
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymyxin B stabilized DNA micelles for sustained antibacterial and antibiofilm activity against P. aeruginosa†
Alexandra
Sousa
a,
Vegard
Borøy
a,
Agnethe
Bæverud
a,
Kjersti
Julin
b,
Annette
Bayer
 c,
Morten
Strøm
d,
Mona
Johannessen
b,
Nataša
Škalko-Basnet
a and
Sybil
Obuobi
*a
c,
Morten
Strøm
d,
Mona
Johannessen
b,
Nataša
Škalko-Basnet
a and
Sybil
Obuobi
*a
aDrug Transport and Delivery Research Group, Department of Pharmacy, UIT The Arctic University of Norway, Tromsø, Norway. E-mail: sybil.obuobi@uit.no
bHost Microbe Interaction Research Group, Department of Medical Biology, UIT The Arctic University of Norway, Tromsø, Norway
cDepartment of Chemistry, University of Tromsø The Arctic University of Norway, Universitetsvegen 57, N-9037 Tromsø, Norway
dNatural Products and Medicinal Chemistry Research Group, Department of Pharmacy, University of Tromsø The Arctic University of Norway, Universitetsvegen 57, N-9037 Tromsø, Norway
First published on 21st July 2023
Abstract
Nucleic acid-based materials showcase an increasing potential for antimicrobial drug delivery. Although numerous reports on drug-loaded DNA nanoparticles outline their pivotal antibacterial activities, their potential as drug delivery systems against bacterial biofilms awaits further studies. Among different oligonucleotide structures, micellar nanocarriers derived from amphiphilic DNA strands are of particular interest due to their spontaneous self-assembly and high biocompatibility. However, their clinical use is hampered by structural instability upon cation depletion. In this work, we used a cationic amphiphilic antibiotic (polymyxin B) to stabilize DNA micelles destined to penetrate P. aeruginosa biofilms and exhibit antibacterial/antibiofilm properties. Our study highlights how the strong affinity of this antibiotic enhances the stability of the micelles and confirms that antibacterial activity of the novel micelles remains intact. Additionally, we show that PMB micelles can penetrate P. aeruginosa biofilms and impact their metabolic activity. Finally, PMB micelles were highly safe and biocompatible, highlighting their possible application against P. aeruginosa biofilm-colonized skin wounds.
 Sybil Obuobi | Dr Sybil Obuobi is an independent researcher at UIT The Arctic University of Norway (Tromsø, Norway). She received her PhD in 2018 from the National University of Singapore (Singapore). She then joined the Department of Pharmacy having won a Marie Curie Individual Fellowship for her project ‘NANOZID’ in 2019. She is currently a project leader funded by Tromsø Research Foundation's Start-up Grant since 2022. Her interests include the development of lipid/nucleic acid/peptide based drug delivery systems and their applications against microbial infections and for use in chronic wound management. She was a recipient of an Otto Bayer Fellowship (2017) and was recently recognized as the Young Researcher of the Year (Faculty of Health, UIT The Arctic University of Norway) in 2021. |
1. Introduction
The fabrication of deoxyribonucleic acids (DNA)-based nanocarriers for therapeutic applications has gained extensive recognition over the past years due to the inherent biocompatibility and programmability of DNA.1–3 Based on Watson–Crick base pairing rules, DNA sequences can be rationally designed to hybridize in a predictable manner and have been shown to self-assemble into various polyhedral nanostructures (e.g., icosahedron,3 tetrahedron4 and crosslinked nanostructures (e.g., dendrimers5 and nanogels)). These nanocarriers have been applied as topical delivery systems where their high compatibility has supported wound healing and minimized skin inflammation.6 Given the ease of functionalization, inorganic/organic materials or lipophilic compounds can be easily incorporated into DNA-based nanocarriers to modulate their physicochemical properties and extend their applications.7–9 For example, lipophilic modifications of DNA sequences generate amphiphilic compounds that self-assemble into micellar structures with a hydrophobic core and hydrophilic corona to improve drug loading. In this context, DNA-based micelles have been synthesized with hydrophobic polymers,10 lipids11 and cholesterol12 units covalently attached to DNA strands. However, these nanosized structures are mainly formed and stabilized due to hydrophobic interactions, ergo have lower stability in biological fluids as they disassemble upon dilution.13 Currently, the majority of DNA micelles in the drug delivery field has been used for cancer applications9,14–16 with little focus on antibacterial applications,11 particularly for biofilm therapy, an area that desperately needs novel therapeutic options.The current threat posed by persistent infections demands a call to action amongst researchers. It is estimated that up to 80% of chronic and recurrent infections in humans are related to biofilm formation, that involves microbes such as Pseudomonas aeruginosa. More alarming is the fact that P. aeruginosa is amongst the World Health Organization (WHO) list of global priority pathogens for which new antibiotics are necessary.17 Bacterial pathogens within biofilms are embedded in a self-produced sticky matrix that provides protection from external threats.18,19 This matrix accounts for higher antimicrobial tolerance as it restricts the penetration and retention of antibiotics.20 The notorious persistence and poor antimicrobial susceptibility of P. aeruginosa biofilms warrants novel drug delivery strategies to enhance penetration, minimize toxicity and preserve the bactericidal effect of last line antibiotics in clinics. Polymyxin B (PMB) is a last line antibiotic used against Gram-negative bacteria such as P. aeruginosa.21 It has a positive net charge at neutral pH, a hydrophilic head group and lipophilic fatty acid tail and acts by altering the permeability of the bacterial membrane.22 Recent studies have reported the development of various anionic, amphiphilic, or lipophilic systems for the delivery of polymyxin B. For instance, phenylboronic hydrogels formulated from amphiphilic triblock copolymers were reported to control the topical delivery of polymyxin B against P. aeruginosa wound infections.23
Herein, we report on the development of DNA micelles for sustained delivery of polymyxin B (PMB micelles) (Fig. 1) and their therapeutic applications against P. aeruginosa biofilms. We demonstrate that the high binding affinity of polymyxin B in DNA micelles promotes stabilization of the DNA micelles against dilution-induced disintegration. A significant effect of antibiotic/salt concentration and ionic strength was observed, thus providing a convenient opportunity to tailor the physical properties of the micelles. The release of polymyxin B was sustained while the preserved bactericidal activity reduced bacterial colonization and metabolic activity of P. aeruginosa biofilms in a dose-dependent manner. Finally, high biocompatibility against dermal cells was demonstrated. Overall, we report for the first time how antibiotics (i.e., polymyxin B) can improve the stability of DNA amphiphiles and the potential benefit of amphiphilic DNA nanocarriers for bacterial biofilm therapy.
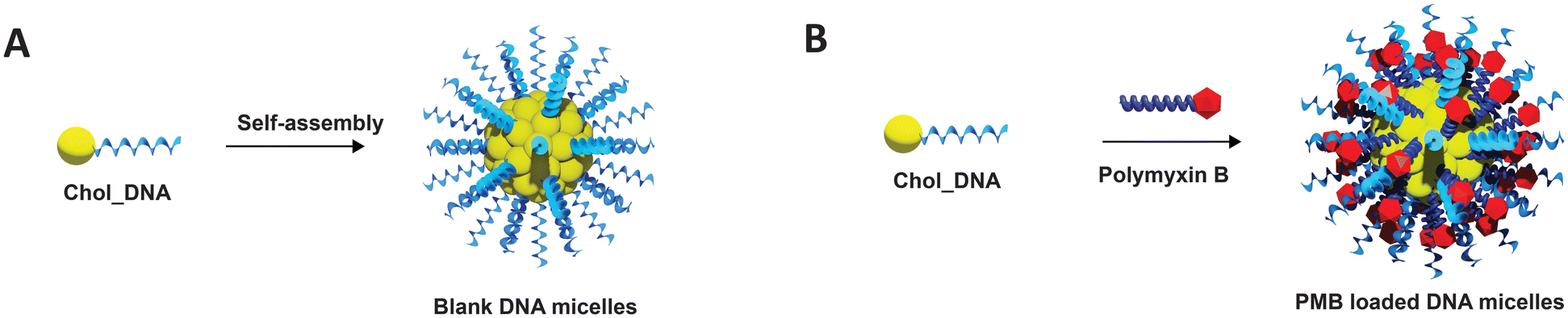 | ||
| Fig. 1 Schematic representation of the fabrication of (A) Blank DNA micelles (ssDNA micelles) and (B) polymyxin B loaded DNA micelles (PMB micelles). | ||
2. Results and discussion
2.1 Characterization and optimization of DNA-based micelles
Towards optimization of the PMB micelles for sustained antibacterial and antibiofilm action against P. aeruginosa, we first confirmed the self-assembly DNA amphiphile via size measurements using dynamic light scattering (DLS). In the absence of thermal annealing, the DNA amphiphiles had a mean size of 248 ± 38 nm and polydispersity index (PDI) of 0.51 ± 0.07 in water. In the presence of micelle buffer (Na+ 5 mM, Mg2+ 2.5 mM; pH 5.6), a significant reduction in size (83 ± 10 nm) was observed without a substantial change in PDI (0.63 ± 0.06) (Fig. 2A). These results reflect the significant role of acidic and ionic strength conditions on micelle formation and agrees with previous reports. A fundamental challenge of cholesterol conjugated DNA amphiphiles is the issue of uncontrolled hydrophobic aggregation.12,24 Additionally, the anionic backbone of DNA has also been reported to modulate the morphology of DNA amphiphiles with a high dependence on solution pH and ionic strength.25 For instance, Zhang et al., visualized the self-assembly of cholesterol-DNA micelles and nanorods via transmission electron microscopy (TEM) and reported on a pH and cations-dependent assembly.24 The authors observed that buffer conditions similar to what is used in our study induced the formation of small sized spherical micelles. While buffers prepared with 10 mM of Na+ and low Mg2+ concentrations generated nanorods, lower salt concentration of 5 mM induced the formation of spherical micelles. At low concentrations, the addition of salt can induce charge screening of the anionic backbone of DNA, which may generate compact and smaller sized micelles. Despite the size reduction, analysis of the PDI of the blank DNA micelles revealed a high PDI which was attributable to a broad size distribution and indicates a system with low morphological homogeneity.26 This was confirmed via TEM imaging as shown in Fig. S1 (ESI†).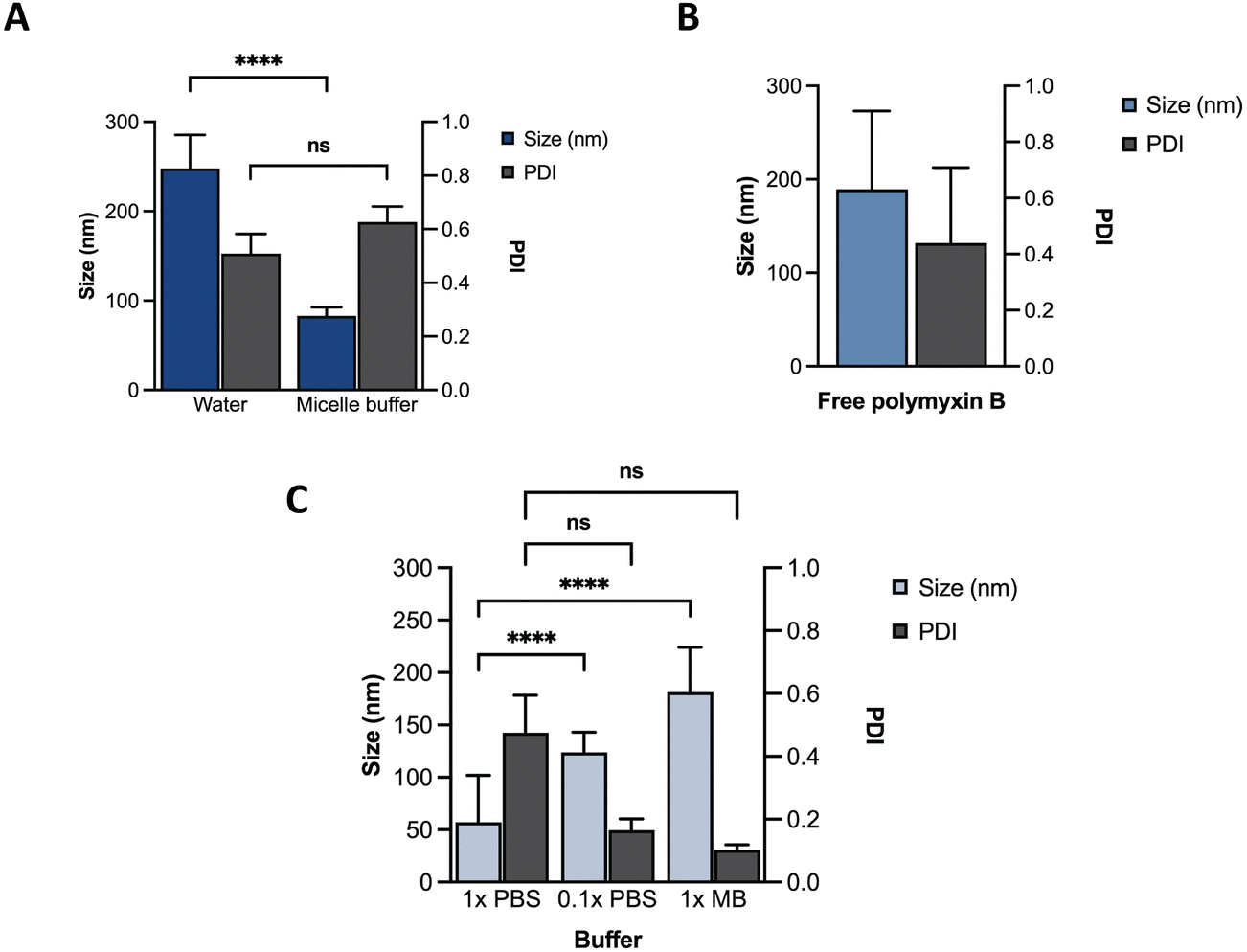 | ||
| Fig. 2 Characterization of different formulations using DLS. Size and PDI of (A) ssDNA micelles in water or micelle buffer; (B) free PMB; (C) PMB micelles in different buffers. | ||
We hypothesized that incorporation of an amphiphilic cationic antibiotic could overcome the heterogeneity of the DNA micelles. Polymyxin B (PMB) is an amphiphile drug used against Gram-negative bacteria and was employed as a model drug for this study. Due to its amphiphilic characteristics, we compared the self-assembly of PMB alone and in the presence of the DNA amphiphile. Simultaneously, we assessed the effect of buffer composition and ionic strength on the PMB micelles. In the absence of the DNA amphiphile, we observed a large size distribution (PDI of 0.44 ± 0.27) and a mean size of 189 ± 84 nm (Fig. 2B). At a concentration of 64 μg mL−1 and in the presence of the cholesterol-modified DNA amphiphile, a reduction in size (182 ± 43 nm) with a narrow/homogeneous size distribution (PDI of 0.10 ± 0.02) was observed. In the presence of PBS (1×), the micelles demonstrated a broad size distribution (PDI of 0.48 ± 0.12) and mean size of 57 ± 45 nm. Conversely, in dilute PBS solution (0.1×), a reduction in particle size (124 ± 19 nm) alongside homogeneous nanoparticles with narrow size distribution (PDI of 0.17 ± 0.04) as depicted in Fig. 2C was observed. The results confirm the impact of buffer composition in controlling the size and size distribution of the PMB micelles. To further explore the effects of the drug on the micelles, we formulated PMB micelles at different drug:DNA ratios and evaluated the impact on size, surface charge and morphology. The tested PMB concentrations were 16, 32 and 64 μg mL−1 and the DNA concentration was kept constant (20 μM). The mean size assessment via DLS showed a size increase in a drug concentration-dependent manner (Fig. 3A), with the lowest size reported for the PMB micelles with 16 μg mL−1 with an average of 22 ± 3 nm (PDI of 0.38 ± 0.05). However, the mean size of micelles with 32 μg mL−1 and 64 μg mL−1 increased to 158 ± 28 nm (PDI of 0.17 ± 0.04) and 182 ± 43 nm (PDI of 0.1 ± 0.02), respectively. For better understanding of surface properties of the carrier, zeta potential measurements were conducted. The surface charge was consistently negative independently of drug concentration (Fig. 3B). These findings suggest that the outermost portion of the micelles is comprised by the oligonucleotides.
 | ||
| Fig. 3 Characterization of different formulations using DLS, agarose gel electrophoresis and TEM. (A) Size and PDI of PMB micelles at different drug:oligonucleotide ratios (constant DNA concentration and increasing drug amounts); (B) zeta potential of PMB micelles at different drug:oligonucleotide ratios (constant DNA concentration and increasing drug amounts); (C) agarose gel electrophoresis of different formulations (Lane 1 – ladder; Lanes 2–5 – PMB micelles with drug concentrations of 128, 64, 32, 16 μg mL−1, respectively; Lane 6 and 7 – ssDNA micelles in water and micelle buffer, respectively; Lane 8 – unmodified ssDNA); (D) absorbance spectra of cholesterol-modified ssDNA, unmodified ssDNA without PMB or unmodified ssDNA and cholesterol-modified ssDNA with PMB and free PMB (E) fluorescence spectra of free dansyl-labelled PMB alone or in the presence of cholesterol-modified ssDNA (PMB micelles) or unmodified ssDNA; (F) TEM images of PMB micelles at 64 μg mL−1. | ||
As additional means to assess the interactions between DNA and drug on the micellar structures, different formulations were loaded on an agarose gel. As shown in Fig. 3C, distinct bands were observed on the DNA ladder (Lane 1) and a single band can be seen on the unmodified ssDNA strand (Lane 8). However, a smear band was generated upon the addition of cholesterol-modified ssDNA sequences both in water and micelle buffer (Lane 6 and 7, respectively). Such patterns have been previously observed in agarose gels for DNA micelles that corresponded to the formation of aggregated micellar structures27 and are attributed to transient interactions between the oligonucleotide strands.12 Despite the similarity in the smear patterns, a single band is seen at the end of each lane, potentially due to the incomplete self-assemble of the blank micelles. This band disappears in the lanes with PMB micelles (Lane 2, 3, 4 and 5) which are fabricated at different PMB concentrations (128, 64, 32 and 16 μg mL−1, respectively). Surprisingly, the patterns observed for PMB micelles at 16 μg mL−1 and plain ssDNA micelles (Lanes 5 and 6, respectively) were very similar. Opposingly, formulations with higher drug concentrations (Lane 2, 3 and 4) showed an intensified smear and the lower band faded. These observations indicate improved control over the self-assemble process, with stronger binding forces, and an increase in size as observed with the DLS measurements. At higher concentrations (128, 64 and 32 μg mL−1), the lower band fades and the smear is more intense, suggesting the presence of interactions between the antibiotic and oligonucleotides that has been associated with transient interactions.12
To study the interactions within the PMB micelles, absorbance measurements were employed to monitor the changes in the DNA molecules upon interacting with PMB. At the absorption maximum of 258 nm, the cholesterol_DNA amphiphile revealed an absorbance reading of 0.38 ± 0.02. In the presence of PMB, we observed a hyperchromic effect which corresponds to a significant increase in the absorbance reading (0.51 ± 0.04) and a red shift of 2 nm in the absorbance maximum to 260 nm. In contrast, unmodified ssDNA revealed an absorbance maximum at 257 nm with an absorbance reading of 0.38 ± 0.02, and the incubation with the PMB led to a hypochromic effect that corresponded with a reduction in absorbance to 0.27 ± 0.02. Additionally, we observed a red shift of 1 nm with an increased absorbance maximum of 258 nm (Fig. 3D). A hypochromic effect may be due to interactions between PMB and the bases of DNA while a hyperchromic effect can be attributable to electrostatic binding or binding that causes uncoiling of the DNA strand to expose the bases.28 Moreover, the observed shift in the spectra profile for the cholesterol_DNA (2 nm shift) reveals greater interactions with PMB than the unmodified_ssDNA sequence (1 nm shift).
Fluorescence spectroscopy was also used to study the binding between PMB and the DNA amphiphile. The dansyl probe displays an altered fluorescence intensity and a shift in the fluorescence emission wavelength following changes in the polarity of its environment.29,30 As shown in Fig. 3E, maximum emission at 560 nm was observed with a fluorescence intensity 7292.5 ± 6.36 for free dansyl_PMB. In the presence of the unmodified ssDNA, an increase in fluorescence intensity to 14480 ± 763.67 was noted alongside a characteristic blue shift in the emission spectrum from 560 nm to 500 nm upon binding to dansyl_PMB. Incubation of the cholesterol_DNA amphiphile with dansyl_PMB exhibited a similar characteristic blue shift in the emission spectra to 500 nm, with a greater increase in the fluorescence intensity (35499.5 ± 81.32). The increased fluorescence for dansyl_PMB in the presence of ssDNA and cholesterol_DNA led to an overall 103 ± 10.71% and 377.7 ± 1.14% increase in fluorescence intensity. The enhanced fluorescence potentially implies that PMB experiences a relatively non-polar environment30 when loaded into PMB micelles and may imply that cholesterol modification also contributes to this interaction. These observations are consistent with our proposed mechanism on the self-assembly of the PMB micelles (Fig. 1) and with previous reports on the interaction between DNA and PMB. Proposed interactions have been attributed to electrostatic interactions (between the cationic antibiotic and the polyanionic phosphate backbone from the DNA), hydrophobic interactions, hydrogen bonds and aromatic stacking.31 Considering that PMB has been reported to also self-assemble into micellar aggregates,32 its lower fluorescence intensity compared to the PMB micelles potentially indicates a more hydrophobic environment likely due to the cholesterol presence. Interestingly, when dansyl_PMB was incubated with a peptide nucleic acid (a synthetic DNA mimic without the anionic phosphate backbone), a reduction in fluorescence intensity (4041.5 ± 184.6) at 560 nm was seen (Fig. S3, ESI†) as opposed to the increase in fluorescence showed when DNA was added, suggesting that the interaction between the PMB and DNA versus PNA differs. These results also could potentially point to the importance of electrostatic interactions between PMB and DNA. Proceeding with the characterization of the formulations, the morphology of the PMB micelles at the highest concentration was visualized using transmission electron microscopy (TEM). The images revealed spherical shape of PMB micelles and sizes in agreement with DLS measurements (Fig. 3e). Images from the ssDNA showed poor micellar formation with different sizes, in agreement with DLS data (Fig. S1, ESI†).
DNA based nanostructures are also rather unstable since they are prone to dilution-induced disassembly, resulting from the depletion of positively charged ions such as magnesium and sodium. Previous work by Liu et al., has shown that the presence of counterions (e.g., sodium and potassium) enhanced the thermal stability of DNA micelles.33 We hypothesized that the blank ssDNA micelles would not withstand ion depletion whilst the incorporation of cationic PMB would contribute to an increased stability (Fig. 4A and B, respectively). We envisioned that this will be observed because of the self-assembly of two oppositely charged amphiphiles (polyanionic DNA amphiphile and cationic PMB) that would form a more stable nanosized spherical complex and significantly protect the nanoparticles against dilution-induced disintegration. To test the stability of the structure, we made serial dilutions of the ssDNA and PMB micelles in water and assessed changes in nanoparticle size and PDI with the depletion of counterions. As expected, ssDNA micelles exhibited a large size increase and broad PDI variation upon serial dilutions (Fig. 4B), indicating their lack of stability against dilution. Similar findings showed the critical micelle concentration of these structures to be 1 μM (observed with TEM images).24 Our studies are aligned with these findings, where the PDI increase was more prominent at 1.25 μM, seen after a 16x dilution (assessed using DLS). On the other hand, a highly stable complex formation for the PMB micelles was observed with no changes in size or PDI of the micelles up to a concentration of 0.63 μM (after 32× dilution) (Fig. 4C).
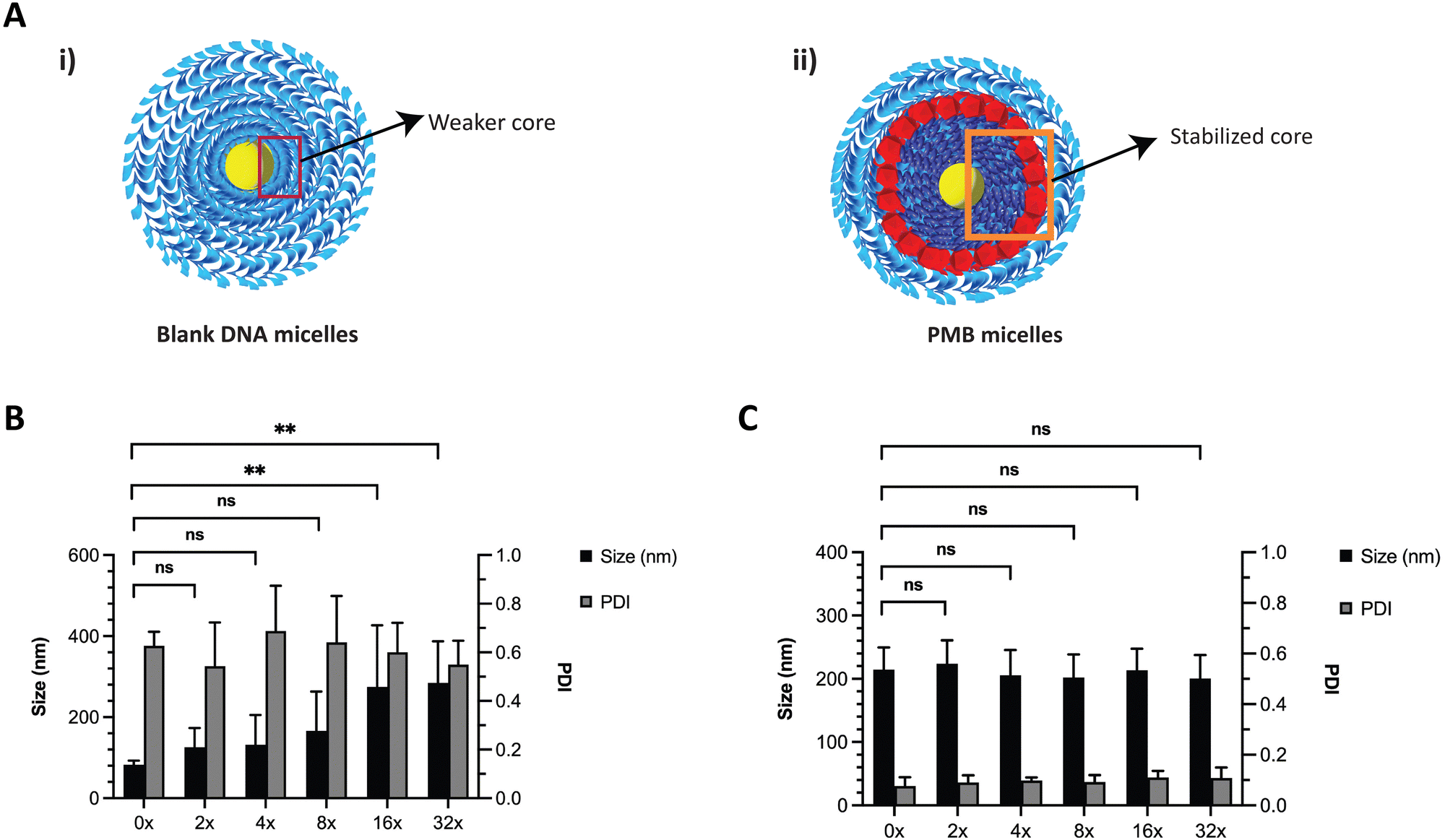 | ||
| Fig. 4 Stability of ssDNA micelles and PMB micelles upon dilution with water, showcasing the enhanced stability upon cation depletion. (A) Schematic representation of interactions in the (i) ssDNA micelles and (ii) PMB loaded DNA micelles (PMB micelles). Size and PDI changes upon serial dilution of (B) ssDNA micelles and (C) PMB micelles. | ||
Additionally, DLS measurements of a 5x dilution of PMB micelles in Mueller–Hinton broth showed that the peak size corresponding to the PMB micelles was still presence, suggesting that dilution in this media didn’t result in their disassembly (data not shown). Moreover, micellar stability over time was also characterized with this technique (Fig. S2, ESI†). After 1 h, ssDNA micelles showed a significant size increase, whereas the PMB micelles took 24 h to reach a size increase with the same magnitude. We then assessed the encapsulation efficiency of PMB in the formulation by determining the affinity of PMB to the DNA micelles. We observed a significantly high drug content of 98.26 ± 1.89% revealing that the drug is complexed with the DNA amphiphile and not freely dispersed in the aqueous media, in agreement to the observed affinity between the drug and oligonucleotides. Whilst this strong affinity proved to be important for DNA micelles’ stabilization, we questioned if these interactions would hinder the release of PMB from the nanocarrier, and therefore diminish its antibacterial activity when incorporated in the micelles. Drug release profile can be of major importance for the performance of drug delivery systems in preventing biofilm formation or biofilm destruction.
Biofilms can be associated with acidic environments (for example oral biofilms34 or respiratory35), and infection sites are also characterized by a pH drop (for example due to the excretion of acidic metabolites by bacteria36). On the other hand, these bacterial structures can also be found in chronic skin wounds, where neutral to alkaline pH is commonly observed.37 Nevertheless, since biofilms can withstand greater pH ranges comparatively to planktonic bacteria38 and the environment pH influences biofilm formation, we were interested in assessing the drug release profile under different pH conditions. Therefore, we assessed the drug release of PMB from the PMB micelles at physiological pH (7.0) and acidic pH (4.5) (Fig. 5A). The release profile of PMB from the PMB micelles was revealed to be independent of pH conditions. In particular, we observed a sustained release profile of PMB from the PMB micelles over 24 h at pH 7.0 (49.9 ± 9.7%) and pH 4.5 (45.5 ± 9.0%) respectively. This is in contrast to other reports where the pH variations are used to trigger drug release.34,37,39–41 We observed a burst release of PMB from the micelles within the first two hours, followed by a sustained release over 24 h. The observed release profile is similar to a previous report where sustained release of the amphotericin B was observed from polycarbonate micelles (48.6 ± 2.1%) and mixed micelle (59.2 ± 1.8%) formulations.42 This release profile is of interest for antibacterial activity as it provides a high antibiotic concentration within a short period of time followed by a sustained release. Micellar nanocarriers frequently carry a lipophilic drug that is accommodated within the hydrophobic core. Under these circumstances, drug release occurs via drug diffusion or micelle disassembly and can be attributed to the impact of dilution, pH or salt content.43 Since the self-assembly of the micelles is initiated and controlled by PMB and because the drug release profile is similar under acidic and neutral pH, we propose that the release mechanism of PMB from the PMB micelles is due to micellar disassembly.
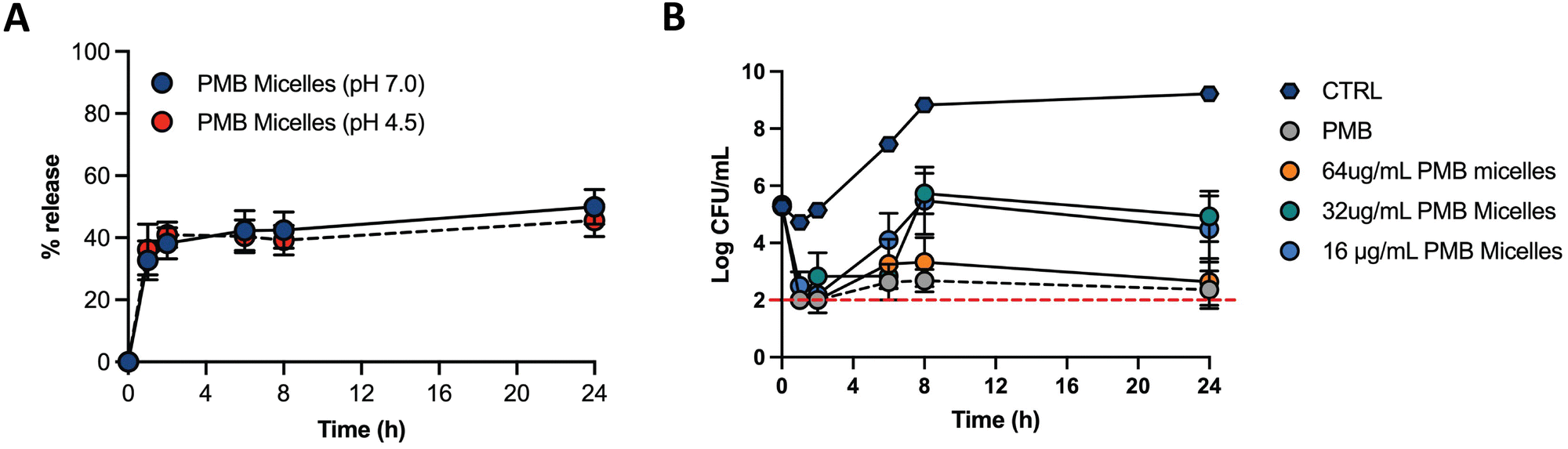 | ||
| Fig. 5 Drug release profile of PMB micelles (64 μg mL−1) at different pH and antibacterial effect of PMB micelles at different drug concentrations. (A) Drug release profile of PMB micelles at neutral and acidic pH (pH 7.0 and pH 4.5); (B) CFU mL−1 reduction of P. aeruginosa over 24 h after exposure to different PMB micelles and free PMB (64 μg mL−1). | ||
2.2 Antibacterial and biofilm inhibitory activity of PMB micelles
The antimicrobial effectiveness of the PMB micelles was assessed against planktonic P. aeruginosa via the microbroth assay to determine the minimum inhibitory concentration (MIC), as well as to evaluate the impact of a micellar formulation in comparison to the free drug (Table S2, ESI†). The MIC was estimated as the loss of visual turbidity and via absorbance readings at OD600. The data obtained indicated effective bactericidal activity of free drug and PMB micelles at 2 μg mL−1 after 18 h. These results confirm that the antibacterial activity of polymyxin B remined preserved even within the micelles. Next, we used time-kill kinetics to determine the impact of drug concentration and time on bacterial growth and death after treatment with the PMB micelles. PMB micelles at different drug concentrations were incubated with planktonic bacteria, and the killing-rate was compared with free PMB at the highest concentration used for micelles fabrication (Fig. 5B). A faster killing rate was observed for PMB micelles loaded with 32–64 μg mL−1 as well as for the free PMB solution (64 μg mL−1). After 1 h of incubation, approximately 3-log reduction in bacterial viability was observed for the free drug and PMB micelles (32–64 μg mL−1 and 64 μg mL−1, respectively). After 2 h, both the free drug and the PMB micelles loaded with 64 μg mL−1 retained a 3-log reduction in CFU of P. aeruginosa. For the PMB micelles at 16–32 μg mL−1 a 3.05–2.46-log reduction in bacteria CFU after 2 h was seen. After 8 h, the effectiveness of the formulations was more pronounced and dose-dependent; a 3.33-log reduction was reported with 64 μg mL−1versus no log reduction for the 32 μg mL−1. The killing kinetics of the free drug was similar to the PMB micelles loaded with the drug in the same concentration. Overall, rapid onset of antibacterial action in micelles loaded with 64 μg mL−1 corresponds to the observed burst release from the PMB micelles within 2 h as demonstrated in Fig. 5A.2.3 Biofilm penetration
To establish the potential antibiofilm application, we hypothesized that the PMB micelles could penetrate the full depth of mature biofilms and efficiently accumulate within P. aeruginosa biofilms, despite their anionic surface properties. To verify our hypothesis, we performed penetration assays via confocal laser scanning microscopy (CLSM). A P. aeruginosa clone (10145GFP) expressing the green fluorescent protein (GFP), was selected for the studies and the penetration assessments were made over 2 h period. The PMB micelles were labelled with rhodamine and biofilm penetration was monitored via a rhodamine-associated bright red signal. We obtained 2D and 3D projection images of the micelle-treated biofilms to illustrate the influence of time on the penetration as well as the overall depth of penetration attainable by the micelles. Control images obtained after treatment with only the buffer confirmed the formation of mature biofilms with dense clusters of P. aeruginosa as shown in Fig. 6A and B. After treatment with the PMB micelles (64 μg mL−1 PMB) for 30 min, a weak signal from the red channel and a dominant green signal representing living bacteria cells was obtained. The z-stack images obtained after 30 minutes showed shallow penetration and retention in the upper layers of the biofilm matrix (Fig. S4, ESI†). In contrast, higher intensity was obtained from the red channel on the 2D images after 2 h (Fig. 6A), which demonstrates that biofilm penetration was highly dependent on incubation time. Additionally, visual examination of the 3D-projections (Fig. 6B) also revealed strong red fluorescence throughout the whole depth of the biofilm matrix, thus confirming that the PMB micelles could achieve deep biofilm penetration. Hydrophobic and cationic properties have been reported to improve biofilm penetration while anionic features have been generally shown to limit biofilm penetration. Our results demonstrate that the PMB micelles can accumulate within biofilms despite their polyanionic properties, potentially due to their amphiphilic properties. Furthermore, we observed no overlap between GFP labelled bacteria cells and the rhodamine labelled DNA micelles within the biofilm which demonstrate that the PMB micelles solely accumulated within the biofilm matrix.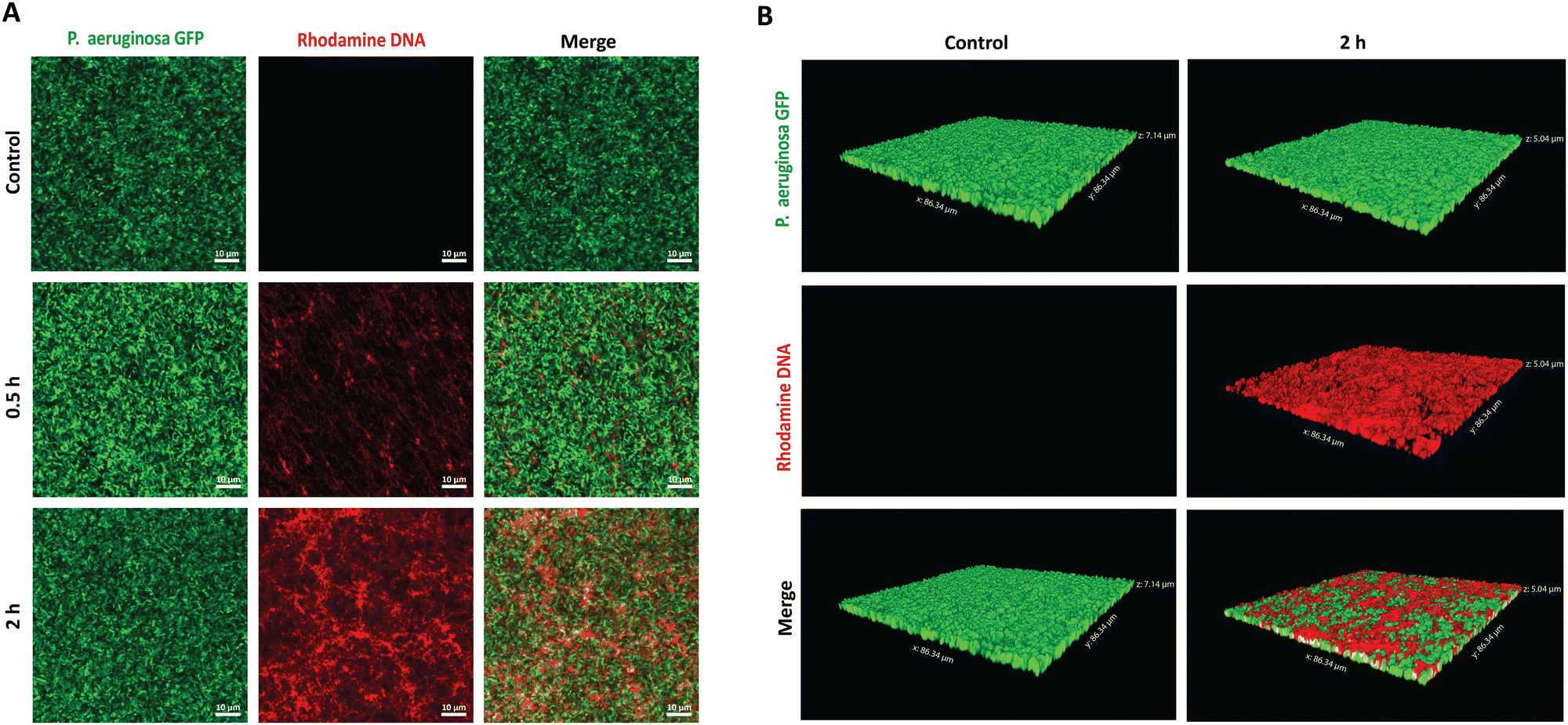 | ||
| Fig. 6 In vitro biofilm penetration evaluation of the PMB micelles. (A) CLSM 2D-images of GFP-labelled P. aeruginosa biofilms after 30 min and 2 h treatment with the PMB micelles (64 μg mL−1). (B) CLSM 3-D projections of the GFP-labelled biofilms after exposure to the PMB micelles (64 μg mL−1) for 2 h. | ||
2.4 Biofilm inhibitory effect of PMB micelles
The potential of the PMB micelles to prevent biofilm attachment was evaluated via biofilm inhibitory assays against the biofilm forming pathogen, P. aeruginosa. For this purpose, we incubated a planktonic bacterial suspension for 16 h with PMB micelles loaded at different drug concentrations and evaluated the effect on the formation of biofilm biomass via crystal violet (CV) staining. As shown in Fig. 7A, biofilm biomass decreased at the highest concentrations of PMB. For micelles loaded with 16 μg mL−1 and 32 μg mL−1, the biofilm mass was 89.91 ± 14.09% and 101.00 ± 21.26% in comparison to the control group (100%), respectively. These results show that there is no significant reduction in biofilm formation for these formulations (p = 0.78 and 0.99, respectively). Conversely, the PMB micelles (64 μg mL−1) reduced the biofilm mass to 14.15 ± 0.74% (p = 0.0002). The effect of free PMB at 64 μg mL−1 also reduced the biofilm mass to 20.56 ± 13.21% (p = 0.73) (Fig. S5, ESI†), results and confirm that the incorporation of PMB in the micelles still retains the antimicrobial activity of the drug and allows for its release to inhibit P. aeruginosa biofilms.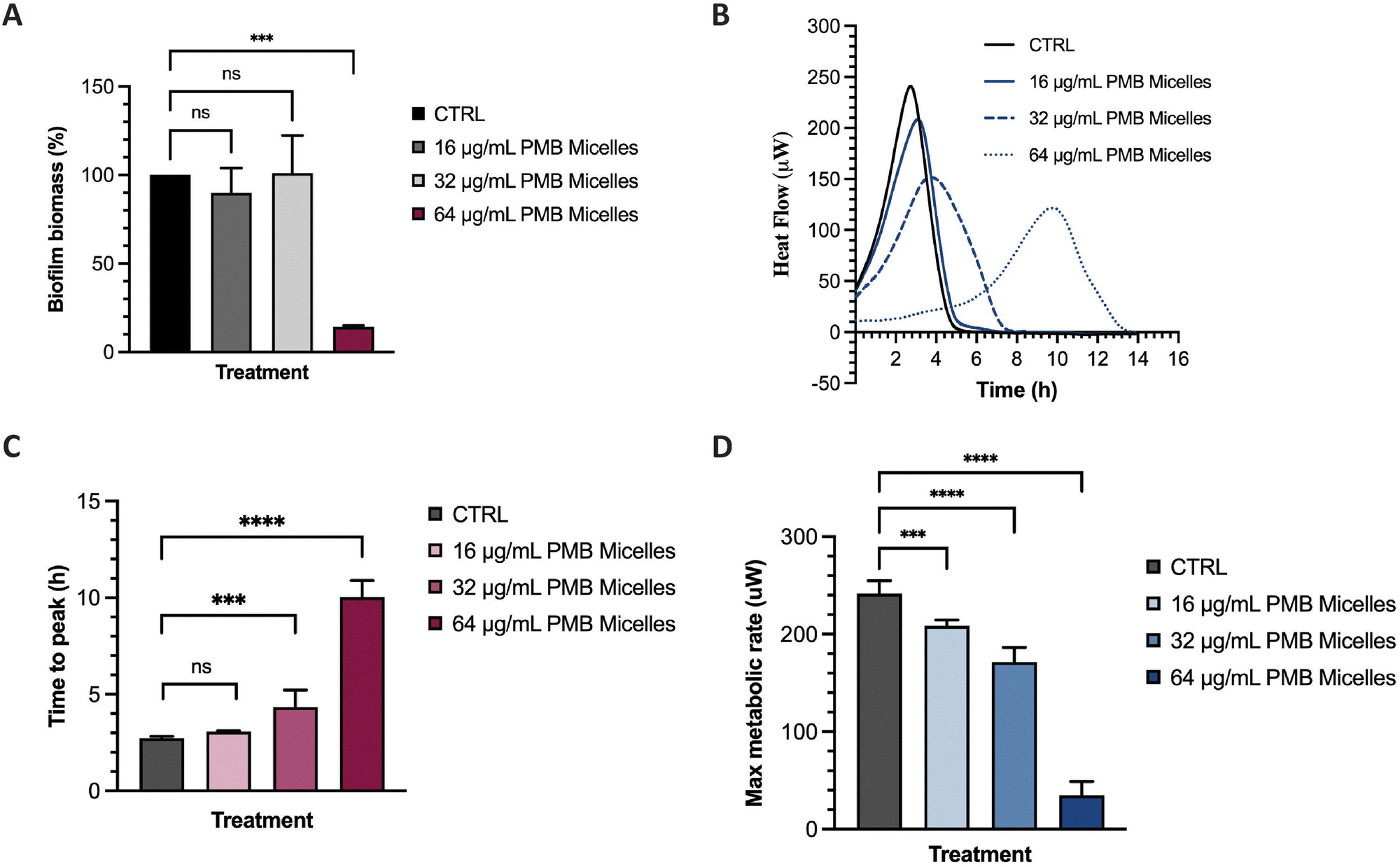 | ||
| Fig. 7 Biofilm inhibition and impact on metabolic activity of PMB micelles in P. aeruginosa. (A) Biofilm inhibition capacity of different PMB micelles; (B) thermogram showing heat flow (μW) vs. time of P. aeruginosa biofilms upon exposure to different PMB micelles; (C) time to peak (h) of treated biofilms; (D) Max metabolic rate (μM) of biofilms exposed to PMB micelles. | ||
Many experimental assays determine bacterial susceptibility to formulations using planktonic bacteria without addressing the same susceptibility profiles in biofilms, therefore the higher tolerance of biofilms, frequently associated with the presence of the extrapolymeric matrix and persister cells, is often disregarded.44 For this reason, after visualizing the high capacity of our micelles to penetrate biofilms, we evaluated the impact of the PMB micelles on pre-formed biofilms using isothermal microcalorimetry. Using this method, we can monitor bacterial metabolism in real time and assess the impact of the formulation on bacteria on their biofilm growth mode. Other studies have demonstrated the feasibility of this technique in evaluating of the antibiofilm activity of antibiotic-loaded nanocarriers.45,46 Isothermal calorimetry measures the energy generated from bacteria and provides an indication on their growth rate, an important criterion to assess the kinetics of bacterial growth.47 Polymyxin B exhibits a bactericidal action by interacting with the lipid A present on the membrane of bacteria and destabilizing the outer membrane, ultimately resulting in leakage of cytoplasmic components.48 Differences in bacterial metabolic activity induced by PMB micelles loaded with different concentrations of PMB was assessed by comparing time to peak, maximum metabolic rate and overall heat flow over time. As shown in Fig. 7B and C, a clear shift in time to peak was obtained for all the formulations in a dose-dependent manner. For PMB micelles loaded with 16 μg mL−1 of drug, a time to peak of 3.07 ± 0.05 h was observed versus the 2.72 ± 0.10 h observed in the control group. A significant delay in the time to peak was obtained in PMB micelles loaded with higher PMB concentrations of 32 μg mL−1 (4.34 ± 0.88 h) and 64 μg mL−1 (10.03 ± 0.87 h). After 6 h incubation with the micellar formulations, differences in the maximum metabolic rate were observed (Fig. 7D) where a significant reduction in the maximum metabolic activity was obtained for micelles loaded with 64 μg mL−1 (34.50 ± 14.32 μW) compared to the control (241.67 ± 13.23 μM). For micelles loaded with 16 μg mL−1 and 32 μg mL−1, a maximum metabolic rate of 208.50 ± 6.15 μM and 171.33 ± 14.92 μM was obtained after 6 h. At the end of each growth curve, there was no sign of residual metabolic activity over the 14 h period which reflects the well-known bactericidal activity of PMB. Moreover, the observed impact of the released drug on the growth kinetics is in agreement with previous work performed with other bactericidal drugs. For instance, in a study by Cirnski and colleagues,49 exposure of A. baumannii to ciprofloxacin led to a lag phase shift and increased time to peak, with a decreased maximum heat flow, supporting the bactericidal effect of ciprofloxacin. The changes observed on the heatflow graph are similar to the graph generated in our study, where the time to peak increased (Fig. 7B and C) and the maximum metabolic rate decreased (Fig. 7D). The profile is different from other compounds such as chloramphenicol, also analyzed in the same study, where the bacteriostatic mechanism of action significantly changed the metabolic activity of the pathogen in the stationary phase. Furthermore, results seem to suggest that the concentration-dependent size increase of the formulations does not affect the ability of the nanocarriers to sustain antibacterial activity and affect biofilm metabolic activity.
2.5 Cytotoxicity of PMB micelles
Having achieved high biofilm penetration, effective biofilm inhibition and significant antibiofilm activity, we sought to further evaluate whether the PMB micelles could serve as safe topical formulations. Previous work has highlighted a strong correlation between nanoparticle toxicity and its physicochemical properties such as the size, surface charge and interaction with the surrounding milieu.50–53 As the system is intended for topical applications, we investigated the impact of the PMB micelles on cell viability against fibroblasts and keratinocytes. These cells are present in different layers of the skin and are involved in the wound healing process.54Cells were incubated with the PMB micelles at concentrations of 16–64 μg mL−1 and compared with the control group after 24 h in human fibroblasts. As shown in Fig. S6 (ESI†), treatment with the free drug up to 64 μg mL−1 resulted in no change in cell viability compared to the control group. Whilst the control group showed 100% cell viability, treatment with PMB micelles at 16 μg mL−1, 32 μg mL−1 and 64 μg mL−1 of drug content led to a 96.61 ± 9.46%, 95.21 ± 7.60% and 102.82 ± 5.55%, respectively, in HDF cells. Cells treated with the free drug at 64 μg mL−1 demonstrated cell viabilities of 92.00 ± 5.33%. Assessments of the morphology of the HDF cells did not show any changes as shown in Fig. 8B. Similarly, no loss in cell viability was obtained for keratinocytes incubated with the PMB micelles. For instance, the cell viability after treatment with 16 μg mL−1 PMB micelles was 101.10 ± 8.38%. Concordantly with the results in the other skin cell type, at the highest concentration of 32 μg mL−1 and 64 μg mL−1, the compatibility was high, and the cells showed viabilities of 98.37 ± 1.71% and 92.00 ± 5.33%, respectively. The high cellular viability of the DNA based nanostructures against cells has been previously reported and our results here confirm the biocompatibility of PMB micelles.33 Standardized tests for cytotoxicity and thresholds are described in ISO 10993-5:2009, which is the international standard issued by the recognized authority on standards.55 This document states that reduction in cell viability by more than 30% is to be considered cytotoxic to the tested cell line. Considering the variation in size following drug encapsulation in the DNA micelles, our results indicate that neither the components of the micellar formulation nor the different sizes of the micelles induce immediate toxicity on important skin cell lines.
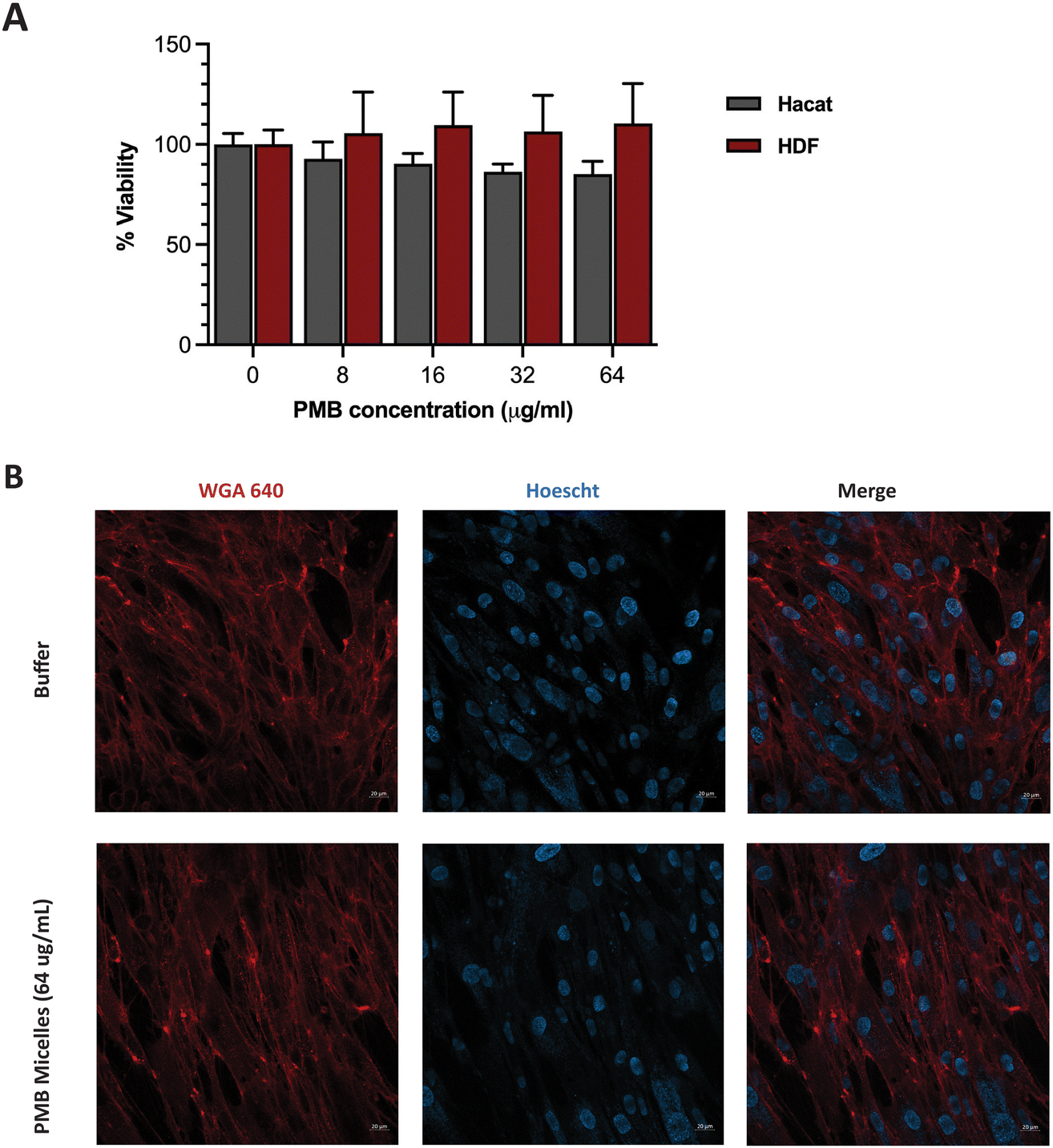 | ||
| Fig. 8 (A) Cell viability assays of the PMB micelles against fibroblast cell line (HDF) and keratinocyte cell line (HaCaT) cells at different concentrations of PMB. (B) Confocal microscopy images of HDF cells treated with buffer and PMB micelles (64 μg mL−1) for 24 h. Scale bar: 20 μm. | ||
3. Conclusions
In this study, cholesterol-modified DNA micelles were stabilized with polymyxin B and assessed for their antibacterial and antibiofilm potentials. The incorporation of PMB into the DNA micelles showed formation of micellar structures with greater control over size distributions and enhanced resistance to serial dilutions in comparison to plain ssDNA micelles. At the highest concentration of PMB, the micelles displayed high encapsulation efficiency and the assessment of the release kinetics indicated a burst release of PMB within first 2 h, followed by a sustained drug release over 24 h. By retaining the bactericidal efficacy of the PMB within the DNA micelles, high depletion of bacteria within the first 2 h and significant reduction after 24 h was observed. The PMB micelles demonstrated an ability to penetrate P. aeruginosa biofilms, inhibit its formation and influence biofilm metabolism by prolonging the time to peak metabolism and reducing the metabolic rate. Furthermore, cell viability data conducted in two important skin cell lines involved in wound healing (fibroblasts and keratinocytes) revealed no evident signs of cell death or change in cell morphology even at the highest PMB concentration, supporting its safe topical use. Overall, PMB stabilized DNA micelles showed clinical potential against P. aeruginosa biofilms and can have extensive application in skin wound infections treatment.4. Experimental
4.1 Materials
Synthetic oligonucleotides (see ESI† Table S1 for sequences and modifications) were obtained from Integrated DNA Technologies, Inc (CA, USA). Pseudomonas aeruginosa (PAO1 and ATCC 10145GFP) was obtained from American Type Culture Collection (DC, USA). Human dermal fibroblasts (HDF) cells were purchased from Lonza (Basel, Switzerland) and Human Immortalized Keratinocytes (HaCaT) were obtained from CLS Cell line service GmbH (Eppelheim Germany). Cation-adjusted Mueller Hinton Broth (MHB), polymyxin B (PMB), crystal violet, trypsin, accutase, methanol, DMSO, CCK-8 and Dulbecco's Modified Eagle's Medium (DMEM) high glucose were purchased from Sigma-Aldrich (MO, USA). Fetal bovine serum and penicillin–streptomycin were purchased from Hyclone Laboratories (UT, USA). DNase-free water was obtained from Thermoscientific Fisher. Phosphate-buffered saline (PBS) solution was purchased from Bio-Rad Laboratories (CA, USA).4.2 Preparation of DNA micelles
The PMB micelles were prepared according to a previously reported protocol with slight modifications.24 Briefly, the PMB micelles and empty micelles were formed through hydrophobic self-assembly of cholesterol conjugated single-stranded DNA and PMB by dilution in RNase free water and three different buffers 1×MB (2.5 mM NaCl, 1.25 mM MgCl2 and 0.05 M sodium acetate, pH 4.5), 1×PBS and 0.1×PBS to a DNA concentration of 20 μM. For polymyxin B encapsulation efficiency, stock solutions were made in buffers and added to a suspension of DNA amphiphile to achieve final concentrations of 8, 16, 32 and 64 μg mL−1.4.3 Micelle characterization
Hydrodynamic diameter (size), polydispersity index (PDI) and zeta potential of the micellar formulations were determined through dynamic light scattering (DLS) technique with the Zetasizer Nano Zen 2600 (Malvern Instruments Ltd, Malvern, UK). Measurements of undiluted micelle suspension were performed at 25 °C and recorded as mean ± standard deviation (SD).The interactions between PMB and either cholesterol-modified ssDNA or unmodified ssDNA were studied by recording changes in fluorescence or UV absorbance spectra. Different solutions were prepared in 1× micelle buffer, namely PMB micelles (via the addition of PMB to cholesterol-modified ssDNA in the final concentrations of 20 μM of DNA and 64 μg mL−1 of PMB), free PMB, a PNA sequence as well as ssDNA micelles (using cholesterol-modified ssDNA) and unmodified ssDNA in the same concentrations as used for PMB micelle synthesis. The solutions were prepared with fluorescently labeled PMB for fluorescent spectra or unlabeled PMB for UV spectra. The measurements were conducted using the plate reader.
The morphology of the micelles was investigated by transmission electron microscopy (TEM). Briefly, the micellar formulations were diluted, stained with UranyLess for 30 seconds and dried for 20 min. The images were obtained a using a transmission electron microscope HT7800 Series (Hitachi High-Tech Corp., Tokyo, Japan).
4.4 Stability against dilution induced disintegration
The stability of the 64 μg mL−1 PMB micelle formulation against dilution-induced degradation was compared to blank micelles via a 2-fold dilution series in DNase free water. The hydrodynamic diameter (size) and polydispersity index (PDI) of the diluted samples were measured with the Zetasizer Nano Zen 2600 (Malvern Instruments Ltd., Malvern, UK) and reported as mean ± standard deviation (SD).4.5 Encapsulation efficiency of DNA micelles
The encapsulation efficiency of the DNA amphiphiles was quantified via separation of free from entrapped PMB. Briefly, fluorescent labeled PMB micelles were added onto Amicon Ultra Centrifugal Filters with a 3K MWCO (Merck KGaA, Darmstadt; Germany) and centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and 25 °C for approximately 20 min on the Biofuge Pico centrifuge (Heraesus Instruments GmbH, Hanau, Germany). The micelles in the donor compartment of the filter were disrupted with methanol (10× dilution) and the removal of the DNA nanostructures was performed using the same method as for the initial separation step. The fluorescence intensity of the filtrate and methanol solution containing unentrapped and entrapped polymyxin B, respectively, were measured using the Spark M10 multimode plate reader (Tecan Trading AG, Switzerland). On the basis of data from the calibration curve, calculations of PMB concentrations were performed and the percentage encapsulation efficiency (EE) was determined using the following equation:
000 rpm and 25 °C for approximately 20 min on the Biofuge Pico centrifuge (Heraesus Instruments GmbH, Hanau, Germany). The micelles in the donor compartment of the filter were disrupted with methanol (10× dilution) and the removal of the DNA nanostructures was performed using the same method as for the initial separation step. The fluorescence intensity of the filtrate and methanol solution containing unentrapped and entrapped polymyxin B, respectively, were measured using the Spark M10 multimode plate reader (Tecan Trading AG, Switzerland). On the basis of data from the calibration curve, calculations of PMB concentrations were performed and the percentage encapsulation efficiency (EE) was determined using the following equation: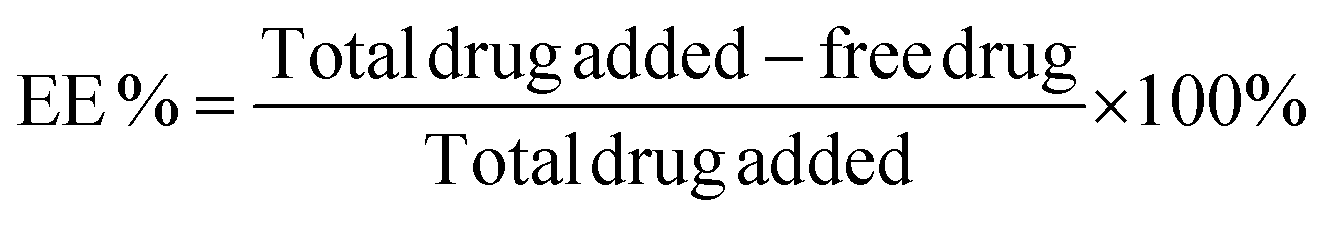
4.6 PMB release assay
In vitro release of Polymyxin B from the micelles was examined in PBS buffer (pH 7.0) and in sodium acetate buffer (pH 4.5) by monitoring changes in fluorescence intensity. Prior to the release assessments, two formulations of 64 μg mL−1 fluorescently labeled PMB micelles were synthesized in the respective release buffers. Pierce Microdialysis devices (Thermo Scientific™, Waltham, MA, USA) with a 3.5 K MWCO cut-off were loaded with 90 μL of micellar formulation or free drug and inserted into light protected Eppendorf tubes containing 1 mL of acceptor media. At pre-determined time, samples (100 μL) were withdrawn from the acceptor compartment and replaced with fresh media to ensure continuous sink conditions. After the last sample collection, the micelles in the donor chamber were disrupted in methanol (10×) and the solution was added to Amicon Ultra Centrifugal Filter with a 3 K MWCO (Merck KGaA, Darmstadt; Germany) and centrifuged at 13000 rpm and 25 °C for 10 min on the Biofuge Pico centrifuge (Heraesus Instruments GmbH, Hanau, Germany) to separate the remaining drug from the cholesterol-modified DNA sequences. The amount of drug released at each time point was quantified via fluorescence intensity readings on a Spark M10 multimode plate reader (Tecan Trading AG, Switzerland) with excitation and emission wavelengths set at 335 and 518 nm, respectively. The results were reported as the percentage mean ± standard deviation (SD) in reference to the total amount of PMB loaded in the formulation prior to the start of the experiment and the cumulative release percentage of polymyxin B was calculated by the following equation: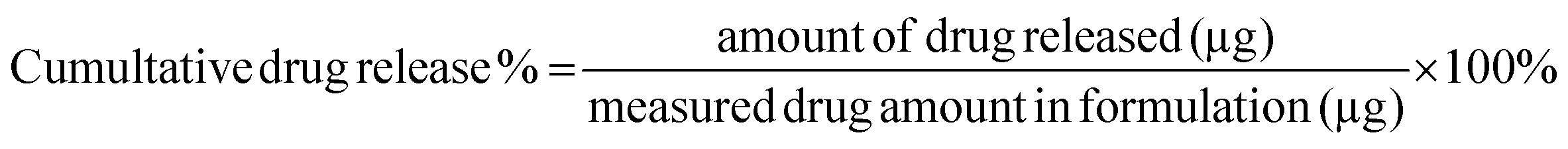
4.7 Biofilm penetration
PMB micelles penetration into biofilms was investigated with Confocal Laser Scanning Microscopy (CLSM). GFP expressing P. aeruginosa was cultivated for 7 hours in LB with 200 μg mL−1 ampicillin at 37 °C and a stirring speed at 100 rpm. The OD600 of the bacteria suspension was adjusted to 0.07 by dilution in LB and aliquots of 200 μL were added to each well of an 8 well chambered slide. The slide was placed light protected in an incubator at 37 °C for 10 h to allow biofilm formation. The supernatant was removed, and the biofilms were treated with 200 μL of rhodamine B labeled PMB micelles or MHB as controls. After 30 min and 2 h treatment, the supernatants were removed and 200 μL media was added prior to imaging. The penetration into biofilms was observed with the Zeiss LSM 800 (Carl Zeiss, Oberkochen, Germany) using a water 25x objective lens.4.8 Antibacterial activity
:150, v/v). The free PMB and PMB micelles were diluted in a two-fold manner in the MHB in 96-well plates, to which the bacterial suspension was added (1:1, v/v). The plates were further incubated for 18 h at 37 °C without shaking. The MIC was determined by visual observation of loss of turbidity and through absorbance measurement at 600 nm.
4.9 Biofilm activity
4. 10 Cell seeding
The cell toxicity/biocompatibility of micellar formulations was investigated using immortalized human keratinocytes (HaCaT). The cells were cultured in cell culture flasks with Dulbecco's Modified Eagle's Medium (DMEM) high glucose (supplemented with 10% w/v fetal bovine serum (FBS) and 1% penicillin–streptomycin infused with serum). At approximately 80% confluency, the culture medium was discarded, and the keratinocytes attached to the flask were washed once with 10 mL phosphate-buffered saline (PBS). The PBS was discarded, and 4 mL Accutase® cell detachment solution was pipetted into the cell culture flask and set to incubate at 37 °C for 15 minutes. After 15 minutes, the cells were agitated by vigorously mixing the solution with a pipette. The flask was then inspected under a microscope to ensure that the cells were properly detached. Thereafter, 10 mL fresh DMEM was added to the cells and thoroughly mixed with a pipette. The cell density of this solution was determined using a Scepter 2.0 handheld automated cell counter. The cells were then seeded in 96 well plates at a cell density of 6000 cells per well and set to incubate at 37 °C for 48 h. The biocompatibility of the PMB micelles was investigated for PMB concentrations between 0.1–12.8 μg mL−1 serial dilution, with free PMB in the same concentrations serving as positive controls and DMEM as a negative control. For the drug-free formulations, a 5× dilution in DMEM was used, with the buffer serving as a positive control and DMEM as negative control. After the 48 h incubation, the residual DMEM was removed and discarded gently. Aliquots (100 μL) of the samples were added in the wells (in triplicates), and the plates were set at 37 °C for another 48 h. The cytotoxicity of the formulations was determined after the 48 h treatment using the colorimetric 3-(4.5-dimethylthiazol-2-yl)-2.5-diphenyltetrazoliumbromide (MTT) assay.4. 11 In vitro cytotoxicity
Biocompatibility of the micellar formulations and 1× MB (control) was determined using either the MTT or CCK-8 assay according to previously described methods58,59 on two cell lines: immortalized human keratinocytes (HaCaT) and human dermal fibroblasts (HDF) after 48 h and 24 h respectively. The cells were cultured in cell culture flasks with Dulbecco's Modified Eagle's Medium (DMEM) high glucose (supplemented with 10% v/v fetal bovine serum (FBS) and 1% penicillin–streptomycin). At 80% confluency, the cell monolayer was washed with phosphate-buffered saline (PBS), which was then discarded and Accutase® or Trypsin was added to the flask containing HaCaT or HDF, respectively, and incubated for 15 minutes or 5 minutes at 37 °C to detach the cells from the flask. Fresh DMEM was added to the cell culture and the solution was pipetted to separate the cells. Cell density was measured using a hand-held automated cell counter device. Afterwards, cells were seeded in 96 well plates at a cell density of 6000 cells per well at 37 °C for 48 h (HaCaT) or 10000 cells per well at 37 °C for 24 h (HDF). The cytotoxicity of the formulation was determined at 8, 16, 32 and 64 μg mL−1 of polymyxin B. Wells containing only DMEM served as negative control and free polymyxin B as a positive control.
4. 12 Statistical analysis
Statistical analysis was conducted with software Graphpad Prism (Version 7.0, GraphPad Software Inc., CA, USA). The student's t-test was used between two groups with statistical difference. For results with three or more groups, one-way Anova or two-way Anova with Turkey's post hoc analysis was performed. Results are presented as mean ± standard deviation and p values of <0.05 were deemed significant.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by Tromsø Forsknings-Stiftelse (TFS) through the TFS starting grant (20_SG_SO) to Dr Obuobi. We also acknowledge research funding support to Alexandra Sousa from CANS Centre for New Antibacterial Strategies (TFS grant no. 18_CANS_AS) at UIT The Arctic University of Norway. We thank The Department of Pharmacy, UIT The Arctic University of Norway for providing research support.References
- H. Kim and M. Kwak, Structures and Applications of Nucleic Acid-Based Micelles for Cancer Therapy, Int. J. Mol. Sci., 2023, 24, 1592 CrossRef CAS.
- R. J. Banga, et al., Cross-Linked Micellar Spherical Nucleic Acids from Thermoresponsive Templates, J. Am. Chem. Soc., 2017, 139, 4278–4281 CrossRef CAS PubMed.
- M. Chang, C. Yang and D. Huang, Aptamer-Conjugated DNA Icosahedral Nanoparticles As a Carrier of Doxorubicin for Cancer Therapy, ACS Nano, 2011, 5, 6156–6163 CrossRef CAS PubMed.
- R. P. Goodman, R. M. Berry and A. J. Turberfield, The single-step synthesis of a DNA tetrahedron, Chem. Commun., 2004, 1372–1373, 10.1039/b402293a.
- Y. Li, et al., Controlled assembly of dendrimer-like DNA, Nat. Mater., 2004, 3, 38–42 CrossRef CAS.
- S. Obuobi, et al., Facile and efficient encapsulation of antimicrobial peptides via crosslinked DNA nanostructures and their application in wound therapy, J. Controlled Release, 2019, 313, 120–130 CrossRef CAS.
- S. Huo, et al., Gold-DNA nanosunflowers for efficient gene silencing with controllable transformation, Sci. Adv., 2019, 5(10), 1–11 Search PubMed.
- S. Pal, Z. Deng, B. Ding, H. Yan and Y. Liu, DNA-Origami-Directed Self-Assembly of Discrete Silver-Nanoparticle Architectures, Angew. Chem., Int. Ed., 2010, 49, 2700–2704 CrossRef CAS.
- F. E. Alemdaroglu, N. C. Alemdaroglu, P. Langguth and A. Herrmann, DNA Block Copolymer Micelles – A Combinatorial Tool for Cancer Nanotechnology, Adv. Mater., 2008, 20, 899–902 CrossRef CAS.
- F. E. Alemdaroglu, N. C. Alemdaroglu, P. Langguth and A. Herrmann, Cellular Uptake of DNA Block Copolymer Micelles with Different Shapes, Macromol. Rapid Commun., 2008, 29, 326–329 CrossRef CAS.
- J. Willem de Vries, et al., DNA nanoparticles for ophthalmic drug delivery, Biomaterials, 2018, 157, 98–106 CrossRef CAS PubMed.
- A. Ohmann, et al., Controlling aggregation of cholesterol-modified DNA nanostructures, Nucleic Acids Res., 2019, 47, 11441–11451 CrossRef CAS.
- J. Hahn, S. F. J. Wickham, W. M. Shih and S. D. Perrault, Addressing the Instability of DNA Nanostructures in Tissue Culture, ACS Nano, 2014, 8, 8765–8775 CrossRef CAS PubMed.
- Y. Wu, K. Sefah, H. Liu, R. Wang and W. Tan, DNA aptamer–micelle as an efficient detection/delivery vehicle toward cancer cells, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5–10 CrossRef CAS PubMed.
- F. Charbgoo, et al., MUC1 aptamer-targeted DNA micelles for dual tumor therapy using doxorubicin and KLA peptide, Nanomed. Nanotechnol. Biol. Med., 2018, 14, 685–697 CrossRef CAS.
- R. J. Banga, et al., Drug-Loaded Polymeric Spherical Nucleic Acids: Enhancing Colloidal Stability and Cellular Uptake of Polymeric Nanoparticles through DNA Surface-Functionalization, Biomacromolecules, 2017, 18, 483–489 CrossRef CAS.
- E. Tacconelli, et al., Global Priority List Of Antibiotic-Resistant Bacteria To Guide Research, Discovery, And Development Of New Antibiotics, W. H. O. Tech., 2017, 1–7 Search PubMed.
- G. Gebreyohannes, A. Nyerere, C. Bii and D. B. Sbhatu, Challenges of intervention, treatment, and antibiotic resistance of biofilm-forming microorganisms, Heliyon, 2019, 5, e02192 CrossRef PubMed.
- D. Sharma, L. Misba and A. U. Khan, Antibiotics versus biofilm: an emerging battleground in microbial communities, Antimicrob. Resist. Infect. Control, 2019, 8, 76 CrossRef.
- A. Brauner, O. Fridman, O. Gefen and N. Q. Balaban, Distinguishing between resistance, tolerance and persistence to antibiotic treatment, Nat. Rev. Microbiol., 2016, 14, 320–330 CrossRef CAS.
- Y. Cai, W. Lee and A. L. Kwa, Polymyxin B versus colistin: an update, Expert Rev. Anti-Infect. Ther., 2015, 13, 1481–1497 CrossRef CAS.
- N. V. Dubashynskaya and Y. A. Skorik, Polymyxin Delivery Systems: Recent Advances and Challenges, Pharmaceuticals, 2020, 13, 83 CrossRef CAS PubMed.
- S. Obuobi, et al., Phenylboronic Acid Functionalized Polycarbonate Hydrogels for Controlled Release of Polymyxin B in Pseudomonas Aeruginosa Infected Burn Wounds, Adv. Healthcare Mater., 2018, 7, 1701388 CrossRef PubMed.
- Y. Zhang, R. Peng, F. Xu and Y. Ke, Hierarchical Self-Assembly of Cholesterol-DNA Nanorods, Bioconjugate Chem., 2019, 30, 1845–1849 CrossRef CAS PubMed.
- A. Gubu, et al., Nucleic acid amphiphiles: synthesis, properties and applications, Mol. Ther.–Nucleic Acids, 2023, 134, 112712 Search PubMed.
- Y. Takechi-Haraya, et al., Current Status and Challenges of Analytical Methods for Evaluation of Size and Surface Modification of Nanoparticle-Based Drug Formulations, AAPS PharmSciTech, 2022, 23, 150 CrossRef CAS PubMed.
- J. Zou, Q. Gao, J. Nie, Y. Zhang and C. Jin, Enzymatic dephosphorylation-triggered self-assembly of DNA amphiphiles, RSC Adv., 2021, 11, 18322–18325 RSC.
- M. Sirajuddin, S. Ali and A. Badshah, Drug-DNA interactions and their study by UV-Visible, fluorescence spectroscopies and cyclic voltametry, J. Photochem. Photobiol., B, 2013, 124, 1–19 CrossRef CAS.
- B. Ding, et al., Origins of cell selectivity of cationic steroid antibiotics, J. Am. Chem. Soc., 2004, 126, 13642–13648 CrossRef CAS PubMed.
- C. J. Thomas, B. P. Gangadhar, N. Surolia and A. Surolia, Kinetics and mechanism of the recognition of endotoxin by polymyxin B, J. Am. Chem. Soc., 1998, 120, 12428–12434 CrossRef CAS.
- L. Kong, Z. Liu, X. Hu and S. Liu, Interaction of polymyxin B with ds-DNA, and determination of DNA or polymyxin B via resonance Rayleigh scattering and resonance non-linear scattering spectra, Microchim. Acta, 2011, 173, 207–213 CrossRef CAS.
- S. Kaewpaiboon and T. Srichana, Formulation Optimization and Stability of Polymyxin B Based on Sodium Deoxycholate Sulfate Micelles, J. Pharm. Sci., 2022, 111, 2249–2257 CrossRef CAS PubMed.
- H. Liu, et al., DNA-Based Micelles: Synthesis, Micellar Properties and Size-Dependent Cell Permeability, Chem. – Eur. J., 2010, 16, 3791–3797 CrossRef CAS PubMed.
- K. H. Min, et al., pH-Responsive mineralized nanoparticles for bacteria-triggered topical release of antibiotics, J. Ind. Eng. Chem., 2019, 71, 210–219 CrossRef CAS.
- Q. Lin, J. M. Pilewski and Y. P. Di, Acidic Microenvironment Determines Antibiotic Susceptibility and Biofilm Formation of Pseudomonas aeruginosa, Front. Microbiol., 2021, 12, 1–15 Search PubMed.
- S. B. Behbahani, et al., pH variation in medical implant biofilms: Causes, measurements, and its implications for antibiotic resistance, Front. Microbiol., 2022, 13, 1–28 Search PubMed.
- J. Wang, et al., PH-Switchable Antimicrobial Nanofiber Networks of Hydrogel Eradicate Biofilm and Rescue Stalled Healing in Chronic Wounds, ACS Nano, 2019, 13, 11686–11697 CrossRef CAS PubMed.
- B. Lr, et al. , The pH of wounds during healing and infection: a descriptive literature review, 2017, 25, 63–69 Search PubMed.
- N. Singh, et al., Dual bioresponsive antibiotic and quorum sensing inhibitor combination nanoparticles for treatment of: Pseudomonas aeruginosa biofilms in vitro and ex vivo, Biomater. Sci., 2019, 7, 4099–4111 RSC.
- D. Hassan, C. A. Omolo, V. O. Fasiku, C. Mocktar and T. Govender, Novel chitosan-based pH-responsive lipid-polymer hybrid nanovesicles (OLA-LPHVs) for delivery of vancomycin against methicillin-resistant Staphylococcus aureus infections, Int. J. Biol. Macromol., 2020, 147, 385–398 CrossRef CAS.
- H. Fullriede, et al., PH-responsive release of chlorhexidine from modified nanoporous silica nanoparticles for dental applications, Bionanomater., 2016, 17, 59–72 CrossRef.
- Y. Wang, et al., Biodegradable functional polycarbonate micelles for controlled release of amphotericin B, Acta Biomater., 2016, 46, 211–220 CrossRef CAS PubMed.
- M. Ghezzi, et al., Polymeric micelles in drug delivery: An insight of the techniques for their characterization and assessment in biorelevant conditions, J. Controlled Release, 2021, 332, 312–336 CrossRef CAS PubMed.
- A. R. Sultan, et al., Real time monitoring of Staphylococcus aureus biofilm sensitivity towards antibiotics with isothermal microcalorimetry, PLoS One, 2022, 17, e0260272 CrossRef CAS PubMed.
- A. Bettencourt, et al., Activity of daptomycin- and vancomycin-loaded poly-epsilon-caprolactone microparticles against mature staphylococcal biofilms, Int. J. Nanomed., 2015, 10, 4351 CrossRef.
- I. Santos Ferreira, et al., Encapsulation in Polymeric Microparticles Improves Daptomycin Activity Against Mature Staphylococci Biofilms—a Thermal and Imaging Study, AAPS PharmSciTech, 2018, 19, 1625–1636 CrossRef CAS PubMed.
- C. Tellapragada, et al., Isothermal microcalorimetry minimal inhibitory concentration testing in extensively drug resistant Gram-negative bacilli: a multicentre study, Clin. Microbiol. Infect., 2020, 26, 1413.e1–1413.e7 CrossRef CAS PubMed.
- M. Vaara, Polymyxins and Their Potential Next Generation as Therapeutic Antibiotics, Front. Microbiol., 2019, 10, 1–6 CrossRef.
- K. Cirnski, J. Coetzee, J. Herrmann and R. Müller, Metabolic Profiling to Determine Bactericidal or Bacteriostatic Effects of New Natural Products using Isothermal Microcalorimetry, J. Visualized Exp., 2020, 164, e61703, DOI:10.3791/61703.
- S. Moghimi, I. Muir, L. Illum, S. Davis and V. Kolb-Bachofen, Coating particles with a block co-polymer (poloxamine-908) suppresses opsonization but permits the activity of dysopsonins in the serum, Biochim. Biophys. Acta, Mol. Cell Res., 1993, 1179, 157–165 CrossRef CAS.
- J. Wolfram, et al., Safety of Nanoparticles in Medicine, Curr. Drug Targets, 2015, 16, 1671–1681 CrossRef CAS PubMed.
- D. Boraschi, et al., Nanoparticles and innate immunity: new perspectives on host defence, Semin. Immunol., 2017, 34, 33–51 CrossRef CAS PubMed.
- L. Ding, et al., Nanotoxicity: The Toxicity Research Progress of Metal and Metal- Containing Nanoparticles, Mini-Rev. Med. Chem., 2015, 15, 529–542 CrossRef CAS.
- A. M. Wojtowicz, et al., The importance of both fibroblasts and keratinocytes in a bilayered living cellular construct used in wound healing, Wound Repair Regen., 2014, 22, 246–255 CrossRef.
- International Organization for Standardization (2009) ISO 10993-5:2009 Biological evaluation of medical devices - Part 5: Tests for in vitro cytotoxicity.
- M. Balouiri, M. Sadiki and S. K. Ibnsouda, Methods for in vitro evaluating antimicrobial activity: A review, J. Pharm. Anal., 2016, 6, 71–79 CrossRef.
- S. Obuobi, et al., Nucleic acid peptide nanogels for the treatment of bacterial keratitis, Nanoscale, 2020, 12, 17411–17425 RSC.
- N. Ahmad, et al., Daunorubicin oral bioavailability enhancement by surface coated natural biodegradable macromolecule chitosan based polymeric nanoparticles, Int. J. Biol. Macromol., 2019, 128, 825–838 CrossRef CAS PubMed.
- J. Cauzzo, M. Nystad, A. M. Holsæter, P. Basnet and N. Škalko-Basnet, Following the Fate of Dye-Containing Liposomes In Vitro, Int. J. Mol. Sci., 2020, 21, 4847 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb00704a |
| This journal is © The Royal Society of Chemistry 2023 |