
Human mini-brains for reconstituting central nervous system disorders
You Jung
Kang
ab,
Yingqi
Xue
ab,
Jae Hee
Shin
ab and
Hansang
Cho
 *abc
*abc
aInstitute of Quantum Biophysics, Sungkyunkwan University, Suwon, Republic of Korea. E-mail: h.cho@g.skku.edu
bDepartment of Biophysics, Sungkyunkwan University, Suwon, Republic of Korea
cDepartment of Intelligent Precision Healthcare Convergence, Sungkyunkwan University, Suwon, Republic of Korea
First published on 16th January 2023
Abstract
Neurological disorders in the central nervous system (CNS) are progressive and irreversible diseases leading to devastating impacts on patients' life as they cause cognitive impairment, dementia, and even loss of essential body functions. The development of effective medicines curing CNS disorders is, however, one of the most ambitious challenges due to the extremely complex functions and structures of the human brain. In this regard, there are unmet needs to develop simplified but physiopathologically-relevant brain models. Recent advances in the microfluidic techniques allow multicellular culture forming miniaturized 3D human brains by aligning parts of brain regions with specific cells serving suitable functions. In this review, we overview designs and strategies of microfluidics-based human mini-brains for reconstituting CNS disorders, particularly Alzheimer's disease (AD), Parkinson's disease (PD), traumatic brain injury (TBI), vascular dementia (VD), and environmental risk factor-driven dementia (ERFD). Afterward, the applications of the mini-brains in the area of medical science are introduced in terms of the clarification of pathogenic mechanisms and identification of promising biomarkers. We also present expanded model systems ranging from the CNS to CNS-connecting organ axes to study the entry pathways of pathological risk factors into the brain. Lastly, the advantages and potential challenges of current model systems are addressed with future perspectives.
1. Introduction
Central nervous system (CNS) diseases are progressive and irreversible disorders, majorly found in the elderly, leading to cognitive impairment, dementia, or even loss of essential body functions.1 Such CNS disorders involve neurodegenerative conditions (Alzheimer's disease (AD), Parkinson's disease (PD)), structural impairment (traumatic brain injury, vascular dementia), and environmental factor-driven dementia.1–6 Given the fact that an aging population becomes dominant in the world, there are high demands on the development of new pharmaceutics targeting neurological disorders.7 The market for the therapeutics was valued at $80 billion in 2021 and is expected to reach 125 billion by 2029.7 Despite the efforts for the development of cures rewinding such progressive neurodegeneration, no successful clinical outcome has been accomplished compared to other therapeutic areas.8,9 The complexity of the human brain, attributed to the interconnected cross-talks among the hundreds of different neural cells, makes it extremely difficult to identify novel therapeutic targets expressed in specific cells.4,10 To decipher the comprehensive pathogenesis in the brain, a number of in vitro and in vivo models representing CNS disorders have been developed.Current in vitro models for CNS diseases highly rely on the simple 2D culture of neuronal cells expressing rare familial mutations found in the specific disease. To develop in vitro AD models, for instance, overexpression of genes associated with familiar AD (FAD) in neural cells or neuroprogenitor cells is the most popular method.11 However, the major drawback of 2D culture is that the monolayer of single- or co-cultured models could alter cellular morphology and polarity as well as local concentration of soluble factors, chaining key cellular responses in disease progression.12 In addition, these models do not involve the immune system, which plays significant roles in the pathology of CNS disorders.13–15 Alternatively, in vivo transgenic mice with mutations in Aβ precursor protein (APP) and/or presenilin (PS) have been gold-standard AD models, which recapitulate not only AD features such as amyloidosis and tauopathy but also innate immunity.11 Since such animal models mimicking human CNS diseases are founded with rare familial forms, other aspects attributed to non-familiar cases are not established yet. Even if the transgenic animal models could reflect damaged human brain structures and reactive innate immunity found in CNS disorders, there is some discrepancy in the pathology due to the different genetic backgrounds.16 To overcome these issues, human multicellular organoids have been suggested and employed to study CNS disorders.17
Among the human multicellular organoids, a brain-on-a-chip, in which neural cells along with other glial cells around neurons are co-cultured together, has been a representative CNS disease model.17,18 Microfluidics, initially employed for handling liquid dynamics, is now advantageous for cell culture due to its ability to supply continuous nutrient and remove waste.17 Furthermore, it enables the formation of stable gradients for soluble factors from cell-to-cell or cell-to-environment interactions that can induce chemotaxis.17,19 Furthermore, this compartmentalized system having multiple chambers connected through several microchannels is capable of multi-cellular culture by aligning multiple parts of brain regions with specific cells.13 Therefore, the major benefit of using compartmentalized microfluidics is the simplification of the extremely complex human brain in terms of its structures and functions depending on the location and disease status.17 In addition, microfluidic devices are very thin and optically transparent, allowing multiple robust and high-throughput analyses in parallel.17
In this review, we explore the details on brain-on-a-chip representing multiple CNS disorders, including AD, PD, TBI, vascular dementia, and environmental factor-driven dementia. Furthermore, we expand our model scopes from the CNS to CNS-connecting organ axis. We particularly focus on (1) the major characteristics of CNS diseases to be implemented into the brain chips, (2) the design and application of the brain chips, (3) advantages and potential challenges, and (4) perspectives at the end.
2. Signatures for modelling CNS disorders
This section provides essential backgrounds regarding key features to be accomplished in models for CNS disorders (Fig. 1). The evaluation criteria are summarized in Table 1.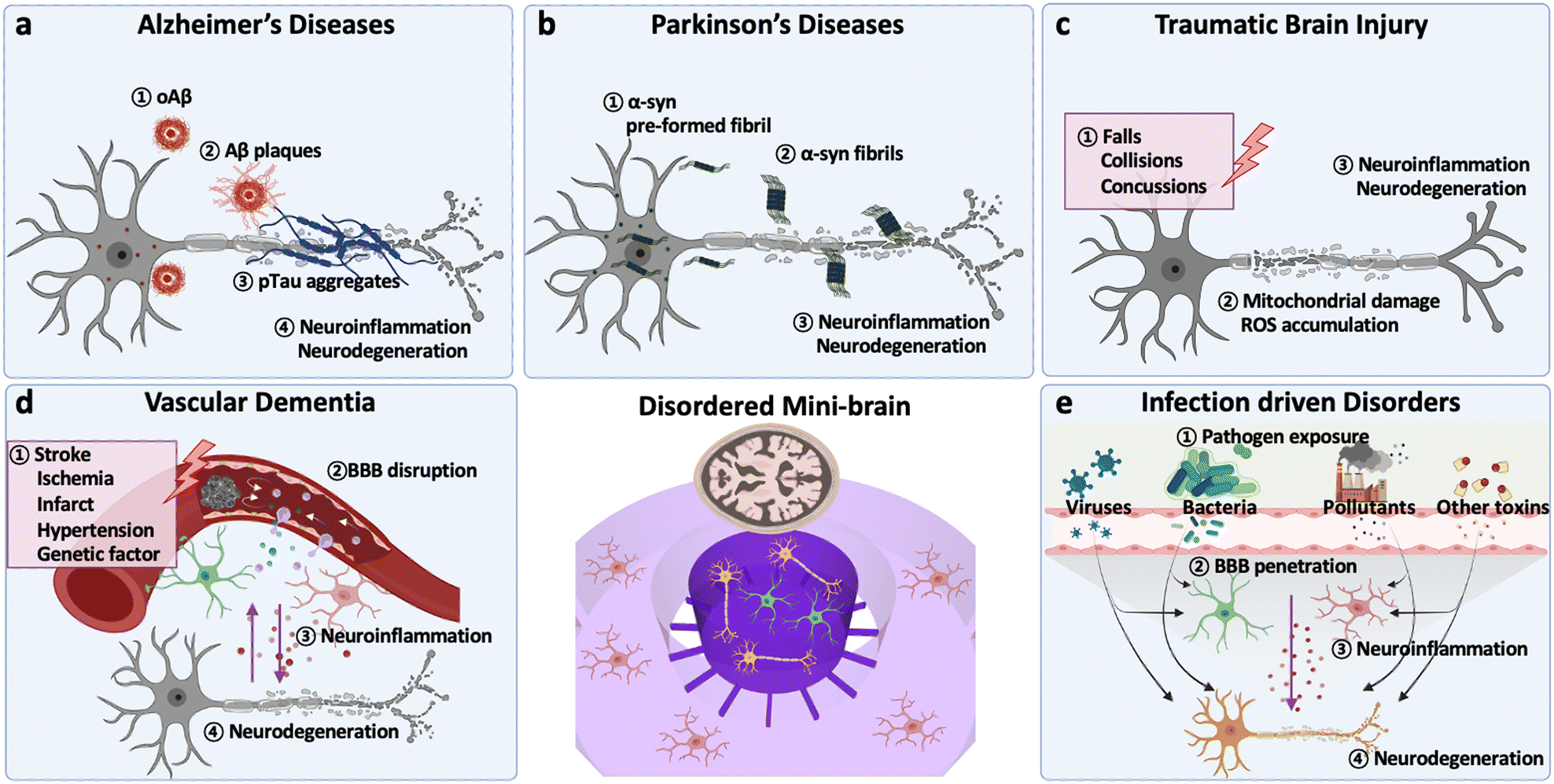 | ||
| Fig. 1 Overview of signatures in major CNS disorders. (a) Alzheimer's disease (AD): in early stages of AD, aggregation of amyloid beta forming amyloid beta oligomers (oAβ) and Aβ plaques activates neuroinflammation. At middle and late stages, deposition of hyperphosphorylated tau protein accelerates neurodegeneration such as synaptic impairment and neuronal loss. (b) Parkinson's disease (PD): deposition of α-syn PFF, an amyloid-like structure forming the aggregate, is the pivotal factor in PD development. Such misfolded α-syn aggregates are neurotoxic and induce neuroinflammation. Also, α-syn PFF can promote tau accumulation, accelerating loss of dopaminergic neurons. (c) Traumatic brain injury (TBI): neurodegeneration in TBI is mainly caused by the secondary damage after the primary external physical brain damage: falls, collisions, and concussions. The secondary damage involves mitochondrial damage and ROS accumulation in neural cells, leading to neuroinflammation and neuronal death. (d) Vascular dementia (VD): vascular dementia is a progressive cognitive disorder caused by the impairment of blood vessels allowing the uncontrolled entrance of pathogens or neurotoxins to the brain. Direct damage on the tight junction of endothelial cells or inflammatory cytokines can cause such vascular impairment and accelerate neurodegeneration. (e) Environmental risk factor-driven disorder (ERFD): exposure to pathogens including viruses, bacteria, pollutants, and neurotoxins may weaken biological barriers and increase inflammation; the inflammation can be further transmitted to the brain and increase neurological loss. Another hypothesis is that such pathogens can directly penetrate the biological barriers and reach to the brain resulting in neuroinflammation followed by neurodegeneration. | ||
| Type of disorder | Major signatures | Validated modelsref | |
|---|---|---|---|
| Alzheimer’s, diseases (AD) | Aβ hypothesis | Aβ42/Aβ40 ratio ↑ | 3xTgAD mouse,71 APP mouse,73 AD iPSC spheroids,74 chip13,75 |
| Aβ oligomers/fibrils | APP mouse,73,76 APP/PS1 spheroids,77 chip13 | ||
| Aβ plaques | 3xTgAD mouse,71 APP mouse,76 5xFAD/TEM2KO mouse,14 hAβ plaque mice78 | ||
| Aβ propagation | Chip19 | ||
| Tauopathy | Phosphorylated tau | 3xTgAD mouse,71 AD iPSC spheroids,74 APP/PS1 spheroids,77 chip13 | |
| PHF/NFT formation | rTg4510 mice,23 APP/PS1 3D AD21, chip13 | ||
| Tau propagation | Chip79,80 | ||
| Neuroinflammation | Astrocyte activation | APP/PS1 mouce,26 chip13 | |
| Microglia activation | 5xFAD/TREM2KO mouse,14 chip,13,14,19 | ||
| Neurodegeneration | Defect in neural functions | Chip,13 spheroid81 | |
| Reduction in neural cells | Chip13,75 | ||
| Parkinson's diseases (PD) | α-syn deposition | α-syn PFF/Lewy body | α-syn rat,82,83 α-syn mouse44,84,85 |
| α-syn/α-syn PFF propagation | α-syn rat,82 chip40,86–88 | ||
| Tauopathy | Tau propagation | α-syn mouse,44,85 chip22,88 | |
| Neuroinflammation | Astrocyte activation | α-syn mouse,44,84,85 chip | |
| Microglia activation | α-syn rat,82,83 α-syn mouse,84 chip40 | ||
| Neurodegeneration | Loss of dopaminergic neurons | α-syn rat,83 α-syn mouse,84,85 PINK1KO mouse,50 PINK1KO rat,52 DJ-1KO mouse,51 DJ-1KO rat,52 LRK2mut mouse,53 chip88 | |
| Motor impairment | α-syn mouse,44,84 PINK1KO rat,52 DJ-1KO rat52 | ||
| Traumatic brain injury (TBI) | Neurochemical alteration | Massive glutamate release | FPI rat,57 CCII mouse56 |
| Calcium/sodium dysregulation | FPI rat57 | ||
| ROS accumulation | FPI rat57 | ||
| Neuroinflammation | Reactive astrocytes | FPI rat,60 aged CCI mouse,62 CCII mouse,56 CCII rat63 | |
| Neurodegeneration | Axonal damage | Aged CCI mouse,62 CCII mouse,56 CCII rat,63 chip89–95 | |
| Reduction in neural cells | FPI rat,57 CCII mouse,56 chip94–97 | ||
| Vascular dementia | Vascular dysfunction | Permeability ↑ | BBB-spheroid,70 chip75,98 |
| Neurochemical alteration | Plasma protein influx | RHRSP rat,68 BCAS/ApoEKO mouse69 | |
| Neuroinflammation | Astrocyte activation | BCAS/ApoEKO mouse69 | |
| Neurodegeneration | Reduction in neural cells | BCAS/ApoEKO mouse,69 chip75,98 | |
| Environmental risk factor-driven disorders (ERFD) | Tauopathy | Phosphorylated tau | 3xTgAD mouse,71,72 chip15,99 |
| Neuroinflammation | Reactive astrocytes | 3xTgAD mouse,71 chip15,99 | |
| M1 type microglia | 3xTgAD mouse,71 chip15,99 | ||
| Neurodegeneration | Defect in neural functions | 3xTgAD mouse,72 chip15 | |
| Reduction in neural cells | Chip100,101 | ||
2.-1. Alzheimer's diseases (AD)
Alzheimer's disease (AD) is known to be the most common cause of dementia typically observed in elderly as it involves progressive and irreversible neurodegeneration (Fig. 1a).20At early stages of AD, one of evident features involves the accumulation of amyloid fibrils and formation of amyloid plaques in the patient brain, which activates the innate immune system.20 In this regard, both in vitro and in vivo AD models are overexpressed FAD mutations, such as APP mutations (KM670/671NL (Swedish), V717I (London), V717F (Indiana), etc.) and/or PS mutations (PS2-N141I, PS1-L166P, PS1-M146L, PS1-I213T, etc.).11 Previous studies have validated that AD models with APP/PS1 mutations could achieve the production of higher Aβ42/Aβ40 ratios, the major forms of Aβ inducing the formation of Aβ plaques.13,21
In addition, the formation of tau aggregates as well as neurofibrillary tangles (NFTs) in the neuronal cells is the major hallmark observed in the patient's biopsy at middle to late stages of AD.11 Tau proteins are supposed to perform roles stabilizing microtubules under normal physiological conditions. When it comes to the AD condition, the tau proteins are hyper-phosphorylated and self-aggregate into paired helical filaments (PHF) followed by NFTs. Wu et al. employed a microfluidic device with murine hippocampal and cortical neurons exposed to NFTs and validated the tau transport through the synapses between two neurons.22 Kuchibhotla et al. showed that in vivo rTg4510 mice, overexpressing the human mutant form of tau (P301L), could form NFTs in the cortical region of the brain.23 Later, a number of studies found a significant correlation between NFT formation and neurodegeneration. For instance, the MAPT transgenic system exhibited a notable axonal degradation and synaptic impairment. Furthermore, tauopathy could decrease the blood–brain barrier (BBB) integrity, increase inflammation, and exacerbate AD pathology consequently.24 Therefore, the formation of NTFs is the second characteristic to be implemented in AD models.
Thirdly, the prominent activation of innate immune cells has been reported in entire stages of AD progression. Among the innate immune cells, astrocytes and microglia, the most abundant glial cells in the CNS, play distinctive roles depending on the severity of AD.25 In early stages of AD, both astrocytes and microglia are known to retain anti-inflammatory phenotypes in response to moderate deposition of Aβ and tau aggregates in the brain.14,25 In later stages of AD, such glial cells are polarized into proinflammatory phenotypes and secrete neurotoxic soluble factors as sources of oxidative stress (hydrogen peroxide (H2O2), nitric oxide (NO)), proinflammatory cytokines (IL1β, IL6, TNFα, IFNγ) and chemokines (CCL1, CCL2, CXCL1).13,25,26 Several studies validated their neurodetrimental roles as the increase of oxidative stress as well as tau accumulation in neurons and the recruitment of other innate immune systems that further exacerbated AD pathology.26,27 Since the glial cells obtain mutations on the AD risk factors (APOE e4, TREM2, etc.) as well,14,28 AD models with astrocytes and microglia can reconstitute the unique characteristics found in sporadic AD (SAD) as well.
Lastly, significant neurodegeneration is a general hallmark found at late stages of AD.25 At this point, AD patients show significant loss of white matter connectivity and cortical thinning with clinical symptoms such as progressive decline in memory, thinking, language, and essential body functions.29 Such loss of functions is attributed to synaptic impairment followed by neuronal loss.29 Previous studies showed that the downregulation of synaptic proteins in AD conditions, such as calsyntenin, GluR, neurexin, syntaxin, and synapsin, led to failure of essential processes of neural cells such as dendritic spine assembly, postsynaptic Ca2+ signaling, synaptic transmission, synaptogenesis, and presynaptic differentiation.30 In addition, Park et al. revealed that the loss of neural functions was closely correlated to the reduction of neural population (MAP2- or Tuj1-positive cells) in in vitro AD models.13 Therefore, the validation of neurodegeneration would be a significant criterion for the development of effective AD models.
2.-2. Parkinson's diseases (PD)
Parkinson's disease (PD) is the second-most common progressive neurodegenerative disorder mainly characterized by motor symptoms of bradykinesia, rigidity, tremor and postural instability (Fig. 1b).31 Some non-motor symptoms, including neuropsychiatric symptoms, sleep disorders, autonomic symptoms and sensory symptoms, are also thought to play an important role in advanced PD and reduced quality of life.32PD is caused by the accumulation of misfolded α-synuclein (α-syn) followed by the loss of dopaminergic neurons in substantia nigra.33,34 α-syn has been identified to induce Lewy bodies in the presynaptic nerve terminals and nucleus, resulting in neurodegeneration.35–37 Growing evidence shows that α-syn fibrils can be transmitted cell-to-cell through receptor-mediated endocytosis as well as fluid-phase endocytosis, causing the recruitment of monomeric α-syn into pathological inclusions.38–40 Recent studies have confirmed Braak's hypothesis that α-syn aggregates can also be transmitted from neuron-to-neuron; yet, the specific molecular mechanism of how α-syn accumulates and leads to PD remains unsolved.41–43 In this regard, the addition of α-syn or genetic mutations/deletions leading to α-syn depositions and dopaminergic neurons loss have been adopted in the development of PD models.
To develop PD conditions, several groups introduced transgenic animal models overexpressing human α-syn or mutants leading to α-syn fibrils in the brain. Giasson et al. proposed a transgenic mouse model overexpressing mutant A53T human α-syn, found in early-onset PD patients, encoded in the MoPrP.Xho vector system so that it is specifically overexpressed in the brain region.44 The transgenic mice expressing mutant A53T successfully presented major PD features, such as the formation of α-syn fibrils, activation of glial cells, and degeneration of motor neurons. However, increasing evidence showed that the overexpression of endogenous α-syn via vector transduction resulted in the formation of α-syn fibrils in limited parts of the brain near injection spots.45 Recently, the injection of α-syn preformed fibrils (PFFs) has been suggested to induce the deposition of α-syn fibrils diffused through the entire brain.
Loss of function mutations observed in early-onset PD patients, such as PARKIN, PTEN-induced putative kinase 1 (PINK1), and DJ-1,46 has been employed to develop PD models. PARKIN encodes an integral ligase in the ubiquitin proteasome system; therefore, loss of function mutations on PARKIN would promote aggregation of α-syn.47 However, there were contradictory results on PARKIN-deficient mice models, inducing mitochondrial dysfunction while no significant PFF formation and dopaminergic neuronal loss.48 In this regard, PARKIN-based models were suggested to co-express with other mutations such as PINK1 and DJ-1, a mitochondrial kinase recruiting PARKIN and a chaperone protein, enhancing PARKIN-mediated mitophagy and PD pathology.49 Recent studies also showed that PINK1 and DJ-1 deficiency models could represent PD signatures in early-onset as mitochondrial dysfunctions and oxidative stress leading to moderate dopaminergic neuronal loss.50–52 For the late-onset PD models, Ramonet et al. introduced transgenic mice expressing the disease-causing mutant of leucine rich repeat kinase2 (LRK2) (G2019S), found in late-onset familiar PD, accomplishing both induction of α-syn depositions and dopaminergic neuronal loss in the aged brain.53
2.-3. Traumatic brain injury (TBI)
Traumatic brain injury (TBI) is the major cause of death and disability worldwide triggered by a sudden, external, and physical damage on the brain (Fig. 1c).54 TBI consists of two phases: the primary damage at the incident involving immediate contusion, laceration, and intracranial hemorrhage and the second delayed damage after the incident including ischemia, edema, and other long-term disabilities in brain functions.54 Recent studies showed that TBI has a high correlation to increase risks of other dementia such as AD and PD due to the second delayed damage, which would alter neurochemical mechanisms and neurodegeneration.55Changes in neurochemical conditions are the major characteristics found in cerebrospinal fluid (CSF) and body fluids obtained from most TBI patients. The prolonged damage on the brain leads to the disruption of membrane integrity and activation of glutamate receptors (iGluRs) and metabotropic glutamate receptors (mGluRs) without control.56,57 Such aberrant activation of glutamate receptors can result in the massive release of glutamates in the brain. Dysfunction in membrane proteins modulating calcium, such as calcium channels and calcium/sodium pumps, is also known to impede the control over calcium homeostasis in the neuronal cells.57 Sun et al. revealed that long-term dysregulation in calcium homeostasis after TBI, induced by fluid percussion injury (FPI), elevated the calcium level and increased necrotic or apoptotic neurons in hippocampal regions of mice.57 Afterward, the excessive glutamate and calcium in the brain are known to impair mitochondrial function, elevate ROS (NO and H2O2), and activate apoptotic cell death in neuronal cells. Therefore, the disruption of membrane integrity followed by increased levels of both glutamate and calcium after the stimulus would be the important criteria to be confirmed in TBI models.
The local changes in neurochemical environments are known to activate resident glial cells and recruit other peripheral immune cells to the damaged brain.56 The changes involve not only the increased glutamate/calcium levels but also proinflammatory cytokines (IL1β, IL-6, IL-12, TNFα, IFNγ, TGFβ) and chemokines (CCL2, CCL3, CCL4, CXCL1, CXCL2) released by damaged neural cells or other glial cells near the TBI regions.55 In this regard, the increased level of proinflammatory factors would offer another evidence for TBI-driven neuroinflammation. Recent studies with mouse models after contusion injury generated by controlled cortical impact (CCI) suggested that the reactivity of astrocytes, measured by the expression level of glial fibrillary acidic protein (GFAP), could be a representative marker for evaluating the severity of TBI.56,58–60 In mild TBI, several studies confirmed that moderate astrogliosis produces neurotropic factors and anti-inflammatory mediators that ameliorate TBI conditions. In addition, they played pivotal roles in the control over homeostasis of the brain by buffering the imbalanced neurotransmitters.61,62 On the other hand, a number of studies with severe TBI models, involving aged animals after CCI or animals after chronic constriction impact injury (CCII), revealed the induction of overly-reactive astrocytes producing proinflammatory mediators that further exacerbated the TBI-driven neuroinflammation followed by neurodegeneration.56,62,63 Thus, the reactivity of astrocytes can be another key feature to be represented in TBI models.
2.-4. Vascular dementia
Vascular dementia (VD), a progressive cognitive decline, is caused by the impaired blood flow in the brain leading to neurodegeneration (Fig. 1d).64 According to Vascular Impairment of Cognition Classification Consensus Study (VICCCS), there are 4 major subtypes of VD: post-stroke dementia (PSD), subcortical ischemic vascular dementia (SIVD), multi-infarct (cortical) dementia, and mixed dementia with other CNS disorders, particularly AD and PD.64Leaky vessels increasing the impaired blood flow passing through the BBB are the common feature in VD. In the normal condition, an intact BBB permits only essential molecules to maintain CNS homeostasis while the damaged BBB found in the aged brain does not. Initial studies validated the leaky vessels by showing morphological changes followed by increased diffusion of endogenous proteins such as albumin and IgG passing through the vessels in brains of animal models.65–67 Other VD animal models exhibiting the plasma leaky feature involve stroke-prone renovascular hypertensive (RHRSP) and bilateral common carotid artery stenosis (BCAS) rats or mice.68,69 The advent of techniques (CT, MRI, PET images) enabled the tracking of plasma albumin concentrations among the entire CSF in brains of human elderly and confirmed the significant passage of albumin from vessels to brain regions. Several in vivo and in vitro BBB models confirmed the increased permeability by showing reduction in tight-junctions (ZO-1, occludin, claudin) and increase of the TEER value or permeability coefficient value of fluorescently-labelled molecules.70 Therefore, the development of a swollen and leaky BBB with increasing permeability of molecules found in CSF has become a gold standard to develop VD models.
Elevation of neuroinflammation, especially near the leaky vessels, is another hallmark reported in many VD cases. Endothelial cells on the damaged BBB are known to release various proinflammatory mediators and activate residential glial cells around them. In the normal case, beneficial astrocytes are activated and secrete growth factors such as vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), and glial cell line-derived neurotrophic factor (GDNF) that maintain the BBB integrity. In addition, such astrocytes release tumor growth factor beta (TGFβ), which guides pericytes around the BBB to enhance BBB tightness. In the aged brain, however, proinflammatory mediators such as IL1β and TNFα induce reactive astrocytes, which decrease BBB tightness and increase neurodegeneration in VD. Moreover, endothelial cells can release proinflammatory chemokines to recruit monocytes and macrophages. These peripheral immune cells then penetrate brain parenchyma and release neurotoxic factors as free radicals and other proinflammatory mediators. In this regard, effective VD models should recapitulate neuroinflammation followed by neuronal damage around the BBB.
2.-5. Environmental risk factor-driven disorders
Environmental risk factors (ERFs), such as pollutants (particulate matter (PM), microplastic particles (MP)), pathogens (bacteria and viruses), and their derivates, have been highlighted recently since epidemiological studies have alarmed us about the high correlation between exposure to ERFs and incident of CNS diseases including AD, PD, and other types of dementia (Fig. 1e).Because ERFs originate from outside of our body, the presence of ERFs in the brain or their entrance mechanisms should be validated prior to testing ERF-driven neurodegeneration. Kang et al. confirmed that PM downregulated tight-junctions on the BBB and decreased BBB integrity resulting in the penetration of PM to the brain regions.15 In addition, a number of studies revealed that external pathogens such as bacteria and viruses as well as their derivates could reach the brain area via the olfactory bulb–brain, gut–brain, or lung–brain axis and increase the risk of CNS disorders. Therefore, models for ERF-driven dementia are required to validate the entry pathway of environmental factors into the brain regions.
The common features found in the brain exposed to either air pollutants or pathogens are the induction of neuroinflammation. Several studies validated that PM induced reactive astrocytes increasing oxidative stress, amyloidosis, and tauopathy, leading to cognitive decline in PM-inhaling mice models or PM-treated in vitro models. Kang et al. revealed that M1 type microglia in brain models polluted with fine PM is the major source of neuroinflammation, which facilitated tauopathy and neurodegeneration.15 Recent studies with AD mice revealed that PM can further induce astrogliosis and microgliosis that would contribute to increase of AD severity significantly.71,72 In addition, Tran et al. validated that conditioned media of oral pathogenic bacteria (Porphyromonas gingivalis) activated both astrogliosis and microgliosis driving neuroinflammation followed by tauopathy in human brain models. Thus, ERF-mediated neuroinflammation leading to neurodegeneration is a criterion to be confirmed in ERF-driven dementia models.
3. Brain-on-a-chip platforms for modelling CNS diseases
3.-1. Alzheimer's disease-on-chip
Several AD models based on microfluidics have been developed and used for investigating the underlying mechanisms of amyloidosis and tauopathy contributing to AD progression. Park et al. introduced a simple AD model by preparing 3D neurospheroids in the microfluidics and validated Aβ-mediated neurodegeneration.81 Wu et al. designed a microfluidic device composed of two somatodendritic and axonal compartments, which were connected through multiple channels so that dendrites and axons of murine hippocampal and cortical neurons from each compartment form synapses at the center of channels.22 Afterward, they added tau aggregates into the somatodendritic compartment and monitored tau transportation from one side to the other. Later, Dujardin et al. employed the same device to culture rat neurons transduced with lentivirus encoding V5-hTau46, which produced tau proteins from the primary neurons (Fig. 2a).79 This study confirmed axonal transport of tau from the primary to the secondary neurons confirming tauopathy through cell-to-cell interaction. Furthermore, Takeda et al. designed a three-chamber system for modelling dual-layered neurons and validated the axonal transport of NFTs from the primary to secondary neurons (Fig. 2b).80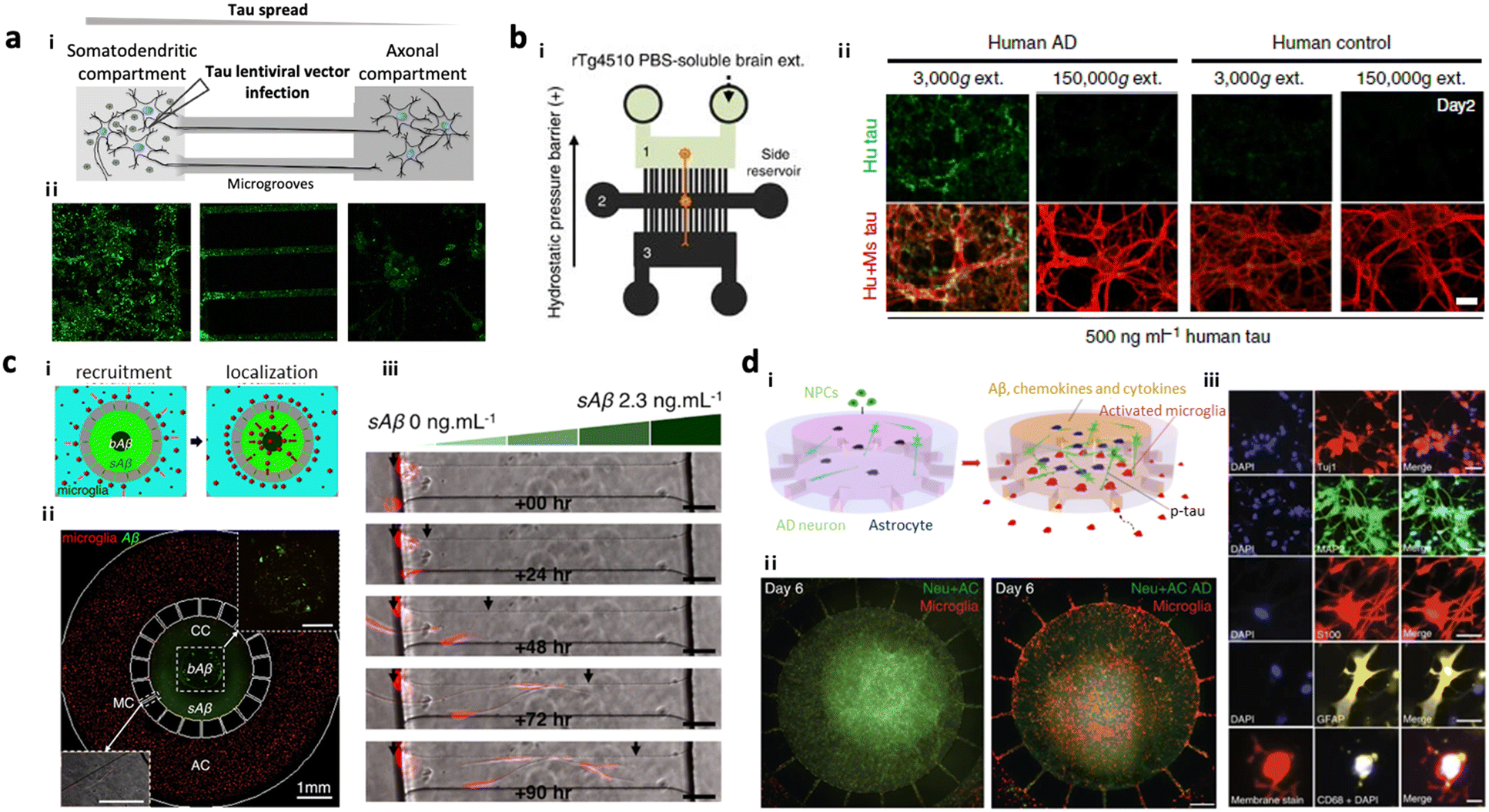 | ||
| Fig. 2 Mini-brains mimicking Alzheimer's disease (AD). (a) 2-Chamber microfluidic system for investigating tau aggregate propagation along axons. i) Scheme of the microfluidic device consisting of two chambers: a somatodendritic chamber (left) with tau-producing rat neuronal cells and an axonal chamber (right) with control neurons, which were interconnected by several microgrooves. Synapses between two types of neurons were formed at the middle of microgrooves. ii) Fluorescence images confirmed that GFP-labelled tau proteins were transported from the somatodendritic chamber to the axonal chamber not by diffusion. Copyright 2014, Springer Nature.79 (b) 3-Chamber microfluidic system for NFT propagation along a multi-step neuronal circuit. i) Scheme of the microfluidic device consisting of three chambers: a somatodendritic chamber (left) with tau-producing mouse neuronal cells, a synapse chamber (middle) connecting two neurons, and an axonal chamber (right) with control neurons. ii) NFTs propagating along the neurons in the first chamber (green) were observed to be taken up and propagated along the neurons in the second chamber toward the third chamber, representing selective propagation from region to region in the brain. Copyright 2015, Springer Nature.80 (c) Microglia migration chip to investigate Aβ response in AD progression. i and ii) Scheme and fluorescence image of the migration chip: the central compartment was surrounded by the annular compartment connected by migration channels. iii) The model discovered that microglia can be activated and recruited by soluble and insoluble Aβ. Copyright 2013, Springer Nature.19 (d) Multicellular 3D AD brain model with an innate immune system on a chip. i) Scheme of model development. This study further improved the microglia migration chip by tri-culturing neurons expressing APP/PS1 and astrocytes in the central chamber as well as microglia in the angular chamber. ii) This AD model found that the interaction between AD neurons and astrocytes produced proinflammatory mediators that activate and recruit microglia promoting neuronal loss. iii) Fluorescence images confirmed the presence of neural cells (Tuj+, MAP2+), reactive astrocytes (S100+, GFAP+) and microglia (CD68+). Copyright 2018, Springer Nature.13 | ||
Neuroinflammation has emerged as another key therapeutic target since the severity of inflammation is closely related to the stage of AD.17,25 The unique feature of innate immune cells in AD is their chemotactic activity in response to AD ques which initiate neuroinflammation.102 The microfluidic system has emerged as an ideal platform to construct such cell-to-cell or cell-to-environment interactions based on chemotaxis, which could contribute to AD progression. Cho et al. introduced a chemotactic microfluidic platform forming stable chemical gradients for AD ques such as soluble Aβ, Aβ fibrils, and conditioned media from AD brain tissues, which stimulated human microglia (Fig. 2c).19 This unique platform allowed stimulated microglia with a specific phenotype to be separated from the heterogeneous population in response to AD ques; however, single-cultured models could not recapitulate other aspects from the multi-cellular crosstalk found in AD pathology.
Recent advances in the technique of cell culture within microfluidics allow the development of multicellular AD models, reproducing physio-pathogenically relevant AD signatures leading to neurodegeneration. For instance, Park et al. constructed a 3D human AD brain model along with an innate immune system around the brain by co-culturing human neuroprogenitor cells expressing APP/PS1 and human adult microglia in the microfluidic device (Fig. 2d).13 It should be noted that the neuroprogenitor cells were later differentiated into neurons and astrocytes producing Aβ; therefore, the model became a tri-culture system. The model involved two compartments with one for the AD brain with AD neurons and astrocytes and another for microglia, which were interconnected with multiple channels allowing microglial recruitment in response to soluble factors from the AD brain compartment. This study revealed that the astrogliosis driven by the interaction between AD neurons and astrocytes in the AD brain could induce M1 type microglia, which further promoted tau accumulation and neuronal death at the end. This model was further advanced with the use of iPSC-derived microglia in order to investigate the critical role of TREM2 in the microglial activity offering neuroprotection in AD conditions.14 In summary, the multicellular AD models in microfluidics were able to reproduce key AD features such as pathological accumulation of Aβ, induction of neuroinflammation, deposition of tau, and neurodegeneration.
3.-2. Parkinson disease-on-chip
To date, several models based on microfluidics have successfully simulated PD progression in terms of spreading of α-syn in the brain through neuron-to-neuron interactions. Freundt et al. employed microfluidics to align two neurons from mice forming a synapse between presynaptic soma and postsynaptic axon and revealed neuron-to-neuron transmission of α-syn PFFs through axonal transport.86 Fernandes et al. employed a microfluidic platform comprising two compartments interconnected by multiple channels with pneumatic valves to control the diffusion of α-syn.40 This study found neuronal transmission of α-syn as well as microglial recruitment in response to the α-syn gradient. Later, Prots et al. seeded neurons and astrocytes expressing α-syn oligomer-forming mutants (E46K and E57K) differentiated from iPSCs of PD patients in the microfluidic device, forming axon-to-soma projection (Fig. 3a).88 This study revealed that accumulation of α-syn oligomers impaired the anterograde mitochondrial axonal transport as well as facilitated tau spreading through neuron-to-neuron interactions. The dysfunction in synucleinopathy led to energy deflects and synaptic impairment at the end. Recently, Mao et al. employed a two layered axon-to-soma chip and revealed a key mediator, lymphocyte-activation gene 3 (LAG3), that bound to α-syn and promoted α-syn transmission through dopaminergic neurons (Fig. 3b).87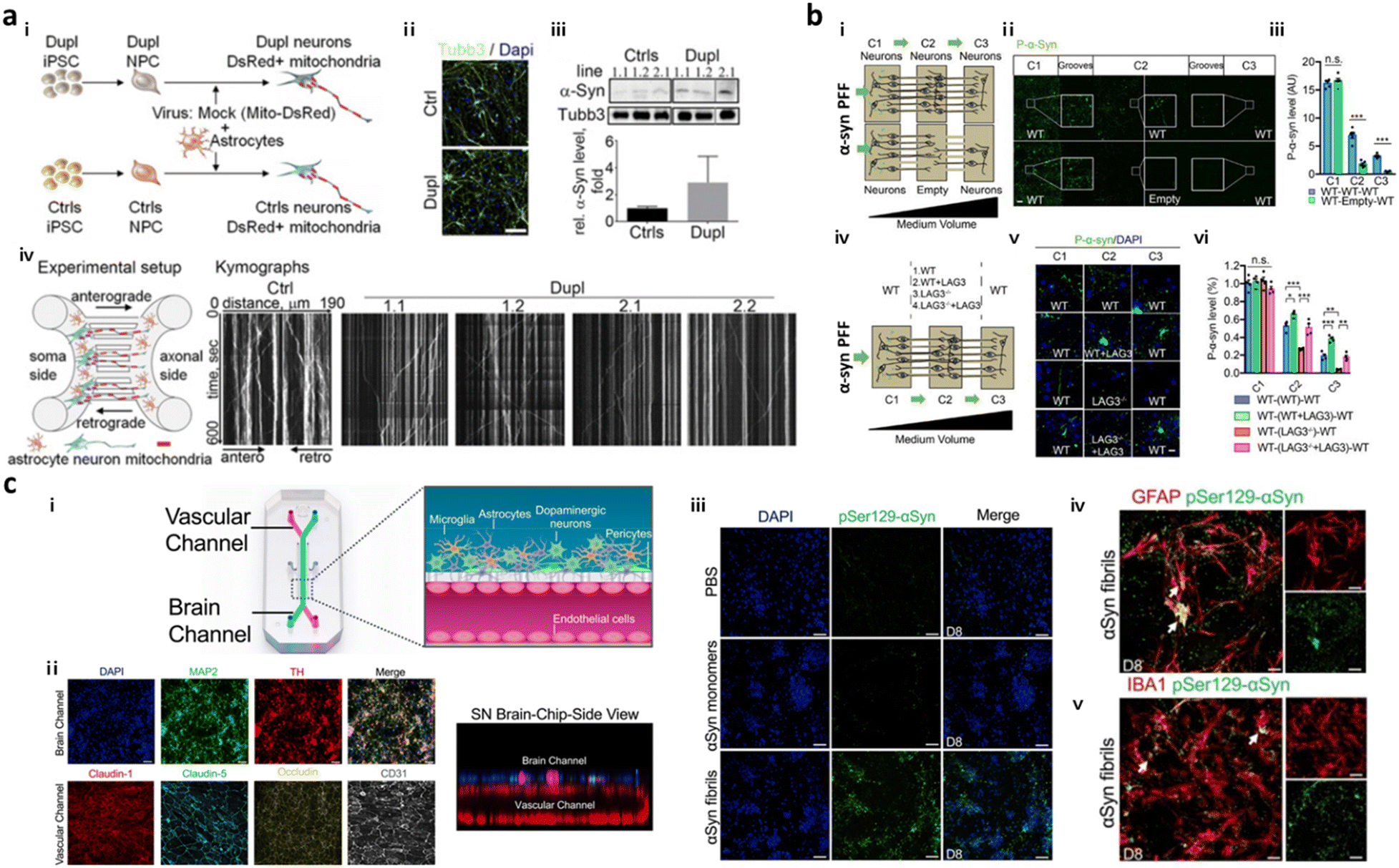 | ||
| Fig. 3 Mini-brains representing Parkinson's disease (PD). (a) Axon-to-soma chip to investigate axonal transport of α-syn. i–iii) Differentiation of neurons from PD-patient driven iPSCs (Dupl) or control iPSCs (Ctrl). Validation of Tubb3 and α-syn expression in Dupl neurons representing the completion of neural differentiation and synucleinopathies. iv) The microfluidic device with two microchannels separated the soma and axonal parts from a single neuron, which could analyse axonal transport in neurons, particularly focusing on α-syn trafficking. This device showed that the α-syn oligomers strongly impaired the axon and triggered synucleinopathies. Copyright 2018, PNAS.88 (b) Double-layered axon-to-soma chip for the study of α-syn transport through the neural network. i) Scheme of the device configuration. ii and iii) Validation of α-syn propagation through axon-to-soma interaction. iv–vi) This study illustrated that LAG3 played an important role in α-synuclein transmission. Copyright 2016, Science.87 (c) Multicellular 3D PD brain-BBB chip. i) Two channels of the PD brain with dopaminergic neurons along with glial cells and blood vascular unit were divided by a PDMS porous membrane allowing direct cell–cell interaction and communication through soluble factors released by cells in the chip. ii) Immunostaining results for functional dopaminergic neurons (TH: tyrosine hydroxylase) and tight-junctions (claudin-1, claudin-5, occludin, CD31) confirmed physiologically relevant conditions of multicellular culture in the same chip. iii–v) This study demonstrated that exogenous α-syn PFFs added to the brain channel (iii) accumulated in neuronal cells and promoted (iv) astrogliosis and (v) microgliosis. Copyright 2021, Springer Nature.98 | ||
Another key signature found in PD is dopaminergic neuronal loss in the brain. Prior to development of PD models representing dopaminergic neuronal degeneration, Moreno et al. introduced a 3D OrganoPlate, a microfluidic platform with a perfusion system allowing for the continuous differentiation of dopaminergic neurons from neuroepithelial stem cells.103 Later, Bolognin et al. employed the same platform for the construction of a PD model in the microfluidics with 3D cultured dopaminergic neurons carrying a pathogenic mutation (LRRK2-G2019S) from PD patient-derived iPSCs. This model validated PD pathology such as mitochondrial dysfunction in dopaminergic neurons leading to neurodegeneration, but did not reproduce another pathology such as α-syn accumulation yet. Pediaditakis et al. developed a substantia nigra brain-chip, culturing iPSC-derived dopaminergic neurons, microglia, astrocytes, and pericytes in the microfluidics (Fig. 3c).98 They added α-syn PFFs to the model to induce the pathological conditions. The exposure of this device to α-syn fibrils led to mitochondrial dysfunction and progressive neuronal death along with the activation of astrocytes and microglia. Given the recent advance in dopaminergic neuronal culture in the microfluidics, there is tremendous potential to develop PD models reproducing both dysfunction of synucleinopathy and loss of dopaminergic neurons in the future.
3.-3. Traumatic brain injury-on-chip
To develop TBI conditions inducing delayed brain damage, a number of studies have designed microfluidic platforms with well-controlled mechanical stimuli, such as fluid shear stress, hydrostatic pressure, compression, and shear strain.Microfluidics equipped with a vacuum aspiration system have been widely applied for inducing axonal injury. TBI models in the two-compartments interconnected by multiple microchannels, in which synapses could form in the center of microchannels, were the most popular configuration as they could apply vacuum aspiration to one side and induce damage only on the axonal part. Taylor et al. found that the vacuum-assisted axonal damage changed groups of mRNA transcripts for cytoskeletal and mitochondrial maintenance.89 Zhang et al. employed a microfluidic device with the same configuration and revealed that collapsin response mediator protein-2 (CRMP2) was the key mediator to facilitate mitochondrial damage followed by axonal degeneration in the TBI models.90 This TBI model was further utilized to discover a promising strategy to induce axonal regeneration after the injury, such as neurotrophins-3, peroxisome proliferator-activated receptor gamma (PPAR-γ), and therapeutic peptide NOGO-66.91,92,104 The vacuum-induced neural injury method is, however, limited in a specific region of the microfluidics due to the high fluidic resistance between the interconnected compartments.
To further expand the injury region of the brain prepared in microfluidics, Rogers et al. cultured neurons and glial cells in a microfluidic device forming neural networks and incubated the entire device in the impact chamber, exposing it to controlled g force (Fig. 4a).95 This study validated the force-dependent post-impact on the neurodegeneration driven by neuroinflammation and oxidative stress. Hosmane et al. introduced an axonal injury micro-compression (AIM) platform, where pneumatic pressure in a range of moderate (<55 kPa) to high (55–95 kPa) was directly applied onto axons (Fig. 4b).93 TBI models prepared in AIM microfluidics precisely reflected the delayed damage presumably due to the rearrangement of neurofilaments and microtubules in neurons. In addition, physical force by stretch and strain motions was applied to develop TBI models. Dollé et al. introduced a pneumatic strain-based injury model in the microfluidics by inducing deformation of the PDMS surface (Fig. 4c). In this platform, neurons straightened their axons on the PDMS membrane, which can be stretched by pressurizing a cavity underneath the PDMS surface.94 The stretching axons increased the mitochondrial membrane potential dramatically which led to axon degeneration.
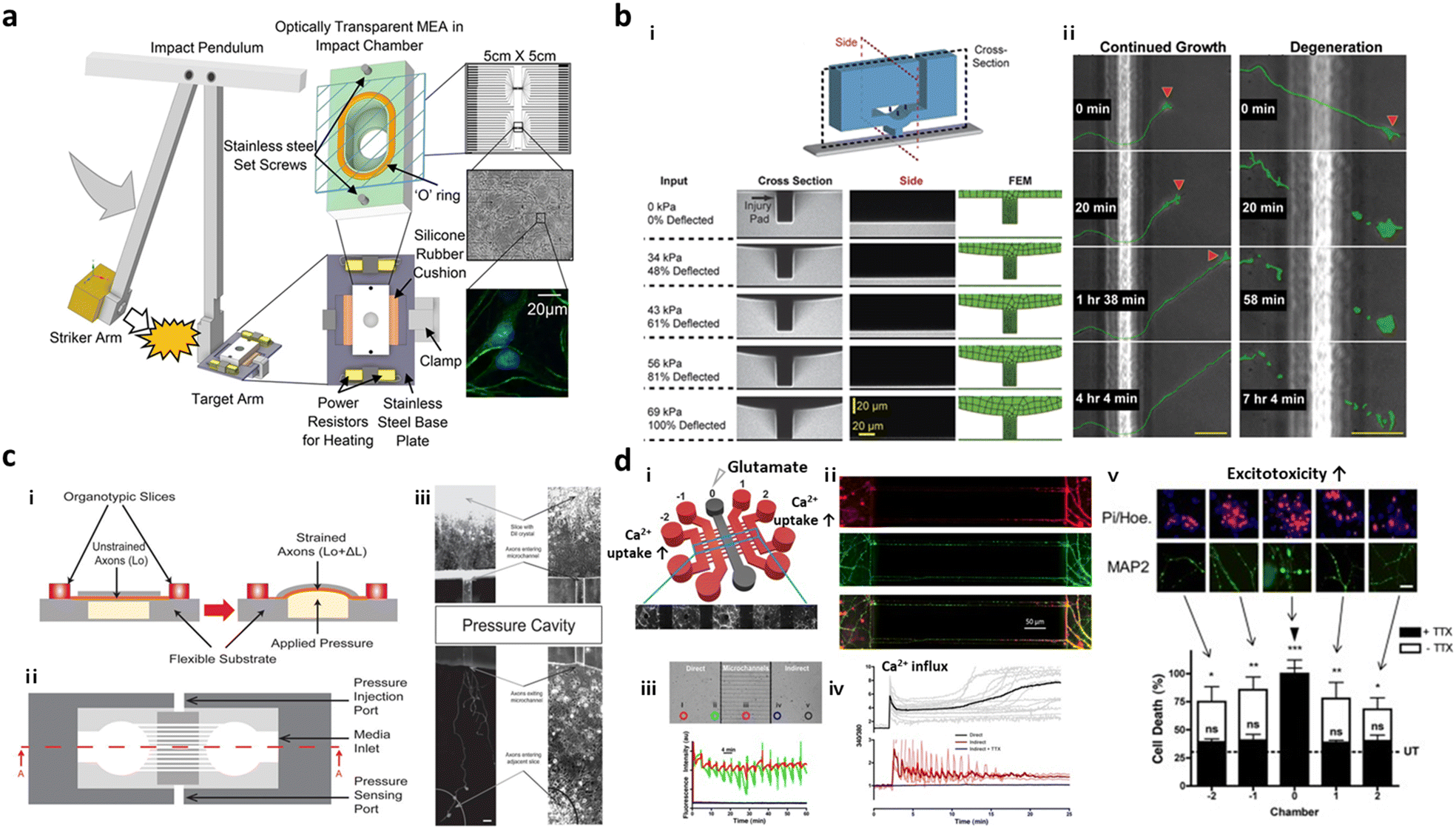 | ||
| Fig. 4 Mini-brains reconstituting traumatic brain injury (TBI). (a) Post-TBI was reproduced by the direct mechanical impact on the chip forming a neural network between neurons and glial cells. To this end, the striker arm hit the target incubator in which cells were cultured and formed neural networks. This TBI-on-a-chip simulated secondary damage on neurons. Copyright 2022, Springer Nature.95 (b) Valve-based microfluidic axon injury micro-compression (AIM) platform. i) The microfluidic device could apply a focal and graded compression on the axon specifically to induce different levels of injury to axons. ii) This study clarified the correlation between varying compressive loads and axon injury leading to neurodegeneration at the end. Copyright 2011, Royal Society of Chemistry.93 (c) Microfluidic chip for applying uniaxial strain injury on axons. i–ii) Neurons were cultured in one chamber and their axons grew through microchannels and reached another chamber. On the basement of the axonal chamber layer, the applied pressure could stretch out the PDMS layer on which axons were grown and induce controlled strains on the axonal part (i, side view; ii, top view). iii) Mitochondria damage followed by axon degeneration was observed after applying pneumatic force on the PDMS surface. Copyright 2014, Royal Society of Chemistry.94 (d) Neural network chip with TBI condition. i) A web-like multi-channel platform was connected to chambers for hippocampal neurons. ii) Axons from each chamber formed a neural network in the multichannel. iii) To induce TBI condition, high concentration of glutamate was added to one chamber. iv and v) This study validated glutamate and calcium transfer from primary to secondary neurons, which promoted neurotoxicity. Copyright 2016, Springer Nature.96 | ||
As described previously, massive influx of neurotransmitters involving glutamates is the major characteristic observed during the secondary damage on neurons. Samson et al. generated neuronal networks in microfluidics, where hippocampal neurons were cultured in 5 chambers and their axons–somas were interconnected by multiple channels (Fig. 4d).96 To induce the secondary damage, they added a high concentration of glutamate at the central chamber, and validated the spread of neuronal damage through cell–cell interactions in the network. Among the receptors for neurotransmitters, the GluN2B type N-methyl-D-aspartate (NMDA) receptor was majorly engaged in the spread of secondary damage induced by the glutamate storm. Charier et al. prepared TBI models in microfluidics by addition of lysed blood or blood clot and validated that NMDA played pivotal roles in damage transmission.97
3.-4. Neurovascular unit-on-chip
Present in vitro VD models simulate the transmission of neurotoxic factors, originating from the neurovascular unit (NVU), into the brain and the initiation of neurodegeneration. NVU chips are majorly composed of the BBB part along with other accessory parts such as pericytes and astrocytes; further advanced NVU chips are connected to brain tissue parts with neuronal cells.NVU chips have described the correlation between the impaired BBB and neurodegeneration in AD progression. Cho et al. created an in vitro 3D capillary-like structured BBB integrated with chemotactic channels, allowing infiltration of neutrophils from the BBB lumen to the brain compartment in response to proinflammatory mediators originating from the brain side (Fig. 5a).105 Later, the 3D tubular BBB was connected to 3D AD brain models cultured with APP/PS1neuroprogenitor cells in the microfluidics (Fig. 5b).75 This study revealed that proinflammatory factors such as Aβ, MMP-2, and IFNγ from AD brains induced ROS accumulation and tight-junction reduction in endothelial cells in the BBB part. Afterward, they discovered that the leaky BBB permitted the increased influx of neurotoxin to the brain part and promoted neuronal damage at the end.
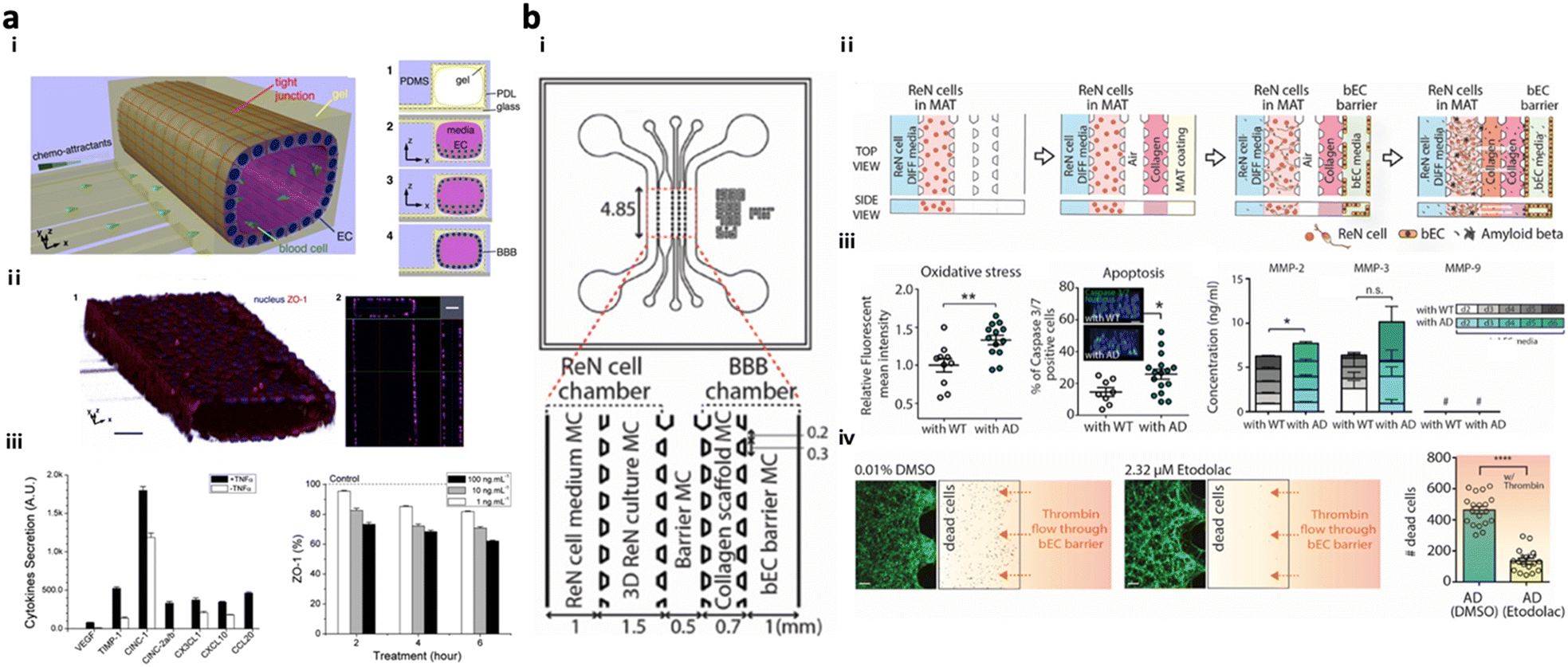 | ||
| Fig. 5 Neurovascular unit-on-chips reconstituting vascular dementia (VD). (a) 3D BBB with an innate immune system. Schematic representation of i) a 3D BBB chip configuration and steps forming the cylindrical monolayer of EC. ii) Immunostaining results confirmed the 3D vascular-like structure of the EC layer and tight-junction formation in the BBB model. iii) Induction of inflammation and iv) reduction of the ZO-1 level in the BBB triggered by TNFα. Copyright 2015, Springer Nature.105 (b) Multicellular 3D AD-BBB chip. i) Schematic illustration of the chip configuration showing the 3D AD brain (left) and 3D BBB (right) connected through multiple channels. ii) Timeline for the model preparation. iii) Validation of AD conditions as induction of oxidative stress, apoptotic cell death, and neuroinflammation in AD models. iv) This study confirmed the therapeutic effects of etodolac, such as passing though the BBB and decreasing apoptotic cell death in AD conditions. Copyright 2019, John Wiley and Sons.75 | ||
In addition, NVU chips clarified that α-syn PFFs could affect BBB integrity in PD progression. Pediaditakis et al. employed microfluidics comprising two compartments of a 3D brain and a cylindrical structured BBB and demonstrated that α-syn PFFs impaired BBB tight-junctions and increased the risk of PD.98 This study also revealed the disruption of BBB integrity after the treatment of α-syn PFFs to the BBB side of the model while no discernible effects by the α-syn monomer were observed, confirming that α-syn PFF was the pathological form of α-syn.
3.-5. Environment risk factor-driven dementia-on-chip
To test potential neurotoxicity driven by ERFs, a number of groups introduced brain models in the chip, which reconstituted the human brain continuously exposed to various pathogens or pollutants. DeOre et al. employed a 3D BBB model, previously described by Partyka et al., to study the entrance mechanism of viruses such as SARS-CoV-2 (Fig. 6a).106,107 This study found that the treatment of SARS-CoV-2 spike proteins activated RhoA and disrupted BBB tightness. Tran et al. developed chip-based brain models exposed to oral bacteria-conditioned media to test the induction of any neurotoxicity driven by the bacteria (Fig. 6b).99 Koo et al. investigated the effects of organophosphate-based compounds (OPs) in the human brain by using a membrane-free tetra-cultured brain on chip, which reconstituted a blood vessel embedded in the brain tissue with innate immune cells.100 The addition of dynamic media flow at the center of the blood vessel part enabled the investigation of not only OP entry mechanisms but also its roles in neurodegeneration. Liu et al. further improved the brain tissue part with iPSC-derived GABAerinc inhibitory neurons and astrocytes exposed to OPs, which simulated an OP-driven neurodegenerative model.101 This study validated the therapeutic efficacy of butyrylcholinesterase, one of the exogenous bioscavengers of OPs. Kang et al. engineered a biomimetic model of a PM-polluted brain in the microfluidics, mimicking proinflammation followed by neurodegeneration driven by reactive astrocytes and microglia under PM conditions (Fig. 6c).15 This model showed the notable recruitment and activation of microglia in response to soluble factors from neurons and astrocytes under long-term exposure to PM. The model found that PM could induce M1 type microglia, which increase pTau accumulation, synaptic impairment, and neuronal loss.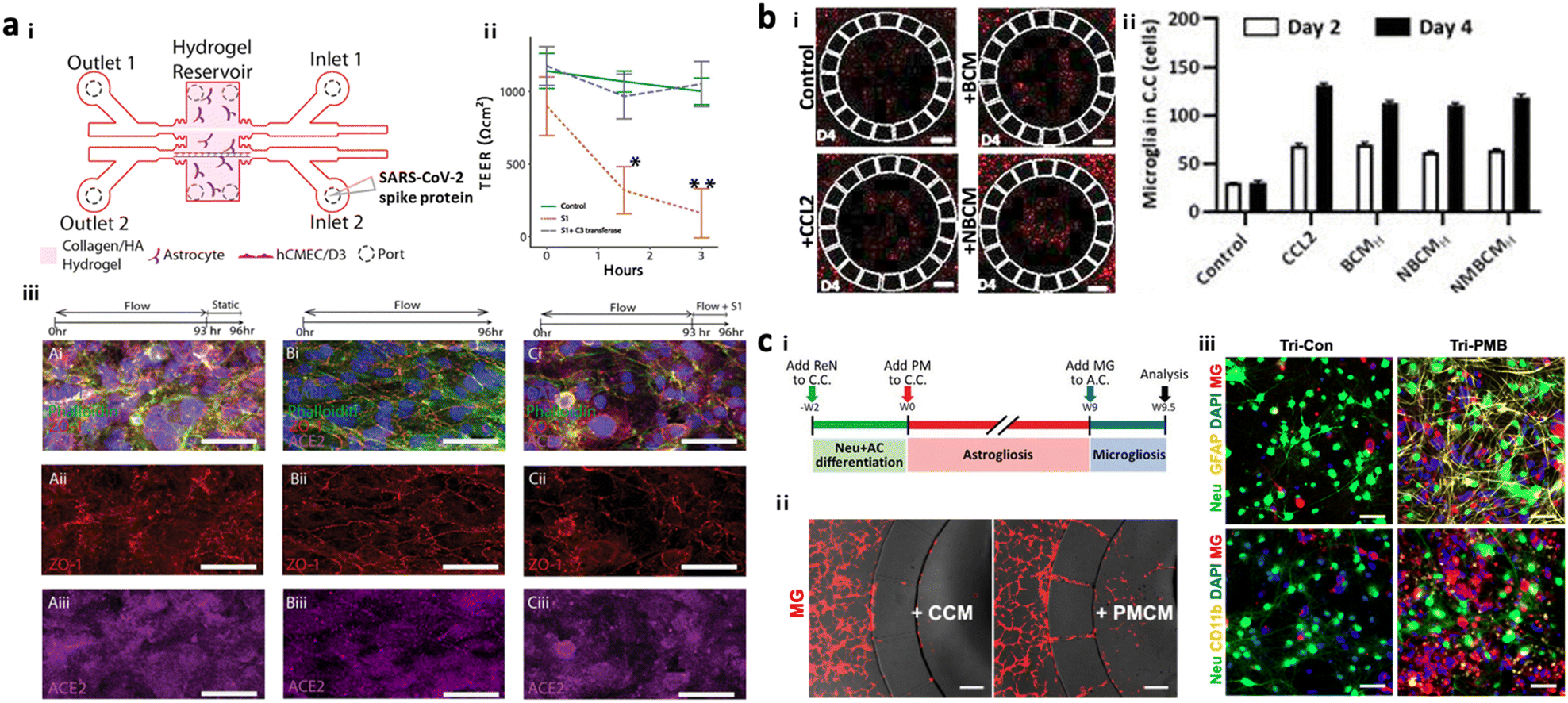 | ||
| Fig. 6 Summary of brain chips for environmental risk factor-driven dementia (EFRD). (a) Infection model of SARS-CoV-2. i) Schematic illustration of the 3D BBB model to investigate the underlying mechanisms of SARS-CoV-2 penetrating the BBB. Cerebral endothelial cells formed a 3D vascular structure and were surrounded by astrocytes in the microfluidic device. Copyright 2017, Elservier.107 ii) TEER measurement validated the reduction of BBB tightness after the treatment of SARS-CoV-2 spike proteins. iii) Immunostaining results confirmed the significant roles of SARS-CoV-2 spike proteins in activating ACE2 and reducing tight-junctions in the BBB. Copyright 2021, Springer Nature.106 (b) Infection model of oral bacteria. i and ii) This study revealed that microglia were recruited to the central chamber containing soluble factors from brain models exposed to bacteria-condition media (NBCM). In this study, neurodegenerative microgliosis and loss of astrocytes are observed after exposure to the oral pathogen, leading to AD. Copyright 2021, MDPI.99 (c) Polluted brain model with particulate matter (PM). i) Timeline for the tri-culture brain model exposed to particulate matter. A biomimetic device was developed to mimic neuroinflammation and neurodegeneration caused by PM. ii) Microglia migrated to the central chamber with co-cultured neurons and astrocytes exposed to PM (PMCM). iii) Fluorescence images confirmed the induction of proinflammatory microglia (CD11b+) leading to the reduction in neural population marked by Neu in the brain model exposed to particulate matter. Copyright 2021, John Wiley & Sons.15 | ||
4. Multi-organ chips for investigating the entry of risk factors to the brain
4.-1. From organ chip to multi-organ chip
In our body, there are multiple paracellular layers comprising epithelium cells and neuronal cells that serve as the first front-line of defence against environmental risk factors such as pathogens or microbiomes. The layers involve epithelial layers for the gut, lung, nasal cavity, cornea, and skin. Therefore, dysregulation of these defensive layers, in terms of lowered tightness or damaged neurons, could allow the invasion of pathogens in our bodies leading to inflammation. Previous studies hypothesized that the accumulation of these risk factors and inflammation originating from these layers could further transmit to the brain region through the circulatory system or nervous system linking a specific organ to the brain.108–110 In this regard, the recent paradigm for organ chips has been expanded to multi-organ chips. Here, we review recently proposed multi-organ chips in terms of key characteristics they achieved and future directions at the end.4.-2. Gut–brain axis
One of the most popular tracks to reach the CNS is the gut–brain axis (GBA), a bidirectional link passing through either the systematic circulatory system or enteric nervous system.108 Many studies have suggested that environmental risk factors or other risk factors accumulated in the gut would penetrate to the brain and promote neurodegeneration diseases such as Parkinson's disease, Alzheimer's disease, and autism spectrum disorders.111–120 Kim et al. came up with a modular microfluidic device, on which co-cultured gut epithelial cells and brain endothelial cells develop the GBA track on a chip (Fig. 7b).121 They measured the permeability of lipopolysaccharides, butyrates, and exosomes derived from the gut to both barriers of the intestinal system and the BBB, demonstrating that such molecules produced in the intestine can be delivered to the brain and affect the microenvironment in the brain. Trapecar et al. further connected the gut–liver–brain together on a physiological model in order to simulate the physiology in the human body.122 They identified that short-chain fatty acids produced by gut microbiome promoted maturation of healthy brain cells but exacerbated neuronal death in PD conditions. Furthermore, they showed that these kinds of opposite effects may be mediated by lipid metabolism instead of the immune system by observing the pathology-related transcriptomic changes after removing immune cells from the platform. Overall, GBA microfluidic platforms have great potential for us to investigate the pathology of neurodegenerative diseases at the molecular level in vitro; however, there is no existing model connecting the gut-to-brain with the enteric nervous system yet.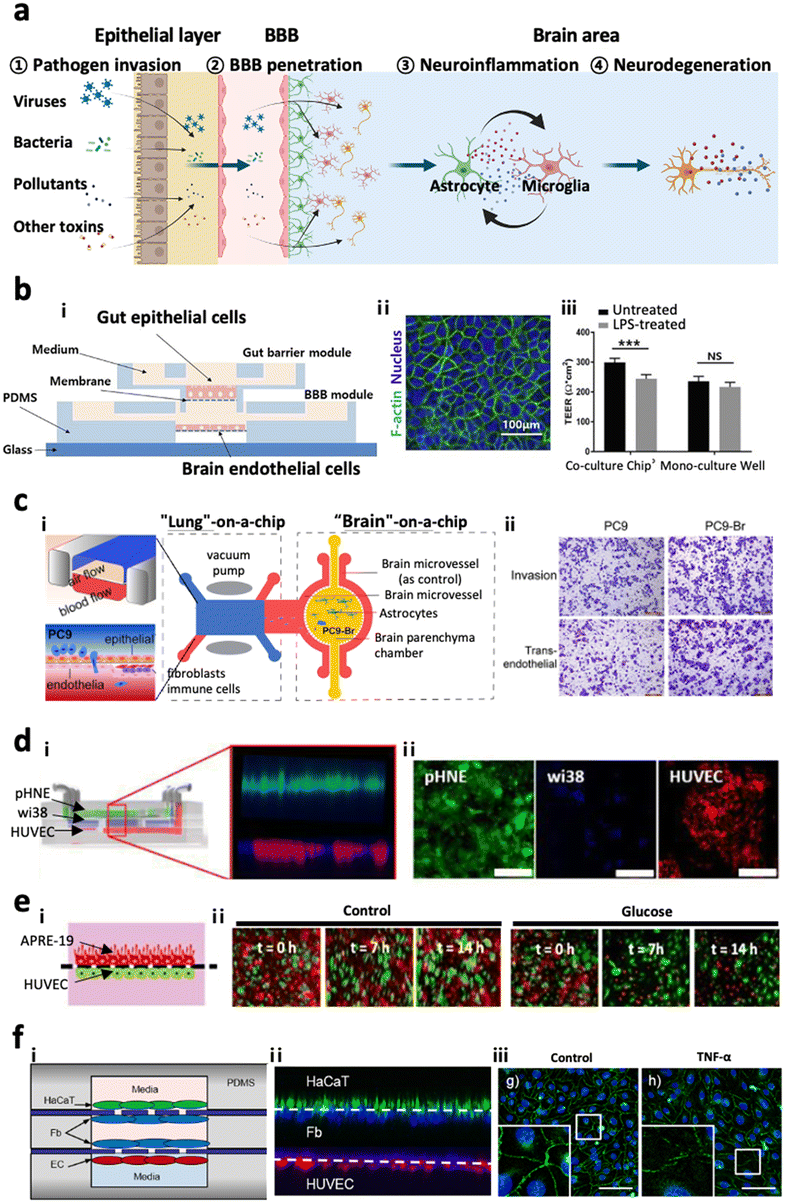 | ||
| Fig. 7 Multi-organs-on-chips as the potential platforms for CNS disorders. (a) Entry pathways of risk factors to the brain passing across two distinctive paracellular barriers. Risk factors such as viruses, bacteria, pollutants, and other toxins can penetrate the first barrier of epithelial layer in organs followed by the second barrier of cerebral endothelial layer of the brain blood vessel, also known as the blood–brain barrier (BBB). Such pathogens reaching the brain can further promote neuroinflammation as well as neurodegeneration leading to dementia. (b) Gut–brain axis (GBA). i) Design of a GBA chip to study the transport of microbial by-products across the gut epithelial layer (top) followed by the BBB layer (bottom). ii) Fluorescence image of F-actin (green) in Caco-2 cells validating the formation of the gut epithelial layer on the top membrane. iii) This model simulated LPS-driven inflammatory response, such as decrease in TEER values of gut and BBB layers. Copyright 2021, Elsevier.121 (c) Lung–brain axis (LBA). i) Scheme of the LBA chip involving lung (top), blood vessel (middle), and brain chambers. ii) This study confirmed that human metastatic lung cells (PC9-Br) in the lung compartment can penetrate into the brain region of the LBA chip. Copyright 2020, Frontiers Media SA.125 (d) Olfactory bulb–brain axis (OBA). i) Nasal mucosa chip to investigate the toxicity of urban particulate matter (UPM) on OBA. ii) Fluorescence images mark the layers of human nasal epithelial cells (pHNE), fibroblast cells (Wi38), and human umbilical vein endothelial cells (HUVEC). Copyright 2019, Springer Nature.126 (e) Eye–brain axis (EBA). i) Eye-BBB chip to investigate the angiogenesis process under hypoxia conditions. Two layers of human retinal pigmental epithelial cells (APRE-19, red) and human endothelial cells (HUVEC, green) facing each other were co-cultured in the microfluidic device. ii) The addition of chemical hypoxia induces HUVEC from the endothelial layer which penetrated and disrupted the epithelial layer. Copyright 2017, Springer Nature.127 (f) Skin–brain axis (SBA). Skin-BBB chip simulating edema-driven inflammation. i) Configuration of the skin–BBB model showing epidermis (green), dermis (blue), and blood vessel (red). ii) Fluorescence images show the configuration of epithelial cells (HaCaT) forming the epidermis, fibroblasts (Fb) forming the dermis, and endothelial cells (HUVEC) forming the blood vessel compartments in the chip. iii) This model confirmed the tight-junction disruption driven by edema-driven TNFα. Copyright 2016, Springer Nature.128 | ||
4.-3. Lung–brain axis
The lung–brain axis (LBA) is assumed to connect the CNS and lung through the autonomic nervous system or circulatory system, by which primary pulmonary diseases can affect the brain and lead to neurodegenerative disorders.109,110 There were initial efforts on the development of functionalized liver models prepared on the chip prior to creating LBA chips. Huh et al. introduced a ‘breathing’ lung model in the chip, simulating breathing-induced physiologically-relevant mechanical force by co-culturing epithelium and endothelial cells on the stretchable PDMS forming an alveolar–capillary barrier.123 This model was employed to closely reconstitute pathogen entry, neutrophil recruitment, and pulmonary edema.124 Xu et al.125 further developed an LBA track on a chip by combining two microfluidics such as lung-on-a-chip and brain-on-a-chip and investigated lung cancer metastasis pathology (Fig. 7c). They isolated the brain metastasis cells from the device and identified that these cells possessed different protein expression profiles and overactive glutathione (GSH) metabolism pathways acquiring drug-resistance ability via endogenous alteration. The microfluidic LBA chips, therefore, have great potential for studying disease pathology as a contribution of lung-derived risk factors to CNS diseases. However, further study is required to develop LBA track on chips to reconstitute the autonomic nerve system.4.-4. Olfactory bulb–brain axis
The olfactory bulb (OB) is anatomically located in the forebrain of humans and vertebrate animals which detects and transduces odorant information to the brain parts such as pyriform cortex, hypothalamus, and amygdala.129 The most distinctive feature of the olfactory bulb is a glomerulus, in which the axons of sensory neurons come into contact with the apical dendrites forming synapses and project odorant signals.129 Recent studies show that inhaled airway risk factors can be accumulated in the brain through the sensory neurons connecting the OB and brain bypassing the lung or blood brain barrier systems.130,131 Other independent studies revealed that the risk factors and other environmental pathogens accumulated in the OB could be transferred to the brain and further propagate neurodegenerative diseases including AD, PD, and dementia.132–134 To clarify the penetration of pathogens to the OB followed by the BBB, Na et al. developed a mucosa gland-on-a-chip, which reconstituted the mucosa layer with primary human nasal epithelial cells (hNECs) exposed to the air way along with blood vessels with human microvessel endothelial cells (hMVECs) underneath of the mucosa layer.135 In between the epithelial layer and endothelial layer, there was an intermediate chamber filled with ECMs that allowed the formation of mucosa glands originating from the epithelial layer that may further develop polys and invade the circulatory system. Later, Byun et al. engineered a similar platform as a mucosa-BBB chip and employed the chip to investigate the underlying mechanisms of PM penetration from mucosa to the BBB layer (Fig. 7d).126 However, further effective models adding the brain component and reconstituting the OB–brain axis are needed to clarify the underlying mechanisms of OB-driven pathogenesis leading to CNS disorders in the future as well.4.-5. Eye–brain axis
Light information from outside can be gathered, processed into electrical signals, and transmitted into the brain by retina, majorly composed of retinal ganglion cells (RGCs).136,137 Recent studies showed that patients with CNS disorders including AD and PD suffer from ophthalmologic symptoms at high frequency.138,139 To support the hypothesis on the eye–brain axis contributing to CNS disorders, microfluidics have been employed. Some initial studies enabled the culture of RGCs in the microfluidics. For instance, Wu et al. cultured RGCs in the microfluidics, which were connected to a 3D pressure-applying adaptor, designed to impose the same pressure on RGCs, and investigated how the RGCs respond to different hydrostatic pressures.140 Later, Chen et al. established a microfluidic chip to co-culture human retinal pigmental epithelial cells (ARPE-19) and human umbilical vein endothelial cells (HUVEC) (Fig. 7e).127 Utilizing this platform, more VEGFs can be secreted under the low glucose and hypoxia conditions, in turn causing the directional migration of HUVEC. Arik et al. further modified this device to develop an outer blood–retinal barrier (oBRB) chip which evidenced that exposure to reactive oxygen species (ROS) may lead to vascular hyperpermeability and induce wet AMD. These microfluidic devices can be applied to study complex eye disease pathology and even utilized for treatment evaluation with precision medicine.4.-6. Skin–brain axis
Psychological hyperstress can cause and aggravate neurogenic inflammation, which may further trigger skin diseases such as atopic dermatitis and psoriasis.141 Meanwhile, the skin can also affect the brain through the transmission of hormones or cytokines to the brain region.141,142 Given the correlation between the skin and brain, there is an increasing demand on the development of skin–brain axis on chips. Wufuer et al. successfully developed a human skin-BBB chip on which human immortalized keratinocytes (HaCaT), fibroblasts, and human umbilical vein endothelial cells (HUVEC) were cultured (Fig. 7f).128 They induced inflammation and edema conditions of the human skin in vitro via perfusing TNFα into the device. Afterward, they validated that dexamethasone, a well-known anti-inflammatory compound, alleviated the skin inflammation and its transmission to the blood vessel. Although no skin–brain microfluidic device has been developed yet, the advance of the cell culture technique and its biomaterials would contribute to the development of skin–brain disease models in the near future.5. Perspectives
Herein, we overview the current state-of-art techniques for developing a multicellular brain organoid on a chip, which precisely represents the pathological conditions of CNS diseases. Microfluidics are powerful platforms to reconstitute CNS diseases due to the following reasons. Firstly, the microfluidic platform can culture cells in long-term periods allowing the development of chronic status found in most CNS diseases as it could supply nutrient and remove waste continuously by modulating the flow of aqueous solution and gas in the device.17 For instance, the perfusion system enables not only continuous supplement of essential factors for cell culture but also exchange of buffers and reagents for multiple analyses. Secondly, capillary channels forming stable gradients for chemotaxis could be applied for the study of infiltrating innate immune cells observed in neuroinflammation, a common signature found in CNS disorders.19 The capillary structures can be further employed to guide the neuronal growth and control the connectivity of neural networks.143 Thirdly, the compartmentalized system having multiple chambers connected through several microchannels could divide a mini-brain into multi-compartments having specific functions and structures.17 This compartmental system further enables the development of multi-organ models, which can expand the scope of the study from the brain to other organs. In this way, the microfluidic-based brain organoid can simplify the complex aspects of the human brain compared to in vivo models while keeping sophisticated aspects. Lastly, microfluidic devices are very thin and optically transparent, allowing multiple robust and high-throughput analyses in parallel compared to other in vitro and in vivo models.17Despite the multiple benefits of using the microfluidic system, there are some limitations on the miniaturized brain models in the chip compared to other types of in vitro models and animal models. The minute amounts of cells in the models limit some analyses requiring at least 1 × 105 cells per sample, such as western blotting, immunoprecipitation, exosome purification, fluorescence-activated cell sorting (FACS), and others. In addition, random absorption of biological molecules on the device surface, commonly composed of PDMS or other polymers, occurs which would reduce the detection power of biomarkers in the models. This would further reduce the delivery efficacy of nutrients or other drug candidates to be tested. We believe that continuous advances in the cell culture technique and biomaterials would resolve such issues in the near future. We also propose future directions to improve microfluidic-based CNS disease models as follows:
• Alzheimer's disease-on-chip: current AD models, adopting the overexpression of Aβ mutants or their precursors to promote Aβ depositions in the chip, have elucidated the underlying mechanisms of FAD-associated cases well, but not SAD.17,144 Recently the advent of AD patient-derived iPSCs could reconstitute pathological signatures driven by risk factors found in SAD such as age, gender, and other environmental factor-driven neurodegeneration;145 however, the standardization of SAD models on chips remains to be solved in the future.
• Parkinson disease-on-chip: here, we introduced examples of microfluidic-based PD models allowing the simplification of intricate biological parameters in the study of PD progression. The risk factors simulated in current PD models on chips are limited to the promotion of α-syn depositions, leading to inflammation and neurodegeneration. Other risk factors such as age, gender, and other disorder conditions increasing the PD incidents should be considered and described in future directions.146
• Traumatic brain injury-on-chip: microfluidic platforms enabled the sophisticated control over mechanical stress on neurons and precisely reconstitute the axonal degeneration followed by damage transmission through neuron-to-neuron networks. Given the fact that dysfunctions in other cellular components such as glial cells maintaining homeostasis in the brain can affect TBI conditions,56,58–60 further improvement such as multi-cellular culture is required to construct pathologically-relevant TBI models for the next generation.
• Neurovascular units-on-chip: VD models on chips have demonstrated the transport of risk factors to the brain region and clarified their contributions to the development of CNS disorders. The use of patient iPSC-derived cells, retaining the inherent pathological conditions, could further improve VD models.147 In addition, the dysfunction in neurovascular coupling, the bi-directional crosstalk between neurons and vessels, should be considered in future VD models.148,149 Other risk factors leading to the dysregulation of cerebral blood flow and neurovascular coupling, such as hypertension, atherosclerosis, and stroke, are needed to be accomplished in the next generation of VD models.150–152
• Environmental risk factor-driven dementia-on-chip: here, we described a few examples of microfluidic platforms for the study of ERFDs. Further studies are needed to develop chip-based platforms connecting the brain to other organs reconstituting not only the entry pathways but also neurodegeneration driven by the external risk factors. In the previous section, we introduced a few examples of promising multi-organ systems providing the entry pathways of external risk factors.
Overall, we envision that brain chips would offer versatile platforms to simulate physio-pathologically relevant CNS disorder conditions and powerful model systems to analyse the underling mechanism of complex disease progression; otherwise, such applications are limited by using conventional in vitro or in vivo models.
Author contributions
H. C. supervised the whole project. Y. J. K. mainly wrote the manuscript. Y. X. and J. H. S. contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation (NRF-2020R1A2C2010285, NRF-I21SS7606036) and the Ministry of Health & Welfare and Ministry of Science and ICT (HU22C0115) through the Korea Health Industry Development Institute and Korea Dementia Research Center to H. C. and NRF-2022R1I1A1A01063094 to YJK.References
- Z. Arvanitakis, R. C. Shah and D. A. Bennett, JAMA, J. Am. Med. Assoc., 2019, 322, 1589–1599 CrossRef PubMed.
- J. Gratwicke, M. Jahanshahi and T. Foltynie, Brain, 2015, 138, 1454–1476 CrossRef PubMed.
- A. Nordström and P. Nordström, PLoS Med., 2018, 15, e1002496 CrossRef PubMed.
- D. E. Pankevich, B. M. Altevogt, J. Dunlop, F. H. Gage and S. E. Hyman, Neuron, 2014, 84, 546–553 CrossRef CAS PubMed.
- R. Peters, N. Ee, J. Peters, A. Booth, I. Mudway and K. J. Anstey, J. Alzheimer's Dis., 2019, 70, S145–s163 CAS.
- T. Erkinjuntti, Stroke Vasc. Neurol., 2002, 91–109 CAS.
- Data Bridge Market Research, Global Neurological Disorder Drugs Market, 2022 Search PubMed.
- A. Abbott, Nature, 2011, 480, 161–162 CrossRef CAS PubMed.
- V. K. Gribkoff and L. K. Kaczmarek, Neuropharmacology, 2017, 120, 11–19 CrossRef CAS PubMed.
- D. S. Bassett and M. S. Gazzaniga, Trends Cognit. Sci., 2011, 15, 200–209 CrossRef PubMed.
- E. Drummond and T. Wisniewski, Acta Neuropathol., 2017, 133, 155–175 CrossRef CAS PubMed.
- M. Kapałczyńska, T. Kolenda, W. Przybyła, M. Zajączkowska, A. Teresiak, V. Filas, M. Ibbs, R. Bliźniak, Ł. Łuczewski and K. Lamperska, Arch. Med. Sci., 2018, 14, 910–919 Search PubMed.
- J. Park, I. Wetzel, I. Marriott, D. Dréau, C. D'Avanzo, D. Y. Kim, R. E. Tanzi and H. Cho, Nat. Neurosci., 2018, 21, 941–951 CrossRef CAS PubMed.
- A. McQuade, Y. J. Kang, J. Hasselmann, A. Jairaman, A. Sotelo, M. Coburn, S. K. Shabestari, J. P. Chadarevian, G. Fote, C. H. Tu, E. Danhash, J. Silva, E. Martinez, C. Cotman, G. A. Prieto, L. M. Thompson, J. S. Steffan, I. Smith, H. Davtyan, M. Cahalan, H. Cho and M. Blurton-Jones, Nat. Commun., 2020, 11, 5370 CrossRef CAS PubMed.
- Y. J. Kang, H.-Y. Tan, C. Y. Lee and H. Cho, Adv. Sci., 2021, 8, 2101251 CrossRef CAS PubMed.
- M. F. Chesselet and S. T. Carmichael, Neurotherapeutics, 2012, 9, 241–244 CrossRef PubMed.
- Y. J. Kang and H. Cho, Organoid, 2021, 1, e5 CrossRef.
- P. M. Holloway, S. Willaime-Morawek, R. Siow, M. Barber, R. M. Owens, A. D. Sharma, W. Rowan, E. Hill and M. Zagnoni, J. Neurosci. Res., 2021, 99, 1276–1307 CrossRef CAS PubMed.
- H. Cho, T. Hashimoto, E. Wong, Y. Hori, L. B. Wood, L. Zhao, K. M. Haigis, B. T. Hyman and D. Irimia, Sci. Rep., 2013, 3, 1823 CrossRef PubMed.
- J. M. Long and D. M. Holtzman, Cell, 2019, 179, 312–339 CrossRef CAS PubMed.
- Y. H. Kim, S. H. Choi, C. D'Avanzo, M. Hebisch, C. Sliwinski, E. Bylykbashi, K. J. Washicosky, J. B. Klee, O. Brüstle, R. E. Tanzi and D. Y. Kim, Nat. Protoc., 2015, 10, 985–1006 CrossRef CAS PubMed.
- J. W. Wu, M. Herman, L. Liu, S. Simoes, C. M. Acker, H. Figueroa, J. I. Steinberg, M. Margittai, R. Kayed, C. Zurzolo, G. Di Paolo and K. E. Duff, J. Biol. Chem., 2013, 288, 1856–1870 CrossRef CAS PubMed.
- K. V. Kuchibhotla, S. Wegmann, K. J. Kopeikina, J. Hawkes, N. Rudinskiy, M. L. Andermann, T. L. Spires-Jones, B. J. Bacskai and B. T. Hyman, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 510–514 CrossRef CAS PubMed.
- A. Michalicova, P. Majerova and A. Kovac, Front. Mol. Neurosci., 2020, 13, 1 CrossRef PubMed.
- Y. J. Kang, Y. N. Diep, M. Tran and H. Cho, Int. J. Mol. Sci., 2020, 21, 9591 CrossRef CAS PubMed.
- H. Chun, H. Im, Y. J. Kang, Y. Kim, J. H. Shin, W. Won, J. Lim, Y. Ju, Y. M. Park, S. Kim, S. E. Lee, J. Lee, J. Woo, Y. Hwang, H. Cho, S. Jo, J.-H. Park, D. Kim, D. Y. Kim, J.-S. Seo, B. J. Gwag, Y. S. Kim, K. D. Park, B.-K. Kaang, H. Cho, H. Ryu and C. J. Lee, Nat. Neurosci., 2020, 23, 1555–1566 CrossRef CAS PubMed.
- A. Nunomura, R. J. Castellani, X. Zhu, P. I. Moreira, G. Perry and M. A. Smith, J. Neuropathol. Exp. Neurol., 2006, 65, 631–641 CrossRef CAS PubMed.
- C. M. Wolfe, N. F. Fitz, K. N. Nam, I. Lefterov and R. Koldamova, Int. J. Mol. Sci., 2018, 20, 81 CrossRef PubMed.
- P. M. Thompson, K. M. Hayashi, G. de Zubicaray, A. L. Janke, S. E. Rose, J. Semple, D. Herman, M. S. Hong, S. S. Dittmer, D. M. Doddrell and A. W. Toga, J. Neurosci., 2003, 23, 994–1005 CrossRef CAS PubMed.
- A. Lleó, R. Núñez-Llaves, D. Alcolea, C. Chiva, D. Balateu-Paños, M. Colom-Cadena, G. Gomez-Giro, L. Muñoz, M. Querol-Vilaseca, J. Pegueroles, L. Rami, A. Lladó, J. L. Molinuevo, M. Tainta, J. Clarimón, T. Spires-Jones, R. Blesa, J. Fortea, P. Martínez-Lage, R. Sánchez-Valle, E. Sabidó, À. Bayés and O. Belbin, Mol. Cell. Proteomics, 2019, 18, 546–560 CrossRef PubMed.
- D. J. Gelb, E. Oliver and S. Gilman, Arch. Neurol., 1999, 56, 33–39 CrossRef CAS PubMed.
- K. R. Chaudhuri, D. G. Healy and A. H. V. Schapira, Lancet Neurol., 2006, 5, 235–245 CrossRef PubMed.
- W. Poewe, K. Seppi, C. M. Tanner, G. M. Halliday, P. Brundin, J. Volkmann, A.-E. Schrag and A. E. Lang, Nat. Rev. Dis. Primers, 2017, 3, 17013 CrossRef PubMed.
- R. Balestrino and A. H. V. Schapira, Eur. J. Neurol., 2020, 27, 27–42 CrossRef CAS PubMed.
- L. Breydo, J. W. Wu and V. N. Uversky, Biochim. Biophys. Acta, 2012, 1822, 261–285 CrossRef CAS PubMed.
- M. Spillantini, M.-Y. Lee, J. Q. Trojanowski and R. M. Goedert, Nature, 1997, 388, 839–840 CrossRef CAS PubMed.
- K. M. Danzer, L. R. Kranich, W. P. Ruf, O. Cagsal-Getkin, A. R. Winslow, L. Zhu, C. R. Vanderburg and P. J. McLean, Mol. Neurodegener., 2012, 7, 42–42 CrossRef CAS PubMed.
- R. J. Karpowicz, Jr., J. Q. Trojanowski and V. M. Lee, Lab. Invest., 2019, 99, 971–981 CrossRef PubMed.
- E. Angot and P. Brundin, Parkinsonism Relat. Disord., 2009, 15, S143–S147 CrossRef PubMed.
- J. T. Fernandes, O. Chutna, V. Chu, J. P. Conde and T. F. Outeiro, Front. Neurosci., 2016, 10, 511 Search PubMed.
- H.-J. Lee, E.-J. Bae and S.-J. Lee, Nat. Rev. Neurol., 2014, 10, 92–98 CrossRef CAS PubMed.
- H. Braak, U. Rub, W. P. Gai and K. Del Tredici, J. Neural Transm., 2003, 110, 517–536 CrossRef CAS PubMed.
- H. A. Lashuel, C. R. Overk, A. Oueslati and E. Masliah, Nat. Rev. Neurosci., 2013, 14, 38–48 CrossRef CAS PubMed.
- B. I. Giasson, J. E. Duda, S. M. Quinn, B. Zhang, J. Q. Trojanowski and V. M. Y. Lee, Neuron, 2002, 34, 521–533 CrossRef CAS PubMed.
- M. Gómez-Benito, N. Granado, P. García-Sanz, A. Michel, M. Dumoulin and R. Moratalla, Front. Pharmacol., 2020, 11, 356 CrossRef PubMed.
- D. A. Olszewska, A. McCarthy, A. I. Soto-Beasley, R. L. Walton, O. A. Ross and T. Lynch, Ir. J. Med. Sci., 2022, 191, 901–907 CrossRef CAS PubMed.
- H. Shimura, M. G. Schlossmacher, N. Hattori, M. P. Frosch, A. Trockenbacher, R. Schneider, Y. Mizuno, K. S. Kosik and D. J. Selkoe, Science, 2001, 293, 263–269 CrossRef CAS PubMed.
- F. A. Perez and R. D. Palmiter, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2174–2179 CrossRef CAS PubMed.
- P. Ge, V. L. Dawson and T. M. Dawson, Mol. Neurodegener., 2020, 15, 20 CrossRef PubMed.
- T. Kitada, A. Pisani, D. R. Porter, H. Yamaguchi, A. Tscherter, G. Martella, P. Bonsi, C. Zhang, E. N. Pothos and J. Shen, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11441–11446 CrossRef CAS PubMed.
- M. S. Goldberg, A. Pisani, M. Haburcak, T. A. Vortherms, T. Kitada, C. Costa, Y. Tong, G. Martella, A. Tscherter, A. Martins, G. Bernardi, B. L. Roth, E. N. Pothos, P. Calabresi and J. Shen, Neuron, 2005, 45, 489–496 CrossRef CAS PubMed.
- K. D. Dave, S. De Silva, N. P. Sheth, S. Ramboz, M. J. Beck, C. Quang, R. C. Switzer, S. O. Ahmad, S. M. Sunkin, D. Walker, X. Cui, D. A. Fisher, A. M. McCoy, K. Gamber, X. Ding, M. S. Goldberg, S. A. Benkovic, M. Haupt, M. A. S. Baptista, B. K. Fiske, T. B. Sherer and M. A. Frasier, Neurobiol. Dis., 2014, 70, 190–203 CrossRef CAS PubMed.
- D. Ramonet, J. P. L. Daher, B. M. Lin, K. Stafa, J. Kim, R. Banerjee, M. Westerlund, O. Pletnikova, L. Glauser, L. Yang, Y. Liu, D. A. Swing, M. F. Beal, J. C. Troncoso, J. M. McCaffery, N. A. Jenkins, N. G. Copeland, D. Galter, B. Thomas, M. K. Lee, T. M. Dawson, V. L. Dawson and D. J. Moore, PLoS One, 2011, 6, e18568 CrossRef CAS PubMed.
- X. Zhang, Y. Chen, L. W. Jenkins, P. M. Kochanek and R. S. Clark, Crit. Care, 2005, 9, 66–75 CrossRef PubMed.
- Y. Zhou, A. Shao, Y. Yao, S. Tu, Y. Deng and J. Zhang, Cell Commun. Signaling, 2020, 18, 62 CrossRef PubMed.
- E. J. Perez, S. A. Tapanes, Z. B. Loris, D. T. Balu, T. J. Sick, J. T. Coyle and D. J. Liebl, J. Clin. Invest., 2017, 127, 3114–3125 CrossRef PubMed.
- D. A. Sun, L. S. Deshpande, S. Sombati, A. Baranova, M. S. Wilson, R. J. Hamm and R. J. DeLorenzo, Eur. J. Neurosci., 2008, 27, 1659–1672 CrossRef PubMed.
- J. E. Burda, A. M. Bernstein and M. V. Sofroniew, Exp. Neurol., 2016, 275, 305–315 CrossRef CAS PubMed.
- K. V. Rao, P. V. Reddy, K. M. Curtis and M. D. Norenberg, J. Neurotrauma, 2011, 28, 371–381 CrossRef PubMed.
- B. L. Bartnik-Olson, U. Oyoyo, D. A. Hovda and R. L. Sutton, J. Neurotrauma, 2010, 27, 2191–2202 CrossRef PubMed.
- D. J. Myer, G. G. Gurkoff, S. M. Lee, D. A. Hovda and M. V. Sofroniew, Brain, 2006, 129, 2761–2772 CrossRef CAS PubMed.
- A. N. Early, A. A. Gorman, L. J. Van Eldik, A. D. Bachstetter and J. M. Morganti, J. Neuroinflammation, 2020, 17, 115 CrossRef CAS PubMed.
- E. Zhao, L. Bai, S. Li, L. Li, Z. Dou, Y. Huang, Y. Li and Y. Lv, Neurotoxic. Res., 2020, 38, 723–732 CrossRef CAS PubMed.
- O. A. Skrobot, J. O'Brien, S. Black, C. Chen, C. DeCarli, T. Erkinjuntti, G. A. Ford, R. N. Kalaria, L. Pantoni, F. Pasquier, G. C. Roman, A. Wallin, P. Sachdev, I. Skoog, F. E. Taragano, J. Kril, M. Cavalieri, K. A. Jellinger, G. G. Kovacs, S. Engelborghs, C. Lafosse, P. H. Bertolucci, S. Brucki, P. Caramelli, T. C. de Toledo Ferraz Alves, C. Bocti, T. Fulop, D. B. Hogan, G. R. Hsiung, A. Kirk, L. Leach, A. Robillard, D. J. Sahlas, Q. Guo, J. Tian, L. Hokkanen, H. Jokinen, S. Benisty, V. Deramecourt, J. Hauw, H. Lenoir, M. Tsatali, M. Tsolaki, U. Sundar, R. F. Coen, A. D. Korczyn, M. Altieri, M. Baldereschi, C. Caltagirone, G. Caravaglios, A. Di Carlo, V. D. I. Piero, G. Gainotti, S. Galluzzi, G. Logroscino, P. Mecocci, D. V. Moretti, A. Padovani, T. Fukui, M. Ihara, T. Mizuno, S. Y. Kim, R. Akinyemi, O. Baiyewu, A. Ogunniyi, A. Szczudlik, A. J. Bastos-Leite, H. Firmino, J. Massano, A. Verdelho, L. S. Kruglov, M. K. Ikram, N. Kandiah, E. Arana, J. Barroso-Ribal, T. Calatayud, A. J. Cruz-Jentoft, S. López-Pousa, P. Martinez-Lage, M. Mataro, A. Börjesson-Hanson, E. Englund, E. J. Laukka, C. Qiu, M. Viitanen, G. J. Biessels, F.-E. de Leeuw, T. den Heijer, L. G. Exalto, L. J. Kappelle, N. D. Prins, E. Richard, B. Schmand, E. van den Berg, W. M. van der Flier, B. Bilgic, L. M. Allan, J. Archer, J. Attems, A. Bayer, D. Blackburn, C. Brayne, R. Bullock, P. J. Connelly, A. Farrant, M. Fish, K. Harkness, P. G. Ince, P. Langhorne, J. Mann, F. E. Matthews, P. Mayer, S. T. Pendlebury, R. Perneczky, R. Peters, D. Smithard, B. C. Stephan, J. E. Swartz, S. Todd, D. J. Werring, S. N. Wijayasiri, G. Wilcock, G. Zamboni, R. Au, S. Borson, A. Bozoki, J. N. Browndyke, M. M. Corrada, P. K. Crane, B. S. Diniz, L. Etcher, H. Fillit, S. M. Greenberg, L. T. Grinberg, S. W. Hurt, M. Lamar, M. Mielke, B. R. Ott, G. Perry, W. J. Powers, C. Ramos-Estebanez, B. Reed, R. O. Roberts, J. R. Romero, A. J. Saykin, S. Seshadri, L. Silbert, Y. Stern, C. Zarow, Y. Ben-Shlomo, A. P. Passmore, S. Love and P. G. Kehoe, Alzheimer's Dementia, 2017, 13, 624–633 CrossRef PubMed.
- M. Hosokawa and M. Ueno, Neurobiol. Aging, 1999, 20, 117–123 CrossRef CAS PubMed.
- A. W. Vorbrodt, D. H. Dobrogowska, M. Ueno and M. Tarnawski, Folia Histochem. Cytobiol., 1995, 33, 229–237 CAS.
- C. Pelegrí, A. M. Canudas, J. del Valle, G. Casadesus, M. A. Smith, A. Camins, M. Pallàs and J. Vilaplana, Mech. Ageing Dev., 2007, 128, 522–528 CrossRef PubMed.
- Y. Fan, L. Lan, L. Zheng, X. Ji, J. Lin, J. Zeng, R. Huang and J. Sun, Metab. Brain Dis., 2015, 30, 1479–1486 CrossRef PubMed.
- E. S. Lee, J. H. Yoon, J. Choi, F. R. Andika, T. Lee and Y. Jeong, J. Cereb. Blood Flow Metab., 2019, 39, 44–57 CrossRef PubMed.
- G. Nzou, R. T. Wicks, E. E. Wicks, S. A. Seale, C. H. Sane, A. Chen, S. V. Murphy, J. D. Jackson and A. J. Atala, Sci. Rep., 2018, 8, 7413 CrossRef PubMed.
- D. Herr, K. Jew, C. Wong, A. Kennell, R. Gelein, D. Chalupa, A. Raab, G. Oberdörster, J. Olschowka, M. K. O'Banion and A. Elder, Neurotoxicology, 2021, 84, 172–183 CrossRef CAS PubMed.
- S.-H. Lee, Y.-H. Chen, C.-C. Chien, Y.-H. Yan, H.-C. Chen, H.-C. Chuang, H.-I. Hsieh, K.-H. Cho, L.-W. Kuo, C. C. K. Chou, M.-J. Chiu, B. L. Tee, T.-F. Chen and T.-J. Cheng, PLoS One, 2021, 16, e0254587 CrossRef CAS PubMed.
- K. Fassbender, M. Simons, C. Bergmann, M. Stroick, D. Lütjohann, P. Keller, H. Runz, S. Kühl, T. Bertsch, K. von Bergmann, M. Hennerici, K. Beyreuther and T. Hartmann, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5856–5861 CrossRef CAS PubMed.
- H.-K. Lee, C. Velazquez Sanchez, M. Chen, P. J. Morin, J. M. Wells, E. B. Hanlon and W. Xia, PLoS One, 2016, 11, e0163072 CrossRef PubMed.
- Y. Shin, S. H. Choi, E. Kim, E. Bylykbashi, J. A. Kim, S. Chung, D. Y. Kim, R. D. Kamm and R. E. Tanzi, Adv. Sci., 2019, 6, 1900962 CrossRef CAS PubMed.
- E. McGowan, F. Pickford, J. Kim, L. Onstead, J. Eriksen, C. Yu, L. Skipper, M. P. Murphy, J. Beard, P. Das, K. Jansen, M. DeLucia, W.-L. Lin, G. Dolios, R. Wang, C. B. Eckman, D. W. Dickson, M. Hutton, J. Hardy and T. Golde, Neuron, 2005, 47, 191–199 CrossRef CAS PubMed.
- M. Jorfi, C. D'Avanzo, R. E. Tanzi, D. Y. Kim and D. Irimia, Sci. Rep., 2018, 8, 2450 CrossRef PubMed.
- C. M. Pedrero-Prieto, A. Flores-Cuadrado, D. Saiz-Sánchez, I. Úbeda-Bañón, J. Frontiñán-Rubio, F. J. Alcaín, L. Mateos-Hernández, J. de la Fuente, M. Durán-Prado, M. Villar, A. Martínez-Marcos and J. R. Peinado, Alzheimer's Res. Ther., 2019, 11, 56 CrossRef CAS PubMed.
- S. Dujardin, K. Lécolle, R. Caillierez, S. Bégard, N. Zommer, C. Lachaud, S. Carrier, N. Dufour, G. Aurégan, J. Winderickx, P. Hantraye, N. Déglon, M. Colin and L. Buée, Acta Neuropathol. Commun., 2014, 2, 14 CrossRef PubMed.
- S. Takeda, S. Wegmann, H. Cho, S. L. DeVos, C. Commins, A. D. Roe, S. B. Nicholls, G. A. Carlson, R. Pitstick, C. K. Nobuhara, I. Costantino, M. P. Frosch, D. J. Müller, D. Irimia and B. T. Hyman, Nat. Commun., 2015, 6, 8490 CrossRef CAS PubMed.
- J. Park, B. K. Lee, G. S. Jeong, J. K. Hyun, C. J. Lee and S.-H. Lee, Lab Chip, 2015, 15, 141–150 RSC.
- P. Thakur, L. S. Breger, M. Lundblad, O. W. Wan, B. Mattsson, K. C. Luk, V. M. Y. Lee, J. Q. Trojanowski and A. Björklund, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E8284–E8293 CrossRef CAS PubMed.
- M. F. Duffy, T. J. Collier, J. R. Patterson, C. J. Kemp, K. C. Luk, M. G. Tansey, K. L. Paumier, N. M. Kanaan, D. L. Fischer, N. K. Polinski, O. L. Barth, J. W. Howe, N. N. Vaikath, N. K. Majbour, O. M. A. El-Agnaf and C. E. Sortwell, J. Neuroinflammation, 2018, 15, 129 CrossRef PubMed.
- W. Cai, D. Feng, M. A. Schwarzschild, P. J. McLean and X. Chen, EBioMedicine, 2018, 29, 13–22 CrossRef PubMed.
- M. W. C. Rousseaux, M. de Haro, C. A. Lasagna-Reeves, A. De Maio, J. Park, P. Jafar-Nejad, I. Al-Ramahi, A. Sharma, L. See, N. Lu, L. Vilanova-Velez, T. J. Klisch, T. F. Westbrook, J. C. Troncoso, J. Botas and H. Y. Zoghbi, eLife, 2016, 5, e19809 CrossRef PubMed.
- E. C. Freundt, N. Maynard, E. K. Clancy, S. Roy, L. Bousset, Y. Sourigues, M. Covert, R. Melki, K. Kirkegaard and M. Brahic, Ann. Neurol., 2012, 72, 517–524 CrossRef CAS PubMed.
- X. Mao, M. T. Ou, S. S. Karuppagounder, T. I. Kam, X. Yin, Y. Xiong, P. Ge, G. E. Umanah, S. Brahmachari, J. H. Shin, H. C. Kang, J. Zhang, J. Xu, R. Chen, H. Park, S. A. Andrabi, S. U. Kang, R. A. Gonçalves, Y. Liang, S. Zhang, C. Qi, S. Lam, J. A. Keiler, J. Tyson, D. Kim, N. Panicker, S. P. Yun, C. J. Workman, D. A. Vignali, V. L. Dawson, H. S. Ko and T. M. Dawson, Science, 2016, 353, aah3374 CrossRef CAS PubMed.
- I. Prots, J. Grosch, R.-M. Brazdis, K. Simmnacher, V. Veber, S. Havlicek, C. Hannappel, F. Krach, M. Krumbiegel, O. Schütz, A. Reis, W. Wrasidlo, D. R. Galasko, T. W. Groemer, E. Masliah, U. Schlötzer-Schrehardt, W. Xiang, J. Winkler and B. Winner, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7813–7818 CrossRef CAS PubMed.
- A. M. Taylor, N. C. Berchtold, V. M. Perreau, C. H. Tu, N. Li Jeon and C. W. Cotman, J. Neurosci., 2009, 29, 4697–4707 CrossRef CAS PubMed.
- J.-N. Zhang, U. Michel, C. Lenz, C. C. Friedel, S. Köster, Z. d'Hedouville, L. Tönges, H. Urlaub, M. Bähr, P. Lingor and J. C. Koch, Sci. Rep., 2016, 6, 37050 CrossRef CAS PubMed.
- J. P. Lezana, S. Y. Dagan, A. Robinson, R. S. Goldstein, M. Fainzilber, F. C. Bronfman and M. Bronfman, Dev. Neurobiol., 2016, 76, 688–701 CrossRef CAS PubMed.
- H. J. Kim, J. W. Park, J. W. Park, J. H. Byun, B. Vahidi, S. W. Rhee and N. L. Jeon, Ann. Biomed. Eng., 2012, 40, 1268–1276 CrossRef PubMed.
- S. Hosmane, A. Fournier, R. Wright, L. Rajbhandari, R. Siddique, I. H. Yang, K. T. Ramesh, A. Venkatesan and N. Thakor, Lab Chip, 2011, 11, 3888–3895 RSC.
- J. P. Dollé, B. Morrison, 3rd, R. S. Schloss and M. L. Yarmush, Lab Chip, 2013, 13, 432–442 RSC.
- E. A. Rogers, T. Beauclair, A. Thyen and R. Shi, Sci. Rep., 2022, 12, 11838 CrossRef CAS PubMed.
- A. J. Samson, G. Robertson, M. Zagnoni and C. N. Connolly, Sci. Rep., 2016, 6, 33746 CrossRef CAS PubMed.
- D. Charier, O. Beauchet, M. Bell, B. Brugg, R. Bartha and C. Annweiler, ACS Chem. Neurosci., 2015, 6, 393–397 CrossRef CAS PubMed.
- I. Pediaditakis, K. R. Kodella, D. V. Manatakis, C. Y. Le, C. D. Hinojosa, W. Tien-Street, E. S. Manolakos, K. Vekrellis, G. A. Hamilton, L. Ewart, L. L. Rubin and K. Karalis, Nat. Commun., 2021, 12, 5907 CrossRef CAS PubMed.
- V. T. A. Tran, Y. J. Kang, H.-K. Kim, H.-R. Kim and H. Cho, Int. J. Mol. Sci., 2021, 22, 6925 CrossRef CAS PubMed.
- Y. Koo, B. T. Hawkins and Y. Yun, Sci. Rep., 2018, 8, 2841 CrossRef PubMed.
- L. Liu, Y. Koo, T. Russell, E. Gay, Y. Li and Y. Yun, PLoS One, 2020, 15, e0230335 CrossRef CAS PubMed.
- C. L. Sokol and A. D. Luster, Cold Spring Harbor Perspect. Biol., 2015, 7, a016303 CrossRef PubMed.
- E. L. Moreno, S. Hachi, K. Hemmer, S. J. Trietsch, A. S. Baumuratov, T. Hankemeier, P. Vulto, J. C. Schwamborn and R. M. Fleming, Lab Chip, 2015, 15, 2419–2428 RSC.
- A. M. Taylor, M. Blurton-Jones, S. W. Rhee, D. H. Cribbs, C. W. Cotman and N. L. Jeon, Nat. Methods, 2005, 2, 599–605 CrossRef CAS PubMed.
- H. Cho, J. H. Seo, K. H. K. Wong, Y. Terasaki, J. Park, K. Bong, K. Arai, E. H. Lo and D. Irimia, Sci. Rep., 2015, 5, 15222 CrossRef CAS PubMed.
- B. J. DeOre, K. A. Tran, A. M. Andrews, S. H. Ramirez and P. A. Galie, J. Neuroimmune Pharmacol., 2021, 16, 722–728 CrossRef PubMed.
- P. P. Partyka, G. A. Godsey, J. R. Galie, M. C. Kosciuk, N. K. Acharya, R. G. Nagele and P. A. Galie, Biomaterials, 2017, 115, 30–39 CrossRef CAS PubMed.
- M. Carabotti, A. Scirocco, M. A. Maselli and C. Severi, Ann. Gastroenterol., 2015, 28, 203–209 Search PubMed.
- C. Li, W. Chen, F. Lin, W. Li, P. Wang, G. Liao and L. Zhang, Cell. Mol. Neurobiol., 2022 DOI:10.1007/s10571-022-01238-z.
- O. Bajinka, L. Simbilyabo, Y. Tan, J. Jabang and S. A. Saleem, Crit. Rev. Microbiol., 2022, 48, 257–269 CrossRef PubMed.
- S. Jo, W. Kang, Y. S. Hwang, S. H. Lee, K. W. Park, M. S. Kim, H. Lee, H. J. Yoon, Y. K. Park, M. Chalita, J. H. Lee, H. Sung, J. Y. Lee, J. W. Bae and S. J. Chung, npj Parkinson's Dis., 2022, 8, 87 CrossRef CAS PubMed.
- F. Baert, C. Matthys, J. Maselyne, C. Van Poucke, E. Van Coillie, B. Bergmans and G. Vlaemynck, npj Parkinson's Dis., 2021, 7, 72 CrossRef CAS PubMed.
- X. H. Qian, X. X. Song, X. L. Liu, S. D. Chen and H. D. Tang, Ageing Res. Rev., 2021, 68, 101317 CrossRef CAS PubMed.
- C. Fulling, T. G. Dinan and J. F. Cryan, Neuron, 2019, 101, 998–1002 CrossRef CAS PubMed.
- C. G. de Theije, J. Wu, S. L. da Silva, P. J. Kamphuis, J. Garssen, S. M. Korte and A. D. Kraneveld, Eur. J. Pharmacol., 2011, 668(Suppl 1), S70–S80 CrossRef CAS PubMed.
- E. A. Mayer, D. Padua and K. Tillisch, BioEssays, 2014, 36, 933–939 CrossRef PubMed.
- A. Fasano, N. P. Visanji, L. W. C. Liu, A. E. Lang and R. F. Pfeiffer, Lancet Neurol., 2015, 14, 625–639 CrossRef CAS PubMed.
- M. G. Cersosimo, G. B. Raina, C. Pecci, A. Pellene, C. R. Calandra, C. Gutiérrez, F. E. Micheli and E. E. Benarroch, J. Neurol., 2013, 260, 1332–1338 CrossRef CAS PubMed.
- K. L. Adams-Carr, J. P. Bestwick, S. Shribman, A. Lees, A. Schrag and A. J. Noyce, J. Neurol., Neurosurg. Psychiatry, 2016, 87, 710–716 CrossRef PubMed.
- T. H. Mertsalmi, V. T. E. Aho, P. A. B. Pereira, L. Paulin, E. Pekkonen, P. Auvinen and F. Scheperjans, Eur. J. Neurol., 2017, 24, 1375–1383 CrossRef CAS PubMed.
- M.-H. Kim, D. Kim and J. H. Sung, J. Ind. Eng. Chem., 2021, 101, 126–134 CrossRef CAS.
- M. A.-O. Trapecar, E. Wogram, D. Svoboda, C. Communal, A. A.-O. Omer, T. Lungjangwa, P. Sphabmixay, J. A.-O. Velazquez, K. Schneider, C. W. Wright, S. A.-O. Mildrum, A. A.-O. Hendricks, S. A.-O. X. Levine, J. A.-O. Muffat, M. J. Lee, D. A.-O. X. Lauffenburger, D. A.-O. Trumper, R. A.-O. Jaenisch and L. A.-O. Griffith, Sci. Adv., 2021, 5, 1707 CrossRef PubMed.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS PubMed.
- D. D. Huh, Ann. Am. Thorac. Soc., 2015, 12(Suppl 1), S42–S44 CrossRef PubMed.
- M. Xu, Y. Wang, W. Duan, S. Xia, S. Wei, W. Liu and Q. Wang, Front. Bioeng. Biotechnol., 2020, 8, 612091 CrossRef PubMed.
- J. Byun, B. Song, K. Lee, B. Kim, H. W. Hwang, M.-R. Ok, H. Jeon, K. Lee, S.-K. Baek, S.-H. Kim, S. J. Oh and T. H. Kim, J. Biol. Eng., 2019, 13, 88 CrossRef PubMed.
- L. J. Chen, S. Ito, H. Kai, K. Nagamine, N. Nagai, M. Nishizawa, T. Abe and H. Kaji, Sci. Rep., 2017, 7, 3538 CrossRef PubMed.
- M. Wufuer, G. Lee, W. Hur, B. Jeon, B. J. Kim, T. H. Choi and S. Lee, Sci. Rep., 2016, 6, 37471 CrossRef CAS PubMed.
- S. Nagayama, R. Homma and F. Imamura, Front. Neural Circuits, 2014, 8, 98 Search PubMed.
- Y. Li, Y. Liu, C. Hu, Q. Chang, Q. Deng, X. Yang and Y. Wu, Environ. Int., 2020, 143, 105598 CrossRef CAS PubMed.
- X. Ji, R. Liu, J. Guo, Y. Li, W. Cheng, Y. Pang, Y. Zheng, R. Zhang and J. Tang, Sci. Total Environ., 2022, 821, 153456 CrossRef CAS PubMed.
- N. Uemura, J. Ueda, T. Yoshihara, M. Ikuno, M. T. Uemura, H. Yamakado, M. Asano, J. Q. Trojanowski and R. Takahashi, Mov. Disord., 2021, 36, 2036–2047 CrossRef CAS PubMed.
- M. M. Esiri and G. K. Wilcock, J. Neurol., Neurosurg. Psychiatry, 1984, 47, 56–60 CrossRef CAS PubMed.
- J. Alves, A. Petrosyan and R. Magalhães, World J. Clin. Cases, 2014, 2, 661–667 CrossRef PubMed.
- K. Na, M. Lee, H.-W. Shin and S. Chung, Lab Chip, 2017, 17, 1578–1584 RSC.
- A. Vlasiuk and H. Asari, PLoS One, 2021, 16, e0254611 CrossRef CAS PubMed.
- A. London, I. Benhar and M. Schwartz, Nat. Rev. Neurol., 2013, 9, 44–53 CrossRef CAS PubMed.
- C. Borm, F. Visser, M. Werkmann, D. de Graaf, D. Putz, K. Seppi, W. Poewe, A. M. M. Vlaar, C. Hoyng, B. R. Bloem, T. Theelen and N. M. de Vries, Neurology, 2020, 94, e1539–e1547 CrossRef PubMed.
- H. J. Kim, J. H. Ryou, K. T. Choi, S. M. Kim, J. T. Kim and D. H. Han, PLoS One, 2022, 17, e0262226 CrossRef CAS PubMed.
- J. Wu, H. K. Mak, Y. K. Chan, C. Lin, C. Kong, C. K. S. Leung and H. C. Shum, Sci. Rep., 2019, 9, 9057 CrossRef PubMed.
- D. R. Fregoso, Y. Hadian, A. C. Gallegos, D. Degovics, J. Maaga, C. E. Keogh, I. Kletenik, M. G. Gareau and R. R. Isseroff, Brain, Behav., Immun.: Health, 2021, 15, 100279 CAS.
- J. A. Mapunda, H. Tibar, W. Regragui and B. Engelhardt, Front. Immunol., 2022, 13, 805657 CrossRef CAS PubMed.
- J. Wan, S. Zhou, H. J. Mea, Y. Guo, H. Ku and B. M. Urbina, Chem. Rev., 2022, 122, 7142–7181 CrossRef CAS PubMed.
- Y. J. Kang, Y. N. Diep, M. Tran and H. Cho, Int. J. Mol. Sci., 2020, 21, 1–34 Search PubMed.
- D. S. Knopman, H. Amieva, R. C. Petersen, G. Chételat, D. M. Holtzman, B. T. Hyman, R. A. Nixon and D. T. Jones, Nat. Rev. Dis. Primers, 2021, 7, 33 CrossRef PubMed.
- C. H. Lin, J. W. Lin, Y. C. Liu, C. H. Chang and R. M. Wu, Parkinsonism Relat. Disord., 2014, 20, 1371–1375 CrossRef PubMed.
- G. D. Vatine, R. Barrile, M. J. Workman, S. Sances, B. K. Barriga, M. Rahnama, S. Barthakur, M. Kasendra, C. Lucchesi, J. Kerns, N. Wen, W. R. Spivia, Z. Chen, J. Van Eyk and C. N. Svendsen, Cell Stem Cell, 2019, 24, 995–1005.e1006 CrossRef CAS PubMed.
- L. Kaplan, B. W. Chow and C. Gu, Nat. Rev. Neurosci., 2020, 21, 416–432 CrossRef CAS PubMed.
- T. Osaki, V. Sivathanu and R. D. Kamm, Sci. Rep., 2018, 8, 5168 CrossRef PubMed.
- S. Tarantini, C. H. T. Tran, G. R. Gordon, Z. Ungvari and A. Csiszar, Exp. Gerontol., 2017, 94, 52–58 CrossRef CAS PubMed.
- O. Shabir, J. Berwick and S. E. Francis, BMC Neurosci., 2018, 19, 62 CrossRef CAS PubMed.
- H. Girouard and C. Iadecola, J. Appl. Physiol., 2006, 100, 328–335 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |