
Designing electrocatalysts for seawater splitting: surface/interface engineering toward enhanced electrocatalytic performance
Bo
Xu
 ab,
Jie
Liang
c,
Xuping
Sun
*c and
Xiaoli
Xiong
*a
ab,
Jie
Liang
c,
Xuping
Sun
*c and
Xiaoli
Xiong
*a
aCollege of Chemistry and Materials Science, Sichuan Normal University, Chengdu 610068, Sichuan, China. E-mail: xiongxiaoli2000@163.com
bCollege of Architecture and Environment, Sichuan University, Chengdu 610065, Sichuan, China
cInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 610054, Sichuan, China. E-mail: xpsun@uestc.edu.cn
First published on 12th April 2023
Abstract
As an ideal large-scale energy conversion/storage technology, electrochemical hydrogen production has great potential as a means of smoothing out the volatility of renewable sources. Electrocatalytic seawater splitting utilizes abundant natural seawater to replace purified water; this has considerable economic and environmental benefits, and will greatly expand the applications scope of water splitting. However, complex compositions existing in natural seawater hinder efficient H2 electrosynthesis, especially chlorides that corrode the catalysts. Advanced surface and interface engineering has been demonstrated to be critical for the construction of efficient and stable electrodes for seawater electrolysis. In the review, we firstly introduce the fundamentals of direct seawater splitting and provide a comprehensive analysis of the basic reactions on electrodes from the perspective of thermodynamics and kinetics. Subsequently, rational design strategies for HER and OER electrocatalysts applied to seawater or chloride-containing electrolytes in terms of catalytic activity, selectivity and corrosion resistance are discussed comprehensively. Moreover, the applications of surface/interface engineering in the performance tuning of seawater electrolytic electrocatalysts are presented. Finally, the current state of the research is presented, along with potential areas for further innovation.
1. Introduction
The dual challenges of energy and environment are forcing a profound shift in global energy consumption structure, replacing traditional fossil energy with renewable energy sources (such as solar, wind and hydropower).1–3 However, the intermittent nature of electricity generation from these renewable sources is compelling the adoption of efficient energy conversion and storage technologies to ensure consistent energy supply.4–6 Among the various energy conversion/storage technologies, hydrogen is considered to be the most promising path due to its high energy density, clean exhausts, easy conversion and mature supporting facilities.7–10 Notably, the current tremendous endeavours in electrochemical water splitting mainly focus on purified water systems, inevitably limiting the spread of their application in areas of fresh water scarcity, such as the Middle East and North Africa.11–14 For this reason, researchers have proposed an electrochemical seawater splitting method that directly uses natural seawater as the hydrogen source, aiming to convert abundant seawater into hydrogen for energy storage.As an emerging technology, low-temperature direct seawater splitting faces fierce competition, including from high-temperature steam electrolysis and desalination-coupled electrolysis.15–17 With mature equipment and stable operation, the desalination-coupled electrolysis technology demonstrates comprehensive advantages for large-scale energy storage projects. However, for numerous small-scale coastal photovoltaic/wind energy storage devices, direct seawater splitting offers a more economically competitive option.11,14,18 By optimizing the electrolytic cell, electrocatalyst and ion exchange membrane to remedy disadvantages in corrosion and stability, extracting hydrogen directly from seawater avoids the disadvantages of the high upfront construction investment and post-operation costs of seawater desalination plants.19–23 Besides, the application of seawater also provides salutary attempts to broaden the use of saline surface water/recycled water, with significant economic and environmental benefits. However, complex solutes also pose a significant challenge to the electrolysis system. Compared with the purified water electrolysis system, the presence of various impurities (such as inorganic ions, insoluble micro-particles and microbes) in natural seawater inevitably has a negative impact on the electrolytic system, especially for the electrodes. Specifically, the competitive adsorption of chlorine ions (Cl−) during the oxygen evolution reaction (OER) blocks the active site on the catalyst; meanwhile, the chlorine-oxidation reaction impedes the OER reaction and forms chlorine-containing products, which accelerate the corrosion or degradation of the electrocatalyst.3,19,24 Besides, the local increase in pH (pH > 9.5) due to electrolysis may lead to insoluble deposits of cations (e.g., Mg2+ and Ca2+) in solution, blocking some active sites and thus reducing the catalytic performance.19,25 Therefore, the rational design for seawater electrolysis catalysts needs to ensure high catalytic activity while taking into account selectivity and stability.
The surface/interface, where the catalytic reactions occur, directly involving the adsorption/desorption of intermediates and transfer of electrons, is crucial for catalytic performance.26,27
Therefore, surface and interface engineering, which directly modulates the surface/interface physicochemical characteristics of the relevant catalyst, is one of the most effective means to enhance the catalytic performance. Through surface and interface engineering, the density of accessible active sites, the electronic conductivity and reaction energy barrier on the surface of catalysts can be accurately modified via adjusting the surface atoms, interfacial stress, bridge bonds or electronic structure, resulting an excellent catalytic performance.28–30 The catalytic selectivity can also be significantly improved by controlling the adsorption behavior of intermediates on the electrocatalyst surface with the help of surface/interface engineering, including defect engineering, heteroatom doping and constructing the heterointerface.31–34 Moreover, the construction of corrosion-resistant layers or buffer structures on the catalyst by means of anion doping is of great significance for dealing with the damage from chloride chemistry in saline water systems.33,34
To date, a series of reviews on seawater electrolysis has been presented that attempt to comprehensively analyze the challenges and outline critical future directions of seawater electrolysis from various perspectives,14,19,20 including electrolyser design,11 membrane selection,21 electrolyte treatment,14,16 and catalyst design.18,23,35,36 Among these, catalyst design has received significant attention as a crucial component. Based on material types, Wang et al.13 sorted out the key points of catalyst activity regulation, while Wu et al.10 proposed activity regulation strategies, and Khatun et al.25 also put forward catalyst design strategies for selectivity and stability. As a multifunctional modulation strategy, there is an extensively growing interest in using surface/interface engineering to enhance the catalytic activity of seawater electrolysis catalysts, but insufficient attention has been paid to the regulation of selectivity and stability. Recently, there have been presentations of reviews specifically focused on surface/interface engineering-regulated electrochemical activity,37,38 and some literature39,40 also partly touches upon selectivity and stability, but these reviews have been less comprehensive. Accordingly, it is timely and necessary to present updates and report on recent advances in surface/interface engineering used for direct seawater electrolysis. This review aims to discuss the regulation strategies based on surface/interfacial engineering, applying to the regulation of catalytic activity, selectivity, and stability of seawater electrolysis catalysts, and provide the reader with a comprehensive overview of seawater electrocatalyst design. Specifically, in the early part of the manuscript, we briefly introduce the fundamentals of electrochemical seawater splitting, along with an explanation of the hydrogen evolution reaction (HER), OER, and side reactions. Subsequently, rational design strategies for HER and OER electrocatalysts applied to seawater or chloride-containing electrolytes in terms of catalytic activity, selectivity and corrosion resistance are discussed comprehensively. Then, effective strategies including morphology design, defect engineering, doping, phase engineering, heterojunction design and wettability engineering are critically discussed (Fig. 1a) along with corresponding catalytic performances, i.e., activity, selectivity and corrosion resistance. Finally, a summary of the current state of the research is presented, along with useful perspectives on potential areas.
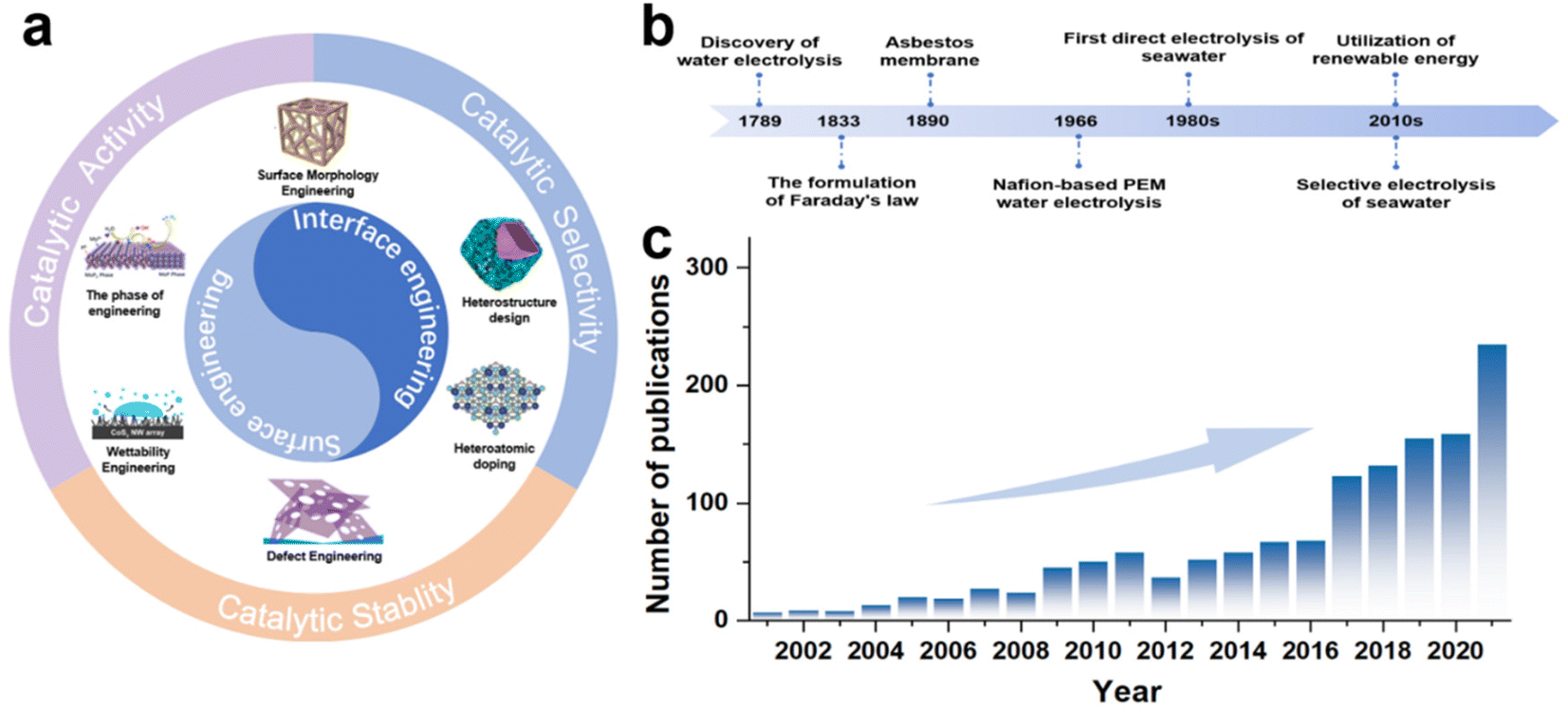 | ||
| Fig. 1 (a) Schematic representation of interface/surface engineering methodologies modulation methods for high-performance HER/OER electrocatalysts. (b) A brief summary of the development of the history. (c) Quantity of SCI articles based on seawater splitting. (The keywords used for a search in the Web of Science were “electrochemical” and “seawater splitting”.) | ||
2. Fundamentals of seawater electrolysis
2.1 Development of seawater electrolysis
In 1789, the first water electrolysis was conducted in a Leyden tank by Paets van Troostwijk and Deiman with an electrostatic machine.41 By 1833, Faraday's law was proposed and provided a quantitative relationship between the electrical energy consumed and the gas produced (Fig. 1b), scientifically defining the meaning of water electrolysis.42,43 Consequently, the first commercial asbestos separator was used for water electrolysis in 1890, followed by General Electric's development of Nafion-based proton exchange membrane (PEM) electrolysis of water in 1966 to meet the special energy needs in the space and military fields.44,45 Subsequently, early investigations on the selective electrolysis of seawater into hydrogen and oxygen were carried out by Bennett, in the early 1980s.46 Meanwhile, for the industrial production of chlorine gas, Trasatti et al. also carried out early studies on the reactivity of different catalyst materials for OER and the chlorine evolution reaction (CER) activity.47 To date, various studies involving electrodes,31,48 ion-exchange membranes11,49 and electrolyzers11,20,50 have been applied to seawater electrolysis, and the number of such research articles has been on the rise since the 2000s (Fig. 1c).2.2 Mechanism of seawater electrolysis
As illustrated in the simplified schematic Fig. 2a, water molecules are split into H2 and O2 when an adequate voltage is applied between the anode and cathode, as eqn (1).51 Therefore, the reaction of electrochemical water splitting is divided into two half-reactions: cathodic HER (eqn (2) and (3)) and anodic OER (eqn (4) and (5)). From the calculation of thermodynamics, the thermodynamic electrical potential of water splitting is only 1.23 V (vs. NHE).52,53 However, the ion transfer rate, conductivity, surface bubble patency, and reaction entropy also are led to a high overpotential.51,54,55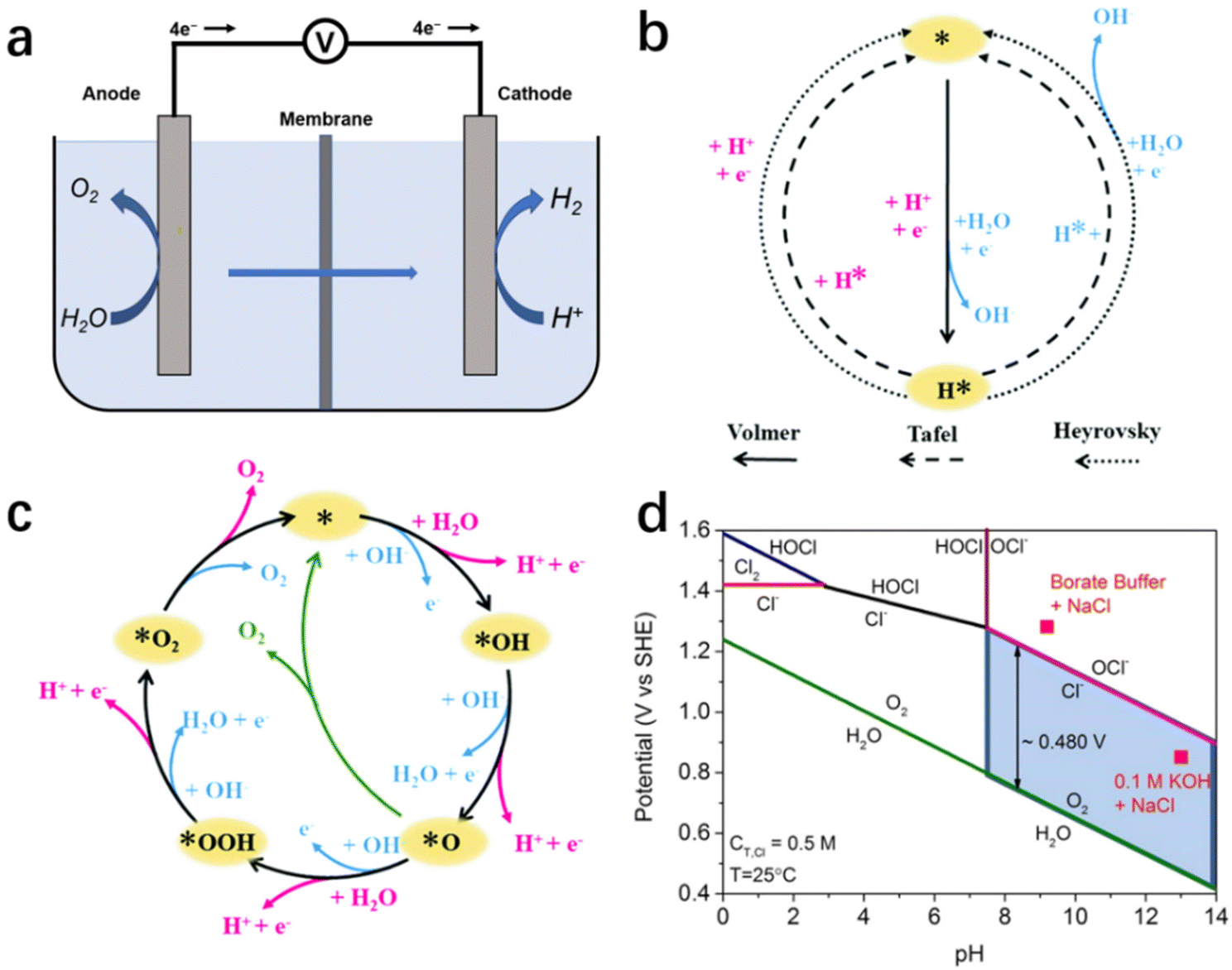 | ||
| Fig. 2 (a) Schematic illustration of electrochemical seawater splitting. Mechanism of HER (b) and OER (c) in alkaline (blue way) and acidic (pink way) solutions.54 Copyright 2021, RSC. (d) A thermodynamically simulated Pourbaix diagram of saltwater containing oxygen and Cl− redox reactions, with a total chlorine content of 0.5 M and a temperature of 25 °C.24 Copyright 2016, Wiley-VCH Verlag GmbH. | ||
Overall reaction
| H2O → H2 + O2 E° = −1.23 V | (1) |
| Acidic: 2H+ + 2e− → H2 E° = 0.00 V | (2) |
| Alkaline: 2H2O + 2e− → H2 + 2OH− E° = −0.83 V | (3) |
Anodic:
| Acidic: 2H2O → 4H+ + O2 + 4e− E° = −1.23 V | (4) |
| Alkaline: 4OH− → 2H2O + O2 + 4e− E° = −0.40 V | (5) |
On the cathode, the HER is a two-electron transfer process involving multiple steps with two reaction mechanisms.55 In electrolytes with different pH values, the relevant reaction mechanism varies, as illustrated in Fig. 2b.54 In an acidic medium, HER has two reaction mechanisms: Volmer–Heyrovsky or Volmer–Tafel.56 In the Volmer step, abundant protons in solution are adsorbed on the surface of the electrode, and obtain electrons to generate adsorbed hydrogen atoms (H*).57 Subsequently, depending on the occupancy on the catalyst surface, the H* form H–H bonds with each other or with the H+ in solution, and desorb hydrogen molecules via the Tafel reaction or Heyrovsky reaction, respectively.54,58 In an alkaline/neutral medium, the reaction mechanism is similar to that of the acidic medium, but the decomposition of water molecules precedes the Volmer step due to the scarcity of protons in solution.59,60 Therefore, the additional energy consumption in cleavage of the H–O bond results a higher overpotential.
As the other half-reaction of water splitting, the oxygen-evolution reaction is a complicated four-electron transfer process (as shown in Fig. 2c), consisting of intricate elementary reactions and involving a variety of intermediates. As a result of the increased number of reaction steps and intermediates, OER is regarded as a kinetic-limited process hindering the efficiency of overall water splitting.55 At present, several different possible OER mechanisms have been proposed in alkaline and acidic media, as the OER pathways are susceptible to the structure/properties of the catalyst's surface. Here, we cite the classical scheme proposed by Nørskov for a brief description, as shown in schematic illustration Fig. 2c.51,54 In alkaline medium, the anodic reaction starts with the adsorption of hydroxide ions on the electrode surface and the formation of the intermediate OH* after discharge.54,58 Subsequently, OH* decomposes to form O*, and the reaction between O* and another adsorbed H2O forms OOH*, which eventually produces O2 and releases. Similar to HER, OER exhibits pH dependence, with the reaction process changing as the pH changes.56,57 In acidic or neutral medium, there is less OH−, H2O is initially electrolyzed to produce OH− and subsequently undergoes a process similar to the previous adsorption of OH*, O* and OOH* and the desorption of O2.57
In natural seawater, the presence of a wide range of substances, mainly including inorganic ions (Cl−, Br−, SO42−, Na+, K+, Mg2+ and Ca2+), microbes and other small particulates, inevitably impacts negatively on the operation of seawater splitting.14,23 Abundant Cl− corrodes electrodes severely by reacting with electron-deficient transition metals and converts to unwanted Cl2/ClO− at the anode.3,14,15 During seawater electrolysis, the chlorine-involved oxidation reaction varies with pH, oxygen/chlorine concentration and temperature.14,23
CER:
| 2Cl− → Cl2 + 2e− E° = 1.36 V, pH = 0 | (6) |
HCFR:
| Cl− + 2OH− → ClO− + H2O + 2e− E° = 0.89 V, pH = 14 | (7) |
2.3 Assessment of and recommendations for electrolysis techniques
Besides the electrode, the selection of seawater electrolysis technologies is also essential for long-term stable operation. Various types of electrolysis cell design suffer from different technical challenges, which directly affect operational stability and investment costs of energy storage.68 Hence, we conduct a discussion on the four mainstream electrolysis technologies applied to seawater splitting. According to the reaction temperature, it can be roughly classified into two categories: low-temperature electrolysis (<100 °C) and high-temperature electrolysis (700–900 °C).19,69Fig. 3a–d depicts these four combinations.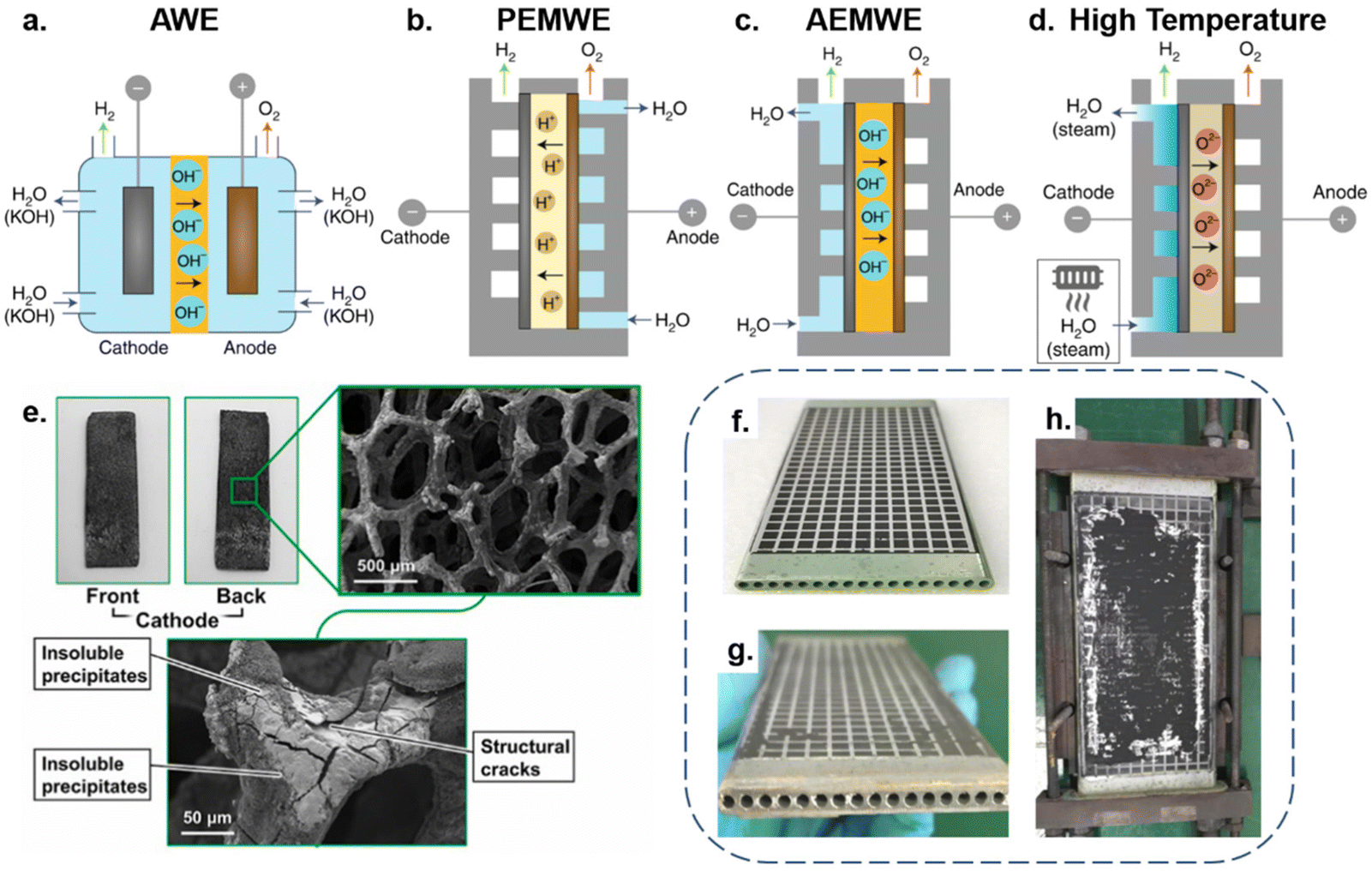 | ||
| Fig. 3 (a) An alkaline water electrolyser (AWE) operates as a 2-compartment cell in which a liquid alkaline electrolyte (typically 20–30% KOH) is pumped around both sides and a porous diaphragm allows hydroxyl ion (OH−) migration while preventing gas crossover. (b) An anion exchange membrane water electrolyser (AEMWE) sandwiches an OH− transporting membrane between the anode and cathode. Water is supplied to the cathode in this cell; however, it is also possible to supply water to the anode or both sides. (c) A proton-exchange membrane water electrolyser (PEMWE) consists of a solid acid electrolyte polymer sandwiched between the anode and cathode. In most cases, water is only fed to the anode. (d) High-temperature solid oxide electrolysis (∼700–1000 °C) water electrolysers. Water evaporates and transports to the cathode as steam to produce H2 while a solid oxide or ceramic membrane transports O2− to the anode.19 Copyright 2019, Springer Nature. (e) Digital photos of CNC-MO and corresponding SEM images after the long-range test.100 Copyright 2021, Elsevier. The Solid Oxide Electrolysis (SOE) Cell digital photo (f) and the corresponding images (g and h) following a 420-hour experiment.79 Copyright 2021, Elsevier. | ||
Among the low-temperature electrolysis technologies, alkaline water electrolysis (AWE) is currently the most mature.70 Typically, it functions in a two-chamber cell separated by a membrane/separator, with single cell voltages usually around 1.7–1.8 V and current densities typically in the range of 100–300 mA cm−2. In this case, the alkaline aqueous solutions are the feedstock, usually NaOH or KOH, and electrolyze water to generate hydrogen and oxygen in the presence of direct current.71 When seawater is supplied to AWE cells, the long-term operation of the electrolysis system will suffer from adverse effects mainly involving free chloride ions and alkaline-earth ions, as shown in Fig. 3e. However, benefiting from the high-pH electrolyte and selective catalyst, the evolution of chloride ions in the AWE cell is readily inhibited. In terms of preventing equipment corrosion the chlor-alkali industry can be referenced, by replacing stainless components that are in contact with seawater with anticorrosive titanium-based components and covering the electrolyser chamber with a Teflon coating to avoid ion etching.19,72 As for physical blockage of the separator membrane by impurities and precipitates, it has proved feasible to maintain membrane activity through recovery procedures.19 Additionally, by leaving the electrolyser at open circuit periodically, the electrodes can regain part of the activity that declines with chloride blocking of the membrane.19,73 Thanks to its simple construction, the AWE unit has a significant cost advantage over other units, with investment costs ranging from roughly 500 to 1000 € per kW.74 Even with the anti-corrosion design, the extra equipment expenses are far less than the investment and running costs of a reverse osmosis facility.75 Compared with the AWE cell, the proton exchange membrane (PEM) electrolyser encounters more obstacles when purified water is replaced by seawater. As shown in Fig. 3b, the purified water is supplied to the anode where water is oxidized to form O2 and H+. Subsequently, protons migrate through the PEM towards the HER catalyst (cathode) to generate hydrogen. To ensure speedy proton transport, electrolysis systems usually operate in highly acidic (pH < 2) and high potential (1.7–1.8 V) conditions.76 However, the electrolysis of seawater places extreme demands on the OER catalyst selectivity design. Furthermore, the highly integrated membrane electrode assembly (MEA) also makes the PEM electrolyser very sensitive to impurities.77 In this regard, dissolved cations (Mg2+ and Ca2+) would accompany protons across the PEM and precipitate Mg(OH)2 and Ca(OH)2 on the cathode side, causing irreversible blistering on the PEM.19,73 Regarding investment costs, the highly sophisticated component design significantly increases the overall manufacturing costs of the PEM, with the associated equipment costing around 600 to 1300 € per kW and with maintenance costs ranging from around 3% to 5%.74 As an emerging technology, the potential of anion exchange membrane (AEM) electrolysers lies in combining the low cost of AWE with the high efficiency of PEM. As shown in Fig. 3c, water molecules are electrolyzed at the cathode to produce H2 and OH−; OH− then migrates across the membrane to the anode, where O2 is formed. However, similar structures usually lead to similar problems. In seawater electrolysis, Cl− in the seawater also accompanies OH− across the AEM membrane.19 Fortunately, the high operating pH of AEM can help minimize Cl− oxidation, alleviating the design difficulties of the OER catalyst. Additionally, the AEM electrolyser possesses the advantages of being quick to start and stop, using less energy, and being better paired with methods for producing renewable energy.78 Consequently, AEM technology will have enormous potential with the development of membrane technology. Being a pre-commercial technology, an accurate assessment of its investment cost is not currently feasible, but it should be comparable with PEM under mature membrane technology circumstances.77
Apart from low-temperature electrolysis technologies, solid oxide electrolysis (SOE), which electrolyzes steam under a high-temperature condition, has also been employed in seawater electrolysis. Differing from the previous technologies, SOE cleverly integrates the seawater purification process into the steam generation process, by directly evaporating the seawater and using the steam as the feedstock for electrolysis at high temperatures.79,80 Hence, the cells operate in essentially the same state as purified water electrolysis, with a high theoretical conversion rate. In the study by Liu et al.,79 their cell operated at 750 °C with a current density of 200 mA cm−2 and achieved a conversion rate of 72.47%. Of note, the long-term seawater electrolysis with SOE is still disturbed by sea salt, as shown in Fig. 3g and h, where pipes of cells are blocked by dissolved sea salt in the steam.79 Besides, the dissolved sea salt may infiltrate the cell along with the resulting steam, causing solid oxide electrode poisoning and blockage of the triple-phase boundaries near the electrode–electrolyte interface.81 Furthermore, salt-laden steam possesses a stronger corrosive effect on the equipment at high temperature. Therefore, further improvement is needed for the existing SOE cell to be directly used in seawater splitting. As a pre-commercial technology, the accurate estimate of investment costs is still unavailable, but may exceed 2000 € per kW.74 Furthermore, as the equivalent balance of the plant components of this technology is subjected to more demanding conditions, the operating costs of SOE will be comparable to or higher than those of PEM or AWE.77
Additionally, desalination coupled with electrolysis is another rival to direct seawater electrolysis. In terms of technical maturity, existing reverse osmosis membrane desalination technology is fairly mature, and the combination with commercialized AWE or PEM has essentially no technical limitations, so the cost is the key issue restricting its widespread application.16,75 Previously, Khan et al.75 estimated desalination expenses as a tiny fraction of seawater-splitting costs based on commercial accounting, with electricity expenditure being the main cost. Nevertheless, this is not suitable for energy storage schemes for renewable energy producers. Since the electricity to keep the electrolysis system running comes from free solar/wind energy that fails to enter the grid and is discarded, seawater desalination will be a major cost in the energy storage segment, both for upfront equipment investment and later operating costs.17,75,82 Thus, it is crucial for future applications to lower the cost of seawater desalination. Considering industrial application, the current unpretreated seawater splitting schemes are less costly, but have difficulty meeting long-term stability requirements mainly caused by precipitate coverage, thus requiring pre-treatment of natural seawater.
2.4 Evaluation of electrocatalytic performance
As there is a complex collection of reactions involving severe reaction conditions and complex reaction paths, the evaluation system for catalysts needs to be based on this as well, with more emphasis on selectivity and stability.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lg|j|. Among them, a is the constant, b is the Tafel slope, and both the overpotential and the current density j take absolute values (i.e., positive values). The magnitude of the Tafel slope value enables the identification of other critical parameters, including electron mobility number, charge mobility coefficient, and reaction rate-determining step. Besides, the exchange current density (j0) is another important index for evaluating the performance of electrocatalysts, which describes the current density at an overpotential of zero under equilibrium conditions, reflecting the electron transfer ability and the difficulty of electrode reaction. According to the Tafel equation, the performance of the electrocatalyst for the target reaction improves with increasing value. Therefore, for a standard electrocatalyst, the lower the overpotential, the smaller the Tafel slope and the higher the exchange current density, and the better the electrocatalytic activity.
lg|j|. Among them, a is the constant, b is the Tafel slope, and both the overpotential and the current density j take absolute values (i.e., positive values). The magnitude of the Tafel slope value enables the identification of other critical parameters, including electron mobility number, charge mobility coefficient, and reaction rate-determining step. Besides, the exchange current density (j0) is another important index for evaluating the performance of electrocatalysts, which describes the current density at an overpotential of zero under equilibrium conditions, reflecting the electron transfer ability and the difficulty of electrode reaction. According to the Tafel equation, the performance of the electrocatalyst for the target reaction improves with increasing value. Therefore, for a standard electrocatalyst, the lower the overpotential, the smaller the Tafel slope and the higher the exchange current density, and the better the electrocatalytic activity.
3. The strategy for catalyst design
Efficiently and inexpensively converting sustainable electrical energy to H2 generation is essential for the electrolysis of seawater, where the catalyst is the key factor. However, poor activity, low selectivity, and degraded catalytic durability are among the difficulties in the design of seawater electrolytic electrodes. Thus, rational designs to compensate for the specific disadvantages of catalysts are highly valuable for catalyst performance enhancement.3.1 Strategies for regulating activity
From the reaction mechanism, the inherent electrical conductivity, the number of active sites, and reaction energy barrier are essential parameters that restrict the catalytic efficiency of electrocatalysts.84,85 A high intrinsic conductivity, as mirrored in a high electron transfer rate, favors effective catalysis.86 Abundant exposed active sites on electrocatalysts ensure adequate contact with the electrolyte reactants. Additionally, regulating the reaction kinetics and surface properties of electrocatalysts to lower the reaction energy barrier is also a significant approach for strengthening electrocatalytic performance.84,87Surface/interface nanoengineering has been applied to the performance tuning of seawater electrolysis catalysts, with excellent results. Tuning the energy band structure of the catalyst improves the intrinsic conductivity of the catalyst, and hence the catalytic activity, with the use of defect engineering, doping, and interfacial construction.88–91 This not only ameliorates the kinetic limitations of some catalysts with low intrinsic conductivity, but also mitigates the effect of poor conductivity in natural seawater.13 Surface morphology engineering aids in the exposure of the active sites by regulating the electrocatalyst morphology and expanding the specific surface area, which increases full contact between the active sites and the electrolyte.91,92 Moreover, mass transfer-friendly structures are built with the help of morphological engineering, which not only speeds up the electrode–electrolyte mass transfer process, but also relieves stress produced by local pH increases when the active site is covered by precipitate.40,85 As for the reaction energy barrier, it is directly associated with the adsorption/desorption behavior between intermediates and the surface/interface of the catalyst. By optimizing the electronic configuration of the catalyst, a suitable free energy of adsorption (ΔG) can be generated, thus inducing the desired electrochemical activity.85,93–95
3.2 Strategies for regulating selectivity
The presence of chloride ions, which are abundantly present in seawater, inevitably affects the HER/OER reaction process.96 Chloride ion adsorption blocks the active site on HER catalysts, and the chloride-involved oxidation reaction competes with the OER, causing electrocatalyst corrosion and additional energy consumption.19,97 Therefore, the catalytic selectivity of the reaction system becomes critical for minimizing the negative effects mentioned above caused by chlorine-involved side reactions. In practice, chlorine-involved oxidation is a complex reaction that varies with reaction conditions such as pH, applied potential and temperature.14,23 According to the Pourbaix diagram (Fig. 2d), OER has a thermodynamic advantage over CER across the entire pH range, especially in the high pH region where the constant potential gap is up to 480 mV.3,24 Of note, with the increasing number of electron transfers, OER occupies a kinetic disadvantage compared with the two-electron transfer chlorine-involved reaction (eqn (4) and (5)), resulting a potential gap decline between OER/CER reactions in practice. This means that an alkaline condition (pH > 7.5) is more suitable for the implement of selective oxidation in seawater splitting, reducing the influence of aggressive chlorine.25 Consequently, controlling electrolyte pH and applied potential has become the most universal method for researchers to selectively regulate seawater electrolysis.3,19 The reaction system is maintained at a high potential gap interval at all times by simply adding a buffer solution to the seawater to deplete the protons formed during OER.19,25 This reduces the occurrence of side reactions. Furthermore, this improves the high selectivity of the catalytic reactions while reducing the damage caused by pH fluctuations during electrolysis to the electrocatalyst.For the design strategy of electrocatalysts, following the design criteria for OER electrocatalysts in alkaline chloride-containing electrolytes as summarized by Strasser and his co-workers, those maintaining an overpotential of <480 mV can theoretically achieve high oxygen/chlorine selectivity in the presence of Cl−, and the activity and stability of electrocatalysts following this criterion will not be compromised.3,24 In contrast, the design of relevant electrocatalysts in acidic electrolytes requires the achievement of extremely high selectivity. Besides the few choices of materials with high intrinsic catalytic selectivity (e.g., MnOx), it is more common to endow a higher selectivity to highly active materials through a rational design strategy.23,35,46 Among the various modulation strategies, surface/interface engineering is the main instrument for catalytic selectivity modulation, mainly including heteroatom doping, defect engineering and heterojunction design.98–101 Through manipulating the physicochemical properties of the catalyst surface/interface, heteroatom doping and defect engineering modulate the binding energy of OER intermediates on the catalyst to achieve a highly selective OER.26,101 On the other hand, appropriate heterojunction design can expose an anion-rich decorative interface to repel chloride ions with the help of negative electric repulsion.101 In addition, composite strategies combining inert slow chloride layers (e.g., graphene, MnOx)97,102 with conventional highly active electrocatalysts also achieve excellent selectivity in chlorine-containing electrolytes.
3.3 Strategies for regulating stability
Apart from the properties mentioned above, the catalytic stability is also a tough challenge in the design of electrodes for seawater electrolysis. In electrolysis of water, the stability of the electrocatalyst is mainly influenced by the electrode potential, electrolyte and gas molecules formed in the reaction.103,104Among them, reasonable potential selection is of great significance for the long-term operation of electrolysis, when the potential window of the catalytic reaction overlaps with the redox potential of the catalyst for a long time, which may cause the dissolution/degeneration of the catalysts.103,105 Meanwhile, with the help of potential window overlap, reconstructing a stable protective layer on the catalyst surface/interface has also been proved to be an effective strategy in seawater electrolysis.106 On top of that, bubbles attached to the electrode surface during electrolysis are a source of concern.67 Specifically, these gas species in the long term adhere to the electrode surface and interact with the catalytic active site, which tends to damage the catalyst surface/interface structure. Therefore, the stability of the catalyst can be improved by regulating the wettability of the catalyst surface/interface to promote gas desorption. Due to the presence of impurities in natural seawater, electrocatalysts not only suffer from the same electrochemical corrosion as that in purified water electrolysis, but also face additional chemical erosion with other ions, resulting in significantly increased damage to the catalysts. Therefore, an anti-corrosion design is critical in the design of electrodes for seawater electrolysis. In this regard, the chemical corrosion mainly originates from free Cl− and oxidative corrosion of aggressive chloride products (e.g., Cl2, HClO or ClO−) generated in the electrolysis reaction.3,14 Although the oxidation products of chloride ions are more oxidative and have a greater corrosion impact, the formation of oxidation products of chloride ions can be largely suppressed by adopting a suitable electrolyte, applied voltage and high selectivity catalyst.61,62 However, the corrosion caused by free Cl− is difficult to avoid, especially for metal-based catalysts. With long-term immersion in seawater, the free Cl− adsorbs on the electrode surface and gradually corrodes the catalyst by forming metal chloride-hydroxides, as in eqn (8)–(10).23,61
Adsorption of Cl− by surface polarization:
| M + Cl− → MClads + e− | (8) |
Dissolution of chloride:
| MClads + Cl− → MClx− | (9) |
| MClx− + OH− → M(OH)x + Cl− | (10) |
112) over conventional highly active electrocatalysts have also demonstrated great potential in improving the catalyst stability. (c) Building of an in situ anti-corrosion layer by reconfiguring the surface/interface of catalyst. Oxidizing the doped S or P heteroatoms in catalysts reconstitutes a sulfate or phosphate passivation layer on the electrocatalyst surface, protecting the anode from Cl−/ClO− corrosion.113,114 (d) Constructing buffer structures to mitigate the damage to the structure by Cl− invasion. Different from the above strategies, this actively dopes Cl− into the lattice to form chlorinated hydroxide to avoid the structural damage and deactivation of the catalyst caused by Cl− infiltration.
4. Surface/interface engineering
4.1 Surface morphology engineering
Generally, the morphology modulation strategies for seawater electrolysis catalysts are similar to those used in applications for purified water, in which a high specific surface area is pursued to expand close contact between the exposed active sites and the electrolyte for a better catalytic performance.85,115 Nevertheless, the mass transfer channels become essential in catalyst design because of the presence of multiple ions or microorganisms in natural seawater, which can adhere to the catalyst surface, thus blocking some of the active sites and reducing catalytic performance.19,63 Therefore, the structures with a large specific surface area and favourable electrolyte diffusion are preferred by researchers in seawater electrolysis.Three-dimensional porous materials with a high surface area, which facilitates the expression of active sites, the adsorption of intermediates, and the release of gases, are a promising material in water splitting. Recently, Li and his co-worker116 adopted the dispersing-etching-holing strategy, synthesizing a RuIrOx (x ≥ 0) nano-netcage catalyst with high activity and durability. In this report, Zeolitic imidazolate frameworks (ZIF-8) were in situ etched and amphoteric ZnO removed (Fig. 4a), forming the three-dimensional porous RuIrOx nano-netcage as depicted in Fig. 4b–3d, which enabled the three-dimensional porous nano-netcage to expose more active sites while making the substrate molecules more accessible. As a result, the nano-netcage performs admirably as an overall water electrolysis catalyst across a wide pH range (0–14), with a potential of just 1.45 V (pH = 0) or 1.47 V (pH = 14) at 10 mA cm−2 (Fig. 4e). Notably, this catalyst is durable for at least 24 h in electrolytes of various pH values, demonstrating good stability (Fig. 4f). Additionally, similar effects have been observed in the hierarchical structure catalysts. In those cases, the massive low-dimensional sub-units on hierarchical catalysts provide numerous active sites for the electrolyte.117,118 Meanwhile, the interlaced trunks and branches of hierarchical nanostructures provide ample free space for mass transfer, and accelerate the bubble release rate.119–121 For example, Sun et al.122 used Cu(OH)2 nanowires as a template loading nanosheet-like Fe–Co sulfide units on the one-dimensional Cu2O/Cu backbone through electrodeposition, forming Fe–Co–S/Cu2O/Cu with a unique hierarchical structure. In practice, this catalytic electrode achieved 50 mA cm−2 in 1 M KOH while requiring only 338 mV overpotential. Exploring the reason, the authors attributed it to the unique structure. In water electrolysis, the Cu2O/Cu backbone covered with Fe–Co sulfide units possesses a large surface area, allowing for more active iron centers to be expressed and accelerating the bubble release rate. Benefiting from the spatial structure that facilitates mass transfer, the Fe–Co–S/Cu2O/Cu catalytic electrode possesses excellent stability, maintaining good catalytic performance after long-term electrolysis in natural seawater, with only slight insoluble precipitate on the catalyst surface. In addition, the core–shell structure is considered as a potential candidate for achieving both high efficiency and high selectivity in seawater electrolysis. The core, composed of metal or alloy, ensures high intrinsic conductivity, and the synergistic effect with the shell provides an effective path for modulating the electronic structure on the surface environment, thus facilitating the adsorption of intermediates in the electrochemical process.123–126 Wu et al.117 synthesized a core–shell structured CoPx@FeOOH as an OER catalyst, which exhibited excellent catalytic activity in seawater electrolysis. Specifically, only 283 mV and 337 mV were required to reach the current density of 100 and 500 mA cm−2, respectively. Investigating the causes, the authors ascribed the excellent OER performances to the core–shell structure, where the core composed of CoP ensures high intrinsic conductivity and the synergistic effect with the shell layer enhances the electron transport in the FeOOH shell. In addition, negatively charged P atoms in the CoPx core can moderate the absorption energy of the FeOOH active sites to OER intermediates, resulting in a higher catalytic selectivity. As a result, the catalyst achieved more than 80 h of continuous testing without any hypochlorite generation in the durability test.
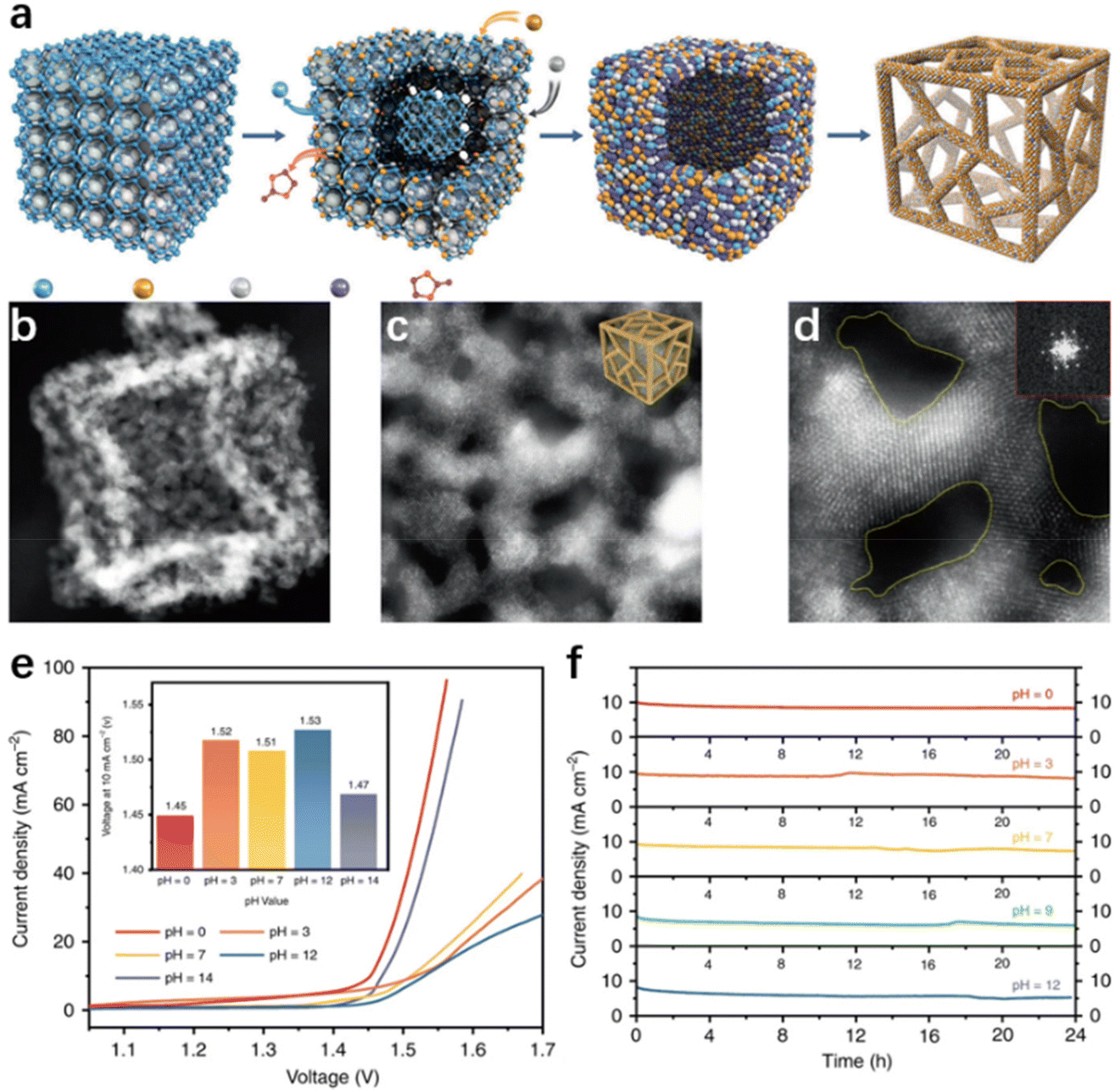 | ||
| Fig. 4 (a) The diagram of the synthetic process. The TEM (b) and HRTEM (d) images of RuIrOx nano-netcages, with a scale bar of 10, 5, and 2 nm. (e) The polarization curves and the voltages of RuIrOx nano-netcages at 10 mA cm−2 (inset) throughout a wide pH range (0–14). (f) The current–time (I–t) curves of RuIrOx nano-netcages for 24 hours at various pH values.116 Copyright 2019, Springer Nature. | ||
Although three-dimensional nanostructures can facilitate mass transfer, the negative impact of the slow diffusion of electrolytes in holes persists. The precipitation of hydroxide is difficult to avoid due to the slowing down of the electrolyte exchange rate in the nanostructure and the local pH surge. Hence, forced acceleration of electrolyte flow by mechanical stirring is necessary. As the result, adequate mechanical strength is required when constructing a three-dimensional structure.
4.2 Defect engineering
Defects directly affect the charge distribution and adsorption/desorption behaviour on the electrocatalyst surface, which can significantly improve the catalytic activity and selectivity.84,127,128 Hence, the construction of defects on the catalyst surface is frequently employed to modulate the specific adsorption behaviour and chemical activity of reactants on the catalyst surface, decreasing the energy barrier in the electrolysis of seawater.84,129Defect-free catalytic surfaces generally exhibit unsatisfactory adsorption/desorption behaviour, while little trace of defects can profoundly alter atomic and/or nanoscale electronic properties to enhance intrinsic activity.129–132 Such defects often act as catalytically active sites to boost the activity, selectivity, and stability of an electrocatalytic material.130,132 Understanding/establishing the defect–activity relations of the catalysts for seawater electrolysis would guide effective design at the atomic level.131,133 In recent research works, introducing various types of surface/interface defects in catalysts achieved greater seawater electrolysis efficiency, encouraging us to summarize these defect designs. In this section, rationally designed defects such as cation vacancies, edges, interfacial dislocations, etc., will be discussed to highlight the vital role of defects in boosting catalytic performance in seawater electrolysis. Zhao et al.134 reported that Co doping increased the number of edges of VS2 (Fig. 5a) and consequently improved the HER activity in simulated seawater. Reducing nanosheet size and doping Co heteroatom reveals numerous edges and sulfur defects as active sites, ensuring adequate contact with the electrolyte. In consequence, the Co–VS2 nanosheets exhibited excellent catalytic performance. After 12 hours of testing in simulated seawater, the Co–VS2 nanosheets showed no obvious current decay (Fig. 5b). Hence, defect construction is still effective for improving the HER performance of catalysts in seawater, but seawater electrolysis at the cathode must operate at industrial-level currents. Meanwhile, reasons for the activity decay after long-term electrolysis (e.g., half a month) are worthy of further investigation.
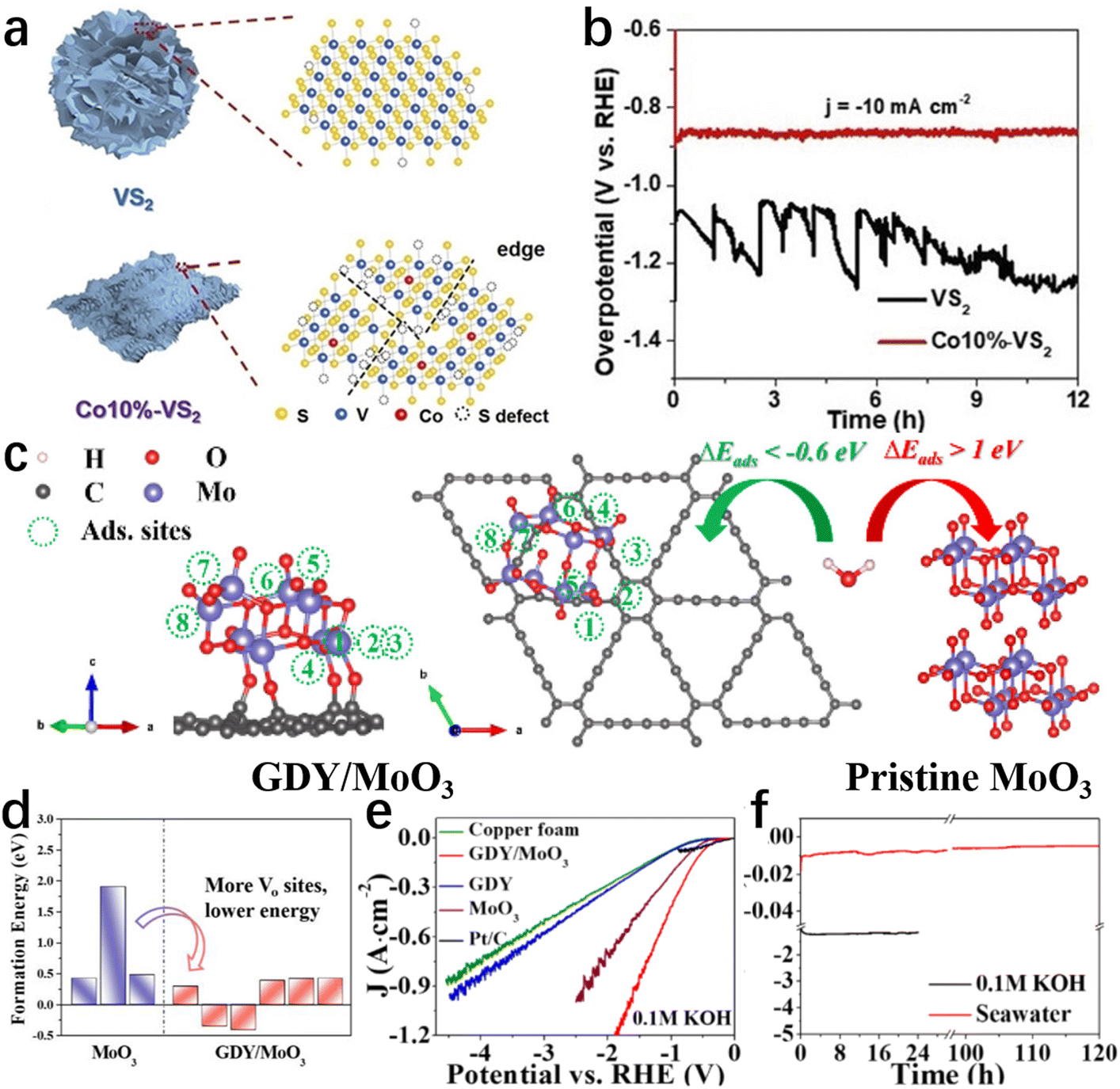 | ||
| Fig. 5 (a) The structure diagram of VS2 and Co10%–VS2. (b) The 12-hour recording of the VS2 and Co10%–VS2 chronopotentiometry curves at a current density of 10 mA cm−2 in seawater.134 Copyright 2021, Wiley-VCH Verlag GmbH. (c) Schematic diagram of H2O adsorption sites on GDY/MoO3 and H2O adsorption on pristine MoO3 and GDY/MoO3 (d) calculated formation energy of oxygen vacancy in pure MoO3 and GDY/MoO3 under O-poor conditions. (e) Polarization curves of different catalysts reacting in seawater. (f) The chronopotentiometry curves of GDY/MoO3 in 0.1 M KOH and natural seawater.138 Copyright 2021, ACS. | ||
Additionally, the construction of surface/interface defects is also widely adopted to improve catalytic performance by enhancing the intrinsic conductivity and lowering the reaction energy barrier.129,135–137 In a typical case, Guo and his colleagues138 developed a graphdiyne/molybdenum oxide coupled material (GDY/MoO3). Depending on the rational design, GDY/MoO3 only requires an overpotential of 170 mV to achieve 10 mA cm−2 in 0.1 M KOH (Fig. 5e). Of note, this catalyst can maintain good activity and stability at high current densities (≥1 A cm−2) in seawater, as shown in Fig. 5f. Among many reasons, as the main active sites of GDY/MoO3, the presence of oxygen vacancies increases the van der Waals gap and narrows the band gap, resulting in better electrochemical kinetics. Meanwhile, benefiting from the formation of dangling bonds,139,140 oxygen vacancies serving as the active sites facilitate the adsorption of H2O molecules with a lower energy barrier (Fig. 5c and d).
Besides, the feasibility of enhancing catalyst selectivity was demonstrated by generating element defects on the catalyst surface to modulate the electronic structure.23,141,142 Recently, Hikaru and colleges obtained Na|MnOx films31 with varying concentrations of oxygen defects by annealing electrodeposited layered Na–MnO2 films at different temperatures. According to the XRD and EXAFS data, oxygen vacancies occur at 200 °C and increase in concentration with temperature, accompanied by a decrease in the valence state of manganese in the oxide. In the selectivity test, Na|MnOx-200 generated by low-temperature annealing is more inclined to undergo CER, indicating that the adsorption and oxidation processes of water molecules (*O + H2O → *OOH + H+ + e−) are kinetically inferior compared with free Cl−. In contrast, Na|MnOx-400 shows an excellent Faraday efficiency of up to 87% in a current-static electrolysis at 10 mA cm−2. Combining the characterization results, the authors attribute high selectivity to the abundance of oxygen vacancies and disordered on the surface. The Na|MnOx-400 film undergoes a different mechanism in OER, namely the recombination of two adjacent oxidation intermediates on the catalyst surface (2*O → 2* + O2), instead of the previous peroxidation pathway. Similar, Vijay and colleagues61 reported synergistic catalysis of elemental doping and oxygen vacancies for enhanced OER selectivity. In their research, the Lead Ruthenate Pyrochlore Oxide (Pb2Ru2O7−x) serves as electrocatalyst for water oxidation in neutral and alkaline seawater. Compared with the benchmark RuO2, the oxygen vacancy-rich Pb2Ru2O7−x electrocatalyst showed higher OER activity and selectivity, which were attributed to the presence of higher concentrations of surface Ru(V) and oxygen vacancies.
Consequently, defect engineering provides a novel approach for the rational design of advanced catalysts for seawater electrolysis, improving catalytic activity and selectivity. However, there are still several challenges to overcome. Firstly, effective methods for controlling the generation of defects effectively and precisely remain scarce. Secondly, the lack of understanding of the mechanisms underlying the reactions that occur on defect-rich electrocatalysts has also hindered their further development. Thirdly, the structural stability of defect-rich electrocatalysts also needs to be improved.
4.3 Heteroatom(s) modification
As another simple and efficient method to modulate the physicochemical properties of catalysts, heteroatom doping is widely employed to enhance the catalytic activity for seawater splitting.143–148 Varying the kind, amount, and location of dopants may offer additional active sites,149,150 higher intrinsic conductivity,150–152 and suitable surface electronic structure for the catalyst,153,154 thus lowering the reaction barrier and optimizing the adsorption and desorption processes of intermediates on the electrocatalyst surface.153–156Previously, heteroatoms were typically included as extra active sites to promote adequate contact with the electrolyte or to modulate the reactivity of the primary metal sites.150,157 Recently, this method has also been employed in direct seawater electrolysis. Ma and colleagues prepared vanadium(V)-doped flower-like CoP/Ni2P heterostructures158 with ultra-low Ru (RuV–CoNiP/NF) composites by impregnating CoV-LDH (Ru–CoV-LDH/NF) with ruthenium phosphide. Acting as catalyst, RuV–CoNiP/NF effectively drives HER (η10 = 28 mV) and OER (η20 = 214 mV) in alkaline media. For electrolysis in alkaline seawater, the required overpotential is only 1.538 V when reaching 20 mA cm−2. Based on the experimental results and theoretical calculation, the authors pointed out that the presence of multivalent-state vanadium as additional active sites is favorable for redox reactions. Moreover, the electric synergistic effect of doped V with other metal atoms further promotes the charge transfer and facilitates accelerated electrochemical kinetics. For intrinsic conductivity modulation, the doping of higher valence metallic heteroatoms may change the electronic structure and narrow the band gap to obtain higher intrinsic conductivity and promote the electron transfer rate, thus improving the catalytic performance of the pristine catalyst.115,159–162 In this regard, Tanveer and his colleagues have made a favourable attempt. They developed a free-standing amorphous porous OER electrocatalyst (Gd–Mn3O4@CuO–Cu(OH)2)163 by embedding Gd-doped Mn3O4 nanosheets into CuO–Cu(OH)2 nanostructure arrays. In their strategy, the doped Gd modifies the electronic structure of Mn3O4 nanosheets by coordinating with the surface oxygen vacancies, improving the carrier mobility and promoting OER intermediate adsorption–desorption. Similarly, Zhang et al. introduced Fe cations into the Co2P bundle of nanorods (BNRs),164 which caused an increase in the density of states on the Fermi energy level, resulting in a higher intrinsic conductivity. Coordinating with the lattice distortion, special coordination environment, and favourable morphology, the Fe–Co2P BNRs outperformed commercial 20% Pt/C catalysts in terms of electrocatalytic activity and stability at high voltages in seawater. Moreover, the incorporation of two or more heteroatoms with different electronegativity can also significantly improve the electrocatalytic activity.165,166 For instance, Yang et al. acquired a 0.5Fe–NiCo2O4@CC electrocatalyst166 by rapidly cooling the heated NiCo–OH@CC precursor in a ferrous sulfate solution. This straightforward quenching approach reconfigures the ideal surface of the NiCo2O4 catalyst while simultaneously achieving surface metal doping and vacancy creation, dramatically enhancing OER activity in alkaline freshwater and seawater electrolytes. In 1 M KOH electrolyte, the electrode demonstrated outstanding OER catalytic activity at a low overpotential of 258 mV. Furthermore, this electrode demonstrated extraordinary activity in alkaline seawater, reaching 10 mA cm−2 at only 293 mV. Density functional theory (DFT) simulations demonstrate a significant electronic synergy between metal cations in the quench-derived catalyst, where metal doping governs the electronic structure, yielding near-optimal adsorption energy and outstanding activity for OER intermediates.
Besides catalytic activity modulation, doping has also been introduced to optimize the selectivity of OER, via altering the surface properties of electrocatalysts and further modulating the adsorption energy of reactants on the active site.3,167–170 Recently, Liu et al. fabricated a Mo-doped Ni3S2171 nanocluster array by the solvothermal method. Benefiting from the introduction of Mo, the Ni–S coordination is significantly promoted, increasing the adsorption of OER intermediates and thus remarkably improving the OER selectivity. In the performance evaluation, the catalyst exhibited excellent catalytic selectivity with no detection of Cl2 during continuous electrolysis for over 180 hours.
Apart from the above applications, related strategies of heteroatom doping have been introduced to protect the anode from Cl−/ClO− corrosion. Based on the principle of corrosion resistance, they are broadly divided into two categories, constructing anti-corrosion layers and buffer structures.33,171–173 Among them, the strategy of constructing anti-corrosion layers has aroused considerable attention. Typically, doping the S or P heteroatoms in transition metal oxides reconstitutes a sulfate or phosphate passivation layer on the electrocatalyst surface, protecting the anode from Cl−/ClO− corrosion.171,172 In this regard, Yang et al.209 introduced P heteroatoms into Ni0.75Fe0.25Se2 precursors by ion exchange. In 0.5 M KOH + seawater electrolytes, the HER and OER of Fe,P-NiSe2 NFs tested almost match the performance of those tested in 1.0 m KOH, as shown in Fig. 6b and c. According to the simulations, the P-doping not only increased the electrical conductivity via accelerating the charge transfer between the active metal centers and oxygen-containing intermediates as depicted in Fig. 6a, but also prevented the Se and the active metallic centers from dissolution during electrochemical tests. Based on analysis of XPS spectra (Fig. 6d and e) of the catalyst after electrolysis, the P-Ni0.75Fe0.25Se2 catalyst works in simulated seawater to form a P–O species oxide passivation layer on the surface, ensuring that the structure remains stable after long-term testing and avoiding the dissolution of Se in seawater. In durability tests, the Fe,P-NiSe2 NFs maintained excellent performance for 200 hours at 1.8 V potential in different electrolytic cells (Fig. 6f). In sharp contrast, the strategy of constructing a buffer structure differs significantly from this in terms of ideas, and actively dopes Cl− into the lattice to form chlorinated hydroxide to avoid the structural damage and deactivation of the catalyst caused by Cl− infiltration. Xu et al.33 doped Cl− into the Co(OH)2 lattice to obtain Co2(OH)3Cl (Fig. 6g). According to the XANES spectra (Fig. 6h and i), the results indicate that lattice Cl− of Co2(OH)3Cl leaches and leaves cavities during the OER, while the electrolyte Cl− preferentially invades the cavity due to the appropriate size. Here, a balance is achieved between lattice Cl− leaching and electrolyte Cl− intrusion, avoiding severe structural damage and consequent catalyst deactivation. Experimentally, after 60000 seconds of operation, Co2(OH)3Cl can retain 99.9% of its initial current density, whereas Co(OH)2 decays 52.7 percent in 7000 seconds (Fig. 6j). Furthermore, the lattice Cl− of Co2(OH)3Cl also contributes to the catalytic activity by optimizing the binding energy between reaction intermediates and adjacent O–Co–O sites. Thus, Co2(OH)3Cl is 45.9 times higher than Co(OH)2 at 1.63 V vs. RHE, reaching a current density of 330.5 mA cm−2.
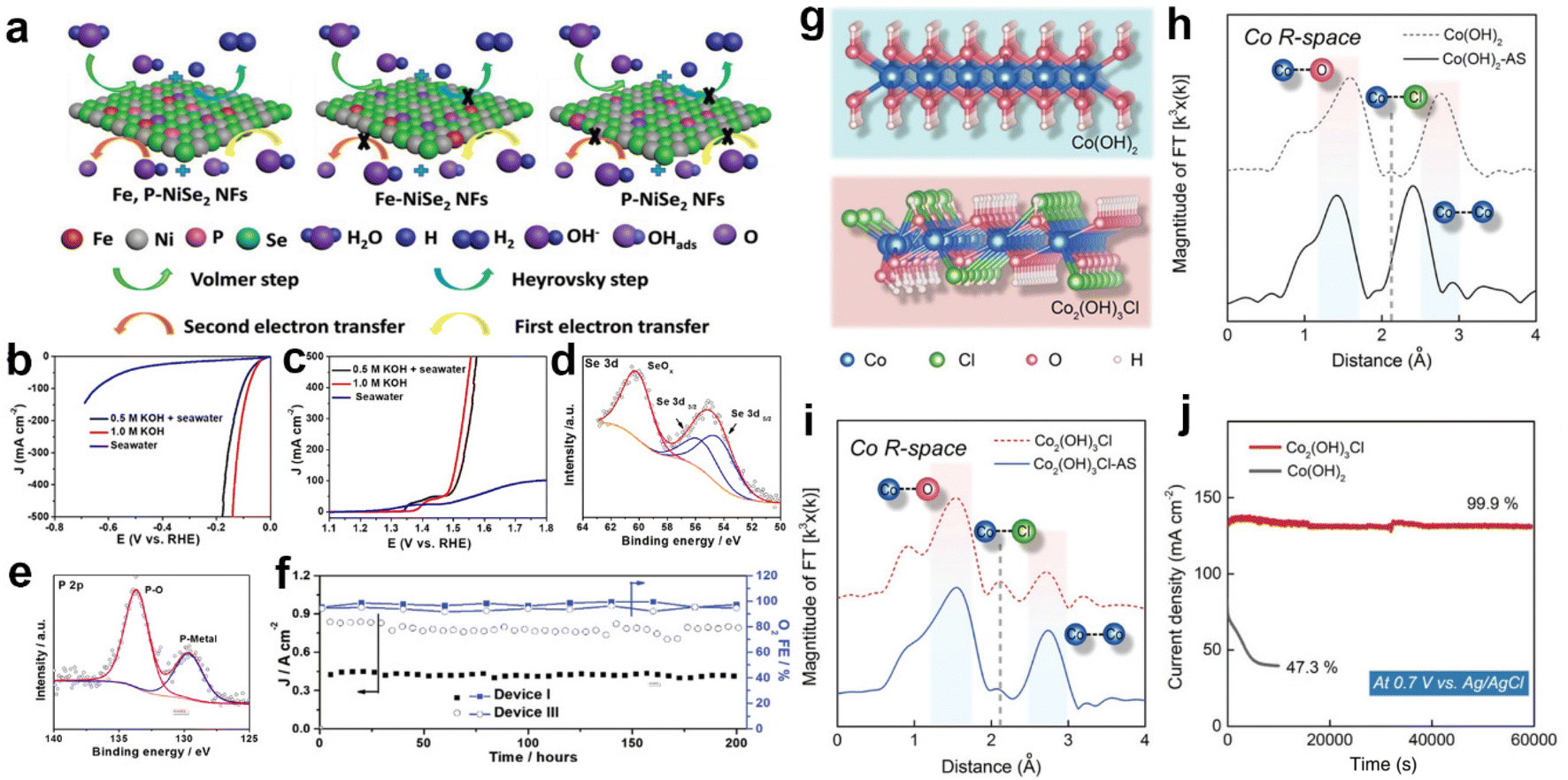 | ||
| Fig. 6 (a) The diagram of the HER and OER reaction pathways on various catalysts. The (b) HER and (c) OER polarization curves of the Fe,P-NiSe2 NFs catalyst test in natural seawater and 0.5 M KOH + seawater. XPS spectra of (d) Se 3d and (e) P 2p for the Fe,P-NiSe2 NFs after OER tests for device III after 200 hours test at Ecell of 1.8 V. (f) Asymmetric device I and symmetric device III seawater electrolyzers maintained an FE of OER over 92% for 200 hours of long-term operation at 1.8 V.209 Copyright 2021, Wiley-VCH Verlag GmbH. (g) Crystal structure of Co(OH)2 and Co2(OH)3Cl. (h) Fourier-transformed Co K-edge EXAFS spectra of Co(OH)2 and Co(OH)2–AS. (i) Fourier-transformed Co K-edge EXAFS spectra of Co2(OH)3Cl and Co2(OH)3Cl–AS. (j) The chronopotentiometry curves of Co(OH)2 and Co2(OH)3Cl in 1.0 M KOH + 0.6 M NaCl, respectively.33 Copyright 2022, Wiley-VCH Verlag GmbH. | ||
From the discussion above, it is clear that heteroatom doping possesses great potential for modulating the electrocatalysts’ performance (catalytic activity, selectivity and corrosion resistance) in seawater splitting. According to the available research, most previous doping strategies for catalytic activity remain effective. Relatively, the progress in selectivity is limited due to the lack of in-depth mechanistic studies, centering around noble metals and a few other metals (e.g., Mn and Ti). Of note, the anti-corrosion layer formed by anion (S, P and Se) doping also exhibits excellent selectivity, caused by electrostatic repulsion. In contrast, the design of buffer structures offers an alternative scheme for maintaining the catalyst's activity, providing a novel approach for followers.
4.4 Crystallographic tailoring
For catalysts, the catalytic performance of many metal-based compounds is greatly influenced by their crystallographic structure.174,175 The surface structural properties of crystals, such as surface facets and surface phases, can affect the coordination environment, the electronic arrangement of atoms, and the surface binding energy of compounds to reaction intermediates.39,176,177 Therefore, surface phase engineering is another effective way to improve electrochemical performance.Under natural conditions, MoSe2 exists thermodynamically stably as a semiconductor phase (2H).178 However, the stable physicochemical properties of 2H MoSe2 in turn limit charge-transfer kinetics and the exposure of active sites on the basal plane, resulting in poor electrocatalytic activity.178–181 In contrast, the metallic phase (1T) of MoSe2 possesses superior hydrophilicity and electrical conductivity, which facilitates charge transfer in the three-phase interface, with more active sites distributed on its edges and basal plane.136,182,183 Hence, phase engineering is used to change the phase of MoSe2 from 2H to 1T. In this regard, a valuable attempt was made by Xu et al.184 Through heteroatom doping, they changed the selenization process of the polyoxometalates precursor (Fig. 7a) and distorted the 1T phase of MoSe2, creating a new defect-rich doped 1T/2H-MoSe2, as shown in Fig. 7b. According to the theoretical results, the doping of transition metals (Fe, Co, or Ni) dramatically lowered ΔG(H2O), especially for Co and Ni. Among them, Co–MoSe2 achieves the best performance of water splitting due to the successful integration of several positive factors, including moderate intrinsic HER activity, abundant effective active sites and fast charge transport. In acidic and simulated seawater, the Co–MoSe2 exhibited low overpotentials of 116 and 274 mV to obtain 10 mA cm−2, respectively (Fig. 7c and d). A similar study was reported by Jiang et al.185 The enhancement in catalyst activity was achieved by transforming orthorhombic Ni–MoO3 into more active monoclinic Ni–MoO2via heat-treatment-induced phase transition under an inert atmosphere. In comparison with Ni–MoO3, the HER overpotential at 10 mA cm−2 drops from 493 mV (1 M KOH) and 818 mV (seawater) to just 234 and 412 mV over Ni–MoO2, respectively.
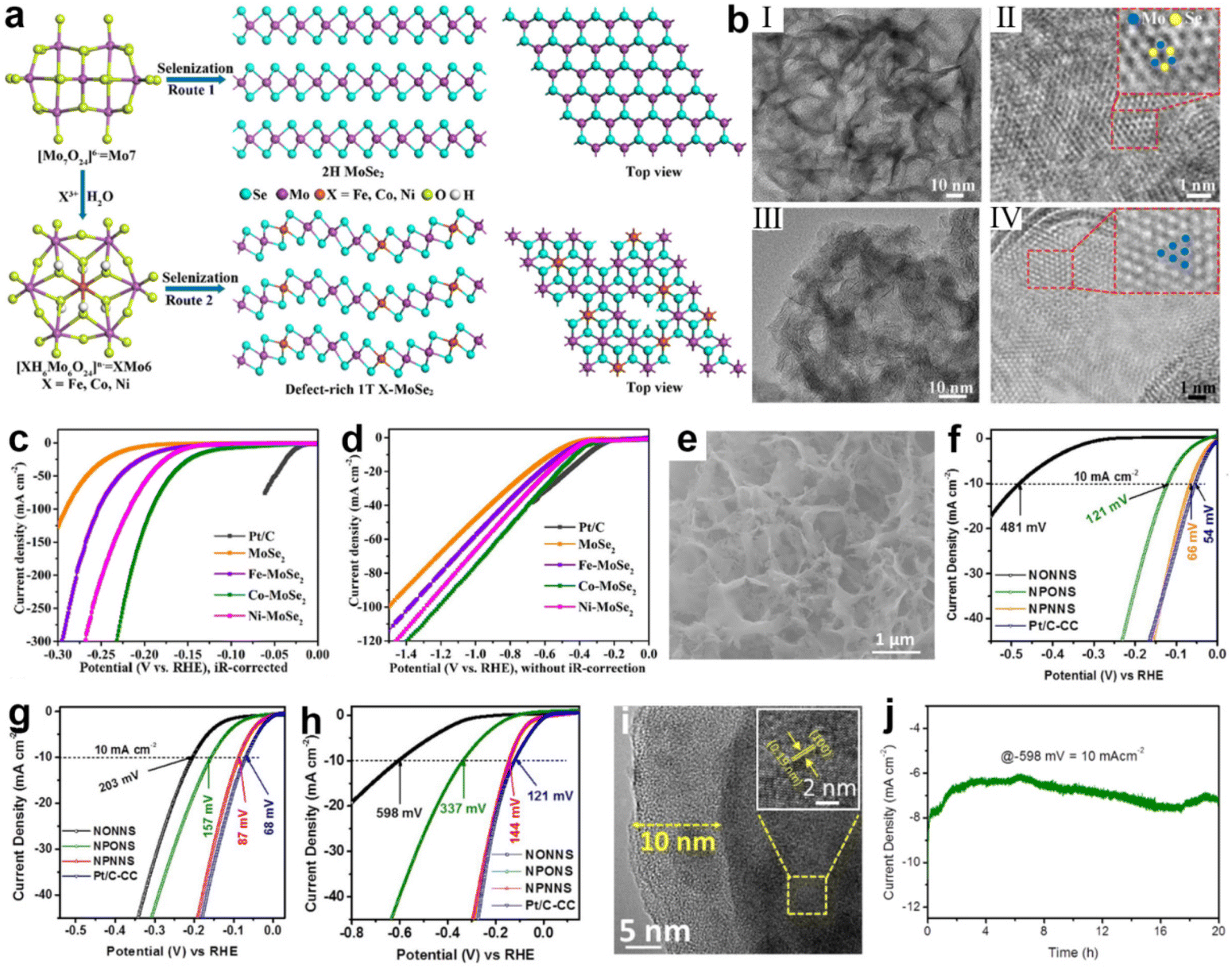 | ||
| Fig. 7 (a) A schematic of the synthesis of defect-rich 1T-MoSe2 nanosheets (route 2 with XMo6) and 2H-MoSe2 nanosheets with a Mo7 precursor. (b) TEM characterizations of 2H-MoSe2 and 1T Co-MoSe2. Low-magnification images of (I) MoSe2 and (III) Co-MoSe2 nanosheets. High-resolution TEM images of (II) MoSe2 and (IV) Co-MoSe2 nanosheets. The blue and yellow balls indicate the Mo and Se atoms, respectively. (c and d) LSV curves of commercial Pt/C, MoSe2 and X-MoSe2 without iR-drop correction in 0.5 M H2SO4 and simulated seawater. Scan rate: 5 mV s−1.184 Copyright 2022, ACS. (e) SEM images of NPONS. (f–h) LSV curves of NONNS, NPONS, NPNNS and Pt/C-CC without iR-drop correction in 0.5 M H2SO4, 1.0 M KOH and seawater. (i) The HRTEM image of NPNNS catalyst after stability test. Inset is the corresponding lattice fringes value. (j) Stability test of the NPONS electrocatalyst at 10 mA cm−2 in seawater.194 Copyright 2019, Elsevier. | ||
In recent years, a number of research efforts have found that amorphous nanomaterials with long-range disorder properties outperform crystalline materials in the field of water splitting.186,187 Interestingly, this disordered structure exposes a large number of active sites on the surface and the high structural flexibility of amorphous nanomaterials allows the reaction to expand into the catalyst volume, and the active sites inside the catalyst are similar to those on the surface, which greatly enhances the catalytic activity.83,188–190 Benefiting from the high degree of structural flexibility endowed by long-range disordered structure, it is easy to realize dynamic surface self-reconfiguration processes that can optimize the surface state and achieve rapid water splitting.191–193 In the study by Tong's group, Ni5P4194 was surface reformed by reaction with gaseous H2O and N2 under high temperature conditions, producing an amorphous coating composed of hydrogen (oxygen) oxides of nickel [Ni2+δOδ(OH)2−δ] (as shown in Fig. 7e). In acidic, alkaline and salty electrolytes, this catalyst obtained a current density of 10 mA cm2 at low overpotentials of 66, 87, and 144 mV (Fig. 6f, g and h), respectively. On analysis of the characterizations, it was found that the presence of Ni2+δOδ(OH)2−δ not only functions as an anti-corrosion layer (Fig. 7i), but also hybridizes with the Ni5P4 to increase the specific surface area of the hybrid electrocatalyst. In addition, theoretical calculations also suggest that the aforementioned hybridization produces favorable electronic interactions and synergistic effects for suppressing P–Hads bonds, which is helpful for facilitating water adsorption and optimizing the free energy of hydrogen adsorption for initiating the catalytic pathway at all pH ranges. In addition, due to the disordered structure, it is easy to dope heteroatoms into the amorphous phase, yielding a synergistic improvement of catalytic activity.136 In a recent report, Riley's group generated an Fe3O4/NiCx composite (NiFe-PBA-gel-cal)195 by calcining the coordination compound precursor NiFe-PBA-gel. After the OER test, the amorphous NiCx network was reconstituted at the oxidation potential to produce NiOOH2−x and wrapped with dispersed Fe3O4 nanoparticles to form a core–shell structure. The shell layer of this structure contained high-valence ions and a significant amount of oxygen defects. According to DFT simulation, the formation of high-valence Ni induces the generation of localized O 2p vacancies, and these vacancies act as electrophilic centers to activate OER redox reactions, greatly enhancing the electrochemical activity of OER. With the aid of 18O in situ isotopic labeling, it was found that the OER reaction pathway on NiFe-PBA-gel-cal is dominated by the LOM pathway due to the high-valent Ni cation and abundant oxygen defects, bypassing the adsorption of oxygen-containing intermediates and facilitating the reaction kinetics. Characterization in HER experiments revealed that the reduction of Fe3+ to Fe2+ in Fe3O4, together with the formation of Ni(OH)2 from NiCx, resulted in strong catalytic performance.
Despite these advances, catalytic performance should still be improved by remarkable progress. First, elaborating advanced and scalable methods to prepare high-purity 1T-phase MoSe2 remains a major challenge. Of note, other polymorphic materials (such as MoS2 or WS2) deserve more attention as well. Besides, amorphous and metastable materials are also difficult to develop due to the lack of understanding of the mechanism. In addition, the chemical instability of amorphous or metastable-phase materials severely hinders their practical applications. Thus, to gain a high degree of chemical and physical stability, more effort should be invested to identify effective methods.
4.5 Heterostructure design
Heterostructure catalysts, consisting of components with multiple phases and with a large number of defects at the boundaries as active sites, will lead to synergistic effects, strong electronic interactions or support effects, resulting in a significant facilitation of charge transfer between the electrocatalyst, electrolyte and gas phase interfaces.87,196–198 Hence, heterostructure catalysts can leverage the benefits of various materials to achieve higher catalytic activity than single-component catalysts.87 Therefore, the construction of heterostructure is considered as the most important method in interface modulation engineering to improve the adsorption and desorption of intermediates on the catalyst interface during electrocatalytic water splitting.199–202In the electrolysis of seawater, heterojunction construction is also commonly utilized because it can optimize the interfacial property and take full advantage of various types of components.62,119 For example, Qiu et al. proposed a design strategy for a multifunctional synergistic catalytic interface and successfully constructed an efficient HER catalyst (CoxMo2−xC/Mxene/NC)203 to achieve effective high-efficiency electrolysis in natural seawater. In this strategy, the highly active CoxMo2−xC/NC is formed by synergistic coupling serving as the active site, and the introduction of Mxene provides sufficient chemical anchor sites for loading of CoxMo2−xC/NC and acts as a conductor, as indicated by Fig. 8a. Theoretical calculations indicate that the collaborative interface between CoxMo2−xC, MXene and nitrogen-doped carbon ensures fast kinetics, lower adsorption–desorption energy barriers of intermediates (H* and H2), and fast charge transfer rates. Precise interfacial engineering ensured good catalytic activity of the electrocatalysts, and the activity of Co0.31Mo1.69C/MXene/NC could exceed that of Pt/C at a current density of 20 mA cm−2 during pH = 2.2–11.2. In seawater, the Co0.31Mo1.69C/MXene/NC catalyst has a Tafel slope very close to that of Pt/C. The ηj = 10 for Co0.31Mo1.69C/MXene/NC, which is 306 mV in the initial scan, is very close to that of Pt/C (297 mV), as shown in Fig. 8d, while its stability is better than that of the Pt/C electrode (Fig. 8e). Similar reports include VS2@V2C nanosheets,204 PF-NiCoP/NF,205etc., all of which exhibit a tremendous improvement compared with single components, respectively. In the design of heterostructures, charge transfer occurs at the interface due to different energy band arrangements with alteration of phases, which facilitates surface/interface electron modulation of the heterogeneous structure.28,206,207 For example, Pan et al.208 synthesized three-dimensional porous hierarchical CoNiP/CoxP multi-phase heterostructures prepared by electrodeposition on nickel foam. Analysing the XPS data of CoNiP/CoxP shows that the strong coupling of CoNiP and CoxP at the heterogeneous interface leads to significant electronic shifts with partial electron transfer from Ni and Co to P. Combined with the theoretical calculations, the high positive charges on Co and Ni atoms can significantly increase the adsorption sites of hydride ions, while the negatively charged P atoms effectively trap protons due to their strong electrostatic affinity, thus promoting hydrogen production. The optimized CoNiP/CoxP multiphase heterojunction was found to have an ultra-low overpotential of 36 mV at 10 mA cm−2, which has excellent ultra-high performance for seawater splitting.
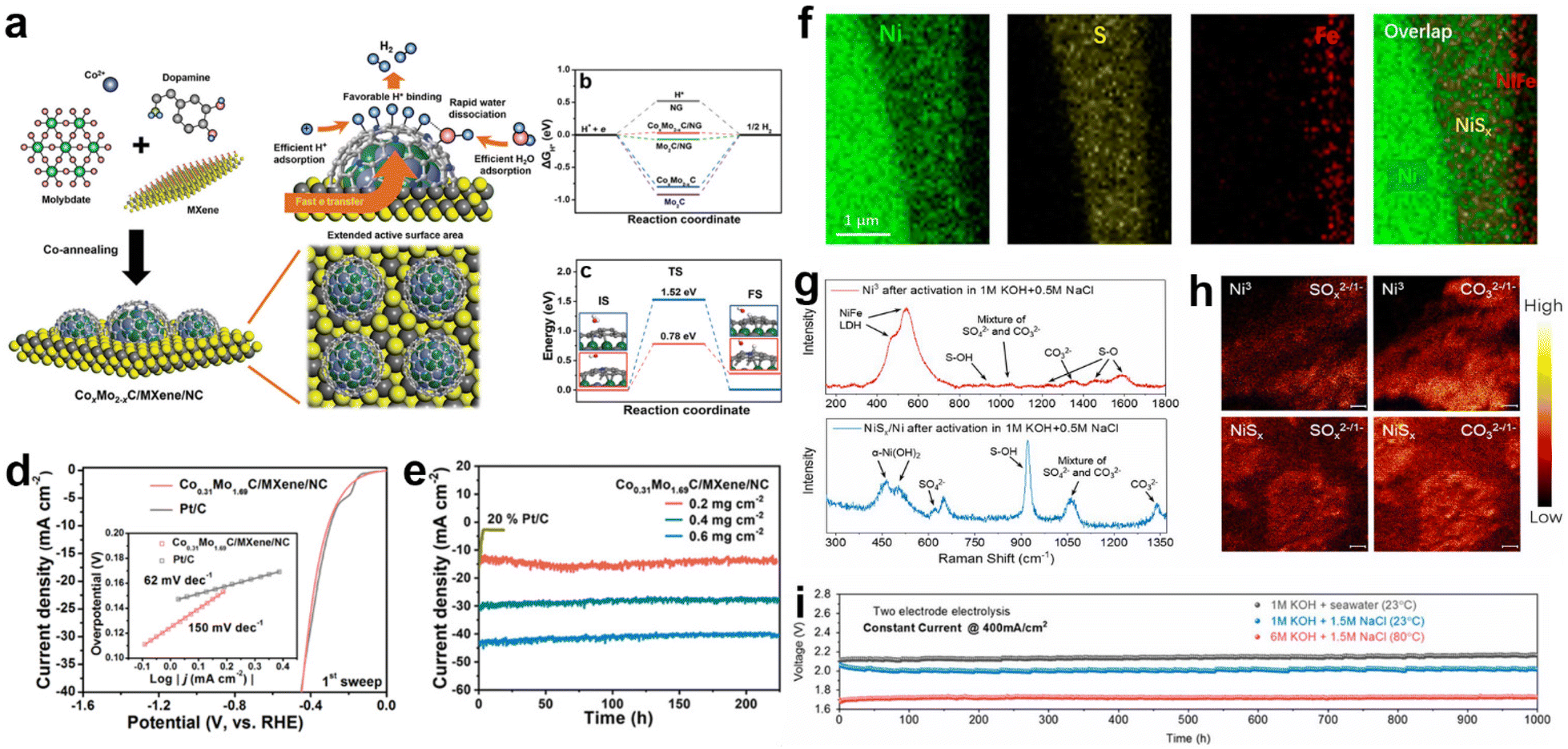 | ||
| Fig. 8 (a) Schematic illustration of synthetic strategy of the CoxMo2−xC/MXene/NC catalyst. (b) Free energy diagrams for HER on the NG, Mo2C, CoxMo2−xC, Mo2C/NG, and CoxMo2−xC/NG at zero potential. (c) UPS spectra of the Co0.31Mo1.69C/MXene/NC and Mo2C/MXene/NC. (d) Polarization curves and Tafel plots (the inset in each panel) of the 1st sweep of the Co0.31Mo1.69C/MXene/C and 20% Pt/C in simulated seawater. (e) Time-dependent current density curves of the Co0.31Mo1.69C/MXene/NC with various mass loading and 20% Pt/C (0.2 mg cm−2) in seawater at a static overpotential of 500 mV for 225 h.203 Copyright 2019, Wiley-VCH Verlag GmbH. (f) Elemental mapping of a cross-section of NiFe/NiSx on an Ni wire in the Ni foam, revealing Ni wire, NiSx, and NiFe layers. (g) The Raman spectra and (h) TOF-SIMS mapping (SOx2−/1− and CO32−/1−) of Ni3 and NiSx/Ni after activation, indicating the LDH phase of the polyatomic anion intercalation and the formation of sulfate species at the LDH/NiSx interface. (i) Durability tests (1000 h) recorded at a constant current of 400 mA cm−2 for the seawater splitting electrolyzer under different conditions.34 Copyright 2019, PNAS. | ||
In terms of regulating selectivity and stability, the selectivity of the heterostructure catalyst can be enhanced by in situ reconstructing a passivation layer, in which the oxidation products (sulfate and carbonate) generated in the passivation layer selectively repel chloride ions.28,209 For example, Sun and co-workers constructed a multistage layered NiFe/NiSx–Ni anode.34 By electrodeposition, the surface-deposited NiFe serves as a highly active OER catalyst in alkaline seawater splitting, while the NiSx layer (Fig. 8f) below provides a conductive intermediate layer and an S source to generate a polyatomic anion-rich anode with cation selectivity that is resistant to chlorine. In the performance evaluation, this passivated Ni3 anode exhibited a high selectivity of about 100% for OER in alkaline-adjusted salty water. This excellent catalytic selectivity is attributed to the chloride-repelling action of the polyatomic anion (SO42− and CO32−)-passivated Ni3 layers (Fig. 8g and h) generated in situ in the passivation layer. Meanwhile, the passivation layer also helps to prevent chloride ions from invading and corroding the underlying structure, thus playing a role in preventing corrosion. In durability testing, the catalyst was operated continuously for over 1000 hours at industrial-grade current densities (0.4 to 1 A cm−2) with only slight degradation, as shown in Fig. 8i. Similar schemes that generate passivation layers to protect the catalyst from free Cl− by interfacial reconstruction include Ni3S2/Co3S4 nanosheets,210 S-NiMoO4@NiFe-LDH211 and Ni2Fe-LDH/FeNi2S4/NF.212 Apart from the previous approach, direct compounding of corrosion-resistant materials with active materials has also proved to be feasible. In their study, Ji and co-workers enabled a highly reactive NiFe LDH composite on corrosion-resistant FeOOH by electrodeposition.172 In the 1.0 M KOH + 0.5 M NaCl electrolyte, the NiFe LDH/FeOOH nanosheets showed superior catalytic activity with a cell voltage of 1.55 V at 10 mA cm−2. Moreover, the NiFe LDH/FeOOH catalysts also showed stability with almost no significant decay when tested at 100 mA cm−2 for 105 h. Tracing the reasons, the authors attribute the excellent activity to the production of NiOOH species and the anti-corrosion properties to the presence of FeOOH. Specifically, the generation of heterojunctions between FeOOH and NiFe LDH allows the optimization of the electronic structure of the interface, while also promoting the formation of active NiOOH species.
Collectively, the construction of heterojunctions exhibits great potential in enhancing seawater electrolysis through improved electron transfer efficiency and optimized reactant adsorption and product desorption processes. However, these aspects still need more attention in the design of catalysts. For activity, the advantage that heterojunctions are easy-to-build 3D hierarchical structures should be used as much as possible to assist electrolyte mass transfer and gas release. As for anti-corrosion, more attention should be paid to the material boundaries that are vulnerable to the medium's corrosive effects during electrolysis.
4.6 Wettability engineering
Wettability, another key physicochemical feature of the catalyst surface, has a significant influence on the mass transfer behavior of electrocatalysts.67,213 In the electrolysis, catalysts with hydrophilic surfaces, as hydrophilicity facilitates catalyst/water interaction and the rapid release of bubbles formed on the surface, are very significant for catalytic activity enhancement, especially at high current densities.39,67 Young's equation (eqn (11)) is usually used to illustrate hydrophilicity and hydrophobicity, where the surface might be categorized as hydrophilic (0° < θ < 90°) or hydrophobic (90° < θ < 180°).87,214| γSV − γSL = γLVcosθ | (11) |
Especially in seawater splitting, the importance of surface wettability regulation is increased due to the weakened electrostatic force in seawater, which results from the presence of high concentrations of cation (e.g., Na+ and Mg2+).67,215,216 Specifically, the weakening of the electrostatic force leads to a more difficult adsorption of reactant on the catalyst surface, manifesting as a stronger hydrophobicity of the catalyst in seawater.67 To guarantee efficient mass transfer and speedy gas release from the reaction process, greater emphasis should be placed on enhancing the wettability of the catalyst in order to soothe the reduced adsorption caused by increased ionic strength. The wettability of catalysts is determined by both chemical composition and surface morphology.213,217 Therefore, it is of great interest to improve the wettability of catalysts through chemical composition control and morphology modulation to reduce the weakening of adsorption due to enhanced ionic strength. Recently, Haik's group218 reported partially amorphous sulfur-doped copper oxide (S–Cu2O–CuO) nanoneedles grown directly on copper foil as OER catalysts in seawater electrolysis. The structural disorder caused by S doping enhances the hydrophilic and aerophobicity of the electrocatalyst and accelerates the ion diffusion process. The improved hydrophilicity improved the reaction kinetics in terms of bubble desorption/separation, while appropriate interaction of H2O with the active site accelerates the ionic and charge kinetics. Combined with other benefits, these highly hydrophilic S–Cu2O–CuO nanoneedles exhibit an excellent OER performance, only requiring an overpotential of 450 mV to obtain high intrinsic activity (1000 mA cm−2) in 1 M KOH. In alkaline seawater, it requires an overpotential of only 420 mV to reach a high current density (500 mA cm−2) and continuously maintains electrolytic seawater for 100 h without hypochlorite production. Furthermore, Ren et al.219 succeeded in modulating catalyst wettability by controlling the surface morphology. By engineering the smooth nickel foam surface (Fig. 9a) as a rough S-doped Ni/Fe(oxy)-hydroxide layer (as shown in Fig. 9b and c), the layer possesses multilayer porosity and good hydrophilic characteristics (Fig. 9d and e), and exhibits excellent OER performance in both alkaline purified water and seawater. The outcomes of its direct utilization as an OER catalyst in alkaline natural seawater are displayed in Fig. 9f. Specifically, the S-doped Ni/Fe(oxy)hydroxide catalyst needs modest overpotentials of 300 and 398 mV to yield current densities of 100 and 500 mA cm2, respectively. Similar findings include CoNiP/CoxP nanosheet208 heterojunctions and CdS/Bi2MoO6 heterojunctions,220 which demonstrate similar pathways for enhancing HER activity.
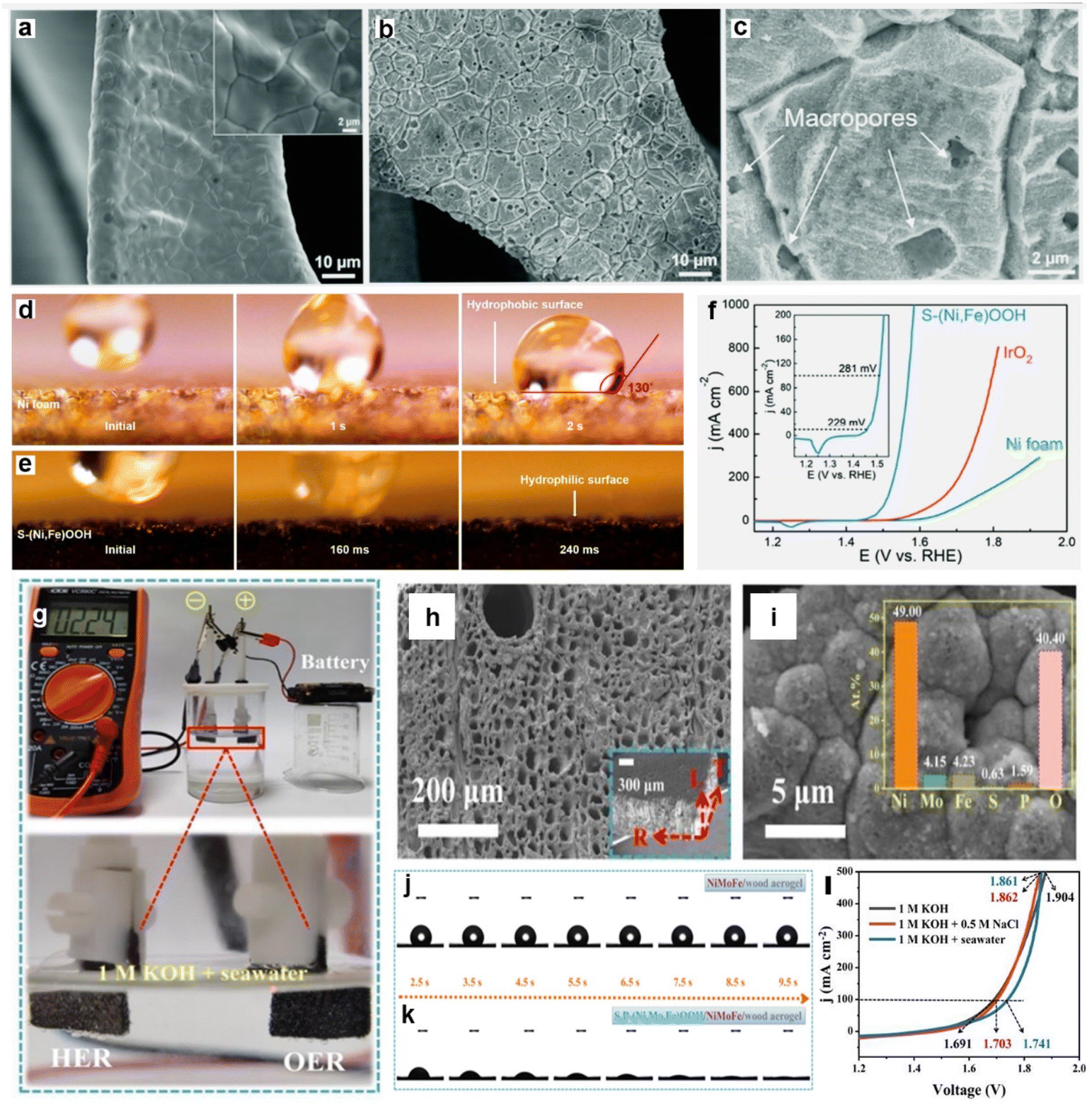 | ||
| Fig. 9 (a–c) SEM images of (a) Ni foam and (b–c) S-(Ni,Fe)OOH at different magnifications. Digital images of a droplet (1 M KOH electrolyte) placed on the surface of (d) commercial Ni foam and (e) S-(Ni,Fe)OOH to investigate the wettability between the electrolyte and the catalyst surface. (f) Polarization curves of the Ni foam, IrO2, and S-(Ni,Fe)OOH electrodes. The inset in (f) shows the polarization curve of S-(Ni,Fe)OOH over a small current density range.219 Copyright 2020, RSC. (g) The photos showing actual two batteries-driven electrolysis (1 cm2 electrodes) of 1 M KOH and seawater under an output voltage of 2.24 V. The SEM images of S,P-(Ni,Mo,Fe)OOH/NiMoP/wood aerogel (h) and the inset picture shows the SEM image of side view of the sample, corresponding EDS mapping images (i), showing compact coverage and homogeneous distribution. Water contact angles of (j) NiMoP/wood aerogel and (k) S,P-(Ni,Mo,Fe)OOH/NiMoP/wood aerogel over time. (l) Polarization curves of the S,P-(Ni,Mo,Fe)OOH/NiMoP/wood aerogel electrolyzer tested in different electrolytes.221 Copyright 2021, Elsevier. | ||
In addition, a composite strategy has been applied to modulate the wettability of catalysts, namely, coupling highly active materials to hydrophilic substrates to improve the hydrophilicity of the overall catalysts.221,222 For example, Yang et al.221 constructed a hierarchical catalyst with highly hydrophilic wood aerogels as the trunk and attached highly active metal phosphides, which generated S,P-(Ni,Mo,Fe)OOH nanolayers on phosphide surfaces after rapid activation (Fig. 9h and i). The layer–layer structure assures a high rough interfacial contact between the wood aerogel/NiMoP and NiMoP/S,P-(Ni,Mo,Fe)OOH, contributing to the bubble release and high electron transfer capacity, as shown in Fig. 9j and k. According to the experimental and theoretical calculations, the open and well-aligned direct multi-channels in the wood aerogel encourage the quick release of bubbles, which generated by S,P-(Ni,Mo,Fe)OOH nanolayer catalyzes the decomposition of water molecules. Of note, the bursting force generated by the rapid release of bubbles moves precipitates/ions away from the active sites on catalyst, significantly improving OER/HER activity in alkaline seawater.
In comparison with other technological advances for facilitating seawater splitting, the progress in wettability engineering has remained very limited. Nevertheless, the high ionic strength in seawater diminishes the electrostatic force of solution, and emphasizes the importance of catalyst surface wettability. At present, studies mainly focus on enhancing the catalytic activity by changing the morphological structure and chemical composition of catalysts, but fundamental evaluations of the influence of ionic effects in solution on the electrolysis of seawater are lacking. These mechanistic studies are extremely valuable to guide subsequent studies on seawater electrolysis.
5. Summary and outlook
Using abundant natural seawater instead of pure water and directly electrolyzing seawater as a hydrogen source has considerable economic and environmental benefits, and has become a research hotspot in recent years. However, the existence of various impurities in natural seawater has a negative impact on the electrolysis system, including Cl− oxidization, scaling, membrane fouling and electrode corrosion. To remove these negative impacts, significant effort has been devoted to the development of efficient and stable electrolytic systems, involving the design of electrolytic cells, Amberplex selection, and electrode synthesis. Considering industrial application, the current unpretreated seawater splitting schemes are less costly, but have difficulty meeting long-term stability requirements mainly caused by precipitate coverage, thus requiring pre-treatment of natural seawater. Although existing desalination pre-treatment schemes can directly remove all solutes from seawater, their high pre-treatment cost limits their wide application in seawater electrolysis. Inspired by the chlor-alkali production process, we propose a lower-cost pre-treatment regimen (such as capacitive deionization coupled seawater splitting) for natural seawater instead of the more costly desalination. The principle of the capacitive deionization (CDI) process relies on the electrical potential difference over a pair of electrodes to remove charged species from the water.64–66 Meanwhile, stronger attractions between the electrodes and multivalent ions enables the selective adsorption of alkaline earth-metal ions (Ca2+, Mg2+) and the softening of the salt-rich water.64–66 Through prefiltration and capacitive selective adsorption, insoluble particles and heteroatoms (Ca2+ and Mg2+) are removed from natural seawater successively, avoiding the generation of hydroxide precipitates and maintaining the stable operation of electrolytic system. On the other hand, the residual inorganic salt ions also enhance the conductivity of the solution and reduce the use of electrolytes. Combined with highly selective seawater electrolysis catalysts to inhibit the oxidation reaction of chlorine, this will enable efficient and low-cost energy storage through seawater electrolysis. Due to the presence of residual chloride ions in the solution, alkaline electrolysis is clearly a preferable option for coupling with capacitive deionization, considering the reduction of chlorine byproduct (HClO/Cl2). Meanwhile, the AWE cells are simple in construction, low in equipment cost, and have a good tolerance to electrolytes. With the aforementioned adjustments to the anti-corrosion design, the AWE cell can accomplish long-term stable seawater electrolysis operation at a cheaper cost. On further progress, with the maturing of hydroxide exchange membrane, the AEM electrolyser will be a superior alternative for a higher energy conversion rate and faster hydrogen generation.In addition, the severe conditions of seawater splitting also demand high requirements in the overall performance of electrocatalysts. Advanced surface and interface engineering has been demonstrated to be critical for the construction of efficient and stable electrodes for seawater electrolysis. Herein, we have summarized various strategies to improve the performance of seawater splitting (Table 1 summarizes the electrochemical performances of the corresponding representative HER and OER catalysts in seawater). The pertinent working mechanisms of these strategies and their unique contributions to catalytic activity, selectivity and corrosion resistance are dissected and summarized as follows.
| Electrocatalyst working electrode | η (mV) (for jb) | Tafel | FE | Reaction medium (pH) | t (h) | Ref. |
|---|---|---|---|---|---|---|
| HER | ||||||
| Co10%–VS2 | 234 mV/10 mA cm−2 | 93 mV dec−1 | — | 1 M PBS | 12 | 134 |
| GDY/MoO3 | 170 mV/10 mA cm−2 | 70 mV dec−1 | — | 0.1 M KOH | 120 | 138 |
| RuV–CoNiP/NF | 81 mV/50 mA cm−2 | 49.6 mV dec−1 | — | 1 M KOH + seawater | 24 | 158 |
| Fe–Co2P BNRs | 771 mV/300 mA cm2 | 95 mV dec−1 | — | Seawater, pH = 7.8 | 100 | 164 |
| Defect-rich doped 1T-MoSe2 nanosheets | 274 mV/10 mA cm−2 | 177 mV dec−1 | — | Seawater | 24 | 184 |
| Ni–MoO3|S600 | 412 mV/10 mA cm−2 | 171 mV dec−1 | — | 1 M KOH + 3.5% NaCl | 24 | 31 |
| Ni2+δOδ(OH)2−δ | 144 mV/10 mA cm−2 | 108 mV dec−1 | 93% | 1 M KOH + seawater | 40 | 194 |
| Co0.31Mo1.69C/MXene/NC | 306 mV/10 mA cm−2 | 76 mV dec−1 | 98% | Seawater, pH = 8.2 | 225 | 203 |
| CoNiP/CoxP | 290 mV/10 mA cm−2 | 70 mV dec−1 | ≈100% | Seawater, pH = 8.19 | 500 | 208 |
| OER | ||||||
| RuIrOx | 233 mV (pH = 0); 250 mV (pH = 14)/10 mA cm−2 | 42 mV dec−1 | — | 0.5 M H2SO4 | >24 | 116 |
| 50 mV dec−1 | 1.0 M KOH | |||||
| Fe–Co–S/Cu2O/Cu | 338 mV/50 mA cm−2 | 111 mV dec−1 | — | 1 M KOH | 70 | 122 |
| CoPx@FeOOH | 283 mV/100 mA cm−2 | 37.6 mV dec−1 | >98% | 1 M KOH seawater | 80 | 117 |
| Na|MnOx | — | 128–139 mV dec−1 | 87% | 0.5 M NaCl | — | 31 |
| RuV–CoNiP/NF | 263 mV/50 mA cm−2 | 53.5 mV dec−1 | 100% | 1 M KOH seawater | 24 | 158 |
| Gd–Mn3O4@CuO–Cu(OH)2 | 400 mV/500 mA cm−2 | 58 mV dec−1 | >97% | 1 M KOH seawater | 75 | 163 |
| 0.5Fe–NiCo2O4@CC | 293 mV/10 mA cm−2 | 76.1 mV dec−1 | — | 1 M KOH seawater | 20 | 166 |
| Mo–Ni3S2 | 291 mV/100 mA cm−2 | 42 mV dec−1 | 100% | 1 M KOH seawater | 500 | 157 |
| Fe,P-NiSe2 NFs | 570 mV/800 mA cm−2 | 48.9 mV dec−1 | 99.5% | 0.5 m KOH + seawater | 200 | 172 |
| Co2(OH)3Cl | 379 mV/100 mA cm−2 | 58.5 mV dec−1 | 99.9% | 1 M KOH + 0.6 M NaCl | 16.7 | 33 |
| NiFe-PBA-gel-cal | 329 mV/100 mA cm−2 | 68.7 mV dec−1 | — | 1 M KOH + 0.5 M NaCl | 60 | 195 |
| NiFe/NiSx–Ni | 160 mV/500 mA cm−2 | — | 100% | 1 M KOH + 0.5 M NaCl | 1000 | 34 |
| NiFe LDH/FeOOH | 286.2 mV/10 mA cm−2 | 69.8 mV dec−1, 1 M KOH | ≈100% | 1 M KOH + 0.5 M NaCl | 105 | 172 |
| S–Cu2O–CuO | 420 mV/500 mA cm−2 | 40 mV dec−1, 1 M KOH | 99% | 1 M KOH + 0.5 M NaCl | 100 | 218 |
| S-(Ni,Fe)OOH | 300 mV/100 mA cm−2 | 48.9 mV dec−1, 1 M KOH | 99% | 1 M KOH seawater | 100 | 219 |
| S,P-(Ni,Mo,Fe)OOH | 420 mV/500 mA cm−2 | 45.5 mV dec−1 | — | 1 M KOH seawater | 30 | 221 |
(1) Rational design of the surface/interface properties of electrocatalysts can significantly enhance three key steps in the electrocatalytic process (mass transfer, charge transfer and surface reaction). Among the various strategies, morphology design, heterojunction design and wettability can enhance the exposure of active sites and improve the mass transfer process. In charge transfer, the introduction of defect engineering, doping and heterojunction construction greatly speed up the transfer of electrons between electrodes and reactants, by adding additional active sites, increasing the intrinsic conductivity and lowering the reaction energy barrier. Tailoring the crystalline surface/surface phase can also significantly improve the surface reaction by primarily exposing more active sites and tuning the adsorption/desorption behavior.
(2) Catalytic selectivity can be significantly enhanced by charge distribution modulation and reconfiguration at the surface/interface. For instance, the placement of defects and the addition of heteroatoms can modify the charge distribution and adsorption/desorption behavior, resulting in improved catalytic selectivity. Moreover, surface reconfiguration can promote the generation of anion-rich passivation layers, and repel chloride ionization with the help of negative electrical repulsion.
(3) Construction of in situ corrosion-resistant layers or buffer structures to improve catalysts’ stability by means of general surface/interface engineering, including heterogeneous phase engineering, heterostructure design and atomic doping.
Despite tremendous advances in enhancing electrocatalytic performance by surface/interfacial engineering, visible challenges and bottlenecks remain at present. Therefore, a deeper understanding of the electrocatalytic reaction mechanism associated with seawater electrolysis is necessary, since it will assist in the development of effective and durable electrode materials. Advanced in situ characterization techniques are necessary to visualize the real-time interaction between the catalyst and electrolyte as well as the dynamic growth of the corrosion-resistant layer, which immediately and unmistakably reflects the process's reaction mechanism. At present, some in situ characterization techniques including Fourier transform infrared spectroscopy (FTIR),223 Raman spectroscopy172,223–225 and synchrotron X-ray absorption spectroscopy (XAS)116 have been introduced to study the mechanism of seawater splitting, but still mainly focus on the adsorption/desorption behavior of intermediates, with few reports that refer to the structural changes/reconstruction of the active site during electrochemical processes. In further investigation, we anticipate that the emerging operando Mössbauer spectroscopy and X-ray emission spectroscopy (XES) will offer complementary and valuable structural information to well-developed related research. Additionally, the simulated calculation of reactants’ chemisorption/desorption behavior on surface structures with the help of computational methods is also an important approach for revealing the catalytic mechanism. Ab initio molecular dynamics simulations and machine learning based on periodic DFT calculations may be able to simulate dynamic processes and contribute to the future understanding of structural design and reaction processes in electrocatalysis. The characterization and simulation results above combined with active machine learning can provide guidance for the design and development of advanced electrocatalytic materials, which will become an increasingly important research direction in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22072015) and Sichuan Department of Science and Technology Program of China (No. 2022ZYFG0312).References
- J. Song, C. Wei, Z. F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- T. Li, T. Hu, L. Dai and C. M. Li, J. Mater. Chem. A, 2020, 8, 23674–23698 RSC.
- J. Yu, B.-Q. Li, C.-X. Zhao and Q. Zhang, Energy Environ. Sci., 2020, 13, 3253–3268 RSC.
- F. Dingenen and S. W. Verbruggen, Renewable Sustainable Energy Rev., 2021, 142, 110866 CrossRef CAS.
- I. Dincer and C. Acar, Int. J. Hydrogen Energy, 2015, 40, 11094–11111 CrossRef CAS.
- F. H. Sobrino, C. R. Monroy and J. L. H. Pérez, Renewable Sustainable Energy Rev., 2010, 14, 772–780 CrossRef CAS.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- Y. Shi, Z. R. Ma, Y. Y. Xiao, Y. C. Yin, W. M. Huang, Z. C. Huang, Y. Z. Zheng, F. Y. Mu, R. Huang, G. Y. Shi, Y. Y. Sun, X. H. Xia and W. Chen, Nat. Commun., 2021, 12, 3021 CrossRef PubMed.
- J. Liu, S. Duan, H. Shi, T. Wang, X. Yang, Y. Huang, G. Wu and Q. Li, Angew. Chem., 2022, 134, e202210753 Search PubMed.
- B. E. Logan, L. Shi and R. Rossi, Joule, 2021, 4, 760–762 CrossRef.
- H. A. Loáiciga, X. Chu, M. Aboutalebi, P. Sarzaeim, B. Zolghadr-Asli and O. Bozorg-Haddad, J. Water Supply: Res. Technol.–AQUA, 2020, 69, 85–98 CrossRef.
- C. Wang, H. Shang, L. Jin, H. Xu and Y. Du, Nanoscale, 2021, 13, 7897–7912 RSC.
- S. Dresp, F. Dionigi, M. Klingenhof and P. Strasser, ACS Energy Lett., 2019, 4, 933–942 CrossRef CAS.
- P. Farràs, P. Strasser and A. J. Cowan, Joule, 2021, 8, 1921–1923 CrossRef.
- J. N. Hausmann, R. Schlögl, P. W. Menezes and M. Driess, Energy Environ. Sci., 2021, 14, 3679–3685 RSC.
- W. Li, H. Tian, L. Ma, Y. Wang, X. Liu and X. Gao, Mater. Adv., 2022, 3, 5598–5644 RSC.
- E. Asghari, M. I. Abdullah, F. Foroughi, J. J. Lamb and B. G. Pollet, Curr. Opin. Electrochem., 2022, 31, 100879 CrossRef CAS.
- W. Tong, M. Forster, F. Dionigi, S. Dresp, R. Sadeghi Erami, P. Strasser, A. J. Cowan and P. Farràs, Nat. Energy, 2020, 5, 367–377 CrossRef CAS.
- A. Kumar, K. R. Phillips, G. P. Thiel, U. Schröder and J. H. Lienhard, Nat. Catal., 2019, 2, 106–113 CrossRef CAS.
- G. A. Lindquist, Q. Xu, S. Z. Oener and S. W. Boettcher, Joule, 2020, 12, 2549–2561 CrossRef.
- W. Zheng, L. Lee and K. Wong, Nanoscale, 2021, 13, 15177–15187 RSC.
- B. Cui, Y. Shi, G. Li, Y. Chen, W. Chen, Y. Deng and W. Hu, Acta Phys.-Chim. Sin., 2021, 38, 2106010 Search PubMed.
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, ChemSusChem, 2016, 9, 962–972 CrossRef CAS PubMed.
- S. Khatun, H. Hirani and P. Roy, J. Mater. Chem. A, 2021, 9, 74–86 RSC.
- X. Liu, P. Wang, X. Liang, Q. Zhang, Z. Wang, Y. Liu, Z. Zheng, Y. Dai and B. Huang, Mater. Today Energy, 2020, 18, 100524 CrossRef CAS.
- Q. Xu, J. Zhang, H. Zhang, L. Zhang, L. Chen, Y. Hu, H. Jiang and C. Li, Energy Environ. Sci., 2021, 14, 5228–5259 RSC.
- P. Chen, Y. Tong, C. Wu and Y. Xie, Acc. Chem. Res., 2018, 51, 2857–2866 CrossRef CAS PubMed.
- Z. Fang, L. Peng, Y. Qian, X. Zhang, Y. Xie, J. J. Cha and G. Yu, J. Am. Chem. Soc., 2018, 140, 5241–5247 CrossRef CAS PubMed.
- Y. Zhu, L. Peng, Z. Fang, C. Yan, X. Zhang and G. Yu, Adv. Mater., 2018, 30, e1706347 CrossRef PubMed.
- H. Abe, A. Murakami, S. Tsunekawa, T. Okada, T. Wakabayashi, M. Yoshida and M. Nakayama, ACS Catal., 2021, 11, 6390–6397 CrossRef CAS.
- S. Jiang, H. Suo, T. Zhang, C. Liao, Y. Wang, Q. Zhao and W. Lai, Catalysts, 2022, 12, 123 CrossRef CAS.
- L. Zhuang, J. Li, K. Wang, Z. Li, M. Zhu and Z. Xu, Adv. Funct. Mater., 2022, 32, 2201127 CrossRef CAS.
- Y. Kuang, M. J. Kenney, Y. Meng, W. H. Hung, Y. Liu, J. E. Huang, R. Prasanna, P. Li, Y. Li, L. Wang, M. C. Lin, M. D. McGehee, X. Sun and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6624–6629 CrossRef CAS PubMed.
- S. Jiang, Y. Liu, H. Qiu, C. Su and Z. Shao, Catalysts, 2022, 12, 261 CrossRef CAS.
- L. Zhuang, S. Li, J. Li, K. Wang, Z. Guan, C. Liang and Z. Xu, Coatings, 2022, 12, 659 CrossRef CAS.
- J. Li, J. Sun, Z. Li and X. Meng, Int. J. Hydrogen Energy, 2022, 47, 29685–29697 CrossRef CAS.
- H. Zhang, Y. Luo, P. K. Chu, Q. Liu, X. Liu, S. Zhang, J. Luo, X. Wang and G. Hu, J. Alloys Compd., 2022, 922, 166113 CrossRef CAS.
- Q. Zhou, L. Liao, H. Zhou, D. Li, D. Tang and F. Yu, Mater. Today Phys., 2022, 26, 100727 CrossRef CAS.
- S. Ke, R. Chen, G. Chen and X. Ma, Energy Fuels, 2021, 35, 12948–12956 CrossRef CAS.
- R. De Levie, J. Electroanal. Chem., 1999, 476, 92–93 CrossRef CAS.
- D. M. Santos, C. A. Sequeira and J. L. Figueiredo, Quim. Nova, 2013, 36, 1176–1193 CrossRef CAS.
- Z. Y. Yu, Y. Duan, X. Y. Feng, X. Yu, M. R. Gao and S. H. Yu, Adv. Mater., 2021, 33, 2007100 CrossRef CAS PubMed.
- M. Paidar, V. Fateev and K. Bouzek, Electrochim. Acta, 2016, 209, 737–756 CrossRef CAS.
- G. G. Stoney, Proc. R. Soc. London, Ser. A, 1909, 82, 172–175 CAS.
- J. E. Bennett, Int. J. Hydrogen Energy, 1980, 5, 401–408 CrossRef CAS.
- S. Trasatti, Electrochim. Acta, 1984, 29, 1503–1512 CrossRef CAS.
- Y. Huang, L. Jiang, X. Liu, T. Tan, H. Liu and J. Wang, Appl. Catal., B, 2021, 299, 120678 CrossRef CAS.
- R. K. Karlsson and A. Cornell, Chem. Rev., 2016, 116, 2982–3028 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- Y. Yan, B. Y. Xia, B. Zhao and X. Wang, J. Mater. Chem. A, 2016, 4, 17587–17603 RSC.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K. Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M. J. Cheng, D. Sokaras, T. C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Norskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- L. Chen, J. T. Ren and Z. Y. Yuan, Green Chem., 2022, 24, 713–747 RSC.
- L. Zhang, H. Zhao, S. Xu, Q. Liu, T. Li, Y. Luo, S. Gao, X. Shi, A. M. Asiri and X. Sun, Small Struct., 2020, 2, 2000048 CrossRef.
- A. B. Laursen, S. Kegnæs, S. Dahl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 5577 RSC.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- C. Weng, J. Ren and Z. Yuan, ChemSusChem, 2020, 13, 3357–3375 CrossRef CAS PubMed.
- J. Wei, M. Zhou, A. Long, Y. Xue, H. Liao, C. Wei and Z. J. Xu, Nano-Micro Lett., 2018, 10, 75 CrossRef CAS PubMed.
- J. Huang, P. Li and S. Chen, J. Phys. Chem. C, 2019, 123, 17325–17334 CrossRef CAS.
- P. Gayen, S. Saha and V. Ramani, ACS Appl. Energy Mater., 2020, 3, 3978–3983 CrossRef CAS.
- J. S. Ko, J. K. Johnson, P. I. Johnson and Z. Xia, ChemCatChem, 2020, 12, 4526–4532 CrossRef CAS.
- Y. S. Park, J. Lee, M. J. Jang, J. Yang, J. Jeong, J. Park, Y. Kim, M. H. Seo, Z. Chen and S. M. Choi, J. Mater. Chem. A, 2021, 9, 9586–9592 RSC.
- E. Liu, L. Lee, S. L. Ong and H. Y. Ng, Water Res., 2020, 183, 116059 CrossRef CAS PubMed.
- F. A. AlMarzooqi, A. A. A. Ghaferi, I. Saadat and N. Hilal, Desalination, 2014, 342, 3–15 CrossRef CAS.
- J. Choi, P. Dorji, H. K. Shon and S. Hong, Desalination, 2019, 449, 118–130 CrossRef CAS.
- F. Zhang, L. Yu, L. Wu, D. Luo and Z. Ren, Trends Chem., 2021, 6, 485–498 CrossRef.
- S. Anwar, F. Khan, Y. Zhang and A. Djire, Int. J. Hydrogen Energy, 2021, 46, 32284–32317 CrossRef CAS.
- D. Ferrero, A. Lanzini, M. Santarelli and P. Leone, Int. J. Hydrogen Energy, 2013, 38, 3523–3536 CrossRef CAS.
- M. David, C. Ocampo-Martínez and R. Sánchez-Peña, J. Energy Storage, 2019, 23, 392–403 CrossRef.
- J. Wang, Y. Gao, H. Kong, J. Kim, S. Choi, F. Ciucci, Y. Hao, S. Yang, Z. Shao and J. Lim, Chem. Soc. Rev., 2020, 49, 9154–9196 RSC.
- L. Zhang, R. Li, H. Zang, H. Tan, Z. Kange, Y. Wang and Y. Li, Energy Environ. Sci., 2021, 14, 6191–6210 RSC.
- T. F. O'Brien, T. V. Bommaraju and F. Hine, Handbook of Chlor-alkali Technology, Volume I: Fundamentals, Springer, US, 2005 Search PubMed.
- A. Buttler and H. Spliethoff, Renewable Sustainable Energy Rev., 2018, 82, 2440–2454 CrossRef CAS.
- M. A. Khan, T. Al-Attas, S. Roy, M. M. Rahman, N. Ghaffour, V. Thangadurai, S. Lartere, J. Hu, P. M. Ajayan and M. G. Kibria, Energy Environ. Sci., 2021, 14, 4831–4839 RSC.
- L. Li, P. Wang, Q. Shao and X. Huang, Adv. Mater., 2021, 33, 2004243 CrossRef CAS PubMed.
- R. d'Amore-Domenech, O. Santiago and T. J. Leo, Renewable Sustainable Energy Rev., 2020, 133, 110166 CrossRef.
- N. Du, C. Roy, R. Peach, M. Turnbull, S. Thiele and C. Bock, Chem. Rev., 2022, 122, 11830–11895 CrossRef CAS PubMed.
- Z. Liu, B. Han, Z. Lu, W. Guan, Y. Li, C. Song, L. Chen and S. C. Singhal, Appl. Energy, 2021, 300, 117439 CrossRef CAS.
- A. Baldinelli, L. Barelli and G. Bidini, J. Cleaner Prod., 2021, 321, 128846 CrossRef CAS.
- C. K. Lim, Q. Liu, J. Zhou, Q. Sun and S. H. Chan, J. Power Sources, 2017, 342, 79–87 CrossRef CAS.
- M. Hoogwijk, D. V. Vuuren, B. D. Vries and W. Turkenburg, Energy, 2007, 32, 1381–1402 CrossRef.
- Z. Wu, X. Lu, S. Zang and X. Lou, Adv. Funct. Mater., 2020, 15, 1910274 CrossRef.
- T. Zhou, N. Zhang, C. Wu and Y. Xie, Energy Environ. Sci., 2020, 13, 1132–1153 RSC.
- R. Ge, J. Huo, M. Sun, M. Zhu, Y. Li, S. Chou and W. Li, Small, 2021, 17, e1903380 CrossRef PubMed.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS PubMed.
- P. Wang and B. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 59593–59617 CrossRef CAS PubMed.
- W. Zang, T. Sun, T. Yang, S. Xi, M. Waqar, Z. Kou, Z. Lyu, Y. P. Feng, J. Wang and S. J. Pennycook, Adv. Mater., 2021, 33, e2003846 CrossRef PubMed.
- L. Zeng, Z. Liu, K. Sun, Y. Chen, J. Zhao, Y. Chen, Y. Pan, Y. Lu, Y. Liu and C. Liu, J. Mater. Chem. A, 2019, 7, 25628–25640 RSC.
- S. Song, Y. Wang, X. Liu, X. Tian, Y. Liu, X. Liu, F. Sun, Y. Yuan, W. Li and J. Zang, Appl. Surf. Sci., 2022, 604, 1544588 Search PubMed.
- X. Sun, P. Yang, S. Wang, J. Hu, P. Chen, H. Xing and W. Zhu, Int. J. Hydrogen Energy, 2022, 47, 28495–28504 CrossRef CAS.
- J. Liang, Q. Liu, T. Li, Y. Luo, S. Lu, X. Shi, F. Zhang, A. M. Asiri and X. Sun, Green Chem., 2021, 23, 2834–2867 RSC.
- M. Caban-Acevedo, M. L. Stone, J. R. Schmidt, J. G. Thomas, Q. Ding, H. C. Chang, M. L. Tsai, J. H. He and S. Jin, Nat. Mater., 2015, 14, 1245–1251 CrossRef CAS PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- C. Xie, D. Yan, W. Chen, Y. Zou, R. Chen, S. Zang, Y. Wang, X. Yao and S. Wang, Mater. Today, 2019, 31, 47–68 CrossRef CAS.
- T. J. Schmidt, U. A. Paulus, H. A. Gasteiger and R. J. Behm, J. Electroanal. Chem., 2001, 508, 41–47 CrossRef CAS.
- K. S. Exner, ChemElectroChem, 2019, 6, 3401–3409 CrossRef CAS.
- Y. Huang, K. D. Seo, K. A. Jannath, D. S. Park and Y. B. Shim, Carbon, 2022, 196, 621–632 CrossRef CAS.
- J. Sun, J. Li, Z. Li, C. Li, G. Ren, Z. Zhang and X. Meng, ACS Sustainable Chem. Eng., 2022, 10, 9980–9990 CrossRef CAS.
- X. Wang, Y. Ling, B. Wu, B. Li, X. Li, J. Lei, N. Li and H. Luo, Nano Energy, 2021, 87, 106160 CrossRef CAS.
- T. Liu, S. Tang, T. Wei, M. Chen, Z. Xie, R. Zhang, Y. Liu and N. Wang, Cell Rep. Phys. Sci., 2022, 3, 100892 CrossRef CAS.
- Y. Y. Ma, C. X. Wu, X. J. Feng, H. Q. Tan, L. K. Yan, Y. Liu, Z. H. Kang, E. B. Wang and Y. G. Li, Energy Environ. Sci., 2017, 10, 788–798 RSC.
- J. Jiang, M. Gao, W. Sheng and Y. Yan, Angew. Chem., 2016, 128, 15466–15471 CrossRef.
- E. Hu, Y. Feng, J. Nai, D. Zhao, Y. Hu and X. Lou, Energy Environ. Sci., 2018, 4, 872–880 RSC.
- J. Staszak-Jirkovský, C. D. Malliakas, P. P. Lopes, M. G. Kanatzidis and N. M. Markovic, Nat. Mater., 2016, 15, 197–203 CrossRef PubMed.
- H. Li, Q. Tang, B. He and P. Yang, J. Mater. Chem. A, 2016, 17, 6513–6520 RSC.
- J. Zheng, Appl. Surf. Sci., 2017, 413, 72–82 CrossRef CAS.
- L. Bigiani, D. Barreca, A. Gasparotto, J. R. Morante and C. Maccatoa, Appl. Catal., B, 2021, 284, 119684 CrossRef CAS.
- X. Zhao, Q. Zhang, X. Huang, L. Ding, W. Yang, C. Wang and Q. Pan, Int. J. Hydrogen Energy, 2022, 47, 2178–2186 CrossRef CAS.
- W. Yu, Z. Chen, Y. Fu, W. Xxiao, T. Ma, B. Dong, Y. Chai, Z. Wu and L. Wang, Appl. Catal., B, 2022, 317, 121762 CrossRef CAS.
- T. Cui, J. Chi, J. Zhu, X. Sun, J. Lai, Z. Li and L. Wang, Appl. Catal., B, 2022, 319, 121950 CrossRef CAS.
- J. G. Vos, T. A. Wezendonk, A. W. Jeremiasse and T. M. Koper, J. Am. Chem. Soc., 2018, 32, 10270–10281 CrossRef PubMed.
- J. Li, T. Yu, K. Wang, Z. Li, J. He, Y. Wang, L. Lei, L. Zhang, M. Zhu, C. Lian, Z. Shao and Z. Xu, Adv. Sci., 2022, 25, 2202387 CrossRef PubMed.
- H. Wang, L. Chen, L. Tan, X. Liu, Y. Wen, W. Hou and T. Zhan, J. Colloid Interface Sci., 2022, 613, 349–358 CrossRef CAS PubMed.
- Y. Liu, C. Xiao, P. Huang, M. Cheng and Y. Xie, Chem, 2018, 4, 1263–1283 CAS.
- Z. Zhuang, Y. Wang, C. Q. Xu, S. Liu, C. Chen, Q. Peng, Z. Zhuang, H. Xiao, Y. Pan, S. Lu, R. Yu, W. C. Cheong, X. Cao, K. Wu, K. Sun, D. Wang, J. Li and Y. Li, Nat. Commun., 2019, 10, 1–10 CrossRef PubMed.
- L. Wu, L. Yu, B. McElhenny, X. Xing, D. Luo, F. Zhang, J. Bao, S. Chen and Z. Ren, Appl. Catal., B, 2021, 294, 120256 CrossRef CAS.
- J. Tian, Q. Liu, A. M. Asiri and X. Sun, J. Am. Chem. Soc., 2014, 136, 7587–7590 CrossRef CAS PubMed.
- P. Liu, B. Chen, C. Liang, W. Yao, Y. Cui, S. Hu, P. Zou, H. Zhang, H. J. Fan and C. Yang, Adv. Mater., 2021, 33, e2007377 CrossRef PubMed.
- M. Yang, C. Zhang, N. Li, D. Luan, L. Yu and X. lou, Adv. Sci., 2022, 9, 2105135 CrossRef CAS PubMed.
- J. Yin, J. Jin, H. Lin, Z. Yin, J. Li, M. Lu, L. Guo, P. Xi, Y. Tang and C. H. Yan, Adv. Sci., 2020, 7, 1903070 CrossRef CAS PubMed.
- J. Sun, P. Song, H. Zhou, L. Lang, X. Shen, Y. Liu, X. Cheng, X. Fu and G. Zhu, Appl. Surf. Sci., 2021, 567, 150757 CrossRef CAS.
- X. Yin, L. Yang and Q. Gao, Nanoscale, 2020, 12, 15944–15969 RSC.
- X. Zhao, S. Yuan, Z. Jin, Q. Zhu, M. Zheng, Q. Jiang, H. Song and J. Duan, Prog. Org. Coat., 2020, 149, 105893 CrossRef CAS.
- H. Huang, H. Jung, C. Y. Park, S. Kim, A. Lee, H. Jun, J. Choi, J. W. Han and J. Lee, Appl. Catal., B, 2022, 315, 121554 CrossRef CAS.
- L. Yu, Q. Zhu, S. Song, B. McElhenny, D. Wang, C. Wu, Z. Qin, J. Bao, Y. Yu, S. Chen and Z. Ren, Nat. Commun., 2019, 10, 5106 CrossRef PubMed.
- Y. Xiao, C. Feng, J. Fu, F. Wang, C. Li, V. F. Kunzelmann, C. M. Jiang, M. Nakabayashi, N. Shibata, I. D. Sharp, K. Domen and Y. Li, Nat. Catal., 2020, 3, 932–940 CrossRef CAS.
- Y. Zhu, X. Zhong, S. Jin, H. Chen, Z. He, Q. Liu and Y. Chen, J. Mater. Chem. A, 2020, 8, 10957–10965 RSC.
- W. Yang, Z. Wang, W. Zhang and S. Guo, Trends Chem., 2019, 1, 259–271 CrossRef CAS.
- Y. Li, M. B. Majewski, S. M. Islam, S. Hao, A. A. Murthy, J. G. DiStefano, E. D. Hanson, Y. Xu, C. Wolverton, M. G. Kanatzidis, M. R. Wasielewski, X. Chen and V. P. Dravid, Nano Lett., 2018, 18, 7104–7110 CrossRef CAS PubMed.
- S. Chandrasekaran, L. Yao, L. Deng, C. Bowen, Y. Zhang, S. Chen, Z. Lin, F. Peng and P. Zhang, Chem. Soc. Rev., 2019, 48, 4178–4280 RSC.
- Y. Wang, J. Zhang, M. S. Balogun, Y. Tong and Y. Huang, Mater. Today Sustainable, 2022, 18, 100118 CrossRef.
- S. Zhang, Y. Si, B. Li, L. Yang, W. Dai and S. Luo, Small, 2021, 17, e2004980 CrossRef PubMed.
- M. Zhao, M. Yang, W. Huang, W. Liao, H. Bian, D. Chen, L. Wang, J. Tang and C. Liu, ChemCatChem, 2021, 13, 2138–2144 CrossRef CAS.
- Y. Yin, Y. Zhang, T. Gao, T. Yao, X. Zhang, J. Han, X. Wang, Z. Zhang, P. Xu, P. Zhang, X. Cao, B. Song and S. Jin, Adv. Mater., 2017, 29, 1700311 CrossRef PubMed.
- J. Xie, S. Li, X. Zhang, J. Zhang, R. Wang, H. Zhang, B. Pan and Y. Xie, Chem. Sci., 2014, 5, 4615–4620 RSC.
- Q. Mao, K. Deng, H. Yu, Y. Xu, Z. Wang, X. Li, L. Wang and H. Wang, Adv. Funct. Mater., 2022, 32, 2201081 CrossRef CAS.
- Y. Yao, Y. Zhu, C. Pan, C. Wang, S. Hu, W. Xiao, X. Chi, Y. Fang, J. Yang, H. Deng, S. Xiao, J. Li, Z. Luo and Y. Guo, J. Am. Chem. Soc., 2021, 143, 8720–8730 CrossRef CAS PubMed.
- Z. Luo, R. Miao, T. D. Huan, I. M. Mosa, A. S. Poyraz, W. Zhong, J. E. Cloud, D. A. Kriz, S. Thanneeru, J. He, Y. Zhang, R. Ramprasad and S. L. Suib, Adv. Energy Mater., 2016, 6, 201600528 Search PubMed.
- R. S. Datta, F. Haque, M. Mohiuddin, B. J. Carey, N. Syed, A. Zavabeti, B. Zhang, H. Khan, K. J. Berean, J. Z. Ou, N. Mahmood, T. Daeneke and K. Kalantar-zadeh, J. Mater. Chem. A, 2017, 5, 24223–24231 RSC.
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Joule, 2018, 2, 2551–2582 CrossRef CAS.
- L. Wu, L. Yu, Q. Zhu, B. McElhenny, F. Zhang, C. Wu, X. Xing, J. Bao, S. Chen and Z. Ren, Nano Energy, 2021, 83, 105838 CrossRef CAS.
- X. Wang, X. Zhou, C. Li, H. Yao, C. Zhang, J. Zhou, R. Xu, L. Chu, H. Wang, M. Gu, H. Jiang and M. Huang, Adv. Mater., 2022, 34, e2204021 CrossRef PubMed.
- Q. Yu, J. Chi, G. Liu, X. Wang, X. Liu, Z. Li, Y. Deng, X. Wang and L. Wang, Sci. China Mater., 2022, 65, 1539–1549 CrossRef CAS.
- T. Marimuthu, R. Yuvakkumar, P. Senthil Kumar, G. Ravi, X. Xu, G. Xu and D. Velauthapillai, Int. J. Hydrogen Energy, 2022, 47, 37171–37182 CrossRef CAS.
- T. Cui, X. Zhai, L. Guo, J. Q. Chi, Y. Zhang, J. Zhu, X. Sun and L. Wang, Chin. J. Catal., 2022, 43, 2202–2211 CrossRef CAS.
- X. Lu, J. Pan, E. Lovell, T. H. Tan, Y. H. Ng and R. Amal, Energy Environ. Sci., 2018, 11, 1898–1910 RSC.
- W. Huang, D. Zhou, G. Qi and X. Liu, Nanotechnology, 2021, 32, 415403 CrossRef CAS PubMed.
- J. Wang, X. Ge, Z. Liu, L. Thia, Y. Yan, W. Xiao and X. Wang, J. Am. Chem. Soc., 2017, 139, 1878–1884 CrossRef CAS PubMed.
- J. Kang, X. Qiu, Q. Hu, J. Zhong, X. Gao, R. Huang, C. Wan, L. M. Liu, X. Duan and L. Guo, Nat. Catal., 2021, 4, 1050–1058 CrossRef CAS.
- P. Ganguly, M. Harb, Z. Cao, L. Cavallo, A. Breen, S. Dervin, D. D. Dionysiou and S. C. Pillai, ACS Energy Lett., 2019, 4, 1687–1709 CrossRef CAS.
- Q. Shi, S. Murcia-López, P. Tang, C. Flox, J. R. Morante, Z. Bian, H. Wang and T. Andreu, ACS Catal., 2018, 8, 3331–3342 CrossRef CAS.
- L. Tian, Z. Li, X. Xu and C. Zhang, J. Mater. Chem. A, 2021, 9, 13459–13470 RSC.
- S. Chandrasekaran, P. Zhang, F. Peng, C. Bowen, J. Huo and L. Deng, J. Mater. Chem. A, 2019, 7, 6161–6172 RSC.
- B. Zhang, J. Wang, J. Liu, L. Zhang, H. Wan, L. Miao and J. Jiang, ACS Catal., 2019, 9, 9332–9338 CrossRef CAS.
- L. Yan, B. Zhang, J. Zhu, Y. Li, P. Tsiakaras and P. K. Shen, Appl. Catal., B, 2020, 265, 118555 CrossRef CAS.
- X. Xu, Y. Pan, Y. Zhong, C. Shi, D. Guan, L. Ge, Z. Hu, Y. Y. Chin, H. J. Lin, C. T. Chen, H. Wang, S. P. Jiang and Z. Shao, Adv. Sci., 2022, 9, e2200530 CrossRef PubMed.
- Q. Ma, H. Jin, F. Xia, H. Xu, J. Zhu, R. Qin, H. Bai, B. Shuai, W. Huang, D. Chen, Z. Li, J. Wu, J. Yu and S. Mu, J. Mater. Chem. A, 2021, 9, 26852–26860 RSC.
- J. Engel and H. L. Tuller, Phys. Chem. Chem. Phys., 2014, 16, 11374–11380 RSC.
- M. Lu, S. Kong, S. Yan, P. Zhou, T. Yu and Z. Zou, J. Mater. Chem. A, 2022, 10, 12391–12399 RSC.
- T. Asefa, Acc. Chem. Res., 2016, 49, 1873–1883 CrossRef CAS PubMed.
- Y. Song, J. Cheng, J. Liu, Q. Ye, X. Gao, J. Lu and Y. Cheng, Appl. Catal., B, 2021, 298, 120488 CrossRef CAS.
- T. Ul Haq, S. Mansour and Y. Haik, ACS Appl. Mater. Interfaces, 2022, 14, 20443–20454 CrossRef CAS PubMed.
- Y. Lin, K. Sun, X. Chen, C. Chen, Y. Pan, X. Li and J. Zhang, J. Energy Chem., 2021, 55, 92–101 CrossRef CAS.
- Y. Liu, X. Hu, B. Huang and Z. Xie, ACS Sustainable Chem. Eng., 2019, 7, 18835–18843 CrossRef CAS.
- J. Yang, Y. Wang, J. Yang, Y. Pang, X. Zhu, Y. Lu, Y. Wu, J. Wang, H. Chen, Z. Kou, Z. Shen, Z. Pan and J. Wang, Small, 2022, 18, 2106187 CrossRef CAS PubMed.
- K. Izumiya, E. Akiyama, H. Habazaki, N. Kumagai, A. Kawashima and K. Hashimoto, Mater. Trans., 1997, 38, 899–905 CrossRef CAS.
- V. Petrykin, K. Macounova, O. A. Shlyakhtin and P. Krtil, Angew. Chem., Int. Ed., 2010, 49, 4813–4815 CrossRef CAS PubMed.
- L. Deng, Y. Liu, G. Zhao, J. Chen, S. He, Y. Zhu, B. Chai and Z. Ren, J. Electroanal. Chem., 2019, 832, 459–466 CrossRef CAS.
- J. Chen, P. Wagner, L. Tong, G. G. Wallace, D. L. Officer and G. F. Swiegers, Angew. Chem., Int. Ed., 2012, 51, 1907–1910 CrossRef CAS PubMed.
- C. Lan, H. Xie, Y. Wu, B. Chen and T. Liu, Energy Fuels, 2022, 36, 2910–2917 CrossRef CAS.
- K. Jiang, W. Liu, W. Lai, M. Wang, Q. Li, Z. Wang, J. Yuan, Y. Deng, J. Bao and H. Ji, Inorg. Chem., 2021, 60, 17371–17378 CrossRef CAS PubMed.
- W. H. Hung, B. Y. Xue, T. M. Lin, S. Y. Lu and I. Y. Tsao, Mater. Today Energy, 2021, 19, 100575 CrossRef CAS.
- Z. Li, C. Chen, E. Zhan, N. Ta, Y. Li and W. Shen, Chem. Commun., 2014, 50, 4469–4471 RSC.
- S. Bai, W. Jiang, Z. Li and Y. Xiong, ChemNanoMat, 2015, 1, 223–239 CrossRef CAS.
- Y. Yu, G. H. Nam, Q. He, X. J. Wu, K. Zhang, Z. Yang, J. Chen, Q. Ma, M. Zhao, Z. Liu, F. R. Ran, X. Wang, H. Li, X. Huang, B. Li, Q. Xiong, Q. Zhang, Z. Liu, L. Gu, Y. Du, W. Huang and H. Zhang, Nat. Chem., 2018, 10, 638–643 CrossRef CAS PubMed.
- J. Zhang, Y. Chen, M. Liu, K. Du, Y. Zhou, Y. Li, Z. Wang and J. Zhang, Nano Res., 2018, 11, 4587–4598 CrossRef CAS.
- J. Zhang, T. Wang, P. Liu, Y. Liu, J. Ma and D. Gao, Electrochim. Acta, 2016, 217, 181–186 CrossRef CAS.
- Y. Zhao, J. Tu, Y. Sun, X. Hu, J. Ning, W. Wang, F. Wang, Y. Xu and L. He, J. Phys. Chem. C, 2018, 122, 26570–26575 CrossRef CAS.
- Y. J. Chung, C. S. Yang, J. T. Lee, G. H. Wu and J. M. Wu, Adv. Energy Mater., 2020, 10, 2002082 CrossRef CAS.
- Z. Lai, Y. Yao, S. Li, L. Ma, Q. Zhang, Y. Ge, W. Zhai, B. Chi, B. Chen, L. Li, L. Wang, Z. Zheng, L. Gu, Y. Du and H. Zhang, Adv. Mater., 2022, 34, e2201194 CrossRef PubMed.
- N. Li, J. Wu, Y. Lu, Z. Zhao, H. Zhang, X. Li, Y.-Z. Zheng and X. Tao, Appl. Catal., B, 2018, 238, 27–37 CrossRef CAS.
- M. Jiang, J. Zhang, M. Wu, W. Jian, H. Xue, T.-W. Ng, C.-S. Lee and J. Xu, J. Mater. Chem. A, 2016, 4, 14949–14953 RSC.
- Y. Xu, Y. Fo, H. Lv, X. Cui, G. Liu, X. Zhou and L. Jiang, ACS Appl. Mater. Interfaces, 2022, 14, 10246–10256 CrossRef CAS PubMed.
- T. Yang, Y. Xu, H. Lv, M. Wang, X. Cui, G. Liu and L. Jiang, ACS Sustainable Chem. Eng., 2021, 9, 13106–13113 CrossRef CAS.
- R. D. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- C. Guo, Y. Shi, S. Lu, Y. Yu and B. Zhang, Chin. J. Catal., 2021, 42, 1287–1296 CrossRef CAS.
- T. Guo, L. Li and Z. Wang, Adv. Energy Mater., 2022, 12, 2200827 CrossRef CAS.
- H. Han, H. Choi, S. Mhin, Y.-R. Hong, K. M. Kim, J. Kwon, G. Ali, K. Y. Chung, M. Je, H. N. Umh, D. H. Lim, K. Davey, S. Z. Qiao, U. Paik and T. Song, Energy Environ. Sci., 2019, 12, 2443–2454 RSC.
- H. Huang, A. Cho, S. Kim, H. Jun, A. Lee, J. W. Han and J. Lee, Adv. Funct. Mater., 2020, 30, 2003889 CrossRef CAS.
- J. Liu, J. Nai, T. You, P. An, J. Zhang, G. Ma, X. Niu, C. Liang, S. Yang and L. Guo, Small, 2018, 14, e1703514 CrossRef PubMed.
- J. Kwon, H. Han, S. Jo, S. Choi, K. Y. Chung, G. Ali, K. Park, U. Paik and T. Song, Adv. Energy Mater., 2021, 11, 2100624 CrossRef CAS.
- Y. Zhou and H. J. Fan, ACS Mater. Lett., 2020, 3, 136–147 CrossRef.
- Y. Huang, L. Hu, R. Liu, Y. Hu, T. Xiong, W. Qiu, M. S. Balogun, A. Pan and Y. Tong, Appl. Catal., B, 2019, 251, 181–194 CrossRef CAS.
- H. Zhang, S. Geng, M. Ouyang, H. Yadegari, F. Xie and D. J. Riley, Adv. Sci., 2022, 9, e2200146 CrossRef PubMed.
- S. Yang, Y. Wang, H. Zhang, Y. Zhang, L. Liu, L. Fang, X. Yang, X. Gu and Y. Wang, J. Catal., 2019, 371, 20–26 CrossRef CAS.
- X. Sun, H. Deng, W. Zhu, Z. Yu, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2016, 55, 1704–1709 CrossRef CAS PubMed.
- Y. Liu, S. Liu, Y. Wang, Q. Zhang, L. Gu, S. Zhao, D. Xu, Y. Li, J. Bao and Z. Dai, J. Am. Chem. Soc., 2018, 140, 2731–2734 CrossRef CAS PubMed.
- S. Bai and Y. Xiong, Chem. Commun., 2015, 51, 10261–10271 RSC.
- X. Jiang, H. Jang, S. Liu, Z. Li, M. G. Kim, C. Li, Q. Qin, X. Liu and J. Cho, Angew. Chem., Int. Ed., 2021, 60, 4110–4116 CrossRef CAS PubMed.
- H. Zhang, J. Wang, F. Qin, H. Liu and C. Wang, Nano Res., 2021, 14, 3489–3496 CrossRef CAS.
- Q. Wang, F. Mi, J. Li, Y. Wu, X. Zhou, G. Ma and S. Ren, Electrochim. Acta, 2021, 370, 137796 CrossRef CAS.
- X. Wu, S. Zhou, Z. Wang, J. Liu, W. Pei, P. Yang, J. Zhao and J. Qiu, Adv. Energy Mater., 2019, 9, 1901333 CrossRef.
- Z. Wang, W. Xu, K. Yu, Y. Feng and Z. Zhu, Nanoscale, 2020, 12, 6176–6187 RSC.
- Q. Lv, J. Han, X. Tan, W. Wang, L. Cao and B. Dong, ACS Appl. Energy Mater., 2019, 2, 3910–3917 CrossRef CAS.
- X. Li, J. Yu, J. Low, Y. Fang, J. Xiao and X. Chen, J. Mater. Chem. A, 2015, 3, 2485–2534 RSC.
- S. Wang, L. Zhao, J. Li, X. Tian, X. Wu and L. Feng, J. Energy Chem., 2022, 66, 483–492 CrossRef CAS.
- D. Liu, H. Ai, M. Chen, P. Zhou, B. Li, D. Liu, X. Du, K. H. Lo, K. W. Ng, S. P. Wang, S. Chen, G. Xing, J. Hu and H. Pan, Small, 2021, 17, e2007557 CrossRef PubMed.
- J. Chang, G. Wang, Z. Yang, B. Li, Q. Wang, R. Kuliiev, N. Orlovskaya, M. Gu, Y. Du, G. Wang and Y. Yang, Adv. Mater., 2021, 33, 2101425 CrossRef CAS PubMed.
- C. Wang, M. Zhu, Z. Cao, P. Zhu, Y. Cao, X. Xu, C. Xu and Z. Yin, Appl. Catal., B, 2021, 291, 120071 CrossRef CAS.
- L. Zhang, J. Liang, L. Yue, K. Dong, J. Li, D. Zhao, Z. Li, S. Sun, Y. Luo, Q. Liu, G. Cui, A. A. Alshehri, X. Guo and X. Sun, Nano Res. Energy, 2022, 1, e9120028 CrossRef.
- L. Tan, J. Yu, C. Wang, H. Wang, X. Liu, H. Gao, L. Xin, D. Liu, W. Hou and T. Zhan, Adv. Funct. Mater., 2022, 32, 2200951 CrossRef CAS.
- A. B. D. Cassie and S. Baxter, Trans. Faraday Soc., 1944, 40, 546–551 RSC.
- H. Li, S. Chen, Y. Zhang, Q. Zhang, X. Jia, Q. Zhang, L. Gu, X. Sun, L. Song and X. Wang, Nat. Commun., 2018, 9, 2452 CrossRef PubMed.
- D. Luo, F. Wang, M. K. Alam, F. Yu, I. K. Mishra, J. Bao, R. C. Willson and Z. Ren, Chem. Mater., 2017, 29, 3454–3460 CrossRef CAS.
- D. Luo, F. Zhang, F. Ding, B. Ren and Z. Ren, Soft Matter, 2019, 15, 7472–7478 RSC.
- R. N. Wenzel, Ind. Eng. Chem., 1936, 28, 988–994 CrossRef CAS.
- T. u. Haq and Y. Haik, Catal. Today, 2022, 400–401, 14–25 CrossRef CAS.
- L. Yu, L. Wu, B. McElhenny, S. Song, D. Luo, F. Zhang, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2020, 13, 3439–3446 RSC.
- B. Ge, C. Li, X. Zhu, W. Li, G. Ren and Z. Zhang, Sep. Purif. Technol., 2022, 286, 120468 CrossRef CAS.
- H. Chen, Y. Zou, J. Li, K. Zhang, Y. Xia, B. Hui and D. Yang, Appl. Catal., B, 2021, 293, 120215 CrossRef CAS.
- C. Ros, S. Murcia-Lopez, X. Garcia, M. Rosado, J. Arbiol, J. Llorca and J. R. Morante, ChemSusChem, 2021, 14, 2872–2881 CrossRef CAS PubMed.
- X. Jiang, Z. Dong, Q. Zhang, G. Xu, J. Lai, Z. Li and L. Wang, J. Mater. Chem. A, 2022, 38, 20571–20579 RSC.
- H. You, D. Wu, D. Si, M. Cao, F. Sun, H. Zhang, H. Wang, T. Liu and R. Cao, J. Am. Chem. Soc., 2022, 21, 9254–9263 CrossRef PubMed.
- S. Duan, Z. Liu, H. Zhuo, T. Wang, J. Liu, L. Wang, J. Liang, J. Han, Y. Huang and Q. Li, Nanoscale, 2020, 42, 21743–21749 RSC.
| This journal is © The Royal Society of Chemistry 2023 |