
Structural transformation of metal–organic frameworks and identification of electrocatalytically active species during the oxygen evolution reaction under neutral conditions†
Xiaoqiang
Liang
 a,
Sen
Wang
a,
Jingyu
Feng
*be,
Zhen
Xu
bf,
Zhenyu
Guo
b,
Hui
Luo
b,
Feng
Zhang
d,
Chen
Wen
c,
Lei
Feng
c,
Chengan
Wan
c and
Maria-Magdalena
Titirici
*b
a,
Sen
Wang
a,
Jingyu
Feng
*be,
Zhen
Xu
bf,
Zhenyu
Guo
b,
Hui
Luo
b,
Feng
Zhang
d,
Chen
Wen
c,
Lei
Feng
c,
Chengan
Wan
c and
Maria-Magdalena
Titirici
*b
aSchool of Environmental and Chemical Engineering, Xi'an Polytechnic University, 710048, Xi'an, PR China
bDepartment of Chemical Engineering, Imperial College London, South Kensington Campus, SW7 2AZ, London, UK. E-mail: j.feng19@imperial.ac.uk; m.titirici@imperial.ac.uk
cBeijing Spacecrafts, 100094, Beijing, PR China
dKey Laboratory of Photochemical Biomaterials and Energy Storage Materials, Heilongjiang Province and College of Chemistry and Chemical Engineering, Harbin Normal University, Harbin 150025, PR China
eSchool of Engineering and Materials Science, Queen Mary University of London, E1 4NS, London, UK
fYusuf Hamied Department of Chemistry, University of Cambridge, CB2 1EW, Cambridge, UK
First published on 7th April 2023
Abstract
The electrocatalytic oxygen evolution reaction (OER) under neutral or near-neutral conditions has attracted research interest due to its environmental friendliness and economic sustainability in comparison with currently available acidic and alkaline conditions. However, it is challenging to identify electrocatalytically active species in the OER procedure under neutral environments due to non-crystalline forms of catalysts. Crystalline metal–organic framework (MOF) materials could provide novel insights into electrocatalytically active species because of their well-defined structures. In this study, we synthesized two isostructural two-dimensional (2D) MOFs [Co(HCi)2(H2O)2·2DMF]n (Co–Ci-2D) and [Ni(HCi)2(H2O)2·2DMF]n (Ni–Ci-2D) (H2Ci = 1H-indazole-5-carboxylic acid, DMF = N,N-dimethyl-formamide) to investigate their OER performance in a neutral environment. Our results indicate that Co–Ci-2D holds a current density of 3.93 mA cm−2 at 1.8 V vs. RHE and an OER durability superior to the benchmark catalyst IrO2. Utilizing the advantages of the structural transformation of MOF materials which are easier to characterize and analyze compared with ill-defined amorphous materials, we found that a mononuclear coordination compound [Co(HCi)2(H2O)4] (Co–Ci-mono-A) and its isomer (Co–Ci-mono-B) were proved to be active species of Co–Ci-2D in the neutral OER process. For Ni–Ci-2D, mononuclear coordination compounds similar to the structures of the cobalt material (Ni–Ci-mono-A and Ni–Ci-mono-B) together with NiHPO4 formed by the precipitation were confirmed as active species for neutral OER catalysis. Additionally, the difference in OER activities between Co–Ci-2D and Ni–Ci-2D, approximately one order of magnitude, can be attributed to changes in bond strength resulting from variations in bond length within coordination octahedra after being treated with the PBS solution. These findings contribute to a better comprehension of the OER procedure in neutral media.
1. Introduction
With the ever-increasing demand for global energy and the rapid depletion of fossil fuels, it has become an urgent task to explore advanced energy conversion and storage technologies with low-cost, high-efficiency and environmentally friendly features, including water electrolyzers,1,2 rechargeable metal–air batteries,3,4 and fuel cells.5,6 Electrochemical water splitting (2H2O → O2 + 2H2) has emerged as one of the most promising solutions to produce low-price and highly pure hydrogen as an attractive alternative to fossil fuels.7–9 As a key half-reaction in water-splitting cells, the oxygen evolution reaction (OER) suffers from sluggish reaction kinetics due to the four proton-coupled electron transfer processes.10,11 To lower the kinetic barrier and expedite the process, significant efforts have been devoted to developing highly active OER electrocatalysts.12,13 Up to now, commercially noble-metal-based materials, e.g. RuO2 and IrO2, were proved to be the most efficient electrocatalysts for OER, but their high cost, scarcity and poor durability are significant obstacles in large-scale practical application.14,15 For this reason, a great deal of research has focused on developing alternative first-row transition metal catalysts that are low cost, Earth-abundant and have high activity,16–18 such as metal oxides/hydroxides,19,20 carbides,21 nitrides,22 phosphides,23,24 sulfides25 and single atoms.26,27 However, most of these reported catalysts need to operate under strongly acidic or alkaline conditions, causing corrosion problems which are neither environmentally friendly nor economical.28,29 Therefore, exploring OER catalysts with use in mild environments is imperative.Unlike extreme pH conditions, OER in neutral or near-neutral media is regarded as an ideal choice for electrochemical water splitting owing to the environmentally benign processes with fewer corrosion issues and a relatively low maintenance cost for the equipment. Recently, considerable research efforts have also been put into designing and synthesizing efficient OER catalysts working in neutral conditions.30–35 For instance, Co–Pi NA/Ti electrodes demonstrate an excellent electrocatalytic activity with a geometric current density of 10 mA cm−2 at an overpotential of 450 mV in 0.1 M phosphate buffered saline (PBS).36 Co–Bi NS/G composite features an attractive OER performance under neutral conditions, which delivers a current density of 10 mA cm−2 driven by an overpotential of 290 mV.37 However, to date, most catalysts operating under neutral conditions are based on amorphous materials, and therefore it is difficult to gain insights into the nature of the active sites during the OER process. Although the mechanism of OER in neutral or near-neutral environments has been proposed previously,38 and is analogical to those of acidic and alkaline electrolytes,39,40 the accurately and universally accepted mechanism remains unclear. Unlike strong acidic and alkaline conditions, the H+ or OH− ions in neutral conditions are far less than in acidic and alkaline conditions, where the dissociation of water molecules under a neutral pH must be considered in the OER steps.41 This extra step results in sluggish kinetics and differentiates the mechanism in pH-neutral conditions from those in acidic and alkaline media. Investigating and identifying active species in such pH conditions could help to further understand the OER steps in neutral conditions.
Crystalline metal–organic frameworks (MOFs) can act as model systems to provide better insights into investigating the OER mechanisms in neutral conditions owing to their well-defined crystal structures and adjustable functionalities.42 Indeed, MOFs with a high electrocatalytic activity have been previously utilized in the OER process.43–50 Nevertheless, most MOFs can quickly react with available molecules/ions in the solutions, being retransformed into new active species to catalyze water oxidation to O2 due to the existence of weak coordination bonds.51–53 Recently, CoOOH54,55 and high-oxidation CoOx![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 56,57 were proved to be critical active species upon the reorganization of Co–MOF materials in alkaline media for the OER. Unfortunately, the active species of MOF materials in neutral or near-neutral solutions forming upon the reorganization of MOFs into new structures in situ remain unknown. This is because of the lack of sufficient hydroxyl ions (OH−) taking part in the reaction to form active sites, as in alkaline solutions. However, MOFs usually show crystal-to-crystal transformations triggered by external stimulation, such as foreign molecules and solvent vapours.58 The process can provide a direct insight into potential changes to their structures, presenting a unique opportunity to better understand the real active species of OER in neutral electrolytes. Therefore, it is essential to determine real active species of reorganizable MOFs during the OER under neutral or near-neutral conditions through a crystal-to-crystal transformation.
56,57 were proved to be critical active species upon the reorganization of Co–MOF materials in alkaline media for the OER. Unfortunately, the active species of MOF materials in neutral or near-neutral solutions forming upon the reorganization of MOFs into new structures in situ remain unknown. This is because of the lack of sufficient hydroxyl ions (OH−) taking part in the reaction to form active sites, as in alkaline solutions. However, MOFs usually show crystal-to-crystal transformations triggered by external stimulation, such as foreign molecules and solvent vapours.58 The process can provide a direct insight into potential changes to their structures, presenting a unique opportunity to better understand the real active species of OER in neutral electrolytes. Therefore, it is essential to determine real active species of reorganizable MOFs during the OER under neutral or near-neutral conditions through a crystal-to-crystal transformation.
In this work, we report two 2D MOF materials with reconfigurable structures [Co(HCi)2(H2O)2·2DMF]n (denoted as Co–Ci-2D) and [Ni(HCi)2(H2O)2·2DMF]n (denoted as Ni–Ci-2D) (H2Ci = 1H-indazole-5-carboxylic acid, DMF = N,N-dimethyl-formamide) along with their OER performances in neutral solution (0.1 M PBS solution). Single crystals of Co–Ci-2D and Ni–Ci-2D were synthesized by the self-assembly reaction of metal salts (Co(NO3)2·6H2O or Ni(NO3)2·6H2O) and organic ligand (H2Ci·HCl) in DMF/H2O/CH3OH mixed solvents. It is found that Co–Ci-2D shows a good OER performance with 3.93 mA cm−2 at 1.8 V vs. RHE and stability. Interestingly, a structural transformation of Co–Ci-2D was observed to undertake an original 2D network to form a mononuclear coordination compound [Co(HCi)2(H2O)4] (denoted as Co–Ci-mono-A) and its isomer (denoted as Co–Ci-mono-B) under the attack of water molecules, which have been confirmed by single-crystal X-ray diffraction (SC-XRD), powder X-ray diffraction (PXRD), thermogravimetric analysis (TGA) and infrared (IR) spectroscopy. More importantly, both the mononuclear coordination compound (Co–Ci-mono-A) and its isomer (Co–Ci-mono-B) were also identified as key active species by PXRD, TGA, XPS (X-ray photoelectron spectroscopy) and XAS (X-ray absorption spectroscopy) measurements for the OER under neutral conditions, which is different from cobalt oxo-/hydroxide active species in a neutral solution.54,55 Similar to Co–Ci-2D, a mononuclear Ni coordination compound and its isomer also serve as key active sites in the OER process. In addition, the NiHPO4 precipitate also participates in the OER reaction as an active component due to significant precipitation between Ni2+ produced by the decomposition of Ni mononuclear compounds and HPO42− from the PBS solution. Finally, it should be emphasized that the distinct OER activities for Co–Ci-2D and Ni–Ci-2D are mainly associated with the different coordination bond strength caused by bond length difference.
2. Results and discussion
2.1. Single-crystal structure of Co–Ci-2D and Ni–Ci-2D
To obtain the structural information of Co–Ci-2D and Ni–Ci-2D, we performed SC-XRD measurements on their single crystals. The SC-XRD results revealed that Co–Ci-2D and Ni–Ci-2D are isostructural, crystallizing in the same monoclinic space group P21/n. As a representative example, the structure of Co–Ci-2D is discussed here in detail. The asymmetric unit of Co–Ci-2D is made up of half one Co(II) cation, one partially deprotonated H2Ci anion (HCi−), one water molecule and one DMF molecule. As illustrated in Fig. 1a, each Co(II) center lies in an octahedral environment surrounded by two nitrogen atoms (N2, N2c) from two different HCi− anions, two carboxylate oxygen atoms (O1a, O1b) from two different HCi− anions and two oxygen atoms (O3, O3c) from water molecules. The HCi− anions as bridging ligands link adjacent Co(II) cations via carboxylate oxygen and pyrazole nitrogen atoms to generate a highly ordered 2D network (Fig. 1b). If each Co(II) ion can be considered as a four-connected node, the 2D layer can be described as a (4, 4) net based on a topological simplification (Fig. S1a†).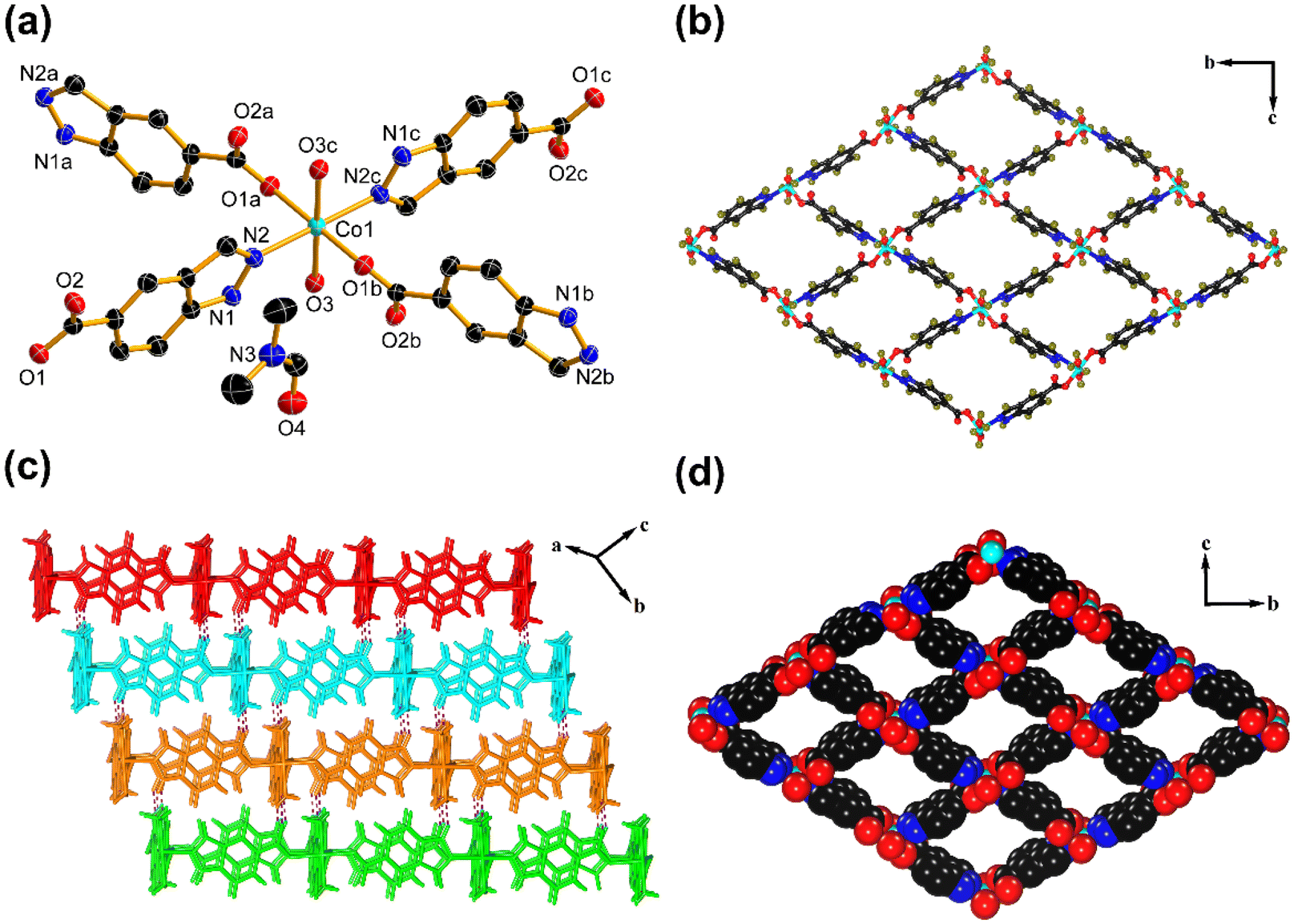 | ||
| Fig. 1 Crystalline structure of Co–Ci-2D. (a) The coordination environment in the asymmetric unit of Co–Ci-2D. Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms have been omitted for clarity. Symmetry codes : (a) 1/2 − x, 1/2 + y, 1/2 − z; (b) −1/2 + x, 3/2 − y, −1/2 + z; (c) −x, 2 − y, −z. (b) The 2D network constructed from the carboxylate–oxygen and nitrogen atoms of the HCi− ligand in Co–Ci-2D. (c) The 3D supramolecular layer formed by N–H⋯O and O–H⋯N hydrogen-bonding interactions in Co–Ci-2D. (d) The cavities with dimensions of 6.9 × 8.3 Å2 in Co–Ci-2D. (Black, C; blue, N; red, O; cyan, Co; dark green, H.) | ||
Furthermore, a hydrogen atom from a pyrazole nitrogen atom donates a hydrogen bond to an uncoordinated carboxylate oxygen atom to form a N–H⋯O hydrogen bond, which joins the above 2D layer to afford a 3D supramolecular framework (Fig. 1c). It is noteworthy that 1D rhomboid channels with the effective dimensions of ca. 6.9 × 8.3 Å2 are found in the 3D supramolecular network (Fig. 1d), and are filled with guest DMF molecules that interact with coordinated water molecules through an O–H⋯O hydrogen-bonding interaction (Fig. S1b†). From PLATON program analysis,59 the solvent-accessible volume was 537.2 Å3 per unit cell, and the pore volume ratio was calculated to be 41.4% after removing these DMF molecules.
2.2. Further structural characterization
Besides the SC-XRD technique, the structures of Co–Ci-2D and Ni–Ci-2D were further confirmed by PXRD, IR, TGA and nitrogen sorption isotherm measurements. As shown in Fig. 2a and b, there is a good match between all major peak positions of the experimental PXRD patterns and those of the simulated ones based on the single-crystal data, indicating that the synthesized bulk samples of Co–Ci-2D and Ni–Ci-2D are impurity-free materials.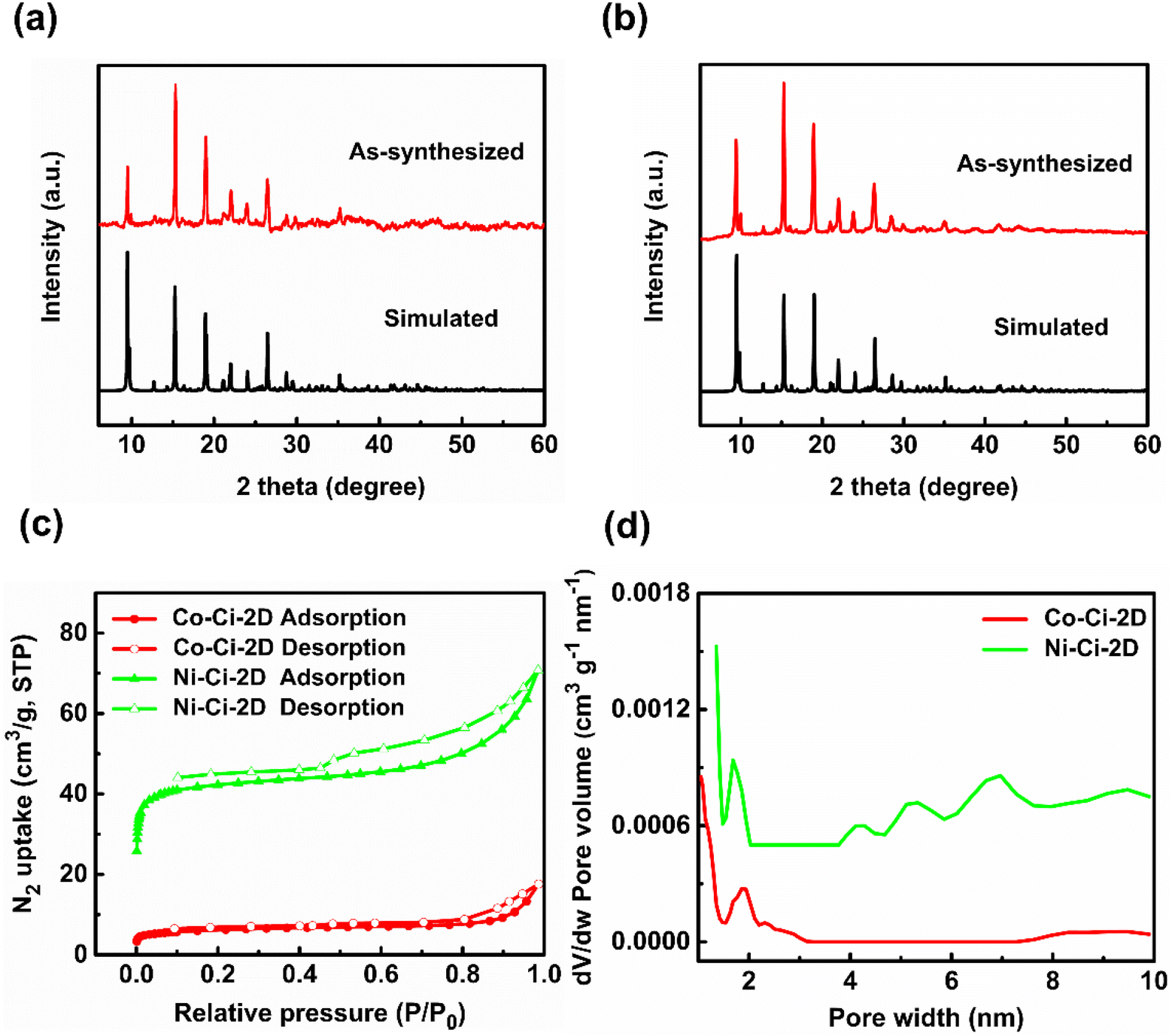 | ||
| Fig. 2 Structural characterization of Co–Ci-2D and Ni–Ci-2D. (a) The PXRD patterns for Co–Ci-2D of a simulation based on single-crystal analysis and as-synthesized bulk crystals. (b) The PXRD patterns for Ni–Ci-2D of a simulation based on single-crystal analysis and as-synthesized bulk crystals. (c) Nitrogen physisorption isotherms of Co–Ci-2D and Ni–Ci-2D at 77 K. (d) Pore size distribution of Co–Ci-2D and Ni–Ci-2D. | ||
The main functional groups and guest molecule were determined by the IR spectrum results. As presented in Fig. S2,† the broad bands centred at 3283 cm−1 for Co–Ci-2D and 3286 cm−1 for Ni–Ci-2D result from the O–H or N–H stretching vibrations from the water molecule and ligand molecule. The peaks at 1658 cm−1 for Co–Ci-2D and 1657 cm−1 for Ni–Ci-2D are attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration from the DMF molecule.60 The characteristic bands of the carboxylate group are observed at 1629 and 1546 cm−1 for the asymmetric stretch, and 1390 and 1298 cm−1 for the symmetric stretch in Co–Ci-2D. Ni–Ci-2D shows similar bands at 1627 and 1548 cm−1 for the asymmetric stretch, and 1349 and 1298 cm−1 for the symmetric stretch. The Δ values between vas(–COO−) and vs(–COO−) are 239 and 248 cm−1 for Co–Ci-2D and 233 and 250 cm−1 for Ni–Ci-2D, suggesting the presence of the carboxylate group with a monodentate mode, which is consistence with the crystal structures as described above.61,62
O vibration from the DMF molecule.60 The characteristic bands of the carboxylate group are observed at 1629 and 1546 cm−1 for the asymmetric stretch, and 1390 and 1298 cm−1 for the symmetric stretch in Co–Ci-2D. Ni–Ci-2D shows similar bands at 1627 and 1548 cm−1 for the asymmetric stretch, and 1349 and 1298 cm−1 for the symmetric stretch. The Δ values between vas(–COO−) and vs(–COO−) are 239 and 248 cm−1 for Co–Ci-2D and 233 and 250 cm−1 for Ni–Ci-2D, suggesting the presence of the carboxylate group with a monodentate mode, which is consistence with the crystal structures as described above.61,62
We examined the thermal stability of Co–Ci-2D and Ni–Ci-2D using the TGA test in the temperature range of 30–800 °C under a N2 atmosphere. TGA data reveal that Co–Ci-2D and Ni–Ci-2D remain intact below 80 °C (Fig. S3†), which is related to the presence of a strong hydrogen-bonding interaction in these materials. Then, the weight losses of 32.49% for Co–Ci-2D and 33.06% for Ni–Ci-2D occur in the range of 81–250 °C, corresponding to the release of one DMF molecule and one water molecule (calculated values: 32.34% and 32.35% respectively). Upon further heating to 300 °C, their host frameworks start to collapse, implying good thermal stability.
To appraise the porosity of Co–Ci-2D and Ni–Ci-2D, we carried out nitrogen sorption isotherm measurements on desolvated samples at 77 K. They show a type IV isotherm with an apparent hysteresis, indicating the presence of micro/mesoporous structures (Fig. 2c). The Brunauer–Emmett–Teller (BET) surface areas for Co–Ci-2D and Ni–Ci-2D are measured to be 22.9 and 165.4 m2 g−1, and the total pore volume for Co–Ci-2D and Ni–Ci-2D is 0.03 and 0.10 m3 g−1, respectively. As shown in Fig. 2d, Co–Ci-2D and Ni–Ci-2D show main micropores at 1.9 and 1.7 nm along with mesopores around 2.3 and 7.0 nm, respectively, further confirming their micro/mesoporous features (Fig. 2d).
2.3. OER performance
Various 3d transition-metal-based materials, including MOFs, can be powerful candidates for the heterogeneous catalysis of OERs.63,64 The as-prepared catalysts were used to explore the electrocatalytic performances of OERs. MOF microcrystalline powders were deposited on glassy carbon electrodes with a catalyst loading of 0.283 mg cm−2 as the working electrodes. Commercial IrO2 catalyst was widely applied as a reference to further compare the OER activity under neutral conditions due to its low overpotential and excellent performance, i.e. electrocatalytic activity and stability, over the whole pH range.31,65 The electrocatalytic properties of Co–Ci-2D, Ni–Ci-2D and IrO2 were measured in 0.1 M phosphate buffered saline (PBS, pH: 6.8) using a typical three-electrode setup. In neutral and near-neutral conditions, the current density at a potential37,66 of 1.8 V vs. RHE or an overpotential67,68 at 1 mA cm−2 was recommended by recent studies as a standard for evaluating performance.After the stabilization of the cyclic voltammetry (CV) scans, the linear sweep voltammetry (LSV) curves on a reversible hydrogen electrode (RHE) scale were firstly collected at a scan rate of 10 mV s−1 with IR correction. As illustrated in Fig. 3a, IrO2 exhibits an excellent activity with a current density of 7.59 mA cm−2 at 1.8 V. Co–Ci-2D is also active for the OER with a current density of 3.93 mA cm−2 at 1.8 V. In contrast, Ni–Ci-2D shows a very poor OER activity with a current density of 0.34 mA cm−2 at 1.8 V. For IrO2 and Co–Ci-2D with better OER activities, IrO2 displays an onset potential of 1.632 V and an overpotential of 410 mV at 1 mA cm−2. Co–Ci-2D shows an onset potential of 1.673 V, and an overpotential of 480 mV at 1 mA cm−2. In contrast, Ni–Ci-2D presents an onset potential of 1.720 V, and an overpotential of 570 mV at 0.34 mA cm−2 due to a very low OER catalytic activity. It is worth mentioning that the overpotential of Co–Ci-2D at 1 mA cm−2 is inferior to that of IrO2 but still superior or comparable to those of some materials documented in the literature (Fig. 3b69–73 and Table S649,66,74–84 in ESI†).
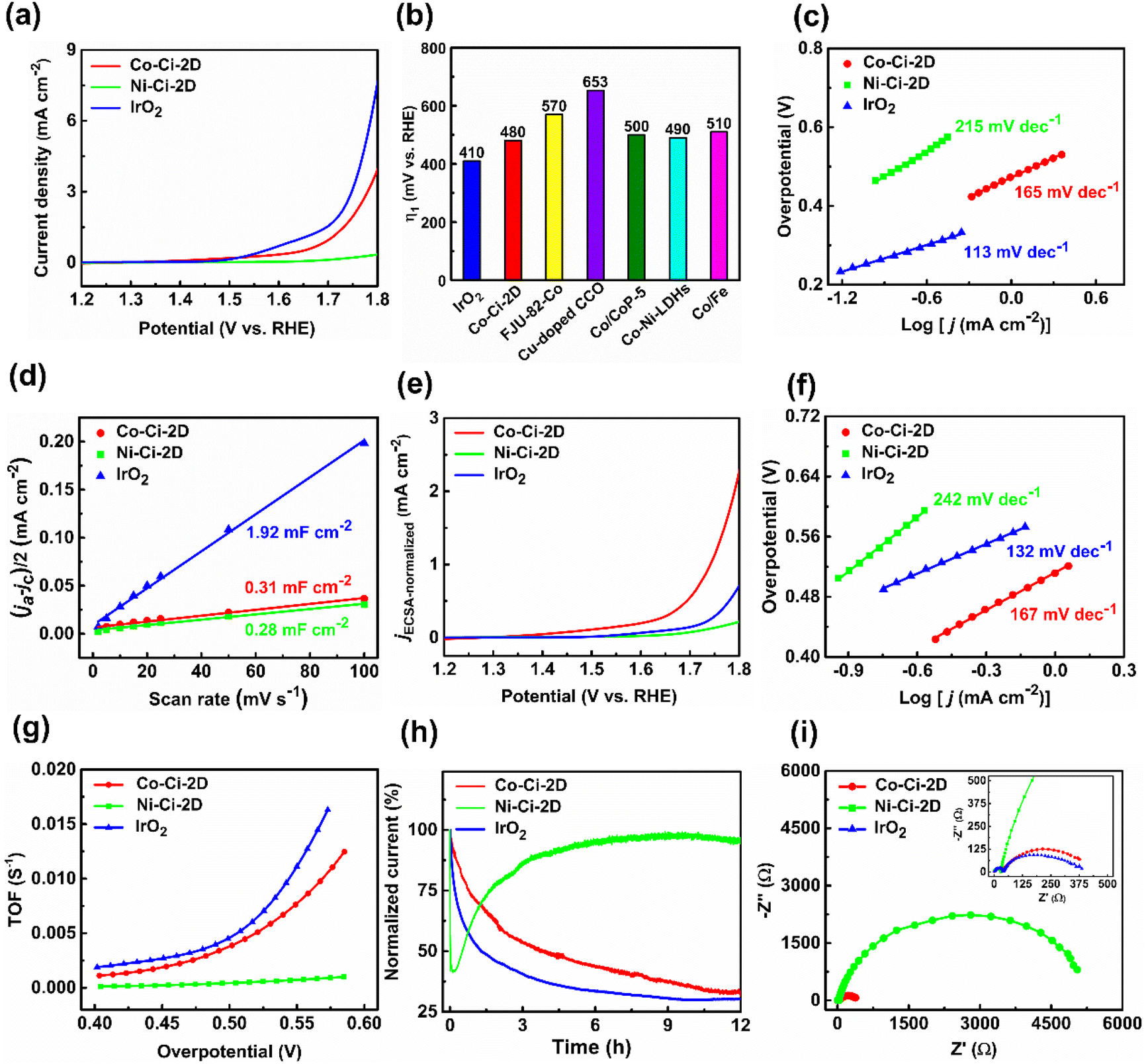 | ||
| Fig. 3 Electrocatalytic OER performances of Co–Ci-2D, Ni–Ci-2D and IrO2. (a) LSV polarization curves of Co–Ci-2D, Ni–Ci-2D and IrO2. (b) Comparison of the overpotential for Co–Ci-2D, IrO2 and other reported materials at a current density of 1 mA cm−2. (c) Tafel plots of Co–Ci-2D, Ni–Ci-2D and IrO2. (d) Double-layer capacitance results determined by the current density at different scan rates for Co–Ci-2D, Ni–Ci-2D and IrO2. (e) LSV polarization curves and (f) Tafel plots normalized by ECSA of the samples. (g) The turnover frequency (TOF) of Co–Ci-2D, Ni–Ci-2D and IrO2 at different potentials. (h) Chronoamperometry curves of Co–Ci-2D, Ni–Ci-2D and IrO2 at 1.76 V. (i) The electrochemical impedance spectroscopy (EIS) of Co–Ci-2D, Ni–Ci-2D and IrO2 at 1.76 V. | ||
The Tafel plots based on LSV curves provide an effective approach to investigating and assessing the OER kinetics of the catalyst. The Tafel slopes of IrO2, Co–Ci-2D and Ni–Ci-2D electrodes are calculated from Fig. 3c to be around 113, 165 and 215 mV per decade, respectively. It should be pointed out that the Tafel slope of Co–Ci-2D falls behind that of the commercial IrO2 but outperforms those of the benchmark catalyst RuO2 (201–245 mV per decade with a loading amount 0.20–0.80 mg cm−2 under similar conditions)85 and Ni–Ci-2D. This indicates that Co–Ci-2D shows a better OER kinetic characteristic among the measured samples.
In order to identify the intrinsic catalytic activity of the samples, the electrochemically active surface area (ECSA) of the catalysts was investigated, which is directly proportional to the double-layer capacitance (Cdl). The relevant CV curves of measured samples with different sweep scan rates in the non-faradaic region were collected to calculate the Cdl value (Fig. S7†).69 As seen from Fig. 3d, IrO2 has a significantly higher Cdl value (1.92 mF cm−2) than those of Co–Ci-2D (0.31 mF cm−2) and Ni–Ci-2D (0.28 mF cm−2). For the as-synthesized sample, Co–Ci-2D produces a larger Cdl value than Ni–Ci-2D, implying more exposed active sites in Co–Ci-2D. On the basis of the obtained Cdl values, the LSV curves and Tafel plots were normalized by ECSA to illustrate the intrinsic activity (see ESI† for detailed procedures). After ECSA normalization, Co–Ci-2D shows the highest electrocatlytic activity (an overpotential of 511 mV at 1 mA cm−2, Fig. 3e) and a lower Tafel slope (167 mV per decade, Fig. 3f), indicating that Co–Ci-2D possesses a good intrinsic performance.
Moreover, the electrocatalytic OER activity of the above catalysts under neutral conditions was further evaluated by the turnover frequency (TOF). As depicted in Fig. 3g, the calculated TOFs of IrO2 and Co–Ci-2D are significantly higher than that of Ni–Ci-2D (see ESI† for further details). For example, the TOF values of IrO2, Co–Ci-2D and Ni–Ci-2D are 0.0045, 0.0038 and 4.3 × 10−4 s−1 at the overpotential of 500 mV, respectively, based on the active center. These results confirm the better electrocatalytic activity of IrO2 and Co–Ci-2D for OERs than that of Ni–Ci-2D.
Besides the electrocatalytic activity, the OER durability is another vital factor for electrocatalysts for practical applications. The OER durability of Co–Ci-2D, Ni–Ci-2D and IrO2 was assessed by chronoamperometry curves at a constant applied potential of 1.76 V vs. RHE (i.e. a current density of ca. 1 mA cm−2 for Co–Ci-2D without IR compensation) in 0.1 M PBS solution. As displayed in Fig. 3h, both Co–Ci-2D and IrO2 exhibit current degradation due to the mass loss of the catalysts under a large potential of 1.76 V for a 12 h testing time. After 12 h of continuous operation, IrO2 displays 30.2% of the current density, but Co–Ci-2D maintains 31.6% of the current density, suggesting that Co–Ci-2D is more stable than IrO2 during the OER procedure. Unlike Co–Ci-2D and IrO2, Ni–Ci-2D shows first decreasing and then increasing trends in current density, which could be related to new active species generated from the decomposition of the Ni–Ci-2D. Surprisingly, 95.9% of the current density remains at the end of 12 h of operation.
Finally, electrochemical impedance spectroscopy (EIS) was applied to measure the charge transfer kinetics. As shown in Fig. 3i, the charge transfer resistances of Co–Ci-2D and IrO2 are 4.05 × 102 and 3.15 × 102 Ω, respectively, which are obviously lower than that of Ni–Ci-2D (5.30 × 103 Ω) at the overpotential of 530 mV. The result suggests an enhanced charge transfer efficiency in Co–Ci-2D and IrO2, which is consistent with the results of the Tafel plots.
On the whole, Co–Ci-2D shows much greater electrocatalytic activity toward the OER than that of Ni–Ci-2D under the same conditions, by about one order of magnitude. Most importantly, it exhibits a durability superior to commercial IrO2 in a neutral solution.
2.4. Water-triggered structural transformation of MOFs
Upon exposure to liquid water or water vapour, some MOFs exhibit crystal-to-crystal structural transformation, leading to the changes in their structures and functionalities.86,87 For catalytic reactions, identifying the active intermediate generated by the structural changes of MOFs will help an in-depth understanding of the detailed catalytic mechanism.88 We tested the structural transformation of Co–Ci-2D and Ni–Ci-2D by soaking the corresponding samples in deionized (DI) water at room temperature. Indeed, both Co–Ci-2D and Ni–Ci-2D exhibit structural changes upon exposure to water as suggested by the PXRD patterns of the water-treated samples (Fig. S13†). The above fact indicates that some new compounds were generated through the reaction of MOFs (Co–Ci-2D or Ni–Ci-2D) and excess water, and that those compounds become the electrocatalytic active sites during the OER. After a careful analysis of the synthetic conditions of these MOF samples, it is found that a small amount of water is actually involved in the reaction to form Co–Ci-2D and Ni–Ci-2D. The amount of water as a reagent plays a significant role in forming compounds with different structures. Hence, we have grown new single crystals by increasing the amount of water in the starting reaction system. Ultimately, we obtained successfully a new single crystal of a different cobalt compound after optimizing the synthetic conditions.The structure of the harvested single crystal (denoted as Co–Ci-mono-A) is a 0 D mononuclear motif in which the Co centre could be described as an octahedral coordination geometry with four oxygen atoms from four water molecules and two nitrogen atoms from the HCi− ligand (Fig. 4a). These isolated units are further linked by O–H⋯O hydrogen bonds between coordinated water molecules and uncoordinated carboxylate groups together with N–H⋯O hydrogen bonds from the pyrazole rings and uncoordinated carboxylate groups into a 3D supramolecular framework (Fig. 4b).
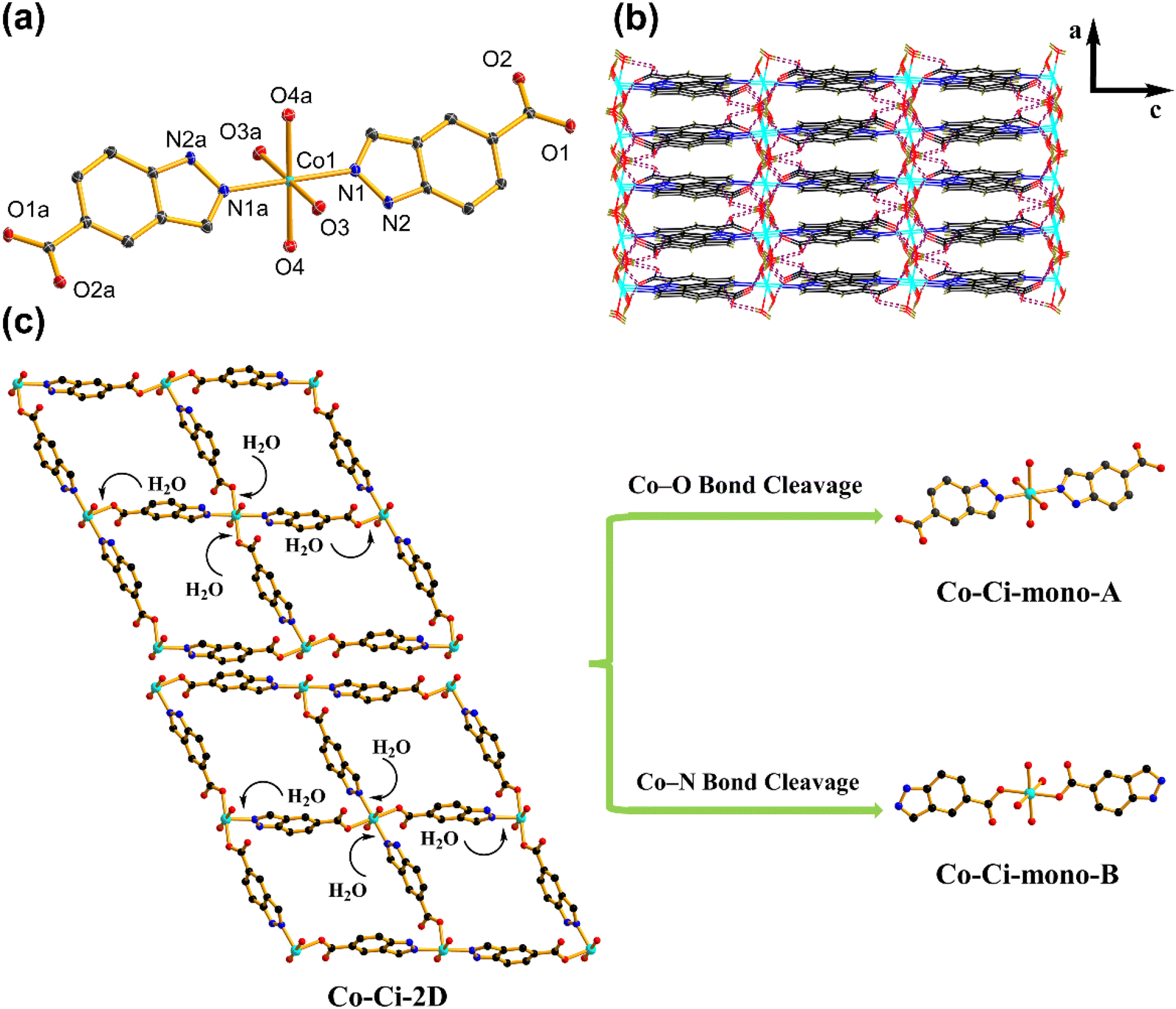 | ||
| Fig. 4 Crystalline structure of Co–Ci-mono-A and structural transformation between the 2D MOF and mononuclear compounds. (a) The asymmetric unit and coordination environment of Co–Ci-mono-A. Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms have been omitted for clarity. Symmetry code: 2 − x, 2 − y, −z. (b) The 3D supramolecular layer formed by N–H⋯O and O–H⋯N hydrogen-bonding interactions in Co–Ci-mono-A. (c) Schematic illustration of Co–Ci-2D changed from the 2D layer to mononuclear compounds under the attack of water molecules. Hydrogen atoms have been omitted for clarity. (Black, C; blue, N; red, O; cyan, Co; dark green, H.) | ||
Furthermore, Co–Ci-mono-A was structurally characterized by PXRD, TGA and IR. The PXRD pattern measured for Co–Ci-mono-A is in good agreement with the simulated one from single-crystal data, indicating the high purity of the as-prepared sample (Fig. S12a†). The results of TGA revealed that there is no noticeable weight loss in Co–Ci-mono-A before 125 °C (Fig. S12b†), higher than that of Co–Ci-2D, suggesting that Co–Ci-mono-A is more stable than Co–Ci-2D in terms of the thermodynamic properties. Subsequent weight loss in the range of 126–203 °C is caused by the elimination of coordinated water molecules (observed, 14.81%; calculated, 15.89%). The IR spectrum of Co–Ci-mono shows a broad band at around 3344 and 3240 cm−1, attributable to the O–H or N–H stretching vibrations from coordinated water molecules and ligand molecules (Fig. S12c†). The characteristic bands of the carboxylate group appear at 1634 and 1539 cm−1 for vas (–COO−) along with 1483 and 1388 cm−1 for vs (–COO−). The Δ values (vas–vs) are both 151 cm−1, indicating that the carboxylate group is an ionic structure rather than an uncoordinated mode, different from that in Co–Ci-2D.61,62
After understanding the structure of Co–Ci-mono-A, we need to solve the next question, which is to explore the relationship among Co–Ci-mono-A, water-treated MOF and Co–Ci-2D samples by means of various characterization techniques. It is easy to observe from the PXRD pattern that some peak positions of the sample of Co–Ci-2D that was water-treated for 2 h are coincident with those of Co–Ci-mono-A, while the rest are new unidentified peaks (Fig. 5a). This implies that there is a mixture of different structures in the Co–Ci-2D water-treated for 2 h. As illustrated in Fig. 5b, the TGA curve revealed that the mixture lost 15.23% of its total weight below 190 °C, similar to the calculated weight loss of Co–Ci-mono-A (15.89%) under the same conditions. In addition, analogue TGA profiles are detected in both the above mixture and the sample of Co–Ci-mono-A upon further heating to 800 °C. All the TGA results show that the rest of the sample of Co–Ci-2D water-treated for 2 h and Co–Ci-mono-A have the same molecular formula, and they could be a pair of isomeric compounds. For the abovementioned mixture, the absorption bands associated with the O–H or N–H stretching vibrations from coordinated water molecules and ligand molecules are displayed at 3326 and 3285 cm−1 (Fig. S15†). The characteristic absorption bands in the spectrum at 1630 and 1545 cm−1, together with 1504, 1382 and 1305 cm−1, are respectively assigned to the asymmetric stretching vibration and the symmetric stretching vibration of the carboxylate group. The Δ values between the two vibrations are 126, 163, 240 and 325 cm−1, respectively, indicating that carboxylate groups with ionic and monodentate modes coexist in the mixture.61,62 Combining the TGA and IR results with the structures of Co–Ci-2D and Co–Ci-mono-A, the analyses are as follows. (1) We can learn from the structural relationship between Co–Ci-2D and Co–Ci-mono-A that Co–Ci-mono-A was generated by the cleavage of two Co–O bonds formed between the Co ion and oxygen atom from the carboxylate group in Co–Ci-2D, where two foreign water molecules participate in the above reaction from two sides in terms of the lower steric-hindrance requirement. According to this idea, we could hypothesize that two Co–N bonds between Co ion and nitrogen atom from the ligand can also be broken to yield the other mononuclear compound in the same way. (2) The isomers determined by TGA further confirmed that the other component is a mononuclear compound similar to Co–Ci-mono-A. (3) The primarily functional groups and different coordination modes monitored by IR spectra also reveal that the cleavage of Co–N or Co–O bonds indeed occurred in Co–Ci-2D, leading to the formation of two mononuclear compounds with the same composition. Based on the analytical results, the other compound (denoted as Co–Ci-mono-B) in the mixture can be speculated, and expressed the structure shown in Fig. 4c.
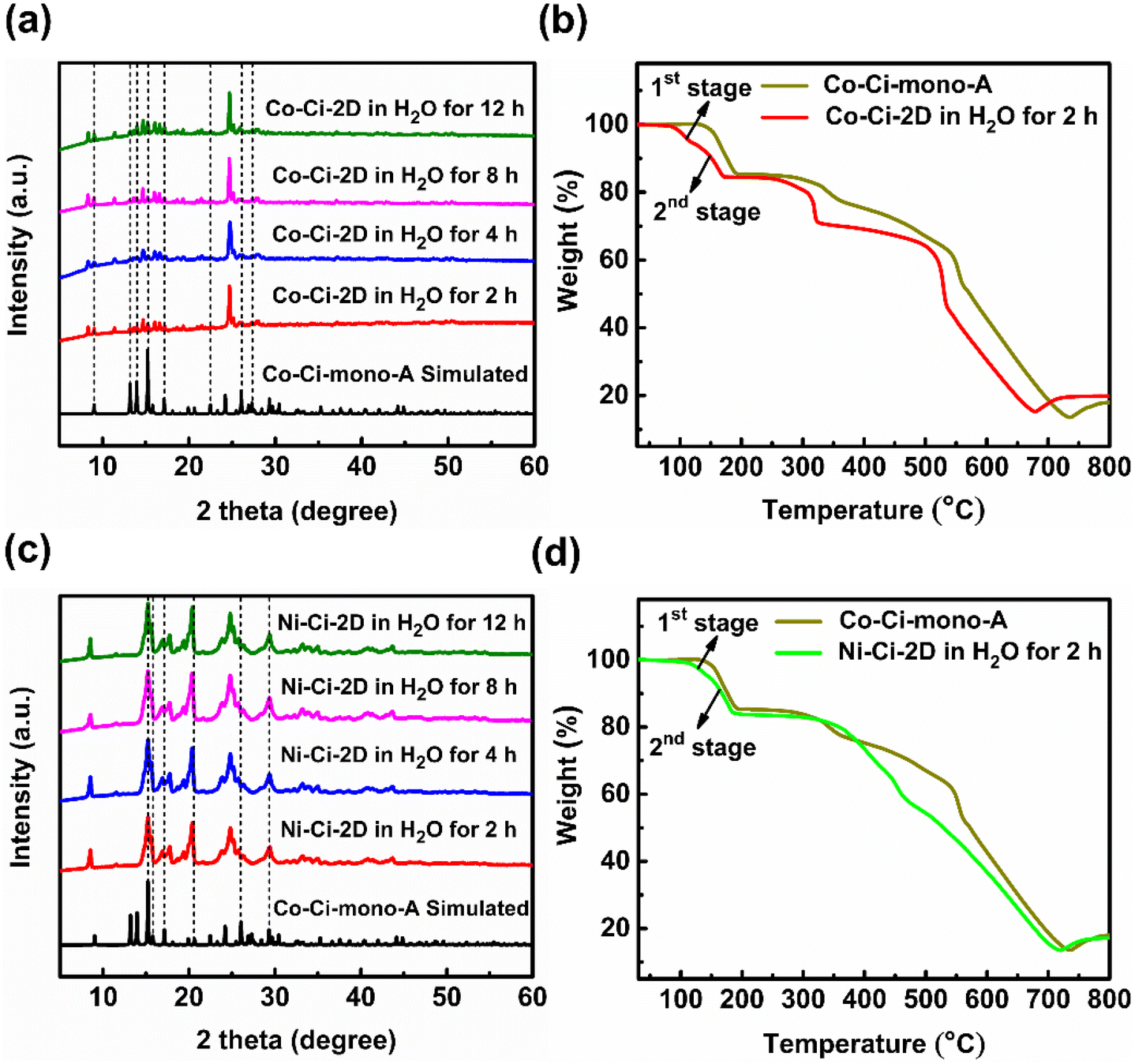 | ||
| Fig. 5 Structural characterization of water-treated Co–Ci-2D and Ni–Ci-2D samples. (a) PXRD patterns for Co–Ci-2D soaked in water and for Co–Ci-mono-A of a simulation based on single-crystal analysis. (b) Thermogravimetric curves for Co–Ci-mono-A and Co–Ci-2D soaked in water for 2 h. (c) PXRD patterns for Ni–Ci-2D soaked in water and for Co–Ci-mono-A of a simulation based on single-crystal analysis. (d) Thermogravimetric curves for Co–Ci-mono-A and Ni–Ci-2D soaked in water for 2 h. | ||
Moreover, we investigated the water resistance and thermal stability of the abovementioned mixture treated with water. Surprisingly, the mixture can also survive in water for at least 10 h due to the appearance of unchanged PXRD patterns of samples water-treated for 2–12 h (Fig. 5a). In other words, the water-treated samples, including Co–Ci-mono-A, are more stable than Co–Ci-2D in water. Compared with the first weight loss of Co–Ci-mono-A, the corresponding weight loss of the sample soaked in water for 2 h is split into two sections, in which one is 5.28% from 75 °C to 124 °C and the other is 9.95% between 125 and 190 °C (Fig. 5b). Regarding the weight loss of Co–Ci-mono-A, the first stage results from the weight loss of Co–Ci-mono-B and the second stage belongs to that of Co–Ci-mono-A, which corresponds to the loss of their coordinated water molecules. The TGA results further present that Co–Ci-mono-A is more thermally stable than Co–Ci-mono-B. It is perhaps important to point out that a weight ratio of Co–Ci-mono-A and Co–Ci-mono-B can be estimated from the TGA curve of the sample soaked in water for 2 h, which is about 1.88/1, signifying that Co–Ci-mono-A is a main product in the water-treated sample. Besides, it is also noteworthy that the results of samples soaked in water for 4 h, 8 h, and 12 h are analogous to that for 2 h, which was determined by identical PXRD patterns, TGA curves and IR spectra (Fig. 5a and S13–S15†).
The structure, composition and stability of the Ni–Ci-2D samples soaked in water are similar to those of water-treated Co–Ci-2D due to the isostructural characteristics of Co–Ci-2D and Ni–Ci-2D, which were characterized and verified by similar PXRD patterns (Fig. 5c and S13†), TGA curves (Fig. 5d and S14†) and IR spectra (Fig. S15†). Compared with the corresponding Co–Ci-2D, the PXRD patterns slightly broaden in water-soaked Ni–Ci-2D samples, which is possibly indicative of a partial reduction of the sample crystallinity. Additionally, the weight ratio of Ni–Ci-mono-A (similar to the structure of Co–Ci-mono-A) and Ni–Ci-mono-B (similar to that of Co–Ci-mono-B) is higher than that of Co–Ci-mono-A and Co–Ci-mono-B for the sample immersed in water for 2 h, which is about is 3.08/1.
2.5. Active species and OER performance
The exploration of active sites in MOF-based catalysts in a real operational environment is crucial to unravel the enhanced OER activity due to possible exchange reactions between ligands and reactive molecules or ions during the OER process. Thus, the samples of Co–Ci-2D and Ni–Ci-2D were initially characterized by PXRD and TGA measurements to identify their active components after being soaked in 0.1 M PBS solution for 2 h. Similar to water-treated samples, the PXRD pattern of treated Co–Ci-2D shows that diffraction peaks of Co–Ci-mono-A were dominant together with other peaks of Co–Ci-mono-B (Fig. 6a and S16a†). The TGA also further indicates that Co–Ci-mono-A (weight percentage: about 70.03%) was a major ingredient in the treated samples (Fig. S16b†). We traced the active species by investigating the structural and chemical evolution of the as-synthesized MOFs after OER tests. Fig. 6a shows PXRD patterns after the OER measurement for 2 h. The broad peaks at 26.49° and 55.45° are assigned to carbon paper,89 and some relatively strong peaks after 30°, such as 31.83° and 45.55°, are related to NaCl from the PBS solution. Beyond that, the rest of the PXRD pattern aligns with characteristic peaks simulated from Co–Ci-mono-A, suggesting that Co–Ci-mono-B has decomposed and residual Co–Ci-mono-A still remains during the OER procedure. In other words, both Co–Ci-mono-B and Co–Ci-mono-A act as active species to catalyze the OER reaction. Like Co–Ci-2D, Ni–Ci-mono-A, whose structure is similar to that of Co–Ci-mono-A, is a main component of the PBS-treated sample of Ni–Ci-2D, which is supported by the fact that an analogical PXRD pattern is observed (Fig. 6b and S17a†) and a weight percentage of Ni–Ci-mono-A calculated from the TGA curve is about 70.45% (Fig. S17b†). In addition, a similar PXRD result is also found in the sample of Ni–Ci-2D measured after the OER test (Fig. 6b), indicating that Ni–Ci-mono-B and Ni–Ci-mono-A are key components during OER. | ||
| Fig. 6 Structural characterization before and after OER measurements. PXRD patterns of Co–Ci-2D (a) and Ni–Ci-2D (b) soaked in 0.10 M PBS solution and after the OER for 2 h. High-resolution XPS spectra of Co 2p (c) and P 2p (e) regions for Co–Ci-2D together with Ni 2p (d) and P 2p (f) regions for Ni–Ci-2D before and after OER. (g) Co K-edge XANES experimental spectra of Co–Ci-2D and PBS-treated Co–Ci-2D. (h) Ni K-edge XANES experimental spectra of Ni–Ci-2D, PBS-treated Ni–Ci-2D, Ni–Ci-2D after the OER for 2 h and Ni(OH)2. (i) The magnitude of the EXAFS FT k2-weight Co K-edge spectra of Co–Ci-2D and PBS-treated Co–Ci-2D. (j) The magnitude of EXAFS FT k2-weight Ni K-edge spectra of Ni–Ci-2D, PBS-treated Ni–Ci-2D and Ni–Ci-2D after the OER for 2 h. (k) Chronoamperometry curves of Co–Ci-2D at 1.76 V. A is Co–Ci-mono-A, B is Co–Ci-mono-B, and B + A is Co–Ci-mono-B and Co–Ci-mono-A. (l) Chronoamperometry curves of Ni–Ci-2D at 1.76 V. | ||
Furthermore, XPS was used to reveal the chemical composition and the state of samples of Co–Ci-2D and Ni–Ci-2D before and after the OER. In pristine Co–Ci-2D, the high-resolution Co 2p spectrum has two main peaks at 781.4 and 797.1 eV together with two distinct satellite peaks at 785.9 and 803.6 eV (Fig. 6c), resulting from the characteristic peaks of the Co 2p3/2 and Co 2p1/2 core levels, respectively.54 For the as-prepared Ni–Ci-2D, the characteristic spin–orbit peaks of Ni2+ are observed in the high-resolution Ni 2p spectrum, in which major peaks of Ni 2p3/2 and Ni 2p1/2 are situated at 856.1, and 873.6 eV and the corresponding satellite peaks are located at 862.1 and 880.1 eV, respectively (Fig. 6d).54 After an OER for 2 h, the binding energy of Co 2p3/2 in the sample of Co–Ci-2D shows no obvious shift (Fig. 6c), unveiling that the surface coordination environment of the treated sample does not change significantly, and it is still a six-coordinated structure, consistent with the crystalline structure of Co–Ci-mono-A. In contrast, the peak of Ni 2p3/2 (856.7 eV) in the sample of Ni–Ci-2D after the OER test shifts to higher energy (ΔE = 0.6 eV, Fig. 6d), revealing that Ni2+ ions bound to atoms with larger electronegativity and subsequently a structural reorganization occurred on the sample surface.90 More startlingly, the high-resolution P 2p spectrum was detected in Co–Ci-2D and Ni–Ci-2D after the OER reactions, originating from the PBS solution (Fig. 6e and f). There are two main peaks at 133.0 eV of P 2p3/2 and 134.2 eV of P 2p1/2 for Co–Ci-2D together with 133.2 eV of P 2p3/2 and 134.5 eV of P 2p1/2 for Ni–Ci-2D after OER, respectively, which can be attributable to HPO4−.91 For Co–Ci-2D, the peak at 133.0 eV of P 2p3/2 is in accordance with the previously reported P 2p3/2 spectrum of Na2HPO4 (133.0 eV), indicating the presence of Na2HPO4, which comes from the PBS solution.92 For Ni–Ci-2D, the P 2p3/2 peak at 133.2 eV is similar to the result of the reported NiHPO4 (133.4 eV),93 which demonstrates that NiHPO4 was produced on the surface in the OER process. Why were different species, such as Na2HPO4 and NiHPO4, detected in Co–Ci-2D and Ni–Ci-2D? The solubility product constant (Ksp) could provide a useful tool for solving the above problem. Based on the theory of solubility product, amorphous CoHPO4 and NiHPO4 belong to the same type of precipitate in which the compound with the lower Ksp prefers to result in the formation of precipitate and vice versa. CoHPO4 has a bulk Ksp of 2.57 × 10−7, which is significantly larger than that of NiHPO4 (1.45 × 10−34).94,95 Thus, Ni2+ ions decomposed from Ni–Ci-2D react with HPO42− ions to generate the precipitate of NiHPO4 and further deposit on the surface of the sample during the OER procedure. Conversely, it is difficult for Co–Ci-2D to form CoHPO4 precipitation due to the presence of a higher Ksp. The decomposed Co2+ ions dissolve in the PBS solution to yield compounds with unidentified structures.
Additionally, to identify the local coordination environments of metal centres in the measured samples, we performed element-selective XAS experiments at the Co and Ni K-edge, including X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopy. As shown in Fig. 6g, pristine Co–Ci-2D and Co–Ci-2D soaked in the PBS solution show very similar XANES profiles, further indicating that the octahedral coordination geometry of the Co2+ centre remains unchanged in the original and treated samples.96Fig. 6i presents the Fourier transforms (FTs) of the EXAFS spectra (R space) of the cobalt samples. The prominent peaks are observed at 1.63 Å (phase-uncorrected distance) for fresh Co–Ci-2D and 1.60 Å (phase-uncorrected distance) for soaked Co–Ci-2D, respectively, attributable to the first coordination shell of Co–O or Co–N in the structures. From the fresh Co–Ci-2D to the PBS-treated Co–Ci-2D (i.e. the mixture of major Co–Ci-mono-A and minor Co–Ci-mono-B), the Co–O/N peaks display a tiny low-R shift of −0.03 Å, indicating that the bond length of Co–O/N becomes shorter in the soaked sample.97 Furthermore, XANES features of Ni–Ci-2D treated in the PBS solution and Ni–Ci-2D after OER are also identical to that of the original Ni–Ci-2D (Fig. 6h), suggesting that the Ni2+ centre is well maintained in an octahedral configuration. Notably, the Ni–Ci-2D sample after the OER also displays an obvious peak at 8364 eV similar to that of Ni(OH)2 (8365 eV), implying, besides coordination compounds, the presence of interactions between the Ni2+ ion and HO− group that arise from the HPO42− ion, likely to be the generation of amorphous NiHPO4 on the surface of the nickel sample after the OER test, which is consistent with XPS measurements. In the EXAFS Fourier transforms (R space), (Fig. 6j), the major peaks of pristine Ni–Ci-2D, PBS-soaked Ni–Ci-2D and Ni–Ci-2D after the OER are located at 1.63, 1.56 and 1.72 Å (phase-uncorrected distance), respectively, which belong to the Ni–N or Ni–O scattering path. Compared with the PBS-immersed sample of Co–Ci-2D, the main peak of the PBS-immersed sample of Ni–Ci-2D shifts to a lower R-value of 0.07 Å, showing that the mean bond length of Ni–O/N gets shorter in the nickel sample treated with the PBS solution.97 In contrast, the dominant peak of Ni–Ci-2D after the OER moves toward an obvious high R-position of 0.09 Å. The phenomenon can be explained by the Ni–O/N bond length of Ni–Ci-2D after the OER being elongated, which is possibly related to the formation of abundant NiHPO4 precipitate.98
Using the combination of SC-XRD, PXRD, TGA, XPS and XANES techniques (Table S5†), we can conclude that Co–Ci-mono-A and Co–Ci-mono-B as active species were involved in the OER process for Co–Ci-2D, and Ni–Ci-mono-B, Ni–Ci-mono-A as well as NiHPO4 precipitate took part in the OER reaction for Ni–Ci-2D. The results are also verified by the chronoamperometry curves of Co–Ci-2D and Ni–Ci-2D (Fig. 6k and l), giving the whole picture of the OER process. For Co–Ci-2D, its time-dependent current density shows a dramatic drop from 0 h to 0.82 h, a moderate decrease in the range of 0.82–2 h, and a gradual decline after 2 h based on the curve with different slopes. Before the electrocatalytic OER, Co–Ci-2D has decomposed into Co–Ci-mono-A and Co–Ci-mono-B because of the pre-treatment of the sample in water (Fig. S20a†). As demonstrated by the TGA measurements, Co–Ci-mono-B is more active than Co–Ci-mono-A in terms of thermodynamic properties. Additionally, PXRD measurements show that Co–Ci-mono-A is only detected in the residual sample after OER for 2 h. As a result, it is quite possible that Co–Ci-mono-B, as a major active component, firstly participates in the OER and proceeds to decompose, corresponding to a dramatic drop in the time-dependent current density curve. And then, the rest Co–Ci-mono-B and Co–Ci-mono-A act as active species to catalyze the OER reaction and further decompose, showing a moderate decrease. Finally, the rest of the Co–Ci-mono-A, as only one catalytic component, completes the eventual OER reaction, presenting a gradual decline due to its higher thermodynamic stability leading to a slow decomposition. Unlike Co–Ci-2D, Ni–Ci-2D exhibits a significant degradation and a slow reduction for 0–0.125 h, along with a continuous increase after 0.125 h in the time-dependent current density curves. Like Co–Ci-2D, the significant and slow reduction of the initial current density is ascribed to the decomposition of Ni–Ci-mono-B and Ni–Ci-mono-A, which act as critical active species to take part in the OER reaction (Fig. S20b†). After the decomposition, NiHPO4 precipitate was generated promptly and coated on the surface of the sample. Next, the newly formed NiHPO4 precipitated together with the remaining Ni–Ci-mono-B and Ni–Ci-mono-A as active species coexist in the sample to accelerate the further OER reaction. As Ni–Ci-mono-B and Ni–Ci-mono-A persistently convert into increasing NiHPO4 precipitate, this NiHPO4 precipitate with a higher OER activity99 continually becomes a major active component in the OER process to the end, ultimately obtaining a continuous increase in current density after 0.125 h.
Finally, it is noteworthy that the current density of Co–Ci-2D is significantly higher than that of Ni–Ci-2D, about one order of magnitude (Table 1). What factors mainly lead to Co–Ci-2D and Ni–Ci-2D exhibiting remarkably distinct OER performances? We can find the answer from the different R-shifts of the EXAFS Fourier transforms of PBS-treated Co–Ci-2D and Ni–Ci-2D, along with the discrepancy in average bond lengths of the fresh Co–Ci-2D and Ni–Ci-2D from SC-XRD results. The SC-XRD analysis confirms that the average bond length of 2.106 Å in the original Co–Ci-2D is longer than that of 2.072 Å in the original Ni–Ci-2D. In addition, compared with the PBS-treated sample of Ni–Ci-2D (−0.07 Å), a smaller negative R-shift can be observed in the PBS-treated sample of Co–Ci-2D (−0.03 Å), representing longer Co–O/N bond lengths than that of Ni–O/N and therefore weaker or looser coordination bonds in the PBS-treated Co–Ci-2D sample than that of the treated Ni–Ci-2D sample. The longer bond length could facilitate the catalyst molecule to bind or eliminate other molecules, becoming more active and possessing a high catalytic performance.100 Therefore, it can be concluded that bond strength differences resulting from the difference in coordination bond length for the treated samples are mainly responsible for the extraordinary variety of OER performances between Co–Ci-2D and Ni–Ci-2D.
| Sample | Current densitya (mA cm−2) | Peak shift in R spaceb (Å) | Feature of coordination bond | Solubility product constant (Ksp)c | Active speciesd |
|---|---|---|---|---|---|
| a Measured from the linear sweep voltammetry (LSV) curves. b Obtained from the Fourier transforms (FTs) of the EXAFS spectra (R space, phase-uncorrected distance). c Originated from the reference. d Identified from SCXRD, PXRD, TGA, XPS and XANES measurements. | |||||
| Co–Ci-2D | 3.93 | −0.03 | Weaker, loose | 2.57 × 10−7 | Co–Ci-mono-A and Co–Ci-mono-B |
| Ni–Ci-2D | 0.34 | −0.07 | Stronger, tight | 1.45 × 10−34 | Ni–Ci-mono-A, Ni–Ci-mono-B and NiHPO4 |
3. Conclusions
In summary, two isostructural 2D MOFs were successfully synthesized through a self-assembly method and applied for an electrocatalytic oxygen evolution reaction in a neutral solution, in which Co–Ci-2D shows a better OER activity than those of the reported materials in the literature. The MOFs in this study convert into the corresponding mononuclear coordination compounds in a water solution. PXRD, TGA, XPS and XANES measurements indicate that a mononuclear compound (Co–Ci-mono-A) and its isomer (Co–Ci-mono-B) act as key active species to participate in the OER of Co–Ci-2D. For Ni–Ci-2D, besides Ni–Ci-mono-A and Ni–Ci-mono-B, similar to the structures of the cobalt sample, NiHPO4 is also identified as an active component in the OER procedure. Moreover, the different OER performances between Co–Ci-2D and Ni–Ci-2D are possibly caused by the alterations in coordination bond strength induced by differences in bond length after being treated by the PBS solution, as evidenced by EXAFS Fourier transforms and SC-XRD analysis. This work provides a novel insight into understanding the active species of the catalyst in neutral solution and opens up exciting new opportunities for the design and preparation of MOF-based electrocatalysts for OER.4. Experimental methods
4.1. Materials
All reagents and chemicals were commercially available and employed without further purification. H2Ci·HCl was purchased from Shanghai Yingrui Biopharma Co., Ltd. IrO2 (99.9%), 0.10 M phosphate buffered saline (PBS, pH = 6.8, 25 °C) from Sigma-Aldrich, carbon black (acetylene, 100% compressed, 99.9+%) and Nafion solution (5 wt%) from Alfa Aesar. Co(NO3)2·6H2O, Ni(NO3)2·6H2O, DMF and MeOH were purchased from Guangdong Guanghua Sci-Tech Co., Ltd.4.2. Experimental detail
4.3. General characterization
The powder X-ray diffraction (PXRD) patterns were determined by a PAnalytical X'Pert Pro X-ray diffractometer equipped with graphite monochromatized Cu Kα radiation (λ = 1.5418 Å) in the 2θ range from 5° to 60° at a scanning speed of 5° min−1. The infrared (IR) samples were prepared as KBr pellets. The corresponding spectra were recorded on a Nicolet 5700 FTIR spectrometer in the 4000–400 cm−1 region. Thermogravimetric analysis (TGA) was carried out under flowing nitrogen in the temperature range of 30–800 °C with a heating rate of 10 °C min−1 by using a NETZSCH TG 209 F3 thermal analyzer. X-ray photoelectron spectroscopy (XPS) analysis was performed on a Thermo Scientific Nexsa XPS system. All of the binding energies were calibrated with respect to the C 1s peak at 284.6 eV as the standard.4.4. Single crystal X-ray crystallography
Suitable single crystals of Co–Ci-2D, Ni–Ci-2D, and Co–Ci-mono-A were selected for indexing and intensity data collection on a Bruker SMART APEX II CCD diffractometer equipped with graphite monochromatized Mo Kα radiation (λ = 0.71073 Å) at 150(2) K. All structures were solved by direct methods with SHELXTL 6.1 and refined by the full-matrix least-squares procedure based on F2 with SHELXL-2014.102,103 All non-hydrogen atoms were allowed the anisotropic thermal motion. The hydrogen atoms were located in their calculated positions and refined by a riding model. Crystallographic data and details of refinements for Co–Ci-2D, Ni–Ci-2D and Co–Ci-mono are listed in Table S1.† Selected bond lengths and angles are given in Table S2.† The corresponding H-bond data are shown in Table S3.† CCDC 2160558–2160560 contains the supplementary crystallographic data for this paper.†4.5. Gas adsorption
The nitrogen adsorption–desorption experiments were carried out on the crystalline samples of Co–Ci-2D and Ni–Ci-2D at 77 K with liquid nitrogen on a Micromeritics Tri-star instrument. Before the measurement, the samples were degassed at 423 K overnight under vacuum. The surface area and pore size distribution were estimated from Brunauer–Emmett–Teller (BET) theory and two-dimensional non-local density functional theory model for heterogeneous surfaces (2D-NLDFT), respectively.4.6. X-Ray absorption spectroscopy sample preparation and analysis
Measurements were performed at Diamond Light Source (I20-EDE) and European Synchrotron Radiation Facility (BM28) in transmission. Samples (∼12 mg) were mixed with cellulose (∼1–2 mg), pressed into 5 mm diameter pellets, and sealed into Kapton tape for the XAS measurements. The data were normalized to the incident intensity and processed using the Athena software package.104 A derivative (E0) value of 8345 eV and 7710 eV corresponds to the first inflection point of the absorption Ni K-edge and Co K-edge, respectively. The measured EXAFS spectra were fitted after subtracting the pre-edge and post-edge background from the overall absorption and then normalizing to the edge step. Subsequently, the χ(k) data of 2.1 to 9.8 A−1 (for Ni edge samples) and 2.8 to 10.7 A−1 (for Co edge samples) were used for the Fourier transformed data using a Henning window (dk = 1.0 Å−1) to separate the extended X-ray absorption fine structure (EXAFS) contributions from different coordination shells around the absorbing Co or Ni atom. Quantitative structural parameters around central atoms were fitted using ARTEMIS software.4.7. Electrocatalytic measurement
The inks used in the electrocatalysis were prepared by the following procedure. Firstly, the samples of Co–Ci-2D, Ni–Ci-2D and IrO2 were milled into a fine powder with a mortar and pestle for 30 min. Then, for Co–Ci-2D and Ni–Ci-2D, 8 mg catalyst and 2 mg carbon black were dispersed in 1400 μL H2O, 528 μL EtOH and 72 μl Nafion (5 wt%) and ultrasonicated for 10 min to form a homogeneous ink. Carbon black was introduced into MOF materials to improve their conductivity.105 In contrast, for IrO2, the ink was prepared by dispersing 8 mg catalyst in a similar mixture solution in the absence of carbon black with further ultrasonication over the same period. Finally, 5 μL ink was slowly dropped onto a polished glassy carbon electrode surface (diameter = 3 mm, geometrical area = 0.07065 cm2 in) at 200 rmp and left to dry at a 700 rpm rate at least for 1 h at room temperature. Electrochemical studies were performed on an RDE system using AUTOLAB PGSTAT 101, and a MULTI AUTOLAB M101 (CH Instruments) with a typical three-electrode cell using a glassy carbon electrode with a diameter of 3 mm as the working electrode, Ag/AgCl (3 M) electrode as the reference electrode and a carbon rod as the counter-electrode. OER tests were carried out in O2-saturated 0.10 M PBS (pH = 6.80) at room temperature. All the potentials are calibrated to the reversible hydrogen electrode (RHE) according to the following equation ERHE = Eexp + EAg/AgCl (3.0 M) + 0.059 pH = Eexp + 0.21 + 0.059 pH and the current density is normalized to the effective geometrical surface area. The continuous cyclic voltammetry (CV) cycling was conducted at a scan rate of 10 mV s−1 from 1.19 to 1.82 V versus RHE. The linear sweep voltammetry (LSV) data were recorded after 15 CV cycles in the PBS medium with a sweep rate of 10 mV s−1 so as to acquire relatively stable curves. All LSV data were compensated with iR-correction due to voltage drop losses caused by the solution resistance. The stability tests were carried out using the chronopotentiometric/chronoamperometric measurements. Electrochemical impedance spectroscopy (EIS) measurements were carried out in a frequency range from 106 Hz to 0.1 Hz by applying an AC voltage with 1.15 V amplitude. The double-layer capacitance (Cdl) was obtained by measuring CV curves in a potential window nearly without faradaic processes at different scan rates of 2, 5, 10, 15, 20, 25, 50, and 100 mV s−1.Author contributions
Xiaoqiang Liang: Investigation, resources, conceptualization, formal analysis, data curation, writing – original draft, writing – review & editing. Sen Wang: Investigation, resources, software. Jingyu Feng: Investigation, resources, data curation, methodology, writing – review & editing, visualization, validation. Zhen Xu: Investigation, data curation, formal analysis, validation. Zhenyu Guo: Investigation, data curation, validation. Hui Luo: Investigation, software. Feng Zhang: Investigation, formal analysis, validation, software. Chen Wen: Methodology, formal analysis, software, project administration. Lei Feng: Software, visualization. Chengan Wan: Methodology, project administration, funding acquisition. Maria-Magdalena Titirici: Validation, writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful for the financial support of the National Natural Science Foundation of China (21401147), the Natural Science Basic Research Plan in Shaanxi Province of China (2019JM-397), and the Special Project of Talent Cultivation in the West Region of China Scholarship Council (201808615077). The authors thank Monica Amboage from I20-EDE beamline (SP28663) at Diamond Light Source and Paul Thompsom from BM28 beamline (MA5108) at the European Synchrotron Radiation Facility to support and assist the measurements.References
- S. Park, Y. Y. Shao, J. Liu and Y. Wang, Oxygen electrocatalysts for water electrolyzers and reversible fuel cells: status and perspective, Energy Environ. Sci., 2012, 5, 9331–9344 RSC.
- N. A. Burton, R. V. Padilla, A. Rose and H. Habibullah, Increasing the efficiency of hydrogen production from solar powered water electrolysis, Renewable Sustainable Energy Rev., 2021, 135, 110255 CrossRef CAS.
- C. Zhang and K. Huang, A comprehensive review on the development of solid-state metal–air batteries operated on oxide-Ion chemistry, Adv. Energy Mater., 2021, 11, 2000630 CrossRef CAS.
- G. Li, X. Wang, J. Fu, J. Li, M. G. Park, Y. Zhang, G. Lui and Z. W. Chen, Pomegranate-inspired design of highly active and durable bifunctional electrocatalysts for rechargeable metal–air batteries, Angew. Chem., Int. Ed., 2016, 55, 4977–4982 CrossRef CAS PubMed.
- Y. Wang, D. F. R. Diaz, K. S. Chen, Z. Wang and X. C. Adroher, Materials, technological status, and fundamentals of PEM fuel cells – A review, Mater. Today, 2020, 32, 178–203 CrossRef CAS.
- S. Ma, M. Lin, T.-E. Lin, T. Lan, X. Liao, F. Maréchal, J. Van herle, Y. Yang, C. Dong and L. Wang, Fuel cell-battery hybrid systems for mobility and off-grid applications: A review, Renewable Sustainable Energy Rev., 2021, 135, 110119 CrossRef.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Combining theory and experiment in electrocatalysis: Insights into materials design, Science, 2017, 355, eaad4998 CrossRef PubMed.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Design of electrocatalysts for oxygen- and hydrogen-involving energy conversion reactions, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- H. Jin, C. Guo, X. Liu, J. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S. Z. Qiao, Emerging two-dimensional nanomaterials for electrocatalysis, Chem. Rev., 2018, 118, 6337–6408 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and S.-H. Yang, A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. García de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. De Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Homogeneously dispersed multimetal oxygen-evolving catalysts, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- X. Li, X. Hao, A. Abudula and G. Guan, Nanostructured catalysts for electrochemical water splitting: Current state and prospects, J. Mater. Chem. A, 2016, 4, 11973–12000 RSC.
- C. C. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, Benchmarking hydrogen evolving reaction and oxygen evolving reaction electrocatalysts for solar water splitting devices, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and S.-H. Yang, Synthesis and activities of rutile IrO2 and RuO2 nanoparticles for oxygen evolution in acid and alkaline solutions, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- E. Antolini, Iridium as catalyst and cocatalyst for oxygen evolution/reduction in acidic polymer electrolyte membrane electrolyzers and fuel cells, ACS Catal., 2014, 4, 1426–1440 CrossRef CAS.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Electrocatalysis for the oxygen evolution reaction: recent development and future perspectives, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- T. Zhang, Y. Zhu and J. Y. Lee, Unconventional noble metal-free catalysts for oxygen evolution in aqueous systems, J. Mater. Chem. A, 2018, 6, 8147–8158 RSC.
- W. Shen, J. Jin, Y. Hu, Y. Hou, J. Yin, Z. Ma, Y.-Q. Zhao and P. Xi, Surface chlorine doped perovskite–type cobaltate lanthanum for water oxidation, Chin. J. Catal., 2022, 43, 1485–1492 CrossRef CAS.
- Y. Huang, R. Yang, G. Anandhababu, J. Xie, J. Lv, X. Zhao, X. Wang, M. Wu, Q. Li and Y. Wang, Cobalt/iron (oxides) heterostructures for efficient oxygen evolution and benzyl alcohol oxidation reactions, ACS Energy Lett., 2018, 3, 1854–1860 CrossRef CAS.
- F. Song and X. Hu, Exfoliation of layered double hydroxides for enhanced oxygen evolution catalysis, Nat. Commun., 2014, 5, 4477 CrossRef CAS PubMed.
- Y. Yu, J. Zhou and Z. Sun, Novel 2D transition-metal carbides: ultrahigh performance electrocatalysts for overall water splitting and oxygen reduction, Adv. Funct. Mater., 2020, 30, 2000570 CrossRef CAS.
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Atomically dispersed iron–nitrogen species as electrocatalysts for bifunctional oxygen evolution and reduction reactions, Angew. Chem., Int. Ed., 2017, 56, 610–614 CrossRef CAS PubMed.
- M. Ledendecker, S. K. Calderýn, C. Papp, H.-P. Steinrîck, M. Antonietti and M. Shalom, The synthesis of nanostructured Ni5P4 films and their use as a non-noble bifunctional electrocatalyst for full water splitting, Angew. Chem., Int. Ed., 2015, 54, 12361–12365 CrossRef CAS PubMed.
- X. Guo, X. Wan, Q. Liu, Y. Li, W. Li and J. Shui, Phosphated IrMo bimetallic cluster for efficient hydrogen evolution reaction, eScience, 2022, 2, 304–310 CrossRef.
- Y. Liu, Q. Li, R. Si, G. D. Li, W. Li, D. P. Liu, D. Wang, L. Sun, Y. Zhang and X. Zou, Coupling sub-nanometric copper clusters with quasi-amorphous cobalt sulfide yields efficient and robust electrocatalysts for water splitting reaction, Adv. Mater., 2017, 29, 1606200 CrossRef PubMed.
- J. Yin, J. Jin, M. Lu, B. Huang, H. Zhang, Y. Peng, P. Xi and C. H. Yan, Iridium single atoms coupling with oxygen vacancies boosts oxygen evolution reaction in acid media, J. Am. Chem. Soc., 2020, 142, 18378–18386 CrossRef CAS PubMed.
- Y. Yang, Y. Yang, Z. Pei, K.-H. Wu, C. Tan, H. Wang, L. Wei, A. Mahmood, C. Yan, J. Dong, S. Zhao and Y. Chen, Recent progress of carbon-supported single-atom catalysts for energy conversion and storage, Matter, 2020, 3, 1442–1476 CrossRef.
- M. D. Symes and L. Cronin, Materials for water splitting, in Materials for a sustainable future, RSC Publishing, Cambridge (UK), 2012, pp. 592–614 Search PubMed.
- M. Chen, Y. Wu, Y. Han, X. Lin, J. Sun, W. Zhang and R. Cao, An iron-based film for highly efficient electrocatalytic oxygen evolution from neutral aqueous solution, ACS Appl. Mater. Interfaces, 2015, 7, 21852–21859 CrossRef CAS PubMed.
- Y. Dong, C. W. Oloman, E. L. Gyenge, J. Su and L. Chen, Transition metal based heterogeneous electrocatalysts for the oxygen evolution reaction at near-neutral pH, Nanoscale, 2020, 12, 9924–9934 RSC.
- P. Li, R. Zhao, H. Chen, H. Wang, P. Wei, H. Huang, Q. Liu, T. Li, X. Shi, Y. Zhang, M. Liu and X. Sun, Recent advances in the development of water oxidation electrocatalysts at mild pH, Small, 2019, 15, 1805103 CrossRef PubMed.
- L. Zeng, K. Sun, Y. Chen, Z. Liu, Y. Chen, Y. Pan, R. Zhao, Y. Liu and C. Liu, Neutral-pH overall water splitting catalyzed efficiently by a hollow and porous structured ternary nickel sulfoselenide electrocatalyst, J. Mater. Chem. A, 2019, 7, 16793–16802 RSC.
- K. Xu, H. Cheng, L. Liu, H. Lv, X. Wu, C. Wu and Y. Xie, Promoting active species generation by electrochemical activation in alkaline media for efficient electrocatalytic oxygen evolution in neutral media, Nano Lett., 2017, 17, 578–583 CrossRef CAS PubMed.
- S. Anantharaj, J. Kennedy and S. Kundu, Microwave-initiated facile formation of Ni3Se4 nanoassemblies for enhanced and stable water splitting in neutral and alkaline media, ACS Appl. Mater. Interfaces, 2017, 9, 8714–8728 CrossRef CAS PubMed.
- L. Cui, D. Liu, S. Hao, F. Qu, G. Du, J. Liu, A. M. Asiri and X. Sun, In situ electrochemical surface derivation of cobalt phosphate from a Co(CO3)0.5(OH)·0.11H2O nanoarray for efficient water oxidation in neutral aqueous solution, Nanoscale, 2017, 9, 3752–3756 RSC.
- L. Xie, R. Zhang, L. Cui, D. Liu, S. Hao, Y. Ma, G. Du, A. M. Asiri and X. Sun, High-performance electrolytic oxygen evolution in neutral media catalyzed by a cobalt phosphate nanoarray, Angew. Chem., Int. Ed., 2017, 56, 1064–1068 CrossRef CAS PubMed.
- P. Chen, K. Xu, T. Zhou, Y. Tong, J. Wu, H. Cheng, X. Lu, H. Ding, C. Wu and Y. Xie, Strong-coupled cobalt borate nanosheets/graphene hybrid as electrocatalyst for water oxidation under both alkaline and neutral conditions, Angew. Chem., Int. Ed., 2016, 55, 2488–2492 CrossRef CAS PubMed.
- Y. Dong, C. W. Oloman, E. L. Gyenge, J. Su and L. Chen, Transition metal based heterogeneous electrocatalysts for oxygen evolution reaction at near-neutral pH, Nanoscale, 2020, 12, 9924–9934 RSC.
- J. Shan, Y. Zheng, B. Shi, K. Davey and S.-Z. Qiao, Regulating electrocatalysts via surface and interface engineering for acidic water electrooxidation, ACS Energy Lett., 2019, 4, 2719–2730 CrossRef CAS.
- X. Li, X. Hao, A. Abudula and G. Guan, Nanostructured catalysts for electrochemical water splitting: Current state and prospects, J. Mater. Chem. A, 2016, 4, 11973–12000 RSC.
- B. Kim, T. Kim, K. Lee and J. Li, Recent advances in transition metal phosphide electrocatalysts for water splitting under neutral pH condition, ChemElectroChem, 2020, 7, 3578–3589 CrossRef CAS.
- L. Kong, M. Zhong, W. Shuang, Y. Xu and X. H. Bu, Electrochemically active sites inside crystalline porous materials for energy storage and conversion, Chem. Soc. Rev., 2020, 49, 2378–2407 RSC.
- P.-Q. Liao, J.-Q. Shen and J.-P. Zhang, Metal–organic frameworks for electrocatalysis, Coord. Chem. Rev., 2018, 373, 22–48 CrossRef CAS.
- Q. Liang, J. Chen, F. Wang and Y. Li, Transition metal-based metal-organic frameworks for oxygen evolution reaction, Coord. Chem. Rev., 2020, 424, 213488 CrossRef CAS.
- Q. Shi, S. Fu, C. Zhu, J. Song, D. Du and Y. Lin, Metal–organic frameworks-based catalysts for electrochemical oxygen evolution, Mater. Horiz., 2019, 6, 684–702 RSC.
- H. B. Aiyappa, J. Masa, C. Andronescu, M. Muhler, R. A. Fischer and W. Schuhmann, MOFs for electrocatalysis: from serendipity to design strategies, Small Methods, 2019, 3, 1800415 CrossRef.
- D. Zhu, M. Qiao, J. Liu, T. Tao and C. Guo, Engineering pristine 2D metal–organic framework nanosheets for electrocatalysis, J. Mater. Chem. A, 2020, 8, 8143–8170 RSC.
- J. Liu, D. Zhu, C. Guo, A. Vasileff and S.-Z. Qiao, Design strategies toward advanced MOF-derived electrocatalysts for energy-conversion reactions, Adv. Energy Mater., 2017, 7, 1700518 CrossRef.
- W. Wang, X. Xu, W. Zhou and Z. Shao, Recent progress in metal-organic frameworks for applications in electrocatalytic and photocatalytic water splitting, Adv. Sci., 2017, 4, 1600371 CrossRef PubMed.
- L. Jiao, Y. Wang, H. L. Jiang and Q. Xu, Metal–organic frameworks as platforms for catalytic applications, Adv. Mater., 2018, 30, 1703663 CrossRef PubMed.
- Y. Yang, Y. Yang, Y. Liu, S. Zhao and Z. Tang, Metal-organic frameworks for electrocatalysis: beyond their derivatives, Small Sci., 2021, 1, 2100015 CrossRef CAS.
- S. Zhao, Y. Yang and Z. Tang, Insight into structural evolution, active site and stability of heterogeneous electrocatalysts, Angew. Chem., Int. Ed., 2022, 61, e202110186 CrossRef CAS PubMed.
- S. Zhao, C. Tan, C. T. He, P. An, F. Xie, S. Jiang, Y. Zhu, K.-H. Wu, B. Zhang, H. Li, J. Zhang, Y. Chen, S. Liu, J. Dong and Z. Tang, Structural transformation of highly active metal–organic framework electrocatalysts during the oxygen evolution reaction, Nat. Energy, 2020, 5, 881–890 CrossRef CAS.
- C.-P. Wang, H.-Y. Liu, G. Bian, X. Gao, S. Zhao, Y. Kang, J. Zhu and X. H. Bu, Metal-layer assisted growth of ultralong quasi-2D MOF nanoarrays on arbitrary substrates for accelerated oxygen evolution, Small, 2019, 15, 1906086 CrossRef CAS PubMed.
- A. Bucci, S. S. Mondal, V. Martin-Diaconescu, A. Shafir and J. Lloret-Fillol, Cobalt amide imidate imidazolate frameworks as highly active oxygen evolution model materials, ACS Appl. Energy Mater., 2019, 2, 8930–8938 CrossRef CAS PubMed.
- J. Jiang, L. Huang, X. Liu and L. Ai, Bioinspired cobalt–citrate metal–organic framework as an efficient electrocatalyst for water oxidation, ACS Appl. Mater. Interfaces, 2017, 9, 7193–7201 CrossRef CAS PubMed.
- S. Dou, C.-L. Dong, Z. Hu, Y.-C. Huang, J.-L. Chen, L. Tao, D. Yan, D. Chen, S. Shen, S. Chou and S. Wang, Atomic-scale CoOx species in metal–organic frameworks for oxygen evolution reaction, Adv. Funct. Mater., 2017, 27, 1702546 CrossRef.
- A. Chaudhary, A. Mohammad and S. M. Mobin, Recent advances in single-crystal-to-single-crystal transformation at the discrete molecular level, Cryst. Growth Des., 2017, 17, 2893–2910 CrossRef CAS.
- A. L. Spek, Single-crystal structure validation with the program PLATON, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- C. Qin, X. Wang, Y. Qi, E. Wang, C. Hu and L. Xu, A novel two-dimensional β-octamolybdate supported alkaline-earth metal complex: [Ba(DMF)2(H2O)]2[Mo8O26]·2DMF, J. Solid State Chem., 2004, 177, 3263–3269 CrossRef CAS.
- V. Zelenak, Z. Vargova and K. Gyoryova, Correlation of infrared spectra of zinc(II) carboxylates with their structures, Spectrochim. Acta, Part A, 2007, 66, 262–272 CrossRef CAS PubMed.
- X. Zhou, J. Dong, Y. Zhu, L. Liu, Y. Jiao, H. Li, Y. Han, K. Davey, Q. Xu, Y. Zheng and S. Z. Qiao, Molecular scalpel to chemically cleave metal–organic frameworks for induced phase transition, J. Am. Chem. Soc., 2021, 143, 6681–6690 CrossRef CAS PubMed.
- S. Anantharaj and V. Aravindan, Developments and perspectives in 3D transition–metal–based electrocatalysts for neutral and near–neutral water electrolysis, Adv. Energy Mater., 2020, 10, 1902666 CrossRef CAS.
- J. Peng, W. Dong, Z. Wang, Y. Meng, W. Liu, P. Song and Z. Liu, Recent advances in 2D transition metal compounds for electrocatalytic full water splitting in neutral media, Mater. Today Adv., 2020, 8, 100081 CrossRef.
- A. Minguzzi, F.-R. F. Fan, A. Vertova, S. Rondinini and A. J. Bard, Dynamic potential–pH diagrams application to electrocatalysts for water oxidation, Chem. Sci., 2012, 3, 217–229 RSC.
- Y.-T. Xu, Z.-M. Ye, J. W. Ye, L. M. Cao, R. K. Huang, J. X. Wu, D.-D. Zhou, X.-F. Zhang, C.-T. He, J.-P. Zhang and X.-M. Chen, Non-3d metal modulation of a cobalt imidazolate framework for excellent electrocatalytic oxygen evolution in neutral media, Angew. Chem., Int. Ed., 2019, 58, 139–143 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- Y. Wu, M. Chen, Y. Han, H. Luo, X. M.-T. Zhang, X. Lin, J. Sun, L. Wang, L. Deng, W. Zhang and R. Cao, Fast and simple preparation of iron–based thin films as highly efficient water–oxidation catalysts in neutral aqueous solution, Angew. Chem., Int. Ed., 2015, 54, 4870–4875 CrossRef CAS PubMed.
- M. Zhang, Q. Lin, W. Wu, Y. Ye, Z. Yao, X. Ma, S. Xiang and Z. Zhang, Isostructural MOFs with higher proton conductivity for improved oxygen evolution reaction performance, ACS Appl. Mater. Interfaces, 2020, 12, 16367–16375 CrossRef CAS PubMed.
- Y. Zhang, B. Cui, O. Derr, Z. Yao, Z. Qin, X. Deng, J. Li and H. Lin, Hierarchical cobalt-based hydroxide microspheres for water oxidation, Nanoscale, 2014, 6, 3376–3383 RSC.
- Z.-H. Xue, H. Su, Q. Y. Yu, B. Zhang, H.-H. Wang, X.-H. Li and J.-S. Chen, Janus Co/CoP nanoparticles as efficient Mott–Schottky electrocatalysts for overall water splitting in wide pH range, Adv. Energy Mater., 2017, 7, 1602355 CrossRef.
- Y. Zhang, B. Cui, C. Zhao, H. Lin and J. Li, Co–Ni layered double hydroxides for water oxidation in neutral electrolyte, Phys. Chem. Chem. Phys., 2013, 15, 7363–7369 RSC.
- M. Aksoy, S. V. K. Nune and F. Karadas, A novel synthetic route for the preparation of an amorphous Co/Fe prussian blue coordination compound with high electrocatalytic water oxidation activity, Inorg. Chem., 2016, 55, 4301–4307 CrossRef CAS PubMed.
- J. Saha, S. Verma, R. Ball, C. Subramaniam and R. Murugavel, Compositional control as the key for achieving highly efficient OER electrocatalysis with cobalt phosphates decorated nanocarbon florets, Small, 2019, 16, 1903334 CrossRef PubMed.
- S. Gutiérrez-Tarriño, J. L. Olloqui-Sariego, J. J. Calvente, G. M. Espallargas, F. Rey, A. Corma and P. Oña-Burgos, Cobalt metal–organic framework based on layered double nanosheets for enhanced electrocatalytic water oxidation in neutral media, J. Am. Chem. Soc., 2020, 142, 19198–19208 CrossRef PubMed.
- G. Givirovskiy, V. Ruuskanen, T. Väkiparta and J. Ahola, Electrocatalytic performance and cell voltage characteristics of 1st-row transition metal phosphate (TM-Pi) catalysts at neutral pH, Mater. Today Energy, 2020, 17, 100426 CrossRef.
- Y. Zhang, B. Cui, Z. Qin, H. Lin and J. Li, Hierarchical wreath-like Au–Co(OH)2 microclusters for water oxidation at neutral pH, Nanoscale, 2013, 5, 6826–6833 RSC.
- Y. Liu, C. Xiao, M. Lyu, Y. Lin, W. Cai, P. Huang, W. Tong, Y. Zou and Y. Xie, Ultrathin Co3S4 nanosheets that synergistically engineer spin states and exposed polyhedra that promote water oxidation under neutral conditions, Angew. Chem., 2015, 127, 11383–11387 CrossRef.
- S. W. Lee, C. Carlton, M. Risch, Y. Surendranath, S. Chen, S. Furutsuki, A. Yamada, D. G. Nocera and Y. Shao-Horn, The nature of lithium battery materials under oxygen evolution reaction conditions, J. Am. Chem. Soc., 2012, 134, 16959–16962 CrossRef CAS PubMed.
- H. S. Ahn and T. D. Tilley, Electrocatalytic water oxidation at neutral pH by a nanostructured Co(PO3)2 anode, Adv. Funct. Mater., 2013, 23, 227–233 CrossRef CAS.
- J. Xie, R. Wang, J. Bao, X. Zhang, H. Zhang, S. Li and Y. Xie, Zirconium trisulfide ultrathin nanosheets as efficient catalysts for water oxidation in both alkaline and neutral solutions, Inorg. Chem. Front., 2014, 1, 751–756 RSC.
- T. W. Kim, M. A. Woo, M. Regis and K.-S. Choi, Electrochemical synthesis of spinel type ZnCo2O4 electrodes for use as oxygen evolution reaction catalysts, J. Phys. Chem. Lett., 2014, 5, 2370–2374 CrossRef CAS PubMed.
- H. Schäfer, S. M. Beladi-Mousavi, L. Walder, J. Wollschläger, O. Kuschel, S. Ichilmann, S. Sadaf, M. Steinhart, K. Küpper and L. Schneider, Surface oxidation of stainless steel: oxygen evolution electrocatalysts with high catalytic activity, ACS Catal., 2015, 5, 2671–2680 CrossRef.
- I. Zaharieva, P. Chernev, M. Risch, K. Klingan, M. Kohlhoff, A. Fischer and H. Dau, Electrosynthesis, functional, and structural characterization of a water-oxidizing manganese oxide, Energy Environ. Sci., 2012, 5, 7081–7089 RSC.
- P. Cai, J. Huang, J. Chen and Z. Wen, Oxygen–containing amorphous cobalt sulfide porous nanocubes as high–activity electrocatalysts for the oxygen evolution reaction in an alkaline/neutral medium, Angew. Chem., Int. Ed., 2017, 56, 4858–4861 CrossRef CAS PubMed.
- S.-Y. Ke and C.-C. Wang, Water-induced reversible SCSC or solid-state structural transformation in coordination polymers, CrystEngComm, 2015, 17, 8776–8785 RSC.
- C. Li, J. Chen, C. Liu and M. Du, Dynamic structural transformations of coordination supramolecular systems upon exogenous stimulation, Chem. Commun., 2015, 51, 2768–2781 RSC.
- H. Ding, H. Liu, W. Chu, C. Wu and Y. Xie, Structural transformation of heterogeneous materials for electrocatalytic oxygen evolution reaction, Chem. Rev., 2021, 121, 13174–13212 CrossRef CAS PubMed.
- X. Su, Y. Wang, J. Zhou, S. Gu, J. Li and S. Zhang, Operando spectroscopic identification of active sites in NiFe prussian blue analogues as electrocatalysts: activation of oxygen atoms for oxygen evolution reaction, J. Am. Chem. Soc., 2018, 140, 11286–11292 CrossRef CAS PubMed.
- Y. Zhan, M. Lu, S. Yang, Z. Liu and J. Y. Lee, The origin of catalytic activity of nickel phosphate for oxygen evolution in alkaline solution and its further enhancement by iron substitution, ChemElectroChem, 2016, 3, 615–621 CrossRef CAS.
- Y. Wang, W. Li, L. Zhang, X. Zhang, B. Tan, J. Hao, J. Zhang, X. Wang, Q. Hu and X. Lu, Amorphous cobalt hydrogen phosphate nanosheets with remarkable electrochemical performances as advanced electrode for supercapacitors, J. Power Sources, 2020, 449, 227487 CrossRef CAS.
- A. G. Karunanayake, C. M. Navarathna, S. R. Gunatilake, M. Crowley, R. Anderson, D. Mohan, F. Perez, C. U. Pittman Jr. and T. Mlsna, Fe3O4 nanoparticles dispersed on douglas fir biochar for phosphate sorption, ACS Appl. Nano Mater., 2019, 2, 3467–3479 CrossRef CAS.
- D. Fa and Y. Miao, Synthesis of NiHPO4–Ni(OH)2 nanowire-assembled bouquets for electrocatalytic oxidation of methanol and urea, J. Appl. Electrochem., 2020, 50, 1091–1099 CrossRef CAS.
- M. Schamel, A. Bernhardt, M. Quade, C. Würkner, U. Gbureck, C. Moseke, M. Gelinsky and A. Lode, Cu2+, Co2+ and Cr3+ doping of a calcium phosphate cement influences materials properties and response of human mesenchymal stromal cells, Mater. Sci. Eng., C, 2017, 73, 99–110 CrossRef CAS PubMed.
- J. Perrone, B. Fourest and E. Giffaut, Sorption of nickel on carbonate fluoroapatites, J. Colloid Interface Sci., 2001, 239, 303–313 CrossRef CAS PubMed.
- H. Zhang, W. Zhou, T. Chen, B. Y. Guan, Z. Li and X. W. Lou, A modular strategy for decorating isolated cobalt atoms into multichannel carbon matrix for electrocatalytic oxygen reduction, Energy Environ. Sci., 2018, 11, 1980–1984 RSC.
- H. Shang, Z. Jiang, D. Zhou, J. Pei, Y. Wang, J. Dong, X. Zheng, J. Zhang and W. Chen, Engineering a metal–organic framework derived Mn–N4–CxSy atomic interface for highly efficient oxygen reduction reaction, Chem. Sci., 2020, 11, 5994–5999 RSC.
- Y. Li, B. Wei, M. Zhu, J. Chen, Q. Jiang, B. Yang, Y. Hou, L. Lei, Z. Li, R. Zhang and Y. Lu, Synergistic effect of atomically dispersed Ni–Zn pair sites for enhanced CO2 electroreduction, Adv. Mater., 2021, 33, 2102212 CrossRef CAS PubMed.
- Z. Wang, Y. Wu, M. Cui, S. Ji, H. Wang, X. Wang, V. Linkov and R. Wang, 1D NiHPO4 nanotubes prepared using dissolution equilibrium as bifunctional electrocatalyst for high-efficiency water splitting, J. Power Sources, 2021, 513, 230543 CrossRef CAS.
- K. Jiang, M. Luo, Z. Liu, M. Peng, D. Chen, Y.-R. Lu, T.-S. Chan, F. M. F. de Groot and Y. Tan, Rational strain engineering of single-atom ruthenium on nanoporous MoS2 for highly efficient hydrogen evolution, Nat. Commun., 2021, 12, 1687 CrossRef CAS PubMed.
- A. A. Garcia-Valdiviaa, A. Garcia-Garciaa, F. Jannusb, A. Zabala-Lekuonac, J. M. Mendez-Arriagad, B. Fernandeze, M. Medina-O′donnellf, G. B. Ramirez-Rodrigueza, J. M. Delgado-Lopeza, L. M. Pastrana-Martineza, J. Cepedac, J. A. Lupianezb, F. J. Reyes-Zuritab and A. Rodriguez-Diegueza, Antiparasitic, anti-inflammatory and cytotoxic activities of 2D coordination polymers based on 1H-indazole-5-carboxylic acid, J. Inorg. Biochem., 2020, 208, 111098 CrossRef PubMed.
- G. M. Sheldrick, SHELXTL Version 6.1, Bruker Analytical X-ray Systems, Inc., Madison, Wisconsin, USA, 2000 Search PubMed.
- G. M. Sheldrick, SHELXL 2014, Program for the Refinement of Crystal Structure, University of Göttingen, Germany, 2014 Search PubMed.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: Data Analysis for X-Ray Absorption Spectroscopy Using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- X.-L. Wang, L.-Z. Dong, M. Qiao, Y. J. Tang, J. Liu, Y. Li, S.-L. Li, J.-X. Su and Y.-Q. Lan, Exploring the performance improvement of the oxygen evolution reaction in a stable bimetal–organic framework system, Angew. Chem., Int. Ed., 2018, 57, 9660–9664 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2160558–2160560. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qi02436e |
| This journal is © the Partner Organisations 2023 |