
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organoboron catalysis for direct amide/peptide bond formation
Masayoshi
Koshizuka†
 a,
Naoya
Takahashi†
a and
Naoyuki
Shimada
*b
a,
Naoya
Takahashi†
a and
Naoyuki
Shimada
*b
aLaboratory of Organic Chemistry for Drug Development and Medical Research Laboratories, Department of Pharmaceutical Sciences, Kitasato University, 5-9-1 Shirokane, Minatao-ku, Tokyo 108-8641, Japan
bLaboratory of Organic Chemistry for Molecular Transformations, Department of Chemistry and the Institute of Natural Sciences, Nihon University, 3-25-40 Sakurajosui, Setagaya-ku, Tokyo 156-8550, Japan. E-mail: shimada.naoyuki@nihon-u.ac.jp
First published on 28th August 2024
Abstract
Amides and peptides are ubiquitous functional groups found in several natural and artificial materials, and they are essential for the advancement of life and material sciences. In particular, their relevance in clinical medicine and drug discovery has increased in recent years. Dehydrative condensation of readily available carboxylic acids with amines is the most “direct” method for amide synthesis; however, this methodology generally requires a stoichiometric amount of condensation agent (coupling reagent). Catalytic direct dehydrative amidation has become an “ideal” methodology for synthesizing amides from the perspective of green chemistry, with water as the only byproduct in principle, high atom efficiency, environmentally friendly, energy saving, and safety. Conversely, organoboron compounds, such as boronic acids, which are widely used in various industries as coupling reagents for Suzuki–Miyaura cross-coupling reactions or pharmaceutical structures, are environmentally friendly molecules that have low toxicity and are easy to handle. Based on the chemical properties of organoboron compounds, they have potential Lewis acidity and the ability to form reversible covalent bonds with dehydration, making them attractive as catalysts. This review explores studies on the development of direct dehydrative amide/peptide bond formation reactions from carboxylic acids using organoboron catalysis, classifying them based on chemical bonding and catalysis over approximately 25 years, from the early developmental days to 2023.
 Masayoshi Koshizuka | Masayoshi Koshizuka was born in Tochigi, Japan. He received his PhD in 2021 from Kitasato University under the supervision of Prof. Naoyuki Shimada and Prof. Kazuishi Makino. Then, he joined the Department of Pharmaceutical Sciences, Kitasato University as an Assistant Professor. His current research interests are focused on the development of phosphorus catalysis and its synthetic applications. |
 Naoya Takahashi | Naoya Takahashi was born in Chiba, Japan. He received his PhD in 2024 from Kitasato University under the supervision of Prof. Naoyuki Shimada and Prof. Kazuishi Makino. He is currently working as a postdoctoral research associate at the University of Hawaii, Manoa, under the guidance of Prof. Marcus A. Tius. His current project focuses on the total synthesis of natural products. |
 Naoyuki Shimada | Naoyuki Shimada was born in Tokyo, Japan. He obtained his B.Eng, and M.Eng. from Nihon University under the guidance of Prof. Shoichi Shimizu. He received his PhD in 2007 from the Hokkaido University under the supervision of Prof. Shunichi Hashimoto. Then, he joined Prof. Tius’ Research Group at the University of Hawaii as a postdoctoral research associate. He returned to Japan and worked as an Assistant Professor at Kitasato University. He moved to the Department of Chemistry, Nihon University, as an Associate Professor in 2022. His research interests include the development of organoboron catalysis and its synthetic applications. |
1. Introduction
Amide bond is an essential chemical linkage found in several natural and non-natural materials, such as pharmaceutical, agrochemical, and functional materials.1 It is particularly useful as a pharmaceutical substructure, with more than 40% of the top-selling 200 small molecule drugs in 2023 containing at least one amide bond.2 Peptide bond, which is the amide bond between amino acids, is found in not only biomolecules of the human body but also several pharmaceuticals, functional materials, cosmetics, and foods. Recent drug discovery has shifted to medium-molecule drugs with molecular weights of approximately 1000–5000, combining the advantages of small-molecule and antibody drugs. Particularly, peptides have attracted considerable attention for next-generation modalities; more than 260 peptides progressed into human clinical trials in 2018.3 The development of an efficient synthetic methodology for amides is essential because amide bond formation reactions account for 50% of chemical reaction processes in drug discovery and development.4 Accordingly, “amide formation avoiding poor atom economy reagents” was voted as the top priority for improvement at the ACS Green Chemistry Institute Pharmaceutical Roundtable (GCIPR) in 2006.5 In 2018, ACS GCIPR identified “general methods for catalytic/sustainable (direct) amide or peptide bond formation” as a key green chemistry research area.6Although amide bond formation has been reported using various starting materials, the most straightforward synthesis involves dehydrative amide condensation from readily available carboxylic acids with amines. However, extremely high temperatures (>160 °C) are generally required to obtain amides from carboxylic acids and amines without an activating agent.7 This is because ammonium salt formation through acid–base interaction is significantly more favorable than dehydration condensation, particularly for highly acidic carboxylic acids such as aromatic carboxylic acids.8 Therefore, to form an amide bond from a carboxylic acid and an amine, it is generally necessary to use a condensation agent (coupling reagent). Although several excellent condensation agents have been developed, there is still room for improvement.9 Particularly, the activation of carboxylic acids requires a stoichiometric amount of a condensation agent, which poses several challenges, including low atom efficiency, high cost, a complicated workup and purification process, and frequent racemization. Regarding peptide bond formation, Merrifield10 established a solid-phase peptide synthesis (SPPS) method using carbonate-based protecting groups; however, the issue of racemization due to the use of stoichiometric amounts of condensation agents persists.
The “ideal” methodology for synthesizing amides from carboxylic acids and amines involves a direct dehydrative amide condensation reaction using catalysts. Direct dehydrative amide condensation reactions using transition metal complex catalysts such as Ti, Zr, Hf, Nb, and polyoxometalate11 have been reported.12 A noteworthy recent study is Yamamoto's report,13 which presents the use of boron catalysts, namely aromatic boronic acids, and represents a milestone in this field. Boron compounds exhibit Lewis acidity due to the vacant p-orbital and thus induce electrophilic activation through interactions with carbonyl compounds that exhibit Lewis basicity. In addition, boron compounds with a B–OH bond react with the hydroxyl functional groups of alcohols or carboxylic acids to form reversible covalent bonds through dehydration.14–16 Both chemical properties exhibited by boron compounds facilitate the catalytic electrophilic activation of carboxylic acids. Furthermore, reactions using carboxylic acids with hydroxyl groups can be applied to substrate-directed molecular transformation.17
Several excellent reviews have been published focusing on general amide synthesis,1 catalytic amide bond formations,12 and organoboron catalysis.14 In this article, we review studies on organoboron catalysis for direct amide/peptide bond formation using dehydrative condensation.
2. Catalytic direct amide bond formation from carboxylic acids
2.1. Direct amide bond formation using boronic acid catalysis
As a pioneering report on catalytic amidation using organoboron compounds, in 1996, Yamamoto and co-workers13 reported the first catalytic amidation using arylboronic acids as a catalyst. Arylboronic acids with electron-withdrawing substituents at the benzene rings are water-, acid-, and base-tolerant Lewis acids. They are also thermally stable and easy to handle in air. Yamamoto13 postulated that the strong Lewis acidity of boron enhances the formation of (acyloxy)boron intermediates and their reactivity with amines. In this context, the catalytic activities of a series of electron-deficient aryl boronic acids for amidation reactions were investigated. As a result, the catalytic amount of boronic acids 4–9 promoted the amide condensation reactions of 4-phenylbutylic acid (1) and 3,5-dimethylpiperidine (2) in toluene under reflux conditions in the presence of molecular sieves (MS) 4A to obtain amide 3 (Scheme 1).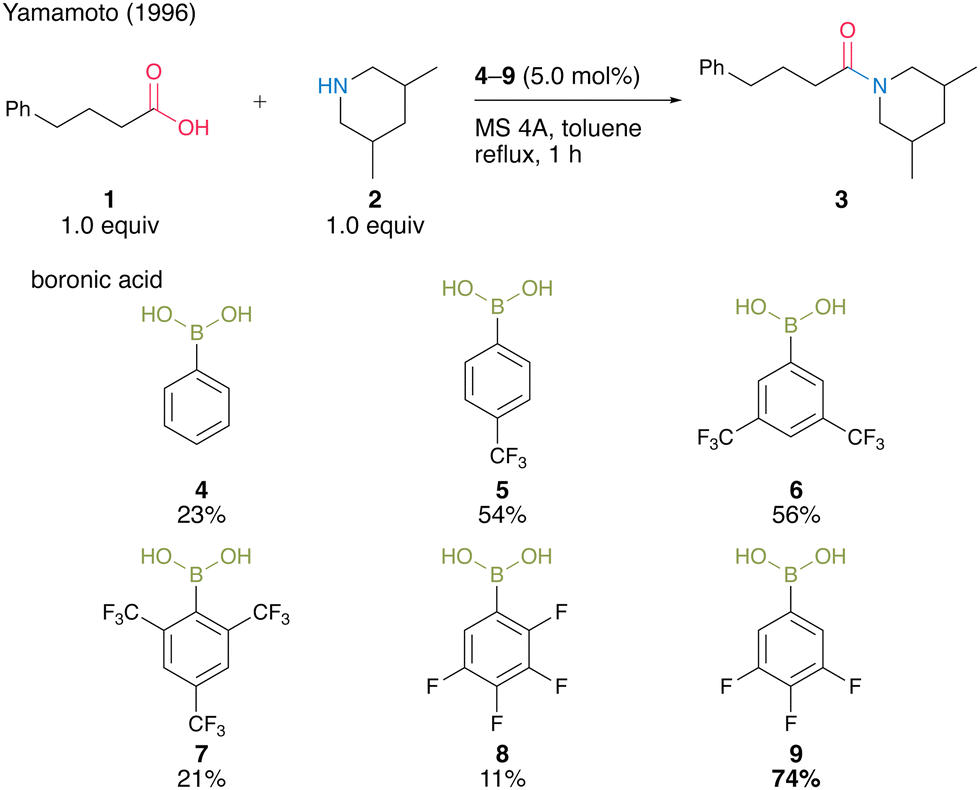 | ||
| Scheme 1 Electron-deficient boronic acid-catalyzed amidation reported by Yamamoto.13 | ||
Among them, 3,4,5-trifluorobenzeneboronic acid (9) was proven to be an effective catalyst for catalytic amidation. A remarkable feature of this catalysis is that the amide can be obtained with a satisfactory level of purity after only a simple aqueous workup. Yamamoto13 proposed a mechanistic insight of amidation reactions using boronic acid catalysts (Fig. 1). First, boronic acid or boroxine 10, which is a dehydrated trimer of boronic acid, reacts with carboxylic acid to form (acyloxy)boron intermediate 11, which is characterized by 1H NMR and IR analysis. In intermediate 11, the electrophilic activation of carboxylic acid was doubly induced by the Lewis acidic boron and intramolecular hydrogen bonding. Then, 11 reacts with an amine to yield the corresponding amide with water as a byproduct and regenerates the catalyst.
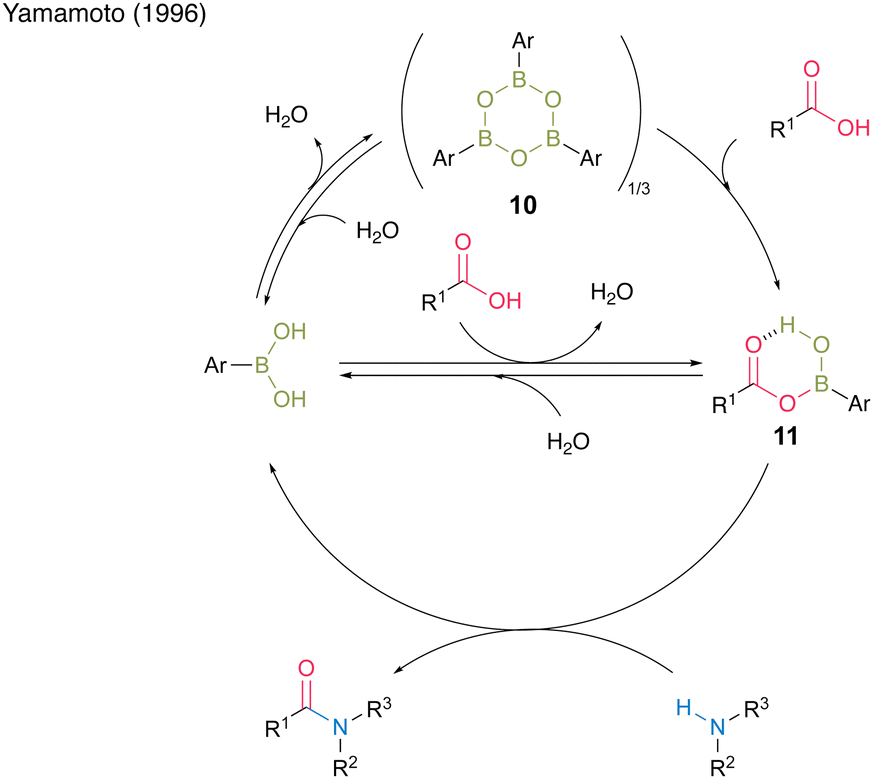 | ||
| Fig. 1 Proposed amidation mechanism reported by Yamamoto.13 | ||
Because the formation of (acyloxy)boron is an equilibrium reaction, the removal of water is crucial. Following Yamamoto's report,13 several scholars subsequently developed their original aromatic boronic acid catalysts to employ in catalytic amidation.
Whiting and co-workers8a,b reported catalytic amidation reactions using N,N-diisopropylbenzylaminoboronic acid 12 and its derivatives 13–15 (Scheme 2). They revealed that the electron density of the aromatic rings affected the catalytic activity and amide formation rate via kinetic studies. Electron-rich boronic acid 13 exhibited nearly half the catalytic activity of electron-deficient boronic acid 14 for the reaction of 4-pheny butyric acid and benzylamine. Furthermore, an increase in the reaction rate was observed for the para-trifluoromethyl-substituted aromatic boronic acid 15 compared with an unsubstituted boronic acid 12. Under optimized azeotropic reflux conditions in toluene or fluorobenzene, unsubstituted boronic acid 12 promoted the catalytic amidation of benzoic acid or aniline, which is a difficult substrate for amidation, to produce the corresponding amides 16–18 in good to high yields.
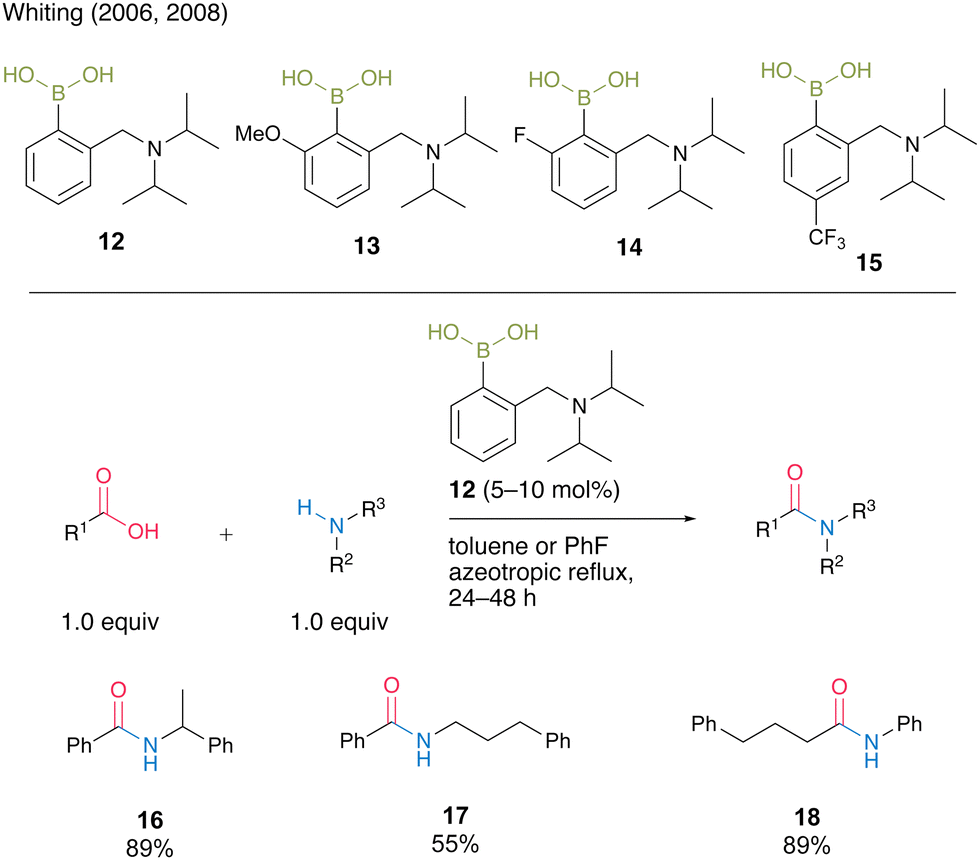 | ||
| Scheme 2 N,N-Diisopropylbenzylaminoboronic acid-catalyzed amidation reported by Whiting.8a,b | ||
In 2008, Whiting's group18 reported asymmetric direct amide synthesis by kinetic amine resolution using chiral ferrocene-derived aminoboronic acid. Using chiral ferrocene 22 in the reaction of benzoic acid (19) with racemic α-alkylamine 20, a catalytic activity similar to that of 12, although by asymmetric induction, was observed. Meanwhile, 23, in which the amine moiety was replaced by N-n-butylbenzimidazole, yielded amide 21 with 41% ee (Scheme 3). The relevance of the mild basicity of benzimidazole in asymmetric induction was suggested because catalyst 23 containing benzimidazole may preferentially select one amine enantiomer that reacts with activated diacyl boronates via hydrogen bonding. By contrast, the more basic N,N-diisopropylamino group of 22 promotes ammonium formation by deprotonation rather than hydrogen bonding, generating amides without inducing chirality. This Whiting's finding is the first successful boronic acid-catalyzed asymmetric dehydrative amidation. However, little progress has been made in this research field.
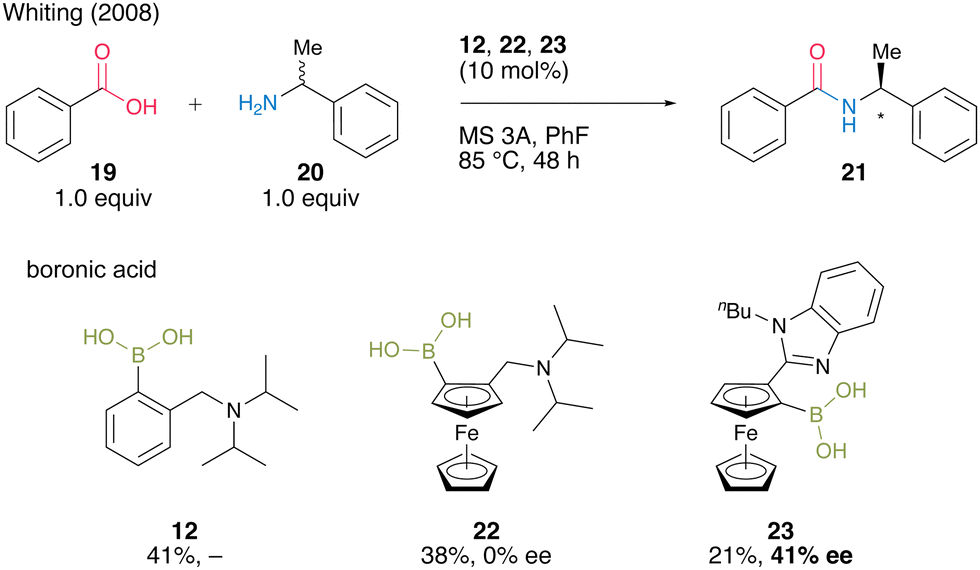 | ||
| Scheme 3 Asymmetric direct amide synthesis via kinetic amine resolution reported by Whiting.18 | ||
In the same year, Hall and co-workers19 reported that the halogen atom at the ortho-position drastically enhances the catalytic activity of aromatic boronic acids in dehydrative amidation. The catalytic activities of functionalized arylboronic acids, including 9, 27–30, in the reaction of phenylacetic acid (24) with benzylamine (25) in the presence of activated MS 4A were surveyed (Scheme 4). As a result, 2-iodophenylboronic acid (30) served as the best catalyst to produce the corresponding amide 26 in the highest yield, although para isomers or o,o′-dihaloarylboronic acids did not exhibit sufficient catalytic activity. These results indicate that an ortho-mono substituent is necessary for boronic acids to achieve satisfactory levels of catalytic activity. This catalysis is advantageous because the reaction proceeded even at room temperature.
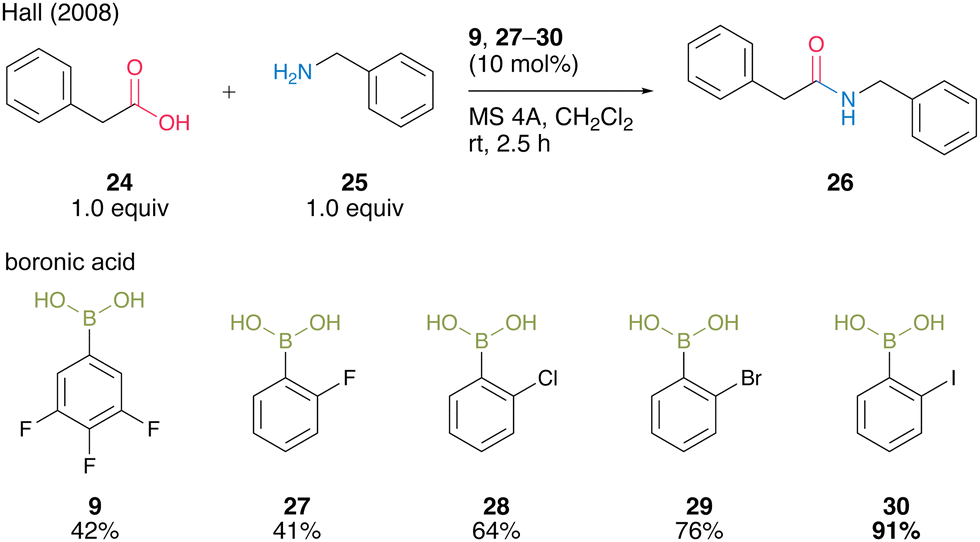 | ||
| Scheme 4 ortho-Halophenylboronic acid-catalyzed amidation reported by Hall.19 | ||
In 2012, the same group developed 5-methoxy-4-iodophenylboronic acid (MIBA, 37), which was designed based on 2-iodophenylboronic acid (30), and revealed that an electron-donating group introduction at the para position of the iodide enhanced the catalytic activity for dehydrative amidation at room temperature (Scheme 5).20 The methoxy group enhanced the electron density of para-iodine, resulting in high catalytic activity. These results indicate that the catalytic activity of aromatic boronic acids is influenced not only by the inductive effect but also by other effects. The proposed catalytic cycle based on density functional theory (DFT) calculations by Marcelli21 is shown in Fig. 2. First, intermediate 39 was formed by the nucleophilic addition of amine to acyl boronate intermediate 38, which was formed by boronic and carboxylic acid reactions. Intermediate 39 exhibits the transition state shown in 40, which promotes the dehydration step of amide bond formation via hydrogen bonding between the electron-rich iodine atom and water molecules and is highly catalytically active.
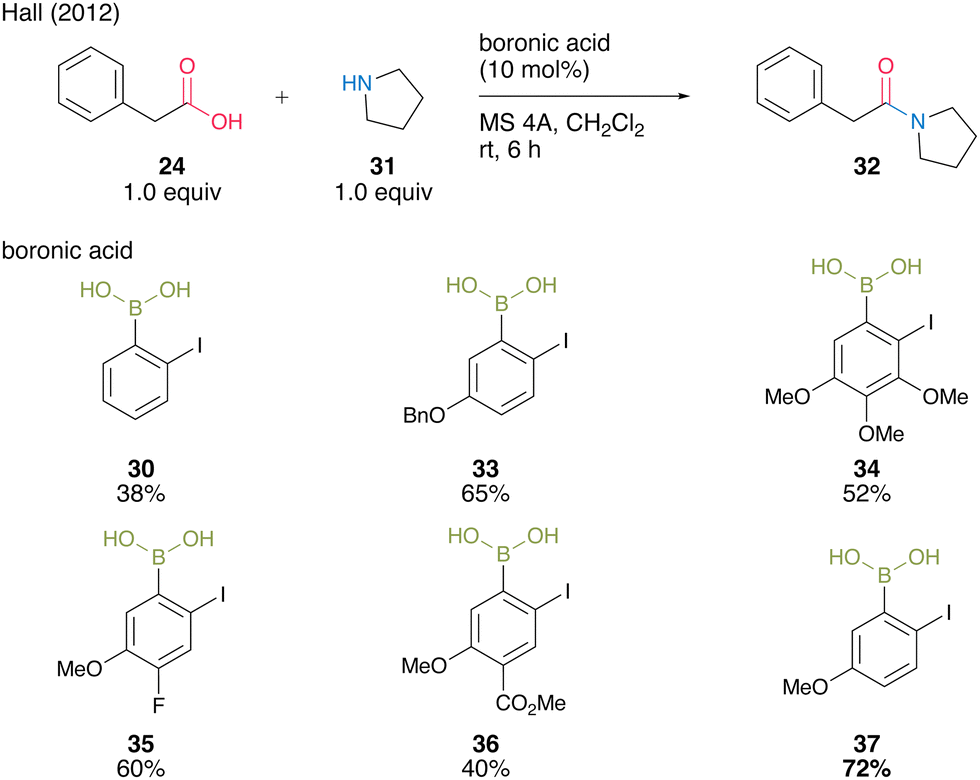 | ||
| Scheme 5 MIBA-catalyzed amidation reported by Hall.20 | ||
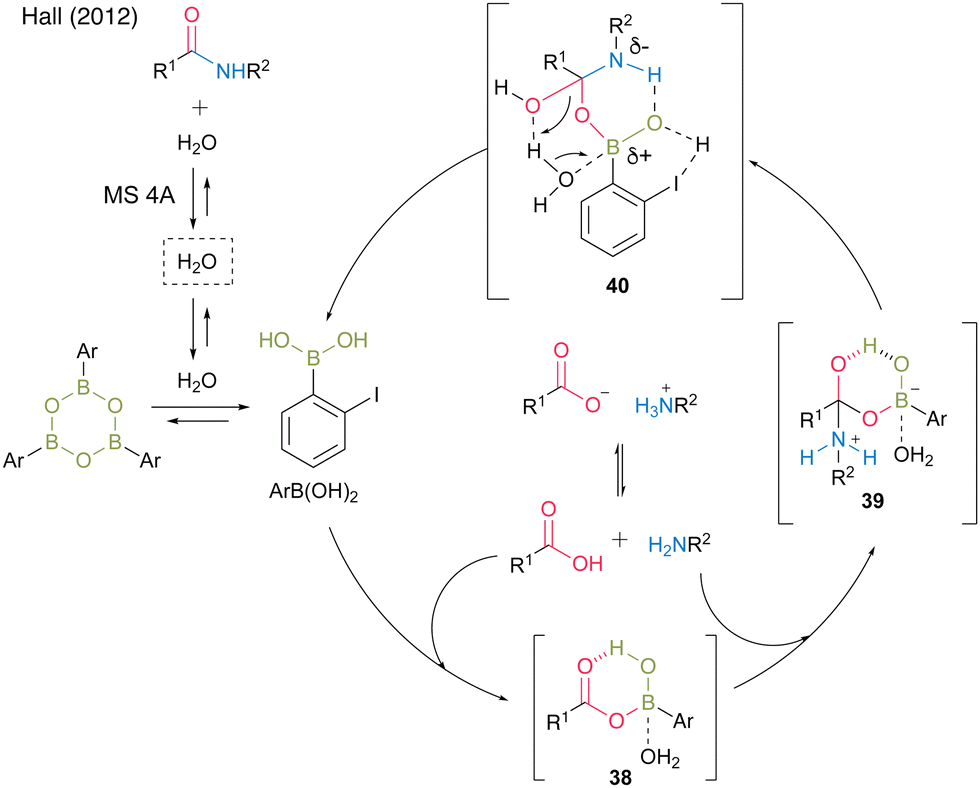 | ||
| Fig. 2 Proposed amidation mechanism reported by Hall.20 | ||
The catalytic activity was assumed to be high by accelerating the dehydration step in amide bond formation via hydrogen bonds between electron-rich iodine atoms and water molecules. As an alternative reaction mechanism, Hall proposed a plausible mechanism via a transition state in which a water molecule in a transition state 40 is replaced by a carboxylic acid. In this reaction, the use of powdered MS 4A dried at 250 °C under high vacuum (<2 mbar) with a Kugelrohr instrument is crucial for obtaining high product yields and reproducibility. MS is thought to play a role in both taking in water and releasing it again as a water reservoir. In addition, a premix of a boronic acid catalyst and carboxylic acid in the presence of MS is extremely effective.
In 2013, Sakakura, Ishihara, and co-workers22 found that alkylboronic acids such as methylboronic acid (43) and n-butylboronic acid (44) are more active catalysts for amidation using α-hydroxycarboxylic acids than arylboronic acid catalysts 9, 12, and 30 (Scheme 6).
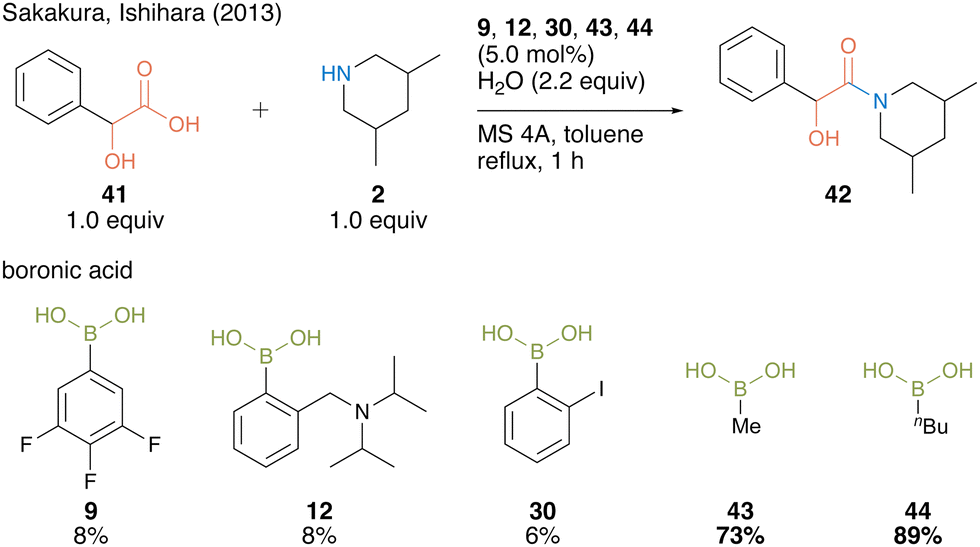 | ||
| Scheme 6 Alkylboronic acid-catalyzed amidation reported by Sakakura and Ishihara.22 | ||
In this catalysis, 2.2 equivalent of water was necessary to obtain a high amide yield because water promotes triboroxine hydrolysis. Based on the X-ray structure of a complex of methylboronic acid, phenylpruvic acid, and 1,2,3,4-tetrahydroquinoline, a cyclic active intermediate was proposed (Fig. 3). Alkylboronic acid and α-hydroxycarboxylic acids formed cyclic acyl boronate 45. Subsequent dehydration led to equilibrium between active species 46 and 47. Although the desired product was obtained by the addition of an amine to 46 or 47, it was assumed that the equilibrium was biased toward 47 rather than toward the more reactive active species 46 while using electron-deficient aromatic boronic acid catalysts. This result indicated that electron-rich alkylboronic acids were prevented from forming inactive 47, providing better amidation results than electron-deficient arylboronic acids. Furthermore, in some cases, the addition of a catalytic amount of benzoic acid improved the product yield by promoting ligand exchange between the product or substrate on the catalyst and benzoic acid.
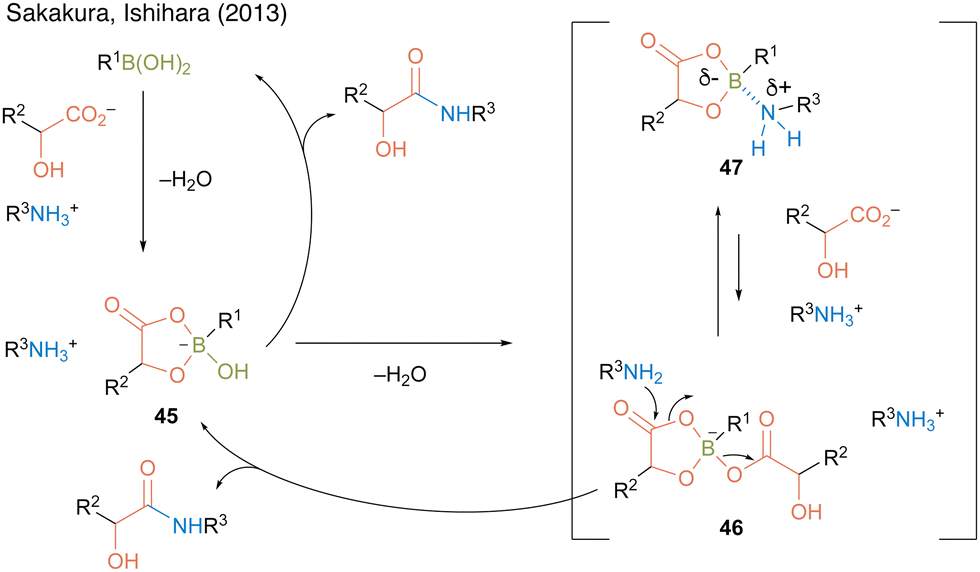 | ||
| Fig. 3 Proposed amidation mechanism reported by Sakakura and Ishihara.22 | ||
In 2015, Tam, Chen and co-workers23 screened a wide range of commercially available aryl boronic acids, including meta-substituted benzeneboronic acid or heteroarylboronic acid and found that 2-furanylboronic acid (48) is an active catalyst for the dehydrative amidation of aliphatic carboxylic acids and primary or secondary amines (Scheme 7). Although bulky or aromatic carboxylic acids were not applicable, amides 26, 49–51 were obtained in high yields even at room temperature in the presence of MS 4A in dichloromethane.
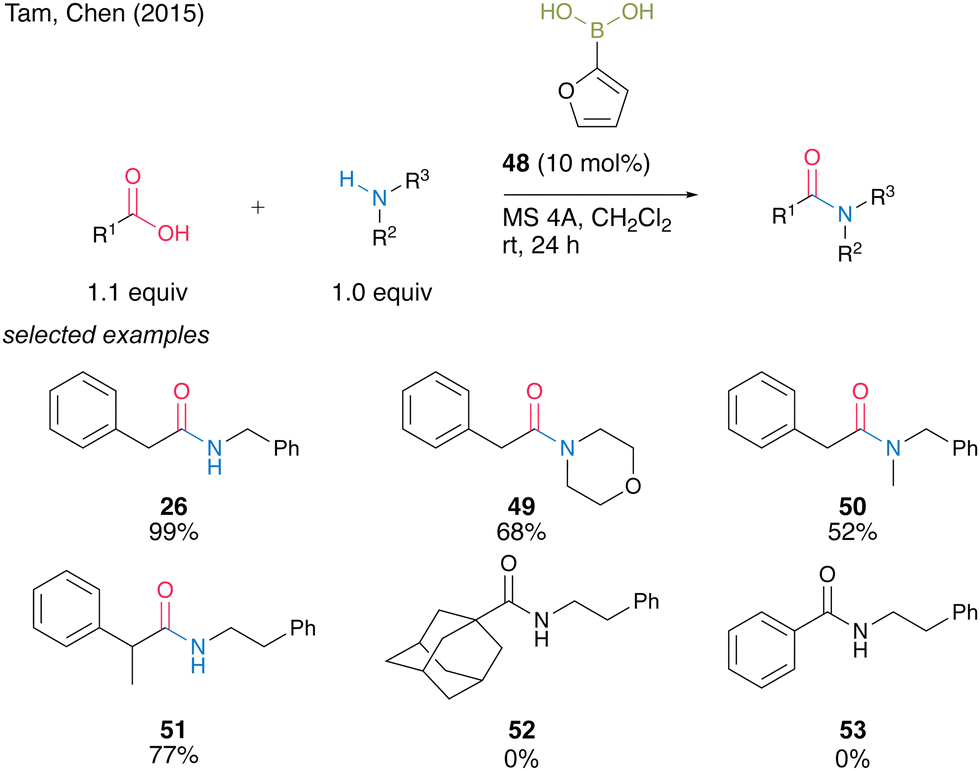 | ||
| Scheme 7 2-Furanylboronic acid-catalyzed amidation reported by Tam and Chen.23 | ||
In the same year, Blanchet and co-workers24 developed a bench-stable 2-(thiophen-2-ylmethyl)phenyl)boronic acid (54). They expected that sulfur atoms would promote active intermediate formation and dehydration after forming amides. Boronic acid 54 was effective for the direct dehydrative amidation of aliphatic and heteroaromatic carboxylic acids at room temperature in the presence of MS 5A in dichloromethane to afford the corresponding amides 55–57 in good to excellent yields (Scheme 8). Although moderate heating at 45 °C was required, acyclic secondary amine was also applicable to obtain tertiary amides 55 in excellent yield.
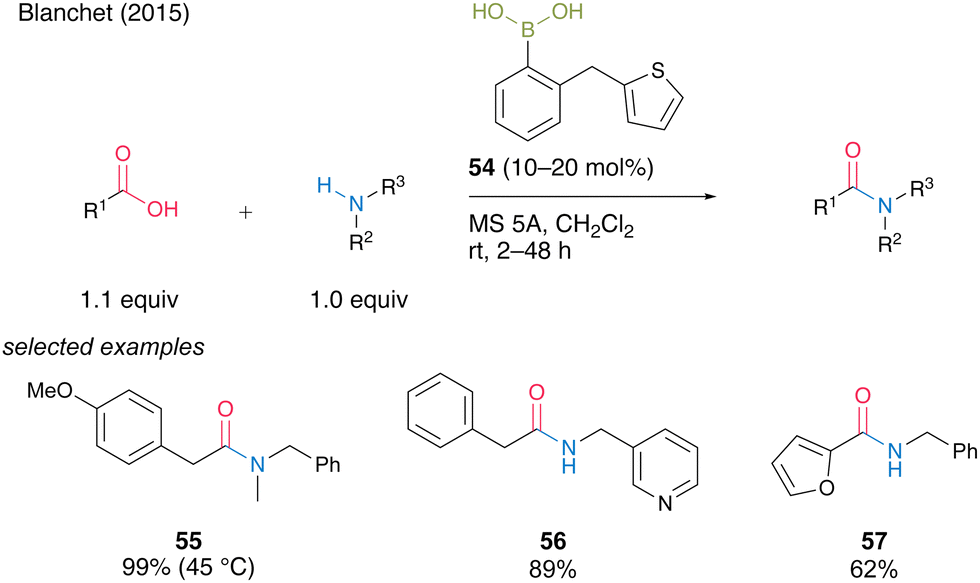 | ||
| Scheme 8 2-(Thiophen-2-ylmethyl)phenylboronic acid-catalyzed amidation reported by Blanchet.24 | ||
In 2016, Ishihara and co-workers25 discovered the electron-deficient 3,5-bis(trifluoromethyl)phenylboronic acid (6) and DMAPO (63) cooperative catalysis for dehydrative amidation. 63 exhibited significantly more effectiveness as an additive than other bases 60–62 and 64 (Scheme 9).
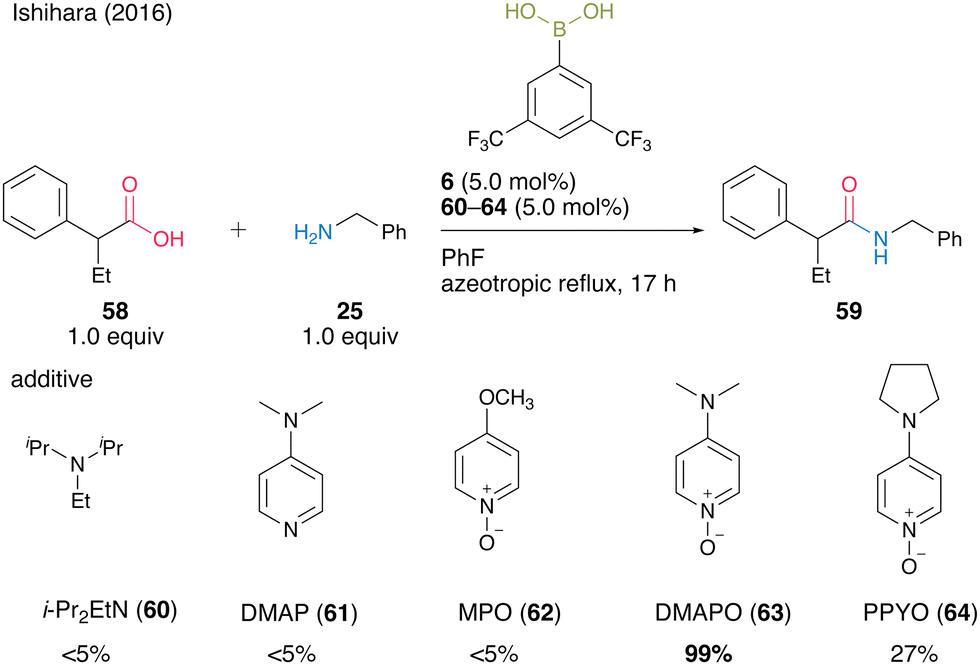 | ||
| Scheme 9 Boronic acid–DMAPO cooperatively catalyzed amidation reported by Ishihara.25 | ||
This cooperative catalysis was useful for the chemoselective amidation of β-substituted acrylic acid 65 with aniline derivative 66 to obtain the desired amide 67. When boronic acid 6 was used without DMAPO (63), the yield and selectivity of 67 were moderate (Scheme 10).
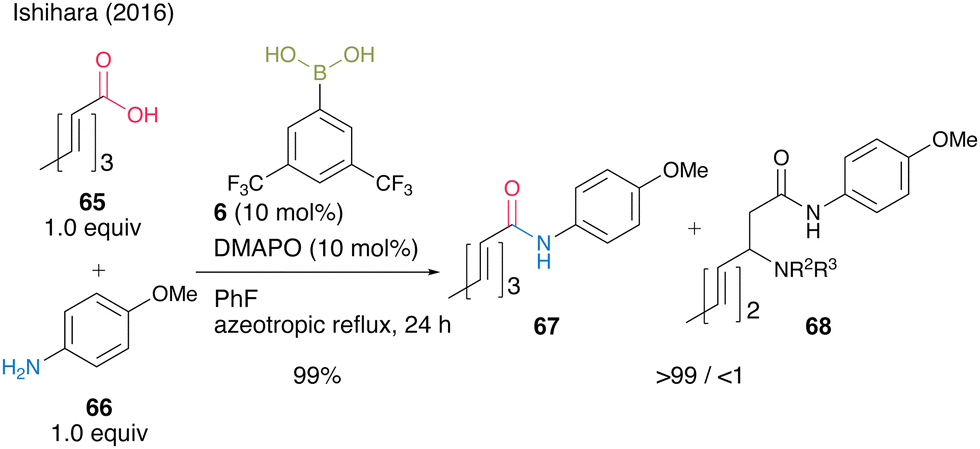 | ||
| Scheme 10 Boronic acid–DMAPO cooperatively catalyzed chemoselective amidation reported by Ishihara.25 | ||
In the presence of a nucleophilic catalyst (Nu) as an additive, the reaction proceeded through a more active intermediate 71via tetrahedral intermediate 70, accelerating amidation. However, when the additive reacted with the boron of the boronic acid catalyst as a Lewis base, inactive intermediate 69 was formed, which might have inhibited amide formation; thus, the choice of additive was crucial in this cooperative catalysis (Fig. 4).
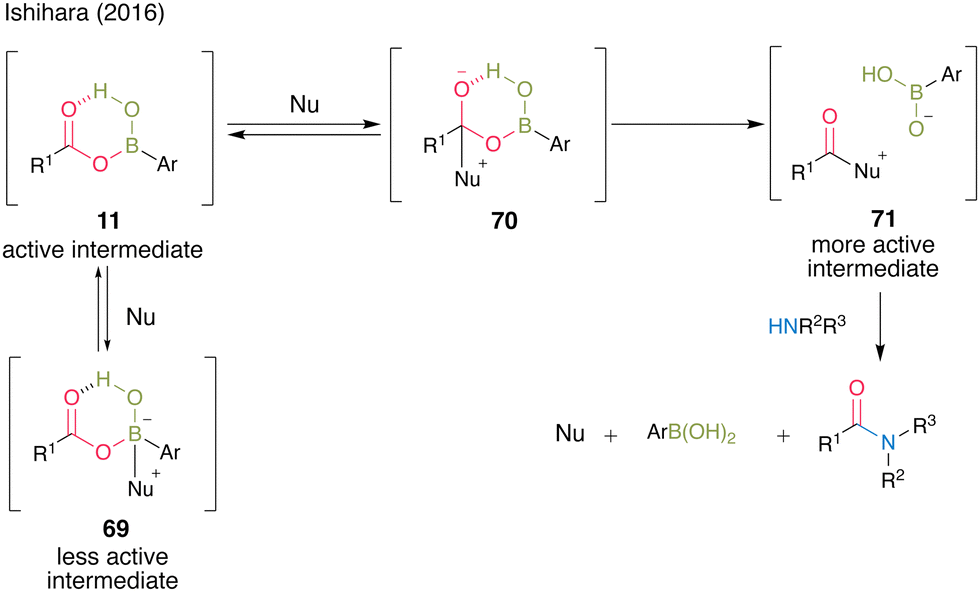 | ||
| Fig. 4 Proposed amidation mechanism reported by Ishihara.25 | ||
Since Yamamoto's pioneering work,13 there have been several reports on improved reaction mechanisms of boronic acid-catalyzed amidation via a monoacyl boronate intermediate 11.20,21,26 In 2018, Whiting et al.27 reported a completely novel reaction mechanism for boronic acid-catalyzed amidation (Fig. 5). They proposed B–O–B motif 72 as a certain active intermediate, which has a unique structure of a dimer of monoacyl boronate intermediate 11 based on the X-ray analysis of intermediates, theoretical studies, and experimental results on the reaction mechanism. In this report, a more reactive B–N–B structure of 73 was also proposed and detected using the 11B NMR spectra derived from a dimeric B–O–B compound with an internal amine. This report marked a turning point in the field of boronic acid-catalyzed dehydrative amidation.
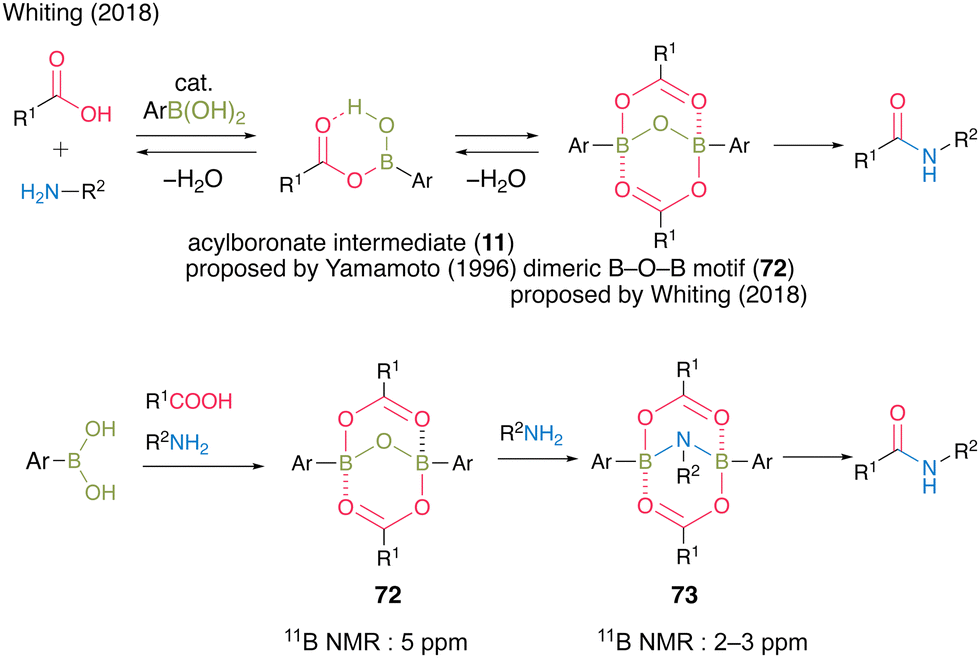 | ||
| Fig. 5 Proposed amidation mechanism reported by Whiting.27 | ||
In 2018, Ishihara and co-workers28 reported catalytic dehydrative amidation using 2,4-bis(trifluoromethyl)boronic acid (74). In this reaction, the bulky ortho-trifluoromethyl group played a vital role in suppressing the coordination of amines to boron atoms in the active intermediate 78, thereby preventing the loss of Lewis acidity, which is crucial for catalytic activity (Fig. 6).
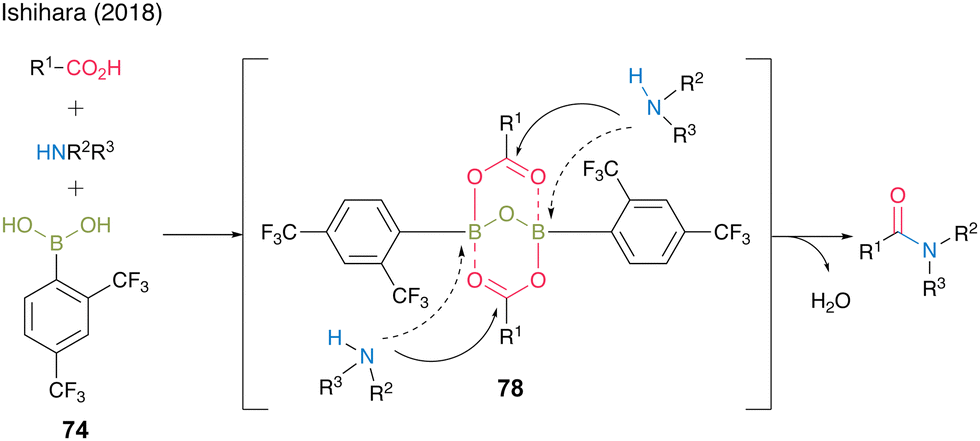 | ||
| Fig. 6 Effect on the ortho-substituent group reported by Ishihara.28 | ||
A dimeric intermediate of acyl boronate was detected by in situ IR analysis and ESI-MS. Furthermore, similar to the findings of Hall,20 a premix of boronic acid and carboxylic acid was effective, ascribed to the initiation step for the formation of an active intermediate containing the B–O–B bond proposed by Whiting.27 This catalyst can be used for reactions at low temperature (25 °C) and bulky substrates to provide the corresponding amides 75 and 76 in high yields. Moreover, racemization was not observed even when α-amino acid derivatives were used as amine substrates to produce amide 77 in 96% yield. (Scheme 11).
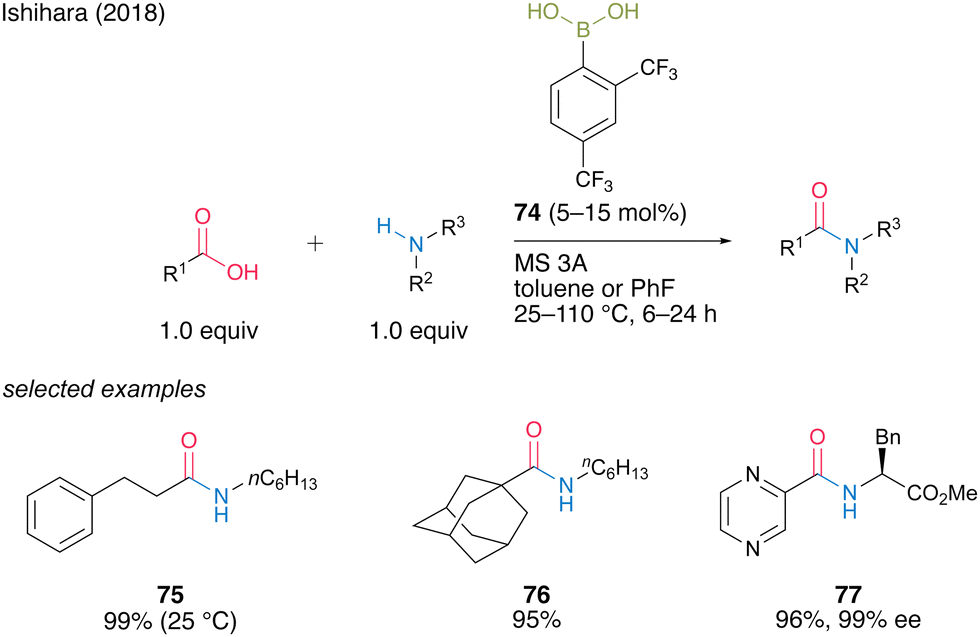 | ||
| Scheme 11 2,4-Bis(trifluoromethyl)phenylboronic acid-catalyzed amidation reported by Ishihara.28 | ||
In 2022, Hall and co-workers29 reported thioether-substituted biphenylboronic acid 80 as an effective amidation catalyst for poorly nucleophilic amines such as aniline derivatives. Most boronic acids for catalytic amidation have been reported to be highly reactive when combined with aliphatic amines; however, few studies have focused on the amidation of aromatic amines. Furthermore, these studies often required very high-temperature conditions. They suggested that 80 could serve as a bifunctional catalyst because the boron atom activated the carboxylic acid and sulfur could serve as a hydrogen-bond acceptor for amine, thereby producing reaction intermediate 81. In the presence of 50 mol% of 80 and 5.0 mol% of DMAPO (63), the amidation of aniline (79) at 85 °C with MS 4A in fluorobenzene was very effective (Scheme 12). This catalysis can be applied to highly electron-deficient aromatic amines, such as 3-cyanoaniline.
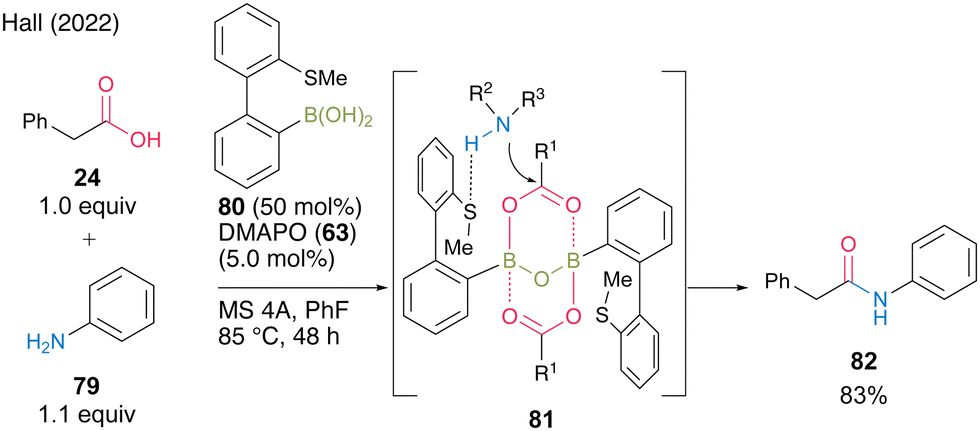 | ||
| Scheme 12 Thioether-substituted biphenylboronic acid-catalyzed amidation reported by Hall.29 | ||
In 2023, Su and co-workers30 screened aromatic boronic acids with heterocyclic structures, including 85–88, and found that 1-thianthrenylboronic acid (88) was an effective catalyst for dehydrative amidation at room temperature in the presence of MS 4A in dichloromethane (Scheme 13). In this catalysis, the sulfur atom of thianthrene was assumed to contribute to the activation of amines via hydrogen bonding or the electrophilic activation of carboxylic acids via the formation of sulfonium ions. Boronic acid 88 is commercially available, easy to handle, and has pharmaceutical benefits.
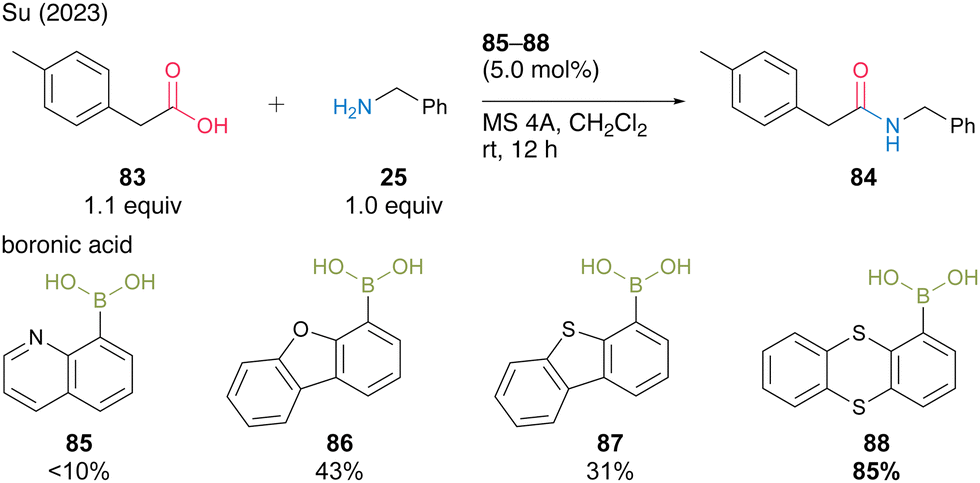 | ||
| Scheme 13 1-Thianthrenylboronic acid-catalyzed amidation reported by Su.30 | ||
In the same year, Blanchet and co-workers31 found that ortho-(sulfonyloxy)benzeneboronic acids, which are aromatic boronic acids containing strong electron-withdrawing groups, were effective for dehydrative amidation at low temperatures in the presence of MS 5A in dichloromethane or under azeotropic reflux conditions in toluene. Based on their original studies using electron-deficient (aryloxybenzene)boronic acid catalysts and the estimated Lewis acidity of boron calculated using DFT, highly electron-deficient ortho-(sulfonyloxy)benzeneboronic acids, such as 2-(methanesulfonyloxy)phenylboronic acid (90) and 2-(trifluoromethanesulfonyloxy)phenylboronic acid (92), were proposed and synthesized (Scheme 14). These catalysts were stable and showed high catalytic activities to produce amides 91 and 94 in excellent yields.
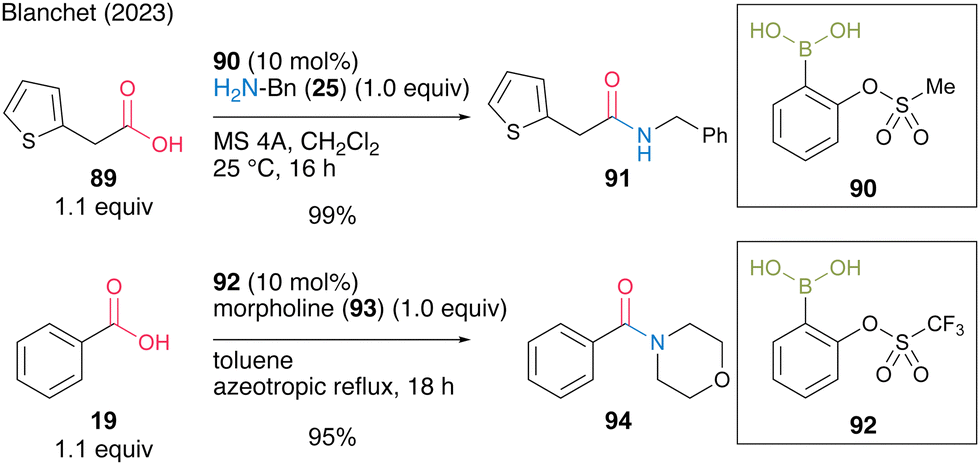 | ||
| Scheme 14 2-Sulfonatobenzeneboronic acid-catalyzed amidation reported by Blanchet.31 | ||
2.2. Direct amide bond formation using recoverable and reusable boronic acid catalysis
From the perspective of improving the practicality of amide synthesis, a recoverable and reusable boronic acid catalyst for the direct dehydrative amidation of carboxylic acids is desired. In this section, we present some successful examples. In 2001, Yamamoto and co-workers32 developed 3,5-bis(perfluorodecyl)phenylboronic acid (96), which has a fluorous alkyl side chain exhibiting properties of electron-deficient and fluorous-layer-supporting functional groups. They successfully developed two different protocols for catalyst recovery and reuse. Catalyst 96 (3.0 mol%) promoted dehydrative amidation under azeotropic reflux conditions in a homogeneous system of a mixed solvent of o-xylene, toluene, and perfluorodecalin (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) (Scheme 15). When the homogeneous mixture was cooled to room temperature, it separated into a heterogeneous organic solvent layer and a catalyst-supported fluorous layer, allowing liquid–liquid separation by decantation. In the reaction to obtain amide 97 from cyclohexanecarboxylic acid (95) and benzylamine (25), the fluorous solvent containing the fluorous catalyst was recovered and reused five times.
:1:1) (Scheme 15). When the homogeneous mixture was cooled to room temperature, it separated into a heterogeneous organic solvent layer and a catalyst-supported fluorous layer, allowing liquid–liquid separation by decantation. In the reaction to obtain amide 97 from cyclohexanecarboxylic acid (95) and benzylamine (25), the fluorous solvent containing the fluorous catalyst was recovered and reused five times.
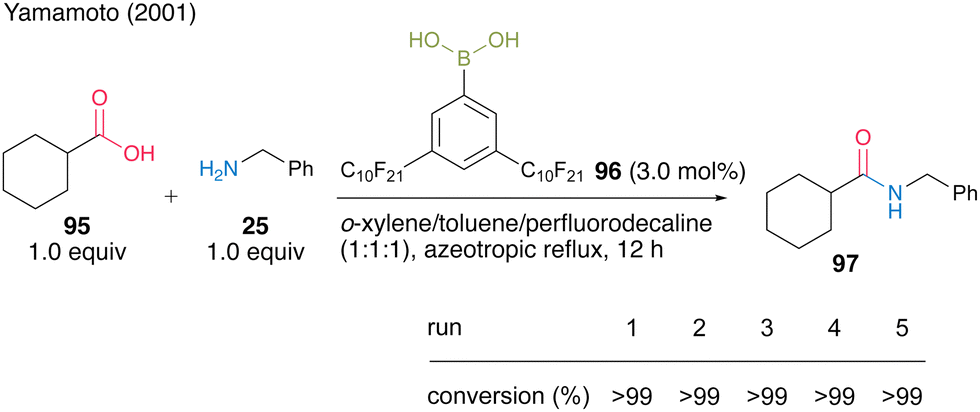 | ||
| Scheme 15 3,5-Bis(perfluorodecyl)phenylboronic acid-catalyzed amidation reported by Yamamoto.32 | ||
Another protocol exploited the property that catalyst 96 is soluble in nonfluorous o-xylene under high-temperature conditions of azeotropic reflux but is insoluble under low-temperature conditions. It was reported that 5.0 mol% of catalyst 96 could be recoverable and reusable 10 times in the same dehydrative amidation of 95 and 25 (Scheme 16).
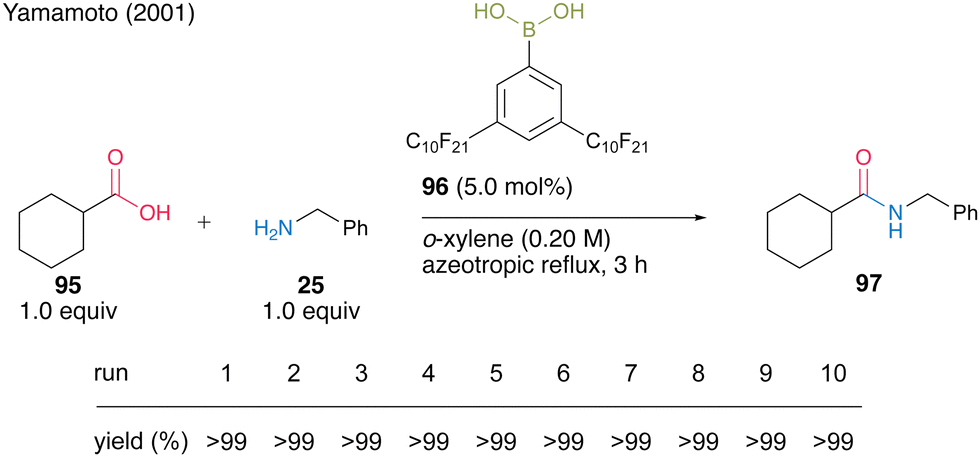 | ||
| Scheme 16 3,5-Bis(perfluorodecyl)phenylboronic acid-catalyzed amidation in nonfluorous solvent reported by Yamamoto.32 | ||
In the same year, Wang and co-workers33 developed an electron-deficient polymer-supported pyridinium boronic acid. A polystyrene-based pyridinium catalyst 98 (no more than 1.0 mol%) prepared from 1,3-propanediol cyclic ester of pyridine-3-boronic acid and Merrifield's resin promoted dehydrative amidation between benzoic acid (19) and benzylamine (25) under reflux conditions using a Soxhlet extractor in toluene (Scheme 17). The solid-phase catalyst 98 exhibited catalytic activity comparable to liquid-phase synthesis using pyridine-3-boronic acid as a catalyst and could be recovered and reused three times. This report is the first successful example of a solid-phase supported aromatic boronic acid catalyst.
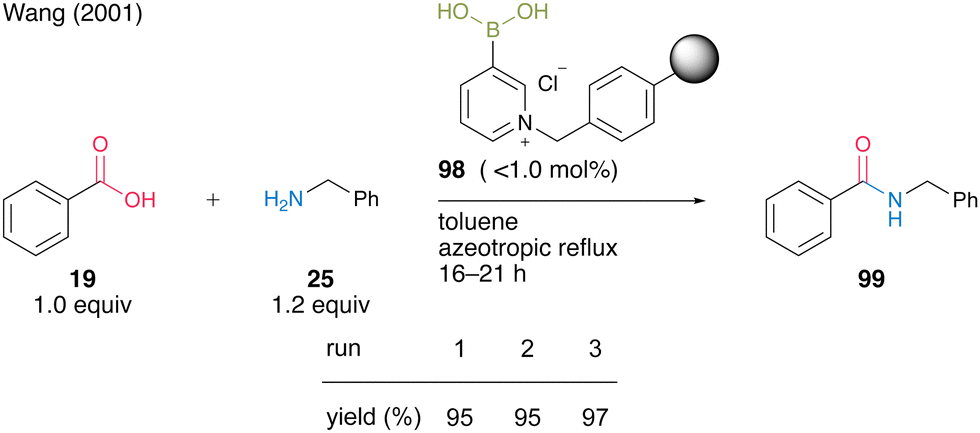 | ||
| Scheme 17 Polymer-supported 3-pyridinium boronic acid-catalyzed amidation reported by Wang.33 | ||
In 2005, Yamamoto and Ishihara's group34 found that 4-pyridineboronic acid is more stable to heat than 3-pyridineboronic acid and 2-pyridineboronic acid, and developed a polystyrene polymer-supported 4-pyridinium boronic acid catalyst. The pyridinium salt-type catalyst exhibits much higher catalytic activity when using a mixed biphasic solvent (5:1) of toluene and 1-ethyl-3-methylimidazolium trifluoromethanesulfonate [emim][OTf] than when using only toluene as the solvent. Unlike Wang's polystyrene polymer-supported 3-pyridinium salt-based boronic acid catalyst, which partially decomposes during the catalyst recovery and reuse process, polystyrene polymer-supported 4-pyridinium salt-based boronic acid catalyst 100 shows high recovery and reusability. Polymer-supported catalyst 100 could be recovered and reused ten times in the dehydrative amidation of an equal mixture of phenylbutanoic acid (1) and benzylamine (25) under azeotropic reflux conditions (Scheme 18). The polymer-supported catalyst can be recovered simply by filtration without any extraction after the reaction was completed.
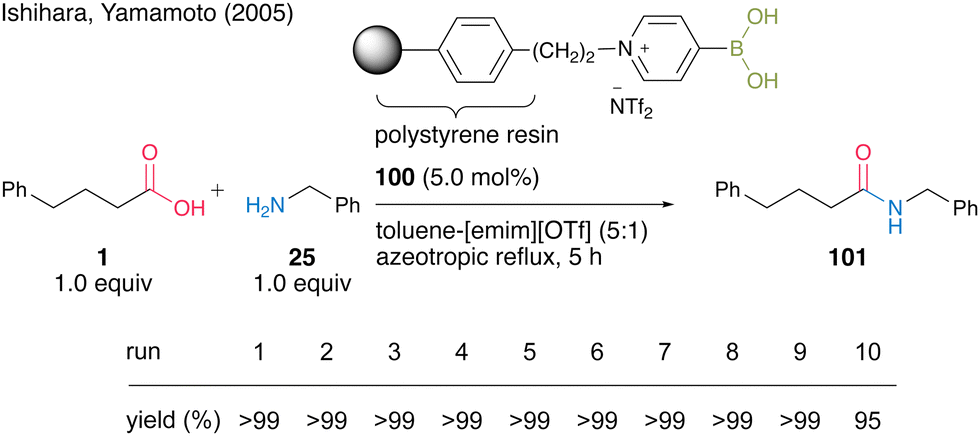 | ||
| Scheme 18 Polymer-supported 4-pyridinium boronic acid-catalyzed amidation reported by Ishihara and Yamamoto.34 | ||
In 2013, Hall35 successfully developed solid-phase-supported 5-alkoxy-2-phenylboronic acid, which is a highly active solid-phase-supported MIBA catalyst.20 Solid-phase-supported catalyst 102, which has a structure in which a polystyrene resin and an aminopropyl ether linker at the 5-position of an aromatic boronic acid are connected by an amide bond, can undergo dehydrative amidation in the presence of MS 4A in dichloromethane even at room temperature, similar to MIBA. It accelerates the reaction and can be recovered by filtration with a polypropylene filter. Polymer catalyst 102 can be recovered by filtration using a polypropylene filter. Amide 26 or 103 can be obtained in high yields using the recovered catalyst (Scheme 19).
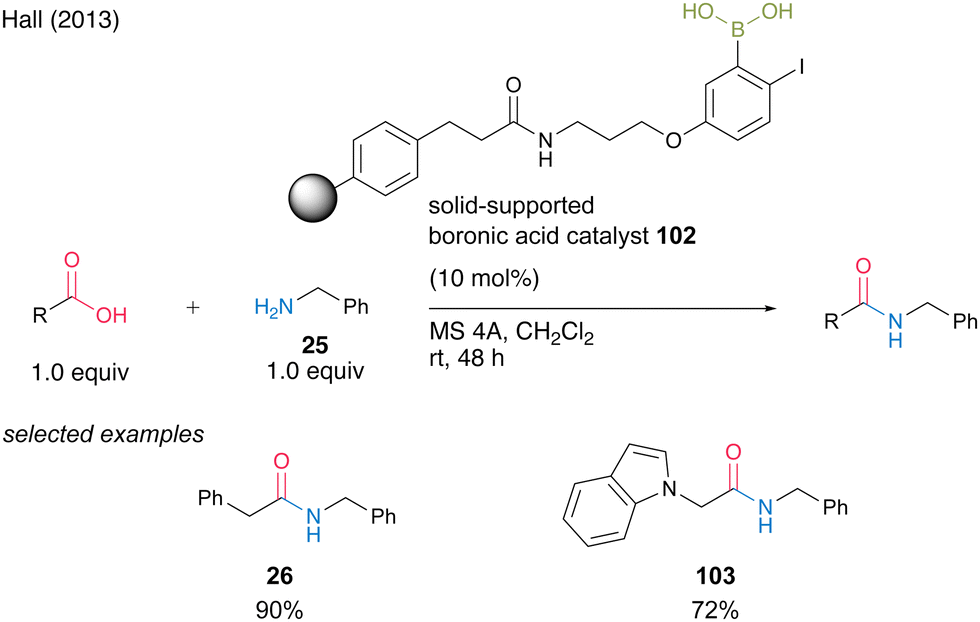 | ||
| Scheme 19 Polymer-supported MIBA-catalyzed amidation reported by Hall.35 | ||
In 2014, Gu and Lee's group36 developed a silica-supported boronic acid catalyst using mesocellular siliceous foam (MCF) as a solid support. They found that capping silanol groups on the silica gel surface with fluoroalkyl groups was effective in improving the catalytic activity and silica-supported catalyst 104 with a trifluoroalkyl capping group showed high catalytic activity for the dehydrative amidation of several aliphatic and aromatic carboxylic acids. In the presence of 5.0 mol% of catalyst, the reaction between phenylacetic acid (24) and benzylamine (25) proceeded to produce amide 26 in high yield under azeotropic reflux conditions in toluene, and the silica-supported boronic acid 104 was recoverable and reusable six times by a simple filtration operation (Scheme 20).
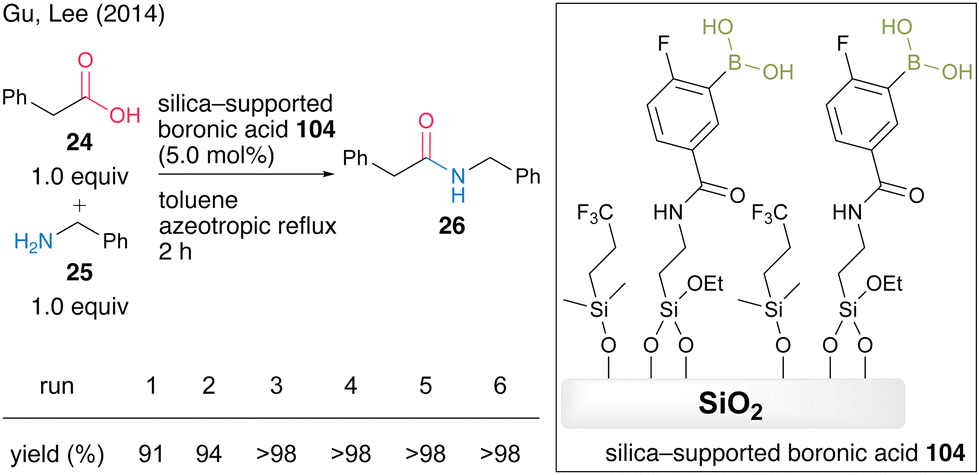 | ||
| Scheme 20 Silica-supported boronic acid-catalyzed amidation reported by Gu and Lee.36 | ||
In 2017, Ishihara and colleagues37 reported two protocols for recoverable and reusable catalytic dehydrative amidation based on the development of homogeneous boronic acid precatalysts by the complexations between a boronic acid and a base. The first protocol uses the formation of an acid–base complex between an electron-deficient aromatic boronic acid and a strongly basic anion-exchange resin. In this reaction, the complex serves as a precatalyst, and the free boronic acid is released into the reaction mixture and exhibits high catalytic activity. As a result of examining various boronic acids, the complex with 3,5-bis(trifluoromethyl)phenylboronic acid (6) produced the best results, and the reaction with phenylpropanoic acid (107) and benzylamine (25) proceeded under azeotropic reflux conditions in fluorobenzene in the presence of precatalyst 106 to produce amide 108 in excellent yield (Scheme 21). After the reaction was completed, the resin-supported boronate 106 was recovered by decantation, and no decrease in catalyst activity was observed even after 10 reuses. Notably, this reaction system is highly practical, as it can be scaled up to 200 mmol in the presence of 1 mmol of precatalyst.
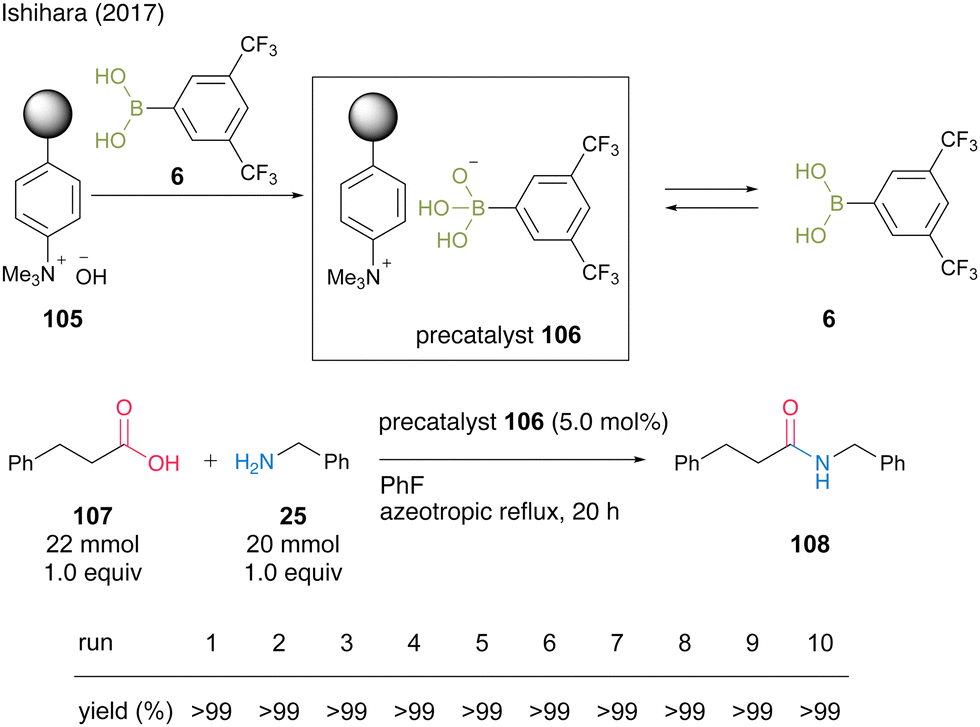 | ||
| Scheme 21 Boronic acid–DOWEXTM complex-catalyzed amidation reported by Ishihara.37 | ||
The second approach uses the formation of acid–base complexes between electron-deficient aromatic boronic acids and DMAP or DMAPO. In this catalysis, the complex serves as a precatalyst, and the boronic acid is liberated during the reaction. Using a complex derived from 3,5-dinitro-4-tolueneboronic acid (109), a precipitated solid suitable for recovery and reuse was generated. Amines DMAP (61) and DMAPO (63) formed complexes with boronic acid 109, which were recoverable and reusable three times in the amidation reaction of benzoic acid (19) and hexylamine (110) (Scheme 22). The complex with DMAPO showed higher activity as a precatalyst, whereas the complex with DMAP showed better recovery efficiency. Notably, the amidation catalytic activity of boronic acid 109 itself was reported to be extremely low.
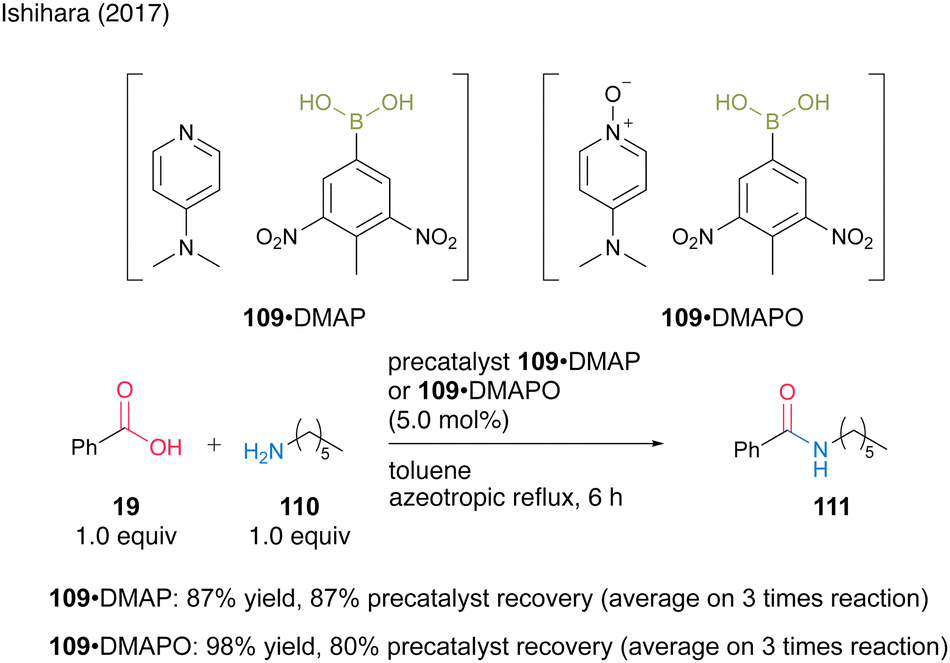 | ||
| Scheme 22 Boronic acid–DMAP(O) complex-catalyzed amidation reported by Ishihara.37 | ||
In 2019, Whiting and Baxendale's group38 developed solid-supported boronic acids via copolymerization. Polystyrene-supported boronic acid catalyst 112 was prepared by the copolymerization of 4-vinylphenylboronic acid, divinylbenzene, and styrene in the presence of AIBN. In the reaction between phenylacetic acid (24) and benzylamine (25), catalyst 112 was recoverable and reusable (Scheme 23). Solid-state 11B-NMR and in situ IR (ReactIR) observations suggested the formation of a dicarboxylate active intermediate containing a B–O–B bond, similar to that previously reported in homogeneous catalysis.27 Furthermore, a preliminary application to the flow reaction using a heat column packed with the catalyst was considered.
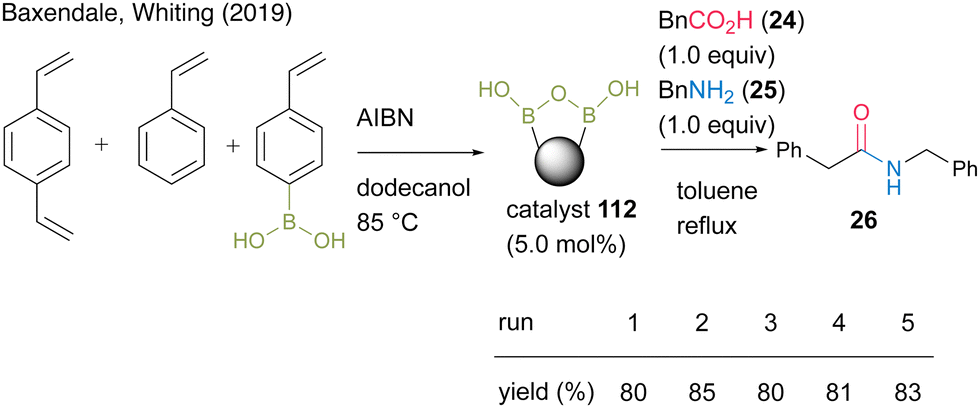 | ||
| Scheme 23 Polymer-supported boronic acid-catalyzed amidation reported by Baxendale and Whiting.38 | ||
Most recently, Lo and Lam's group39 developed catalyst 113 by incorporating a perfluoroalkyl group (C8F17) at the end of the side chain of aromatic boronic acid at the ortho-position and reported a catalytic dehydrative amidation of carboxylic acids with microwave irradiation under neat conditions. The product and catalyst can be easily separated from the crude product via fluorous solid-phase extraction (SPE). For the reaction between phenylacetic acid (24) and benzylamine (25), catalyst 113 could be recovered and reused four times (Scheme 24).
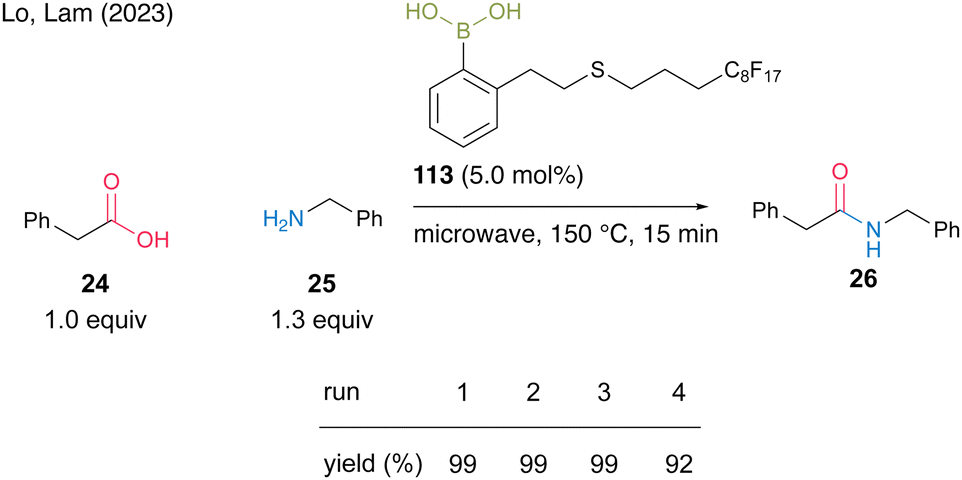 | ||
| Scheme 24 Fluorous boronic acid-catalyzed amidation reported by Lam.39 | ||
2.3. Direct amide bond formation using other monoboron catalysis
In 2006, Ishihara and Yamamoto40 reported catalytic amidation with 4,5,6,7-tetrachlorobenzo[d][1,3,2]dioxaborole (114) prepared using tetrachlorocatechol and BH3·SMe2. 114 showed superior catalytic activity to 3,5-bis-(trifluoromethyl) phenylboronic acid (6) for the amidation of sterically hindered aliphatic and aromatic carboxylic acids, although 114 presented slightly lower catalytic activity than 6 for the amidation of less hindered carboxylic acid substrates (Scheme 25). Catalysis using 114 proceeds in toluene or o-xylene under azeotropic reflux conditions.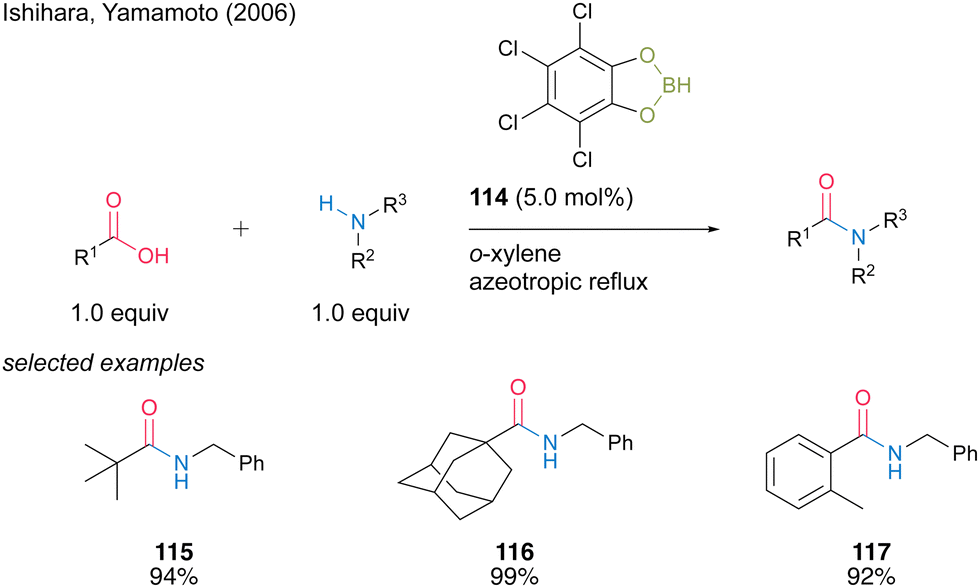 | ||
| Scheme 25 4,5,6,7-Tetrachlorobenzo[d][1,3,2]dioxaborole-catalysed amidation reported by Ishihara.40 | ||
In 2017, Sheppard and co-workers41 found that B(OCH2CF3)3 (118) was an effective catalyst for dehydrative amidation reaction under azeotropic reflux conditions in tert-amyl methyl ether (TAME) (Scheme 26).42 In this reaction, unreacted carboxylic acids, amines, and catalysts can be easily removed by simple filtration using three mixed resin scavengers, i.e., poly hydroxylated Amberlite IRA743 as a boron scavenger, acidic Amberlyst 15 as an amine scavenger, and basic Amberlyst A-26(OH form) as an acid scavenger, to produce the corresponding amides. The catalytic protocol using B(OCH2CF3)3 (118) offers significant improvements in terms of a large reduction of overall waste during amide bond formation and shows a low process mass intensity (PMI)43 value of 5 for 119. Furthermore, this reaction is suitable for manufacturing active pharmaceutical ingredient precursors such as 120 and 121.
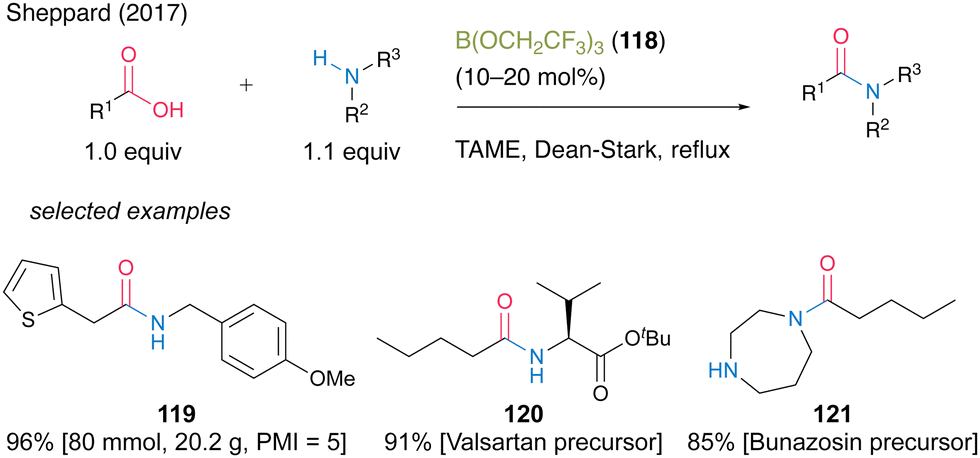 | ||
| Scheme 26 B(OCH2CF3)3-catalyzed amidation reported by Sheppard.41 | ||
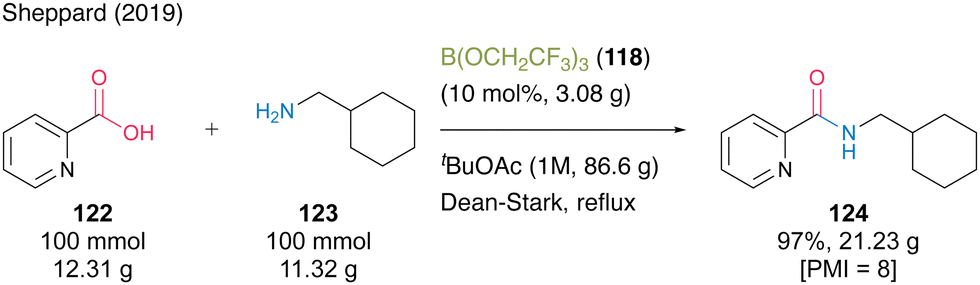
In 2019, the same group reported that tert-butyl acetate (tBuOAc) was more suitable for B(OCH2CF3)3 (118)-catalyzed dehydrative amidation than TAME.44 This catalysis demonstrated a wide substrate scope, and the corresponding amides were obtained using a solid-phase workup procedure. The catalytic efficiency was demonstrated using a 100-mmol scale reaction of the synthesis of 2-picolinic acid (122) and cyclohexanemethylamine (123) in the presence of 10 mol% of 118 in tBuOAc under Dean–Stark reflux conditions (Scheme 27). The desired amide 124 was obtained in 97% yield (21.23 g) with a PMI of 8. This is one of the few successful examples of large-scale synthesis using organoboron-catalyzed amidation.
 | ||
| Scheme 27 B(OCH2CF3)3-catalyzed large-scale amidation reported by Sheppard.44 | ||
2.4. Direct amide bond formation using multiboron catalysis
In 2017, Shibasaki, Kumagai and co-workers45 developed multiboron containing six-membered heterocycles, 1,3-dioxa-5-aza-2,4,6-triborinane (DATB) ring systems. DATB derivatives are unique compounds with a stable m-terphenyl scaffold, and their boron atoms exhibit peculiar Lewis acidity. The catalytic performance of DATB was investigated, and its catalytic activity was superior to that of commercially available boronic acid or metal catalysts, especially in the reaction of bulky carboxylic acids to produce 126 in excellent yield. DATB (125) maintained excellent catalytic activity even in the amidation of aromatic carboxylic acids, which have low reactivity, and the corresponding amide 127 can be obtained in high yield even in the presence of only 0.5 mol% of the catalyst. Furthermore, aniline derivatives are applicable as amine substrates, affording 128 in high yield. It is also shown that pharmaceuticals that were previously synthesized using stoichiometric amounts of activating reagents can now be synthesized by catalytic amidation using 125. DATB demonstrated not only a wide range of substrate scope but also its great level of functional group tolerance. DATB catalysis was promoted under mild conditions; thus, the degradation of optical purity of α-chiral carboxylic acid or α-chiral amines did not occur for 129.In 2019, the same group also developed a novel DATB derivative containing a pyrimidine ring.46 They found a reliable and rapid synthetic route to install boron atoms into the 4,6-diarylpyrimidine scaffold using a pyrimidine ring as a directing group (Scheme 28). The previous synthetic protocol of DATB required stepwise synthesis of the m-terphenyl scaffold to introduce two boron atoms properly and it caused difficulty in functionalizing or introducing arbitrary substituents into the skeletons. By contrast, the pyrimidine ring introduction allows the effective and sufficient synthesis of the derivatives without chromatographic purification. Although the catalytic performance of functionalized Pym-DATB (130) derivatives slightly decreased, the most accessible unsubstituted one exhibited sufficient catalytic activity for the amidation of a wide range of substrates.
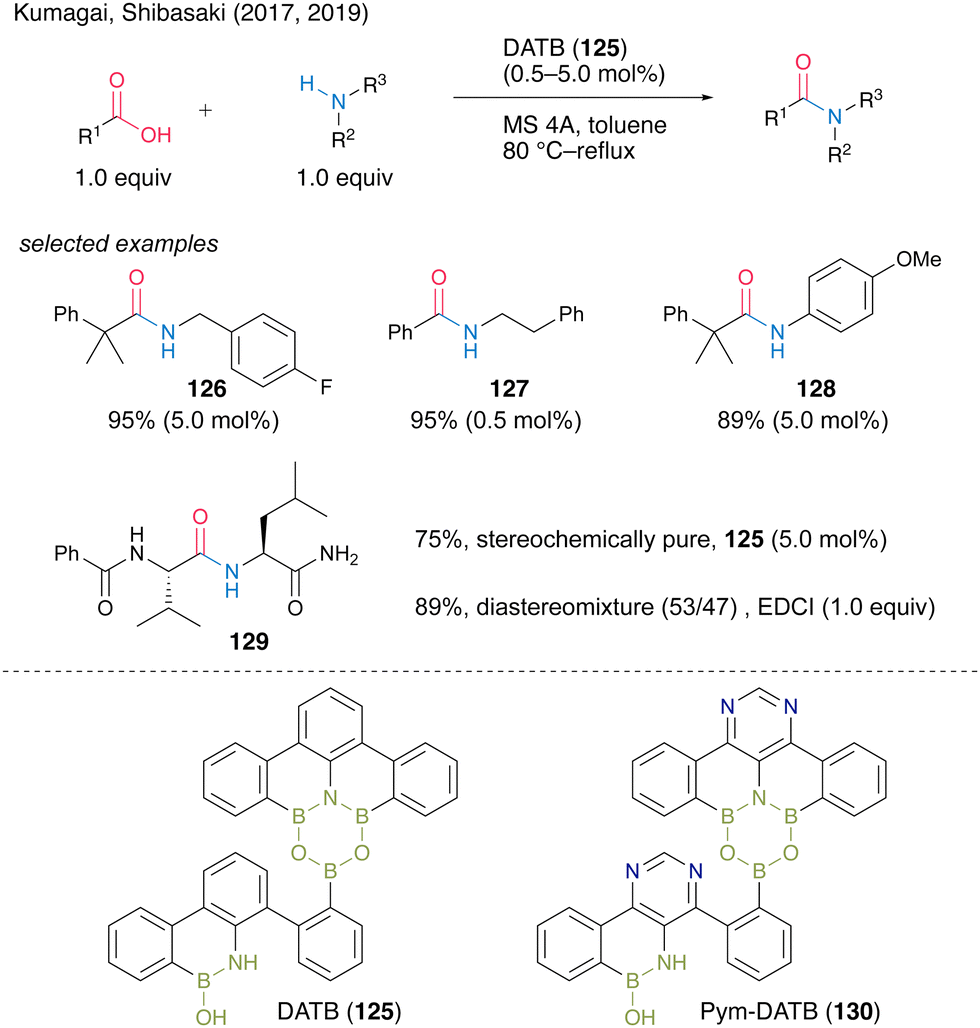 | ||
| Scheme 28 DATB-catalyed amidation reported by Kumagai, Shibasaki.45,46 | ||
Recently, Kumagai47 reported an improved two-step synthetic route for DATB containing a m-terphenyl scaffold without chromatographic purification. In this protocol, 2,6-dibromoaniline (131) and 1,2-phenylenediboronic acid (132) were converted to a dimeric spiroborate salt (133) as a thermodynamically stable intermediate under Suzuki–Miyaura coupling conditions (Scheme 29). The intermediate (133) was converted to DATB (125) derivatives with acidic hydrolysis under high-temperature conditions. Although this method requires slight modifications of the reaction conditions for each substrate, it enables the gram-scale synthesis of various DATB derivatives.
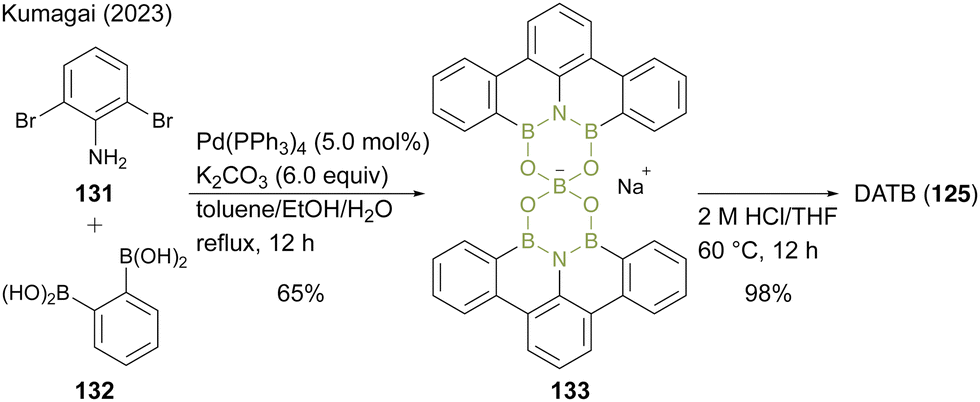 | ||
| Scheme 29 Improved synthesis of DATB reported by Kumagai.47 | ||
The plausible catalytic cycle for DATB-catalyzed direct amidation was proposed based on NMR (Fig. 7).45 First, a kinetically and thermodynamically favored complex (134) was produced by the reaction of DATB with an amine substrate. Next, intermediate 135 was formed by harnessing two B–O bonds, thereby electrophilically activating the carboxylic acid functional group via the interaction of two boron Lewis acidic atoms. Amine was then attached to the carboxylic acid carbonyl group to form intermediate 136. Subsequently, intermediate 136 collapsed, simultaneously forming the corresponding amide product and water as a clean byproduct and then regenerating the DATB catalyst.
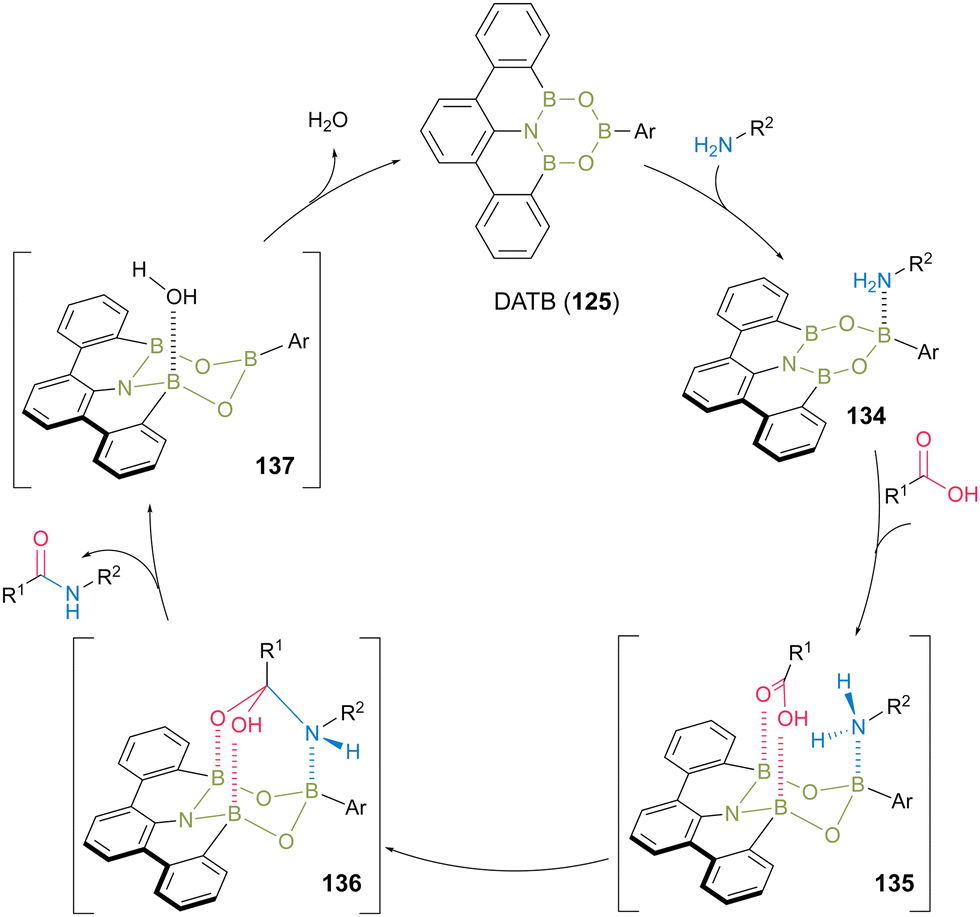 | ||
| Fig. 7 Proposed amidation mechanism reported by Kumagai and Shibasaki.45 | ||
Recently, Kumagai, Noda, and co-workers48 developed a novel diboron 139 with the N(BOH)2 structure and found it usefulness as an amidation catalyst (Scheme 30). This is the first isolable N(BOH)2 compound, and it showed superior catalytic activity to DATB (125) in terms of turnover frequency for dehydrative amidation. In fact, for the reaction between acid 1 and amine 20 in toluene, the use of 1.0 mol% of 139 resulted in a higher product yield than using 5.0 mol% DATB (125).
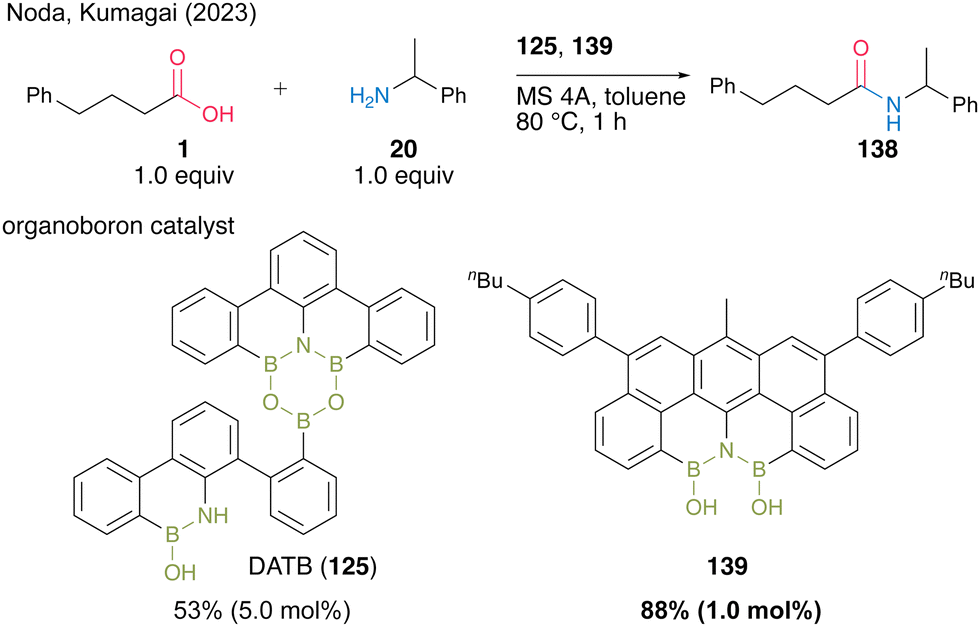 | ||
| Scheme 30 N(BOH)2-containing diboron-catalyzed amidation reported by Noda and Kumagai.48 | ||
In 2018, the Saito group49 reported a successful catalytic amidation of aromatic carboxylic acids using catalysts of the X2B–BX2 structure. In general, aromatic carboxylic acids are inert because they form stable salts with amines. They hypothesized that 140, which undergoes protonation, promotes the generation of free amines and carboxylates and that the B–B moiety electrophilically activates carboxylate derived from aromatic carboxylic acids, thereby accelerating the amidation reaction through intermediate 141. Based on the screening of catalysts with diboron structures (142–144), commercially available catalysts—tetrakis(dimethylamido)diboron (142) and tetrahydroxydiboron (143)—demonstrated catalytic efficiency in the direct amidation of aromatic carboxylic acids, affording amide 99 in high yield (Fig. 8).
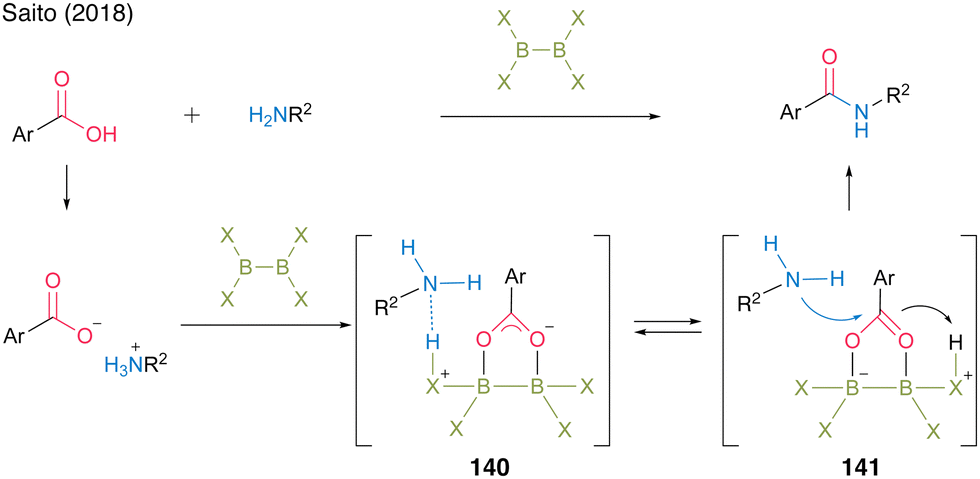 | ||
| Fig. 8 Proposed amidation mechanism reported by Saito.49 | ||
In most cases, using aromatic carboxylic acids as substrates, catalysts 142 and 143 demonstrate far superior catalytic activity compared with boric acid (B(OH)315a,b) (Scheme 31).
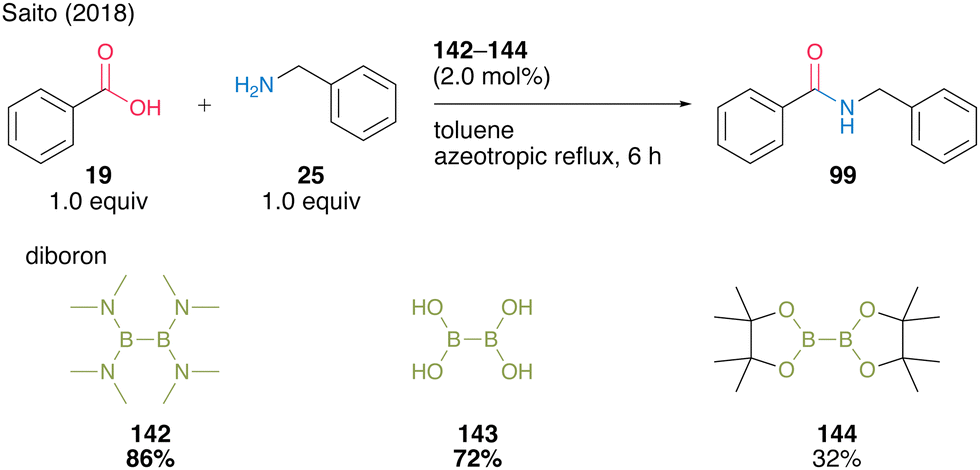 | ||
| Scheme 31 Diboron-catalyzed amidation reported by Saito.49 | ||
In 2019, Shimada and co-workers50 designed a hydroxy-directed catalytic amidation of α- or β-hydroxycarboxylic acids using a catalyst containing preorganized B–O–B motif 145 based on the structure of an active intermediate in the revised amidation mechanism reported by Whiting.27 They hypothesized that the B–OH moiety of 145 recognizes the hydroxyl group to form intermediate 146via covalent B–O bonds and then the two boron atoms in the B–O–B motif promotes the electrophilic activation of carboxylic acid in a bidentate manner to form reactive cyclic intermediate 147, thereby accelerating the reaction to produce α- or β-hydroxy amides (Fig. 9).
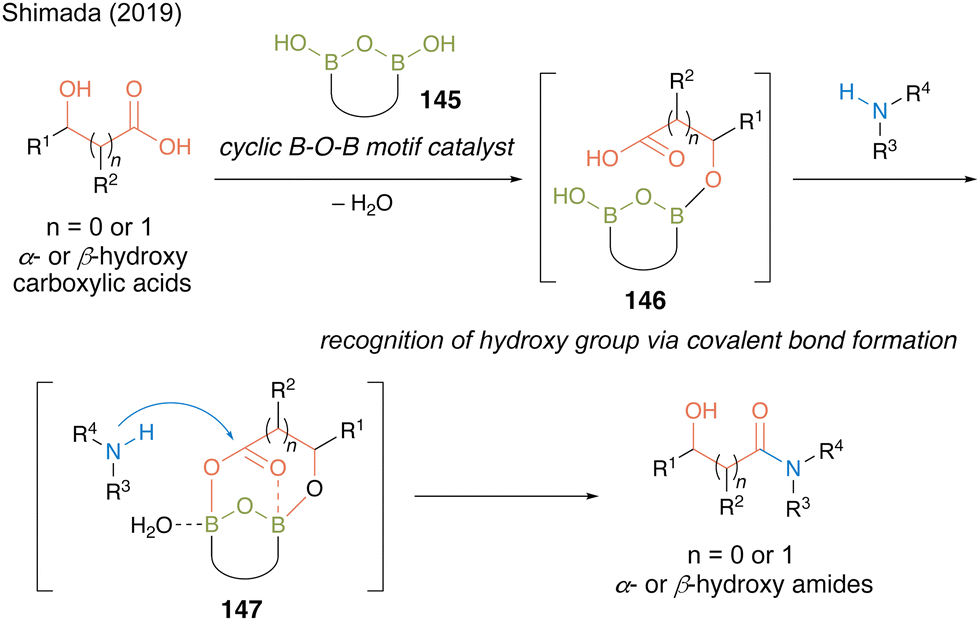 | ||
| Fig. 9 Design of hydroxy-directed catalytic amidation reported by Shimada.50 | ||
The reaction of 3-hydroxy-3-phenylpropanoic acid (148) with N-benzyl-N-methylamine (149) in the presence of 1.0 mol% of biphenyl-based diboronic acid anhydride 150 proceeded smoothly in toluene at 110 °C within 4 h without any azeotropic dehydration protocol or a dehydrating agent, producing β-hydroxyamide 153 in 70% yield (Scheme 32). The yield improved to 87% when 1.0 mol% of catalyst 151, which has two bromine atoms at both ortho-positions of the boron atom based on Hall19,20 reports, was used. Furthermore, amide 153 was obtained in quantitative yields when 152 namely DBAA (diboronic acid anhydride), which has additional bromine substituents at the para position, was used, and a high yield of 98% was maintained even when the catalytic amount was reduced to only 0.2 mol%. The ortho-bromine atom contributed to not only the electron-withdrawing effect but also the stabilization effect of the catalyst B–O–B motif and inactivity suppression due to amine coordination.
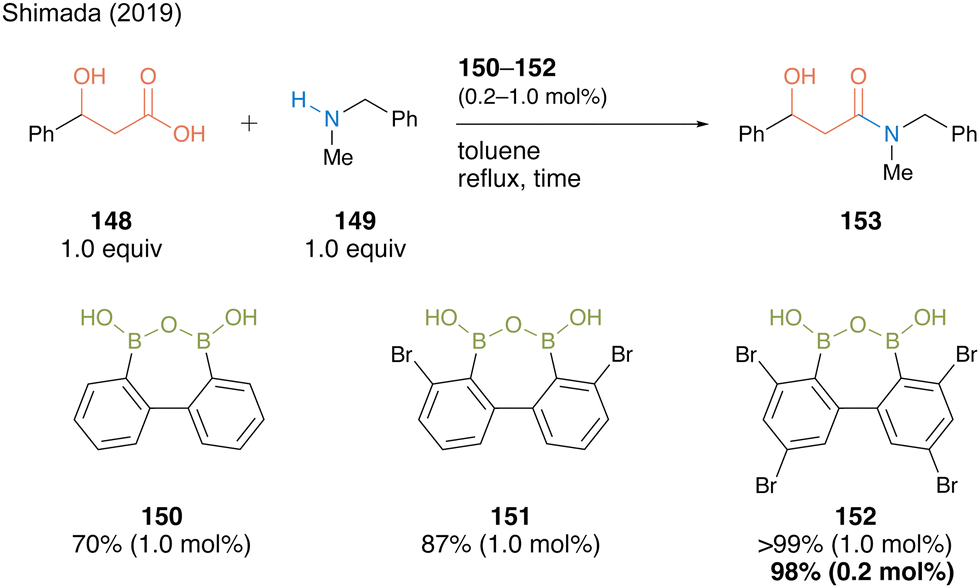 | ||
| Scheme 32 DBAA-catalyzed amidation reported by Shimada.50 | ||
This substrate-directed catalytic dehydrative amidation proceeded through an effective electrophilic activation of carboxylate by the multiboron B–O–B motif of the acyl boronate intermediate 154via covalent bond formation of hydroxy groups of carboxylic acid substrates with the catalyst. The proposed mechanism was supported by the detection of intermediates by ESI-MS and 11B-NMR analyses (Fig. 10).
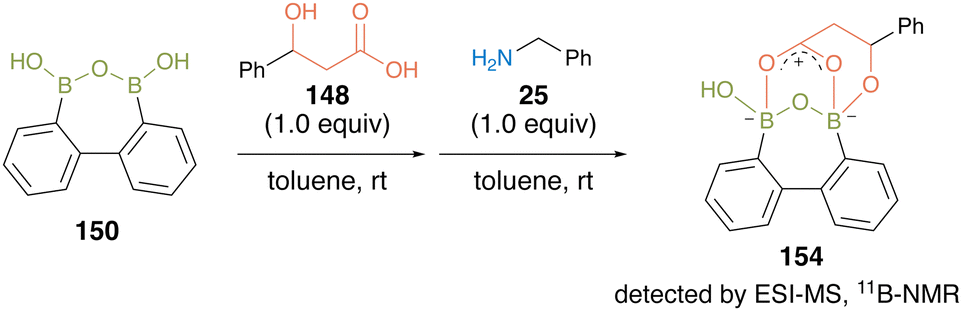 | ||
| Fig. 10 Mechanistic insights reported by Shimada.50 | ||
DBAA (152) showed exceptionally high catalytic activity for dehydrative amidation of α- or β-carboxylic acids and amines. Notably, DBAA catalysis does not require strict dehydration operations, such as adding a drying agent or azeotropic reflux conditions. Indeed, high catalytic performance is maintained even in a mixed solvent of toluene/water (19:1), providing a corresponding β-hydroxy amide 153 in excellent yield with 1.0 mol% of the catalyst (Scheme 33). For amide 155, the catalytic amount of DBAA (152) could decrease to 0.01 mol% and turnover number (TON) recorded 7500, which is noteworthy for an organoboron catalysis. The synthetic efficiency of DBAA catalysis was demonstrated by one-step gram-scale synthesis of tropicamide (156), a known a mydriatic drug, from readily available tropic acid with 4-(ethylaminomethyl)pyridine at a Lewis basic site with 0.5 mol% of the catalyst.
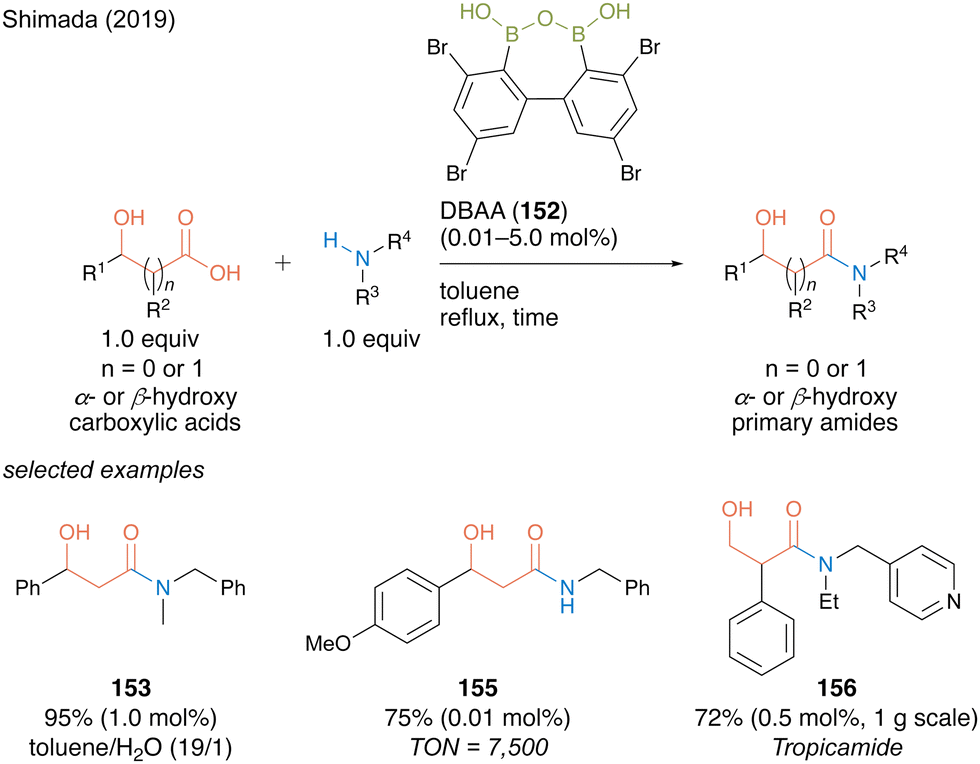 | ||
| Scheme 33 DBAA-catalyzed amidation reported by Shimada.50 | ||
In 2020, the same group applied dehydrative amidation using DBAA (152) for the catalytic synthesis of Weinreb amides.51 The N-methyl-N-methoxy amine (157) has poor nucleophilicity compared to ordinally secondly amines due to the electron-withdrawing oxygen substituted in the amino group, thereby posing challenges for dehydrative amidation. Nevertheless, in the presence of a catalytic amount of DBAA (152), the reaction of carboxylic acid with 3.0 equivalents of 157 proceeded smoothly to produce the corresponding Weinreb amide in high to excellent yields. This was the first successful catalytic direct synthesis of useful Weinreb amide from carboxylic acid. Because the only byproduct of this reaction was water, the resulting Weinreb amide could be used in the next reaction step without purification. Indeed, by exploiting the convenience of this operation, the concise synthesis of various bioactive α-hydroxy ketone natural products (acyloin natural products) 158–161 was achieved using a one-pot two-step sequence including DBAA-catalyzed Weinreb amide synthesis followed by a reaction of a Grignard reagent addition after switching the solvent (Scheme 34).
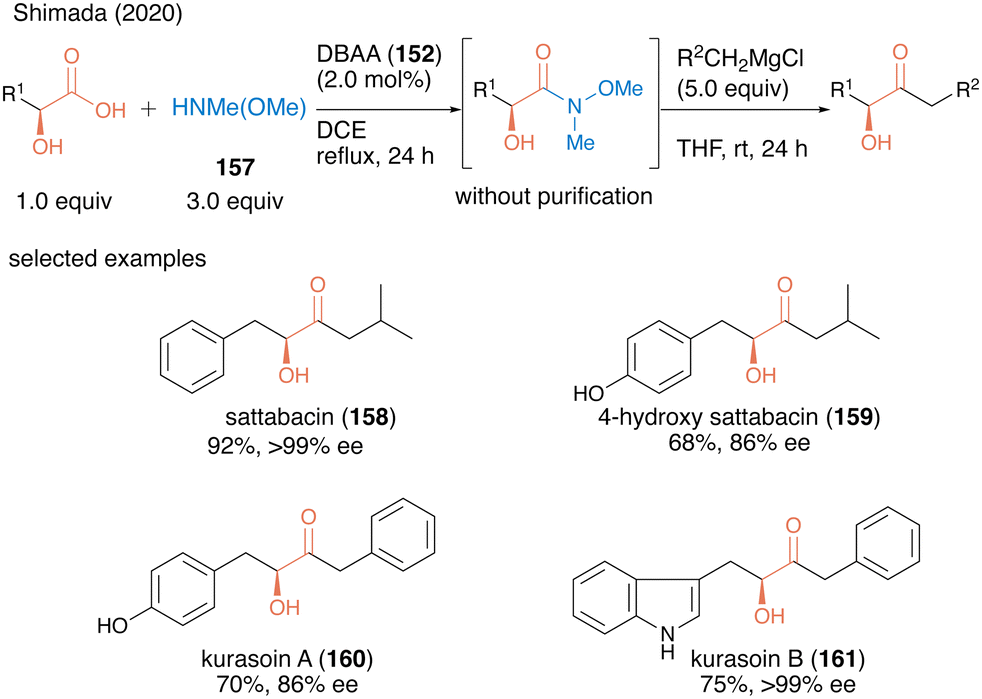 | ||
| Scheme 34 DBAA-catalyzed Weinreb amide synthesis reported by Shimada.51 | ||
In 2021, a facile synthetic protocol of Garner's aldehyde (164), which is useful as a chiral building block, was achieved using a three-step sequence on a gram scale based on a similar DBAA-catalyzed Weinreb amide synthesis from N-Boc protected serine 162 as the chiral starting material.52 The optical purity of Weinreb amide 163 was high and the α-position of the carbonyl group was not racemized (Scheme 35). Notably, no racemization was observed at the α-position of the carbonyl group, yielding Garner's aldehyde with high optical purity.
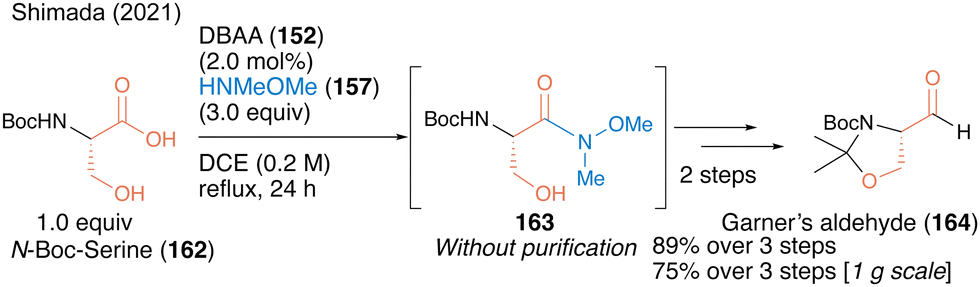 | ||
| Scheme 35 DBAA-catalyzed serine-derived Weinreb amide synthesis reported by Shimada.52 | ||
In 2023, the same group53 reported catalytic primary amide synthesis using an aqueous ammonia solution (167) as an amine substrate. Although the primary amide is a key structure for several bioactive compounds, no successful synthesis using carboxylic acid as a substrate was observed. The fact that DBAA promotes catalytic amidation without any dehydrative reagent or azeotropic reflux conditions facilitated the expansion of the catalytic synthesis using an aqueous ammonia solution. Using 10 mol% DBAA, the reaction between α-hydroxycarboxylic acid and aqueous ammonia solution (167) proceeded smoothly under reflux conditions in chlorobenzene and the corresponding primary amides 168 and 169 were obtained in high yields (Scheme 36). The synthetic efficiency of the protocol was demonstrated by the derivatization of bioactive molecules, such as octopamine (171), aegeline (173), and tembamide (174), by β-aminoalcohol 170 or 172 prepared by reducing the synthetic primary amides.
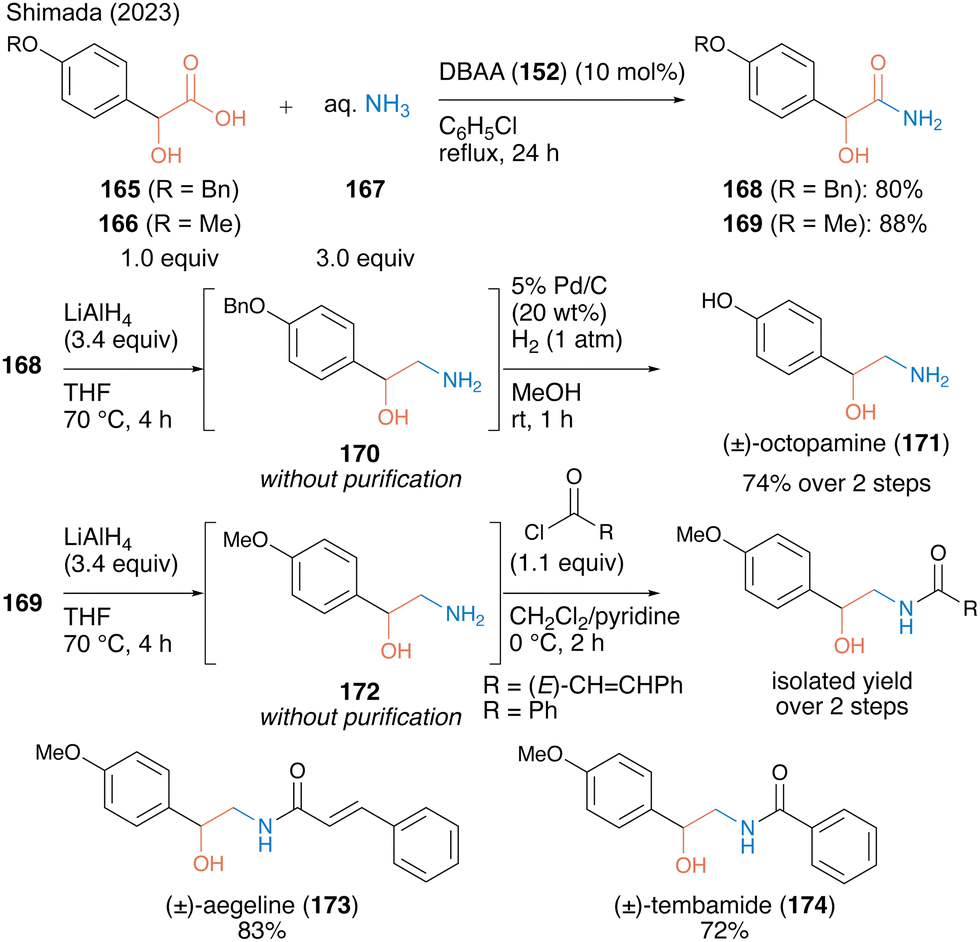 | ||
| Scheme 36 DBAA-catalyzed primary amide synthesis reported by Shimada.53 | ||
3. Catalytic direct peptide bond formation from α-amino acids
3.1. Direct peptide bond formation using boronic acid catalysis
The development of catalytic amidation has been well studied. However, there was little example to establish catalytic peptides synthesis using α-amino acids as carboxylic acid substrates. In 2013, Whiting and co-workers54 reported the first example of direct amidation using α-amino acids as both carboxylic acids and amines, respectively. First, they found that the electron-deficient o-nitrophenylboronic acid (175) was an effective catalyst for amidation using α-amino acids as carboxylic acids, and the resulting amides retained the optical purity at the α-position of carbonyl groups. Nonpolar solvents such as fluorobenzene were suitable for this reaction and the reaction proceeded smoothly in the presence of MS 3A. However, the steric hindrance of amines had a negative impact on the product yields. Next, they found that α-amino acid hydrogen chloride salts could be used for dipeptide synthesis as amine substrates in the presence of Hünig's base by in situ generation of free amine functional groups. Thus, electron-deficient arylboronic acids were effective in the presence of MS 3A in an electron-deficient and non-polar solvent for amidation using amino acids as carboxylic acids or amines. However, it has been difficult to apply a simple catalyst system using a single boronic acid for the direct access of dipeptide derivatives. As a result of continuing challenges, they found that the use of the mixture of two catalysts, o-nitrophenylboronic acid (175), and o-methylphenylboronic acid (176), which have different electronic factors, was effective for the direct dipeptide synthesis. Although this binary arylboronic acid catalyst systems required 50 mol% each of them to obtain dipeptides, the corresponding dipeptides 177–179 were afforded in moderate yields (Scheme 37).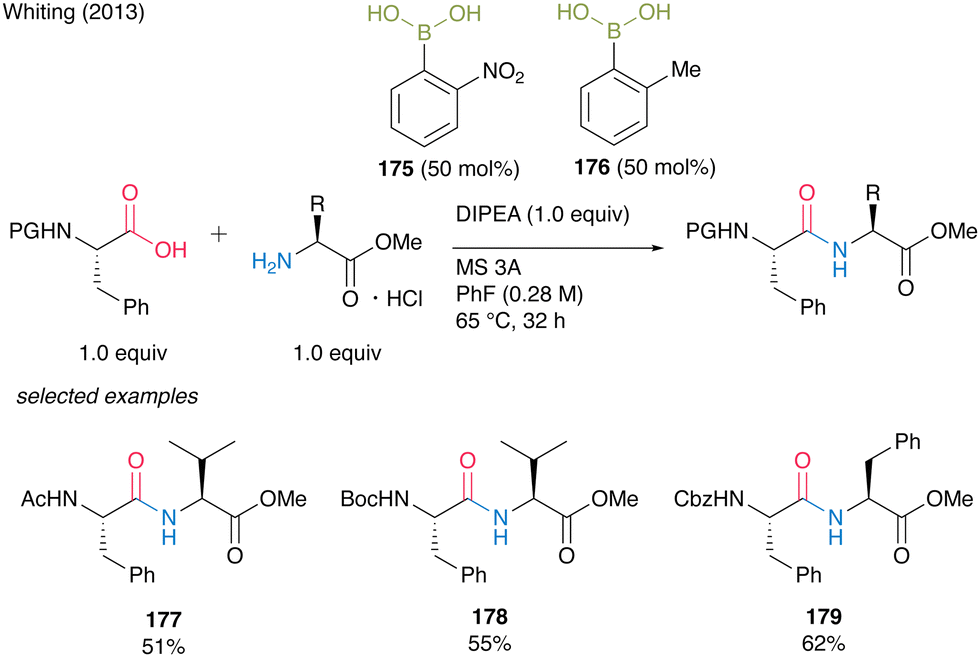 | ||
| Scheme 37 Peptide bond formation using arylboronic acids reported by Whiting.54 | ||
Certainly, these results indicate that boronic acids have room for improvement in catalytic efficiency; however, they showed that peptide bonds can be formed by boronic acid catalysis without using stoichiometric amounts of condensation reagents. Whiting's report became the milestone of peptide synthesis using boronic acids.
Thereafter, during the optimization study for catalytic amide synthesis using boronic acids with sulfur atoms, Blanchet and co-workers24 found that (2-(thiophen-2-ylmethyl)phenyl)boronic acid (54) was effective in generating catalytic peptide bond formation. The boronic acid catalyzed the coupling of each equimolar amount of Boc-Phe-OH (180) and free H-Val-OMe (181) in the presence of MS 5A in fluorobenzene at 65 °C (Scheme 38). Although there is room for improvement in catalytic efficiency (TON = 2) in only one preliminary case, the catalysis can achieve dipeptide synthesis with minimal epimerization. Thus, improving catalytic activity remains a major issue in applying aromatic boronic acids to catalyze peptide bond formation.
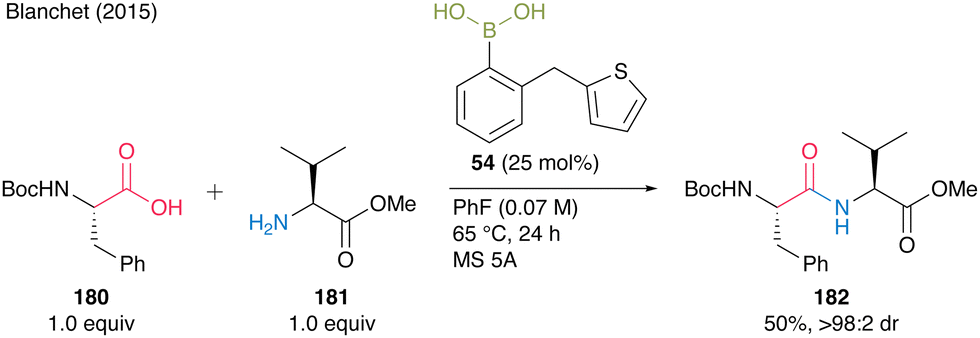 | ||
| Scheme 38 2-(Thiophen-2-ylmethyl)phenylboronic acid-catalyzed peptide bond formation reported by Blanchet.24 | ||
In 2015, Hall55 found that free, relatively acidic NH functional groups in carboxylic acids had a significant negative impact on catalytic direct peptide bond formation using their proprietary MIBA catalyst. They suggested that the inhibition of the reaction occurred because boronic acids could form 5-membered complexes with monoprotected α-amino acids derivatives through covalent bond formation with NH. The amidation of Fmoc-Leu-OH (183) and benzylamine (25) did not proceed; however, the amidation of N-methylated Fmoc-Leu-OH (184) and benzylamine (25) did (Scheme 39). Therefore, doubly protected α-amino acids such as N-phthaloyl α-amino acids or amine surrogates like α-azido and α-amino acid substrates were designed to avoid the formation of inactive complex 187, leading to inhibition during boronic acid catalysis.
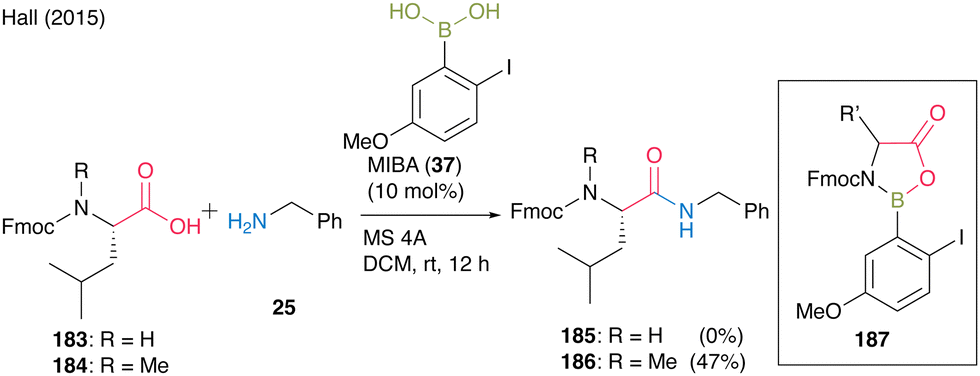 | ||
| Scheme 39 MIBA-catalyzed peptide bond formation reported by Hall.55 | ||
Indeed, a catalytic amount of MIBA (37) accelerated dipeptide 188–190 formation in moderate to good yields (28–60%) when N-phthaloyl α-amino acids were used as the carboxylic acid substrate, although steric bulkiness still negatively influenced the yield (Scheme 40).
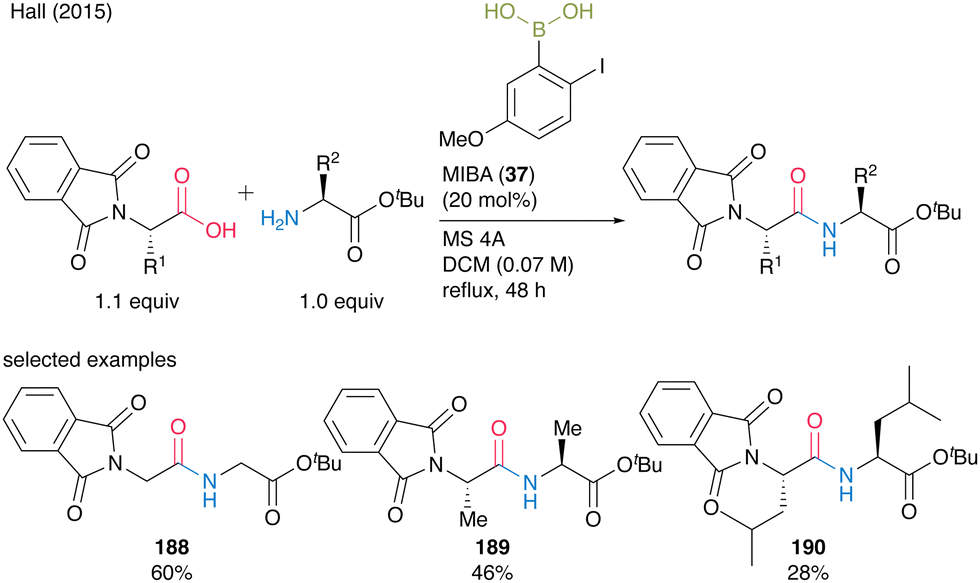 | ||
| Scheme 40 MIBA-catalyzed peptide bond formation of N-phthaloyl amino acids reported by Hall.55 | ||
When α-azido acids were used as carboxylic substrates, toluene was a more suitable solvent than dichloromethane, and dipeptides 191–193 were obtained in good yields. MIBA (37) was also applicable to the multigram-scale synthesis of N-Boc β-alanine-derived dipeptides with carbamate-protecting groups (Scheme 41).
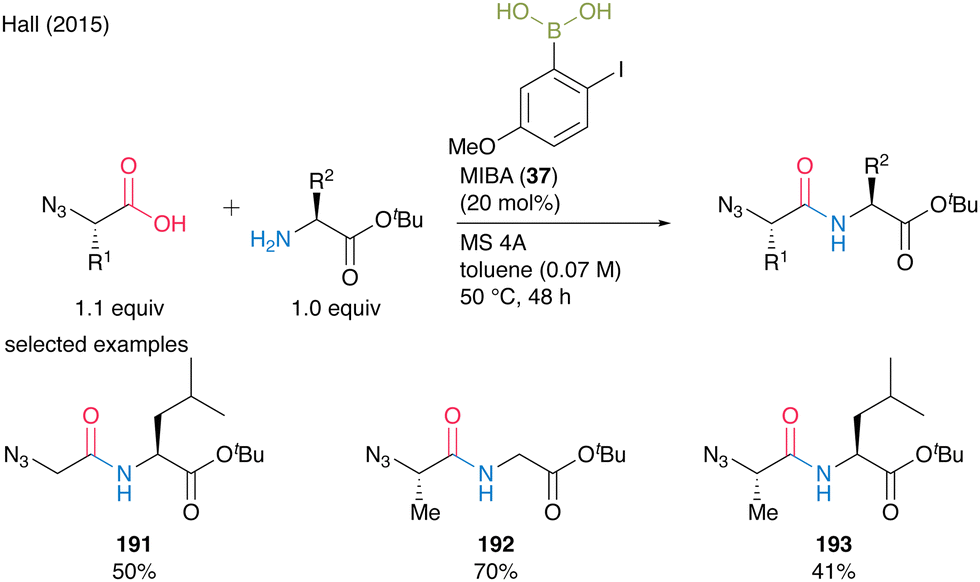 | ||
| Scheme 41 MIBA-catalyzed peptide bond formation of a-azido acids reported by Hall.55 | ||
In 2018, Ishihara and co-workers28 expanded the 2,4-bis(trifluoromethyl)phenyboronic acid (74)-catalyzed amidation reaction of carboxylic acids to α-peptide synthesis. Based on the idea that the electron-deficient N-protecting group weakens the nucleophilicity of the amino moiety of α-amino acids and prevents the construction of an inert five-membered complex derived from an arylboronic acid and an α-amino acid, N-trifluoroacetyl-protected α-amino acids were designed. Direct peptide bond formation catalyzed by 15 mol% of 2,4-bis(trifluoromethyl)phenylboronic acid (74) produced dipeptides 194–199 with minimum epimerization (Scheme 42).
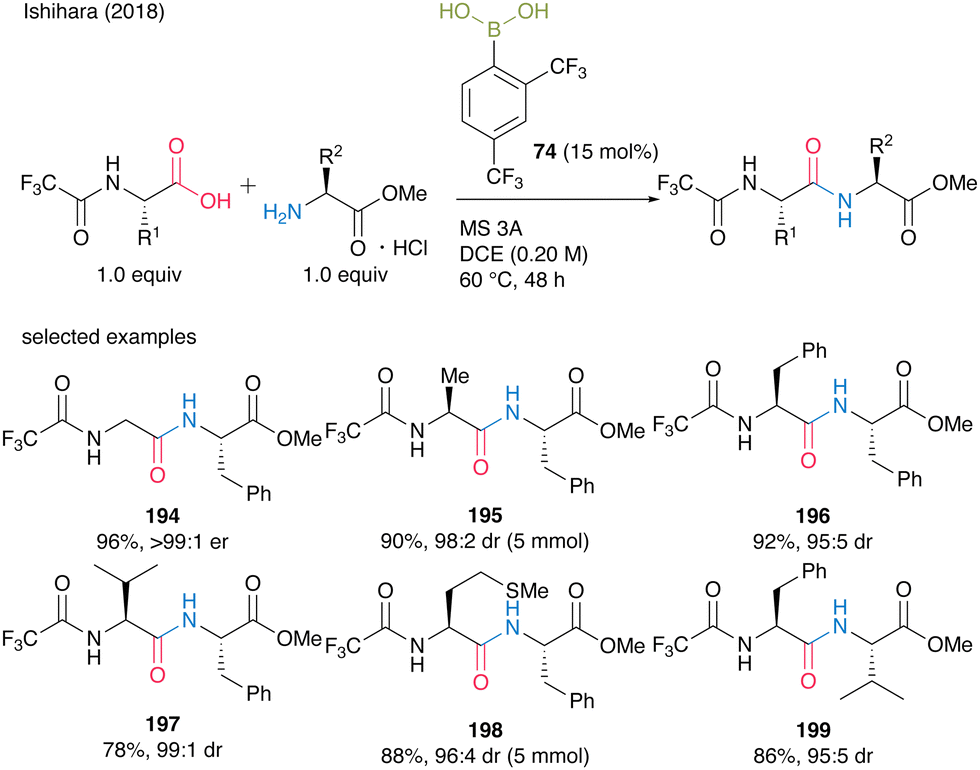 | ||
| Scheme 42 2,4-Bis(trifluoromethyl)phenylboronic acid-catalyzed peptide bond formation reported by Ishihara.28 | ||
Notably, N-trifluoromethyl-masked dipeptides 200 can be chemoselectively deprotected to produce 201 using methanolic hydrogen chloride (Scheme 43).
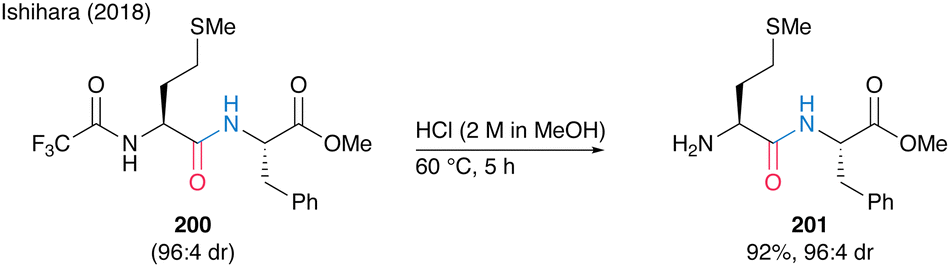 | ||
| Scheme 43 Selective deprotection of N-trifluoro acetamide reported by Ishihara.28 | ||
As described above, several scholars have reported organoboronic acid catalyzed direct dipeptide syntheses derived from α-amino acids with devised protecting groups on the nitrogen. However, the difficulty of applying frequently used carbamate-protecting groups to catalytic peptide synthesis using organoboronic acids still remains a severe issue. Recently, the application of catalytic peptide synthesis using a mono organoboron catalyst has been reported for limited examples,30,39,56 but the catalytic activity still needs to be improved.
3.2. Direct peptide bond formation using borate ester catalysis
In 2017, Sheppard's group41 reported a peptide synthesis of α-amino acids bearing a carbamate group that exhibits efficient catalytic turnover. They confirmed that a commercially available borate ester B(OCH2CF3)3 (118) was the most effective catalyst for the reaction under azeotropic reflux conditions with Dean–Stark apparatus in tert-amyl methyl ether (TAME). The reaction produced dipeptides or tripeptides in high to excellent yields (82–96%) with a catalytic amount of B(OCH2CF3)3 (118, 10–20 mol%) without epimerization (Scheme 44).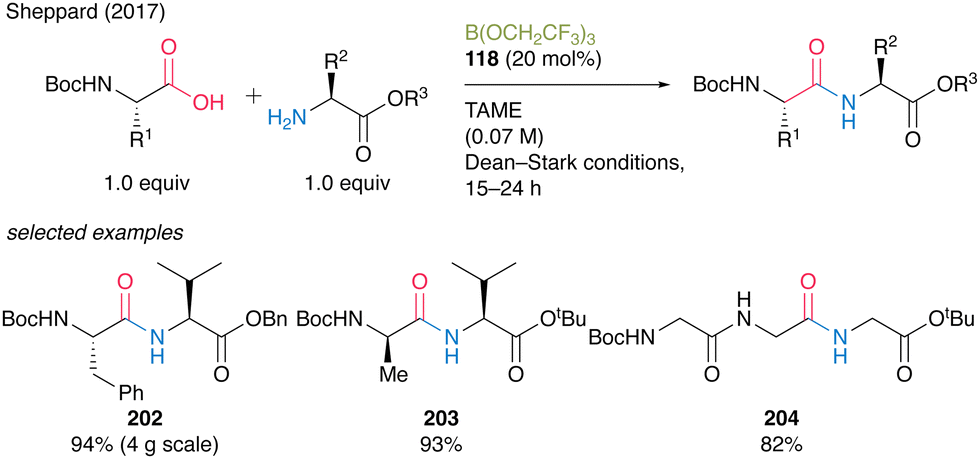 | ||
| Scheme 44 B(OCH2CF3)3-catalyzed peptide bond formation reported by Sheppard.41 | ||
A practical advantage of this protocol is the simple purification procedure of the target peptides 202–204 by filtration using scavenger resins to remove unreacted α-amino acid substrates and catalysts. Surprisingly, this environmentally benign catalysis can be applicable to the chemoselective amidation of unprotected α-amino acids with amine, although its application to peptide bond formation is limited.57
3.3. Direct peptide bond formation using multiboron catalysis
Recently, a series of successful reports of peptide synthesis using multinuclear organoboron catalysts have been published. In 2018, Kumagai and Shibasaki group58 reported DATB (125)-catalyzed peptide bond formation reactions. For this catalysis, commercially available α-amino acid hydrogen chloride salts could be directly used in the presence of MS 4A. Although dipeptides often construct oxazolones and promote epimerization during peptide bond formation, electrophilic activation with DATB (125) is not stringent, thereby making amidation of dipeptides as carboxylic substrates to produce tripeptides 206–208 in high yields without epimerization (Scheme 45).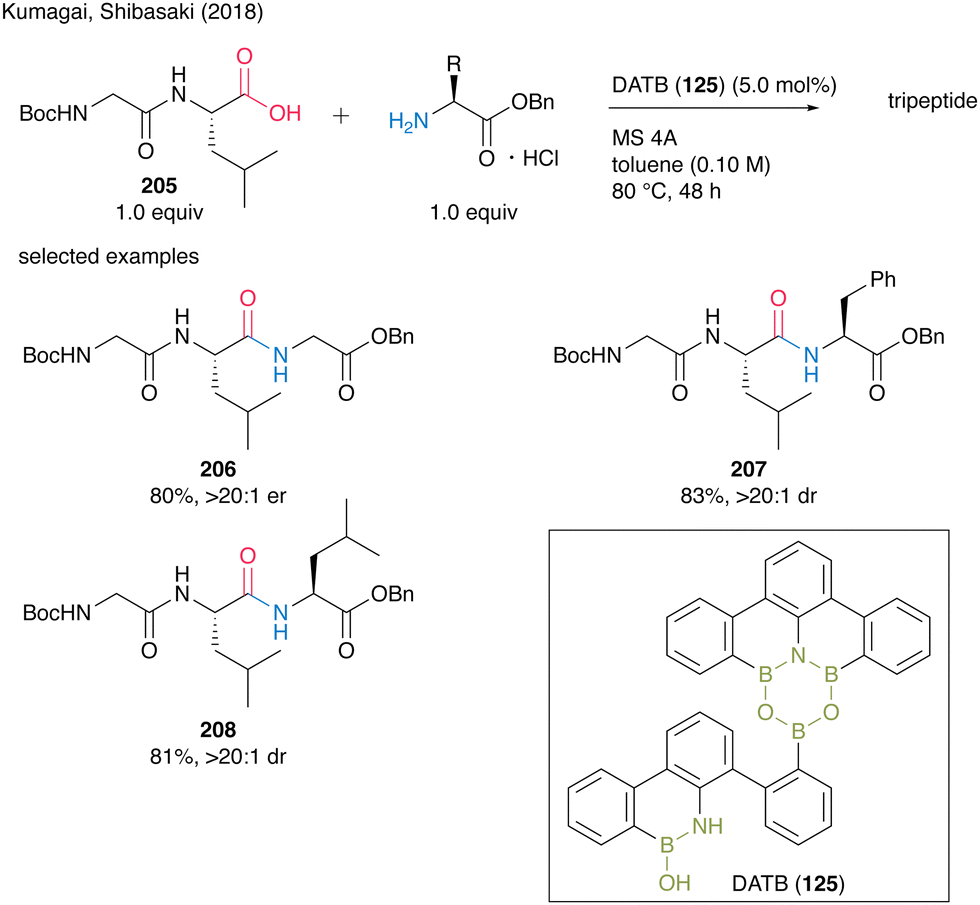 | ||
| Scheme 45 DATB-catalyzed peptide bond formation reported by Kumagai and Shibasaki.58 | ||
They also demonstrated catalytic peptide synthesis using Fmoc-protected α-amino acids as carboxylic acid substrates, suggesting future application for solid phase peptide synthesis (SPPS). Indeed, the reaction catalyzed by DATB (125) proceeded smoothly to convert both nonfunctionalized and functionalized natural α-amino acids, affording the corresponding peptides 209–214 without epimerization in most cases (Scheme 46).
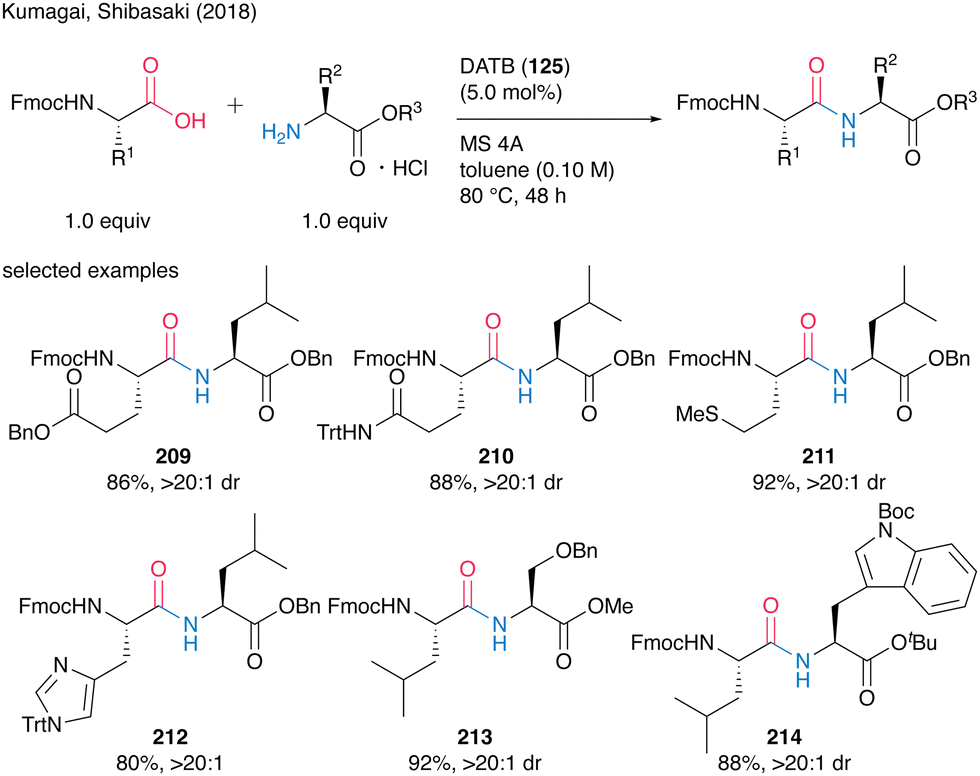 | ||
| Scheme 46 DATB-catalyzed peptide bond formation of Fmoc-protected amino acids reported by Kumagai and Shibasaki.58 | ||
Although the highly reactive carboxylic acid substrates exhibited partial epimerization at 80 °C, the corresponding dipeptides 215–217 were obtained without notable stereochemical erosion via coupling reaction at a lower temperature of 50 °C (Scheme 47).
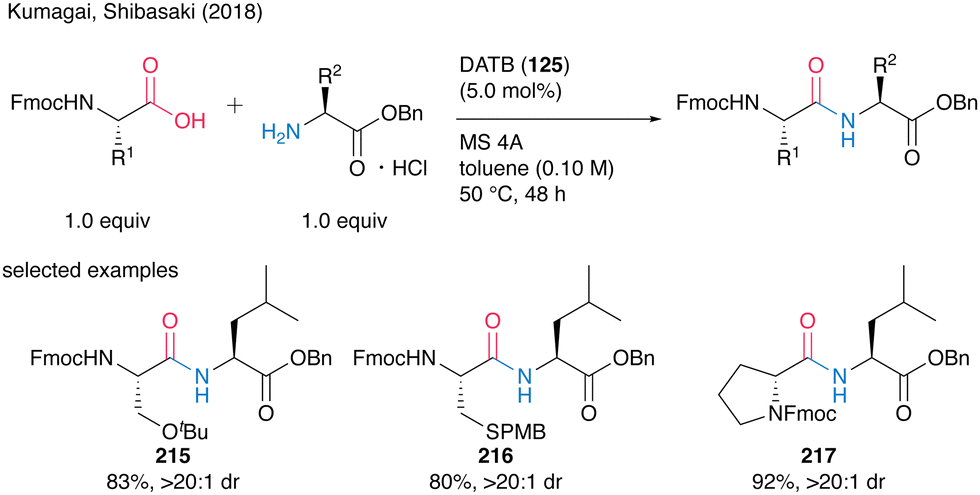 | ||
| Scheme 47 DATB-catalyzed peptide bond formation at lower temperatures reported by Kumagai and Shibasaki.58 | ||
The synthetic efficiency of DATB (125)-catalyzed peptide bond formation was highlighted by the convergent chemical synthesis of biologically active pentapeptides OGP(10–14) and H-Tyr-Gly-Phe-Gly-Gly-OH (Scheme 48). They achieved an efficient synthesis of the target peptide by repeating the catalytic protocol four times, including dipeptide and tripeptide fragment synthesis processes. Notably, 0.5 mol% DATB (125) in dipeptide synthesis of Fmoc-Gly-Gly-OtBu (220) by the coupling of Fmoc-Gly-OH (219) with H-Gly-OtBu·HCl (218·HCl) recorded a TON of 180. The deprotection of Fmoc-Tyr(tBu)-Gly-Phe-Gly-Gly-OtBu (226) formed by DATB-catalyzed coupling of tripeptide and dipeptide led to the synthesis of OGP(10–14).
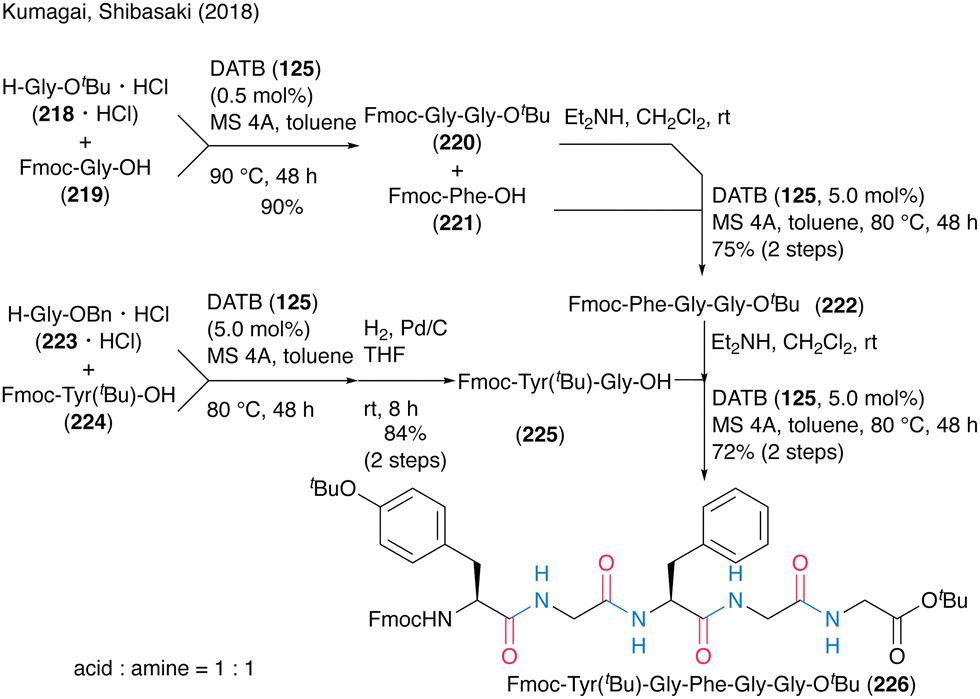 | ||
| Scheme 48 DATB-catalyzed oligopeptide synthesis reported by Kumagai and Shibasaki.58 | ||
In 2020, Takemoto and co-workers59 developed a catalytic direct peptide bond formation reaction using a multiboron catalyst possessing pseudo-active B–C–B moieties. Based on the structure of the active intermediate reported by Whiting et al.,27 they assumed that a catalyst possessing gem-diboronic acid (gem-DBA) structure would be more effective in amidation of amino acids with lower reactivity than simple carboxylic acids. Initially, catalytic amidation of α-amino acid 180 with simple amine 25 was investigated using 228 or 229 as a catalyst, demonstrating that the bulkiness of both the substrate and center atom of the catalyst significantly influenced product yields (Scheme 49).
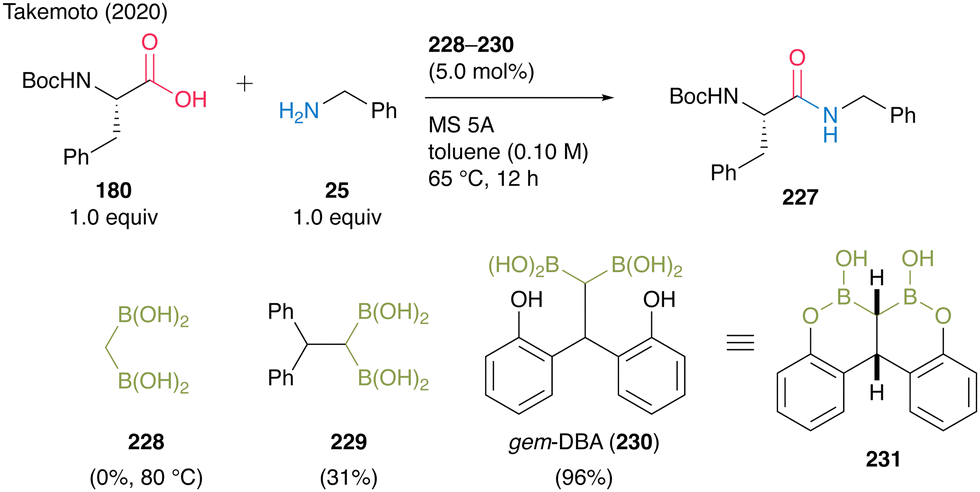 | ||
| Scheme 49 gem-DBA-catalyzed peptide bond formation reported by Takemoto.59 | ||
Moreover, gem-DBA (230), which has a phenolic hydroxyl group, exhibits high catalytic activity. The in situ cyclocondensation of 230 to 231 reduces the bulkiness around the carbonyl group of the electrophile, enhancing attack by nucleophiles to promote catalytic efficiency. Owing to the covalent bond formation between the linker and boron atoms, gem-DBA effectively causes electrophilic activation of two carboxylic acid moieties (Fig. 11).
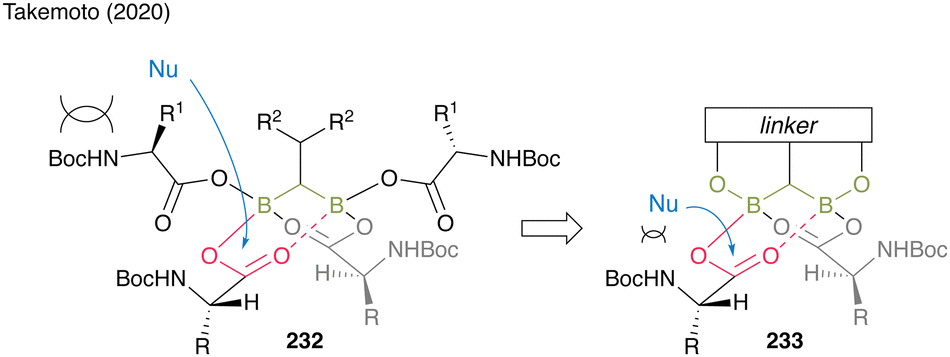 | ||
| Fig. 11 Design of the gem-DBA catalyst reported by Takemoto.59 | ||
Notably, the reaction of N-trifluoroacetyl-protected α-amino acid 234 with 235·HCl proceeded to produce dipeptide 236 at room temperature (Scheme 50). This is the first successful unprecedented catalytic peptide synthesis at room temperature.
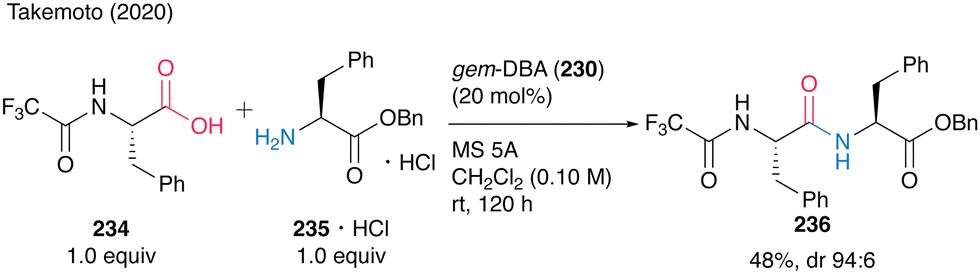 | ||
| Scheme 50 gem-DBA-catalyzed peptide bond formation at room temperature reported by Takemoto.59 | ||
This catalysis tolerated a wide range of functionalized α-amino acids incorporating α-side chains, such as alkynes, sulfur atoms, and amide moieties, to produce dipeptides 237–242 in moderate to high yields with minimum epimerization (Scheme 51). Cyclopropane, unprotected thiol, and NH-free indole functional groups also survived during the catalysis. Commercially available amine hydrogen chloride salts could be used as amine substrates without coexisting bases in the presence of MS 5A, but the hydrogen bromide and tosylate salts of amines could not be used.
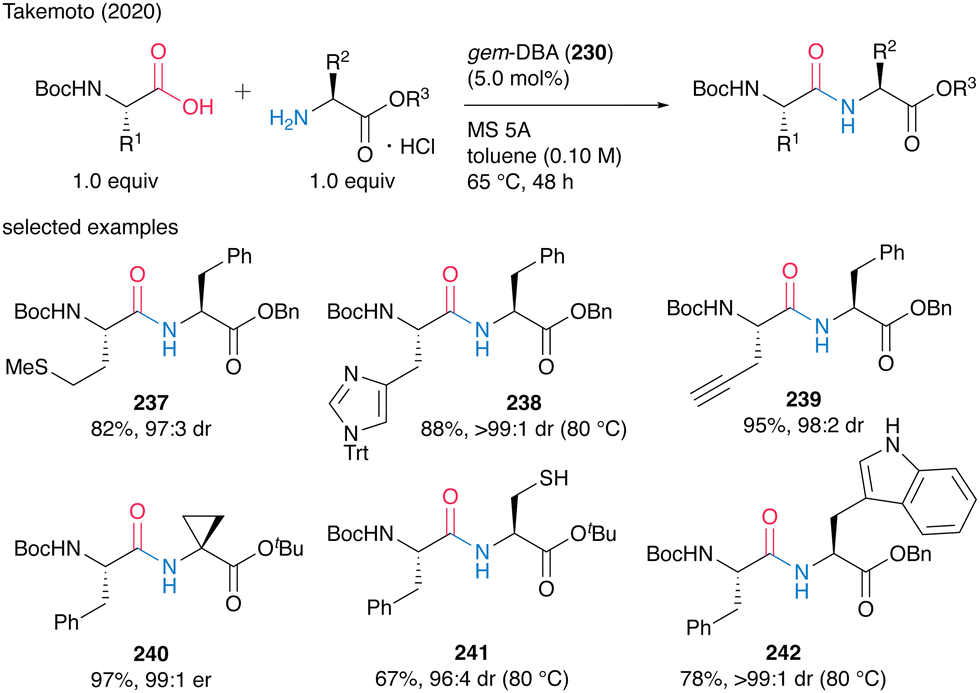 | ||
| Scheme 51 gem-DBA-catalyzed dipeptide synthesis reported by Takemoto.59 | ||
The gem-DBA (230)-catalyzed peptide bond formation could be applied to the synthesis of oligopeptide 247 (Scheme 52). Dipeptides 244 and 246 were prepared by catalytic peptide synthesis using gem-DBA. To avoid cyclization during deprotection, the tert-butyl ester protecting group was suitable for dipeptide C-terminus. The deprotection of 244 and 246 was followed by gem-DBA (230)-catalyzed coupling to produce the corresponding tetrapeptide 247 in 57%. This Fmoc-based gem-DBA catalytic method is thought to be suitable for application in SPPS.
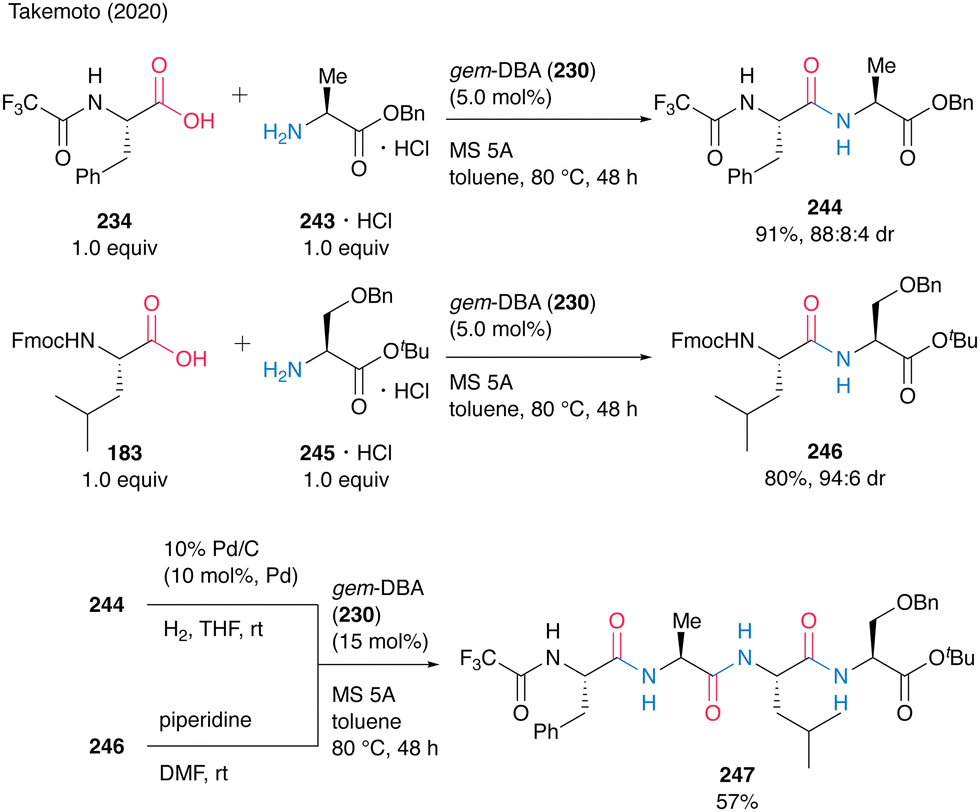 | ||
| Scheme 52 gem-DBA-catalyzed oligopeptide synthesis reported by Takemoto.59 | ||
To gain mechanistic insight, they treated gem-DBA (230) in CDCl3 with MS 5A and obtained 231, and the bearing cis-configuration structure was determined by X-ray crystallography (Scheme 53). When intermediate 248 was treated with an equimolar amount of Boc-Phe-OH and proton sponge, the negative ESI-MS value of the mixture represented a corresponding structure of a dehydrative complex 231 that was assumed to be caused by a unique bidentate carboxylate activation by gem-DBA. This result indicated that gem-DBA (230) forms anion-binding with carboxylate anions, unlike the usual arylboronic acids.
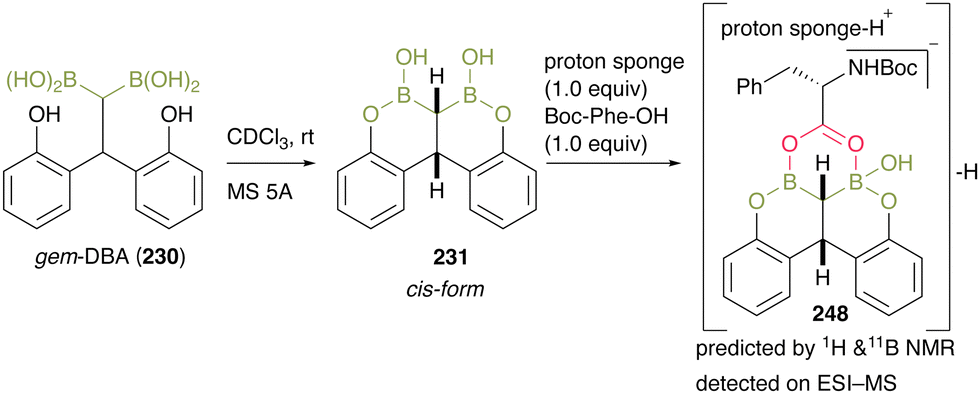 | ||
| Scheme 53 Mechanistic insights reported by Takemoto.59 | ||
In the same year, Shimada and co-workers60 reported DBAA (152) as an effective catalyst for the hydroxy-directed dehydrative peptide bond formation of β-hydroxy-α-amino acids such as serine, threonine, and β-hydroxy valine as carboxylic acid substrates, and various amino acid esters were suitable for this reaction as amine substrates, affording the corresponding dipeptides 249–255 in high to excellent yields (Scheme 54).
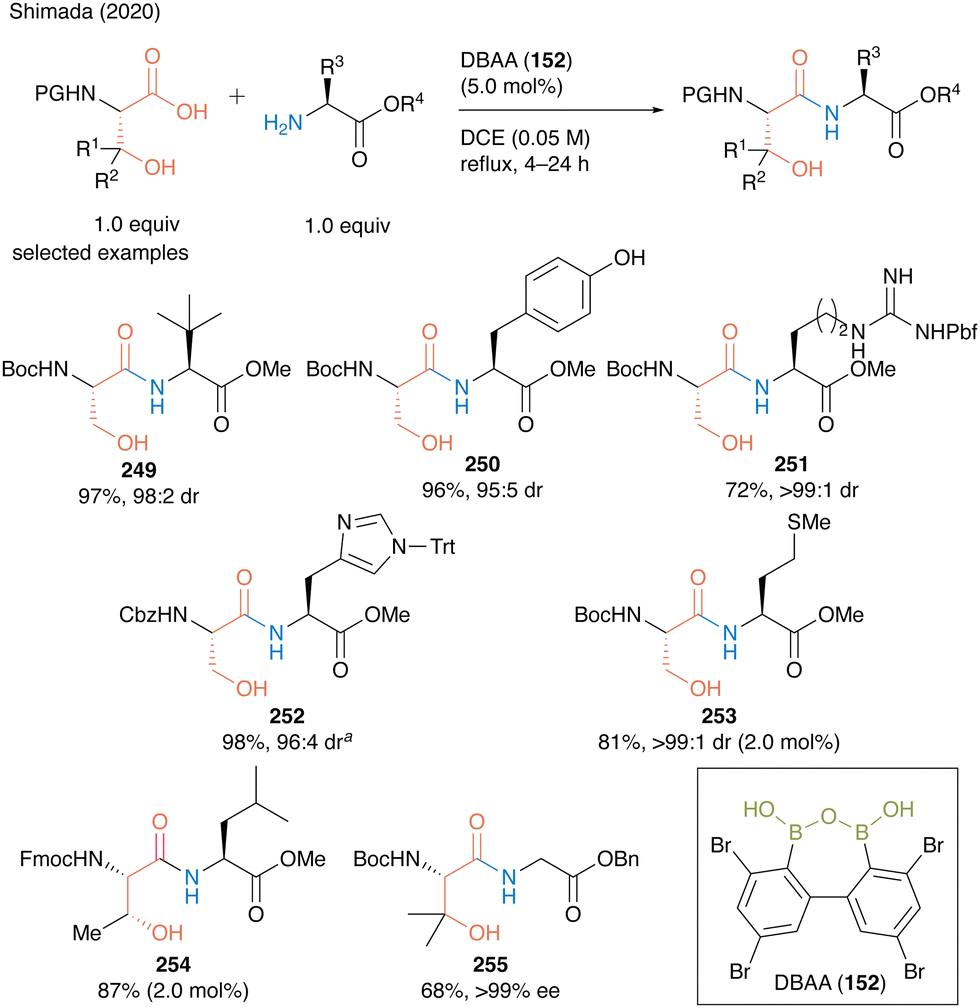 | ||
| Scheme 54 DBAA-catalyzed peptide bond formation reported by Shimada: aPerformed with HCl salt of amino ester (1.0 equiv.) in the presence of MS 4A.60 | ||
Notably, this DBAA catalysis proceeded smoothly without azeotropic reflux using Dean–Stark apparatus or dehydrative reagents, and the desired dipeptides were obtained with minimal epimerization. DBAA (152) can selectively activate α-amino acids containing hydroxy group at the β-position. Indeed, dehydrative condensation between serine derivative 162 and aspartic acid derivative 256 containing a free carboxyl group occurred with only a hydroxycarboxylic acid moiety, selectively producing the serine-derived dipeptide 257 (Scheme 55).
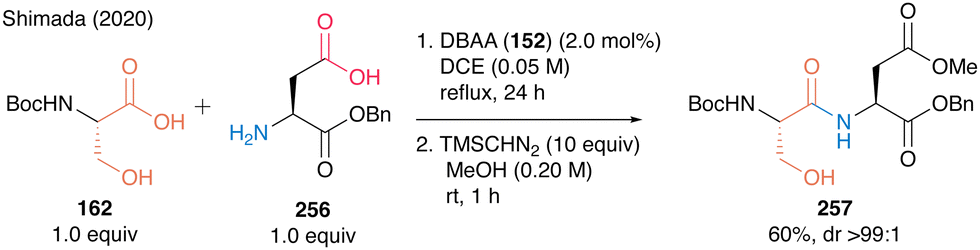 | ||
| Scheme 55 DBAA-catalyzed dipeptide synthesis reported by Shimada.60 | ||
This chemoselectivity in DBAA catalysis was highlighted by the competition reaction of Cbz-Ser-OH (258) and Cbz-Val-OH (259) with H-Gly-OtBu (218), generating dipeptide Cbz-Ser-Gly-OtBu (260) as a sole product in high yield (Scheme 56). These results show that DBAA (152)-catalyzed hydroxy-directed peptide bond formation enables selective peptide coupling of β-hydroxy-α-amino acid residue at its C-terminus.
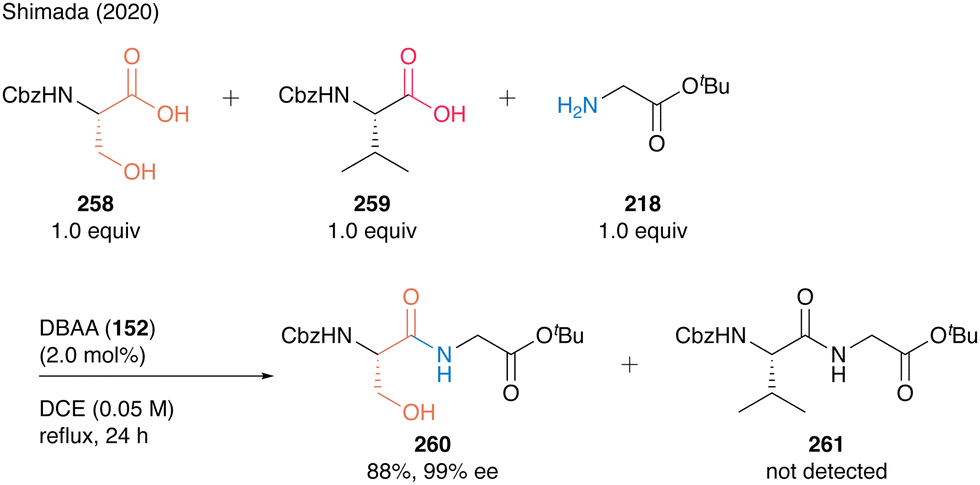 | ||
| Scheme 56 DBAA-catalyzed chemoselective peptide bond formation reported by Shimada.60 | ||
Tripeptide 263 was synthesized using DBAA-catalyzed peptide bond formation of 262 as a carboxylic substrate without any epimerization at the α-position of serine moieties (Scheme 57).
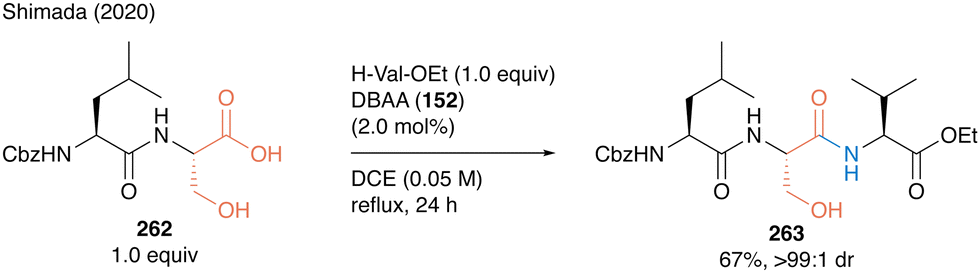 | ||
| Scheme 57 DBAA-catalyzed tripeptide synthesis reported by Shimada.60 | ||
In 2023, Shimada and co-workers also reported a concise synthesis of serine- or threonine-derived 2,5-diketopiperazines (2,5-DKPs) (Scheme 58).61 In catalytic peptide synthesis using DBAA, water is the only byproduct so that the crude product can be used for the next reaction without any purification. A three-step sequential reaction comprising catalytic peptide synthesis, deprotection of the nitrogen-protecting group, and subsequent intramolecular condensation reaction, affording 2,5-diketopiperazines 264–266 in high to excellent yields without the purification of each synthetic intermediate. The synthetic efficiency of the protocol was demonstrated by the synthesis of bioactive molecules.
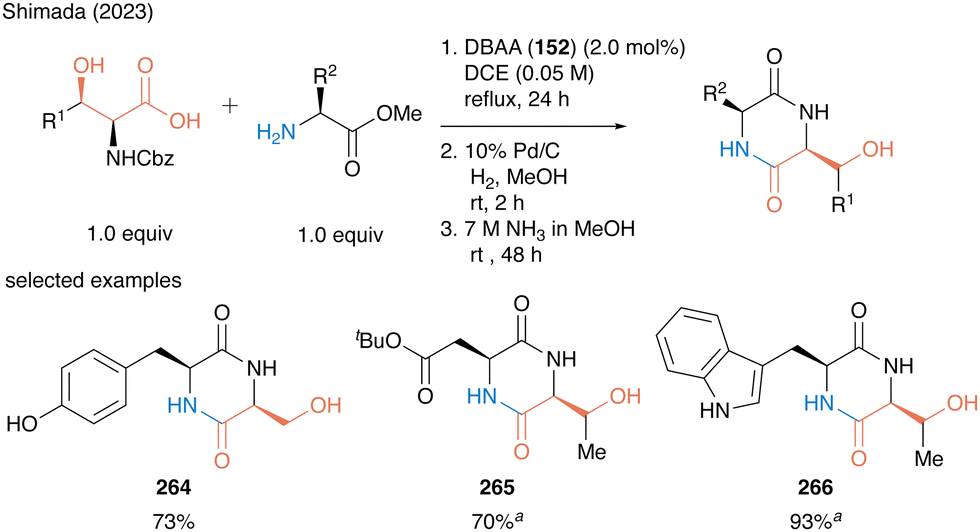 | ||
Scheme 58 Concise synthesis of 2,5-diketopiperazines using DBAA-catalyzed peptide bond formation reported by Shimada.61a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Performed with HCl salt of amino ester (1.0 equiv.) in the presence of MS 4A. Performed with HCl salt of amino ester (1.0 equiv.) in the presence of MS 4A. | ||
4. Conclusions
In this article, we review boron-catalyzed direct dehydrative amide bond and peptide bond formation reactions of carboxylic acids. Boron compounds are attractive as catalysts because of their ability to form reversible covalent bonds via dehydration and their potential Lewis acid properties. Dehydrative amide condensation using boron compounds as catalysts, which have low toxicity and are easy to handle, can become a practical and environmentally friendly method to replace conventional condensation reactions using stoichiometric amounts of reagents.Historically, this research field began with the development of dehydrative amidation using electron-deficient aromatic boronic acids. After the discovery of the effect of substituents at the ortho-position of aromatic boronic acids, several boronic acid catalysts have been developed. In parallel with these efforts, the development of recoverable and reusable boronic acid catalysts has been investigated and reusable catalysts supported on polystyrene, MCF, fluorous, and ion-exchange resins have been discovered. The direct dehydrative amide bond formation reaction of carboxylic acids with amines was expected to be applied to direct peptide bond formation reactions between amino acids. However, it was not easy to develop a practical boronic acid catalysis. The report of an improved mechanism for aromatic boronic acid-catalyzed dehydrative amidation that supports the existence of an active intermediate with a B–O–B bond was the turning point in this research field. In the past five years, several excellent multiorganoboron catalysts have been developed and research on direct dehydrative peptide bond formation reactions has rapidly progressed.
Recent developments in this field have been remarkable; however, some issues still need to be addressed in terms of catalytic activity (reducing the amount of the catalyst), energy saving (lowering reaction temperature), substrate limitations (wide range of substrate application), ease of operation (no dehydration operation), ease of catalyst availability, and catalyst reusability. To overcome the current situation and realize truly practical catalytic amide and peptide bond formation that can be used in several industries, researchers are required to continue creating novel catalysts.
Author contributions
M. K. and N. T. wrote the initial draft. N. S. proofread and revised the manuscript. M. K. and N. T. contributed equally to this work.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was partially supported by JSPS KAKENHI Grant Number 22K06506 for Scientific Research (C) (N. S.), Grant Number 24K18255 for Early-Career Scientists (M. K.), Takeda Science Foundation for Pharmaceutical Research Grants (N. S.), UBE Foundation (N. S.), Takahashi Industrial and Economic Research Foundation (N. S.), and Fukuoka Naohiko Memorial Foundation (N. S.), Nihon University for Research Grant-in-Aid for Early-Career Scientists (N. S.).Notes and references
- (a) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed; (b) R. M. de Figueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS PubMed; (c) A. Ojeda-Porras and D. Gamba-Sánchez, J. Org. Chem., 2016, 81, 11548–11555 CrossRef CAS PubMed.
- (a) Top 200 Small Molecule Drugs by Retail 2023 Sales in 2023 The Poster. https://bpb-us-e2.wpmucdn.com/sites.arizona.edu/dist/9/130/files/2024/05/2023Top200SmallMoleculePosterV5.pdf (accessed 2024-06-19); (b) Njardarson Web site. https://sites.arizona.edu/njardarson-lab/(accessed 2024-06-19); (c) N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS PubMed.
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082–5103 RSC.
- H. Charville, D. Jackson, G. Hodges and A. Whiting, Chem. Commun., 2010, 46, 1813–1823 RSC.
- (a) K. Arnold, B. Davies, R. L. Giles, C. Grosjean, G. E. Smith and A. Whiting, Adv. Synth. Catal., 2006, 348, 813–820 CrossRef CAS; (b) K. Arnold, A. S. Batsanov, B. Davies and A. Whiting, Green Chem., 2008, 10, 124–134 RSC; (c) H. Charville, D. A. Jackson, G. Hodges, A. Whiting and M. R. Wilson, Eur. J. Org. Chem., 2011, 5981–5990 CrossRef CAS.
- (a) J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS; (b) J. Magano, Org. Process Res. Dev., 2022, 26, 1562–1689 CrossRef CAS.
- (a) R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS; (b) M. Amblard, J.-A. Fehrentz, J. Martinez and G. Subra, Mol. Biotechnol., 2006, 33, 239–254 CrossRef CAS PubMed; (c) I. Coin, M. Beyermann and M. Bienert, Nat. Protoc., 2007, 2, 3247–3256 CrossRef CAS PubMed; (d) R. Behrendt, P. W. J. White and J. Offer, J. Pept. Sci., 2016, 22, 4–27 CrossRef CAS PubMed.
- (a) A. Wang, Y. Xie, J. Wang, D. Shi and H. Yu, Chem. Commun., 2022, 58, 1127–1130 RSC; (b) Y. Zhang, F. de Azambuja and T. N. Parac-Vogt, Catal. Sci. Technol., 2022, 12, 3190–3201 RSC.
- (a) K. Ishihara, Tetrahedron, 2009, 65, 1085–1109 CrossRef CAS; (b) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC; (c) S. Roy, S. Roy and G. W. Gribble, Tetrahedron, 2012, 68, 9867–9923 CrossRef CAS; (d) R. M. Lanigan and T. D. Sheppard, Eur. J. Org. Chem., 2013, 7453–7465 CrossRef CAS; (e) H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714–2742 RSC; (f) M. T. Sabatini, L. T. Boulton, H. F. Sheddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef CAS; (g) X. Wang, Nat. Catal., 2019, 2, 98–102 CrossRef; (h) M. Tosorovic and D. M. Perrin, Pept. Sci., 2020, 112, e24210 CrossRef; (i) W. Muramatsu, T. Hattori and H. Yamamoto, Chem. Commun., 2021, 57, 6346–6359 RSC; (j) K. Pedrood, S. Bahadorikhalili, V. Lotfi, B. Larijani and M. Mahdavi, Mol. Diversity, 2022, 26, 1311–1344 CrossRef CAS PubMed; (k) A. Taussat, R. M. de Figueiredo and J.-M. Campagne, Catalysts, 2023, 13, 366 CrossRef CAS.
- K. Ishihara, S. Ohara and H. Yamamoto, J. Org. Chem., 1996, 61, 4196–4197 CrossRef CAS PubMed.
- (a) E. Dimitrijević and M. S. Taylor, ACS Catal., 2013, 3, 945–962 CrossRef; (b) M. S. Taylor, Acc. Chem. Res., 2015, 48, 295–305 CrossRef CAS PubMed; (c) D. G. Hall, Chem. Soc. Rev., 2019, 48, 3475–3496 RSC; (d) K. Hollanders, B. U. W. Maes and S. Ballet, Synthesis, 2019, 2261–2277 CAS; (e) M. Sawamura and Y. Shimizu, Eur. J. Org. Chem., 2023, e202201249 CrossRef CAS; (f) P. Acosta-Guzman, A. Ojeda-Porras and D. Gamba-Sanchez, Adv. Synth. Catal., 2023, 365, 4359–4391 CrossRef CAS; (g) B. J. Graham and R. T. Raines, J. Org. Chem., 2024, 89, 2069–2089 CrossRef CAS PubMed.
- For selected examples of dehydrative amidation using boric acid and its derivative as catalytsts, see: (a) P. W. Tang, Org. Synth., 2005, 81, 262–272 CrossRef CAS; (b) R. K. Mylavarapu, K. GCM, N. Kolla, R. Veeramalla, P. Koilkonda, A. Bhattacharya and R. Bandichhor, Org. Process Res. Dev., 2007, 11, 1065–1068 CrossRef CAS; (c) S. A. Ghorpade, D. N. Sawant and N. Sekar, Tetrahedron, 2018, 74, 6954–6958 CrossRef CAS.
- Dehydrative amidations using silicon reagents or a silanol catalyst were reported. For a review, see: (a) J. J. Davies, D. C. Braddock and P. D. Lickiss, Org. Biomol. Chem., 2021, 19, 6746–6760 RSC For selected recent examples, see: ; (b) W. Muramatsu and H. Yamamoto, J. Am. Chem. Soc., 2019, 141, 18926–18931 CrossRef CAS PubMed; (c) E. Morisset, A. Chardon., J. Rouden and J. Blanchet, Eur. J. Org. Chem., 2020, 388–392 CrossRef CAS; (d) W. Muramatsu, C. Manthena, E. Nakashima and H. Yamamoto, ACS Catal., 2020, 10, 9594–9603 CrossRef CAS; (e) M. C. D'Amaral, N. Jamkhou and M. J. Adler, Green Chem., 2021, 23, 288–295 RSC; (f) W. Muramatsu and H. Yamamoto, J. Am. Chem. Soc., 2021, 143, 6792–6797 CrossRef CAS PubMed; (g) J. J. Davies, D. C. Braddock and P. D. Lickiss, Org. Biomol. Chem., 2021, 19, 6746–6760 RSC; (h) T. Lainer, F. Czerny and M. Haas, Org. Biomol. Chem., 2022, 20, 3717–3720 RSC; (i) D. C. Braddock, J. J. Davies and P. D. Lickiss, Org. Lett., 2022, 24, 1175–1179 CrossRef CAS PubMed; (j) T. Hattori and H. Yamamoto, J. Am. Chem. Soc., 2022, 144, 1758–1765 CrossRef CAS PubMed; (k) W. Muramatsu and H. Yamamoto, Org. Lett., 2022, 24, 7194–7199 CrossRef CAS PubMed; (l) D. C. Braddock, B. C. Rowley, P. D. Lickiss, S. J. Fussell, R. Qamar, D. Pugh, H. S. Rzepa and A. J. P. White, J. Org. Chem., 2023, 88, 9853–9869 CrossRef CAS PubMed; (m) T. Nobuta, N. Tsuchiya, Y. Suto and N. Yamagiwa, Org. Biomol. Chem., 2024, 22, 703–707 RSC.
- (a) A. H. Hoveyda, D. A. Evans and G. C. Fu, Chem. Rev., 1993, 93, 1307–1370 CrossRef CAS; (b) S. Bhadra and H. Yamamoto, Chem. Rev., 2018, 118, 3391–3446 CrossRef CAS PubMed; (c) T. Sawano and H. Yamamoto, J. Org. Chem., 2018, 83, 4889–4904 CrossRef CAS PubMed; (d) W. Muramatsu, T. Hattori and H. Yamamoto, Bull. Chem. Soc. Jpn., 2020, 93, 759–767 CrossRef CAS.
- K. Arnold, B. Davies, D. Hérault and A. Whiting, Angew. Chem., Int. Ed., 2008, 47, 2673–2676 CrossRef CAS PubMed.
- R. M. Al-Zoubi, O. Marion and D. G. Hall, Angew. Chem., Int. Ed., 2008, 47, 2876–2879 CrossRef CAS PubMed.
- N. Gernigon, R. M. Al-Zoubi and D. G. Hall, J. Org. Chem., 2012, 77, 8386–8400 CrossRef CAS PubMed.
- T. Marcelli, Angew. Chem., Int. Ed., 2010, 49, 6840–6843 CrossRef CAS PubMed.
- R. Yamashita, A. Sakakura and K. Ishihara, Org. Lett., 2013, 15, 3654–3657 CrossRef CAS PubMed.
- E. K. W. Tam, Rita, L. Y. Liu and A. Chen, Eur. J. Org. Chem., 2015, 1100–1107 CrossRef CAS.
- T. M. El Dine, W. Erb, Y. Berhault, J. Rouden and J. Blanchet, J. Org. Chem., 2015, 80, 4532–4544 CrossRef PubMed.
- K. Ishihara and Y. Lu, Chem. Sci., 2016, 7, 1276–1280 RSC.
- C. Wang, H.-Z. Yu, Y. Fu and Q.-X. Guo, Org. Biomol. Chem., 2013, 11, 2140–2146 RSC.
- S. Arkhipenko, M. T. Sabatini, A. S. Batsanov, V. Karaluka, T. D. Sheppard, H. S. Rzepa and A. Whiting, Chem. Sci., 2018, 9, 1058–1072 RSC.
- K. Wang, Y. Lu and K. Ishihara, Chem. Commun., 2018, 54, 5410–5413 RSC.
- J. Zhou, M. Paladino and D. G. Hall, Eur. J. Org. Chem., 2022, e202201050 CrossRef CAS.
- B. Pan, D.-M. Huang, H.-T. Sun, S.-N. Song and X.-B. Su, J. Org. Chem., 2023, 88, 2832–2840 CrossRef CAS PubMed.
- F. Almetwali, J. Rouden and J. Blanchet, Eur. J. Org. Chem., 2023, e202300720 CrossRef CAS.
- K. Ishihara, S. Kondo and H. Yamamoto, Synlett, 2001, 1371–1374 CrossRef CAS.
- R. Latta, G. Springsteen and B. Wang, Synthesis, 2001, 1611–1613 CrossRef CAS.
- T. Maki, K. Ishihara and H. Yamamoto, Org. Lett., 2005, 7, 5043–5046 CrossRef CAS PubMed.
- N. Gernigon, H. Zheng and D. G. Hall, Tetrahedron Lett., 2013, 54, 4475–4478 CrossRef CAS.
- L. Gu, J. Lim, J. L. Cheong and S. S. Lee, Chem. Commun., 2014, 50, 7017–7019 RSC.
- Y. Lu, K. Wang and K. Ishihara, Asian J. Org. Chem., 2017, 6, 1191–1194 CrossRef CAS.
- Y. Du, T. Barber, S. E. Lim, H. S. Rzepa, I. R. Baxendale and A. Whiting, Chem. Commun., 2019, 55, 2916–2919 RSC.
- K. T. Fridianto, Y.-P. Wen, L.-C. Lo and Y. Lam, RSC Adv., 2023, 13, 17420–17426 RSC.
- T. Maki, K. Ishihara and H. Yamamoto, Org. Lett., 2006, 8, 1431–1434 CrossRef CAS PubMed.
- M. T. Sabatini, L. T. Boulton and T. D. Sheppard, Sci. Adv., 2017, 3, e1701028 CrossRef PubMed.
- Sheppard reported B(OCH2CF3)3 (118)-mediated dehydrative amidations, see: (a) P. Starkov and T. D. Sheppard, Org. Biomol. Chem., 2011, 9, 1320–1323 RSC; (b) R. M. Lanigan, P. Starkov and T. D. Sheppard, J. Org. Chem., 2013, 78, 4512–4523 CrossRef CAS PubMed; (c) V. Karaluka, R. M. Lanigan, P. M. Murray, M. Badland and T. D. Sheppard, Org. Biomol. Chem., 2015, 13, 10888–10894 RSC; (d) R. M. Lanigan, V. Karaluka, M. T. Sabatini, P. Starkov, M. Badland, L. Boulton and T. D. Sheppard, Chem. Commun., 2016, 52, 8846–8849 RSC.
- C. Jimenez–Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef.
- C. E. Coomber, V. Laserna, L. T. Martin, P. D. Smith, H. C. Hailes, M. J. Porter and T. D. Sheppard, Org. Biomol. Chem., 2019, 17, 6465–6469 RSC.
- H. Noda, M. Furutachi, Y. Asada, M. Shibasaki and N. Kumagai, Nat. Chem., 2017, 9, 571–577 CrossRef CAS PubMed.
- C. R. Opie, H. Noda, M. Shibasaki and N. Kumagai, Chem. – Eur. J., 2019, 25, 4648–4653 CrossRef CAS PubMed.
- R. Tsutsumi, N. Kashiwagi and N. Kumagai, J. Org. Chem., 2023, 88, 6247–6251 CrossRef CAS PubMed.
- C. R. Opie, H. Noda, M. Shibasaki and N. Kumagai, Org. Lett., 2023, 25, 694–697 CrossRef CAS PubMed.
- D. N. Sawant, D. B. Bagal, S. Ogawa, K. Selvam and S. Saito, Org. Lett., 2018, 20, 4397–4400 CrossRef CAS PubMed.
- N. Shimada, M. Hirata, M. Koshizuka, N. Ohse, R. Kaito and K. Makino, Org. Lett., 2019, 21, 4303–4308 CrossRef CAS PubMed.
- N. Shimada, N. Takahashi, N. Ohse, M. Koshizuka and K. Makino, Chem. Commun., 2020, 56, 13145–13148 RSC.
- N. Shimada, N. Ohse, N. Takahashi, S. Urata, M. Koshizuka and K. Makino, Synlett, 2021, 1024–1028 CrossRef CAS.
- N. Takahashi, H. Iwasawa, T. Kinashi, K. Makino and N. Shimada, Chem. Commun., 2023, 59, 7391–7394 RSC.
- S. Liu, Y. Yang, X. Liu, F. K. Ferdousi, A. S. Batsanov and A. Whiting, Eur. J. Org. Chem., 2013, 5692–5700 CrossRef CAS.
- S. Fatemi, N. Gernigon and D. G. Hall, Green Chem., 2015, 17, 4016–4028 RSC.
- T. M. El Dine, J. Rouden and J. Blanchet, Chem. Commun., 2015, 51, 16084–16087 RSC.
- M. T. Sabatini, V. Karaluka, R. M. Lanigan, L. T. Boulton, M. Badland and T. D. Sheppard, Chem. – Eur. J., 2018, 24, 7033–7043 CrossRef CAS PubMed.
- Z. Liu, H. Noda, M. Shibasaki and N. Kumagai, Org. Lett., 2018, 20, 612–615 CrossRef CAS PubMed.
- K. Michigami, T. Sakaguchi and Y. Takemoto, ACS Catal., 2020, 10, 683–688 CrossRef CAS.
- M. Koshizuka, K. Makino and N. Shimada, Org. Lett., 2020, 22, 8658–8664 CrossRef CAS PubMed.
- M. Koshizuka, K. Shinoda, K. Makino and N. Shimada, J. Org. Chem., 2023, 88, 6901–6910 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |