
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTherapy and diagnosis of Alzheimer's disease: from discrete metal complexes to metal–organic frameworks
Amalia
García-García
 ab,
Sara
Rojas
*a and
Antonio
Rodríguez-Diéguez
*a
ab,
Sara
Rojas
*a and
Antonio
Rodríguez-Diéguez
*a
aDepartment of Inorganic Chemistry, Faculty of Science, University of Granada, Av. Fuentenueva S/N, 18071 Granada, Spain. E-mail: srojas@ugr.es; antonio5@ugr.es
bCentro de Química del Instituto de Ciencias, Benemérita Universidad Autónoma de Puebla, 18 sur & Av. San Claudio, Col. San Manuel, 72570 Puebla, Mexico
First published on 8th June 2023
Abstract
Alzheimer's disease (AD) is a neurodegenerative disorder affecting 44 million people worldwide. Although many issues (pathogenesis, genetics, clinical features, and pathological aspects) are still unknown, this disease is characterized by noticeable hallmarks such as the formation of β-amyloid plaques, hyperphosphorylation of tau proteins, the overproduction of reactive oxygen species, and the reduction of acetylcholine levels. There is still no cure for AD and the current treatments are aimed at regulating the cholinesterase levels, attenuating symptoms temporarily rather than preventing the AD progression. In this context, coordination compounds are regarded as a promissing tool in AD treatment and/or diagnosis. Coordination compounds (discrete or polymeric) possess several features that make them an interesting option for developing new drugs for AD (good biocompatibility, porosity, synergetic effects of ligand–metal, fluorescence, particle size, homogeneity, monodispersity, etc.). This review discusses the recent progress in the development of novel discrete metal complexes and metal–organic frameworks (MOFs) for the treatment, diagnosis and theragnosis of AD. These advanced therapies for AD treatment are organized according to the target: Aβ peptides, hyperphosphorylated tau proteins, synaptic dysfunction, and mitochondrial failure with subsequent oxidative stress.
 Amalia García-García | Dr Amalia García-García currently has a postdoctoral contract Margarita Salas at both the Benemérita Universidad Autónoma de Puebla in the Applied Bioinorganic Laboratory and the University of Granada in the Biochemistry and Electronics as Sensing Technologies group. She obtained her PhD from the University of Granada in 2021 by working on the synthesis of new coordination compounds with biomedical applications. Her research mainly focuses on the design and synthesis of new coordination metal-complexes with potential applications against worldwide illnesses such as diabetes mellitus, cancer and Alzheimer's disease. |
 Sara Rojas | Dr Sara Rojas obtained her PhD from the University of Granada (in 2014) with a work focus on the synthesis of porous materials and their application as drug delivery systems. After finishing her PhD, she continued her studies at the Institute Lavoisier in Versailles (France, 2016) and the IMDEA Energy institute (Spain, 2018) under a framework of a Marie Curie Individual Fellowship and Atracción del Talento grant, respectively. In 2021, she joined the Biochemistry and Electronics as Sensing Technologies group first as Juan de la Cierva Incorporación fellow and now as Ramón y Cajal. Her research focuses on the development of porous materials for biomedical and environmental applications. |
 Antonio Rodríguez-Diéguez | Prof. Antonio Rodríguez-Diéguez completed his PhD studies in Chemistry at the University of Granada in 2005. Since 2008, he has worked as a Leading Researcher of different projects funded by the Junta de Andalucía and Ministry of Science and Technology and as a Professor at the Inorganic Chemistry Department of the University of Granada. His research interests have included crystalline analysis by XR diffraction, the improvement of solvothermal methods, studies of magnetic and luminescence properties and, recently, the synthesis of metal–organic frameworks for therapeutic applications. |
Introduction
Alzheimer's disease (AD), the most common form of dementia, is an irreversible and progressive neurodegenerative disorder, and a global public health issue, according to the World Health Organization (WHO).1 Given the current aging population, by 2050, the prevalence of dementia is estimated to increase up to 166%,2 and therefore, the number of patients with AD is predicted to increase too.3 As there is no effective treatment and the disease's etiology and pathology remain largely unknown, nowadays, AD is the 5th cause of mortality worldwide, affecting about 44 million people in 2020.4 This means a global societal cost of 1.3 trillion $ in 2019, which is expected to surpass 2.8 trillion $ by 2030.1 Considering these numbers, one could suggest that AD is an epidemic.Although the progress in understanding the disease, there are still some controversies, mainly in AD pathologies and the development of efficient therapies. In 1907, Alois Alzheimer reported the first case of AD in a 51 year-old woman in the insane asylum of Frankfurt am Main (Germany). Clinical observation revealed that the symptoms were so odd that it could not be classified as any known disorder. The patient developed a rapid loss of memory, disorientation, mood change, and hallucinations. However, her walk, hands, and reflexes functioned correctly. Interestingly, the post-mortem examination of the brain showed important atrophy and abnormal vascular tissues. Moreover, neurons exhibited fibrils at the surface and inside them that disintegrated cells.5
Now, we know that AD is first characterized by a progressive deterioration of the capacity to recall new information, as neurons involved in forming new memories are the first to be damaged. As time passes, other symptoms appear such as memory loss, confusion, trouble understanding, changes in mood, increased anxiety, and difficulty in speaking and writing.6 These symptoms might begin 20 years or more after the onset of brain changes.7,8
Pathology of AD
Although Alois Alzheimer reported the presence of abnormal fibrils in the brain of her demented patient, it was not until the end of the 20th century that the pathological hallmarks of AD were clearly identified. As in many neurodegenerative disorders, AD is also caused by the misfolding and irreversible aggregation of specific proteins, β-amyloid (Aβ) and tau, which forms amyloid plaques and neurofibrillary tangles, respectively.9,10Amyloid plaques
In an attempt to explain the formation of these abnormalities in the brain, Hardy and Higgins proposed the “amyloid cascade hypothesis” in 1992.11 This idea suggests that the deposition of Aβ proteins is the cause of Alzheimer's pathology, whereas the abnormal hyperphosphorylation of tau proteins and thus, neurofibrillary tangle formation, cell loss, vascular damage, and dementia are consequences of the deposition.12Aβ peptides are natural proteolytic products of the transmembrane amyloid precursor protein (APP), and consist of 36–43 amino acids, with Aβ40 being the most prevalent one.14 However, depending on the involved secretase enzyme, two pathways to cleave the APP are possible: the amyloidogenic pathway and the non-amyloidogenic pathway (Fig. 1). In the non-amyloidogenic process, the APP is first cleaved by α-secretase, giving rise to a soluble ectodomain sAPPα and an 83-residue carboxy-terminal fragment (C83). Subsequently, γ-secretase digests C83 into a soluble extracellular peptide (p3) and an APP intracellular domain (AICD). On the other hand, the amyloidogenic pathway begins with the catalysis of the APP by β-secretase 1 (BACE-1), generating the soluble ectodomains sAPPβ and C99. The following cleavage of C99 by γ-secretase yields the AICD and Aβ peptides of different lengths, increasing the ratio of Aβ42/Aβ40, with Aβ42 being more toxic than Aβ40. Although the reasons of these abnormalities are still unknown, it is believed that in familial AD (ca. 5–10% of AD cases)15 mutations in the gene encoding APP or the γ-secretase subunit proteins presenilins 1 and 2 lead to an increased ratio between longer and shorter forms of Aβ.16 However, in sporadic AD (ca. 70% of AD cases), γ-secretase displays normal activity.
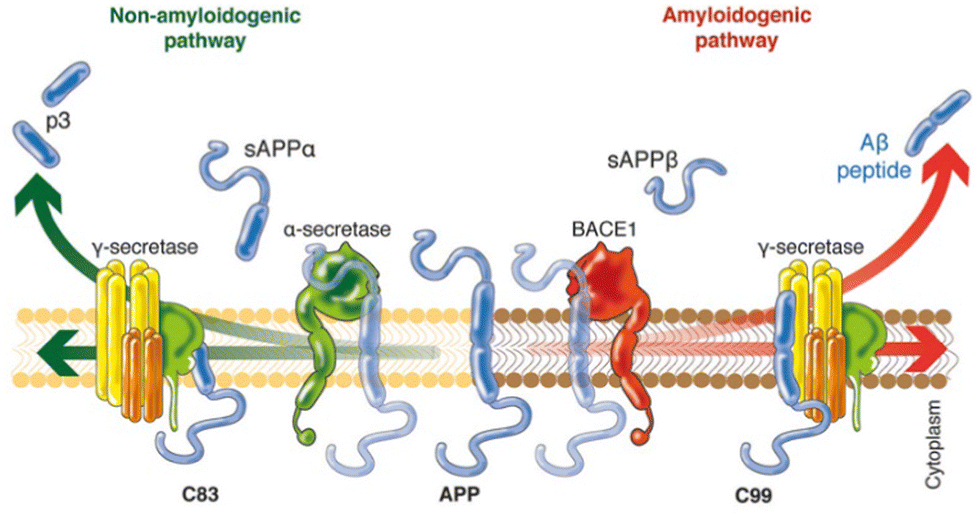 | ||
| Fig. 1 Schematic representation of the two different pathways to cleave the APP. In the non-amyloidogenic pathway (left), the APP is cleaved in the middle by α-secretase, generating the soluble sAPPα and C83. The amyloidogenic pathway (right) is promoted by β-secretase 1 (BACE1) and γ-secretase, giving rise to shorter Aβ species that tends to aggregate. Adapted from ref. 13. Reproduced with permission. | ||
At this point, some controversies appear around the amyloid cascade hypothesis, since many individuals present amyloid plaques without AD symptoms; or the fact that soluble Aβ aggregates, rather than insoluble fibrillar aggregates, are thought to be the main factor responsible for the neurotoxic properties.17
Neurofibrillary tangles
The other neuropathogenic feature in AD is the presence of intracellular neurofibrillary tangles, which are formed by abnormal hyperphosphorylated and aggregated tau proteins in the hippocampus. In healthy brains, tau proteins confer stability to the microtubules of the neuronal axons. However, in AD, tau is hyperphosphorylated, reducing its ability to bind microtubules and self-associate into paired helical filaments, leading to neuronal death.18Metals and neurotransmitters
Apart from amyloid plaques and neurofibrillary tangles, other anomalies linked to AD are altered levels of metals and neurotransmitters. Copper, zinc, and iron are essential trace elements involved in many metabolic pathways,19 and they are also implicated in neurodegenerative processes. These metal ions are found in the amyloid plaques at high concentrations, and in vitro studies revealed that they promote Aβ aggregation and the formation of reactive oxygen species (ROS), which play a key role in AD.20–22 On the other hand, neurochemical studies suggest that synapses containing acetylcholine, glutamate, and serotonin in the neocortex and hippocampus are predominantly affected in AD.23 In presynaptic neurons, choline-acetyltransferase (ChAT) catalyzes the synthesis of acetylcholine (ACh) from choline and acetyl-coenzyme A. Then, ACh is released into the synaptic cleft, where it is bonded to the postsynaptic neuron. Finally, ACh is hydrolyzed in the synaptic cleft by acetylcholinesterase (AChE), which regulates its concentration at the synapse (Fig. 2). Furthermore, in a less-specific pathway, ACh is also hydrolyzed by butyrylcholinesterase (BuChE). However, in AD, the neurotransmitter acetylcholine is gradually lost in the end-stage of the disease.24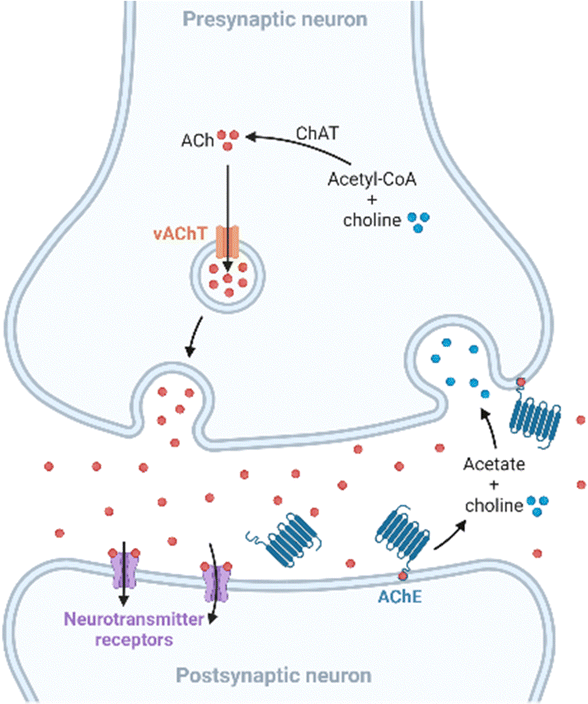 | ||
| Fig. 2 Scheme of the cholinergic synapse. In the presynaptic neuron, choline-acetyltransferase (ChAT) catalyzes the formation of acetylcholine (ACh) from acetyl-coenzyme A and choline. Then, ACh is released into the synaptic cleft by a vesicular ACh transporter (vAChT), where ACh binds to receptors in the postsynaptic neuron. Finally, ACh is hydrolyzed in the synaptic cleft by acetylcholinesterase (AChE) into choline and acetate. | ||
Treatments
Since AD discovery, there is no effective treatment that delays or stops its progression. However, a total of 121 new agents are undergoing clinical trials.25 Nowadays, there are only a few common medications approved by the US Food and Drug Administration (FDA): three cholinesterase inhibitors (donepezil, rivastigmine, and galantamine), one glutamate regulator (memantine), and their combination (memantine with donepezil). However, these drugs temporarily attenuate symptoms but do not alter the course of the disease,26 and they produce side effects associated with high dosages and long periods of treatment.27 From 2021, twenty years after the last approved drug, two new treatments were approved: first aducanumab and later lecanemab.28,29 These two monoclonal antibodies are the first disease-modifying therapies since they are able to reduce Aβ plaques and block their formation, respectively.30,31 Both have been authorized by the FDA via the accelerated approval pathway, and they are only prescribed for early AD.32,33Regarding the AD's drug approval, the G8 leaders set a goal for developing effective drugs to treat or prevent AD by 2025,34 and more efforts are being made to investigate other types of treatments, since the use/efficacy of aducanumab and other approved drugs is limited. Currently, the main target is to inhibit brain Aβ production and aggregation. Several strategies are focused on researching β-secretase inhibitors, the use of immunotherapy through antibodies that bind to Aβ plaques, tau inhibitors, anti-inflammatory drugs, and agents that chelate copper and iron. Mainly, organic molecules and small peptides are being widely studied for this purpose.35,36 Following this trend, coordination compounds could offer advantages as therapeutic agents compared to organic molecules, as the organic moiety can be widely modified to tune its selectivity and reactivity for a particular target, whereas metal ions offer different geometries and variable oxidation states.37 For example, whereas an organic molecule has limited geometry, an octahedral metal center is capable of forming many stereoisomers and, thus, achieving higher protein-binding affinity and selectivity. In addition, these materials could undergo ligand exchange for better reactivity with biomolecular targets (i.e., cisplatin), exhibit photophysical activity, create a 3D porous structure for drug delivery, and possess luminescent and magnetic properties in addition to biological activity.38,39
In this regard, coordination compounds have been approved by the FDA in the treatment and diagnosis of different diseases. Generally, the activity of metal-containing compounds is associated with high toxicity. However, there is a small frontier between the pharmacological efficacy and the toxicity of drugs. Aside from the drug delivery and disposition of the active ingredient, the activity of metal-containing compounds depends on many factors, such as the oxidation state of the metal, coordination geometry, and functional groups of the organic ligands. For example, mercury and its coordination compounds are believed very toxic, but some complexes such as thiomersal and merbromin are utilized as preservatives for vaccines and antiseptic agents, respectively. To date, many metal complexes based on the elements magnesium, iron, zirconium, palladium, platinum, mercury, and bismuth, among others, have been clinically approved in the US and/or the EU for a wide range of medical uses.38 Some examples are cisplatin and carboplatin (cancer),40 sodium stibogluconate and meglumine antimoniate (leishmaniasis),41 and auranofin (rheumatoid arthritis).42 Anyway, the use of biocompatible metals such as zinc, magnesium, and copper, and structures with the controlled delivery of their components is recommendable. With respect to AD, the application of coordination compounds as therapeutic agents is a rather new research field (since 2008), particularly focused on the aggregation and deposition of Aβ proteins, abnormal processing of the APP, formation of neurofibrillary tangles with hyperphosphorylated tau proteins, synaptic degeneration, and neuronal cell death.23
Although chemotherapy is demonstrated to be an effective method in the treatment of AD, it has some disadvantages, such as the lack of specificity, significant side effects and the development of acquired resistance to the drug. Importantly, compared to other areas of the organism, the treatment of brain diseases is presently unsuccessful mostly due to the difficulty in crossing the blood–brain barrier (BBB).43 The BBB is formed by the brain capillary endothelium and excludes from the brain ca. 100% of large-molecule neurotherapeutics and >98% of small-molecule drugs.44 Furthermore, to date, all the approved FDA drugs have been marked as oral formulations (donepezil, galantamine, and memantine) and one of them has been marked as a transdermal formulation (rivastigmine). So, it is necessary to use high doses to reach the brain, after overcoming oral barriers of adsorption, hepatic metabolism, distribution and finally traversing the BBB. In general, only low molecular weight lipophilic drugs can cross the BBB. However, some solutions are suggested, such as the application of vascular activating compounds that increase the permeability of the BBB, designing drugs with fat solubility, and direct drug delivery to the cerebrospinal fluid (CSF).45 In this regard, nanotechnology has provided superior opportunities in the treatment and/or diagnosis of central nervous system diseases. Nanocarriers might be able to cross the BBB, as they can be easily modified and functionalized, increasing the drug concentration in the brain. Furthermore, they have been proposed for theragnosis as they can act as both imaging and therapeutic agents simultaneously.15
Among all the proposed nanocarriers (polymeric nanoparticles, lipids, liposomes, nanoemulsions, microemulsions, and dendrimers), metal–organic frameworks (MOFs) are an emerging class of materials which have attracted interest in AD treatment and/or diagnosis due to their outstanding properties: (i) large specific surface area and pore volume, associated with high sorption capacities, (ii) biocompatibility, (iii) good stability profiles under physiological conditions, (iv) tunable particle size, and (v) can be used as imaging agents.46,47 Considering their plasticity, different strategies have been proposed in the application of MOFs for AD treatment, like the suppression of Aβ aggregation, as antioxidative agents or in promoting nerve regeneration.
While there are few reviews about metal-based drugs in AD treatment48–50 and MOFs in AD diagnosis have already been documented,51–53 the number of reports in this area has sharply increased (34 and 81% published within the last 4 years, according to the Web of Science, February 2023, (i) “coordination” “compounds” “Alzheimer”, and (ii) “metal” “organic” “framework” “Alzheimer”, respectively).
This review will discuss all the developments of coordination compounds, including outstanding discrete compounds and MOFs, published as treatment and/or diagnosis in AD. In order to give a broader picture of opportunities and disadvantages of the use of each compound/material, details of each structure and its properties are also included.
Discrete coordination compounds in AD treatment
Along this review, the reported coordination compounds in AD treatment are organized according to the target: Aβ peptides, hyperphosphorylated tau proteins, synaptic dysfunction, and mitochondrial failure with subsequent oxidative stress.14 A summary is shown in Table 1.| Coordination compound | Target | Methods | Effectivity | Year ref. |
|---|---|---|---|---|
| AChE, acetylcholinesterase; BPS, 4,7-diphenyl-[1,10]phenanthroline disulfonate; bpy, 2,2′-bipyridine; BuChE, butyrylcholinesterase; CD, circular dichroism; DFT, density functional theory; DMSO, dimethyl sulfoxide; dpp, 2,3-bis(2-pyridyl)pyrazine; EPR, electron paramagnetic resonance; ESI-MS, electrospray ionization mass spectra; Flav, flavanones; glz, gliclazide; gtsm, glyoxalbis(N(4)-methylthiosemicarbazonato); HPLC, high-performance liquid chromatography; LDH, lactic dehydrogenase; LTP, long-term potentiation; ϕ-MePy, 2-phenyl-5-methyl-pyridine; NMR, nuclear magnetic resonance; np, non-peripheral; p, peripheral; Pc, phthalocyanines; Phen, 1,10-phenanthroline; ϕ-Phen, 4,7-diphenyl-1,10-phenanthroline; ROS, reactive oxygen species; TEM, transmission electronic microscopy; ThT, thioflavin T; thz, 1,3-thiazole; 8-BQ, 8-(1H-benzoimidazol-2-yl)-quinoline. | ||||
| [Phen-PtCl2] | Aβ42 and Aβ40 | Spectroscopic techniques, ThT studies, in vitro, and in vivo | IC50: 0.3–0.6 μM for ROS inhibition | 200854 |
| Cell viability: 80–100% (5 and 10 μM, Aβ42 10 μM; BL6J × 129sv mouse cortical neurons) | ||||
| LTP: 150–160% (4 μM, Aβ42 2 μM) | ||||
| [PtII(8-BQ)Cl2] | Aβ42 | Spectroscopic techniques, ThT studies, and in vitro | Cell viability: 94% (10 μM, Aβ42 10 μM; BL6J × 129sv mouse cortical neurons) | 201355 |
| LTP: 133% (2 μM, Aβ42 2 μM) | ||||
| [PtIV(8-BQ)Cl4] | In vivo | Reduction of 40% in Aβ42 levels and 26% in Aβ plaques (Tg2576 mice) | ||
| [Pt(BPS)Cl2] | Aβ16 and Aβ42 | Spectroscopic techniques | — | 201356 |
| [Pt(ϕ-MePy)(DMSO)Cl] | Aβ28 | Mass spectrometry and NMR spectroscopy | — | 201257 |
| [Pt(ϕ-MePy)(DMSO)Cl] | Aβ28 | ThT studies, mass spectrometry, and NMR spectroscopy | — | 201358 |
| [Pt(Phen)Cl2] | ||||
| [Pt(ϕ-Phen)Cl2] | ||||
| [Pt(Phen)Cl2] | Aβ16 | HPLC, ESI-MS and EPR spectroscopy | — | 201359 |
| fac-[Ru(CO)3Cl2-(N1-thz)] | Aβ28 | NMR and CD spectroscopies | — | 201060 |
| NAMI A | Aβ42 | In vitro | LDH released: 20% (PMRU20 40 μM), >50% (NAMI A and KP1019 40 μM), 90% Aβ42 (E17 embryos rat neocortex) | 201361 |
| KP1019 | ||||
| PMRU20 | ||||
| [(bpy)2RuII(dpp)PtIICl2]Cl2 | Aβ42 | Quantum chemical calculations and in vitro (ThT studies and western blot) | — | 201062 |
| M(Flav)2 and M(X)(Flav) (M = CuII and ZnII; X: bpy or Phen) | AChE and BuChE | In vitro | 60–80% Inhibition for BuChE and >90% for AChE | 201663 |
| Phthalocyanine ZnII and GaIII complexes | AChE and BuChE | In vitro | IC50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19.24–52.21 μM for BuChE, and 51.32–92.31 μM for AChE :19.24–52.21 μM for BuChE, and 51.32–92.31 μM for AChE |
202164 |
| ZnPcp/ZnPcnp | AChE and BuChE | In silico calculation and in vitro | Best IC50 values: AChE: MgPcnp 25.16 μM BuChE: MgPcnp 30.02 μM | 202265 |
| MgPcp/MgPcnp | ||||
| Amino acid Schiff base ZnII complexes | AChE and BuChE | Molecular docking calculations and in vitro | IC50:4.73–11.25 μM for BuChE, and 7.24–18.06 μM for AChE |
202266 |
| [Pd(glz)2] | AChE, BuChE, and Aβ42 | In vitro and in vivo | IC50:27 ± 1 μM for BuChE |
202267 |
| [CuII(gtsm)] | Aβ and tau | In vitro and in vivo | LTP: 121.4% (1 μM, Aβ42 2 μM) | 200968 |
| 48% Improved cognitive performance (10 mg kg−1 body weight; APP/PS1 transgenic AD mice) | ||||
| Macrocyclic platiniferous chelators | Aβ40 | In vitro and in vivo | Cell viability: 65–81% in the MII-Aβ system (Aβ40 10 μM; C57BL6/J mice cortical neurons) | 201269 |
Inhibition of β-amyloid aggregation
Aβ peptides show high affinity for binding metal ions that can modulate peptide aggregation and toxicity. As previously mentioned, metals ions such as CuII, ZnII, and FeIII interact with Aβ via three histidine residues (6, 13, and 14) modulating the aggregation and toxicity of Aβ producing reactive oxygen species (ROS). For this reason, most of the reported complexes in AD treatment are centered on inhibiting Aβ aggregation by coordinating with histidine residues.70,71 In 2008, Barnham et al. reported the first discrete coordination compounds active against AD, with the general formula [Phen-PtCl2] (Phen: phenanthroline derivatives; Fig. 3).54 These compounds, considered as analogues of cisplatin as they are based on labile chloride ligands and 1,10-phenanthroline derivatives, bind to histidine residues of Aβ42 acting as potent inhibitors of Aβ aggregation and neurotoxicity. In addition, the ability of these compounds to inhibit the metal-dependent biochemical and cellular actions of Aβ was studied. The authors demonstrated that all prepared compounds can inhibit Aβ:CuII-mediated redox chemistry with a half maximal inhibitory concentration (IC50) of 0.3–0.6 μM.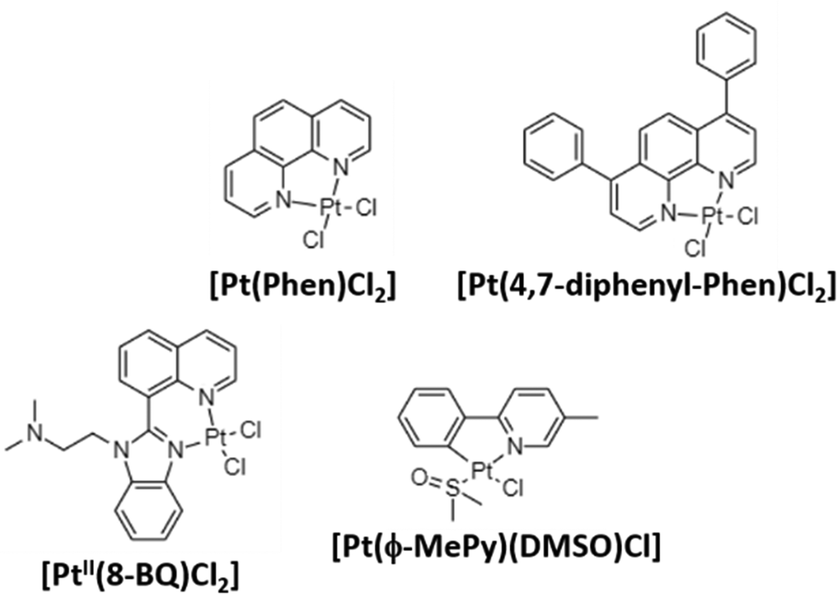 | ||
| Fig. 3 Some reported PtII complexes against Aβ aggregation from Barnham,54 Kenche,55 and Sasaki et al.57 | ||
Following the same trend as for Pt-drugs in cancer treatment, in 2013, novel platinum(II) and (IV) complexes based on 8-(1H-benzoimidazol-2-yl)-quinoline (8-BQ) were obtained showing high solubility and stability in aqueous media.55 In neuronal cell cultures treated with Aβ42 (10 μM, cell viability 77%), compound 4 ([PtII(N,N-dimethyl-2-[2-(quinolin-8-yl)-1H-benzimidazol-1-yl]ethanamine)Cl2], 10 μM) significantly inhibited Aβ aggregation and toxicity in neurons, achieving a 94% cell viability. Finally, in a way to prepare orally administered anti-amyloid compounds, the PtIV complex [PtIV(N,N-dimethyl-2-[2-(quinolin-8-yl)-1H-benzimidazol-1-yl]thanamine)Cl4] was synthesized as a prodrug. It is known that the substitution reactions for PtIV complexes are kinetically very slow, and therefore, stable under acidic environments of the stomach. Then, PtIV complexes can be reduced by natural reductants (i.e., glutathione), and become active. After the oral administration of the PtIV compound to the APP/PS1 mouse (used as the model of AD), a 40% reduction in Aβ42 levels was found, demonstrating the potential of PtIV compounds in AD treatment.
The same year, Streltsov et al. reported a novel platinum complex, [Pt(BPS)Cl2] (BPS: 4,7-diphenyl-[1,10]phenanthroline disulfonate), also active against the inhibition of Aβ42 aggregation by binding to histidine residues.56 Through different experimental techniques (X-ray absorption fine structure spectroscopy-XAS, mass spectrometry-MS and dynamic light scattering-DLS) and density functional theory-DFT modelling, the authors demonstrated the interaction of [Pt(BPS)Cl2] and Aβ42 through histidines 6 and 14, while the non-active cisplatin coordinates predominantly at the S atom of methionine 35. Inspired by Barnham's compounds, Sasaki et al. explored the reactivity of a new cyclometallated PtII complex, [Pt(ϕ-MePy)(DMSO)Cl] (ϕ-MePy: 2-phenyl-5-methyl-pyridine, DMSO: dimethyl sulfoxide),57 with Aβ28, since this peptide includes both the metal binding histidine residues and an hydrophobic part (residues 17–21) responsible for the nucleation process of Aβ aggregation. Compared to the Barnham [Pt(Phen)Cl2] compound, this new complex is able to bind to Aβ28 and to modify the CuII coordination site in the Aβ:CuII adduct. Afterwards, the same group investigated the ability of Barnham's and Sasaki's PtII compounds (Fig. 3) to interfere in ZnII coordination to the Aβ28 peptide, and CuII-induced ROS production,58 but no significant inhibition in Aβ:CuII was induced in all the tested PtII compounds. On the other hand, regarding the ZnII-induced Aβ28 aggregation, the fibril formation depends on the tested compound, decreasing in the following order: [Pt(Phen)Cl2] and [Pt(ϕ-Phen)Cl2] (no active) > [Pt(bpy)Cl2] (bpy: 2,2-bipyridine) > [Pt(ϕ-MePy)(DMSO)Cl] and [Pt(DMSO)2Cl2] (more potent inhibitors). These differences were attributed to the lability of the linkers, since the more active compounds possess labile ligands (DMSO is more labile than chloride ligands), and thanks to the C–Pt bond in [Pt(ϕ-MePy)(DMSO)Cl], as well due to a strong trans effect.
In a similar work, Ma and coworkers also investigated the interaction between [Pt(Phen)Cl2] and the adducts Aβ:CuII and Aβ:ZnII, but using Aβ16.59 Their results showed that [Pt(Phen)Cl2] preferentially targeted CuII/ZnII binding sites and changed their binding modes, releasing metal ions under some conditions (i.e., acidic media). As PtII is less reactive than CuII and ZnII, the chelation of [Pt(Phen)Cl2] might hamper the Aβ aggregation. Again, following a similar line of research by using PtII and PtIV, RuII compounds have also been studied as Aβ aggregation inhibitors. Authors selected this metal as it is well-stablished that ruthenium compounds are on the average far less toxic than platinum-based ones. Thus, in this work, the complex fac-[Ru(CO)3Cl2(N1-thz)] (thz: 1,3-thiazole) was tested against Aβ28.60 The studies revealed that {Ru(CO)3}2+ formed a stable adduct with Aβ28 coordinating with His13 and His14. Following this trend, the same group carried out in vitro assays against the AD cellular model with two well-known anticancer RuIII compounds (NAMI A or [ImH][RuCl4(DMSO)(Im)], KP1019 or [IndH]trans-[RuCl4(Ind)2], and PMRU20 or [2-AmThH]trans-[RuCl4(2-AmTh)2] (Im: imidazole; Ind: indazole; 2-AmTh: 2-aminothiazolium).61 The activity against AD was measured through the released lactic dehydrogenase (LDH) which is normally delivered during cell damage or death.72 Aβ42 produced almost 100% of released LDH compared with the control group, whereas PMRU20 (40 μM) reduced this release to 20%. Furthermore, PMRU20 showed a significant neuroprotective activity; as long as at the same concentration, NAMI A and KP1019 were poorly (ca. 80%) or nearly inactive (ca. 95%).
During their research on inhibiting the Aβ42 peptide aggregation, Kumar et al. obtained a RuII–PtII binuclear metal complex, [(bpy)2RuII(dpp)PtIICl2]Cl2 (dpp: 2,3-bis(2-pyridyl)pyrazine).62 First, authors demonstrated through in silico calculations that the binuclear complex exhibited higher binding affinities in terms of energetic properties than the mononuclear complex [(dpp)PtIICl2], suggesting that a second metal ion could improve the Aβ inhibition behavior. When tested in vitro using the primary mouse neuronal cell culture, this compound is able to inhibit Aβ42 aggregation in a similar manner to Barnham's compound with an IC50 of ∼4 μM. Finally, western blot tests demonstrated the absence of fibrils and oligomeric species of Aβ, whereas Barnham's compound did not prevent the formation of intermediates of aggregation, demonstrating a different way of interaction.
Cholinesterases inhibition
A different mechanism to target AD is to inhibit cholinesterases, mainly acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) in order to avoid the reduction of acetylcholine levels. The research here is mainly focused on the use of different previously reported active organic moieties (flavanones, phthalocyanines, Schiff bases, and gliclazide; Fig. 4), and their coordination with different metals (Cu, Zn, Ga, and Pd), to obtain novel metallodrugs against AD.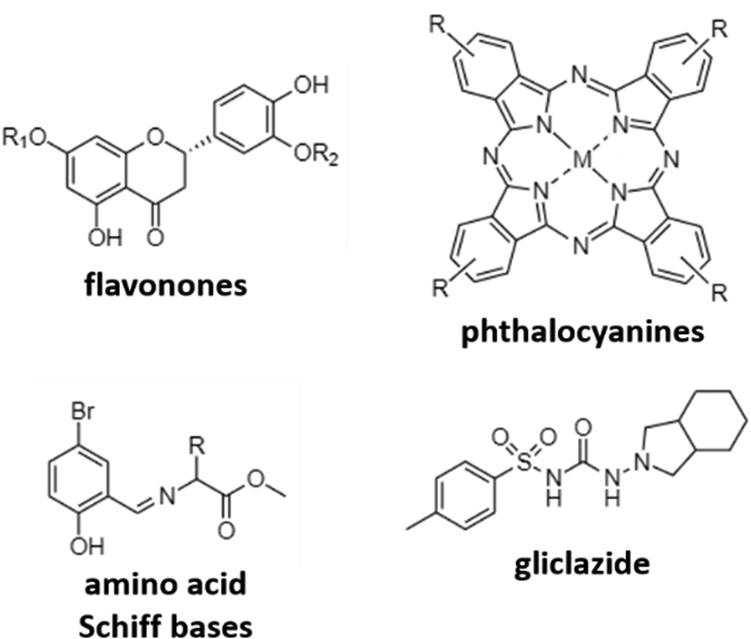 | ||
| Fig. 4 Organic moieties used to obtain new coordination compounds as cholinesterase inhibitors. | ||
In 2016, Sarria et al. reported seventeen CuII and ZnII complexes based on flavanone derivatives (hesperidin, hesperetin, naringin, and naringenin), known for their neuroprotective effects, and studied their activity against AChE and BuChE.63 Compared with the parent flavanones, nine of these complexes constituted potent, reversible, and selective anti-cholinesterase activity. Comparatively, CuII complexes possessed higher inhibitory potency against AChE than the corresponding ZnII complexes. Authors attributed these differences to the d9 electron configuration of CuII, which left two free empty orbitals for interaction with electron donor ligands, such as N atoms of histidine residues in AChE. In contrast, the d10 electron configuration of ZnII imposes the tetrahedral geometry with no free empty orbitals to coordination. Furthermore, CuII complexes containing ancillary ligands (bpy or Phen) showed enhanced inhibitory activity, probably due to the coordination sphere of CuII that favors the π–π stacking interactions. Phthalocyanines are interesting synthetic molecules similar to the heme group with pharmacological and biological applications.73–76 Based on these molecules, Günsel and coworkers obtained ZnII and GaIII complexes and their water-soluble hydrochloride derivatives, and tested their inhibitory activity against AChE and BuChE.64 All complexes showed high BuChE inhibitory activity, with IC50 values in the range of 19.24–52.21 μM, and 51.32–92.31 μM for BuChE and AChE, respectively. These results were further improved compared to those obtained for tacrine (178.03 and 141.34 μM, respectively), which is the first drug introduced into the United States market to manage AD and used here as the standard inhibitor. In a different study using again phthalocyanines, Yalazan et al. obtained ZnII and MgII quinoline-fused phthalocyanine (Pc) compounds (ZnPcp/ZnPcnp and MgPcp/MgPcnp; p: peripheral, np: non-peripheral) in order to combine interesting therapeutic properties with spectroscopic characteristics.65 Among them, MgPcnp exhibited the highest inhibitory potential against AChE and BuChE with IC50 values of 25.16 and 30.02 μM, respectively. In silico calculations showed that MgPcnp had the highest HOMO value and the lowest ΔE energy value, demonstrating that it is the most reactive species. This fact together with its high solubility might explain its great inhibitory effect compared to the other synthesized compounds. However, although MgPcnp demonstrated to be efficient in the inhibition of AChE, it presented a lower inhibitory effect than galantamine (an approved drug in the treatment of AD and used as a positive control in this study).
Due to their interesting biological properties, other organic moieties used in the preparation of metallodrugs to treat AD are Schiff bases. In 2021, Şenocak et al. synthesized some compounds based on ZnII and amino acid Schiff bases and tested them against AChE and BuChE.66 First, amino acid Schiff bases were prepared using 5-bromo-2-hydroxybenzaldehyde and amino acid methyl esters (isoleucine, phenylalanine, and methionine). Once synthesized, the potential inhibition of Zn-based metallodrugs to cholinesterases was explored. The synthesized compounds were found to be highly efficient against BuChE and AChE, obtaining IC50 values in the range of 4.73–11.25 μM and 7.24–18.06 μM, respectively, surpassing the inhibitory activity of tacrine.
Recently, our research group published a complex based on palladium and the antidiabetic drug gliclazide (glz), [Pd(glz)2].67 Originally, we selected glz as the organic moiety in the preparation of AD's metallodrugs since several factors associate Diabetes mellitus with AD: (i) it is a risk factor for Alzheimer's disease, (ii) it has a similar molecular mechanism of action, and (iii) it is associated with protein misfolding.77,78 Thus, the potential of [Pd(glz)2] against AD was explored, obtaining the preferential inhibition of BuChE against AChE (91 vs. 58% inhibition), with an IC50 value of 27 μM for BuChE, as it is capable of interacting with both the free enzyme and the enzyme–substrate complex. Remarkably, its potential activity in vivo was also evaluated using Caenorhabditis elegans as the animal AD model. Aside from the absence of lethal toxicity, [Pd(glz)2] was able to increase the percentage of non-paralyzed worms by interacting with Aβ42 peptides and inhibiting its aggregation, activity probably associated with the planar structure of the complex (as previously reported for other metallic complexes).
Effects in metal chelation
Aβ plaques have abnormally high concentrations of metals such as Cu, Zn, and Fe, which promote the Aβ peptide aggregation. In agreement with these observations, some researchers considered the development of novel metalorganic drugs using known compounds that target abnormal metal accumulation. For example, thiosemicarbazones and bis-thiosemicarbazones have a wide range of pharmacological activities linked to their ability to chelate transition metals.79 These compounds elevate intracellular levels of metals, resulting in the activation of phosphoinositol-3-kinase (PI3K) and the inhibition of glycogen synthase kinase 3 (GSK3), which lead to the reduction of neurotoxic Aβ and tau phosphorylation. In addition, although further research studies are needed, some studies show that these compounds could cross the BBB.80,81 One example is the complex [CuII(gtsm)] (gtsm: glyoxalbis(N(4)-methylthiosemicarbazonato), which promotes the expression of Aβ-degrading pathways (Fig. 5).82,83 Both, in vitro and in vivo studies, demonstrated that [CuII(gtsm)] increases intracellular CuII, which is reduced to CuI, leading to copper dissociation from the ligand and increasing Cu bioavailability. This induces the cellular pathways that inhibit GSK3β, which regulates the accumulation of Aβ peptides and tau phosphorylation. Therefore, this compound is a promising therapy for AD as it attenuates both the Aβ- and tau-mediated pathologies.68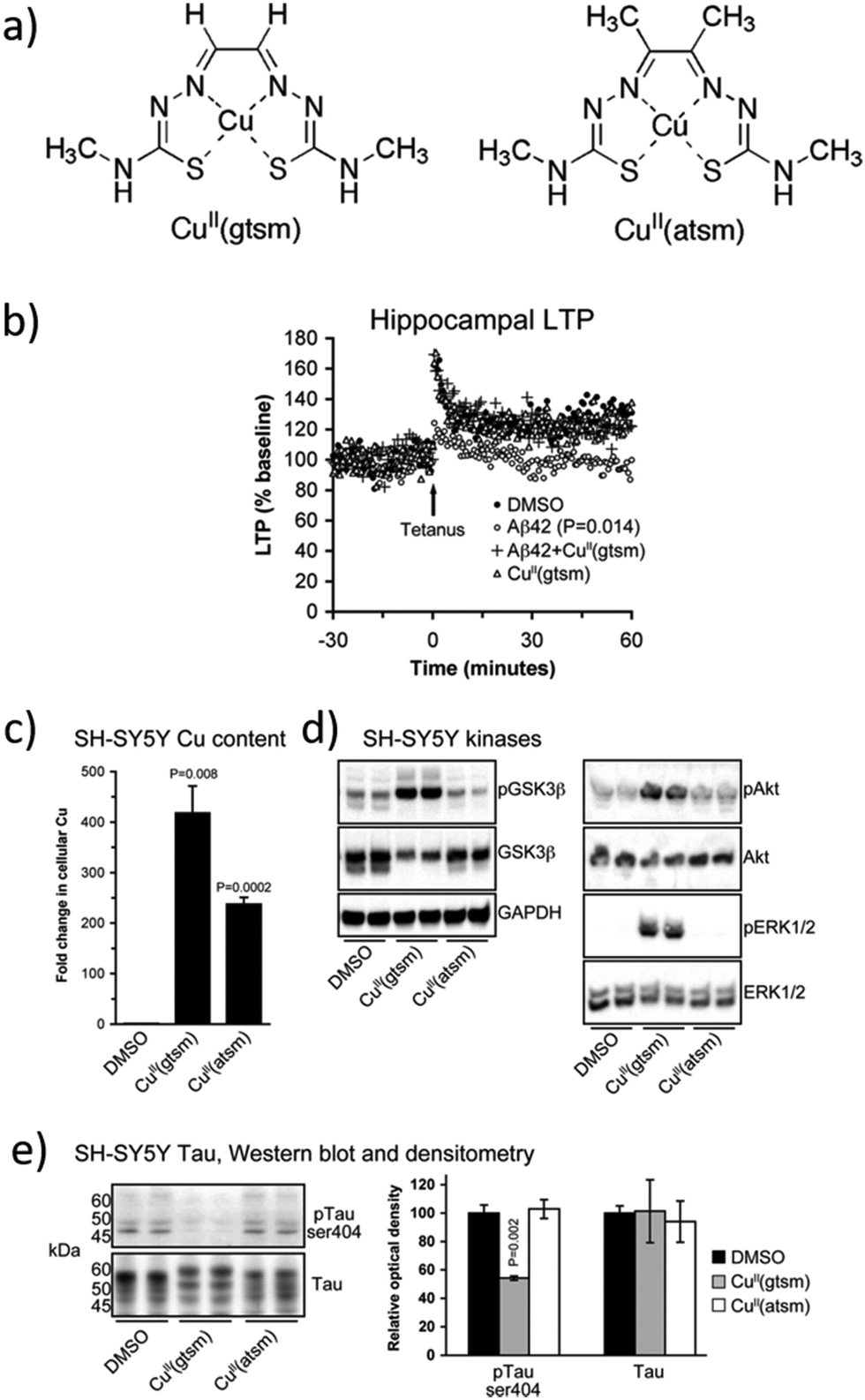 | ||
| Fig. 5 (a) Structures of [CuII(gtsm)] and the control [CuII(atsm)]. (b) Whereas Aβ42 inhibits the hippocampal long-term potentiation (LTP) by 28%, this is prevented when treated with [CuII(gtsm)]. (c) In vitro, [CuII(gtsm)] increases the cellular levels of Cu. (d) Western blot analyses show that [CuII(gtsm)] induces the phosphorylation of kinases GSK3β, Akt, and ERK1/2. (e) [CuII(gtsm)] inhibits tau phosphorylation at ser404 without affecting the abundance of total tau. Adapted from Ref. 68. Reproduced with permission. | ||
In 2015, Wang et al. developed macrocyclic platiniferous chelators containing cyclen (1,4,7,10-tetraazacyclododecane) that bind to metals such as ZnII and CuII, and the unit [Pt(bpy)Cl2] that coordinates to histidine residues of Aβ40.69,84 When tested in the cortical neuronal cell of C57BL6/J mice, these compounds demonstrated the inhibition of Aβ40 aggregation induced by ZnII or CuII and the production of ROS by the CuII–Aβ complex. Furthermore, when tested in vivo, these compounds reduced the extent of Aβ aggregation in brain homogenates of transgenic mice. The authors argue that these powerful inhibitions might be promoted by their bifunctional active characters rather than their individual units. In addition, the results were compared with cisplatin, showing that, in contrast, it aggravated the metal-induced Aβ aggregation as cisplatin coordinates with methionine rather than the metal binding site of Aβ formed by histidine residues.
Finally, a brief mention to combat AD through metal (Cu, Zn, Fe) chelation with purely organic moieties (organic drugs85–87 and small peptides)88,89 is made. One outstanding example is the effective use of quinoline derivatives like clioquinol and 8-hydroxyquinoline which have entered clinical trials for AD.90,91
MOFs as alternative platform in AD treatment
MOFs can adsorb a wide range of molecules. Particularly in biomedicine, MOFs have been used to control the dose and delivery kinetics of previously adsorbed active ingredients (drugs, gases, etc.),46 or as adsorbent detoxifying agents in the treatment of oral overdoses.92,93 Very recently, some research groups have focused their efforts in the application of MOFs in AD treatment (Table 2). One of the first examples is the work of Mensinger et al. who originally studied the Aβ40 peptide adsorption capability of seven archetypal MOFs (HKUST-1, MIL-53, MIL-69, MIL-88B, MIL-100(Fe), MIL-101(Cr)NDC, and UiO-66).94 All MOFs, except MIL-100(Fe), exhibited complete (100%) and quick (10 min) adsorption of Aβ40. Furthermore, authors checked that the intact peptide can be desorbed from HKUST-1 and UiO-66, suggesting that it is adsorbed rather than being hydrolyzed. Although MOFs were demonstrated to be excellent adsorbents of Aβ40, in this work some important issues related to the MOF particle size and their potential passage through the BBB to obtain the peptide have not been considered.| MOF or MOF composite | Target | Methods | Effectivity | Year ref. |
|---|---|---|---|---|
| DFT, density functional theory; PMOF, porphyrinic metal–organic framework; ROS, reactive oxygen species; TEM, transmission electronic microscopy; ThT, thioflavin T. | ||||
| HKUST-1, UiO-66, MIL-53, MIL-69, MIL-88B, MIL-100(Fe) and MIL-101(Cr)NDC | Aβ40 adsorption | In vitro and gel electrophoresis | — | 202094 |
| PCN-224 | Aβ42 photooxygenation | ThT studies, TEM, gel electrophoresis and in vitro | Cell viability: 90% under 650 nm light (PC12 cells) | 201895 |
| PMOFs | Aβ40 photooxygenation CuII chelator | Spectroscopic techniques, DFT calculations and in vivo | Hf-MOF results: ROS production: 173 PL intensity | 201996 |
| Cu-chelating: 0.5 g g−1 | ||||
| Aβ enrichment: 4.1 μmol mg−1 | ||||
| CeRMS | Enhance neurogenesis and reduce ROS | In vitro and in vivo | — | 202097 |
Another approach in the use of MOFs in AD treatment is through the oxygenation of Aβ as a way to suppress its aggregation and neurotoxicity. In 2018, Wang et al. proposed the porphyrinic ZrIV metal–organic framework (PCN-224) as a platform in the inhibition of Aβ aggregation by near-infrared (NIR)-activated photooxygenation.95 The use of porphyrinic ligands give rise MOFs with great porosity that enhances the accessibility of oxygen and subsequently, the photo-induced 1O2 generation. First, the synthesis of PCN-224 was improved to obtain small nanoparticles, as it is known that to cross the BBB the nanoparticle size should be within 10–100 nm. Field-emission scanning electron microscopy (FESEM) images demonstrated that the PCN-224 nanoparticle average size was around 70 nm. However, no data about the particle size of PCN-224 in solutions (water, phosphate buffer saline-PBS, etc.) were reported, which is mandatory to discard the potential aggregation of nanoparticles before their possible administration. Results on the photoinduced inhibitory effect of PCN-224 in the aggregation of Aβ42 peptides showed that after irradiation under 650 nm light, thioflavin T (used to monitor the aggregation degree) fluorescence intensity remained low; hence, the aggregation of Aβ42 was inhibited. Finally, in vitro studies in PC12 cells (from transplantable rat pheochromocytoma) showed an increase in cell viability from 40 to 90% when Aβ42 was incubated with PCN-224 under 650 nm light irradiation. This fact could be explained since the physical adhesion of Aβ42 to the PCN-224 surface may reduce the active sites in Aβ, and therefore, its aggregation, increasing the efficiency of photooxygenation.
In a further study, Yu and coworkers reported the ability of four already known porphyrinic based MOFs (PMOFs) as CuII chelating (as porphyrin is a good chelator) and photooxidation agents for inhibiting Aβ aggregation.96 Among the PMOFs studied (Zr-MOF, Al-MOF, Ni-MOF, and Hf-MOF), the Hf-MOF was the best Aβ oxidant because of its great ability for inducing ROS generation, and the efficient removal of CuII (0.5 g of Cu per g of the PMOF). In addition, thanks to its porosity, the Hf-MOF enriched Aβ at a concentration of 4.1 μmol mg−1, which can minimize the side effects of 1O2 to normal cells, and its oxidation occurred even after binding with CuII. In the presence of the Hf-MOF, CuII-induced Aβ aggregation was reduced and completely suppressed when light was irradiated. Afterwards, the Hf-MOF was modified with a Aβ-targeting peptide LPFFD (Lys-Pro-Phe-Phe-Asp), thus enhancing specificity to Aβ and photooxidation efficiency. This modification increased the percentage of oxidized Aβ (87% vs 66% without the LPFFD), and the reduced cytotoxicity of Aβ and CuII-induced Aβ aggregation. Finally, in vivo studies verified the inhibition effects of the LPFFD-modified Hf-MOF by a commonly used AD transgenic C. elegans model (Fig. 6).
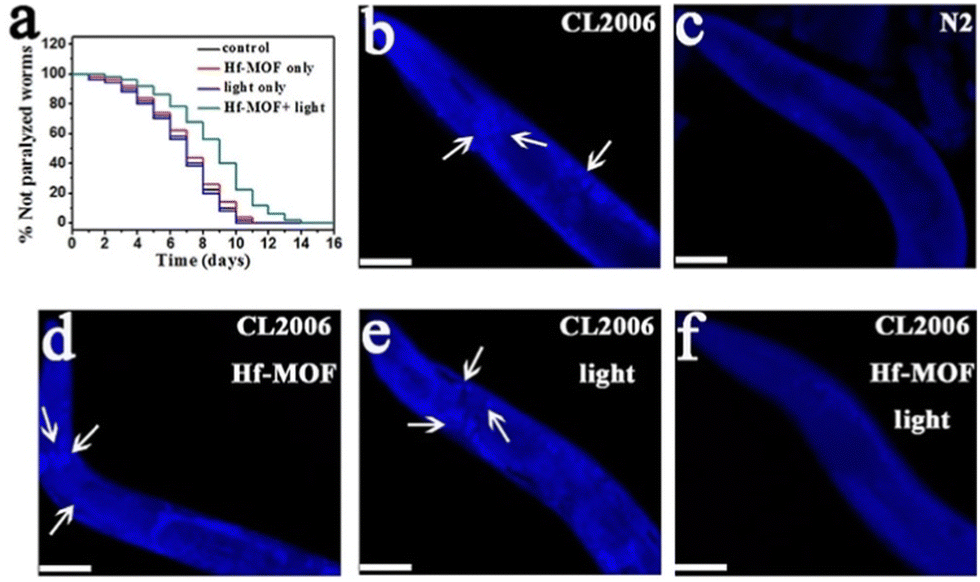 | ||
| Fig. 6 Effect of the Hf-MOF in C. elegans. (a) Kaplan–Meier survival curves of the transgenic strain AD CL2006 show a delay in the Aβ-triggered paralysis of worms when treated with the Hf-MOF under light. (b–f) Images of the worm's head region of CL2006 (arrows indicate Aβ deposits): (b) transgenic CL2006 worm, (c) Bristol N2 worm (wild type), (d) CL2006 treated with the Hf-MOF, (e) CL2006 with light, and (f) CL2006 treated with the Hf-MOF under light. Reproduced from ref. 96 with permission. | ||
In another study, the same research group modulated the neuron differentiation and amelioration of oxidative stress by integrating antioxidative nanozymes (ceria) into a MOF for synergically enhanced neurogenesis, which contributes to self-repair after neuron loss.97 The main issue is that the high level of oxidative stress in AD promotes the neural stem cell (NSC) death and neuronal apoptosis. The nanoplatform, abbreviated as CeRMS (CeNPs/RA@MIL-100/siSOX9), contains Ce (for the elimination of ROS based on the electron transfer CeIII–CeIV), and MIL-100, which also possesses an antioxidative effect, loaded with siSOX9 (the regulator of the SOX9 protein and an essential transcriptor factor beneficial to gliogenesis) and retinoic acid (RA, the regulator in the differentiation of the NSC to neurons). First, the RA release was studied in H2O2 (1 mM). Under these conditions, CeRMS was destroyed and 40% of RA was released after 15 min of treatment. Furthermore, the ability of CeRMS to modulate the NSC differentiation was tested. Whereas other systems without RA, such as CeM, CeRM, and CeMS, used here as controls, differentiated NSC into glial cells, CeRMS, containing RA, completely differentiated the NSC into neurons. Additionally, under both in vitro and in vivo conditions, CeRMS reduced the intracellular levels of ROS better than ascorbic acid and, consequently, it decreased the number of apoptotic cells caused by Aβ42 aggregation. Overall, this study provides an original synergic strategy, promoting nerve regeneration while protecting from oxidative stress.
MOFs in the detection of the AD
AD progresses asymptomatically for years (up to 20 years depending on age, sex, genetic predisposition, etc.) before the appearance of symptoms.98 Thus, the early diagnosis of AD might have a major impact on the progress and efficient timely treatment of the disease. As previously mentioned in the Introduction section, AD begins with the deposition of Aβ, followed by the appearance of tau tangles, and then by cognitive impairment.99 Initially, the AD diagnosis was a result of neurological, psychiatric and clinical examinations, neuropsychological tests, and laboratory studies.100 It was in 2011, and based on neuroimaging techniques and CSF analysis, when the National Institute on Ageing and Alzheimer's Association (NIA-AA) workgroup incorporated into the clinical criteria AD pathology biomarkers, such as Aβ deposition in the brain, Aβ42, and the pathologic tau level in the CSF. Currently, three biomarkers have been well-established and validated to diagnose AD: Aβ1–42, total tau, and phosphor-tau-181.101Aside from high concentrations of metal ions (i.e., Cu, Zn, and Fe) in the brain of AD patients, biomolecules such as glutathione (GSH), a key regulator for the redox balance of biological processes;102 or protein kinases, responsible for the catalysis in the phosphorylation of protein substrates or specific peptides;103 have been proposed as biomarkers. However, only amyloid plaques and tau tangles are specific features that define AD as a unique neurodegenerative disease among other disorders.104 In this review, only the use of MOFs in the detection of Aβ and tau is discussed in detail (Table 3).
| MOF | Biomarker | Detection method | Type of sample | Linear range LOD | Year ref. |
|---|---|---|---|---|---|
| ABEI, N-(aminobutyl)-N-(ethylisoluminol); AβO, β-amyloid oligomer; aCSF, artificial cerebrospinal fluid; BHQ-1, black hole quencher; bpy, 2,2′-bipyridine; BTC, 1,3,5-benzenetricarboxylate; CV, cyclic voltammetry; DPV, differential pulse voltammetry; ECL, electrochemiluminescence; Fc, ferrocene; g-C3N4, graphite carbon nitride; H-USM, highly doped upconversion nanoparticles-SiO2@MOF; LOD, limit of detection; MB, methylene blue; NPs, nanoparticles; PANI, polyaniline; PPy, polypyrrole; RET, resonance energy transfer; SWV, square wave voltametric; ThT, thioflavine T; TMBDA, N,N,N′,N′-tetramethyl-1,4-butanediamine. | |||||
| Fc@IR-MOF-3 | Aβ proteins | Electrochemical | Human serum | 10−4–100 ng mL−1 | 2017105 |
| 0.03 pg mL−1 | |||||
| TMBDA-MIL-100(Fe) | Choline | Photoluminescence | Buffer solution | 0.5–10 μM | 2018107 |
| 27 nM | |||||
| Acetylcholine | 0.1–10 μM | ||||
| 36 nM | |||||
| AuNPs/Cu-MOF | AβO | DPV | aCSF | 1 nM–2 μM | 2018108 |
| 0.45 nM | |||||
| Cu-BTC/Tb | Aβ peptide | Fluorescence | Blood | 1–550 nM | 2018109 |
| 0.3 nM | |||||
| Fc@ZIF-8 | AβO | UV-vis and CV | Real samples | 10−5–102 μM | 2019110 |
| 10−5 μM | |||||
| Ru@MOF | Aβ proteins | ECL-RET | Buffer solution | 10−5–500 ng mL−1 | 2019111 |
| 3.9 fg mL−1 | |||||
| Co-MOFs/ABEI | Aβ proteins | ECL | Human serum | 10 fg mL−1–100 ng mL−1 | 2019106 |
| Fe3O4@PPy-Au | 3 fg mL−1 | ||||
| UiO-66@PANI-MB | Aβ proteins | SWV | Serum | 10 fg mL−1–100 ng mL−1 | 2019112 |
| Cu-Al2O3-g-C3N4-Pd | 3.3 fg mL−1 | ||||
| [Ru(bpy)3][Zn2(C2O4)3] + Au@NiFe | Aβ proteins | ECL | aCSF | 10−4–100 ng mL−1 | 2019113 |
| MOF | 13.8 fg mL−1 | ||||
| Pd NPs@MIL-53(Al)-NH2 | Aβ proteins | ECL | Aβ solutions | 10 fg mL−1–50 ng mL−1 | 2019114 |
| 3.4 fg mL−1 | |||||
| Ru@MIL-101(Al) | AβO | Fluorescence | Blood | 1–10000 pM |
2020115 |
| 0.3 pM | |||||
| Ru(bpy)32+/NH2-UiO-66 and MIL-101@Au–MoS2 | Aβ proteins | ECL | Buffer solution | 0.00001–50 ng mL−1 | 2020116 |
| 3.27 fg mL−1 | |||||
| MOF-armored-anti-DNA antibody | AβO | Fluorescence | Blood | 0.001–100 ng mL−1 | 2020117 |
| 0.4 pg mL−1 | |||||
| ThT@Er-MOF | Presenilin 1 | Fluorescence | CSF | 0–40 nM | 2020118 |
| 0.142 nM | |||||
| 0–33 nM | |||||
| Aβ protein | Fluorescence | 0.517 nM | |||
| Acetylcholine | Photoluminescence | 0–10 nM | |||
| 0.03226 nM | |||||
| AuNPs@CuMOF | AβO | DPV | aCSF | 0.5–500 fM | 2021119 |
| 0.25 fM | |||||
| H-USM-SiO2@ZIF-8/BHQ-1 | AβO | Fluorescence | AβO solutions | 0.01–10 μM | 2021120 |
| 28.4 pM | |||||
| AuNPs@MIL-88(Fe)-NH2 | AβO | ECL | CSF | 0.1–10 pM | 2021121 |
| 71 fM | |||||
In the first example of the potential of MOFs in Aβ detection reported by Han et al.,105 the postsynthetic modification of the Zn based material IR-MOF-3 was used as the key to access to this specialized application. Particularly, the active ferrocene (Fc, frequently used as the signal tag) is covalently combined in the porous MOF through an amination reaction (Fig. 7a), leading to a composite with high-content Fc signal units well distributed with enhanced electrochemical activity. Furthermore, the encapsulation of Fc into the MOF confers some stability and avoids the leakage of the signal molecule. In a further step, the outer surface of the Fc@IR-MOF-3 composite was modified with gold nanoparticles (AuNPs) as platforms to label with detection antibodies (Ab2). The stability of Fc@IR-MOF-3 was assessed under extreme conditions, such as strong acid/base and polar organic solvents, through the relative electrochemical activity of Fc (Fig. 7b). More than 95–97% of the Fc activity was retained when using a pH range from 3 to 10, and different solvents. These results were further better than the ones obtained in the free Fc, where only 15–34% of the Fc activity was retained, respectively, suggesting that the interaction between Fc and the MOF improves the robustness of the signal molecule. The analytical performance of the immunosensor was tested in PBS, achieving a detection limit of 0.03 pg mL−1. Furthermore, the selectivity of the immunoassay for the detection of target Aβ was analyzed against potential interfering substances (human serum albumin, thrombin, immunoglobulin, etc.). Despite the high concentration of the potential interfering substances (100 ng mL−1) compared with Aβ (10 ng mL−1), no differences were observed in the square wave voltammetry (SWV) responses. Thus, the Fc@IR-MOF-3 composite could selectively discriminate the target Aβ from other non-target proteins. Finally, the authors confirmed the possible application of the designed immunosensor in human serum samples, obtaining recoveries of 90.0 to 110.3%, which prove the potential of this material for the detection of Aβ in real samples.
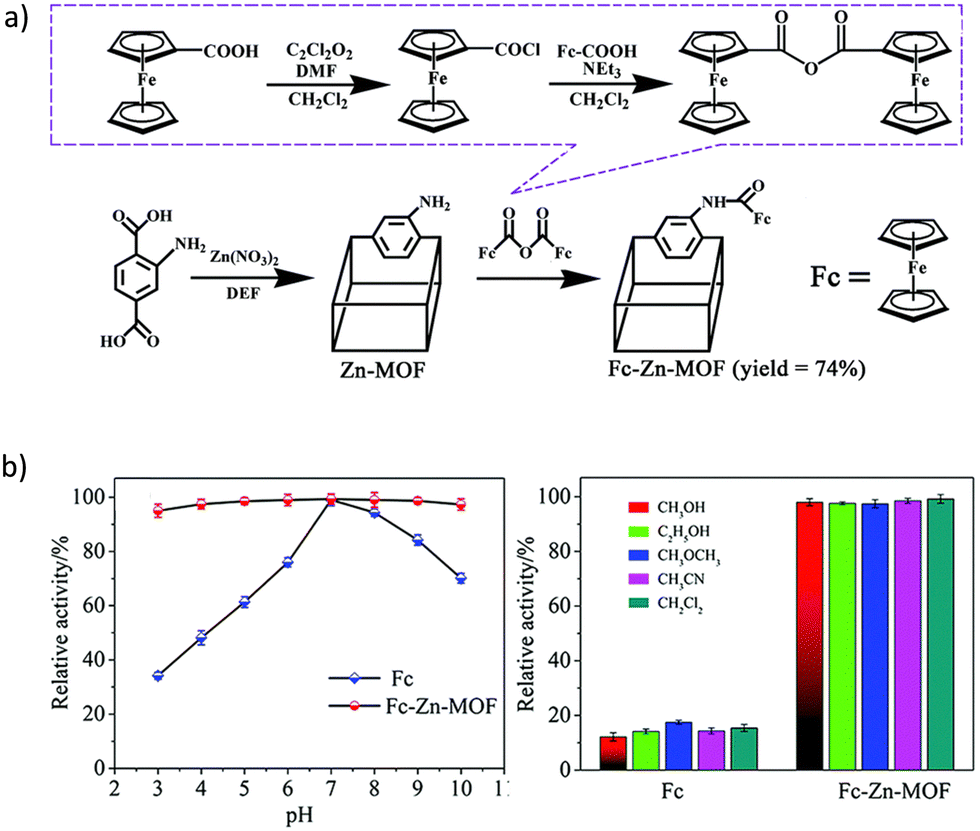 | ||
| Fig. 7 (a) Schematic diagram of the post-synthetic modification of IR-MOF-3 with Fc. (b) Electrochemical activities of Fc and Fc@IR-MOF-3 at different pHs (3 to 10) and in PBS containing 50% of selected organic solvents. Reproduced from Ref. 105 with permission from the Royal Society of Chemistry. | ||
In a subsequent report and using a one-pot synthetic method, an analog of luminol (N-(aminobutyl)-N-(ethylisoluminol), named ABEI) was incorporated into a Co-based MOF.106 The potential of luminol and its derivatives in electrochemiluminescence (ECL) lies in their low oxidation potential, high luminous efficiency, and nontoxicity. The obtained Co-MOF/ABEI was decorated with AuNPs as platforms to label with detection antibodies. Incremental ECL intensities were obtained with the increasing Aβ42 concentration, achieving a detection limit (0.003 pg mL−1) ten times better that the first MOF reported for this application. The feasibility of this proposed immunoassay in AD diagnosis was tested in human serum samples, obtaining excellent recoveries (96.0–104.0%).
The sensitivity towards Aβ oligomers (AβO) was again improved in a further study where an aptasensor was prepared based on a luminescence nanoscaled lanthanum-based MOF.117 The system was designed based on the MOF-armored single-stranded DNA antibody (anti-DNA antibody@MOF) and aptamer magnetic beads (Apt-Fe3O4) as capture probes. Again, the strategy followed in the preparation of the signal probe (anti-DNA antibody@MOF) was a bottle around the ship mechanism. The mineralization of the L-MOF around the anti-DNA antibody forms an exoskeletal cage, potentially protecting the encapsulated antibody. This AβO assay demonstrated high sensibility and selectivity, with a detection limit of 0.4 pg mL−1, even in samples with chief components in blood (K+, Na+, glucose, immunoglobulin, etc.). Compared with other studies on AβO detection based on MOFs, here the reusability of the aptasensor was investigated. The aptamer was easily regenerated from the complex AβO/Apt-Fe3O4 by heating at 50 °C. The aptasensor could be reused 10 times while maintaining its activity. Furthermore, after 7 weeks at room temperature, the aptasensor retained ca. 80% of its fluorescence intensity. This advantage in the long-term preservation activity can potentially solve the problems of manufacturing cost aptasensors.
In a recent study, ZIF-8 loaded with black hole quencher molecules (BHQ-1) was used as a coating for the efficient inactivation of the fluorescence of highly doped upconversion nanoparticles (H-UCNPs) in NH2–SiO2 microspheres.120 BHQ-1 loaded ZIF-8 exhibited an excellent quenching efficiency for upconversion emission. In contrast, when different concentrations of AβO are present, the upconversion luminescence was recovered due to the decomposition of ZIF-8 and the release of BHQ-1. Authors confirmed that after adding AβO, the competitive binding of ZnII and amino acids of AβO will induce the disassembly of ZIF-8. However, considering that ZIF-8 particles are rather unstable in the serum and most common buffers,122–124 a crucial and deep study of the ZIF-8 composite stability in different biological media or in the presence of other competitive biomolecules is necessary for the potential application of this composite material in real samples.
Sensing and treatment for AD (theragnosis)
Theragnosis is the combination of diagnosis and therapy in an effective formulation. Since it is a term fairly new, there is growing interest in this field. Currently, theragnosis is extensively studied in cancer by using mainly nanoparticles and nanocomposites.125–127 However, in AD, only few works have been carried out in theragnosis by using both discrete coordination compounds and MOFs (Table 4).| Complex | Target | Methods | Sensing | Treatment | Year ref. |
|---|---|---|---|---|---|
| bzq, 7,8-benzoquinoline; CeONP, ceria nanoparticles; DMF, N,N-dimethylformamide; H2cdda, 4,4′-(9H-carbazole-2,7-diyl)dibenzoic acid; Ksv, metal ion quenching coefficient; MB, methylene blue; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NOTA, 1,4,7-triazacyclononane-1,4,7-triacetic acid; PCM, 1-tetradecamol; PDA, polydopamine; phq, 2-phenylquinoline; ppy, 2-phenylpyridine; Res, resveratrol; ThT, thioflavin T. | |||||
| [Ir(ppy)2(H2O)2]3+ | Aβ40 | In vitro (ThT fluorescence and mass spectrometry) | [Ir(ppy)2(H2O)2]3+ | RhIII complex decrease Aβ40 length from 6 to 1.3 μm | 2011128 |
| [Rh(ppy)2(H2O)2]3+ | |||||
| [Ir(bzq)2(H2O)2]3+ | 56-fold with Aβ40 monomer and 134-fold with Aβ40 fibrils | IC50 25–50 μM | |||
| [Ir(phq)2(H2O)2]3+ | |||||
| Fe-MIL-88B-NH2-NOTA-DMK6240/MB | Tau protein | In vitro (MTT and ThT methods) and in vivo | — | 63% inhibition of tau aggregation | 2020129 |
| [Tb4(cdda)6(H2O)4](DMF) | Cu2+ ions, nf-κb, akt, and cytokines | In vitro and in vivo | K sv = 2,021.8 M−1 | nf-κb and akt: ∼1 of relative expression. | 2020130 |
| IL-1: 100–125 ng mL−1 | |||||
| IL-6: 140–160 ng mL−1 | |||||
| CeONP-Res-PCM@ZIF-8/PDA | Aβ oligomers | In vitro | 95% of quenching efficiency | 67.23% Res release | 2021132 |
| Detection limit 3.2 nM | 93.87% antioxidant effect | ||||
In 2011, Man et al. reported the first example of coordination compounds as inhibitors of Aβ aggregation and luminescent probes for Aβ40 peptides ([Ir(ppy)2(H2O)2]3+, [Rh(ppy)2(H2O)2]3+, [Ir(bzq)2(H2O)2]3+, and [Ir(phq)2(H2O)2]3+; ppy: 2-phenylpyridine, bzq: 7,8-benzoquinoline, and phq: 2-phenylquinoline).128 They inhibited Aβ40 aggregation in vitro since the labile H2O ligands are replaced by histidine residues and the aromatic ligand form stacking interactions with the peptide. Interestingly, complexes with extended aromatic ligands (i.e., bzq and phq) were less active, probably due to the impediment of the metal center to coordinate with histidine residues. Among them, the RhIII complex showed the highest inhibitory activity with a complex to Aβ40 monomer ratio of 1:10, thus subsequent in vitro experiments were carried out. The cytotoxicity study against neuroblastoma cells showed an IC50 value between 25 and 50 μM, higher than the concentration needed for complete Aβ40 aggregation inhibition. Finally, the luminescence properties of the IrIII complexes showed an increased emission in the presence of histidine solution compared to aqueous buffer.
Methylene blue (MB) is a well-studied tau aggregation inhibitor, but it is quickly dispersed after crossing the BBB, resulting in low bioavailability. In this regard, Zhao and coworkers synthesized the first MOF containing MB focused on theragnosis against hyperphosphorylated tau proteins.129 This nanoscale MOF based on Fe-MIL-88B-NH2 was obtained as a multifunctional material that integrates magnetic imaging (from Fe atoms), targeted therapy, and drug delivery. The nanoplatform Fe-MIL-88B-NH2-NOTA-DMK6240/MB was prepared by post-synthetic modification of the surface of Fe-MIL-88B-NH2 (acts as a nanocarrier and a good magnetic resonance imaging-MRI contrast agent) with NOTA (1,4,7-triazacyclononane-1,4,7-triacetic acid, a macrocyclic linker that binds DMK6240 to the surface), and DMK6240 (hyperphosphorylated tau-directing unit). Finally, MB was loaded into the NMOF, achieving a total particle size of 190–240 nm. After verifying the hyperphosphorylated tau-target ability, MRI capability, and the lack of toxicity, the inhibition of tau aggregation was tested. The nanoplatform could produce 63% of inhibition in tau aggregation and had a neuroprotective effect reaching 80% of cell survival. In addition, learning and memory ability tests in vivo indicated that the ability of targeting and the inhibition of tau hyperphosphorylation contributed to improve memory loss in AD model rats. Finally, the degradation assays of the NMOF showed that both Fe and ligand concentrations reached a maximum on the 7th day and then were gradually reduced, demonstrating its biodegradability.
In a subsequent work, a lanthanide 2D polymer based on TbIII was studied due to its interesting luminescence properties as a CuII sensor.130 Here, the compound [Tb4(cdda)6(H2O)4](DMF) (H2cdda: 4,4′-(9H-carbazole-2,7-diyl)dibenzoic acid, DMF: N,N-dimethylformamide) was incubated with several transition metals and the photoluminescence was measured. Whereas all metal ions just improved or reduced the luminescence strength of the complex, CuII exhibited a strong quenching action with a quenching coefficient of 2021.8 M−1, higher than other Ln-MOF sensors. This effect is a result of the smaller CuII radius, which allows the easy interaction with the N atom of the ligand cdda and, therefore, decreases the ligands’ antenna efficiency and reduces the energy transfer of the compound. On the other hand, since the inflammatory response on neurons could lead to AD, the anti-inflammatory effect of the compound was tested. After injecting Aβ in rats, the nf-κb and akt levels were much higher than the control, but when the polymer was administered, these levels were lowered to normal concentrations. Furthermore, this compound also reduced the levels of the cytokines IL-1 and IL-6 in the CSF, which are responsible for the decrease in the production of the inflammatory response.
In the most recent work about theragnosis in AD, Yan et al. synthesized a nanocomposite as a platform for the detection and inhibition of AβO fibrillation.131 The base of the nanostructure is ZIF-8, since it is a good nanocarrier in biomedicine and has fluorescence quenching properties. The system, CeONP-Res-PCM@ZIF-8/PDA, contains polydopamine (PDA) that enhances the binding ability of ZIF-8 towards biomolecules and acts as a photothermal agent with strong NIR absorption. The nanostructure was loaded with resveratrol (Res) which has a therapeutic effect for AD, 1-tetradecamol (PCM) that acts as a thermal switch to NIR-control Res release, and CeONP that functions as the ROS scavenger based on the electron transfer CeIII–CeIV. The nanostructure had a particle size of 106 nm, a surface area of 163.82 m2 g−1 and a pore size of <1 nm. In vitro measurements demonstrated that the compound doped with a specific-AβO aptamer showed high fluorescence in the presence of AβO over Aβ monomers and fibrils. In addition, the fluorescence was maintained in SH-SY5Y cells, demonstrating the cell membrane permeability of the nanocomposite. On the other hand, under NIR irradiation, the loading amount of Res depended on the irradiation temperature from 13.9% at 37 °C to 67.23% at 42 °C. The antioxidant activity of the nanocomposite reached 93.87% due to the synergetic effects of ZIF-8, PDA, and CeONP. Finally, the nanostructure was found to both inhibit Aβ monomer aggregation and disaggregate Aβ fibrils. These effects were magnified thanks to the anti-aggregation ability of Res and ZIF-8, the photothermal activity of PDA, and the chemotherapeutic effect of Res. In living cells, studies confirmed the ability of the composite to bring about Aβ aggregation inhibition, the reduction of ROS levels, and the inhibition of neuronal apoptosis under NIR irradiation.
Final considerations
Disease modifying target strategies for AD are still under extensive research. Recent results from clinical trials on anti-Aβ immunotherapy approaches have indeed suggested that merely reducing the amyloid burden in the brain of patients suffering from AD is insufficient to improve cognitive function. Currently, it appears as though that amyloid deposition may begin up to 20 years before a patient presents any clinical symptoms of AD. Thus, many researchers believe that treatment with anti-amyloid drugs in the mild to moderate stage of the disease is too late, and that for anti-amyloid approaches to be successful, treatment must begin early in the disease process.55Although great progress has been made in the last few years, a deeper understanding of the biological mechanisms of AD is necessary and will contribute to developing more effective therapeutic strategies, since just targeting protein deposits only ameliorates some of the neurological symptoms.19 Recent studies have integrated multiple new features such as novel biomarkers, new neuropsychological outcomes, treatment of earlier populations, and innovative trial designs. We believe that the future treatment could be determined by the use of different specific agents for every patient, accompanied by particular biomarkers, following a theragnostic approach. Lastly, therapeutic nanomedicine has started to build momentum also in the field, for example improving at the same time diagnosis, prevention, and treatment of various neurological disorders, including AD. Considering the versatility of coordination compounds, its smart functionalization offers the opportunity of obtaining a plethora of configurations, overcoming the main limitations associated with conventional drugs made up by organic molecules. In fact, here, many of the reviewed materials demonstrate that the derived coordination compounds are more active than metals and ligands separately.
Regarding the reported coordination compounds for AD treatment, most of them are discrete complexes, mainly showing two drawbacks: (i) any report evaluates how to eliminate the formed adduct of a complex with the Aβ peptide, and (ii) there is not a standard protocol in the activity of compounds against AD (i.e., the use of different Aβ target peptides), making difficult the comparison between the proposed drugs, as is demonstrated by the reports of Collin and Ma et al.58,59
Despite MOFs being considered promising candidates in AD theragnosis, they have been mainly described in the detection of the illness, rather than its treatment. The important parameters related to their safety use and efficiency, like the particle size and biocompatibility, should be improved. The particle size of MOFs is a limiting factor for some administration routes. Particularly in AD where the permeation of BBB is the target, MOFs should be synthesized at a size smaller than 100 nm, and stability studies under physiological conditions should be performed in order to obtain stable, homogeneous, and monodispersed MOFs. To date, only the MOF MIL-88B-(CH3)4 has been detected in the rats’ brain after intravenous administration of high doses (up to 220 mg Kg−1), suggesting a bypass of the BBB with no evidence of severe cerebral toxicity.133 In summary, the BBB is still a great obstacle for therapeutic agents against AD, and in spite of all reported compounds, in most cases, the permeability of these materials into the brain is not fully studied.
On the other hand, and prior to the potential use of MOFs in AD, the potential toxicity of these materials should be considered. Biocompatibility, and particularly, their interaction between MOFs and living structures, and its resulting effect in biological environments must be studied. Biocompatibility is mainly affected by the chemical composition, structure, morphology, size, and surface properties of MOFs. Therefore, and considering all the above, huge efforts should be made in the preparation of novel biocompatible and nanosized MOFs, or at least to modify the preparation methods to obtain nanosized and stable materials from already known biocompatible MOFs.
Some ambitious and promising examples of discrete metal complexes and MOFs in the treatment, diagnosis and theragnosis of AD have been recently reported. Considering the novelty of this research field and the versatility of coordination compounds, it is expected that they will play an important role in the future development of multifunctional therapies and/or diagnosis. Significant progress is now being achieved in the diversification of MOF bioapplications, which will aid in the continuous exploration of efficient AD treatments.
Abbreviations
| Aβ | β-Amyloid |
| AβO | Aβ oligomers |
| ABEI | N-(Aminobutyl)-N-(ethyli-soluminol) |
| ACh | Acetylcholine |
| AChE | Acetylcholinesterase |
| aCSF | Artificial cerebrospinal fluid |
| AD | Alzheimer's disease |
| AICD | APP intracellular domain |
| APP | Amyloid precursor protein |
| Apt | Aptamer |
| BACE-1 | β-Secretase 1 |
| BBB | Blood–brain barrier |
| BHQ | Black hole quencher |
| BPS | 4,7-Diphenyl-[1,10]phenanthroline disulfonate |
| bpy | 2,2-Bipyridine |
| BTB | 1,3,5-Tris(4-carboxyphenyl)benzene |
| BuChE | Butyrylcholinesterase |
| bzq | 7,8-Benzoquinoline |
| CD | Circular dichroism |
| CeONP | Ceria nanoparticle |
| ChAT | Choline-acetyltransferase |
| CSF | Cerebrospinal fluid |
| CV | Cyclic voltammetry |
| cyclen | 1,4,7,10-Tetraazacyclododecane |
| DFT | Density functional theory |
| DLS | Dynamic light scattering |
| DMF | N,N-Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| dpp | 2,3-Bis(2-pyridyl)pyrazine |
| DPV | Differential pulse voltammetry |
| ECL | Electrochemiluminescence |
| EPR | Electron paramagnetic resonance |
| ESI-MS | Electrospray ionization mass spectra |
| Fc | Ferrocene |
| FDA | US food and drug administration |
| FESEM | Field-emission scanning electron microscopy |
| Flav | Flavanones |
| glz | Gliclazide |
| GSH | Glutathione |
| GSK3 | Glycogen synthase kinase 3 |
| gtsm | Glyoxalbis(N(4)-methylthiosemicarbazonato) |
| H2bpydc | 2,2′-Bipyridine-5,5′-dicarboxylic acid |
| H2cdda | 4,4′-(9H-carbazole-2,7-diyl)dibenzoic acid |
| HPLC | High-performance liquid chromatography |
| H-UCNP | Highly doped upconversion nanoparticle |
| H-USM | Highly doped upconversion nanoparticles-SiO2@MOF |
| IC50 | Half maximal inhibitory concentration |
| Im | Imidazole |
| Ind | Indazole |
| Ksv | Metal ion quenching coefficient |
| LDH | Lactic dehydrogenase |
| LTP | Long-term potentiation |
| MALDI-TOF MS | Matrix-assisted laser desorption ionization time-of-flight mass spectrometry |
| MAPT/τ | Microtubule-associated protein τ |
| MB | Methylene blue |
| ϕ-Mepy | 2-Phenyl-5-methyl-pyridine |
| MOF | Metal–organic framework |
| MRI | Magnetic resonance imaging |
| MS | Mass spectrometry |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NIA-AA | National institute on ageing and Alzheimer's association |
| NIR | Near-infrared |
| NMOF | Nanoscale MOF |
| NMR | Nuclear magnetic resonance |
| NOTA | 1,4,7-Triazacyclononane-1,4,7-triacetic acid |
| np | Non-peripheral |
| NP | Nanoparticle |
| NSC | Neural stem cells |
| p | Peripheral |
| PBS | Phosphate buffer saline |
| Pc | Phthalocyanine |
| PCM | 1-Tetradecamol |
| PDA | Polydopamine |
| Phen | Phenanthroline |
| ϕ-Phen | 4,7-Diphenyl-1,10-phenanthroline |
| phq | 2-Phenylquinoline |
| PI3K | Phosphoinositol-3-kinase |
| PMOF | Porphyrinic based MOF |
| ppy | 2-Phenylpyridine |
| PPy | Polypyrrole |
| RA | Retinoic acid |
| RET | Resonance energy transfer |
| Res | Resveratrol |
| ROS | Reactive oxygen species |
| SWV | Square wave voltammetry |
| TEM | Transmission electronic microscopy |
| ThT | Thioflavin T |
| thz | 1,3-Thiazole |
| vAChT | Vesicular ACh transporter |
| WHO | World health organization |
| XAS | X-ray absorption fine structure spectroscopy |
| 2-AmTh | 2-aminothiazolium |
| 8-BQ | 8-(1H-Benzoimidazol-2-yl)quinoline |
Author contributions
All authors contributed equally to this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the MOFSEIDON project (PID2019-104228RB-100, MCI/AEI/FEDER) and the AgroMOFs project (TED2021-132440B-I00) funded by MCIN/AEI/10.13039/501100011033 and by NextGenerationEU/PRTR and Margarita Salas contract 401 funded by Ministerio de Universidades and Next Generation. B- FQM-394, ProyExcel_00105 and ProyExcel_00386 were funded from la Consejería de Universidad, Investigación e Innovación de la Junta de Andalucía. S.R. is grateful for the grant (RYC2021-032522-I) funded by MCIN/AEI/10.13039/501100011033 and for El FSE invierte en tu futuro. In memory of our grandfathers and grandmothers, with love.Notes and references
- World Health Organization, Dementia, https://www.who.int/news-room/fact-sheets/detail/dementia, (accessed 18 January 2023).
- GBD 2019 Dementia Forecasting Collaborators, The Lancet Public Health, 2022, 7, e105–e125.
- Alzheimer's Association, Alzheimer's Disease Facts and Figures, https://www.alz.org/alzheimers-dementia/facts-figures, (accessed 1 January 2023).
- P. Mirzayi, P. Shobeiri, A. Kalantari, G. Perry and N. Rezaei, Mol. Brain, 2022, 15, 1–14 CrossRef.
- R. A. Stelzmann, H. N. Schnitzlein and F. R. Murtagh, Clin. Anat., 1995, 8, 429–431 CrossRef.
- A. Association, Alzheimer's Dementia, 2016, 12, 459–509 CrossRef.
- J. L. Price and J. C. Morris, Ann. Neurol., 1999, 45, 358–368 CrossRef CAS.
- R. J. Bateman, C. Xiong, T. L. S. Benzinger, A. M. Fagan, A. Goate, N. C. Fox, D. S. Marcus, N. J. Cairns, X. Xie, T. M. Blazey, D. M. Holtzman, A. Santacruz, V. Buckles, A. Oliver, K. Moulder, P. S. Aisen, B. Ghetti, W. E. Klunk, E. McDade, R. N. Martins, C. L. Masters, R. Mayeux, J. M. Ringman, M. N. Rossor, P. R. Schofield, R. A. Sperling, S. Salloway and J. C. Morris, N. Engl. J. Med., 2012, 367, 795–804 CrossRef CAS PubMed.
- C. Ferrari and S. Sorbi, Physiol. Rev., 2021, 101, 1047–1081 CrossRef CAS PubMed.
- M. Jucker and L. C. Walker, Ann. Neurol., 2011, 70, 532–540 CrossRef CAS PubMed.
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184–185 CrossRef CAS PubMed.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS PubMed.
- J. M. Zolezzi, S. Bastías-Candia, M. J. Santos and N. C. Inestrosa, Front. Aging Neurosci., 2014, 6, 1–12 Search PubMed.
- H. W. Querfurth and F. M. LaFerla, N. Engl. J. Med., 2010, 362, 329–344 CrossRef CAS.
- M. M. Wen, N. S. El-Salamouni, W. M. El-Refaie, H. A. Hazzah, M. M. Ali, G. Tosi, R. M. Farid, M. J. Blanco-Prieto, N. Billa and A. S. Hanafy, J. Controlled Release, 2017, 245, 95–107 CrossRef CAS PubMed.
- Y. Zhou, Y. Sun, Q.-H. Ma and Y. Liu, Cell Stress, 2018, 2, 161 CrossRef PubMed.
- R. Ricciarelli and E. Fedele, Curr. Neuropharmacol., 2017, 15, 935 Search PubMed.
- C. L. Masters, R. Cappai, K. J. Barnham and V. L. Villemagne, J. Neurochem., 2006, 97, 1700–1725 CrossRef CAS PubMed.
- H. Kozlowski, A. Janicka-Klos, J. Brasun, E. Gaggelli, D. Valensin and G. Valensin, Coord. Chem. Rev., 2009, 253, 2665–2685 CrossRef CAS.
- P. Faller and C. Hureau, Dalton Trans., 2009, 1080–1094 RSC.
- S. Rivera-Mancía, I. Pérez-Neri, C. Ríos, L. Tristán-López, L. Rivera-Espinosa and S. Montes, Chem.-Biol. Interact., 2010, 186, 184–199 CrossRef PubMed.
- K. J. Barnham and A. I. Bush, Curr. Opin. Chem. Biol., 2008, 12, 222–228 CrossRef CAS PubMed.
- J. Grutzendler and J. C. Morris, Drugs, 2001, 61, 41–52 CrossRef CAS PubMed.
- H. Soreq and S. Seidman, Nat. Rev. Neurosci., 2001, 2, 294–302 CrossRef CAS.
- J. Cummings, G. Lee, A. Ritter, M. Sabbagh and K. Zhong, Alzheimer's Dementia, 2020, 6, 1–29 Search PubMed.
- X. Fang, J. Zhang, R. J. Roman and F. Fan, GeroScience, 2022, 44, 1879–1883 CrossRef.
- L. S. Schneider, Dialogues Clin. Neurosci., 2000, 2, 111–128 CrossRef.
- FDA Grants Accelerated Approval for Alzheimer's Drug, https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug, (accessed 1 January 2023).
- FDA Grants Accelerated Approval for Alzheimer's Disease Treatment, https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-disease-treatment, (accessed 15 May 2023).
- J. Sevigny, P. Chiao, T. Bussière, P. H. Weinreb, L. Williams, M. Maier, R. Dunstan, S. Salloway, T. Chen, Y. Ling, J. O’Gorman, F. Qian, M. Arastu, M. Li, S. Chollate, M. S. Brennan, O. Quintero-Monzon, R. H. Scannevin, H. M. Arnold, T. Engber, K. Rhodes, J. Ferrero, Y. Hang, A. Mikulskis, J. Grimm, C. Hock, R. M. Nitsch and A. Sandrock, Nature, 2016, 537, 50–56 CrossRef CAS PubMed.
- C. H. van Dyck, C. J. Swanson, P. Aisen, R. J. Bateman, C. Chen, M. Gee, M. Kanekiyo, D. Li, L. Reyderman, S. Cohen, L. Froelich, S. Katayama, M. Sabbagh, B. Vellas, D. Watson, S. Dhadda, M. Irizarry, L. D. Kramer and T. Iwatsubo, N. Engl. J. Med., 2023, 388, 9–21 CrossRef CAS PubMed.
- J. Cummings, P. Aisen, L. G. Apostolova, A. Atri, S. Salloway and M. Weiner, J. Prev. Alzheimer's Dis., 2021, 8, 398–410 CAS.
- D. A. Wolk, G. D. Rabinovici and B. C. Dickerson, JAMA Neurology, 2023, 80, 429–430 CrossRef PubMed.
- J. Cummings, P. S. Aisen, B. DuBois, L. Frölich, C. R. Jack, R. W. Jones, J. C. Morris, J. Raskin, S. A. Dowsett and P. Scheltens, Alzheimer's Res. Ther., 2016, 8, 1–12 CrossRef.
- K. Blennow, M. J. de Leon and H. Zetterberg, The Lancet, 2006, 368, 387–403 CrossRef CAS.
- M. P. Mattson, Nature, 2004, 430, 631–639 CrossRef CAS PubMed.
- D.-L. Ma, H.-Z. He, K.-H. Leung, D. S.-H. Chan and C.-H. Leung, Angew. Chem., Int. Ed., 2013, 52, 7666–7682 CrossRef CAS PubMed.
- J. Karges, R. W. Stokes and S. M. Cohen, Trends Chem., 2021, 3, 534 Search PubMed.
- K. L. Haas and K. J. Franz, Chem. Rev., 2009, 109, 4921–4960 CrossRef CAS PubMed.
- S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS PubMed.
- F. Frézard, C. Demicheli and R. R. Ribeiro, Molecules, 2009, 14, 2317–2336 CrossRef PubMed.
- E. V. Capparelli, R. Bricker-Ford, M. J. Rogers, J. H. McKerrow and S. L. Reed, Antimicrob. Agents Chemother., 2017, 61, 1–8 CrossRef PubMed.
- D. M. Teleanu, C. Chircov, A. M. Grumezescu, A. Volceanov and R. I. Teleanu, Pharmaceutics, 2018, 10, 269 CrossRef CAS PubMed.
- W. M. Pardridge, Neurotherapeutics, 2005, 2, 3–14 CrossRef.
- M. B. Alahri, R. Arshadizadeh, M. Raeisi, M. Khatami, M. S. Sajadi, W. K. Abdelbasset, R. Akhmadeev and S. Iravani, Inorg. Chem. Commun., 2021, 134, 108997 CrossRef.
- S. Rojas, A. Arenas-Vivo and P. Horcajada, Coord. Chem. Rev., 2019, 388, 22–226 CrossRef.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- D. Valensin, C. Gabbiani and L. Messori, Coord. Chem. Rev., 2012, 256, 2357–2366 CrossRef CAS.
- D. J. Hayne, S. Lim and P. S. Donnelly, Chem. Soc. Rev., 2014, 43, 6701–6715 RSC.
- J. L. Hickey and P. S. Donnelly, Coord. Chem. Rev., 2012, 256, 2367–2380 CrossRef CAS.
- Z. Liao, J. Zhang, E. Yu and Y. Cui, Polyhedron, 2018, 151, 554–567 CrossRef CAS.
- F. Zeng, K. Peng, L. Han and J. Yang, ACS Biomater. Sci. Eng., 2021, 7, 3573–3585 CrossRef CAS.
- J. P. Leite, F. Figueira, R. F. Mendes, F. A. A. Paz and L. Gales, ACS Sens., 2023, 8, 1033–1053 CrossRef CAS PubMed.
- K. J. Barnham, V. B. Kenche, G. D. Ciccotosto, D. P. Smith, D. J. Tew, X. Liu, K. Perez, G. A. Cranston, T. J. Johanssen, I. Volitakis, A. I. Bush, C. L. Masters, A. R. White, J. P. Smith, R. A. Cherny and R. Cappai, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6813–6818 CrossRef CAS PubMed.
- V. B. Kenche, L. W. Hung, K. Perez, I. Volitakes, G. Ciccotosto, J. Kwok, N. Critch, N. Sherratt, M. Cortes, V. Lal, C. L. Masters, K. Murakami, R. Cappai, P. A. Adlard and K. J. Barnham, Angew. Chem., Int. Ed., 2013, 52, 3374–3378 CrossRef CAS PubMed.
- V. A. Streltsov, V. C. Epa, S. A. James, Q. I. Churches, J. M. Caine, V. B. Kenche and K. J. Barnham, Chem. Commun., 2013, 49, 11364–11366 RSC.
- I. Sasaki, C. Bijani, S. Ladeira, V. Bourdon, P. Faller and C. Hureau, Dalton Trans., 2012, 41, 6404–6407 RSC.
- F. Collin, I. Sasaki, H. Eury, P. Faller and C. Hureau, Chem. Commun., 2013, 49, 2130–2132 RSC.
- G. Ma, E. Wang, H. Wei, K. Wei, P. Zhu and Y. Liu, Metallomics, 2013, 5, 879–887 CrossRef CAS PubMed.
- D. Valensin, P. Anzini, E. Gaggelli, N. Gaggelli, G. Tamasi, R. Cini, C. Gabbiani, E. Michelucci, L. Messori, H. Kozlowski and G. Valensin, Inorg. Chem., 2010, 49, 4720–4722 CrossRef CAS PubMed.
- L. Messori, M. Camarri, T. Ferraro, C. Gabbiani and D. Franceschini, ACS Med. Chem. Lett., 2013, 4, 329–332 CrossRef CAS PubMed.
- A. Kumar, L. Moody, J. F. Olaivar, N. A. Lewis, R. L. Khade, A. A. Holder, Y. Zhang and V. Rangachari, ACS Chem. Neurosci., 2010, 1, 691–701 CrossRef CAS PubMed.
- A. L. F. Sarria, A. F. L. Vilela, B. M. Frugeri, J. B. Fernandes, R. M. Carlos, M. F. das, G. F. da Silva, Q. B. Cass and C. L. Cardoso, J. Inorg. Biochem., 2016, 164, 141–149 CrossRef CAS PubMed.
- A. Günsel, P. Taslimi, G. Y. Atmaca, A. T. Bilgiçli, H. Pişkin, Y. Ceylan, A. Erdoğmuş, M. N. Yarasir and İ. Gülçin, J. Mol. Struct., 2021, 1237, 1–13 CrossRef.
- H. Yalazan, B. Tüzün, D. Akkaya, B. Barut, H. Kantekin and S. Yıldırmış, Appl. Organomet. Chem., 2022, 36, 1–19 CrossRef.
- A. Şenocak, N. A. Taş, P. Taslimi, B. Tüzün, A. Aydin and A. Karadağ, J. Biochem. Mol. Toxicol., 2022, 36, 1–9 CrossRef PubMed.
- A. García-García, S. Rojas, L. Rivas-García, M. D. Navarro-Hortal, J. M. Romero-Márquez, J. G. Fernández-Bolaños, D. Choquesillo-Lazarte, A. Salinas-Castillo, Ó. López, J. L. Quiles and A. Rodríguez-Diéguez, Chem. Commun., 2022, 58, 1514–1517 RSC.
- P. J. Crouch, W. H. Lin, P. A. Adlard, M. Cortes, V. Lal, G. Filiz, K. A. Perez, M. Nurjono, A. Caragounis, T. Du, K. Laughton, I. Volitakis, A. I. Bush, Q.-X. Li, C. L. Masters, R. Cappai, R. A. Cherny, P. S. Donnelly, A. R. White and K. J. Barnham, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 381–386 CrossRef CAS PubMed.
- X. Wang, X. Wang, C. Zhang, Y. Jiao and Z. Guo, Chem. Sci., 2012, 3, 1304–1312 RSC.
- D. P. Smith, D. G. Smith, C. C. Curtain, J. F. Boas, J. R. Pilbrow, G. D. Ciccotosto, T. L. Lau, D. J. Tew, K. Perez, J. D. Wade, A. I. Bush, S. C. Drew, F. Separovic, C. L. Masters, R. Cappai and K. J. Barnham, J. Biol. Chem., 2006, 281, 15145–15154 CrossRef CAS PubMed.
- A. I. Bush, Trends Neurosci., 2003, 26, 207–214 CrossRef CAS PubMed.
- M. Drent, N. A. M. Cobben, R. F. Henderson, E. F. M. Wouters and M. Van Dieijen-Visser, Eur. Respir. J., 1996, 9, 1736–1742 CrossRef CAS PubMed.
- J. B. Pereira, E. F. A. Carvalho, M. A. F. Faustino, R. Fernandes, M. G. P. M. S. Neves, J. A. S. Cavaleiro, N. C. M. Gomes, Â. Cunha, A. Almeida and J. P. C. Tomé, Photochem. Photobiol., 2012, 88, 537–547 CrossRef CAS PubMed.
- Z. Jiang, J. Shao, T. Yang, J. Wang and L. Jia, J. Pharm. Biomed. Anal., 2014, 87, 98–104 CrossRef CAS PubMed.
- A. Günsel, A. Kobyaoğlu, A. T. Bilgiçli, B. Tüzün, B. Tosun, G. Arabaci and M. N. Yarasir, J. Mol. Struct., 2020, 1200, 1–10 CrossRef.
- A. Günsel, G. Y. Atmaca, P. Taslimi, A. T. Bilgiçli, İ. Gülçin, A. Erdoğmuş and M. N. Yarasir, J. Photochem. Photobiol., A, 2020, 396, 1–9 CrossRef.
- S. Okabayashi, N. Shimozawa, Y. Yasutomi, K. Yanagisawa and N. Kimura, PLoS ONE, 2015, 10, 1–19 CrossRef PubMed.
- K. Mittal, R. J. Mani and D. P. Katare, Sci. Rep., 2016, 6, 1–8 CrossRef PubMed.
- B. M. Paterson and P. S. Donnelly, Chem. Soc. Rev., 2011, 40, 3005–3018 RSC.
- M. A. Green, D. L. Klippenstein and J. R. Tennison, J. Nucl. Med., 1988, 29, 1549–1557 CAS.
- Y. Fujibayashi, H. Taniuchi, Y. Yonekura, H. Ohtani, J. Konishi and A. Yokoyama, J. Nucl. Med., 1997, 38, 1155–1160 CAS.
- K. A. Price, A. Caragounis, B. M. Paterson, G. Filiz, I. Volitakis, C. L. Masters, K. J. Barnham, P. S. Donnelly, P. J. Crouch and A. R. White, J. Med. Chem., 2009, 52, 6606–6620 CrossRef CAS PubMed.
- P. S. Donnelly, A. Caragounis, T. Du, K. M. Laughton, I. Volitakis, R. A. Cherny, R. A. Sharples, A. F. Hill, Q.-X. Li, C. L. Masters, K. J. Barnham and A. R. White, J. Biol. Chem., 2008, 283, 4568–4577 CrossRef CAS PubMed.
- X. Wang, X. Wang and Z. Guo, Acc. Chem. Res., 2015, 48, 2622–2631 CrossRef CAS PubMed.
- S. Samanta, K. Rajasekhar, V. Babagond and T. Govindaraju, ACS Chem. Neurosci., 2019, 10, 3611–3621 CrossRef CAS PubMed.
- K. Rajasekhar, K. Mehta and T. Govindaraju, ACS Chem. Neurosci., 2018, 9, 1432–1440 CrossRef CAS PubMed.
- K. Rajasekhar, S. Samanta, V. Bagoband, N. A. Murugan and T. Govindaraju, iScience, 2020, 23, 101005 CrossRef CAS PubMed.
- M. Okafor, P. Gonzalez, P. Ronot, I. El Masoudi, A. Boos, S. Ory, S. Chasserot-Golaz, S. Gasman, L. Raibaut, C. Hureau, N. Vitale and P. Faller, Chem. Sci., 2022, 13, 11829–11840 RSC.
- K. Rajasekhar, C. Madhu and T. Govindaraju, ACS Chem. Neurosci., 2016, 7, 1300–1310 CrossRef CAS PubMed.
- M. Di Vaira, C. Bazzicalupi, P. Orioli, L. Messori, B. Bruni and P. Zatta, Inorg. Chem., 2004, 43, 3795–3797 CrossRef CAS PubMed.
- H. Chen, J. Mi, S. Li, Z. Liu, J. Yang, R. Chen, Y. Wang, Y. Ban, Y. Zhou, W. Dong and Z. Sang, J. Enzyme Inhib. Med. Chem., 2023, 38, 2169682 CrossRef PubMed.
- S. Rojas, T. Baati, L. Njim, L. Manchego, F. Neffati, N. Abdejelil, S. Saguem, C. Serre, M. F. M. F. Najjar, A. Zakhama, P. Horcajada, N. Abdeljelil, S. Saguem, C. Serre, M. F. M. F. Najjar, A. Zakhama and P. Horcajada, J. Am. Chem. Soc., 2018, 140, 9581–9586 CrossRef CAS PubMed.
- S. Rojas, N. Guillou and P. Horcajada, ACS Appl. Mater. Interfaces, 2019, 11, 22188–22193 CrossRef CAS PubMed.
- Z. L. Mensinger, B. L. Cook and E. L. Wilson, ACS Omega, 2020, 5, 32969–32974 CrossRef CAS PubMed.
- J. Wang, Y. Fan, Y. Tan, X. Zhao, Y. Zhang, C. Cheng and M. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 36615–36621 CrossRef CAS PubMed.
- D. Yu, Y. Guan, F. Bai, Z. Du, N. Gao, J. Ren and X. Qu, Chem. – Eur. J., 2019, 25, 3489–3495 CrossRef CAS PubMed.
- D. Yu, M. Ma, Z. Liu, Z. Pi, X. Du, J. Ren and X. Qu, Biomaterials, 2020, 255, 1–10 CrossRef PubMed.
- L. Vermunt, S. A. M. Sikkes, A. van den Hout, R. Handels, I. Bos, W. M. van der Flier, S. Kern, P. J. Ousset, P. Maruff, I. Skoog, F. R. J. Verhey, Y. Freund-Levi, M. Tsolaki, Å. K. Wallin, M. Olde Rikkert, H. Soininen, L. Spiru, H. Zetterberg, K. Blennow, P. Scheltens, G. Muniz-Terrera and P. J. Visser, Alzheimer's Dementia, 2019, 15, 888–898 CrossRef PubMed.
- J. Luo, F. Agboola, E. Grant, C. L. Masters, M. S. Albert, S. C. Johnson, E. M. McDade, J. Vöglein, A. M. Fagan, T. Benzinger, P. Massoumzadeh, J. Hassenstab, R. J. Bateman, J. C. Morris, R. J. Perrin, J. Chhatwal, M. Jucker, B. Ghetti, C. Cruchaga, N. R. Graff-Radford, P. R. Schofield, H. Mori and C. Xiong, Neurology, 2020, 95, e3104–e3116 CrossRef CAS PubMed.
- G. McKhann, D. Drachman, M. Folstein, R. Katzman, D. Price and E. M. Stadlan, Neurology, 1984, 34, 939–944 CrossRef CAS PubMed.
- C. Humpel, Trends Biotechnol., 2011, 29, 26–32 CrossRef CAS PubMed.
- J. Zhu, T. Xia, Y. Cui, Y. Yang and G. Qian, J. Solid State Chem., 2019, 270, 317–323 CrossRef CAS.
- J. Bai, L. Liu, C. Jia, Z. Liu, S. Gao, Y. Han and H. Yan, ACS Appl. Bio Mater., 2019, 2, 6021–6028 CrossRef CAS PubMed.
- L. C. Walker, D. G. Lynn and Y. O. Chernoff, Prion, 2018, 12, 261–265 CrossRef PubMed.
- J. Han, M. Zhang, G. Chen, Y. Zhang, Q. Wei, Y. Zhuo, G. Xie, R. Yuan and S. Chen, J. Mater. Chem. B, 2017, 5, 8330–8336 RSC.
- C. Wang, N. Zhang, Y. Li, L. Yang, D. Wei, T. Yan, H. Ju, B. Du and Q. Wei, Sens. Actuators, B, 2019, 291, 319–328 CrossRef CAS.
- A. H. Valekar, B. S. Batule, M. Il Kim, K.-H. Cho, D.-Y. Hong, U.-H. Lee, J.-S. Chang, H. G. Park and Y. K. Hwang, Biosens. Bioelectron., 2018, 100, 161–168 CrossRef CAS PubMed.
- Y. Zhou, C. Li, X. Li, X. Zhu, B. Ye and M. Xu, Anal. Methods, 2018, 10, 4430–4437 RSC.
- B. Liu, H. Shen, Y. Hao, X. Zhu, S. Li, Y. Huang, P. Qu and M. Xu, Anal. Chem., 2018, 90, 12449–12455 CrossRef CAS PubMed.
- J. Qin, M. Cho and Y. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 11743–11748 CrossRef CAS PubMed.
- Y. Wang, Y. Zhang, H. Sha, X. Xiong and N. Jia, ACS Appl. Mater. Interfaces, 2019, 11, 36299–36306 CrossRef CAS PubMed.
- J. Miao, X. Li, Y. Li, X. Dong, G. Zhao, J. Fang, Q. Wei and W. Cao, Anal. Chim. Acta, 2019, 1089, 48–55 CrossRef CAS PubMed.
- G. Zhao, Y. Wang, X. Li, Q. Yue, X. Dong, B. Du, W. Cao and Q. Wei, Anal. Chem., 2019, 91, 1989–1996 CrossRef CAS PubMed.
- J. Fang, G. Zhao, X. Dong, X. Li, J. Miao, Q. Wei and W. Cao, Biosens. Bioelectron., 2019, 142, 1–6 CrossRef PubMed.
- H.-X. Ren, Y.-B. Miao and Y. Zhang, Microchim. Acta, 2020, 187, 1–8 CrossRef PubMed.
- X. Dong, G. Zhao, X. Li, J. Fang, J. Miao, Q. Wei and W. Cao, Talanta, 2020, 208, 1–7 CrossRef PubMed.
- H.-X. Ren, Q. Zhong, Y.-B. Miao, X.-W. Wen, G.-Y. Wu, H.-L. Wang and Y. Zhang, Microchim. Acta, 2020, 187, 1–8 CrossRef PubMed.
- X. Z. Wang, J. Du, N. N. Xiao, Y. Zhang, L. Fei, J. D. LaCoste, Z. Huang, Q. Wang, X. R. Wang and B. Ding, Analyst, 2020, 145, 4646–4663 RSC.
- X. Wang, L. Li, X. Gu, B. Yu and M. Jiang, Microchim. Acta, 2021, 188, 1–11 CrossRef PubMed.
- W.-K. Fang, L. Liu, L. Zhang, D. Liu, Y. Liu and H.-W. Tang, Anal. Chem., 2021, 93, 12447–12455 CrossRef CAS PubMed.
- L. Yin, Y. Wang, R. Tan, H. Li and Y. Tu, Microchim. Acta, 2021, 188, 1–8 CrossRef PubMed.
- A. S. Spitsyna, A. S. Poryvaev, N. E. Sannikova, A. A. Yazikova, I. A. Kirilyuk, S. A. Dobrynin, O. A. Chinak, M. V. Fedin and O. A. Krumkacheva, Molecules, 2022, 27, 1–13 CrossRef PubMed.
- M. de, J. Velásquez-Hernández, R. Ricco, F. Carraro, F. T. Limpoco, M. Linares-Moreau, E. Leitner, H. Wiltsche, J. Rattenberger, H. Schröttner, P. Frühwirt, E. M. Stadler, G. Gescheidt, H. Amenitsch, C. J. Doonan and P. Falcaro, CrystEngComm, 2019, 21, 4538–4544 RSC.
- A. S. Poryvaev, A. A. Yazikova, D. M. Polyukhov, O. A. Chinak, V. A. Richter, O. A. Krumkacheva and M. V. Fedin, J. Phys. Chem. C, 2021, 125, 15606–15613 CrossRef CAS.
- J. H. Ryu, H. Koo, I. C. Sun, S. H. Yuk, K. Choi, K. Kim and I. C. Kwon, Adv. Drug Delivery Rev., 2012, 64, 1447–1458 CrossRef CAS PubMed.
- K. Kim, J. H. Kim, H. Park, Y. S. Kim, K. Park, H. Nam, S. Lee, J. H. Park, R. W. Park, I. S. Kim, K. Choi, S. Y. Kim, K. Park and I. C. Kwon, J. Controlled Release, 2010, 146, 219–227 CrossRef CAS PubMed.
- D. E. Lee, H. Koo, I. C. Sun, J. H. Ryu, K. Kim and I. C. Kwon, Chem. Soc. Rev., 2012, 41, 2656–2672 RSC.
- B. Y.-W. Man, H.-M. Chan, C.-H. Leung, D. S.-H. Chan, L.-P. Bai, Z.-H. Jiang, H.-W. Li and D.-L. Ma, Chem. Sci., 2011, 2, 917–921 RSC.
- J. Zhao, F. Yin, L. Ji, C. Wang, C. Shi, X. Liu, H. Yang, X. Wang and L. Kong, ACS Appl. Mater. Interfaces, 2020, 12, 44447–44458 CrossRef CAS PubMed.
- R.-Q. Song, X. Ma and G.-Y. Song, J. Chin. Chem. Soc., 2020, 67, 1462–1469 CrossRef CAS.
- X. Yan, Y. Pan, L. Ji, J. Gu, Y. Hu, Y. Xia, C. Li, X. Zhou, D. Yang and Y. Yu, Anal. Chem., 2021, 93, 13823–13834 CrossRef CAS PubMed.
- X. Yan, Y. Pan, L. Ji, J. Gu, Y. Hu, Y. Xia, C. Li, X. Zhou, D. Yang and Y. Yu, Anal. Chem., 2021, 93, 13823–13834 CrossRef CAS PubMed.
- T. Baati, L. Njim, F. Neffati, A. Kerkeni, M. Bouttemi, R. Gref, M. F. Najjar, A. Zakhama, P. Couvreur, C. Serre and P. Horcajada, Chem. Sci., 2013, 4, 1597–1607 RSC.
| This journal is © The Royal Society of Chemistry 2023 |