
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiosynthesis of the fungal nonribosomal peptide penilumamide A and biochemical characterization of a pterin-specific adenylation domain†
Stephanie C.
Heard
a,
Katharine L.
Diehl
b and
Jaclyn M.
Winter
 *a
*a
aDepartment of Pharmacology and Toxicology, College of Pharmacy, University of Utah, Salt Lake City, UT 84112, USA. E-mail: jaclyn.winter@utah.edu; Tel: +1 (801) 585-7117
bDepartment of Medicinal Chemistry, College of Pharmacy, University of Utah, Salt Lake City, UT 84112, USA
First published on 6th September 2023
Abstract
We report the characterization of the penilumamide biosynthetic cluster from Aspergillus flavipes CNL-338. In vitro reconstitution experiments demonstrated that three nonribosomal peptide synthetases are required for constructing the tripeptide and studies with dissected adenylation domains allowed for the first biochemical characterization of a domain that selects a pterin-derived building block.
In the post-genomic era, natural product discovery has been enhanced through various genome mining and bioinformatic platforms. Strains that are likely to generate unique chemistry are prioritized due to our ability to identify putative biosynthetic clusters and predict building blocks and potential downstream modifications. Natural products produced by marine-derived microorganisms continue to be a treasure trove of novel bioactive small molecules,1–3 including those of the well-established nonribosomal peptide class. Although the catalytic domains and mechanisms for peptide biosynthesis are similar between bacterial and fungal nonribosomal peptide synthetases (NRPSs), the ability to predict substrate specificity of building blocks in fungal systems is lacking compared to bacterial counterparts.4 Thus, to enhance our ability to accurately predict nonribosomal peptides in fungal genomes, additional NPRS machinery needs to be biochemically characterized, especially those with selectivity for unprecedented nonproteinogenic building blocks.
In recent years, unique lumazine-containing nonribosomal peptides have been isolated from marine-derived Aspergillus strains (Fig. 1). Penilumamide A (1), the first in its class, is a tripeptide containing a distinctive 1,3-dimethyl-lumazine-6-carboxylic acid functional group,5 and this unique pterin-derived moiety is found in penilumamide analogs, usually exhibiting either 1-N-methylation or N,N-dimethylation. Additional structural differences within this family of compounds includes variation in proteinogenic amino acid incorporation at the second position, as well as three different oxidation states of methionine, and incorporation of different aniline-derived C-terminal units such as anthranilic acid, methyl anthranilate, anthranilamide, or 2-aminophenyl isocyanide.6–11 Despite the number of lumazine-containing peptides that have been isolated, there have been no biosynthetic investigations of the respective nonribosomal peptide machinery, and more importantly, no reports on adenylation domains with preference for lumazine- or pterin-derived building blocks. Herein, we report the biosynthetic pathway for 1, detailed bioinformatic investigation into the biosynthetic machinery, and the biochemical characterization of reconstituted NRPSs and corresponding adenylation domain substrate specificities. Extensive bioinformatic analyses support all findings and to the best of our knowledge, this is the first biochemical characterization of a fungal adenylation domain with native preference for methionine as opposed to substrate promiscuity.12 This is also the first report of an adenylation domain that activates a pterin-derived building block.
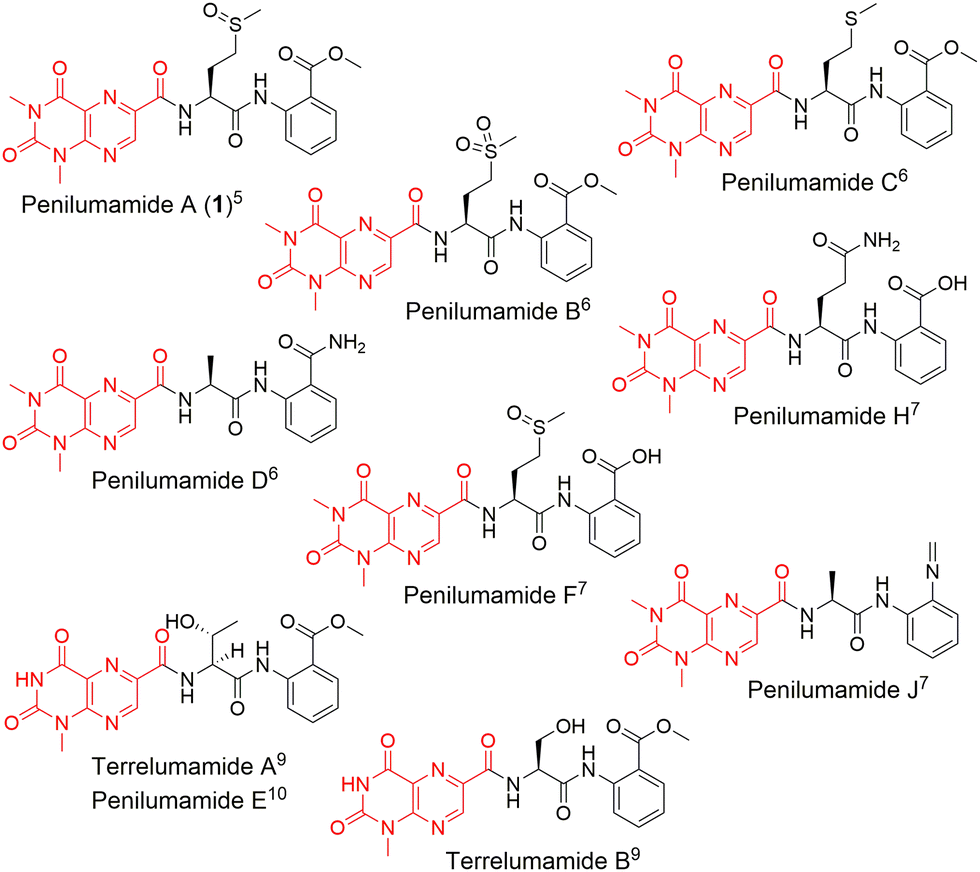 | ||
| Fig. 1 Structures of penilumamide A and other related lumazine-containing peptides. The lumazine-derived moieties are highlighted in red. | ||
Nonribosomal peptides are a well-studied class of natural products that are typically assembled by large modular synthetases in an assembly line-like fashion. These megasynthetases provide a biosynthetic template where each module is typically responsible for the activation, incorporation and modification of proteinogenic or non-proteinogenic amino acid building blocks. Each module is made up of a minimal set of catalytic domains, namely adenylation, thiolation and condensation domains.13 Adenylation (A) domains are responsible for selecting and activating building blocks, which then get loaded onto the phosphopantetheine moiety of a thiolation (T) domain. The tethered acyl substrate can then be delivered to the condensation (C) domain for extension with the upstream nascent peptide.
To identify the biosynthetic gene cluster responsible for 1, the 33 Mbp genome of Aspergillus flavipes CNL-338‡ was sequenced and assembled using SOAPdenovo2 and IDBA-UD software programs.14,15 Initial automated annotation was carried out using antiSMASH,16 which revealed 51 biosynthetic clusters, of which 23 are nonribosomal peptide-related. Additional genome mining using Blast+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 and manual annotation of the genes upstream and downstream of each NRPS locus identified a 30 kb biosynthetic cluster, named the plm cluster, containing three NRPS genes encoding four modules and eight genes dedicated to pterin biosynthesis (Fig. 2A, B and Table S3, ESI†).§ The eight non-NRPS genes within the plm biosynthetic cluster are hypothesized to convert guanosine triphosphate (GTP) to the final modified 1,3- dimethyllumazine-6-carboxylic acid unit, with the first two enzymatic steps predicted to be analogous to microbial folate biosynthesis. It should be noted that the GTP cyclohydrolase-encoding plmC and dihydroneopterin aldolase-encoding plmG are duplicates of genes required for primary metabolism.
17 and manual annotation of the genes upstream and downstream of each NRPS locus identified a 30 kb biosynthetic cluster, named the plm cluster, containing three NRPS genes encoding four modules and eight genes dedicated to pterin biosynthesis (Fig. 2A, B and Table S3, ESI†).§ The eight non-NRPS genes within the plm biosynthetic cluster are hypothesized to convert guanosine triphosphate (GTP) to the final modified 1,3- dimethyllumazine-6-carboxylic acid unit, with the first two enzymatic steps predicted to be analogous to microbial folate biosynthesis. It should be noted that the GTP cyclohydrolase-encoding plmC and dihydroneopterin aldolase-encoding plmG are duplicates of genes required for primary metabolism.
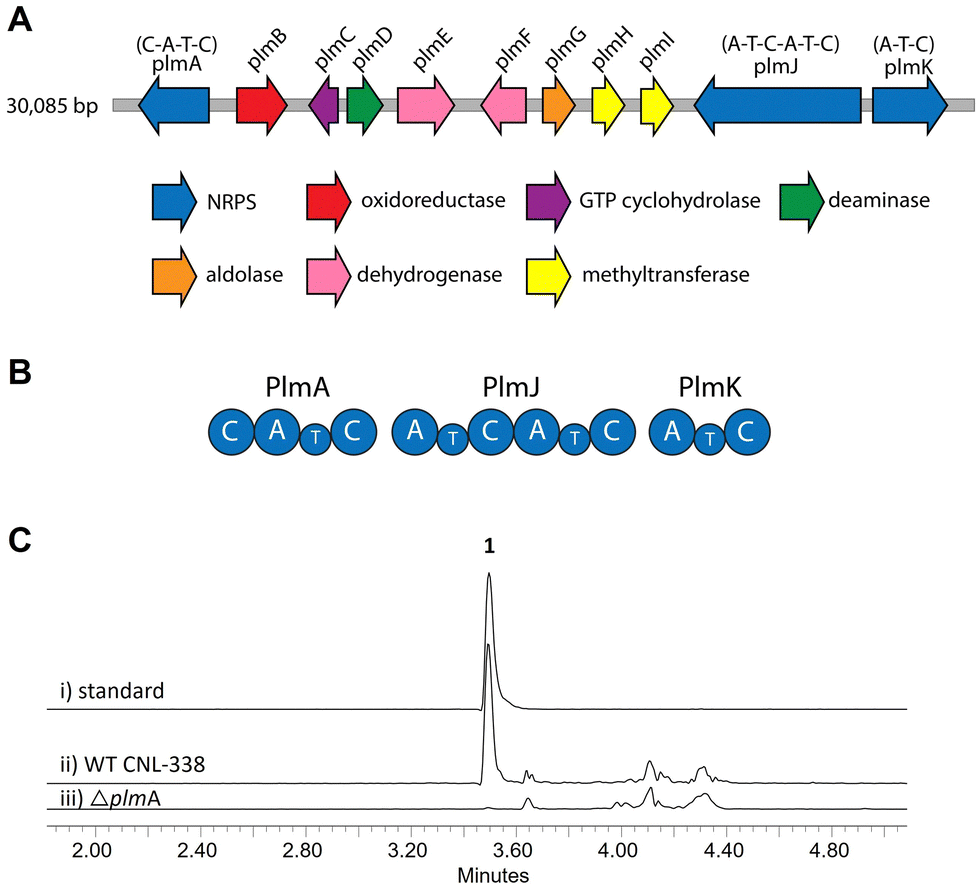 | ||
| Fig. 2 Organization and verification of the plm gene cluster in A. flavipes CNL-338. (A) The plm cluster encoding the machinery responsible for penilumamide A (1) production. The open reading frames are color-coded based on proposed function (Table S3, ESI†). (B) Organization of the Plm NRPSs. Domain organization of PlmA, PlmJ and PlmK consists of adenylation (A), thiolation (T) and condensation (C) domains. (C) LC-MS analysis (EIC traces = 517 m/z) of (i) a standard of 1 compared to crude extracts of (ii) wildtype (WT) A. flavipes CNL-338 or (iii) inactivation of plmA. | ||
Inactivation of plmA, coding for a monomodular NRPS, confirmed the cluster's role in synthesizing 1 (Fig. 2C and Fig. S3, ESI†).18,19 Interestingly, while penilumamide A is a linear tripeptide, closer inspection of the three NRPSs, PlmA, PlmJ and PlmK, revealed a total of four modules, indicative of a tetrapeptide product. Thus, the activities of the monomodular PlmA and PlmK and dimodular PlmJ were reconstituted in vitro to verify if all three NPRSs were required for assembly of 1. plmA, plmJ and plmK were solubly expressed as recombinant C-terminal hexahistidyl-tagged proteins from Saccharomyces cerevisiae BJ5464-NpgA20 (Fig. S4, ESI†). Combinations of PlmA, PlmK and fractions enriched with PlmJ were incubated with pterine-6-carboxylic acid, L-methionine and anthranilic acid before analysis by liquid chromatography-mass spectrometry (LC-MS). It should be noted that pterine-6-carboxylic acid was used as a commercially available alternative to the highly functionalized 1,3-dimethyllumazine-6-carboxylic acid building block incorporated by A. flavipes CNL-338, and no other tailoring enzymes were present in the in vitro assays. Thus, the expected product is the demethylated, pterin-containing penilumamide derivative 2 instead of 1, and 2 was only observed when all three NRPSs, totalling four modules, were incubated together (Fig. 3A).
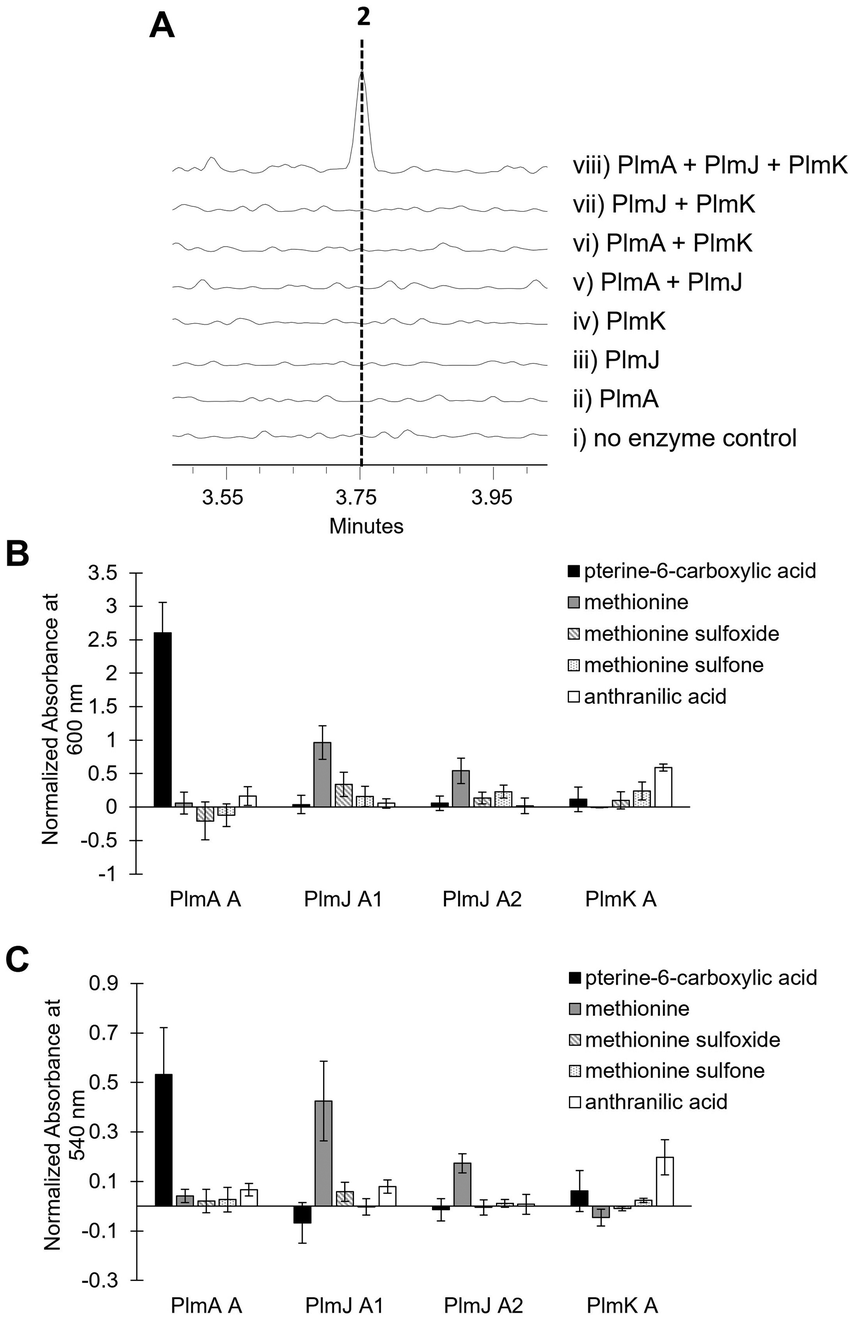 | ||
| Fig. 3 In vitro reconstitution of demethyl-pterin-penilumamide (2) and adenylation assays to determine substrate specificity of the four dissected adenylation domains found in PlmA, PlmJ and PlmK. (A) LC-MS analysis (EIC = 458 m/z) of 2 when the pterine-6-carboxylic acid, L-methionine and anthranilic acid building blocks were (i) incubated together; (ii) incubated with PlmA; (iii) incubated with PlmJ; (iv) incubated with PlmK; (v) incubated with PlmA and PlmJ; (vi) incubated with PlmA and PlmK; (vii) incubated with PlmJ and PlmK; and (viii) incubated with PlmA, PlmJ and PlmK. (B) Adenylation activity was determined first through the malachite green/phosphate detection method.22 Activity was measured in triplicate as absorbance at 600 nm and normalized to boiled enzyme controls for each substrate. (C) Adenylation activity was verified with the hydroxylamine release/iron complex method.23 Activity was measured in triplicate as absorbance at 540 nm and normalized to boiled enzyme controls for each substrate. | ||
As the in vitro reconstitution assays showed that PlmA, PlmJ and PlmK were required for the production of 2, detailed bioinformatic investigations of the signature motifs in each domain of the three NRPSs was carried out to determine if any modules could be inactive (Fig. S8, ESI†). All four A domains and all four T domains in PlmA, PlmJ and PlmK contain the required catalytic residues and were predicted to be active (Tables S5 and S7, ESI†). Of the five C domains, all were predicted to be active with the exception of the N-terminal C domain in PlmA (PlmA C1), which is truncated, lacking key catalytic residues, and thus predicted to be inactive (Table S9, ESI†). As C domains have been posited as secondary gatekeepers to NRPS biosynthetic pathways,21 downstream of the primary selectivity of A domains, a maximum-likelihood phylogenetic tree was constructed to aid in predicting the biosynthetic function of the Plm C domains and perhaps module order (Table S8 and Fig. S10, ESI†). While the C domain in PlmK was shown to clade with terminal anthranilate-incorporating domains, the second C domain in PlmJ (PlmJ C2) clades with C domains that couple L-amino acid donors to anthranilate acceptors. The plm biosynthetic cluster contains no thioesterase, reductive, or otherwise obvious offloading domain; however, due to the C-terminus of 1 being modified to the anthranilic acid methyl ester, it is likely that the C domain embedded in PlmK hydrolyzes 2 as the free carboxylic acid which is then methylated, as in crocacin biosynthesis.24 Thus, the order of NRPSs in the biosynthesis of 1 likely goes from PlmA to PlmJ to PlmK. To validate this order, and as all four A domains were predicted to be active, we investigated the substrate specificities of the individual A domains.
Adenylation domain sequences were excised from all three Plm NRPSs based on predicted domain boundaries25 and expressed as N-terminal octahistidine-tagged proteins from Escherichia coli BL21 (DE3) (Fig. S5, ESI†). As there are several routine biochemical methods that can be used to probe adenylation activity, we chose two complementary colorimetric methods: a PPi release assay and a hydroxylamine-trapping assay. First, using an established phosphate detection method,22 A domain activation was indirectly measured using formic acid, anthranilic acid, pterine-6-carboxylic acid, L-methionine sulfoxide, L-methionine sulfone and the full panel of 20 proteinogenic amino acids (Fig. 3B and Fig. S6, ESI†). From these assays, the A domain in PlmA showed preference for pterine-6-carboxylic acid, whereas the second A domain in PlmJ (PlmJ A2) activated methionine, and the A domain in PlmK was specific for anthranilic acid. Unexpectedly, the first A domain of PlmJ (PlmJ A1) was also found to activate L-methionine with slightly higher activity than PlmJ A2, though only one methionine residue is present in 1. This pattern of adenylation activity was confirmed using a well-established method involving direct detection of hydroxylamine-trapped aminoacyl-adenylates23 (Fig. 3C and Fig. S7, ESI†). Altogether, these A domain results support the order of Plm NRPS modules predicted by C domain analysis.
As all four Plm A domains were biochemically characterized, the amino acid “specificity codes” of each domain were determined and compared to other A domains to interrogate patterns denoting substrate selectivity (Table S5, ESI†). It is well established in bacterial NRPSs that their substrate preferences can often be rationalized by 10 key amino acid residues that line the binding pocket of the A domain and act as a fingerprint for selectivity.26,27 This work is the first report of an A domain, PlmA A, activating a pterin-derived building block; its code DVMVLLMITK appears most similar to tryptophan-loading fungal A domains such as AnaPS A2 from acetylaszonalenin biosynthesis.28,29 The A domain in PlmK, which was demonstrated through in vitro assays to activate anthranilic acid, is comparable to anthranilate-activating fungal A domains30 such as PsyC A from psychrophilin biosynthesis.31 Both A domains of PlmJ were found to activate L-methionine, the first time this proteinogenic amino acid has been biochemically characterized in an Ascomycota-derived fungal NRPS system. Other native methionine-incorporating A domains, such as NpsP8 from napsamycin biosynthesis,32 SsaO from sansanmycin biosynthesis,33 and JahA A3 from jahnellamide biosynthesis34 have been predicted but lack biochemical confirmation. In malpinin biosynthesis, modules 1, 3 and 4 of MalA activate hydrophobic amino acids and were found to have relaxed substrate promiscuity towards L-methionine; however, methionine-containing malpinins were not identified as native natural products and were only observed upon media supplementation.12
The specificity codes for PlmJ A1 and PlmJ A2 were determined to be SIVIVTAGTK and DVVLLLSSTK, respectively. Due to the proclivity of pterins to photosensitize neighbouring amino acid residues,35,36 it is likely that PlmJ incorporates L-methionine which then spontaneously oxidizes to the sulfoxide either while tethered as a dipeptide or after hydrolytic release of the tripeptide. This is supported by the preference for both PlmJ A1 and A2 to activate unoxidized methionine substrates (Fig. 3B and C). Of note, PlmJ A1 lacks the highly conserved aspartate in the first position of its code, which is replaced with a serine. Interestingly, this change has been observed in fungal fumaric acid-activating A domains.37,38 However, penilumamide A does not contain this type of nonproteinogenic amino acid, nor any derivative isolated to date.
From detailed bioinformatic and biochemical investigations, the dimodular PlmJ contributes only one methionine residue to penilumamide A, suggesting the first module may be skipped or the C domain may be inactive. In previous studies on fungal NRPS module skipping,39,40 two possible mechanisms were suggested: either complete A–T–C module skipping via condensation by the upstream C domain, or chain transfer via the T domain of the skipped module (Fig. 4B). Based on our bioinformatic analyses (Fig. S8 and Table S9, ESI†), although PlmJ C1 maintains the required catalytic histidine, its predicted function as an anthranilate-donating C domain does not correlate with the preference of PlmJ A1 for L-methionine observed in the adenylation assays. Furthermore, the function of PlmJ A1 was incorrectly predicted, as it was shown to clade with a conserved group of anthranilate-activating A domains (Fig. S10, ESI†). Moving beyond primary sequence alignments to interrogate the structures of the four intact Plm C domains, homology models were generated using AlphaFold241 and compared to the standalone C domain VibH.42 PlmJ C1 showed the lowest RMSD value when aligned with VibH (10.312), and when its HHxxxDR motif was modeled into VibH, the side chain of R131 protruded into the substrate channel, likely blocking it (Fig. S12, ESI†). Arginine has previously been observed in the motif of the Oxy enzyme-recruiting X domain Tcp12 C243 (Table S9, ESI†) as an H2R mutation, indicating PlmJ C1 might be involved in the docking of tailoring enzymes such as the deaminase PlmD, or the methyltransferases PlmH or PlmI. All other modules and domains in PlmA, PlmJ and PlmK have multiple lines of evidence supporting their proposed roles, making the first module of PlmJ incongruous in the pathway. Therefore, we propose that the first module of PlmJ is not involved in chain extension and possibly skipped due to a structurally inactive C domain. However, it is not yet unequivocally determined which mechanism of module skipping is occurring, since PlmJ T1 maintains the active site serine that would be required for phosphopantetheinylation and subsequent chain transfer (Fig. 4B and Table S7, ESI†). Future work will aim to untangle the mechanistic possibilities, including independent mutagenesis of the PlmJ T1 and T2 domains, which may decrease yield of 1 or abolish production entirely.
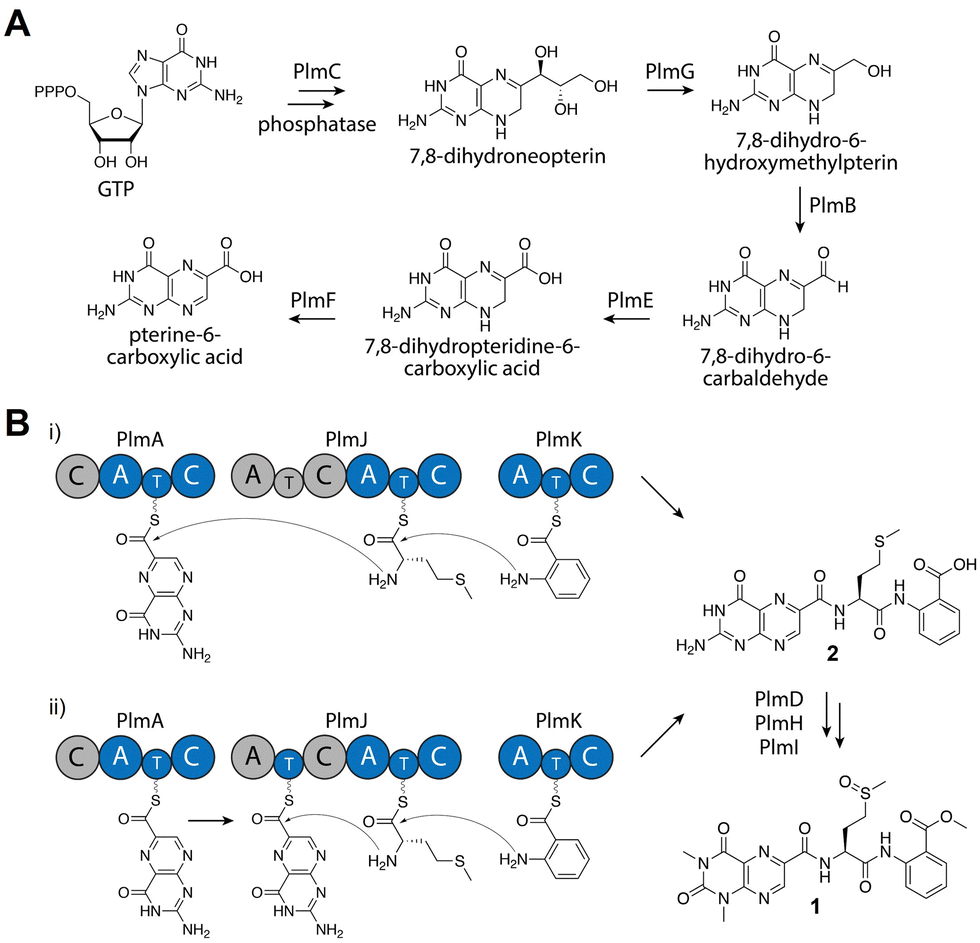 | ||
| Fig. 4 Proposed biosynthesis of penilumamide A (1). (A) Biosynthesis of the pterine-6-carboxylic acid building block. (B) Assembly of the tripeptide can proceed through two possible mechanisms: entire A–T–C module skipping (i) or chain transfer via the T domain of the skipped module (ii). Based on bioinformatic analyses, gray domains are proposed to be inactive or skipped. | ||
Based on bioinformatic data and in vitro biochemical assays, we propose that PlmA, which contains a truncated and inactive C domain at its N-terminus, activates and incorporates the unique pterin-derived building block, which originates from GTP via the GMC oxidoreductase PlmB, the GTP cyclohydrolase I PlmC, the aldehyde dehydrogenase PlmE, the FAD-dependent oxidoreductase PlmF, and the dihydroneopterin aldolase PlmG (Fig. 4A). PlmJ then contributes a single L-methionine, with extension catalyzed by the C domain of PlmA, and anthranilic acid is loaded onto the T domain of PlmK before the terminal C domain of PlmJ couples it with the dipeptide (Fig. 4B(i)). The tripeptide 2 is then offloaded from PlmK via hydrolysis, followed by deamination and methylation reactions facilitated by PlmD, and PlmH and PlmI, respectively, to afford 1. However, we can’t rule out the possibility that deamination or N,N-dimethylation of pterin occurs while the peptide is tethered to a thiolation domain, perhaps mediated by protein–protein interactions with PlmJ C1. Interestingly, the Plm NRPSs do not contain annotated docking domains for inter-module communication as determined by HMMER. It is currently unknown how PlmA, PlmJ and PlmK associate together to generate the tripeptide, but this could provide an opportunity for engineering of NRPS assembly lines. Previous efforts to generate non-native peptides in vivo have revealed that the C domain-A domain interface provides a flexible region for insertion of didomains or entire modules.44 Further, in vitro reconstitution of single NRPS modules has been successful when larger polypeptides are split at the C/A domain interface.45 Because the Plm NRPSs lack this docking region and are already organized into A–T–C modules, they may be more tolerant of non-native protein–protein interactions.
In summary, we have identified and characterized the first biosynthetic cluster responsible for synthesizing a lumazine-containing natural product, penilumamide A, from the marine-derived fungus A. flavipes CNL-338. Using gene inactivation experiments and in vitro reconstitution assays, we have shown that all three Plm NRPSs, encoding four modules, are required for the biosynthesis of the tripeptide, suggesting potential module skipping. Through detailed in vitro biochemical characterization assays, we determined the substrate specificity of the four Plm NRPS adenylation domains, with bioinformatic analyses revealing the first characterized “specificity codes” for native methionine- and pterin-activating A domains. Altogether, this knowledge can be applied to other fungal NRPS systems and assist with bioinformatic-based predictions. Furthermore, penilumamides exhibit promising therapeutic potential as insulin-sensitizing agents for the treatment of type II diabetes mellitus.8,9 Understanding how subtle variations in chemical structure affect bioactivity will improve our bioengineering toolkit and expedite the development of penilumamides for clinical use.
S. C. H. contributed to experiment conceptualization, investigation, validation, data analysis, visualization, and manuscript writing at all stages. K. L. D. contributed to experiment conceptualization, methodology, and helpful discussions. J. M. W. contributed to experiment conceptualization, data analysis, funding acquisition, project administration, resources, supervision, and manuscript writing at all stages.
S. C. H. thanks ARUP Laboratories and the Skaggs Foundation for graduate research fellowships. This work was supported by the Gordon and Betty Moore Foundation (GBMF7621, https://doi.org/10.37807/GBMF7621) and in part by the National Institutes of Health (1R01AI155694) to J. M. W.
Conflicts of interest
The authors declare there are no conflicts of interest.Notes and references
- K.-H. Altmann, Chimia, 2017, 71, 646–652 CrossRef CAS PubMed.
- J. Wiese and J. F. Imhoff, Drug Dev. Res., 2019, 80, 24–27 CrossRef CAS PubMed.
- A. R. Carroll, B. R. Copp, R. A. Davis, R. A. Keyzers and M. R. Prinsep, Nat. Prod. Rep., 2021, 38, 362–413 RSC.
- N. P. Keller, Nat. Rev. Microbiol., 2019, 17, 167–180 CrossRef CAS PubMed.
- S. W. Meyer, T. F. Mordhorst, C. Lee, P. R. Jensen, W. Fenical and M. Köck, Org. Biomol. Chem., 2010, 8, 2158–2163 RSC.
- M. Chen, C.-L. Shao, X.-M. Fu, C.-J. Kong, Z.-G. She and C.-Y. Wang, J. Nat. Prod., 2014, 77, 1601–1606 CrossRef CAS PubMed.
- C. Wang, X. Wu, H. Bai, K. A. U. Zaman, S. Hou, J. Saito, S. Wongwiwatthananukit, K. S. Kim and S. Cao, J. Nat. Prod., 2020, 83, 2233–2240 CrossRef CAS PubMed.
- W. Chen, C. Chen, J. Long, S. Lan, X. Lin, S. Liao, B. Yang, X. Zhou, J. Wang and Y. Liu, J. Antibiot., 2021, 74, 156–159 CrossRef CAS PubMed.
- M. You, L. Liao, S. H. Hong, W. Park, D. I. Kwon, J. Lee, M. Noh, D.-C. Oh, K.-B. Oh and J. Shin, Mar. Drugs, 2015, 13, 1290–1303 CrossRef CAS PubMed.
- B. Chaiyosang, K. Kanokmedhakul, J. Boonmak, S. Youngme, V. Kukongviriyapan, K. Soytong and S. Kanokmedhakul, Nat. Prod. Res., 2016, 30, 1017–1024 CrossRef CAS PubMed.
- C.-J. Zheng, L.-Y. Wu, X.-B. Li, X.-M. Song, Z.-G. Niu, X.-P. Song, G.-Y. Chen and C.-Y. Wang, Helv. Chim. Acta, 2015, 98, 368–373 CrossRef CAS.
- J. M. Wurlitzer, A. Stanišić, S. Ziethe, P. M. Jordan, K. Günther, O. Werz, H. Kries and M. Gressler, Chem. Sci., 2022, 13, 9091–9101 RSC.
- C. T. Walsh, Nat. Prod. Rep., 2016, 33, 127–135 RSC.
- R. Luo, B. Liu, Y. Xie, Z. Li, W. Huang, J. Yuan, G. He, Y. Chen, Q. Pan, Y. Liu, J. Tang, G. Wu, H. Zhang, Y. Shi, Y. Liu, C. Yu, B. Wang, Y. Lu, C. Han, D. W. Cheung, S.-M. Yiu, S. Peng, Z. Xiaoqian, G. Liu, X. Liao, Y. Li, H. Yang, J. Wang, T.-W. Lam and J. Wang, Gigascience, 2012, 1, 18 CrossRef PubMed.
- Y. Peng, H. C. M. Leung, S. M. Yiu and F. Y. L. Chin, Bioinformatics, 2012, 28, 1420–1428 CrossRef CAS PubMed.
- K. Blin, S. Shaw, A. M. Kloosterman, Z. Charlop-Powers, G. P. van Wezel, M. H. Medema and T. Weber, Nucleic Acids Res., 2021, 49, W29–W35 CrossRef CAS PubMed.
- C. Camacho, G. Coulouris, V. Avagyan, N. Ma, J. Papadopoulos, K. Bealer and T. L. Madden, BMC Bioinf., 2009, 10, 421 CrossRef PubMed.
- I. Shibuya, K. Gomi, Y. Iimura, K. Takahashi, G. Tamura and S. Hara, Agric. Biol. Chem., 1990, 54, 1905–1914 CAS.
- E. Szewczyk, T. Nayak, C. E. Oakley, H. Edgerton, Y. Xiong, N. Taheri-Talesh, S. A. Osmani, B. R. Oakley and B. Oakley, Nat. Protoc., 2006, 1, 3111–3120 CrossRef CAS PubMed.
- S. Kawai, W. Hashimoto and K. Murata, Bioeng. Bugs, 2010, 1, 395–403 CrossRef PubMed.
- T. Izoré, Y. T. Candace Ho, J. A. Kaczmarski, A. Gavriilidou, K. H. Chow, D. L. Steer, R. J. A. Goode, R. B. Schittenhelm, J. Tailhades, M. Tosin, G. L. Challis, E. H. Krenske, N. Ziemert, C. J. Jackson and M. J. Cryle, Nat. Commun., 2021, 12, 2511 CrossRef PubMed.
- T. J. McQuade, A. D. Shallop, A. Sheoran, J. E. Delproposto, O. V. Tsodikov and S. Garneau-Tsodikova, Anal. Biochem., 2009, 386, 244–250 CrossRef CAS PubMed.
- N. Kadi and G. L. Challis, Methods Enzymol., 2009, 458, 431–457 CAS.
- S. Müller, S. Rachid, T. Hoffmann, F. Surup, C. Volz, N. Zaburannyi and R. Müller, Chem. Biol., 2014, 21, 855–865 CrossRef PubMed.
- J. Mistry, S. Chuguransky, L. Williams, M. Qureshi, G. A. Salazar, E. L. L. Sonnhammer, S. C. E. Tosatto, L. Paladin, S. Raj, L. J. Richardson, R. D. Finn and A. Bateman, Nucleic Acids Res., 2021, 49, D412–D419 CrossRef CAS PubMed.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef CAS PubMed.
- G. L. Challis, J. Ravel and C. A. Townsend, Chem. Biol., 2000, 7, 211–224 CrossRef CAS PubMed.
- W.-B. Yin, A. Grundmann, J. Cheng and S.-M. Li, J. Biol. Chem., 2009, 284, 100–109 CrossRef CAS PubMed.
- X. Gao, S. W. Haynes, B. D. Ames, P. Wang, L. P. Vien, C. T. Walsh and Y. Tang, Nat. Chem. Biol., 2012, 8, 823–830 CrossRef CAS PubMed.
- B. D. Ames and C. T. Walsh, Biochemistry, 2010, 49, 3351–3365 CrossRef CAS PubMed.
- M. Zhao, H.-C. Lin and Y. Tang, J. Antibiot., 2016, 69, 571–573 CrossRef CAS PubMed.
- L. Kaysser, X. Tang, E. Wemakor, K. Sedding, S. Hennig, S. Siebenberg and B. Gust, ChemBioChem, 2011, 12, 477–487 CrossRef CAS PubMed.
- Q. Li, L. Wang, Y. Xie, S. Wang, R. Chen and B. Hong, J. Bacteriol., 2013, 195, 2232–2243 CrossRef CAS PubMed.
- A. Plaza, K. Viehrig, R. Garcia and R. Müller, Org. Lett., 2013, 15, 5882–5885 CrossRef CAS PubMed.
- D. I. Pattison, A. S. Rahmanto and M. J. Davies, Photochem. Photobiol. Sci., 2012, 11, 38–53 CrossRef CAS PubMed.
- M. L. Dantola, L. O. Reid, C. Castaño, C. Lorente, E. Oliveros and A. H. Thomas, Pteridines, 2017, 28, 105–114 CrossRef CAS.
- W. Steinchen, G. Lackner, S. Yasmin, M. Schrettl, H.-M. Dahse, H. Haas and D. Hoffmeister, Appl. Environ. Microbiol., 2013, 79, 6670–6676 CrossRef CAS PubMed.
- D. Kalb, T. Heinekamp, G. Lackner, D. H. Scharf, H.-M. Dahse, A. A. Brakhage and D. Hoffmeister, Appl. Environ. Microbiol., 2015, 81, 1594–1600 CrossRef PubMed.
- S. C. Wenzel, P. Meiser, T. M. Binz, T. Mahmud and R. Müller, Angew. Chem., Int. Ed., 2006, 45, 2296–2301 CrossRef CAS PubMed.
- T. Degenkolb, R. Karimi Aghcheh, R. Dieckmann, T. Neuhof, S. E. Baker, I. S. Druzhinina, C. P. Kubicek, H. Brückner and H. von Döhren, Chem. Biodiversity, 2012, 9, 499–535 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- T. A. Keating, C. G. Marshall, C. T. Walsh and A. E. Keating, Nat. Struct. Biol., 2002, 9, 522–526 CAS.
- K. Haslinger, M. Peschke, C. Brieke, E. Maximowitsch and M. J. Cryle, Nature, 2015, 521, 105–109 CrossRef CAS PubMed.
- K. A. J. Bozhüyük, F. Fleischhacker, A. Linck, F. Wesche, A. Tietze, C.-P. Niesert and H. B. Bode, Nat. Chem., 2018, 10, 275–281 CrossRef PubMed.
- M. Kaniusaite, J. Tailhades, E. A. Marschall, R. J. A. Goode, R. B. Schittenhelm and M. J. Cryle, Chem. Sci., 2019, 10, 9466–9482 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, supplementary tables and figures, and phylogenetic analyses of enzyme domains. See DOI: https://doi.org/10.1039/d3cb00088e |
| ‡ We thank Professor William Fenical from Scripps Institution of Oceanography for providing A. flavipes CNL-338. |
| § The penilumamide biosynthetic cluster is deposited under the GenBank accession number ON297683. The sequenced fungal internal transcribed spacer region for Aspergillus flavipes CNL-338 is deposited under accession number MT579592. |
| This journal is © The Royal Society of Chemistry 2023 |