
Isoreticular design of flexible Zn-based tetracarboxylate MOFs†
Bodo
Felsner
 *a,
Volodymyr
Bon
a,
Christopher
Bachetzky
b,
Eike
Brunner
b and
Stefan
Kaskel
a
*a,
Volodymyr
Bon
a,
Christopher
Bachetzky
b,
Eike
Brunner
b and
Stefan
Kaskel
a
aDepartment for Inorganic Chemistry I, Technical University Dresden, Bergstraße 66, 01069 Dresden, Germany. E-mail: bodo.felsner@tu-dresden.de
bDepartment for Bioanalytical Chemistry, Technical University Dresden, Bergstraße 66, 01069 Dresden, Germany
First published on 25th April 2023
Abstract
Predictive design of guest-responsive frameworks on the molecular level is still a major challenge for experimental and theoretical chemists. The isoreticular approach is one of the most promising approaches for designing MOFs with desired textural properties and framework topology. In this work we successfully applied the isoreticular approach for the design of the new flexible frameworks DUT-180 and DUT-190, constructed by square planar or octahedral metal nodes and a tetracarboxylate ligand. The flexibility mechanism in DUT-190 was explored using in situ PXRD and NMR in parallel to physisorption of subcritical gases such as nitrogen (77 K), carbon dioxide (195 K) and xenon (206 K). Methane physisorption experiments, conducted in a broad temperature range indicate a softening of the DUT-190 framework compared to its analogue DUT-13. These experimental and computational findings can be used as a roadmap for discovery of new flexible frameworks with desired micromechanical properties.
Introduction
Flexible MOFs are bistable porous solids,1–3 discussed for a wide range of adsorption-related applications, such as natural gas storage,4,5 gas separation,6–8 sensors9–11 and actuators.12,13 In general, flexibility in MOFs either originates from inorganic building units, conformational polymorphism of organic ligands, framework topology or a combination of these factors.14 For example, paddle-wheel dimers, typically formed in Cu(II) and Zn(II) MOFs usually contribute to flexibility in pillared layer MOFs.15 Introduction of conformational labile moieties such as amide groups, long alkyl chains or tertiary amines in the ligand may induce flexibility of the resulting MOF even if the framework topology is considered as rigid.16–18 Finally, certain framework topologies are known to induce flexibility in MOFs.19 The combination of these factors may provide an empirical tool for the design of flexible frameworks for desired applications. The coordination lability of a d10 system renders Zn-based MOFs as ideal model systems, able to form various coordination geometries ranging from tetrahedral to octahedral. In MOF chemistry, Zn-MOFs are usually based on Zn2 paddle-wheel dimers with typical representatives being Zn2(bdc)2dabco,20 fu-MOFs,16 Zn(2,6-ndc)2dabco21 and others,22 typically showing metal cluster induced flexibility. In contrast, the octahedral secondary building unit (SBU) Zn4O(CO2)6 is known in the literature as a rigid cluster with octahedral ligand environment, producing MOFs with ultra-high porosity.23,24 The absence of an inversion center and the presence of a mirror symmetry plane in the Zn4O(CO2)6 node prohibits the distortion of the cluster.25,26 Combination of paddle-wheel units with linear or quasilinear ligands result in 2D square grids, which can be further interconnected in the third direction while direct interconnection of octahedral Zn4O(CO2)6 nodes result in the IRMOF series in which flexibility could only be induced by entropic contribution of the substituents.27The use of tetratopic ligands in the design of MOF structures is advantageous in terms of topological diversity and flexibility. Thus, the combination of paddle-wheels with tetracarboxylates leads to a variety of framework topologies, resulting either in series of rigid frameworks with ultrahigh porosity, such as the NOTT and MFM series (nbo topology),27 or flexible frameworks like DUT-10 (lvt), DUT-11(pts), DUT-12 (ssb)28 or the DUT-49 series (fcu-a).29–31 The combination of 6- and 4-connected nodes may result in 54 different topological types, most commonly cor, she and ith-a nets. In our recent studies the tetratopic carboxylate ligands H4benztb (N,N,N′,N′-benzidine tetrabenzoic acid) and H4BBCDC (9,9′-([1,1′-biphenyl]-4,4′-diyl)bis(9H-carbazole-3,6-dicarboxylic acid)) were extensively used for the design of highly porous MOFs, based on Zn- or Cu-paddle-wheels and Zn4O SBUs.18,28,32 Apart from the similar composition, minor differences in the molecular structure of H4benztb and H4BBCDC result in different angles between the carboxylates and the degree of conformational lability being increased, which is advantageous for the design of flexible MOFs with ligand-based flexibility. In case of H4BBCDC the planar geometry of carbazole moieties reduces the rotational degree of freedom of the molecule, which can switch between staggered and eclipsed conformation, which in combination with buckling of biphenyl bridges may lead to the isotropic contraction of the cubic DUT-49 framework.32 In case of H4benztb, an additional degree of freedom originates from distortion of the tert-amine geometry and rotation of phenyl rings.33 Therefore, in most of the cases the resulting highly porous MOFs show a flexible behavior with exception of Zn4O(benztb)(BTB)2/3, known as DUT-25, in which the framework was stabilized by implementation of an additional tritopic ligand.32 The combination of square-planar paddle-wheel nodes with H4benztb led to variety of structures such as DUT-10 (lvt), DUT-11 (pts) and DUT-12 (ssb).28 From these three nets, DUT-10 shows guest-induced flexibility upon removal of the solvent molecules from the pores resulting in a crystalline phase with yet not resolved structure. These results are in line with the study by Sarkisov et al., who proposed a mechanical model, based on rigid elements, connected by hinges, which rank lvt topology among potentially flexible topologies even if rigid building blocks are used. Directing the synthesis towards the formation of Zn4O clusters results in the formation of the Zn4O(benztb)3/2 framework, also known as DUT-13.18 Depending on how the framework is simplified, cor-a or ttu-a topology can be assigned.34 The framework can be desolvated using supercritical CO2 drying and shows remarkable capacity for hydrogen at 77 K. Although both framework topologies and metal nodes are assumed as rigid, the conformational lability of the ligand induces adsorption-induced breathing in DUT-13, extensively studied using in situ PXRDs, applied in parallel to physisorption of nitrogen (77 K) and n-butane (273 K).33
The isoreticular approach is well-established in MOF chemistry and is based on a variation of ligand length, stiffness, or functionality. This approach was successfully applied for the design of ultrahigh porosity in many open frameworks.23 In terms of flexibility, the isoreticular approach was successfully applied to control the pore size and ligand stiffness in the DUT-49 series, which enormously helped to understand and control Negative Gas Adsorption (NGA).30,31
Following this approach, we synthesized the new ligand H4tpdatb (p-terphenyl-4,4′′-diamin tetrabenzoate), an elongated version of H4benztb, applied to the design of new MOFs. An extra phenyl ring, built in the spacer of the MOF, contributes to the length of the ligand, but should not strongly influence the conformation lability. Therefore, isoreticular frameworks to DUT-10 and DUT-13 are expected when combined with paddle-wheels or Zn4O clusters, respectively.
Herein we report on the synthesis and characterization of the flexible frameworks Zn2(tpdatb)(H2O)2 (DUT-180) and Zn4O(tpdatb)3/2 (DUT-190), both showing guest-induced flexibility. While DUT-180 contracts upon the supercritical desolvation to form a thermodynamically more stable cp (contracted pore) phase, DUT-190 shows adsorption induced breathing transitions upon physisorption of nitrogen (77 K), methane (111 K) and carbon dioxide (195 K). The mechanisms of structural transformations were studied by in situ PXRD and in situ129Xe NMR and the stiffness of the framework was evaluated in methane physisorption experiments and compared with DUT-13.
Results and discussion
Synthesis, crystal structure and porosity
DUT-180 crystallizes as long yellow needles, suitable for single crystal X-ray diffraction studies. The crystal structure of the DMF-filled DUT-180 crystallized in orthorhombic space group Imma. The asymmetric unit contains one zinc atom, one fourth of the ligand molecule and one oxygen from the coordinated solvent, forming an open 3D framework. The framework structure is based on zinc paddle-wheel dimers, interconnected by the tetratopic linker tpdatb4− in the eclipsed conformation. The axial positions of the zinc paddle-wheel are coordinated by oxygen, originating from solvents like DMF or water. Both, inorganic building unit and linker, topologically can be simplified as square nodes therefore forming a (4,4)-network in lvt-a topology. In a more detailed view also the lil-a topology can be derived from the structure splitting the linker square into two separate triangles with the nitrogen atoms as branch points shown in Fig. 1a.34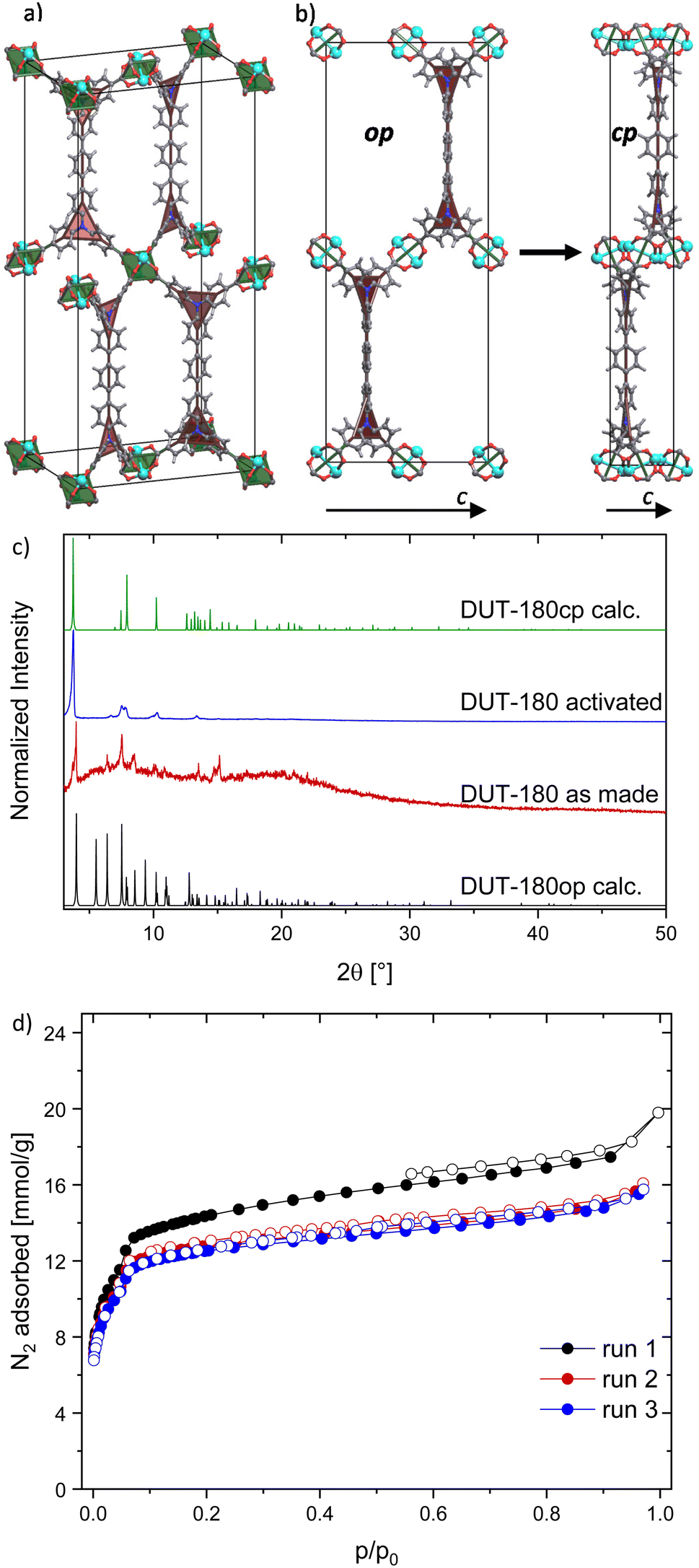 | ||
| Fig. 1 Crystal structure and characterization of DUT-180: (a) unit cell of DUT-180op derived from SC XRD and (b) unit cell contraction of DUT-180op to cp viewed along the crystallographic a axis, with underlying topology representation: green squares representing Zn-paddle-wheels and red triangles representing flexible nitrogen branches in the linker, (c) PXRD of DUT-180 as made and activated in comparison to calculated patterns of op and cp phase, (d) 3 runs of DUT-180 N2 adsorption isotherms (77 K) without reactivation after initial supercritical drying. | ||
DUT-180 is a topological and isoreticular analogue to DUT-10 with an extended spacer in the linker. In the crystal structure, the spacers are oriented along crystallographic b axis, which is reflected by the unit cell parameters of DUT-180. The one-dimensional elongation of the linker leads to a unit cell elongation only in b axis from 35.883 Å in DUT-10 to 44.43 Å.28 Similar to DUT-10, the as made structure of DUT-180 represents an open framework with 83.2% of solvent accessible void and intersecting channels, running along crystallographic a, b and c directions. The size of the hexagonal channels along a and c direction is partially defined by the spacer length and are therefore different in DUT-10 and DUT-180 structures. The rhomboidal channel along b direction stays the same size as in DUT-10. Increasing of the pore size in the DUT-180op (op – open pore) structure is accompanied by increasing the pore accessibility and porosity. The diameter of free included sphere increases from 11.93 Å in DUT-10 to 12.88 Å in DUT-180, total pore volume from 1.65 cm3 g−1 in DUT-10 to 1.85 cm3 g−1 in DUT-180. The pore size distribution, calculated for both structures, indicates only minor increase of the pore diameter of the largest pore from 13.4 to 14.6 Å (Fig. S25, ESI†).
Upon activation by supercritical CO2 a contraction of the structure is indicated by PXRD pattern changes (Fig. 1b and c). PXRD patterns could be successfully indexed and the crystal structure of DUT-180cp was solved and refined using the Rietveld method. Interestingly, the crystal structure of the cp phase stays in the same space group symmetry and shows a strong contraction in crystallographic c direction, while a and b are only slightly stretched upon the transition (exact values see Table S1, ESI†). Since the crystals have the shape of needles and PXRD samples were measured in transmission geometry, preferred orientation leads to some missing reflexes (e.g. (011) at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ = 5.5° in DUT-180op) or reflexes with reduced intensity (e.g. (101) at 2θ = 6.4° in DUT-180op) in comparison to the theoretical patterns. Additionally, the strong contraction of the c axis of DUT-180cp can only be observed indirectly by the resulting stretching of the a and b axes, since nearly all observed reflexes have hkl values with l = 0.
θ = 5.5° in DUT-180op) or reflexes with reduced intensity (e.g. (101) at 2θ = 6.4° in DUT-180op) in comparison to the theoretical patterns. Additionally, the strong contraction of the c axis of DUT-180cp can only be observed indirectly by the resulting stretching of the a and b axes, since nearly all observed reflexes have hkl values with l = 0.
The porosity of the resulting DUT-180cp phase was evaluated in a nitrogen physisorption experiment at 77 K showing a type Ib isotherm and a moderate uptake of 15 to 18 mmol g−1 over three runs without further activation (Fig. 1d). This is about 1.5 times more than in DUT-10.28 The pore volume determined from the isotherm of the first run is 0.61 cm3 g−1 (p/p0 = 0.91) and at the other runs it is 0.54 cm3 g−1 (p/p0 = 0.95) which is slightly more than the calculated accessible volume from Zeo++ for DUT-180cp (0.45 cm3 g−1) but far less than calculated for DUT-180op (1.85 cm3 g−1), indirectly confirming the crystal structure of the cp phase. The drop of pore volume after the first run indicates that a minor part of the sample was still in the op phase before the completion of the first adsorption cycle. The powder of DUT-180cp was soaked in DMF in order to prove the reversibility of the switching (Fig. S6,† ESI). However, no phase transition to the op phase was observed, which points out the thermodynamic stability of DUT-180cp and a high kinetic barrier for the transformation.
DUT-190: Modification of the synthesis conditions leads to the precipitation of rhombohedral crystals suitable for single crystal X-ray diffraction. The resulting compound with the composition Zn4O(tpdatb)3/2, further denoted as DUT-190, crystallized in the trigonal space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c. The crystal structure is constructed from octahedral Zn4O6+ clusters, interconnected by tpdatb4− in staggered conformation resulting in a 3D open framework with a structure isoreticular to DUT-13 (ttu-a topology) with an elongation occurring in the crystallographic c axis (Fig. 2a). This axis is elongated by 26.44 Å to a total length of 141.34 Å, while only minor changes can be observed in the a and b axes (increase from 25.68 Å to 25.76 Å). Further analysis of the crystal structure reveals two types of pores: the small pore is formed by two zinc metal clusters connected by three linker molecules; the large pore is built up from eight metal clusters ordered in a hexagonal bipyramidal arrangement connected by six H4tpdatb linkers. Since both pores are limited by pore walls in a and b direction, the elongation of c axis leads to a 1-dimensional extension of both cages, which are becoming more ellipsoidal than the ones in DUT-13.18,33 Analysis of the solvent accessible void reveals 83.5% of free space in the unit cell. However, the comparison of the pore size distribution of DUT-13 and DUT-190 indicates similar values and, therefore, similar adsorption properties are expected (Fig. S27, ESI†).
c. The crystal structure is constructed from octahedral Zn4O6+ clusters, interconnected by tpdatb4− in staggered conformation resulting in a 3D open framework with a structure isoreticular to DUT-13 (ttu-a topology) with an elongation occurring in the crystallographic c axis (Fig. 2a). This axis is elongated by 26.44 Å to a total length of 141.34 Å, while only minor changes can be observed in the a and b axes (increase from 25.68 Å to 25.76 Å). Further analysis of the crystal structure reveals two types of pores: the small pore is formed by two zinc metal clusters connected by three linker molecules; the large pore is built up from eight metal clusters ordered in a hexagonal bipyramidal arrangement connected by six H4tpdatb linkers. Since both pores are limited by pore walls in a and b direction, the elongation of c axis leads to a 1-dimensional extension of both cages, which are becoming more ellipsoidal than the ones in DUT-13.18,33 Analysis of the solvent accessible void reveals 83.5% of free space in the unit cell. However, the comparison of the pore size distribution of DUT-13 and DUT-190 indicates similar values and, therefore, similar adsorption properties are expected (Fig. S27, ESI†).
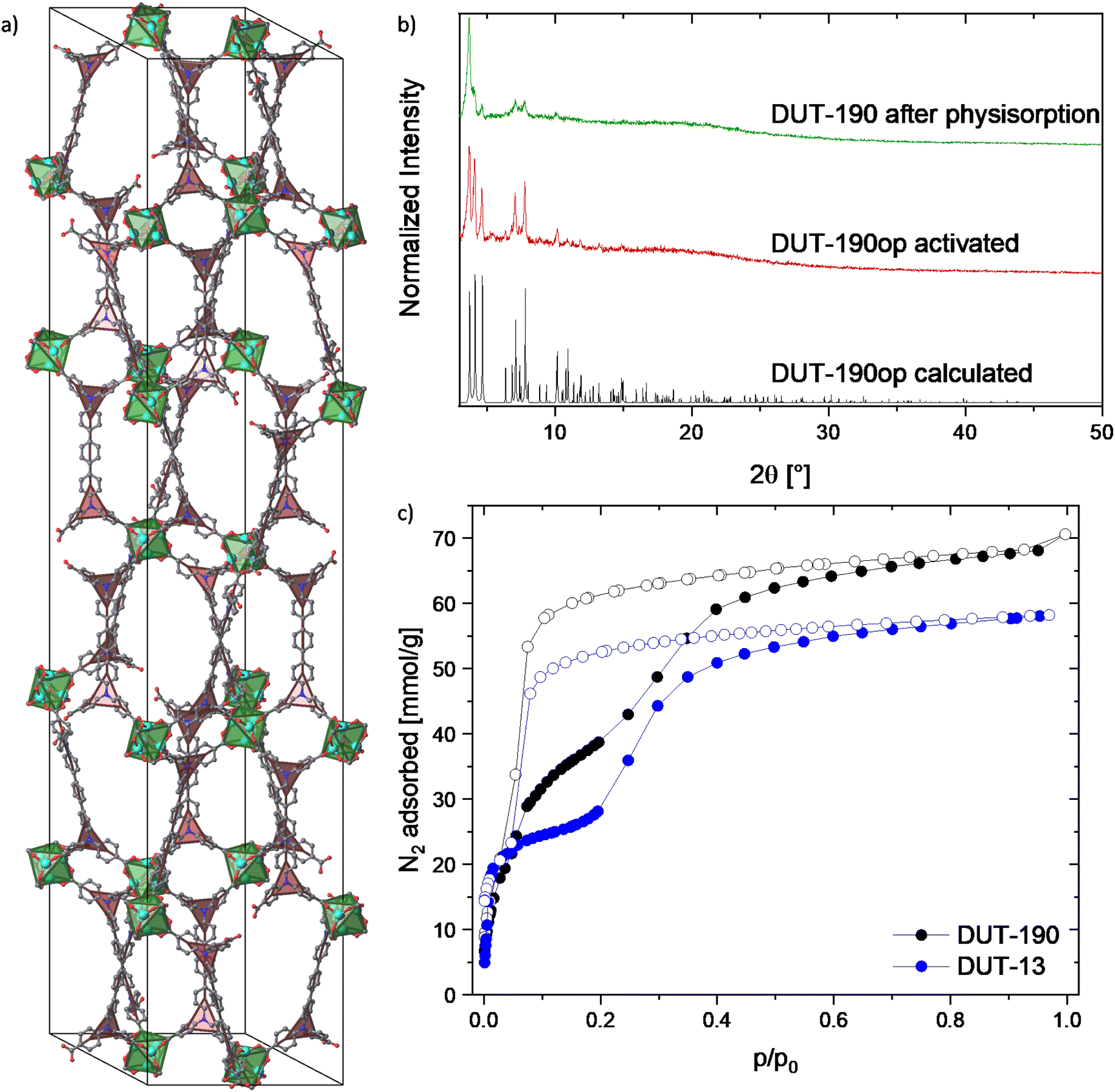 | ||
| Fig. 2 Crystal structure and characterization of DUT-190op: (a) unit cell of DUT-190op without H atoms and with underlying topology representation: green octahedra representing Zn4O(CO2)6-clusters and red triangles representing flexible nitrogen branches in the linker, (b) PXRD patterns of DUT-190op calculated from crystal structure, activated by supercritical CO2 and after nitrogen physisorption, (c) nitrogen physisorption isotherms of DUT-190 and DUT-13 for comparison5 measured at 77 K. | ||
The PXRD pattern of the supercritically activated sample of DUT-190 matches well to the calculated pattern generated from the single crystal structure. Even though the MOF does not seem to retain its flexibility after the first run of nitrogen adsorption, the PXRD pattern after adsorption still shows the main peaks of the op phase (Fig. 2b and c). Nitrogen physisorption at 77 K shows a broad hysteresis between the adsorption and desorption branches in the pressure range p/p0 = 0.05–0.5, which is a first indication of structural dynamics in DUT-190. At p/p0 = 0.05 and 30 mmol g−1 nitrogen loading the slope of the isotherm suddenly reduces showing a quasi-linear slope until p/p0 = 0.4. The plateau is not as flat as in DUT-13, where a flat plateau can be seen up to a pressure of p/p0 = 0.2.18,33 This indicates a lower energy barrier between cp and op phases in the range of loadings and may lead to an incomplete adsorption-induced contraction of DUT-190. The isotherm reaches the final plateau at p/p0 = 0.5 and shows an adsorption capacity of 68.10 mmol g−1, exceeding the recently reported values of DUT-13 by 17.2%18,33 and is close to theoretically calculated values for DUT-190op (72.14 mmol g−1). Desorption of nitrogen starts at p/p0 = 0.1 and shows a steep course. DUT-190 has a pore volume of 2.36 cm3 g−1 at 0.95 p/p0, which is again close to theoretical values calculated using Zeo++ (2.501 cm3 g−1). On another DUT-190 sample, a second adsorption cycle was conducted after the first one without further reactivation of the sample. In the second cycle a reversible isotherm showing a reduced uptake of 25.03 mmol g−1 nitrogen was observed (Fig. S13, ESI†). PXRD patterns, measured on the sample after two physisorption cycles show reduction of the sample crystallinity, but no changes of reflection positions (Fig. 2b). This observation validates the crystallite size dependency, observed in many flexible frameworks,35,36 leading to the contraction of the part of the sample containing large crystallites and can be explained by the relatively low adsorption enthalpy of nitrogen on the one hand and a broad crystallite size distribution on the other hand. Similar effects were also observed in other highly porous flexible MOFs of the DUT-49 series.
In situ PXRD during N2 and CO2 physisorption
In order to follow the adsorption-induced structural dynamics and get deeper mechanistic insights in the flexibility of the framework, in situ PXRD patterns were measured in parallel to physisorption of nitrogen (77 K, Fig. 3) and CO2 (195 K, Fig. S23, ESI†). In situ PXRD experiments were conducted using a home-built instrumentation, based on a laboratory powder X-ray diffractometer, equipped with an X-ray transparent adsorption chamber. Visual analysis of PXRD pattern indicates no significant changes of reflection positions in PXRD patterns, measured at different loadings. The changes of intensities can originate from the nitrogen molecules in the pores and changes in the absorption coefficient of the MOF. However, no clear evidence of a phase transition could be derived from PXRD patterns, measured upon nitrogen and carbon dioxide physisorption. Since PXRD patterns, measured on the CO2-loaded sample show lower crystallinity compared to nitrogen-loaded samples, the latter were further analyzed. The profile of PXRD patterns could be analyzed by a Le Bail fit and the unit cell parameters of DUT-190op could be extracted (Fig. 3). Using these data, the adsorption stress on the framework upon adsorption and desorption was estimated. In the adsorption branch at low pressures the a and b parameters slightly contract from 25.78 Å in vacuum to 25.54 Å at p/p0 = 0.1. At higher pressures the values return to 25.71 Å at p/p0 = 0.95. Different trend is observed for c axis, which increases upon from 141.47 Å in vacuum to 142.41 Å at p/p0 = 0.09. In the pressure range p/p0 = 0.2–0.3 the minimum with c = 141.3 Å is observed, followed by increasing to 141.99 Å at p/p0 = 0.95. Such observations clearly indicate the anisotropy of mechanical properties and point out on the probable direction of the structural contraction. After this point the reverse trend was observed until the numbers stabilized at p/p0 = 0.35, which is the point of the hysteresis closing. Interestingly, some reflections at 2θ = 6.4°, 8.9°, 9.3° and 2θ > 11° are vanishing in the PXRD patterns, measured in the hysteresis range. This may indicate the formation of a disordered contracted phase, which is ordered only in selected directions. The second hypothesis assumes that reflections of the disordered DUT-190cp nearly ideally overlap with reflections of the DUT-190op phase (Fig. S24, ESI†). In order to check this hypothesis, we simulated a DUT-190cp phase with the assumption that DUT-190 shows a contraction mechanism similar to DUT-13. Profile fitting shows good overlap of measured and simulated PXRD (Fig. S22, ESI†). In this situation PXRD cannot provide the unambiguous answer to the question on the mechanism of contraction and additional techniques, sensitive to host–guest and guest–guest interactions should be employed.
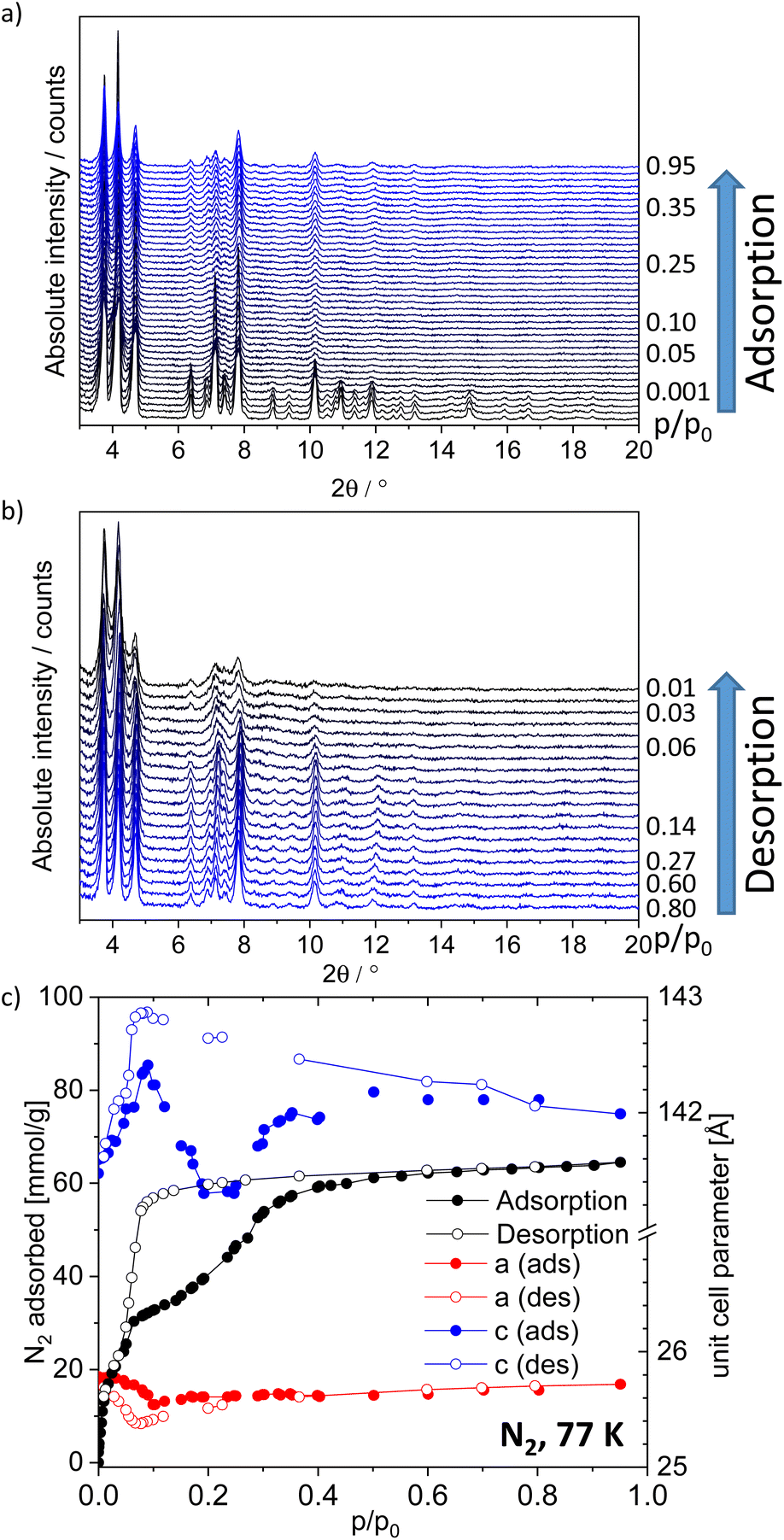 | ||
| Fig. 3 In situ PXRD patterns in parallel to nitrogen physisorption at 77 K; (a) during adsorption; (b) during desorption. (c) Nitrogen physisorption isotherm of the in situ experiment and change of unit cell parameters a and c of DUT-190op. | ||
Physisorption at various temperatures and SEM
To further investigate the flexibility, methane adsorption in a temperature range of 111 K to 180 K was performed similarly to DUT-13 (Fig. S15–S17, ESI†). In comparison to DUT-13 showing a wide hysteresis up to 140 K, which is then decreasing over at 150 K and 160 K until no flexibility is observed at 170 K,33 DUT-190 shows an unchanged wide hysteresis over the whole temperature range. This indicates that the additional phenyl ring in the linker promotes increased flexibility in the network even at higher temperatures and goes along with the findings from nitrogen adsorption experiments. At the boiling point of methane an uptake of 48.2 mmol g−1 could be reached. Additionally, CO2 adsorption at various temperatures was performed (Fig. S19, ESI†) showing a hysteresis up to 250 K. At the sublimation point of CO2 an uptake of 20.8 mmol g−1 could be reached (Fig. S18, ESI†).To confirm the macroscopic changes upon breathing in DUT-190, SEM (Fig. S9, ESI†) images were recorded on the supercritically dried sample (crystal size 2 to 40 μm) and on the sample after nitrogen adsorption. In both cases the images show crystals of the same shape and size, but in the sample after nitrogen physisorption the crystal surface shows defects in the form of cracks and grains, which may cause the loss of crystallinity that can be observed in the PXRD after nitrogen physisorption (Fig. 2b). Furthermore, similar to other subcritical gases, physisorption of xenon at 200 K showed typical hysteresis between adsorption and desorption branches reaching an uptake of 17.3 mmol g−1 in saturation (Fig. S20, ESI†).
129Xe NMR
In situ 129Xenon NMR was used to further investigate the adsorption-induced structural dynamics of DUT-190. The 129Xe NMR spectra recorded at 206 K are shown in Fig. 4a. The chemical shift of xenon depends, among other things, on both the pressure and the chemical environment,37,38 which makes in situ NMR a promising method for studying structural changes of DUT-190. NMR spectra were measured at pressures up to 6.5 bar, the xenon boiling pressure at this temperature.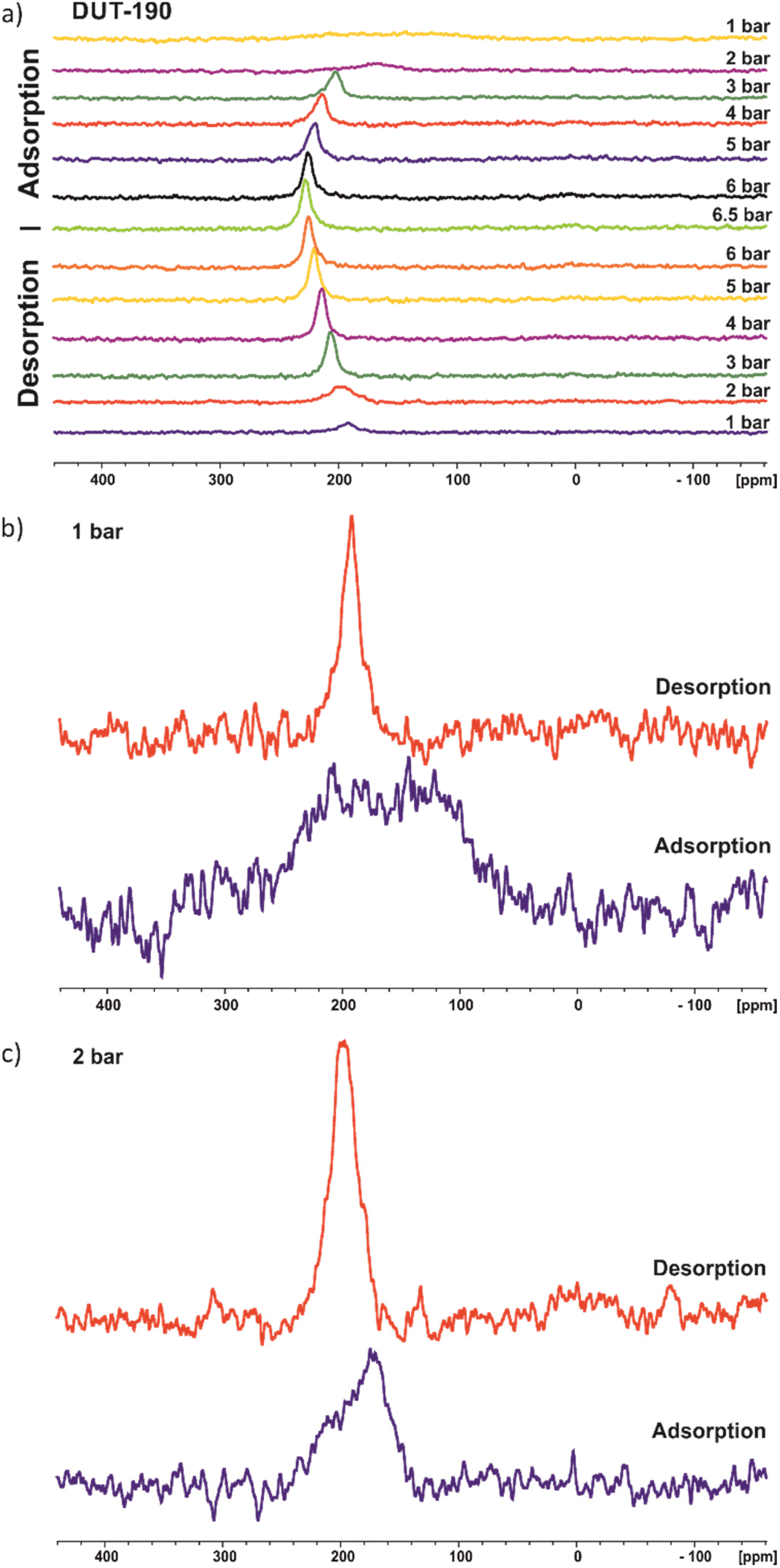 | ||
| Fig. 4 129Xe NMR spectra of DUT-190 during adsorption and desorption measured at 206 K; (a) full adsorption and desorption cycle; (b) for 1 bar; (c) for 2 bar. | ||
Although DUT-190 has pores with two different pore sizes, only one signal from adsorbed xenon is seen. This shows rapid xenon exchange between both types of pores because two signals would otherwise be expected. An increase in chemical shift can be seen during adsorption until it reaches a plateau at high pressures. This is a typical behavior for xenon in porous materials and is due to the increasing density of adsorbed xenon. During desorption, the spectra show a hysteresis at pressures of 3 bar and below. This is particularly evident at 1 bar. During the adsorption, only a very broad signal at 157(±7) ppm with a FWHM (full width at half maximum) of ca. 140 ppm is visible. In contrast, a much narrower signal at 192(±1) ppm with an FWHM of only 16 ppm is observed during desorption (Fig. 4b). The broad xenon signal during adsorption could indicate immobilization of xenon within the pores or structural heterogeneity of the MOF. However, a more Lorentzian line shape would be expected if the broadening is due to the immobilization of xenon in contrast to the observed signal in Fig. 4b especially for 1 bar (adsorption). Therefore, the data rather support the hypothesis of a structural heterogeneity of the sample at the beginning of the adsorption experiment. This heterogeneity may originate from different sized pores or multiple adsorption sites due to a disordered contracted phase as also indicated by in situ PXRD. As already expected from the xenon adsorption isotherm, xenon desorption is still incomplete at 1 bar.
Experimental procedures
General methods
All chemicals and solvents used in the syntheses were at least of reagent grade and were used without further purification.1 H and 13 C NMR spectra were acquired on a Bruker Avance III 500 spectrometer (500 MHz and 125.8 MHz for 1H and 13C, respectively). All 1H and 13C NMR spectra are reported in parts per million (ppm) downfield of TMS and were measured relative to the residual signals of the solvents at 7.26 ppm (CDCl3) or 2.50 ppm (DMSO-d6).33
In situ 129 Xe NMR experiments were carried out on a Bruker Avance 300 spectrometer (1H resonance frequency of 300.13 MHz, magnetic field of 7.04 T) equipped with a liquid-state NMR probe. A home-built in situ apparatus was used which allows temperature and pressure adjustment.39,40 After each pressure change, a waiting time of at least 15 minutes was applied to allow the system to be equilibrated before the measurement. The spectra were referenced using the pressure-dependence of the 129Xe chemical shift.41 Extrapolation to zero pressure is defined as “0 ppm”.
Attenuated total reflection infrared (ATR-IR) spectra were recorded in a wave number range of 4000 cm−1 to 600 cm−1 using a BRUKER Vertex 70 infrared spectrometer.
Elemental Analysis (C, H, N) was performed using a Vario MICRO Cube Elementar device.
PXRD patterns were collected in transmission geometry with a STOE STADI P diffractometer operated at 40 kV and 30 mA with monochromatic Cu-Kα1 (λ = 0.15405 nm) radiation, a scan speed of 120 s per step and a detector step size of 2Θ = 6°. The samples were placed between non-diffracting adhesive tape. “As made” samples were analyzed while suspended in DMF. Desolvated samples were prepared in a glove box under argon atmosphere. Theoretical PXRD patterns were calculated based on crystal structures using the Mercury 3.9 software package.
Thermogravimetric analysis (TG) and differential thermal analysis (DTA) were carried out in air using a Netzsch STA 409 C/CD thermal analyzer with a heating rate of 5 K min−1.
SEM images of the DUT-190 samples were taken with secondary electrons in a HITACHI SU8020 microscope using 2.0 kV acceleration voltage, a working distance of 12.6 to 12.9 mm and different magnifications. The samples were prepared on a sticky carbon sample holder. To avoid degradation upon exposure to air, the samples were prepared in a glove box under argon atmosphere.
Nitrogen physisorption was measured at 77 K on a QUANTACHROME Quadrasorb SI Automated Surface Area and Pore Size Analyser.
Methane, carbon dioxide and xenon adsorption isotherms were collected using a home-built system constructed from a volumetric BELSORP-HP (Microtrac MRB) device and a closed cycle helium cryostat DE-202D (ARS-Cryo) in the self-made high-pressure adsorption cell based on a 1/2 inch VCR-support. The adsorption system is designed for static physisorption experiments in the temperature range of 4 to 300 K and a pressure range of 0.01 to 8000 kPa. The BELSORP-HP instrument is equipped with a turbomolecular pump and possesses a temperature-controlled standard volume (Vs) of 20.663 cm3. The dead volume of the system was determined using helium gas with 99.999% purity. Methane (99.999%), carbon dioxide (99.9995%) and xenon (99.998%) were used in high-pressure gas adsorption experiments. All adsorption isotherms were measured using equilibrium conditions of 0.1% of pressure change within 500 s.
Single crystal X-ray diffraction
Crystal structures of as synthesized DMF-filled phases of DUT-180 and DUT-190 were determined by means of single crystal X-ray diffraction experiments. Suitable single crystals were prepared in the glass capillaries with d = 0.5 mm with small amount of the mother liquid to avoid the desorption of guests upon the experiment. The diffraction images were collected at 100 K using a Bruker KAPPA APEX2 diffractometer, equipped with a long focus X-ray tube with Mo-anode and graphite monochromator (λ = 0.71073 Å) and an APEX2 CCD-detector. The unit cell parameters were determined based on three short scans, performed on different crystal orientations. The data collection strategy was optimized using the APEX3 software and collected images were integrated using the SAINT software. Further instrument- and sample-related intensity corrections were applied using the SADABS software. The crystal structures were solved by direct methods implemented in the SHELXS-2018/3 program.42 The obtained model was refined by full matrix least-squares on F2 using SHELXL-2018/3.42 All non-hydrogen atoms were refined in the anisotropic approximation. Hydrogen atoms were refined in geometrically calculated positions using a “riding model” with Uiso(H) = 1.2Uiso(C). Anisotropic displacement parameters of the carbon atoms of the dabco molecule were treated by using SIMU and DELU instructions in the refinement. The positions of disordered solvent molecules in the pores could not be determined from the Fourier electron density maps, and therefore, the SQUEEZE routine was applied to reduce the contribution of the solvent molecules on the structure factors.43 As a result, 3587 and 21003 electrons were removed from 14873 Å3 of solvent accessible void in DUT-180 and 67905 Å3 of solvent accessible void in DUT-190 correspondingly. CCDC 2247742, 2247743 and 2247744 contain the supplementary crystallographic data for DUT-180cp, DUT-180op and DUT-190op.
888(16) Å3, z = 4, d = 0.334 gcm−3, μ = 0.282 mm−1, F(000) = 1832, 2θmax = 25.07°, 47731/8195/150 reflections collected/unique/parameter, Rint = 0.1557, R1(I>2σ) = 0.0485, wR2(all) = 0.1530, S = 1.001.
c, a = 25.7659(7) Å, c = 141.344(4) Å, V = 81264(5) Å3, z = 12, d = 0.339 gcm−3, μ = 0.365 mm−1, F(000) = 8412, 2θmax = 25.12°, 392276/16126/286 reflections collected/unique/parameter, Rint = 0.228, R1(I>2σ) = 0.0768, wR2(all) = 0.3450, S = 1.014.
Crystal structure determination of the contracted phases of DUT-180 and DUT-190
The crystal structure of the contracted phase of DUT-180cp was determined from PXRD patterns using the Reflex tool of the Materials Studio 5.0 software.44 Indexing of PXRD patterns using suggested orthorhombic crystal symmetry and analysis of intensities resulted in Imma space group, which is the same space group as was determined for DUT-180op phase. The structural model was obtained using geometric optimization of the DUT-180op structure using UFF force field and unit cell parameters, obtained in the indexing. The PXRD profile was refined using a Pawley fit, followed by a Rietveld refinement of the crystal structure. Zn atoms, carboxylate groups and phenyl groups of the ligand molecules were handled as rigid bodies in the refinement in order to reduce the number of parameters. The Rietveld plot is given in Fig. S21 (ESI†). The PXRD pattern of DUT-190cp was measured in situ in parallel to nitrogen physisorption at 77 K. Since the number of observed reflections was not sufficient for ab initio indexing of the PXRD pattern, we hypothesized a similar stimuli-induced behavior of the DUT-13 and DUT-190 structures considering the similarity of the op structures and adsorption properties. We constructed the corresponding structural model and performed a Pawley fit of the PXRD showing good matching of the simulated unit cell (Fig. S22, ESI†).θrange = 3–50°, profile function Thompson–Cox–Hastings, U = 0.462, V = 0.242, W = 0.002, X = 0.753, Y = 0.101, refined motion groups/degree of freedom 20/20, Rp = 0.0824, Rwp = 0.1023.
960.7(8) Å3, Z = 4, λ = 1.54059 Å, T = 77 K, 2θrange = 3–30°, profile function Thompson–Cox–Hastings, U = 0.01, V = −0.001, W = 0.002, X = 0.000, Y = 0.155(2), Rp = 0.0361, Rwp = 0.0491.
In situ PXRD in parallel to gas physisorption
In situ PXRD on DUT-190 in parallel to adsorption and desorption of N2 at 77 K and CO2 at 195 K was measured using a customized setup, based on laboratory powder X-ray diffractometer Empyrean-2 (PANALYTICAL GmbH), equipped with a closed-cycle helium cryostat (ARS DE-102) and a home-built in situ adsorption chamber, connected to a volumetric adsorption instrument BELSORP-max (Microtrac MRB). A TTL trigger was used for establishing the communication between BELSORP-max and Empyrean software and ensure the measurement of the adsorption isotherm and PXRD pattern data collection in a fully automated mode in the pre-defined points of the isotherm. A parallel linear Cu K-α1 X-ray beam (λ = 1.54059 Å), gained after passing the hybrid 2xGe(220) monochromator, 4 mm mask, and primary divergence and secondary antiscatter slits with 1/4° opening was used for data collection. Receiving slits with 1/4° opening and a Pixcel-3D detector in 1D scanning mode (255 active channels) were used as diffracted beam optics. The diffraction experiments were performed using ω–2θ scans in transmission geometry in the range of 2θ = 3–50°.
Synthesis procedures
:1 mixture of pentane and diethyl ether or wash the crude product using diethyl ether until only a colorless filtrate is obtained. A dark yellow powder with a yield of 87.5% can be gained. Rf (TLC) = 0.76 in pentane/diethyl ether 1:1. 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.95 (d, J = 8.8 Hz, 8H), 7.68 (s, 4H), 7.61 (d, J = 8.6 Hz, 4H), 7.21 (d, J = 8.6 Hz, 4H), 7.15 (d, J = 8.8 Hz, 8H), 4.37 (q, J = 7.1 Hz, 8H), 1.39 (t, J = 7.1 Hz, 12H). 13C NMR (126 MHz, CDCl3) δ (ppm) = 166.30, 150.99, 145.64, 139.23, 137.36, 131.17, 128.36, 127.40, 126.56, 124.91, 122.84, 67.24, 60.95, 14.55. NMR Spectra and assignments of all atoms can be seen in Fig. S2, ESI.† Elemental Analysis for C54H48N2O8·0.5CH2Cl2, calculated: C 72.68%, H 5.49%, N 3.10%; found: C 72.85%, H 5.50%, N 3.31%. IR ν/cm−1 = 3396 (w), 3066 (w), 3033 (w), 2978 (w), 2958 (w), 2933 (w), 2904 (w), 2871 (w), 1709 (s), 1613 (w), 1595 (s), 1528 (w), 1506 (m), 1488 (m), 1462 (w), 1446 (w), 1428 (w), 1415 (w), 1389 (w), 1365 (w), 1321 (m), 1265 (s), 1174 (m), 1101 (m), 1016 (w), 1004 (w), 971 (w), 924 (w), 874 (w), 847 (w), 816 (w), 782 (w), 766 (m), 735 (w), 709 (w), 696 (w), 683 (w), 671 (w), 640 (w), 632 (w).
Conclusions
We successfully applied the isoreticular approach to design the new flexible frameworks DUT-180 (lil-a) and DUT-190 (ttu-a) based on zinc paddle-wheel and Zn4O6+ metal nodes, respectively. Similar to DUT-10, the isoreticular framework irreversibly contracts upon desolvation to a less porous, but more thermodynamically stable DUT-180cp phase. In DUT-190, elongation of the backbone of the organic ligand does not influence the pore size. By showing a broad hysteresis, indicating high framework flexibility in the form of a breathing mechanism, a similar behavior to DUT-13 upon physisorption of subcritical fluids is observed. In situ PXRD, measured in parallel to nitrogen physisorption at 77 K, does not show significant changes in reflection positions indicating either complete amorphization of the contracted phase or overlapping of the patterns. In situ129Xe NMR shows a hysteresis between signals measured in adsorption and desorption branches and confirms the framework flexibility. Physisorption of methane on DUT-190 in a broad temperature range indicates that elongation of the ligand backbone strongly influences the micromechanics of the breathing transition. This approach can be further used for the targeted design of breathing frameworks with similar flexibility mechanisms. Further systematic alteration of the micromechanical properties of the tetracarboxylate ligands may result in flexible frameworks with desired stress/strain response, required for specific applications.Author contributions
B. Felsner: investigation, visualization, writing – original draft; Volodymyr Bon: investigation, visualization, writing – original draft; Christopher Bachetzky: investigation (NMR), writing – original draft; Eike Brunner: supervision (NMR), writing – review & editing; Stefan Kaskel: conceptualization, supervision, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement No. 742743). V. B. acknowledges the financial support by the Federal Ministry of Education and Research (Project TIMESWITCH No 05K21OD1). B. F. acknowledges the financial support by the Graduate Academy of the Technical University Dresden. E. B. and S. K. gratefully acknowledge financial support from DFG (FOR 2433). We acknowledge the NMR measurement staff of the Organic Chemistry Department and the Elementary Analysis staff of the Inorganic Molecular Chemistry Department of the Technical University Dresden as well as F. Schwotzer and N. Bönisch for SEM measurements and our TG lab for TGA.References
- S. Horike, S. Shimomura and S. Kitagawa, Soft porous crystals, Nat. Chem., 2009, 1, 695–704 CrossRef CAS.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Flexible metal–organic frameworks, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- S. Krause, N. Hosono and S. Kitagawa, Chemistry of Soft Porous Crystals: Structural Dynamics and Gas Adsorption Properties, Angew. Chem., Int. Ed., 2020, 59, 15325–15341 CrossRef CAS PubMed.
- J. A. Mason, J. Oktawiec, M. K. Taylor, M. R. Hudson, J. Rodriguez, J. E. Bachman, M. I. Gonzalez, A. Cervellino, A. Guagliardi, C. M. Brown, P. L. Llewellyn, N. Masciocchi and J. R. Long, Methane storage in flexible metal-organic frameworks with intrinsic thermal management, Nature, 2015, 527, 357–361 CrossRef CAS PubMed.
- T. Kundu, B. B. Shah, L. Bolinois and D. Zhao, Functionalization-Induced Breathing Control in Metal–Organic Frameworks for Methane Storage with High Deliverable Capacity, Chem. Mater., 2019, 31, 2842–2847 CrossRef CAS.
- M. K. Taylor, T. Runčevski, J. Oktawiec, J. E. Bachman, R. L. Siegelman, H. Jiang, J. A. Mason, J. D. Tarver and J. R. Long, Near-Perfect CO2/CH4 Selectivity Achieved through Reversible Guest Templating in the Flexible Metal-Organic Framework Co(bdp), J. Am. Chem. Soc., 2018, 140, 10324–10331 CrossRef CAS PubMed.
- M. Sin, N. Kavoosi, M. Rauche, J. Pallmann, S. Paasch, I. Senkovska, S. Kaskel and E. Brunner, In Situ 13C NMR Spectroscopy Study of CO2/CH4 Mixture Adsorption by Metal–Organic Frameworks: Does Flexibility Influence Selectivity?, Langmuir, 2019, 35, 3162–3170 CrossRef CAS PubMed.
- K. Roztocki, M. Rauche, V. Bon, S. Kaskel, E. Brunner and D. Matoga, Combining In Situ Techniques (XRD, IR, and 13C NMR) and Gas Adsorption Measurements Reveals CO2-Induced Structural Transitions and High CO2/CH4 Selectivity for a Flexible Metal-Organic Framework JUK-8, ACS Appl. Mater. Interfaces, 2021, 13, 28503–28513 CrossRef CAS PubMed.
- P. Freund, L. Mielewczyk, M. Rauche, I. Senkovska, S. Ehrling, E. Brunner and S. Kaskel, MIL-53(Al)/Carbon Films for CO 2 -Sensing at High Pressure, ACS Sustainable Chem. Eng., 2019, 7, 4012–4018 CrossRef CAS.
- P. Freund, I. Senkovska and S. Kaskel, Switchable Conductive MOF-Nanocarbon Composite Coatings as Threshold Sensing Architectures, ACS Appl. Mater. Interfaces, 2017, 9, 43782–43789 CrossRef CAS PubMed.
- X. Zhang, Q. Zhang, D. Yue, J. Zhang, J. Wang, B. Li, Y. Yang, Y. Cui and G. Qian, Chemical Sensing: Flexible Metal-Organic Framework-Based Mixed-Matrix Membranes: A New Platform for H 2 S Sensors (Small 37/2018), Small, 2018, 14, 1870168 CrossRef.
- P. Freund, I. Senkovska, B. Zheng, V. Bon, B. Krause, G. Maurin and S. Kaskel, The force of MOFs: the potential of switchable metal-organic frameworks as solvent stimulated actuators, Chem. Commun., 2020, 56, 7411–7414 RSC.
- V. Bon, S. Krause, I. Senkovska, N. Grimm, D. Wallacher, D. M. Többens and S. Kaskel, Massive Pressure Amplification by Stimulated Contraction of Mesoporous Frameworks**, Angew. Chem., Int. Ed., 2021, 60, 11735–11739 CrossRef CAS PubMed.
- G. Férey, Giant flexibility of crystallized organic–inorganic porous solids: facts, reasons, effects and applications, New J. Chem., 2016, 40, 3950–3967 RSC.
- O. Kozachuk, K. Khaletskaya, M. Halbherr, A. Bétard, M. Meilikhov, R. W. Seidel, B. Jee, A. Pöppl and R. A. Fischer, Microporous Mixed–Metal Layer–Pillared [Zn 1− x Cu x (bdc)(dabco) 0.5 ] MOFs: Preparation and Characterization, Eur. J. Inorg. Chem., 2012, 2012, 1688–1695 CrossRef CAS.
- I. Schwedler, S. Henke, M. T. Wharmby, S. R. Bajpe, A. K. Cheetham and R. A. Fischer, Mixed-linker solid solutions of functionalized pillared-layer MOFs - adjusting structural flexibility, gas sorption, and thermal responsiveness, Dalton Trans., 2016, 45, 4230–4241 RSC.
- S. Henke, A. Schneemann, A. Wütscher and R. A. Fischer, Directing the breathing behavior of pillared-layered metal-organic frameworks via a systematic library of functionalized linkers bearing flexible substituents, J. Am. Chem. Soc., 2012, 134, 9464–9474 CrossRef CAS PubMed.
- R. Grünker, I. Senkovska, R. Biedermann, N. Klein, M. R. Lohe, P. Müller and S. Kaskel, A highly porous flexible Metal-Organic Framework with corundum topology, Chem. Commun., 2011, 47, 490–492 RSC.
- L. Sarkisov, R. L. Martin, M. Haranczyk and B. Smit, On the flexibility of metal-organic frameworks, J. Am. Chem. Soc., 2014, 136, 2228–2231 CrossRef CAS PubMed.
- D. N. Dybtsev, H. Chun and K. Kim, Rigid and Flexible: A Highly Porous Metal–Organic Framework with Unusual Guest-Dependent Dynamic Behavior, Angew. Chem., 2004, 116, 5143–5146 CrossRef.
- L. Abylgazina, I. Senkovska, S. Ehrling, V. Bon, P. St. Petkov, J. D. Evans, S. Krylova, A. Krylov and S. Kaskel, Tailoring adsorption induced switchability of a pillared layer MOF by crystal size engineering, CrystEngComm, 2021, 23, 538–549 RSC.
- J. Seo, C. Bonneau, R. Matsuda, M. Takata and S. Kitagawa, Soft Secondary Building Unit: Dynamic Bond Rearrangement on Multinuclear Core of Porous Coordination Polymers in Gas Media, J. Am. Chem. Soc., 2011, 133, 9005–9013 CrossRef CAS.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Ultrahigh porosity in metal-organic frameworks, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- I. M. Hönicke, I. Senkovska, V. Bon, I. A. Baburin, N. Bönisch, S. Raschke, J. D. Evans and S. Kaskel, Balancing Mechanical Stability and Ultrahigh Porosity in Crystalline Framework Materials, Angew. Chem., Int. Ed., 2018, 57, 13780–13783 CrossRef PubMed.
- C. Serre, S. Bourrelly, A. Vimont, N. A. Ramsahye, G. Maurin, P. L. Llewellyn, M. Daturi, Y. Filinchuk, O. Leynaud, P. Barnes and G. Férey, An Explanation for the Very Large Breathing Effect of a Metal–Organic Framework during CO2 Adsorption, Adv. Mater., 2007, 19, 2246–2251 CrossRef CAS.
- G. Férey and C. Serre, Large breathing effects in three-dimensional porous hybrid matter: facts, analyses, rules and consequences, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC.
- R. Pallach, J. Keupp, K. Terlinden, L. Frentzel-Beyme, M. Kloß, A. Machalica, J. Kotschy, S. K. Vasa, P. A. Chater, C. Sternemann, M. T. Wharmby, R. Linser, R. Schmid and S. Henke, Frustrated flexibility in metal-organic frameworks, Nat. Commun., 2021, 12, 4097 CrossRef CAS PubMed.
- R. Grünker, I. Senkovska, R. Biedermann, N. Klein, A. Klausch, I. A. Baburin, U. Mueller and S. Kaskel, Topological Diversity, Adsorption and Fluorescence Properties of MOFs Based on a Tetracarboxylate Ligand, Eur. J. Inorg. Chem., 2010, 2010, 3835–3841 CrossRef.
- S. Krause, V. Bon, I. Senkovska, U. Stoeck, D. Wallacher, D. M. Többens, S. Zander, R. S. Pillai, G. Maurin, F.-X. Coudert and S. Kaskel, A pressure-amplifying framework material with negative gas adsorption transitions, Nature, 2016, 532, 348–352 CrossRef CAS PubMed.
- S. Krause, J. D. Evans, V. Bon, I. Senkovska, P. Iacomi, F. Kolbe, S. Ehrling, E. Troschke, J. Getzschmann, D. M. Többens, A. Franz, D. Wallacher, P. G. Yot, G. Maurin, E. Brunner, P. L. Llewellyn, F.-X. Coudert and S. Kaskel, Towards general network architecture design criteria for negative gas adsorption transitions in ultraporous frameworks, Nat. Commun., 2019, 10 DOI:10.1038/s41467-019-11565-3.
- S. Krause, J. D. Evans, V. Bon, I. Senkovska, S. Ehrling, P. Iacomi, D. M. Többens, D. Wallacher, M. S. Weiss, B. Zheng, P. G. Yot, G. Maurin, P. L. Llewellyn, F.-X. Coudert and S. Kaskel, Engineering micromechanics of soft porous crystals for negative gas adsorption, Chem. Sci., 2020, 11, 9468–9479 RSC.
- R. Grünker, V. Bon, A. Heerwig, N. Klein, P. Müller, U. Stoeck, I. A. Baburin, U. Mueller, I. Senkovska and S. Kaskel, Dye Encapsulation Inside a New Mesoporous Metal-Organic Framework for Multifunctional Solvatochromic-Response Function, Chem. – Eur. J., 2012, 18, 13299–13303 CrossRef PubMed.
- B. Felsner, V. Bon, J. D. Evans, F. Schwotzer, R. Grünker, I. Senkovska and S. Kaskel, Unraveling the Guest-Induced Switchability in the Metal-Organic Framework DUT-13(Zn)*, Chem. – Eur. J., 2021, 27, 9708–9715 CrossRef CAS PubMed.
- M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Topological Analysis of Metal–Organic Frameworks with Polytopic Linkers and/or Multiple Building Units and the Minimal Transitivity Principle, Chem. Rev., 2014, 114, 1343–1370 CrossRef CAS.
- Y. Sakata, S. Furukawa, M. Kondo, K. Hirai, N. Horike, Y. Takashima, H. Uehara, N. Louvain, M. Meilikhov, T. Tsuruoka, S. Isoda, W. Kosaka, O. Sakata and S. Kitagawa, Shape-memory nanopores induced in coordination frameworks by crystal downsizing, Science, 2013, 339, 193–196 CrossRef CAS.
- S. Krause, V. Bon, I. Senkovska, D. M. Többens, D. Wallacher, R. S. Pillai, G. Maurin and S. Kaskel, The effect of crystallite size on pressure amplification in switchable porous solids, Nat. Commun., 2018, 9, 1573 CrossRef PubMed.
- T. Ito and J. Fraissard, 129Xe NMR study of xenon adsorbed on Y zeolites, J. Chem. Phys., 1998, 76, 5225 CrossRef.
- J. Fraissard and T. Ito, 129Xe n.m.r. study of adsorbed xenon: A new method for studying zeolites and metal-zeolites, Zeolites, 1988, 8, 350–361 CrossRef CAS.
- H. C. Hoffmann, B. Assfour, F. Epperlein, N. Klein, S. Paasch, I. Senkovska, S. Kaskel, G. Seifert and E. Brunner, High-Pressure in Situ 129Xe NMR Spectroscopy and Computer Simulations of Breathing Transitions in the Metal–Organic Framework Ni2(2,6-ndc)2(dabco) (DUT-8(Ni)), J. Am. Chem. Soc., 2011, 133, 8681–8690 CrossRef CAS PubMed.
- F. Kolbe, S. Krause, V. Bon, I. Senkovska, S. Kaskel and E. Brunner, High-Pressure in Situ 129Xe NMR Spectroscopy: Insights into Switching Mechanisms of Flexible Metal–Organic Frameworks Isoreticular to DUT-49, Chem. Mater., 2019, 31, 6193–6201 CrossRef CAS PubMed.
- A. K. Jameson, C. J. Jameson and H. S. Gutowsky, Density Dependence of 129Xe Chemical Shifts in Mixtures of Xenon and Other Gases, J. Chem. Phys., 2003, 53, 2310 CrossRef.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- Materials Studio 5.0, Accelrys, San Diego, USA, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC numbers 2247742–2247744. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi00480e |
| This journal is © the Partner Organisations 2023 |