
Toxicity and efficacy of green tea catechin derivative-based micellar nanocomplexes for anticancer protein delivery†
Sijing
Xiong
a,
Susi
Tan
 ab,
Peng
Huang
a,
Yao
Li
a,
Joo Eun
Chung‡
a,
Motoichi
Kurisawa‡
a,
Daniele
Zink
*ac and
Jackie Y.
Ying
*abd
ab,
Peng
Huang
a,
Yao
Li
a,
Joo Eun
Chung‡
a,
Motoichi
Kurisawa‡
a,
Daniele
Zink
*ac and
Jackie Y.
Ying
*abd
aInstitute of Bioengineering and Nanotechnology, Agency for Science, Technology and Research (A*STAR), 31 Biopolis Way, The Nanos, 138669, Singapore
bNanoBio Lab, Institute of Materials Research and Engineering, A*STAR, 31 Biopolis Way, The Nanos, 138669, Singapore. E-mail: jyying@imre.a-star.edu.sg
cSingapore Institute of Food and Biotechnology Innovation, A*STAR, 31 Biopolis Way, The Nanos, 138669, Singapore. E-mail: daniele.zink@outlook.com
dNanoBio Lab, A*STAR Infectious Diseases Labs, A*STAR, 31 Biopolis Way, The Nanos, 138669, Singapore
First published on 12th May 2023
Abstract
Toxicity towards non-tumor cells during anticancer therapy can be reduced by using nanoscale systems for anticancer drug delivery. Usually only the loaded drug has anticancer activity. Recently, micellar nanocomplexes (MNCs) comprising green tea catechin derivatives for the delivery of the anticancer proteins, such as Herceptin, have been developed. Herceptin as well as the MNCs without the drug were effective against HER2/neu-overexpressing human tumor cells and had synergistic anticancer effects in vitro and in vivo. It remained unclear which kinds of negative effects the MNCs had on tumor cells exactly, and which of their components mediated them. Also, it was unclear if MNC has any toxicity effects on the normal cells of vital human organ systems. Herein we examined the effects of Herceptin-MNCs and their individual components on human breast cancer cells and on normal primary human endothelial and kidney proximal tubular cells. We applied a novel in vitro model that predicts nephrotoxicity in humans with high accuracy, as well as high-content screening and microfluidic mono- and co-culture models to thoroughly address effects on various cell types. The results showed that MNCs alone were profoundly toxic for breast cancer cells, and induced apoptosis regardless of HER2/neu expression levels. Apoptosis was induced by both green tea catechin derivatives contained within MNCs. In contrast, MNCs were not toxic for normal human cells, and the probability was low that MNCs would be nephrotoxic in humans. Together, the results supported the hypothesis that green tea catechin derivative-based MNCs could improve efficacy and safety of therapies with anticancer proteins.
Introduction
Numerous nano-drug carriers have been developed to improve the pharmacokinetics and biodistribution of drugs over the past three decades.1–5 However, most carriers are just excipients for drug delivery without therapeutic effects, and their drug loading capacities were usually around 10% (w/w).6–8 This in turn leads to the need for repeated injections to attain efficacy, and the use of large amount of carriers can give rise to problems of toxicity, metabolism and elimination.9–11 Toxicity is a critical factor to be considered while evaluating the clinical potential of a novel nano-drug carrier. The kidney being one of the major organs of excretion is often affected due to its heavy exposure to all circulating compounds.12,13 Therefore, nephrotoxicity can be considered as a main limiting factor for the advancement of anticancer treatment.14,15 The main target in the kidney is renal proximal tubular cells (PTCs), which are injured by various types of drugs, environmental chemicals, herbal compounds and other xenobiotics.16–18 As PTCs can be damaged by a wide range of different compounds including anticancer agents, which can lead to kidney injury and serious adverse effects, it is important to address PTC toxicity of novel anticancer agents.Vascular endothelial cells, which build up the inner lining of blood vessels, are also heavily exposed to all circulating compounds and can be damaged by anticancer therapy. Endothelial injury can lead to vital organ damage, thrombosis and vasculitis.19,20 Therefore, it is important to address potential endothelial toxicity of novel anticancer agents, especially with respect to those agents that would be expected to be cleared slowly from the circulation.
A novel drug delivery system has been developed with epigallocatechin-3-O-gallate (EGCG)-derivatives. The micellar nanocomplexes (MNCs) of ECGC derivative were able to stably encapsulate anticancer drugs, and achieve tumor-selective delivery, and dramatically increase therapeutic effect by synergism between the carrier and the drug, compared to the drug alone.21,22 EGCG, a major ingredient of green tea, has been shown to possess anticancer effects, antioxidant effects, and DNA-protective effects, etc.23–26 The MNCs were formed by complexation of oligomerized EGCG (OEGCG) with the anticancer protein, Herceptin (trastuzumab) to form the core, followed by complexation of poly(ethylene glycol)–EGCG conjugate (PEG–EGCG) to form the shell.21 The resulting monodispersed spherical MNCs have a hydrodynamic diameter of ∼90 nm. OEGCG compacted and stabilized protein drug molecules by complexation, and PEG–EGCG shielded the core complex to sequester from environmental challenge during circulation in the body. This high-performance drug carrier has drawn attention as a promising approach to overcome the limitations of potential toxicity and insufficient drug delivery of conventional drug carriers.27,28
Herceptin is a humanized monoclonal antibody against the human epidermal growth factor receptor (HER2/neu).29 Herceptin induces regression of HER2/neu-overexpressing breast tumor tissue, and is not effective against tumors that do not overexpress HER2/neu. It has been shown that MNCs loaded with bovine serum albumin (BSA-MNCs) instead of Herceptin were also effective against HER2/neu-overexpressing human cancer cells.21 However, Herceptin-loaded MNCs (Herceptin-MNCs) were more effective in vitro and in a mouse xenograft model in vivo due to synergistic effects of Herceptin and MNCs. While it was an exciting finding that the MNCs were not just a vehicle for the anticancer protein, but also contributed anticancer activity, the type of effects MNCs and their different components had on cancer cells need to be further studied. In addition, any toxic effects MNCs might have on normal human cells from vital organ systems should be examined to ascertain if these novel carriers have potential for clinical applications.
Herein we investigated the effects of MNCs and their individual compounds on human breast cancer cells, and on normal human primary endothelial cells and human primary renal proximal tubular cells (HPTCs). Recently, in vitro models became available that predict PTC toxicity in humans with high accuracy,30–38 and we have applied models developed in our laboratory in this study. Various other effects on the different human cell types were also investigated by applying high-content screening and microfluidic models.
Experimental
Cell culture
HPTCs were either purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) or isolated from nephrectomy samples obtained from the Tissue Repository of the National University Hospital (NUH, Singapore). HPTCs were cultivated as described previously with epithelial cell basal medium (ATCC) supplemented with renal epithelial cell growth kit (ATCC) and 1% penicillin/streptomycin (Thermo Fisher Scientific, Singapore).33 Human umbilical vein endothelial cells (HUVECs) were purchased from ScienCell Research Laboratories (Carlsbad, California, USA), and were cultivated in complete endothelial cell medium (ScienCell Research Laboratories). For all experimental work, pools (three different donors each) of passage 4 HPTCs or HUVECs were used. BT-474 and MCF7 cells (ATCC) were cultivated in RPMI 1640 medium (Lonza, Basel, Switzerland) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Thermo Fisher Scientific, Singapore). In all experiments with static cultures, cells were cultivated for 3 days (HUVECs and HPTCs were organized into confluent monolayers after 3 days) before compound treatment for 72 hours.MNC preparation
OEGCG and PEG–EGCG were synthesized and assembled into MNCs as described previously.21 Herceptin was purchased from Roche Singapore Pte Ltd (Singapore). MNCs were synthesized as monodispersed spherical complexes with a hydrodynamic diameter of ∼90 nm.Quantification of cell numbers, RELA and γH2AX levels by high-content screening (HCS)
Cells were seeded at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per cm2 (BT-474, MCF7 and HUVECs) or 50000 cells per cm2 (HPTCs) in quadruplicates in 384-well black plates with transparent bottom (Greiner, Kremsmünster, Austria). After compound treatment, immunostaining of RELA and γH2AX, counterstaining of nuclei and cells, and image acquisition by HCS were performed as described previously.35 Briefly, after fixation with phosphate-buffered formaldehyde (3.7%, 10 minutes at room temperature), cells were immunostained with a primary anti-RELA antibody (Abcam, Cambridge, MA, USA) or anti-γH2AX (phospho S139) antibody (Abcam, Cambridge, UK), and a fluorescein isothiocyanate (FITC)-conjugated secondary antibody (Life Technologies, Carlsbad, CA, USA). Nuclei and entire cells were stained with 4′,6-diamidino-2-phenylindole (DAPI; Merck Millipore, Darmstadt, Germany) and whole cell stain (WCS, Thermo Fisher Scientific, Waltham, MA, USA). Image acquisition was performed with the ImageXpressMICRO high-content analysis system equipped with MetaXpress Image Acquisition and Analysis Software version 5.0 (Molecular Devices, Sunnyvale, CA, USA). 9 images or 16 images (apoptosis-related experiments) per well with 4 wells (replicates) per condition were captured with a 20× objective. The multi wavelength cell scoring module was used for determining numbers of cell nuclei, and nuclear RELA and γH2AX-specific fluorescence intensities.
000 cells per cm2 (BT-474, MCF7 and HUVECs) or 50000 cells per cm2 (HPTCs) in quadruplicates in 384-well black plates with transparent bottom (Greiner, Kremsmünster, Austria). After compound treatment, immunostaining of RELA and γH2AX, counterstaining of nuclei and cells, and image acquisition by HCS were performed as described previously.35 Briefly, after fixation with phosphate-buffered formaldehyde (3.7%, 10 minutes at room temperature), cells were immunostained with a primary anti-RELA antibody (Abcam, Cambridge, MA, USA) or anti-γH2AX (phospho S139) antibody (Abcam, Cambridge, UK), and a fluorescein isothiocyanate (FITC)-conjugated secondary antibody (Life Technologies, Carlsbad, CA, USA). Nuclei and entire cells were stained with 4′,6-diamidino-2-phenylindole (DAPI; Merck Millipore, Darmstadt, Germany) and whole cell stain (WCS, Thermo Fisher Scientific, Waltham, MA, USA). Image acquisition was performed with the ImageXpressMICRO high-content analysis system equipped with MetaXpress Image Acquisition and Analysis Software version 5.0 (Molecular Devices, Sunnyvale, CA, USA). 9 images or 16 images (apoptosis-related experiments) per well with 4 wells (replicates) per condition were captured with a 20× objective. The multi wavelength cell scoring module was used for determining numbers of cell nuclei, and nuclear RELA and γH2AX-specific fluorescence intensities.
Quantification of apoptotic cells
HPTCs, HUVECs and BT-474 and MCF7 cells were seeded in quadruplicates in 384-well black plates with transparent bottom. Compromised and apoptotic cells were detected with the Apoptotic/Necrotic/Healthy Cells Detection Kit (PromoKine, Heidelberg, Germany). Arsenic(III) oxide (25 μg mL−1) was used as positive control, and vehicle controls were included on each plate. Imaging and analysis of stained cells were performed by HCS and subsequent image analysis (MetaXpress Image Acquisition and Analysis Software version 5.0).Determination of compound-induced interleukin 6 (IL6) and chemokine (C–X–C motif) ligand 8 (CXCL8) expression levels
Cells were seeded in triplicate in 24-well plates (same densities as for HCS experiments). After compound exposure, quantitative real-time reverse transcription polymerase chain reaction (qPCR) was used to determine IL6 and CXCL8 (formerly called IL8) expression levels with the 2−ΔΔCT method.31–33 Results were normalized to two endogenous controls (glyceraldehyde 3-phosphate dehydrogenase and peptidylprolyl isomerase A) as described previously.31Microfluidics
HUVECs and BT-474 cells were seeded into microchannels (μ-Slide I0.8 Luer, Ibidi GmbH, Munich, Germany) at 96000 cells per cm2 per cell type. Cells were cultivated overnight, before perfusion with cell culture medium for 3 days at 1 dyn per cm2 with an Ismatec 4-channel peristaltic pump (Cole-Parmer GmbH, Wertheim, Germany). Details on the set up are presented in Fig. S1.† Cells were perfused for 7 days with cell culture medium only, or with cell culture medium containing 0.5% dimethyl sulfoxide (DMSO, vehicle control), Herceptin (500 μg ml−1), BSA-MNCs or Herceptin-MNCs (500 μg ml−1 BSA or Herceptin; 24 μg ml−1 OEGCG, 260 μg ml−1 PEG–EGCG), respectively. A closed circuit was used, and compound-loaded medium was completely exchanged after 3 days. Mouse anti-CD31 and rabbit anti-HER2/neu primary antibodies (Abcam, Cambridge, UK) were used in combination with fluorochrome-conjugated secondary antibodies for immunostaining after fixation on day 7. The area covered by BT-474 spheroids was measured with ImageJ.39 The methodology was similar to monocultures with the exception that a single type of human primary cells and no cancer cells were used. BT-474 conditioned medium was obtained from static BT-474 monocultures that were incubated with complete endothelial cell medium for 3 days.
Statistics
Statistical analyses were performed with the unpaired two-tailed Student's t-test using Microsoft Office Excel 2010 (Microsoft, Redmond, Washington, DC, USA). Normal distribution was confirmed using SigmaStat 3.5 (Systat Software Inc., Chicago, IL, USA).Results and discussion
MNCs alone specifically eliminate cancer cells by inducing apoptosis
Herceptin was complexed with OEGCG and PEG–EGCG to form Herceptin-MNCs with a core consisting of Herceptin/OEGCG complexes surrounded by a PEG–EGCG shell (Fig. S2†). Herceptin is an FDA-approved humanized monoclonal antibody against the HER2/neu receptor that induces regression of HER2-overexpressing metastatic breast cancer tumors. BSA-MNCs were used as drug-free controls. The resulting MNCs were monodispersed spherical complexes with a hydrodynamic diameter of ∼90 nm (Fig. S3†). Herceptin-MNCs and BSA-MNCs contained the same protein dose and were applied at doses of up to 1000 μg ml−1 Herceptin or BSA (corresponded to molar concentrations of 6.87 μM Herceptin and 15.05 μM BSA) (Fig. 1). At maximal protein doses, the other MNC compounds PEG–EGCG and OEGCG had concentrations of 520 μg ml−1 (200 μM) and 48 μg ml−1 (100 μM), respectively (Fig. 1). MNCs and their individual compounds tested were also at higher concentration ranges to ensure that any potential toxic effects would not be missed. The maximal human serum concentration (CMax) of Herceptin is 109 μg ml−1 (https://www.medsafe.govt.nz/profs/datasheet/h/Herceptininf.pdf). Thus, the maximal Herceptin concentration tested was ∼9.2-fold the human CMax.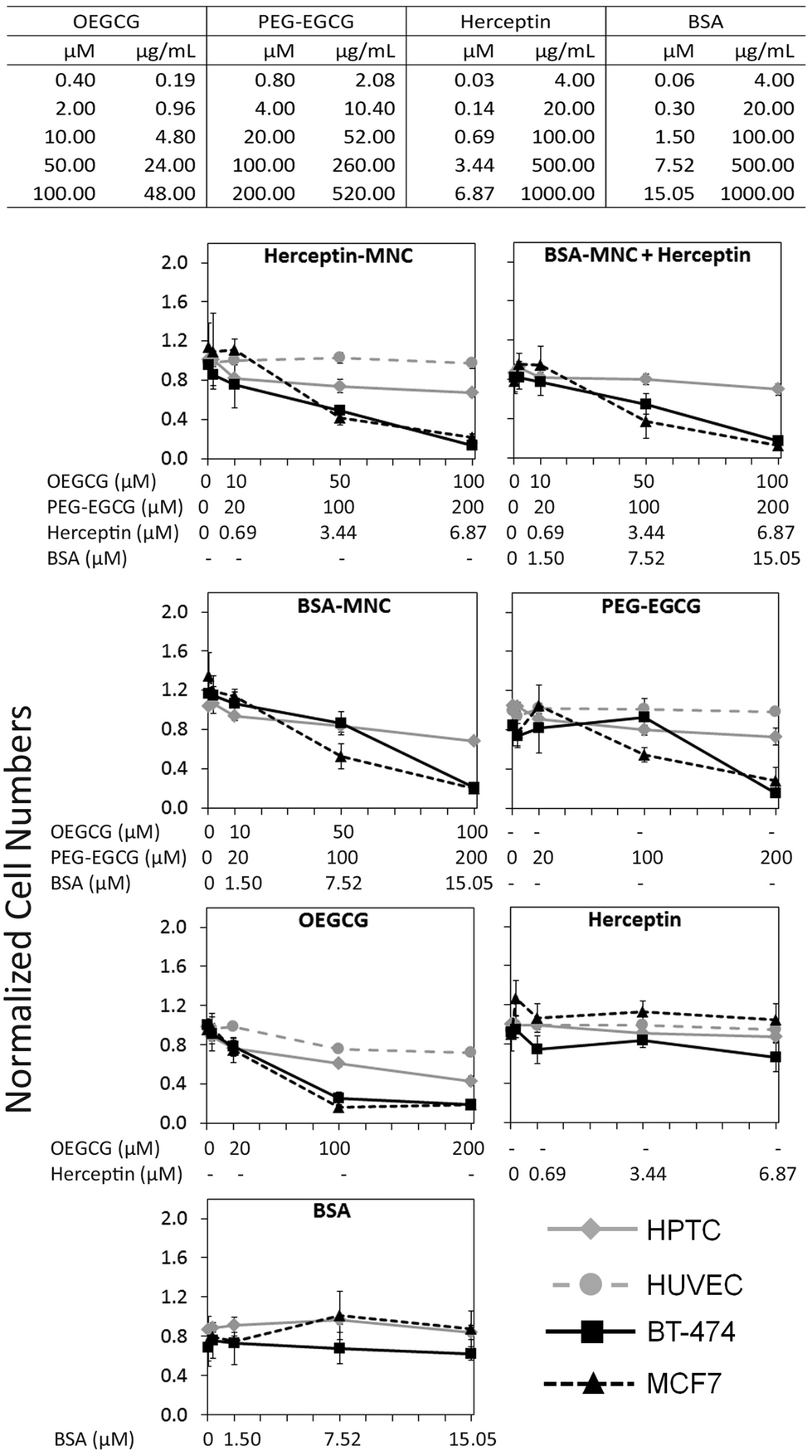 | ||
| Fig. 1 Concentration-dependent effects on cell numbers. Tested compound concentrations are listed in the table (top) in μg ml−1 and μM. Each row represents the concentrations of all different compounds in a Herceptin-MNC or BSA-MNC, respectively, at the indicated protein concentration. HPTCs, HUVECs, BT-474 cells and MCF7 cells were treated with MNCs or their individual compounds as indicated at the top of each diagram. When MNCs were tested, all compounds contained within the MNCs and their respective concentrations were listed below the graph (x-axis). Cell numbers were determined by HCS based on the numbers of DAPI-stained nuclei. All results were normalized to vehicle controls (n = 4, mean ± standard deviation (s.d.)). | ||
The effects of Herceptin-MNCs and BSA-MNCs and their individual compounds were tested on BT-474 cells (HER2/neu-overexpressing human breast cancer cell line), MCF7 cells (human breast cancer cell line not overexpressing HER2/neu), HPTCs and HUVECs. First, we tested the effects on cell numbers (Fig. 1), and only minor effects on HPTCs and HUVECs were observed. Even at maximal MNC concentrations or equivalent concentrations of individual MNC compounds, the numbers of HPTCs and HUVECs were at least ∼70–80% of the vehicle control cell numbers (Fig. 1). In contrast, Herceptin-MNCs and BSA-MNCs reduced the numbers of BT-474 and MCF7 cells in a concentration-dependent manner. Cancer cell numbers were reduced by ∼80% at maximal MNC concentrations. These results suggested that MNCs were specifically toxic for cancer cells irrespective of their HER2/neu expression levels.
When green tea catechin-derived compounds were tested individually, both PEG–EGCG and OEGCG reduced the numbers of BT-474 and MCF7 cells by up to ∼80% in a concentration-dependent manner (Fig. 1). Both compounds were tested at similar molar concentrations for comparison, and the results obtained under these conditions revealed that OEGCG was more toxic for cancer cells than PEG–EGCG (Fig. 1). However, it should be noted that in MNCs, the molar concentration of OEGCG was 2-fold lower than the concentration of PEG–EGCG, and the OEGCG concentration was only 100 μM at the maximal MNC concentration tested (Fig. 1).
Any marked negative effects of Herceptin alone on cell numbers were not observed. Also, at the highest concentrations, the numbers of BT-474 cells, HPTCs and HUVECs were still ∼80% and above that for the vehicle control (Fig. 1). These relatively mild effects were consistent with previous results obtained with BT-474 cells and other HER/neu-overexpressing cancer cell lines.21 The mechanisms of action of Herceptin are not fully understood, but immune cell-mediated toxicity is important in vivo,13,29 which does not occur under in vitro conditions.
Further inspection on the HCS image data revealed pronounced phenotypic changes such as cell rounding and shrinking of BT-474 and MCF7 cells when tested with the positive control (puromycin), Herceptin-MNCs, BSA-MNCs, BSA-MNCs and Herceptin, or PEG–EGCG at an equivalent concentration (Fig. 2). Similar pronounced phenotypic changes were observed on HPTCs and HUVECs with respect to the positive control (puromycin), but not MNCs or any of their compounds (Fig. 2 and S4†). Some milder effects on HUVECs were seen at the highest MNC-equivalent concentration of OEGCG (100 μM), where some vacuolization in the cytoplasm occurred (Fig. S4†).
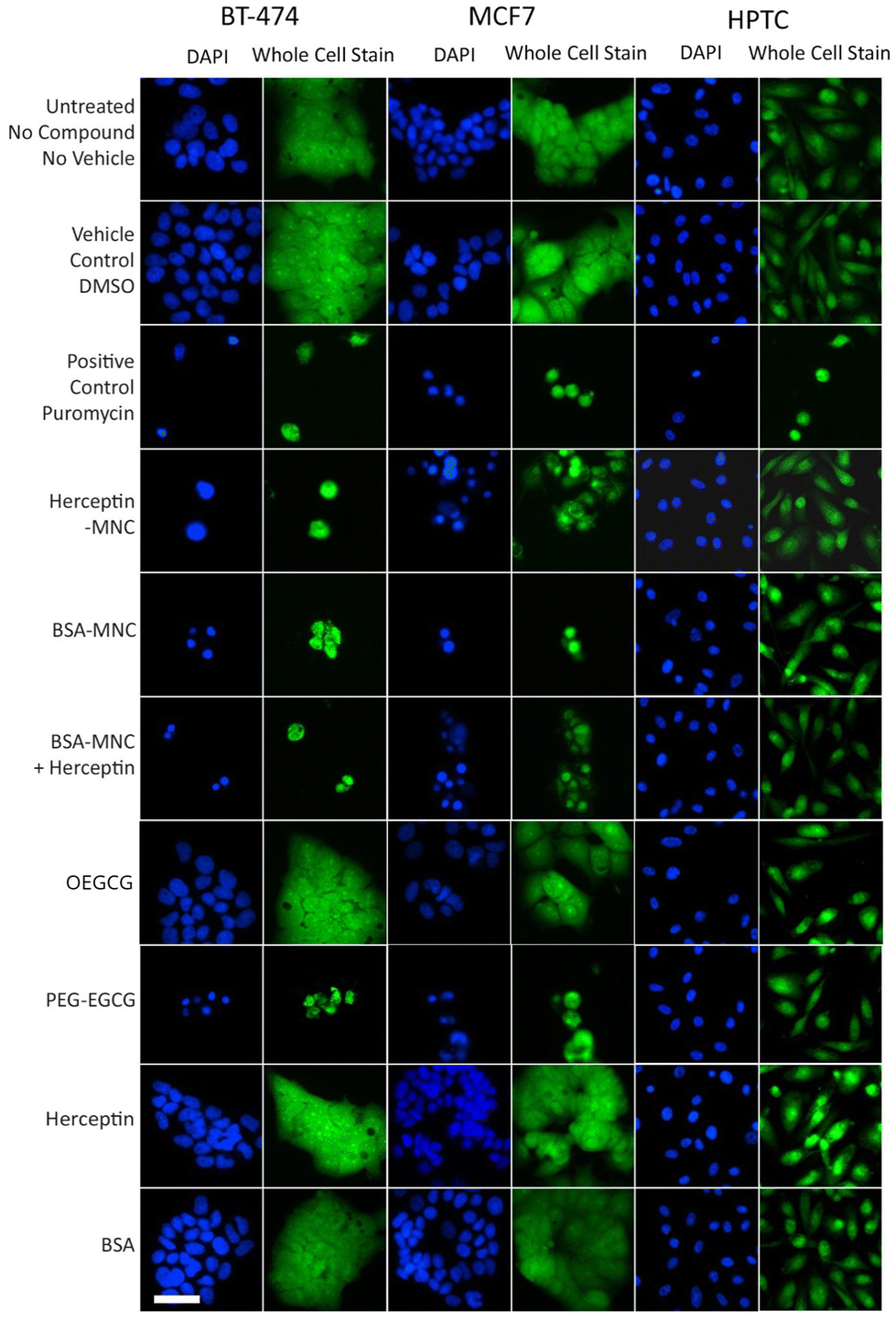 | ||
| Fig. 2 MNC-induced cellular changes. BT-474 cells, MCF7 cells and HPTCs were treated with MNCs or their individual compounds as indicated (left) before imaging by HCS. For each cell type, DAPI-stained nuclei (blue) and entire cells (green) are displayed. Free or MNC-complexed Herceptin and BSA were applied at 1000 μg ml−1 and other compounds were introduced in equivalent concentrations (see Fig. 1). 0.5% DMSO and 100 μg ml−1 puromycin were used as vehicle and positive controls, respectively. Scale bar: 50 μm. | ||
In order to address the mechanism leading to the pronounced negative effects on cancer cells, non-compromised cells (stained by Hoechst 33342 only) and compromised cells (stained by Hoechst 33342 and ethidium homodimer III) were examined (Fig. 3). Compromised cells positive for annexin V were classified as apoptotic cells. Almost all compromised cells were positive for annexin V, and therefore represented apoptotic cells (Fig. 3). High percentages of apoptotic cells were observed in BT-474 and MCF7 cells treated with Herceptin-MNCs or BSA-MNCs (Fig. 3). The results also revealed that both green tea catechin-derived compounds, OEGCG and PEG–EGCG, induced apoptosis in cancer cells (Fig. 3). The numbers of apoptotic cells were significantly higher in PEG–EGCG-treated than OEGCG-treated cancer cells, showing that anticancer cell effects were mainly mediated by PEG–EGCG (Fig. 3). Both compounds were tested at MNC-equivalent concentrations, and the molar concentration of PEG–EGCG was 2-fold higher than the concentration of OEGCG (see Fig. 1). In contrast, only low numbers of compromised and apoptotic cells were observed in similarly treated HPTC and HUVEC cells (Fig. 3).
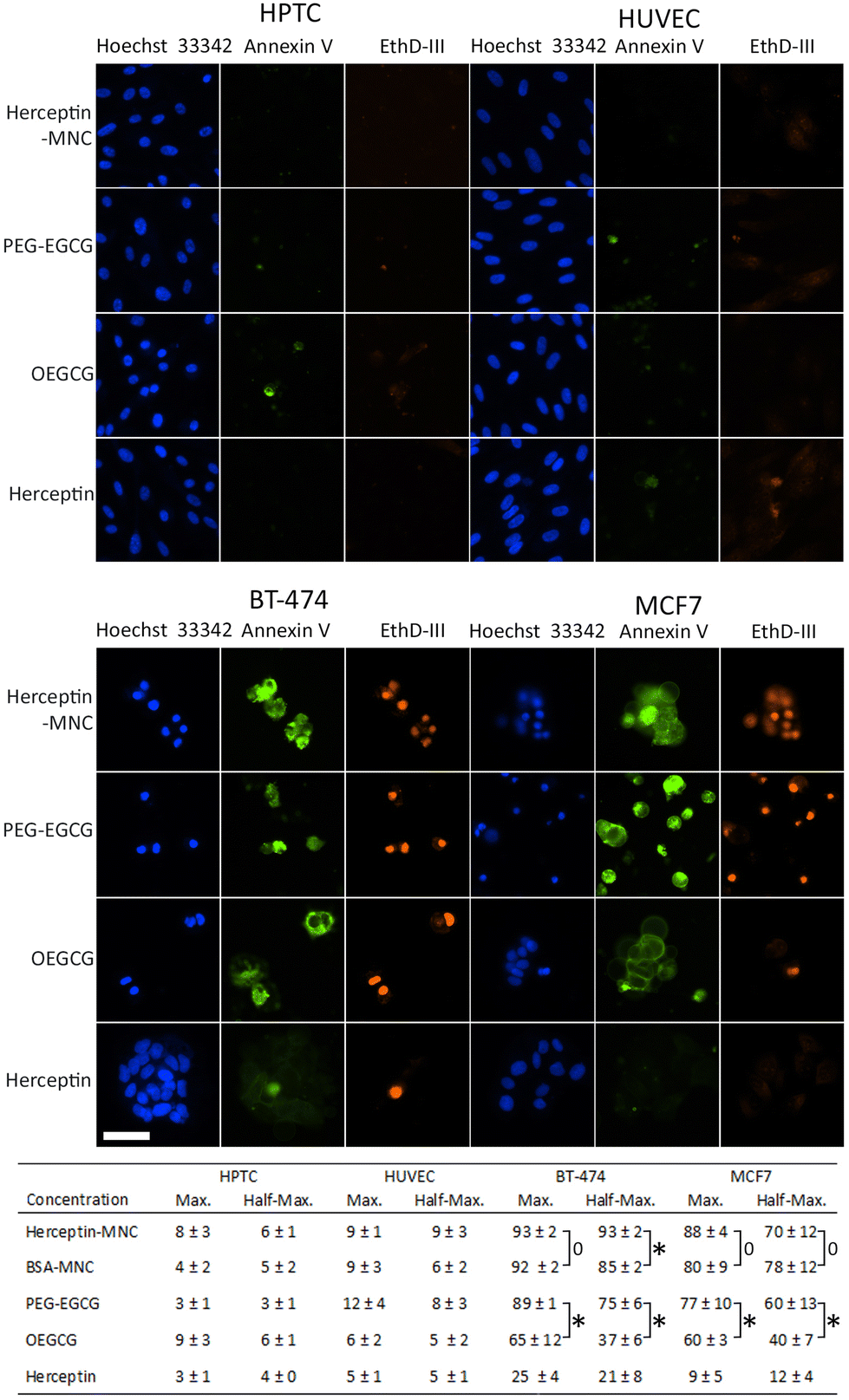 | ||
| Fig. 3 Induction of apoptosis. The images show the different cell types as indicated (top) after treatment with Herceptin-MNCs or their individual compounds (left). Herceptin-MNCs were used at a maximal Herceptin dose of 1000 μg ml−1, and individual compounds were used at equivalent concentrations. Hoechst 33342 (blue), annexin V-specific immunofluorescence (green) and ethidium homodimer III (EthD-III; red) fluorescence are shown. Scale bar: 50 μm. The table (bottom) shows the relative average percentages (n = 4, mean ± s.d.; total cell numbers were set to 100%) of apoptotic cells (positive for all 3 markers) after treatment with the indicated compounds (left) at maximal concentrations (MNCs with 1000 μg ml−1 BSA or Herceptin or equivalent compound concentrations) or half-maximal concentrations (corresponding to 500 μg ml−1 protein). Non-significant differences (p > 0.05) were indicated by a bracket labeled with 0, whereas significant differences (p < 0.05) were labeled with asterisks. | ||
The results also showed significantly higher numbers of apoptotic cells in BT-474 cultures treated with half-maximal concentrations of Herceptin-MNCs (compared to BSA-MNCs; Fig. 3). This would be consistent with a synergistic effect of Herceptin and MNCs in BT-474 cells. A similar synergism would not be expected in the case of MCF7 cells, which did not overexpress HER2/neu, and did not show such synergism. At maximal concentrations, apoptosis was already induced in >90% of BT-474 cells by the BSA-MNC alone, and this high efficiency probably explains why no further increase of apoptotic cells was observed when the Herceptin-MNC was applied (Fig. 3).
Together, the results showed that MNCs specifically eliminated cancer cells, but not normal human cells, by inducing apoptosis. Cancer cell-specific induction of apoptosis was mediated by both green tea catechin-derived compounds contained within MNCs. PEG–EGCG was less toxic for cancer cells than OEGCG, but mainly mediated the anticancer cell effects of the MNCs due to its higher concentration in MNCs.
Induction of pro-inflammatory cytokines/chemokines and of γH2AX formation
To address potential sublethal MNC effects, we investigated the induction of IL6 and CXCL8. Compound-induced upregulation of at least one of these major pro-inflammatory cytokines/chemokines in HPTCs is highly indicative of PTC toxicity, and predicts PTC toxicity in humans with a test balanced accuracy of ∼80%.31–34Fig. 4 shows that neither Herceptin-MNCs, nor any of their compounds, led to significant up-regulation of IL6 or CXCL8 in HPTCs. Based on these results obtained with the thoroughly prevalidated IL6/CXCL8 induction method,31–34 Herceptin-MNCs and their individual compounds were predicted to be not toxic for renal PTCs in humans with a probability of ∼80%.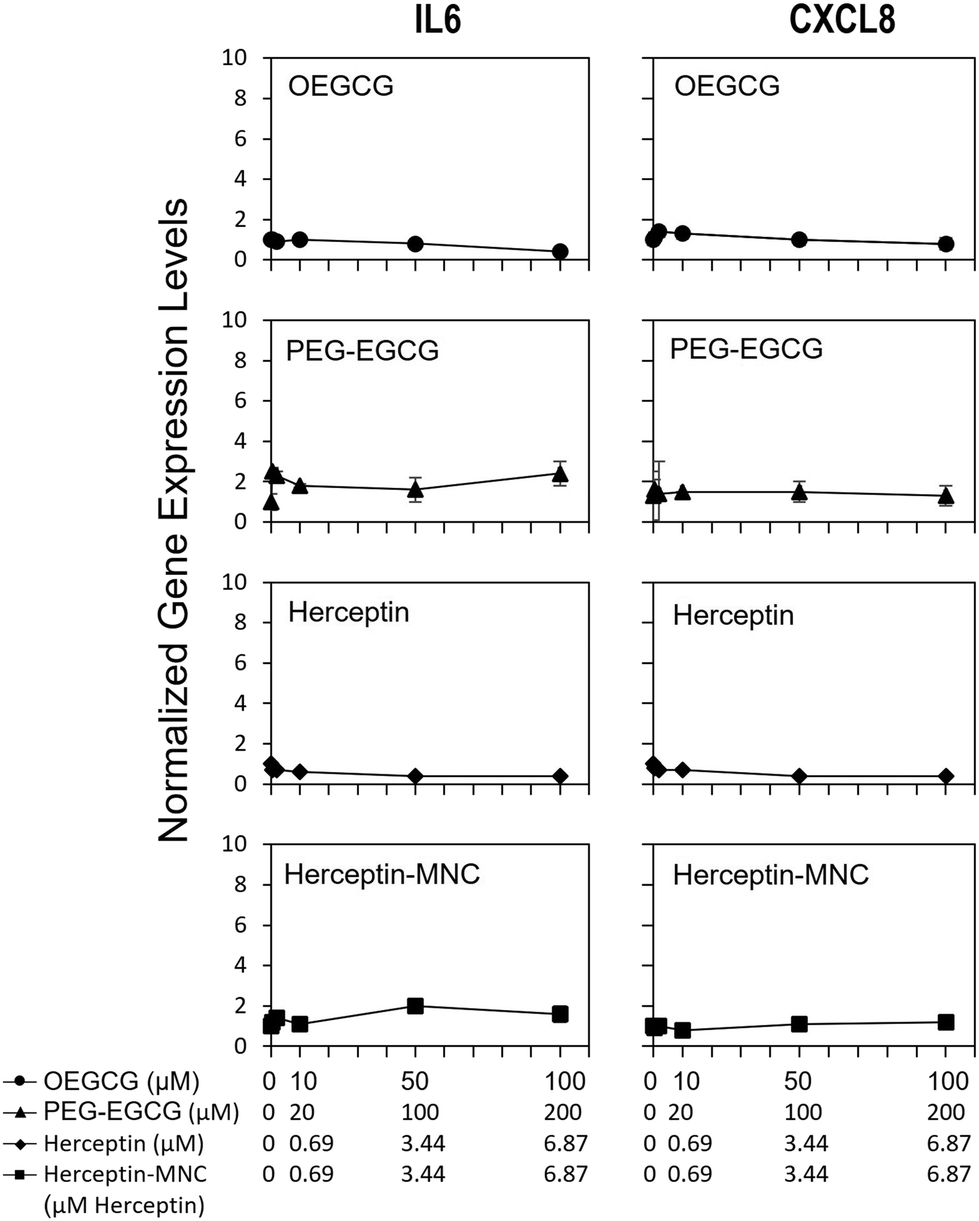 | ||
| Fig. 4 IL6 and CXCL8 expression levels. HPTC cells were treated with Herceptin-MNCs or their individual compounds. Compound concentrations are displayed on the y-axis. IL6 and CXCL8 expression levels were determined by qPCR. All results were normalized to vehicle controls (n = 3, mean ± s.d., *p < 0.05 relative to vehicle controls). | ||
Next, we investigated potential induction of a DNA damage response (DDR) by addressing nuclear levels of the Ser139 phyosphorylation of histone 2AX (γH2AX). Compound-induced increase of γH2AX is a sensitive indicator of genotoxicity.40–42 Once activated by critical DNA damage such as double-strand breaks (DSBs), γH2AX is rapidly accumulated over megabase domains at the site of DSB.43 Antibodies to γH2AX then allow visualization of individual DSB in cell nuclei. A DDR can also be induced indirectly by compound-induced cellular stress and other stressors.44,45 PTC-damaging compounds typically increase nuclear levels of γH2AX in HPTC, which is predictive with respect to PTC toxicity in humans.35
We found that Herceptin-MNCs and BSA-MNCs and their individual compounds induced no or only minor increases of γH2AX (Fig. S5 and S6†). At least 2-fold and significant changes were considered as relevant. None of the results met these criteria. These results indicated no direct or indirect genotoxicity, and also absence of DDR induction by other stressors. The results were in agreement with the prediction based on the results of the IL6/CXCL8 induction method (Fig. 4) that the probability was low that MNCs or their individual compounds would damage PTC in humans (γH2AX increase is a predictor PTC-specific toxicity in humans).35 Overall, the results on pro-inflammatory cytokine/chemokine and DDR induction were in agreement with the previous results indicating very low toxicity for normal human cells and predicted a low probability for PTC toxicity in humans.
Assessment of MNC toxicity and efficacy with microfluidic models
We used a microfluidic endothelial/tumor spheroid co-culture system to investigate MNC-induced effects under more physiological conditions (Fig. 5). BSA-MNC, Herceptin-MNC, and free Herceptin were tested at a protein dose of 500 μg ml−1, which corresponded to 4.6-fold the human Herceptin CMax value of 109 μg ml−1.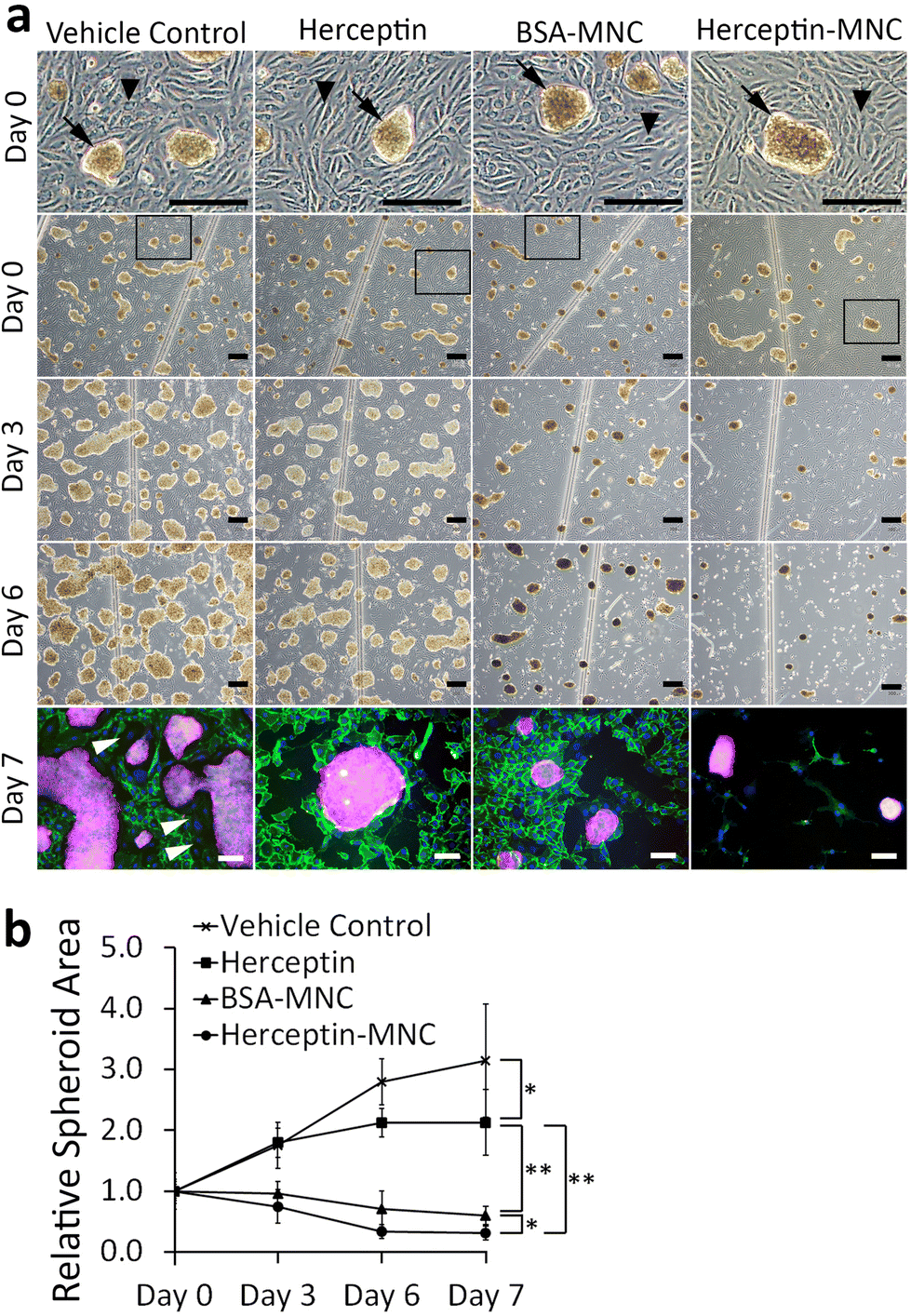 | ||
| Fig. 5 MNC- and Herceptin-induced effects on BT-474/HUVEC microfluidic co-cultures. (a) The panels show phase contrast images of BT-474/HUVEC co-cultures on day 0 (start of perfusion), day 3 and day 6 of perfusion. The top row (day 0) shows enlargements from the images below (day 0), and the enlarged areas are indicated by black frames. Black arrowheads indicate the endothelial HUVEC monolayer, and black arrows point to BT-474 tumor spheroids growing on top of the endothelium. Co-cultures were perfused with 0.5% DMSO (vehicle control), Herceptin (500 μg ml−1), BSA-MNCs and Herceptin-MNCs (500 μg ml−1 BSA or Herceptin) as indicated. Scale bars: 300 μm. On day 7, co-cultures were imaged by epifluorescence after immunostaining of platelet endothelial cell adhesion molecule (CD31; green) and HER2/neu (red; cell nuclei: blue). White arrowheads point to some of the gaps in the HUVEC endothelium. Scale bars: 100 μm. (b) The areas covered by tumor spheroids were determined on day 0, 3, 6 and 7. All data from day 3–7 were normalized to the results from respective samples on day 0, which were set to 1 (n = 3, mean ± s.d., *p < 0.05; **p < 0.01). | ||
In microfluidic co-cultures of HUVECs and BT-474 cells, three-dimensional tumor spheroids were formed on an initially intact and confluent endothelium (Fig. 5; day 0). Tumors interact with the local vasculature in vivo, and lead to endothelial disruption and formation of gaps in the endothelium.46,47 This causes the characteristic tumor vessel leakiness, which is important for drug delivery.48,49 In this co-culture model, the endothelium was disrupted within the culture period of 7 days, and gap formation occurred in particular in the vicinity of tumor spheroids (Fig. 5; day 3–7). To further address whether the endothelial disruption observed here was a recapitulation of typical tumor-associated processes, or related to potential toxicity of MNCs, we investigated microfluidic co-cultures that did not contain any MNCs or the vehicle (Fig. S7†). The endothelium was also disrupted in these untreated co-cultures. These results suggested that tumor cell-induced endothelial disruption was recapitulated in the microfluidic co-culture system.
Next, we addressed tumor-specific toxicity by quantifying the area covered by tumor spheroids during the cultivation period (Fig. 5b). Herceptin significantly reduced the size of tumor spheroids in comparison to vehicle controls. However, the area occupied by tumor spheroids still increased by ∼2 folds during the cultivation period and did not shrink, suggesting that Herceptin alone had mainly growth-inhibitory effects. In contrast, BSA-MNCs and Herceptin-MNCs induced significant (P < 0.05) shrinkage of the area occupied by tumor spheroids (Fig. 5), consistent with the result that MNCs induced apoptosis of cancer cells (Fig. 3). Herceptin-MNCs were significantly more effective than BSA-MNCs (Fig. 5), in agreement with synergistic effects of Herceptin and MNCs.
Notably, endothelial cells were efficiently eliminated in co-cultures treated with Herceptin-MNCs (Fig. 5; day 3–7). Interestingly, HUVEC monocultures perfused with Herceptin-MNCs for 7 days remained intact, and HUVECs remained arranged in a confluent, regularly patterned endothelium with normal membrane localization of CD31 (Fig. 6a). In contrast, perfusion of HUVEC monocultures with BT-474-conditioned medium led to endothelial disruption and gap formation (Fig. 6b and c). As Herceptin-MNCs showed no obvious negative effects on HUVECs, endothelial cell death in co-cultures was probably due to enhanced secretion of stressors and mediators of inflammation by compromised BT-474 cells treated with Herceptin-MNCs. Furthermore, BT-474-conditioned medium induced γH2AX formation in endothelial cell nuclei (Fig. 6b and c). This was consistent with previous reports showing induction of nuclear γH2AX formation by tumor cell-conditioned medium in vitro,50 and by cancer growth at distant sites in vivo.51
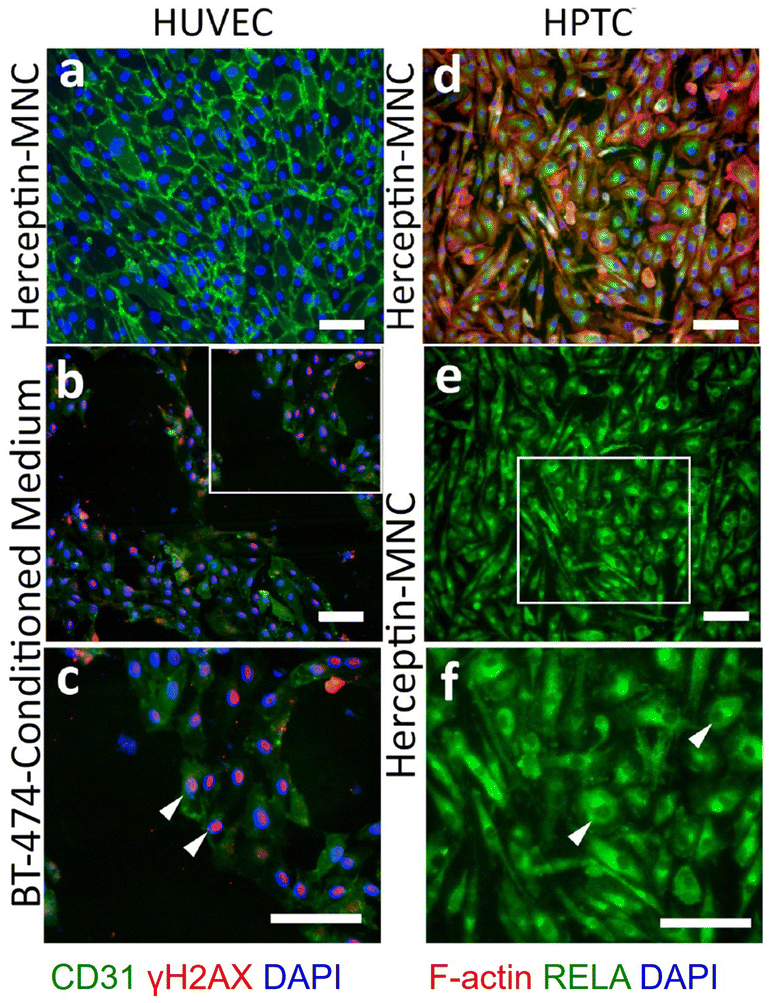 | ||
| Fig. 6 Effects on microfluidic HUVEC and HPTC monocultures. Microfluidic monocultures of HUVECs and HPTCs were perfused for 7 days with Herceptin-MNCs (500 μg ml−1 Herceptin) (a, d, e and f). Alternatively, HUVECs were perfused with BT-474-conditioned medium (b and c). The boxed area in panel (b) is shown enlarged in panel (c). CD31 and γH2AX were detected by immunostaining in HUVEC cultures on day 7. White arrowheads in (c) point at some nuclei with intense γH2AX-specific fluorescence. (d) F-actin and RELA were detected in HPTC cultures on day 7. Panel (e) shows only the RELA-specific fluorescence of the image shown in (d), and the boxed area is shown enlarged in (f). Arrowheads in (f) point to some cells with RELA-depleted nuclei (“dark holes” in intensely stained cytoplasm). Scale bars: 100 μm. | ||
Together, these results showed that normal physiological interactions between endothelial and tumor cells were recapitulated in the microfluidic co-culture system. Furthermore, the results indicated no or very low toxicity of Herceptin-MNCs for normal human endothelial cells, in agreement with the previous results.
HPTCs remained organized in a regular, confluent monolayer after 7 days of perfusion with Herceptin-MNCs (Fig. 6d). Nuclei remained depleted in RELA (p65), a subunit of the nuclear factor NF-κB complex (Fig. 6e and f), indicating no activation of the canonical NF-κB pathway, which is a main regulator of inflammation.52,53 Nuclear translocation of RELA is required for up-regulation of IL6 and CXCL8.54–56 The lack of nuclear RELA enrichment observed here (Fig. 6e and f) was in agreement with the lack of IL6 and CXCL8 upregulation in HPTCs (Fig. 4). Altogether, the results provided no evidence for triggering of pro-inflammatory or cellular stress-related processes by Herceptin-MNCs in normal human cells, even after prolonged exposure for 7 days in a sensitive microfluidic system.
Conclusions
In summary, we have investigated the toxicity and efficacy of green tea catechin derivative-based MNCs for delivery of anticancer protein drugs, such as Herceptin. Various effects on HER2/neu overexpressing and non-overexpressing human breast cancer cells as well as HUVECs and HPTCs were analyzed with static in vitro models, as well as with microfluidic mono- and co-culture models. The MNCs and their individual compounds were predicted to not damage renal proximal tubules in humans with a probability of ∼80%. In addition, direct toxicity for HUVECs was not observed, as indicated by the low pro-inflammatory cytokine/chemokine and DDR induction. In contrast, MNCs were profoundly toxic for tumor cells due to apoptosis induced by both EGCG-derived compounds, OEGCG and PEG–EGCG. This is in good agreement with numerous studies demonstrating the ability of EGCG to induce cancer cell death without damaging surrounding healthy cells by modulating various cellular pathways.21,57 One of the mechanisms involves EGCG's ability to chelate metal ions, specifically copper ions, which are known to be highly raised in cancer cells and results in DNA breakage.58 As effective MNCs could also be generated with other anticancer proteins, such as interferon α, MNC-based systems would be attractive for developing agents with improved efficacy against a broad range of solid tumors and reduced toxicity for normal tissues.Author contributions
D. Z. and J. Y. Y. conceptualized the study. J. Y. Y., D. Z. and M. K. supervised the research. S. X., P. H., Y. L. and J. E. C. performed the experiments. S. X., S. T., P. H., Y. L., J. E. C., D. Z. and J. Y. Y. contributed to data analysis. S. X., S. T., P. H., D. Z. and J. Y. Y. wrote the manuscript. All authors reviewed the manuscript.Conflicts of interest
The authors (S. T., J. E. C., M. K., J. Y. Y.) have filed a patent related to the design of Herceptin-MNC and licensed this technology to a startup company, GreenT Biomed Pte. Ltd. co-founded by J. Y. Y.Acknowledgements
This work is supported by the Health and Biomedical Sciences Industry Alignment Fund Pre-Positioning (IAF-PP) (Grant No. H20C6a0034). We thank Institute of Bioengineering and Nanotechnology, NanoBio Lab, Institute of Materials Research and Engineering, and Singapore Institute of Food and Biotechnology Innovation for supporting this research under A*STAR, Singapore.References
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- J. Panyam and V. Labhasetwar, Adv. Drug Delivery Rev., 2003, 55, 329–347 CrossRef CAS PubMed.
- K. Miyata, R. J. Christie and K. Kataoka, React. Funct. Polym., 2011, 71, 227–234 CrossRef CAS.
- A. A. Halwani, Pharmaceutics, 2022, 14, 106–126 CrossRef CAS PubMed.
- C. Li, J. Wang, Y. Wang, H. Gao, G. Wei, Y. Huang, H. Yu, Y. Gan, Y. Wang, L. Mei, H. Chen, H. Hu, Z. Zhang and Y. Jin, Acta Pharm. Sin. B, 2019, 9, 1145–1162 CrossRef PubMed.
- K. S. Chu, A. N. Schorzman, M. C. Finniss, C. J. Bowerman, L. Peng, J. C. Luft, A. J. Madden, A. Z. Wang, W. C. Zamboni and J. M. DeSimone, Biomaterials, 2013, 34, 8424–8429 CrossRef CAS PubMed.
- K. S. Soppimath, T. M. Aminabhavi, A. R. Kulkarni and W. E. Rudzinski, J. Controlled Release, 2001, 70, 1–20 CrossRef CAS PubMed.
- S. Shen, Y. Wu, Y. Liu and D. Wu, Int. J. Nanomed., 2017, 12, 4085–4109 CrossRef CAS PubMed.
- Y. Bae and K. Kataoka, Adv. Drug Delivery Rev., 2009, 61, 768–784 CrossRef CAS PubMed.
- S. Sharma, R. Parveen and B. P. Chatterji, Curr. Pathobiol. Rep., 2021, 9, 133–144 CrossRef CAS PubMed.
- H. Parhiz, M. Khoshnejad, J. W. Myerson, E. Hood, P. N. Patel, J. S. Brenner and V. R. Muzykantov, Adv. Drug Delivery Rev., 2018, 130, 90–112 CrossRef CAS PubMed.
- H. Y. Tiong, P. Huang, S. Xiong, Y. Li, A. Vathsala and D. Zink, Mol. Pharm., 2014, 11, 1933–1948 CrossRef CAS PubMed.
- N. Lameire, Clin. Kidney J., 2014, 7, 11–22 CrossRef CAS PubMed.
- J. Małyszko, K. Kozłowska, L. Kozłowski and J. Małyszko, Nephrol., Dial., Transplant., 2016, 32, 924–936 Search PubMed.
- M. L. C. Santos, B. B. de Brito, F. A. F. da Silva, A. Botelho and F. F. de Melo, World J. Clin. Oncol., 2020, 11, 190–204 CrossRef PubMed.
- T. R. Van Vleet and R. G. Schnellmann, Semin. Nephrol., 2003, 23, 500–508 CrossRef CAS PubMed.
- S. Li, J. Zhao, R. Huang, T. Steiner, M. Bourner, M. Mitchell, D. C. Thompson, B. Zhao and M. Xia, Curr. Chem. Genomics Transl. Med., 2017, 11, 19–30 CrossRef CAS PubMed.
- A. M. Hall, F. Trepiccione and R. J. Unwin, Pediatr. Nephrol., 2022, 37, 973–982 CrossRef PubMed.
- J. Herrmann, E. H. Yang, C. A. Iliescu, M. Cilingiroglu, K. Charitakis, A. Hakeem, K. Toutouzas, M. A. Leesar, C. L. Grines and K. Marmagkiolis, Circulation, 2016, 133, 1272–1289 CrossRef CAS PubMed.
- Z. Elyaspour, M. J. Zibaeenezhad, M. Razmkhah and I. Razeghian-Jahromi, Clin. Appl. Thromb./Hemostasis, 2021, 27, 1–11 Search PubMed.
- J. E. Chung, S. Tan, S. J. Gao, N. Yongvongsoontorn, S. H. Kim, J. H. Lee, H. S. Choi, H. Yano, L. Zhuo, M. Kurisawa and J. Y. Ying, Nat. Nanotechnol., 2014, 9, 907–912 CrossRef CAS PubMed.
- N. Yongvongsoontorn, J. E. Chung, S. J. Gao, K. H. Bae, A. Yamashita, M.-H. Tan, J. Y. Ying and M. Kurisawa, ACS Nano, 2019, 13, 7591–7602 CrossRef CAS PubMed.
- A. Bordoni, S. Hrelia, C. Angeloni, E. Giordano, C. Guarnieri, C. M. Caldarera and P. L. Biagi, J. Nutr. Biochem., 2002, 13, 103–111 CrossRef CAS PubMed.
- J. Jankun, S. H. Selman, R. Swiercz and E. Skrzypczak-Jankun, Nature, 1997, 387, 561–561 CrossRef CAS PubMed.
- K. Nakagawa, M. Ninomiya, T. Okubo, N. Aoi, L. R. Juneja, M. Kim, K. Yamanaka and T. Miyazawa, J. Agric. Food Chem., 1999, 47, 3967–3973 CrossRef CAS PubMed.
- S. Y. Chong, H. Y. Chiang, T. H. Chen, Y. J. Liang and Y. C. Lo, Sci. Rep., 2019, 9, 3842 CrossRef PubMed.
- K. H. Bae, F. Lai, J. Mong, A. Niibori-Nambu, K. H. Chan, Z. Her, M. Osato, M.-H. Tan, Q. Chen and M. Kurisawa, J. Nanobiotechnol., 2022, 20, 481 CrossRef CAS PubMed.
- K. Liang, J. E. Chung, S. J. Gao, N. Yongvongsoontorn and M. Kurisawa, Adv. Mater., 2018, 30, 1706963 CrossRef PubMed.
- T. Vu and F. X. Claret, Front. Oncol., 2012, 2, 62 Search PubMed.
- D. Zink, J. K. C. Chuah and J. Y. Ying, Trends Mol. Med., 2020, 26, 570–582 CrossRef CAS PubMed.
- K. Kandasamy, J. K. Chuah, R. Su, P. Huang, K. G. Eng, S. Xiong, Y. Li, C. S. Chia, L. H. Loo and D. Zink, Sci. Rep., 2015, 5, 12337 CrossRef PubMed.
- Y. Li, K. Kandasamy, J. K. C. Chuah, Y. N. Lam, W. S. Toh, Z. Y. Oo and D. Zink, Mol. Pharm., 2014, 11, 1982–1990 CrossRef CAS PubMed.
- Y. Li, Z. Y. Oo, S. Y. Chang, P. Huang, K. G. Eng, J. L. Zeng, A. J. Kaestli, B. Gopalan, K. Kandasamy, F. Tasnim and D. Zink, Toxicol. Res., 2013, 2, 352–362 CrossRef CAS.
- R. Su, Y. Li, D. Zink and L. H. Loo, BMC Bioinf., 2014, 15(Suppl. 16), S16 CrossRef PubMed.
- R. Su, S. Xiong, D. Zink and L. H. Loo, Arch. Toxicol., 2015, 90, 2793–2808 CrossRef PubMed.
- J. G. Hengstler, A.-K. Sjögren, D. Zink and J. J. Hornberg, Arch. Toxicol., 2020, 94, 353–356 CrossRef CAS PubMed.
- L.-H. Loo and D. Zink, Altern. Lab. Anim., 2017, 45, 241–252 CrossRef PubMed.
- A. R. Irvine, D. van Berlo, R. Shekhani and R. Masereeuw, Curr. Opin. Toxicol., 2021, 27, 18–26 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- T. Nikolova, M. Dvorak, F. Jung, I. Adam, E. Kramer, A. Gerhold-Ay and B. Kaina, Toxicol. Sci., 2014, 140, 103–117 CrossRef CAS PubMed.
- A. Wilson, P. Grabowski, J. Elloway, S. Ling, J. Stott and A. Doherty, Sci. Rep., 2021, 11, 2535 CrossRef CAS PubMed.
- B. Kopp, L. Khoury and M. Audebert, Arch. Toxicol., 2019, 93, 2103–2114 CrossRef CAS PubMed.
- M. Shrivastav, L. P. De Haro and J. A. Nickoloff, Cell Res., 2008, 18, 134–147 CrossRef CAS PubMed.
- C. E. Redon, A. J. Nakamura, O. A. Martin, P. R. Parekh, U. S. Weyemi and W. M. Bonner, Aging, 2011, 3, 168–174 CrossRef CAS PubMed.
- J. van Stuijvenberg, P. Proksch and G. Fritz, Bioorg. Med. Chem., 2020, 28, 115279 CrossRef CAS PubMed.
- H. Hashizume, P. Baluk, S. Morikawa, J. W. McLean, G. Thurston, S. Roberge, R. K. Jain and D. M. McDonald, Am. J. Pathol., 2000, 156, 1363–1380 CrossRef CAS PubMed.
- K. Hida and N. Maishi, Oral Sci. Int., 2018, 15, 1–6 CrossRef.
- Y. Malam, M. Loizidou and A. M. Seifalian, Trends Pharmacol. Sci., 2009, 30, 592–599 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- J. S. Dickey, B. J. Baird, C. E. Redon, M. V. Sokolov, O. A. Sedelnikova and W. M. Bonner, Carcinogenesis, 2009, 30, 1686–1695 CrossRef CAS PubMed.
- C. E. Redon, J. S. Dickey, A. J. Nakamura, I. G. Kareva, D. Naf, S. Nowsheen, T. B. Kryston, W. M. Bonner, A. G. Georgakilas and O. A. Sedelnikova, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17992–17997 CrossRef CAS PubMed.
- T. Lawrence, Cold Spring Harbor Perspect. Biol., 2009, 1, a001651 Search PubMed.
- A. Oeckinghaus and S. Ghosh, Cold Spring Harbor Perspect. Biol., 2009, 1, a000034 Search PubMed.
- C. Kunsch and C. A. Rosen, Mol. Cell Biol., 1993, 13, 6137–6146 CAS.
- T. Matsusaka, K. Fujikawa, Y. Nishio, N. Mukaida, K. Matsushima, T. Kishimoto and S. Akira, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 10193–10197 CrossRef CAS PubMed.
- A. R. Brasier, Cardiovasc. Res., 2010, 86, 211–218 CrossRef CAS PubMed.
- M. Farhan, Int. J. Mol. Sci., 2022, 23, 10713 CrossRef CAS PubMed.
- M. Farhan, A. Rizvi, I. Naseem, S. M. Hadi and A. Ahmad, Tumor Biol., 2015, 36, 8861–8867 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm01969h |
| ‡ Current address: Graduate School of Advanced Science and Technology, Japan Advanced Institute of Science and Technology (JAIST), 1-1 Asahidai, Nomi, Ishikawa 923-1292, Japan. |
| This journal is © The Royal Society of Chemistry 2023 |