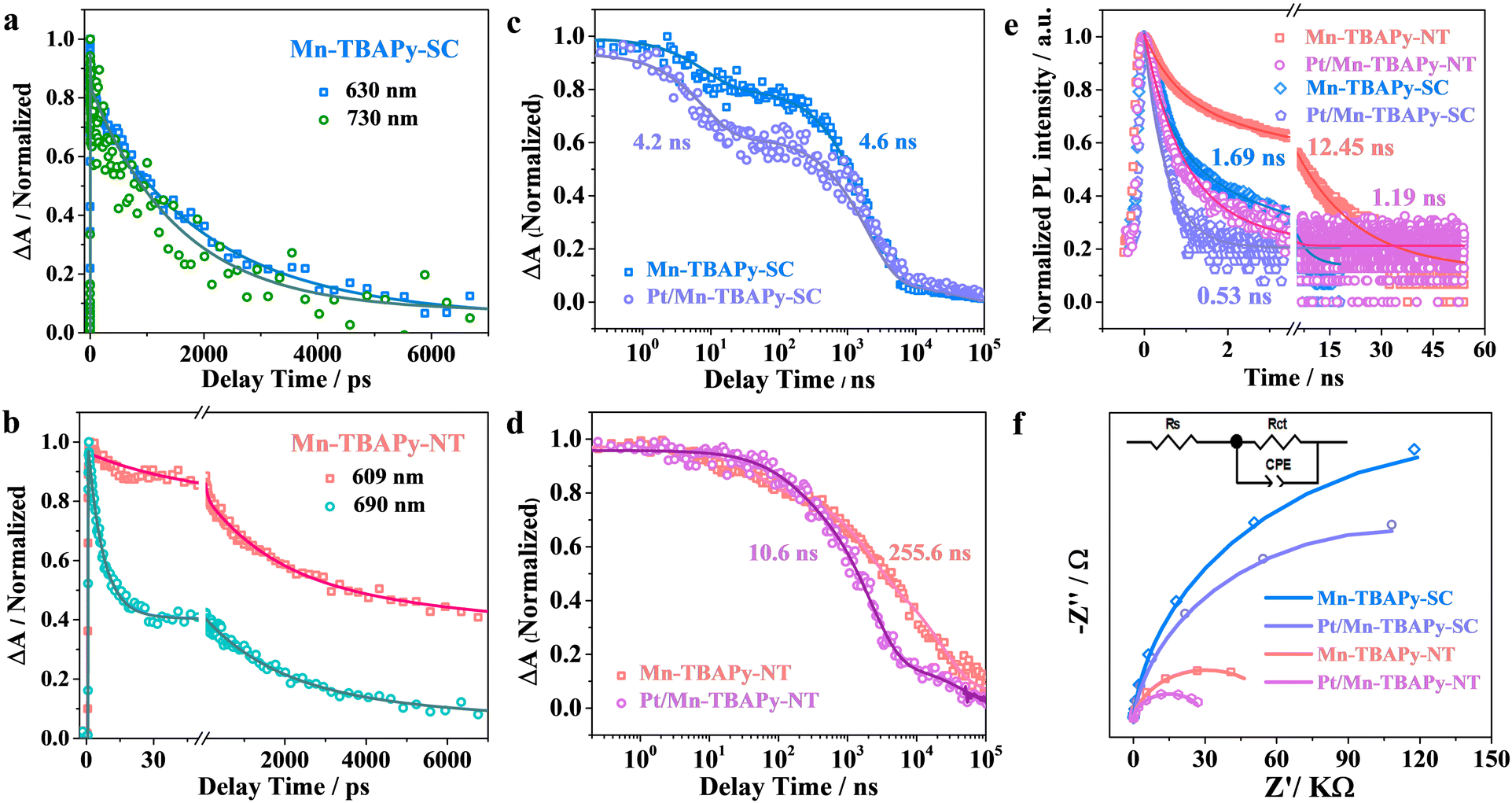 | ||
| Fig. 3 Comparison of charge separation properties over Mn-TBAPy-SC, Mn-TBAPy-NT, Pt/Mn-TBAPy-SC, and Pt/Mn-TBAPy-NT samples. (a) and (b) Normalized fs-TA kinetics probed at different indicated wavelengths. (c) and (d) Normalized TA decay kinetics on a sub-picosecond to microsecond time window for (c) Mn-TBAPy-SC and Pt/Mn-TBAPy-SC probed at 630 nm, and (d) Mn-TBAPy-NT and Pt/Mn-TBAPy-NT probed at 609 nm. (e) Time-resolved fluorescence decay kinetics, and (f) electrochemical impedance spectroscopy (EIS) Nyquist plots. | ||
Moreover, Pt-modified Mn-TBAPy-NT photocatalysts exhibit a markedly accelerated recovery of TA kinetics compared with Pt-modified Mn-TBAPy-SC according to the fs-TA and ns-TA experimental results (Fig. 3c, d and Fig. S15, S16, Tables S3, S4, ESI†), and a faster decay of carriers is also observed according to an analysis of their photoluminescence (PL) (Fig. 3e and Table S5, ESI†), demonstrating an additional promotion of the efficient extraction of electrons from the ICS state of Mn-TBAPy-NT. In short, the long lifetime of the ICS state is the key parameter for effective charge separation in the photocatalytic H2 evolution process, and incorporation of Pt cocatalyst further fosters charge separation.
To further understand the charge-transfer behavior of the 1D Mn-TBAPy-NT nanotubes, photoelectrochemical assays were executed. The 1D Mn-TBAPy-NT nanotubes exhibit significantly lower resistance to charge transfer than does Mn-TBAPy-SC (Fig. 3f and Table S6, ESI†), with a similar result found for the Pt-modified Mn-TBAPy-NT and Mn-TBAPy-SC samples, indicating a more effective charge-transfer process for the 1D Mn-TBAPy-NT nanotubes. Meanwhile, Hall measurements were taken to evaluate the carrier density in Mn-TBAPy-NT, showing it to be about 2 times higher than the carrier density of Mn-TBAPy-SC (Table S7, ESI†). All the above results taken together reveal a more efficient charge separation and enhanced charge transport for the 1D Mn-TBAPy-NT material than for Mn-TBAPy-SC, features expected to be quite responsible for the markedly promoted photocatalytic hydrogen evolution rate of 1D Mn-TBAPy-NT.
To understand the better charge separation displayed by the Mn-TBAPy-NT nanotubes than by Mn-TBAPy-SC, we further performed X-ray absorption fine structure spectroscopy (XAFS), specifically to investigate possible differences in electronic structure and atomic coordination between the Mn-TBAPy-NT nanotubes and Mn-TBAPy-SC. As shown in Fig. 4a, b and Fig. S17, Table S8 (ESI†), the Mn absorption edge shows a slightly positive shift from Mn-TBAPy-SC to Mn-TBAPy-NT according to the corresponding X-ray absorption near-edge spectroscopy (XANES) analysis (Fig. 4a). A similar peak shift of the Mn 2p binding energy is observed in their X-ray photoelectron spectra (XPS) as well (Fig. S18, ESI†), leading to a difference between their average Mn valences. The Fourier-transform curve of the Mn K-edge EXAFS data of the Mn-TBAPy-NT nanotubes shows a dominant broad Mn–O peak at 1.6 Å, and the Mn-TBAPy-NT nanotubes show a slightly decreased coordination number relative to Mn-TBAPy-SC (Fig. 4b and Table S8, ESI†). The slight difference between the samples may be attributed to the effect of strain on the 1D MOF nanotubes, specifically due to separate compression inside and stretching outside of the nanotubes, as widely observed during the process of rolling up 2D nanosheets of inorganic nanomaterials into 1D nanotubes.53,54 Furthermore, an obvious difference between Mn-TBAPy-NT and Mn-TBAPy-SC is observed in their Raman spectra peak positions relating to the Mn–O coordination stretching55 (Fig. 4c), and this difference demonstrates a change between the local Mn environment of Mn-TBAPy-NT and that of Mn-TBAPy-SC, as can be further supported by EPR results (Fig. S19, ESI†). It is worth noting that the strain effect has been recently demonstrated to favor charge separation and benefit photocatalysis,56,57 which is here also expected to be responsible for the significantly promoted charge separation and transfer observed in the nanotube. The significantly shortened distance for charge transfer due to the thinness of the nanotubes may provide another benefit for the photocatalysis.
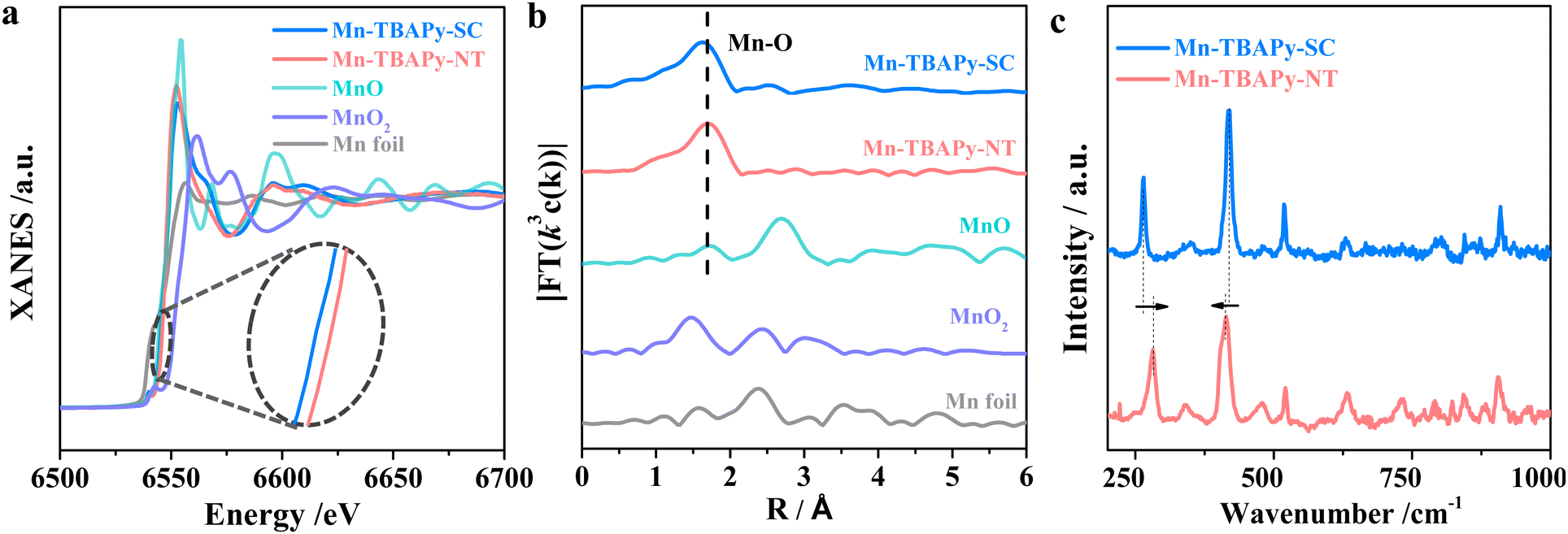 | ||
| Fig. 4 Characterizations of the effect of strain on the 1D Mn-TBAPy-NT nanotubes with a Mn-TBAPy-SC single crystal as a reference: (a) Mn K-edge XANES of the MOFs and reference samples (inset: zoomed-in image), (b) Fourier-transform curves of the Mn K-edge EXAFS spectra of the indicated MOFs and reference samples, and (c) Raman spectra of Mn-TBAPy-NT and Mn-TBAPy-SC measured with 785-nm-wavelength light excitation. | ||
Conclusions
In summary, we for the first time synthesized 1D Mn-TBAPy-NT MOF nanotubes displaying good stability in water and able to absorb a wide range of wavelengths of visible light for promising photocatalytic water splitting, based on which the AQE of hydrogen evolution on the optimized platinum-modified Mn-TBAPy-NT photocatalyst reaches 11.7% at 420 ± 10 nm, higher than that of the state-of-the-art water-stable MOF-based photocatalysts. The strain effect and thinness of the nanotube wall originating from the special 1D nanotube structure of Mn-TBAPy-NT are proposed to favor charge separation, leading to the observation of a long-lived internal charge-separated (ICS) state, and hence highly responsible for the significantly promoted hydrogen evolution relative to that of the single crystal. This work not only proves the feasibility of 1D nanotubes in promoting photocatalytic performances, but also provides an alternative strategy of promoting charge separation of MOF-type photocatalysts.Author contributions
F. Z. conceived and designed the experiments. L. L. carried out preparations, activity test, catalyst characterizations and wrote the first draft. Y. X. carried out the TA measurements and analysis. W. F. carried out the XAFS measurements and analysis. N. Y. and C. J. discussed the synthesis of nanotubes. S. J. assisted the TA measurements and analysis. Y. X. X. G. and F. Z. directed the work and revised the manuscript. L. L. and Y. X. contributed equally to this work. All authors discussed the results and contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22332005, 22261160369), International Partnership Program of Chinese Academy of Sciences (121421KYSB20190025), Dalian supports high-level talent innovation and entrepreneurship projects (2020RD06), Natural Science Foundation of Liaoning Province (2023-MS-002). The authors gratefully acknowledge the staff at the 1W1B beamline of the Beijing Synchrotron Radiation Facility (BSRF), Beijing, China, for providing beam time.Notes and references
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- S. Navalón, A. Dhakshinamoorthy, M. Álvaro, B. Ferrer and H. García, Chem. Rev., 2022, 123, 445–490 CrossRef PubMed.
- K. Sun, Y. Qian and H.-L. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202217565 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- N. Kornienko, J. Z. Zhang, K. K. Sakimoto, P. Yang and E. Reisner, Nat. Nanotechnol., 2018, 13, 890–899 CrossRef CAS PubMed.
- T. Hisatomi and K. Domen, Nat. Catal., 2019, 2, 387–399 CrossRef CAS.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- Y. Guo, Q. Zhou, B. Zhu, C. Y. Tang and Y. Zhu, EES Catal., 2023, 1, 333–352 RSC.
- P. Strobel, M. Riedel, J. Ristein and L. Ley, Nature, 2004, 430, 439–441 CrossRef CAS PubMed.
- X. Tao, Y. Gao, S. Wang, X. Wang, Y. Liu, Y. Zhao, F. Fan, M. Dupuis, R. Li and C. Li, Adv. Energy Mater., 2019, 9, 1803951 CrossRef.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- R. Li, F. Zhang, D. Wang, J. Yang, M. Li, J. Zhu, X. Zhou, H. Han and C. Li, Nat. Commun., 2013, 4, 1432 CrossRef PubMed.
- Y. Qi, J. Zhang, Y. Kong, Y. Zhao, S. Chen, D. Li, W. Liu, Y. Chen, T. Xie, J. Cui, C. Li, K. Domen and F. Zhang, Nat. Commun., 2022, 13, 484 CrossRef CAS PubMed.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS PubMed.
- J. Chen, Y. Xiao, N. Wang, X. Kang, D. Wang, C. Wang, J. Liu, Y. Jiang and H. Fu, Sci. China Mater., 2023, 66, 3165–3175 CrossRef CAS.
- P. Zhang, T. Wang, X. Chang and J. Gong, Acc. Chem. Res., 2016, 49, 911–921 CrossRef CAS PubMed.
- B. Dong, J. Cui, Y. Qi and F. Zhang, Adv. Mater., 2021, 33, 2004697 CrossRef CAS PubMed.
- W. Liu, P. Fu, Y. Zhang, H. Xu, H. Wang and M. Xing, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2218813120 CrossRef CAS PubMed.
- T. Zhai, X. Fang, L. Li, Y. Bando and D. Golberg, Nanoscale, 2010, 2, 168–187 RSC.
- Y. Li, X.-Y. Yang, Y. Feng, Z.-Y. Yuan and B.-L. Su, Crit. Rev. Solid State Mater. Sci., 2012, 37, 1–74 CrossRef.
- S. Liu, Z.-R. Tang, Y. Sun, J. C. Colmenares and Y.-J. Xu, Chem. Soc. Rev., 2015, 44, 5053–5075 RSC.
- M. Ge, Q. Li, C. Cao, J. Huang, S. Li, S. Zhang, Z. Chen, K. Zhang, S. S. Al-Deyab and Y. Lai, Adv. Sci., 2017, 4, 1600152 CrossRef PubMed.
- N. Yang, R. Chen, C. Ni, D. Li, Q. Sun, L. Liu, Y. Qi, S. Jin, X. Wang, F. Fan, C. Li and F. Zhang, J. Energy Chem., 2022, 72, 326–332 CrossRef CAS.
- J. Gao, Q. Huang, Y. Wu, Y.-Q. Lan and B. Chen, Adv. Sustainable Syst., 2021, 2, 2100033 CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974 CrossRef CAS PubMed.
- S.-L. Li and Q. Xu, Energy Environ. Sci., 2013, 6, 1656–1683 RSC.
- H.-C. J. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- S. Wang and X. Wang, Small, 2015, 11, 3097–3112 CrossRef CAS PubMed.
- J. Zhu, P.-Z. Li, W. Guo, Y. Zhao and R. Zou, Chem. Soc. Rev., 2018, 359, 80–101 CAS.
- Y. Xiao, Y. Qi, X. Wang, X. Wang, F. Zhang and C. Li, Adv. Mater., 2018, 30, 1803401 CrossRef PubMed.
- G. Lan, Y.-Y. Zhu, S. S. Veroneau, Z. Xu, D. Micheroni and W. Lin, J. Am. Chem. Soc., 2018, 140, 5326–5329 CrossRef CAS PubMed.
- Y. Shi, A.-F. Yang, C.-S. Cao and B. Zhao, Coord. Chem. Rev., 2019, 390, 50–75 CrossRef CAS.
- Y. Song, Z. Li, Y. Zhu, X. Feng, J. S. Chen, M. Kaufmann, C. Wang and W. Lin, J. Am. Chem. Soc., 2019, 141, 12219–12223 CrossRef CAS PubMed.
- A. Cadiau, N. Kolobov, S. Srinivasan, M. G. Goesten, H. Haspel, A. V. Bavykina, M. R. Tchalala, P. Maity, A. Goryachev, A. S. Poryvaev, M. Eddaoudi, M. V. Fedin, O. F. Mohammed and J. Gascon, Angew. Chem., Int. Ed., 2020, 59, 13468–13472 CrossRef CAS PubMed.
- X. Feng, Y. Pi, Y. Song, C. Brzezinski, Z. Xu, Z. Li and W. Lin, J. Am. Chem. Soc., 2020, 142, 690–695 CrossRef CAS PubMed.
- L. Liu, S. Du, X. Guo, Y. Xiao, Z. Yin, N. Yang, Y. Bao, X. Zhu, S. Jin, Z. Feng and F. Zhang, J. Am. Chem. Soc., 2022, 144, 2747–2754 CrossRef CAS PubMed.
- S. Naghdi, A. Cherevan, A. Giesriegl, R. Guillet-Nicolas, S. Biswas, T. Gupta, J. Wang, T. Haunold, B. C. Bayer, G. Rupprechter, M. C. Toroker, F. Kleitz and D. Eder, Nat. Commun., 2022, 13, 282 CrossRef CAS PubMed.
- Y. Liu, C.-H. Liu, T. Debnath, Y. Wang, D. Pohl, L. V. Besteiro, D. M. Meira, S. Huang, F. Yang, B. Rellinghaus, M. Chaker, D. F. Perepichka and D. Ma, Nat. Commun., 2023, 14, 541 CrossRef CAS PubMed.
- B. Xia, Y. Yang, Y. Zhang, Y. Xia, M. Jaroniec, J. Yu, J. Ran and S.-Z. Qiao, Chem. Eng. J., 2022, 431, 133944 CrossRef CAS.
- T. He, B. Ni, S. Zhang, Y. Gong, H. Wang, L. Gu, J. Zhuang, W. Hu and X. Wang, Small, 2018, 14, 1703929 CrossRef PubMed.
- Q. Zuo, T. Liu, C. Chen, Y. Ji, X. Gong, Y. Mai and Y. Zhou, Angew. Chem., Int. Ed., 2019, 58, 10198–10203 CrossRef CAS PubMed.
- Y. Xiao, X. Guo, T. Yang, J. Liu, X. Liu, Y. Xiao, L. Liu, T. Liu, S. Ye, J. Jiang, F. Zhang and C. Li, Sci. China: Chem., 2020, 63, 1756–1760 CrossRef CAS.
- X. Wang and Y. D. Li, Chem. – Eur. J., 2003, 9, 5627–5635 CrossRef CAS PubMed.
- X. Wang, X. M. Sun, D. P. Yu, B. S. Zou and Y. D. Li, Adv. Mater., 2003, 15, 1442–1445 CrossRef CAS.
- B. Zhao, Z. Wan, Y. Liu, J. Xu, X. Yang, D. Shen, Z. Zhang, C. Guo, Q. Qian, J. Li, R. Wu, Z. Lin, X. Yan, B. Li, Z. Zhang, H. Ma, B. Li, X. Chen, Y. Qiao, I. Shakir, Z. Almutairi, F. Wei, Y. Zhang, X. Pan, Y. Huang, Y. Ping, X. Duan and X. Duan, Nature, 2021, 591, 385–390 CrossRef CAS PubMed.
- Y. Xiao, J. Liu, J. Leng, Z. Yin, Y. Yin, F. Zhang, C. Sun and S. Jin, ACS Energy Lett., 2022, 7, 2323–2330 CrossRef CAS.
- X. Cui, Z. Kong, E. Gao, D. Huang, Y. Hao, H. Shen, C.-A. Di, Z. Xu, J. Zheng and D. Zhu, Nat. Commun., 2018, 9, 385–390 CrossRef PubMed.
- J. An, Y. Peng and Q. Zhang, J. Membr. Sci., 2018, 53, 15530–15540 CAS.
- I. Strauss, A. Mundstock, D. Hinrichs, R. Himstedt, A. Knebel, C. Reinhardt, D. Dorfs and J. Caro, Angew. Chem., Int. Ed., 2018, 57, 7434–7439 CrossRef CAS PubMed.
- Y. Miao, Y. Zhao, S. Zhang, R. Shi and T. Zhang, Adv. Mater., 2022, 34, 2200868 CrossRef CAS PubMed.
- B. Zhou, S. Xu, L. Wu, M. Li, Y. Chong, Y. Qiu, G. Chen, Y. Zhao, C. Feng, D. Ye and K. Yan, Small, 2023, 19, 2302058 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00007b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |