
Conducting polymers with redox active pendant groups: their application progress as organic electrode materials for rechargeable batteries
Er-Tai
Liu†
a,
Shi-Lin
Mei†
a,
Xian-He
Chen
a and
Chang-Jiang
Yao
 *ab
*ab
aState Key Laboratory of Explosion Science and Technology, School of Mechatronical Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: cjyao@bit.edu.cn
bBeijing Institute of Technology Chongqing Innovation Center, Chongqing, 401120, China
First published on 9th June 2022
Abstract
Conducting redox polymers (CRPs) composed of a conducting polymer backbone and covalently linked redox active pendant groups have attracted increasing interest as electrode materials for rechargeable metal batteries. Combining the intrinsic properties of conducting polymers and redox active polymers, CRPs would possibly bring high charge capacity, fast charge transport, and stable cycling provided that the individual merits of the backbone and the redox active groups can be preserved and act in a synergistic manner. Thus, the application of CRPs requires good matching of the redox chemistry of the pendant group and the CP backbone. In this review, the fundamentals of CRPs are first introduced. Second, the application advances of CRPs as active electrode materials in monovalent metal-ion/sulfur (e.g. Li-ion/S, Na-ion/S) batteries and multivalent-ion/S (e.g. Zn-ion/S) batteries are summarized based on their specific redox reactions. Perspectives on pressing challenges and further research opportunities regarding CRP-based electrode materials are also discussed. This review provides concepts of rational design of CRPs to develop highly efficient organic electrodes and promote their practical applications.
Er-Tai Liu received his BS degree from the North University of China in 2019. He is currently a MS candidate at the Beijing Institute of Technology under the supervision of Prof. Chang-Jiang Yao. His research interests are focused on the design and synthesis of novel organic polymeric materials for lithium ions and lithium–sulfur batteries. |
Shi-Lin Mei received his PhD degree in 2017 with Prof. Matthias Ballauff at the Humboldt University of Berlin, Germany. He worked on his postdoctoral research on energy storage materials in the Department for Electrochemical Energy Storage, Helmholtz-Zentrum Berlin für Materialien und Energie GmbH from 2017. Since 2020 he has been an associate professor at the Beijing Institute of Technology. His research interests are focused on functional hybrid nanomaterials for energy storage. |
 Chang-Jiang Yao | Chang-Jiang Yao obtained his PhD degree from the Institute of Chemistry, Chinese Academy of Sciences (ICCAS), in 2013. Then he worked as an assistant professor in the same institute for two years. After research experience in Regensburg University (Germany), Wuerzburg University (Germany) and Nanyang Technology University (Singapore), he joined the Beijing Institute of Technology as a professor in 2019. His current research interest is focused on organic/polymer functional materials for energy and photo/electronic applications. |
1. Introduction
People are rushing to produce renewable energy because of growing environmental concerns and the gradual depletion of fossil resources. Electrochemical energy storage is more suited to the current needs of human society than wind and solar energies, which cannot provide a consistent and sustained energy supply.1 This strategy has resulted in the widespread use of various batteries in electric vehicles and portable and wearable devices. Rechargeable lithium batteries are a mature and dependable energy storage technology capable of providing stable electric energy and undergoing continuous repeated charging and discharging processes.2 Due to the unique embedding mechanism of lithium ion batteries, their practical specific capacity is limited, which is unable to meet the needs of people.3 Optimization techniques for lithium-ion batteries or new batteries with high energy density, long cycle life and high safety have been studied.4–11 Lithium–sulfur batteries are considered as a popular alternative to lithium batteries due to their high theoretical discharge capacity, high energy density and environmental friendliness.12 However, the low conductivity caused by sulfur, the shuttle effect and volume change in charging and discharging processes limit the practical application of lithium–sulfur batteries.13–15 In addition, the aqueous battery has become a new alternative to the traditional battery because of its advantages of lower cost, higher power density and significantly reduced content of toxic and flammable organic matter.9 However, the aqueous battery also suffers from lithium dendrite generation and by-product generation in the reaction process, which leads to low efficiency and poor cycle stability.16–18Electrode materials greatly affect the capacity, density and cost of battery equipment, so it is of scientific and practical significance to explore more optimized electrode materials.19,20 To date, organic materials have been emerging as a possible substitution for inorganic electrode materials due to their intrinsic advantages of low cost and density, earth-abundant, and tunable chemical properties.21,22 Many kinds of organic materials have been explored including carbonyl-based compounds and polymers for Li-ion batteries,23,24 organosulfur compounds and polymers for Li–S batteries,25 and radical-based polymers for both Li-ion and Li–S batteries.26 Among the well-developed organic electrode materials, small organic molecules are widely used, but usually show poor cycling performance due to the dissolution of active materials.27 Applying suitable organic polymers is considered one of the most promising strategies for overcoming the above challenges.28–30 In particular, the use of conducting polymers (CPs) provides opportunities for exploring a new generation of battery devices.31
In simple terms, conducting polymers are polymers that have highly π conjugated polymeric chains. By doping, conducting polymers can achieve conductivity comparable to that of metals or metal semiconductors, but often do not store energy. In addition to the conducting framework, the functional groups involved in the main chain or the side chain which are usually redox-active can largely enrich the electrochemistry of rechargeable batteries. Such polymers are referred to as conducting redox polymers (CRPs). Their good conductivity plus reversible electrochemical doping/de-doping capability largely enriched the family of organic electrode materials and have been growing as encouraging substances for common polymers. Compared to extensive research on CPs (up to 900 papers published per year), an emerging study of CRPs in a battery field is far from sufficient (less than 100 publications per year) (Fig. 1a). Therefore, the rational design and synthesis of CRPs for battery applications is highly desired, which can be inspired by the systematic summary of the recent advances in this field. To date, such a review is still missing.
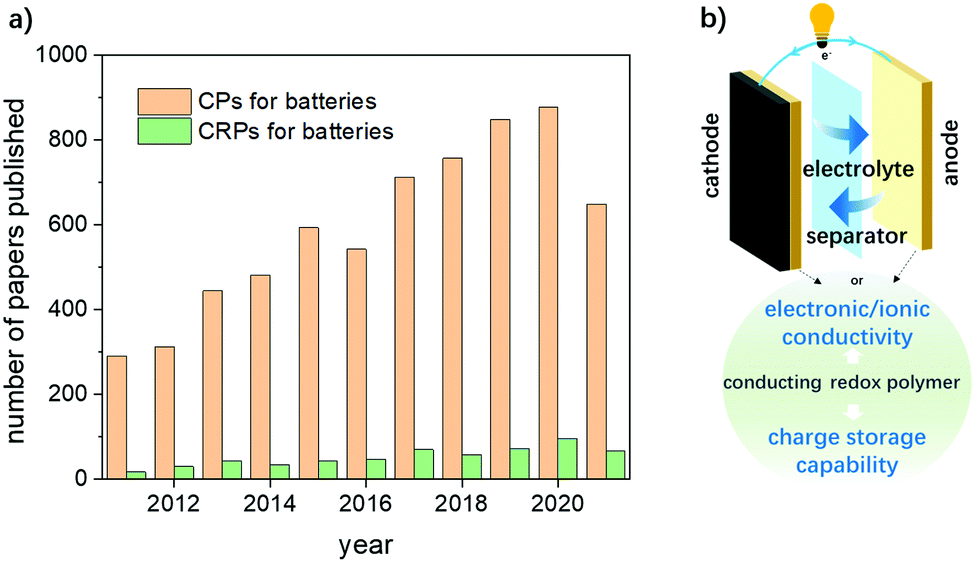 | ||
| Fig. 1 (a) Number of papers (article, review and meeting) published CPs and CRPs for rechargeable batteries during the period of 2011–2021 (10 years). Source: Web of Science database. The data were updated on May 2022. (b) The function of CRPs as organic electrode materials. | ||
The combination of conducting frameworks from CPs and energy storage capability of redox active groups helps in solving at least three major problems of organic electrode materials: the low electronic conductivity, the dissolution of the active material, and the limited charge storage capacity (Fig. 1b). In addition, existing research on organic electrode materials usually involves the use of more conducting agents to improve the poor conductivity of insulating polymers, which can induce additional inactive mass and result in lower energy density of the batteries. The use of conducting polymers can efficiently address such issues taking advantage of their intrinsic good conductivity. Because of the strong designability of the molecular structure, different redox pendant groups with desirable charge storage capacity or shuttle effects inhibiting functions can be designed, which can largely improve the electrochemical performances of batteries. Conventional organic redox active groups (e.g. carbonyls, radicals, and organic sulfur compounds as aforementioned) have been successfully integrated with conducting polymers and applied as efficient organic electrode materials for rechargeable batteries.22,23,25,26,32 By virtue of their good conductivity and high charge storage capacity, CRPs have demonstrated their great potential for practical applications in the future.
In this review, we summarize the applications of conducting polymers with different types of redox-active groups as organic electrode materials based on their synergistic effect of good conductivity and energy storage capability. Rechargeable batteries including but not limited to lithium batteries have been discussed according to the different functions of the applied polymer materials. Additionally, the current challenges, possible solutions, and potential future research directions on such materials for batteries are proposed. Notably, special emphasis will be conducted on CRPs with intrinsic conductivity and well-designed redox active pendant groups. Therefore, extrinsically conducting polymers and metal organic frameworks (MOFs) will not be discussed in detail in spite of extensive studies of this sort.
2. Conducting polymers
Conducting polymers were applied in modern research in 1977. Early attempts to use them for organic batteries as active materials soon emerged, and cyclic conjugated polymers were widely studied, such as poly(aniline), poly(pyrrole), poly(thiophene), poly(p-phenylene), and poly(carbazole).33–36 Through continuous research and development, fruitful results have been achieved in the synthesis and characterization of polymers and their applications in electronics, biology and other fields.37In general, conducting polymers include intrinsically conducting polymers and extrinsically conducting polymers. The conductivity of extrinsically conducting polymers is due to the presence of externally added conducting elements such as carbon or metallic materials. Intrinsically conducting polymers can be further divided into conjugated conducting polymers and doped conducting polymers. Conjugated polymer chains with alternating single and double bonds allow easy movement of delocalized π electrons, and thus can generate conductivity through directional polarization of an external electric field. However, in the intrinsic state stage, its conductivity is usually between an insulator and a semiconductor, and the conductivity needs to be improved by “doping”. In the doping process, the polymers are either oxidized (removal of electrons) or reduced (addition of electrons) so that polymer chains carry the resonating charge. Doped conducting polymers can be further classified as p-doped (oxidative doping) and n-doped types. The oxidation reaction causes the main chain to lose electrons, and the polymer becomes positively charged, attracting anions to bind to the polymer chain and thus remains electrically neutral, which is called p-type doping. In contrast, the reduction reaction makes the main chain negatively charged, attracts cations to bind, and becomes n-type doping. It should be noted that the doping rate of conducting polymers is usually high and their band structure changes greatly before and after doping.
Some of the most widely used conducting polymers involve polyacetylene (PA), polythiophene (PT), poly[3,4-(ethylenedioxy)thiophene] (PEDOT), polypyrrole (PPy), polyphenylene, and polyaniline (PANi). These polymers and their derivatives have been extensively studied in biomedical applications, energy storage systems, and so on. In recent years, a new class of porous materials consisting of organic building blocks via dynamic covalent bonds, namely covalent organic frameworks (COFs), have emerged as promising materials for electrochemical energy storage.38,39 Those with intrinsic good conductivity, which generally arises from the conjugated structure of the organic building blocks, show great potential in applications as organic electrode materials due to their tunable chemistry, tailorable structures, and well-defined pores. With these features, rational design of targeted functionalities can be realized, such as facilitating the penetration of electrolytes and enhancing the ion transport.40–42 Thus, some of the COFs with a well-defined porous structure and good conductivity have attracted increasing interest in the field of electrochemical energy storage.
3. Redox-active groups
In addition to the above-mentioned conducting polymers, there is another polymer that is widely used in the field of electrochemical energy storage, that is, redox polymers.33,43–45 On the one hand, redox polymers are the polymer that contains a redox group in the backbone; on the other hand, there is a class of redox polymers defined as consisting of redox active groups fixed on a non-conducting polymer network.46–51 Moreover, the advantages offered by conducting polymers and redox centers can be combined through designing the redox components to adhere to a conducting polymer backbone. Apart from the redox functional group being able to provide a certain amount of capacity, this combination helps in solving the problem of dissolution of the active substance and low electronic conductivity.Up to now, the intensively studied redox active groups for batteries include carbonyls,52–54 radicals,55,56 and organic sulfur compounds.57 Carbonyl groups are formed by SP2 carbon and oxygen through double bond junctions, exhibiting different theoretical capacities ranging from 120 to 600 mA h g−1 depending on the molecular structure.58 Thus, carbonyl organic polymers have been widely used as anode materials for batteries.26,58,59 For example, quinone and imide-based redox groups are widely used in carbonyl polymer electrode materials.60–64 In addition, it is essential to study the matching of the doping voltage range between redox groups and conducting polymers. Emanuelsson et al.65 covalently connected hydroquinone and dimethoxy-substituted benzoquinone groups to the polypyrrole main chain, respectively, for the battery cathode and anode.
At present, carbonyl materials are divided into three categories according to the mechanism, as shown in Fig. 2.58 In group I, compounds such as 1,2-diones and vicinal carbonyls are used to form stable enolates after reduction. This enolate, like phenanthraquinones, can be further stabilized by the surrounding aromatic systems. Group II includes aromatic carbonyl derivatives. Because the carbonyl groups are closely connected to an aromatic core, delocalization disperses the negative charge. The third collection of compounds (group III) consists of quinone substructures that are identical to those in groups I and II, but their main stabilizing factor is the creation of an additional aromatic system after reduction.
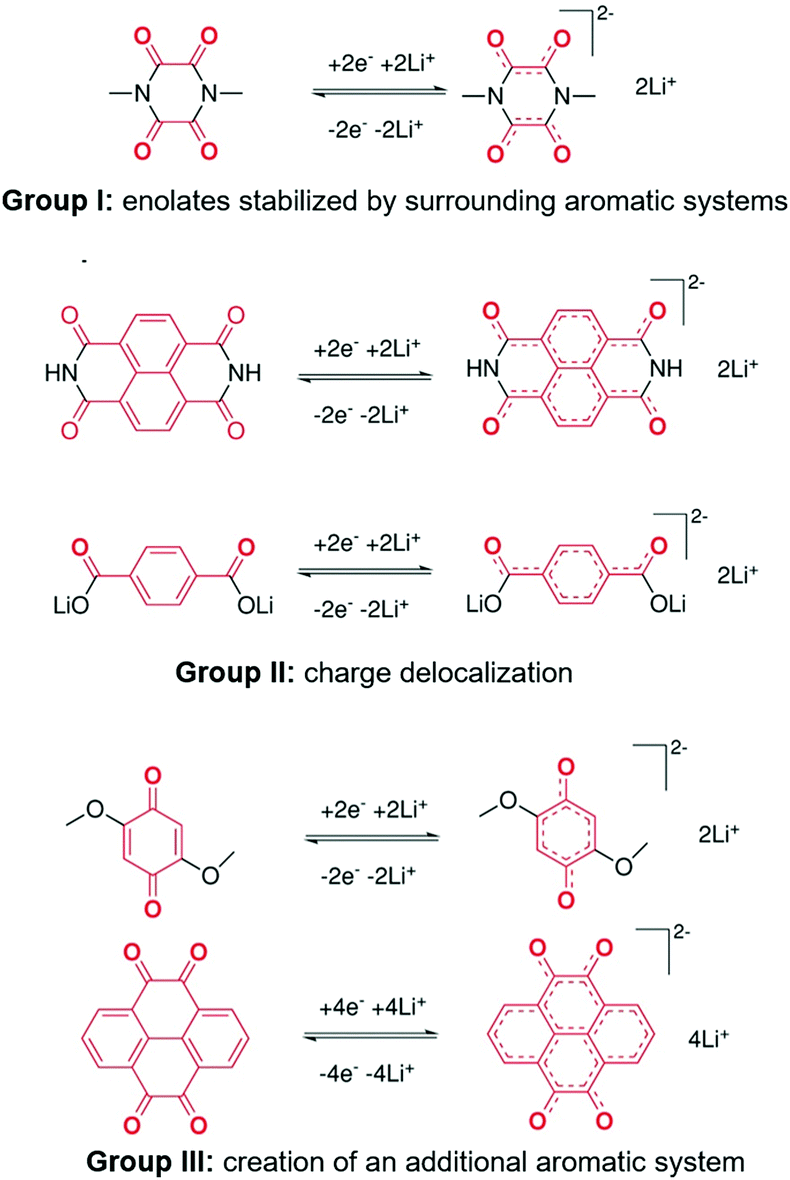 | ||
| Fig. 2 Classification of three carbonyl-based compounds.58 Reprinted with permission from ref. 58 Copyright 2015, Wiley-VCH. | ||
Moreover, the electrochemical stabilization mechanism is derived from the additional aromatic system after the carbonyl unit reduction reaction.66,67 However, aromatic systems with four anions often have high energy configurations, resulting in undesirable side effects and even structural damage and decomposition.23,68,69
In addition to carbonyls, stable organic radical moieties have attracted a lot of attention due to their high charge transfer reactions and the possibility to form n-type and p-type radical polymers depending on the functional pendant group.70,71 In 2002, Nakahara et al. reported the first work on the use of an organic radical for charge storage in lithium batteries.72 Since then, more studies on polymers carrying radicals such as nitroxide have been reported by Nishide et al.55,56,73–75 The most striking features of such nitroxide radical-containing polymers include the efficient electron hopping, which allows a fast electron transfer rate and quantitative redox reactions occurring under a constant electronic potential. Using these organic radical polymers, batteries show fast charge–discharge rates, stable cycling, and high power density.
In previous studies for Li-ion batteries, organic electrodes were prepared by covalently connecting aforementioned redox-active functional groups with a conducting polymer backbone.55,56,76–81 In the field of lithium–sulfur batteries, functional groups such as hydroxyl and amine groups have been used to optimize carbon materials to increase their binding sites to polysulfides for sulfur-fixation through chemisorption.82–86 However, the number of nitrogen atoms and functional groups doped by the carbon material itself is limited. Moreover, the interactions between sulfur/polysulfides and the surfaces of host materials are often interpreted to be weak van der Waals forces, electrostatic adsorption, and Lewis-acid/base interactions.87,88 In contrast, some functional groups can be cross-linked with a S atom by covalent bonds. Through the S–S bond cleavage/dimerization, the disulfide bond is used to provide charge storage. For example, a benzene-free and vinyl-free molecule, azulene, has been reported by Zhuang et al. to form an organosulfur polymer for Li–S batteries.89 Azulene can react with a liquid sulfur diradical chain upon heating due to the high reactivity of azulene. As a result, high sulfur loading has been achieved through inverse vulcanization. Improved cycling stability has been demonstrated due to the formation of the covalent bond between sulfur and azulene. Nevertheless, the kinetics of such a reaction is poor in the case of non-conducting polymers, resulting in considerable anodic and cathodic peak separation.90 In spite of the successful employment of organic sulfur polymers containing S–S bonds in the main chain as organic cathodes, the introduction/connection of more active groups to the conducting polymer main chains are to be further explored.91,92
4. Applications of CRPs in different batteries
4.1 Li-ion batteries
As a popular organic energy storage material, conducting polymers exhibit an intrinsic electronic conductivity when electrochemically doped cations (such as lithium ions and magnesium ions), which contributes to the charge transfer at a higher charge and discharge rate. In addition, the polymers themselves are slightly or insoluble in liquid electrolytes, making conducting polymer electrode materials more stable than small molecule-based electrodes. However, in practical applications, conducting polymer electrodes are faced with the problems of excessive additives and low battery capacity. This is mainly due to its low lithium-ion conductivity and limited functionality, which hinders its practical application in batteries.To date, researchers have been paying increasing attention to the effect of functional groups of conducting polymers on the battery storage performance, including the solubility and processing capacity of the polymer, the electronic and ionic conductivities of the conjugated polymer electrode, and the nature of the electrochemical interaction between lithium ions and the conjugated skeleton/redox-active side groups. Some of the selected examples are summarized in Table 1.
| Polymer name | Molecular structure | Electrode formulation/mass loading | Electrochemical performance | Battery type | Ref. | ||
|---|---|---|---|---|---|---|---|
| Specific capacity (mA h g−1) | Capacity retention (%) | Coulombic efficiency | |||||
| Poly(DTP-TEMPO) |
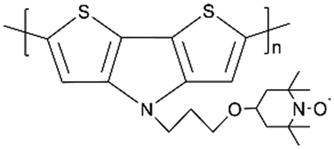
|
N/A | 64.9 (2 μA cm−2) | 70% (200 cycles) | 90% (0.005 mA cm−2) | LIBs | 93 |
| PNDI-T2EG |
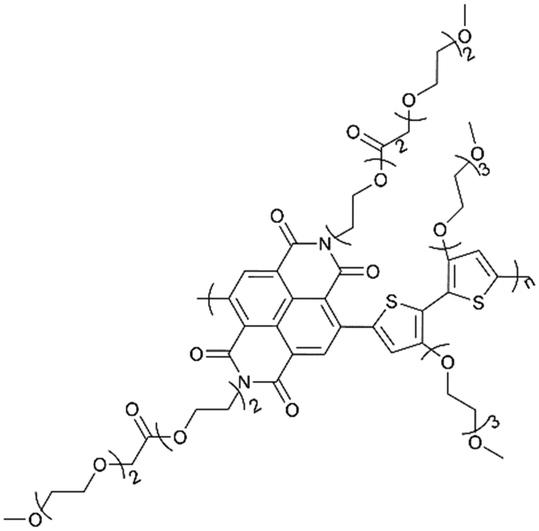
|
60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 (active material:conducting carbon)/1.28 mg cm−2 :40 (active material:conducting carbon)/1.28 mg cm−2 |
34.9 (1 C) | 97.4% (1 C 14 days) | ∼100% (20 C) | 95 | |
| 2DPP-OD-HEX |
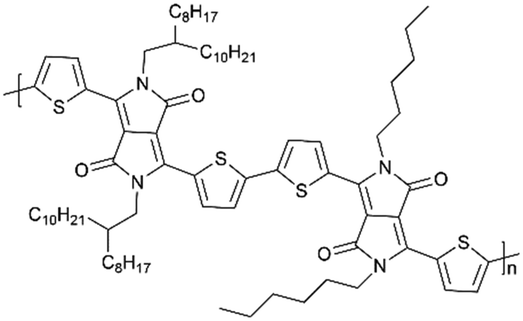
|
60:40 (active material: conducting carbon)/0.3 mg cm−2 |
40 | ∼94% (400 mA g−1 1000 cycles) | 100% (1000 cycles) | 117 | |
| 2DPP-OD-TEG |
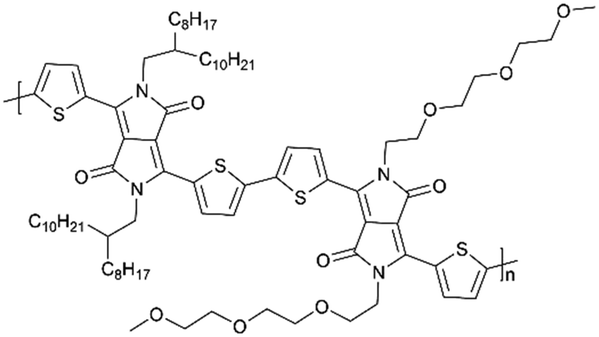
|
35 | ∼90% (400 mA g−1 1000 cycles) | ∼97.5% (1000 cycles) | |||
| Poly-Q(CF3)2-PEDOT |
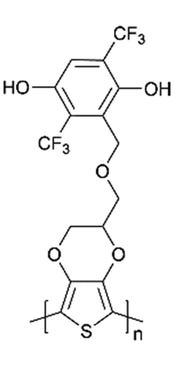
|
Freestanding/0.15 mg cm−2 | 35 | 67% (200 cycles) | N/A | 111 | |
| PDTAn |
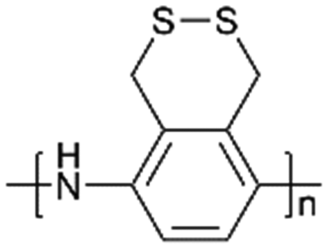
|
40:40:20 (active material:carbon: binder) |
225 | 80% | N/A | LSBs | 137 |
| Carbyne polysulfide |
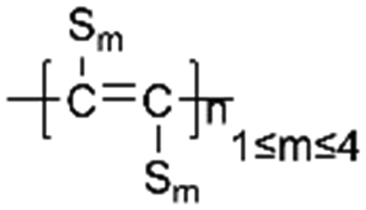
|
70:20:10 (active material:acetylene black:PTFE)/2 mg cm−2 |
960 (0.1 C 200 cycles) | 96% | N/A | 139 | |
| PANI |
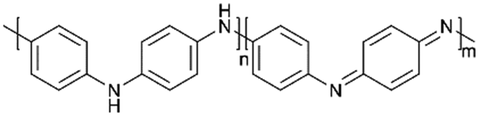
|
80:10:10 (active composite (1:4):super P:PVDF) |
837 (0.1 C) | 76% (1 C, 500 cycles) | >90% | 144 | |
| S-P3HT |
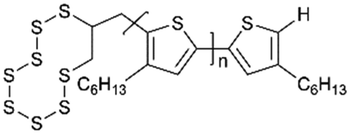
|
70:25:5 (S:(P3HT + conducting carbon):polyethylene)/1 mg cm−2 |
739.41 (1 C) | ∼70% (0.5 C 100 cycles) | >90% | 140 | |
| poly(2-vinyl,1,4-phenylene sulfide) |
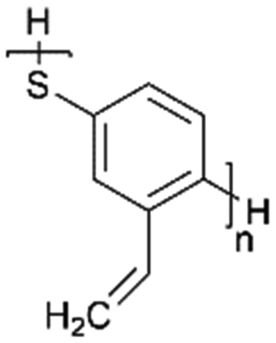
|
N/A | 413 (1 C) | N/A | N/A | 141 | |
| S-PAMT |
|
80:10:10 (cp(S-PMAT):PVDF:conducting carbon black)/1.5 mg cm−2 |
1240 (0.1 C) | 66.9% (2 C, 1000 cycles) | ∼100% (1000 cycles) | 142 | |
| PTFHQS |
|
70:20:10 (organosulfur copolymer: acetylene black:PVDF)/2 mg cm−2 |
906 (0.5 C) | 87% (0.5 C, 600 cycles) | 99% | 143 | |
| pEP(NQ)E |
|
10 mg cm−2 | 76 (anode) | 99% (1 C, 50 cycles) | 99% (1 C, 50 cycles) | Aqueous Battery | 21 |
| N/A | 76 | 98% (100 cycles) | 100% | Polymer–air secondary battery | 178 | ||
| Poly(NQ-EPE) |
|
N/A | 62 (4.5 C) | 80% (4.5 C, 500 cycles) | ∼100% | All-organic proton battery | 179 |
| Poly(Q2H2-EPE) |
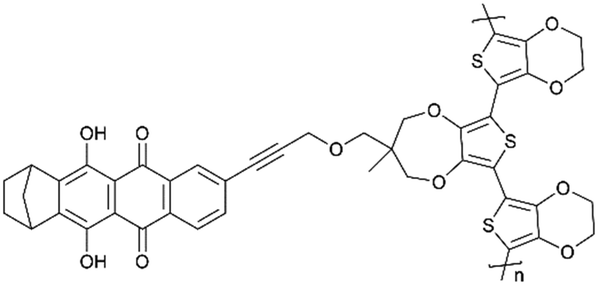
|
N/A | |||||
:2,3-d]pyrrole (DTP) moieties, it had a two-step energy storage mechanism, in which poly(DTP) was oxidized before TEMPO radicals were oxidized (Fig. 3b). This polymer had a discharge capacity of 64.9 mA h g−1 at 2 μA cm−2, which is 47.4% of its theoretical capacity. In comparison with poly(DTP) without the radical, this performance was poor. This occurrence demonstrated that an internal electron transfer occurred between two redox groups. The conducting polymer backbone transferred an electron to a free radical upon charging, rendering the polymer inactive. Furthermore, this behavior had resulted in a decrease in the Coulombic efficiency.
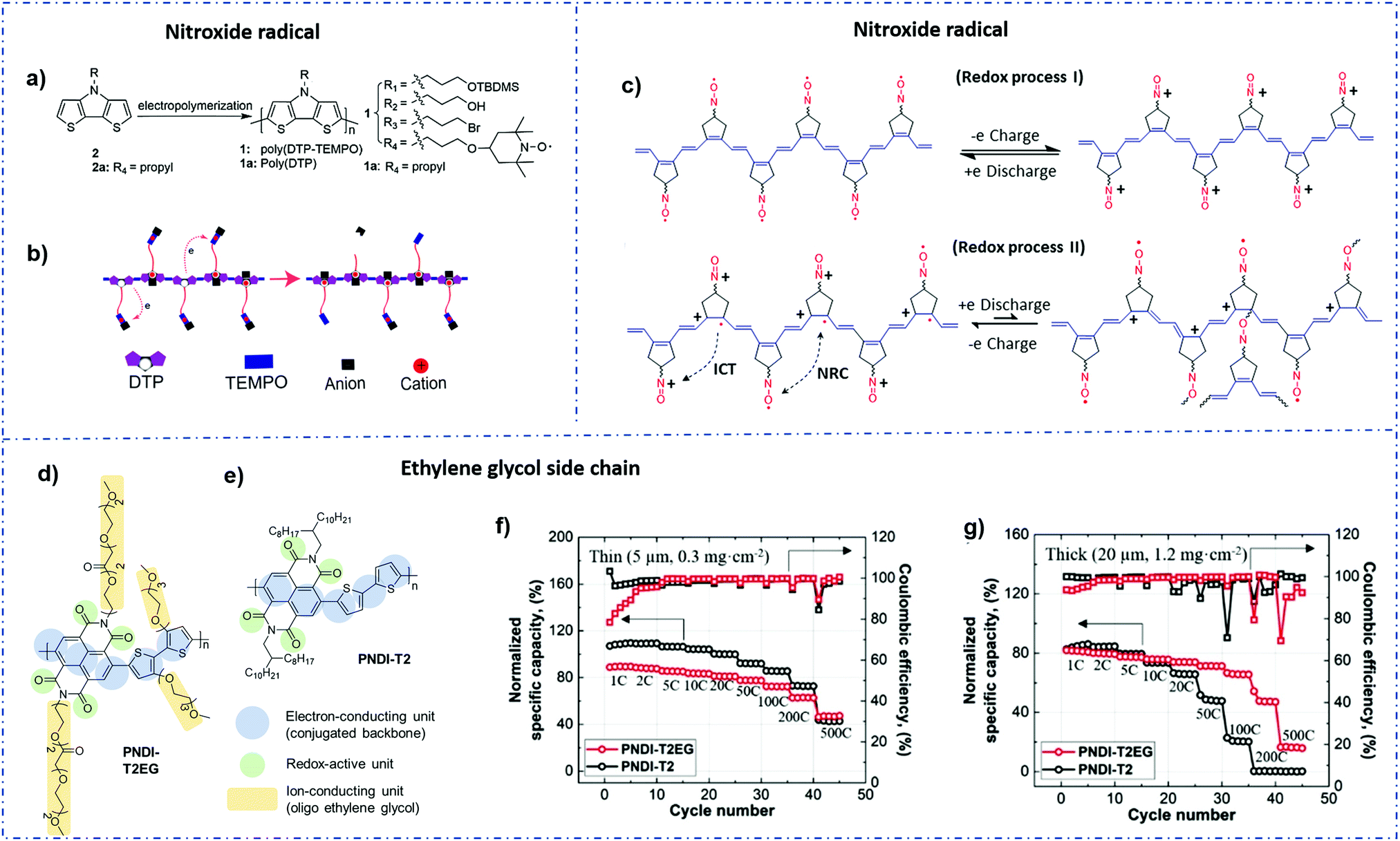 | ||
| Fig. 3 (a) Synthesis of poly(DTP-TEMPO). (b) Schematic illustration of internal electron transfer during the OCP relaxation process.93 Reprinted with permission from ref. 93 Copyright 2018, the American Chemical Society. (c) Schematic illustration of the charge transfer mechanism at high current densities and narrow potential windows.94 Reprinted with permission from ref. 94 Copyright 2019, the American Chemical Society. (d and e) Chemical structures of PNDI-T2 and PNDI-T2EG and the structural functionalities. (f and g) Normalized specific capacity and Coulombic efficiency with an increasing cycle number for thin (5 μm, 0.32 mg cm−2) and thick (20 μm, 1.28 mg cm−2) PNDI-T2EG and PNDI-T2 electrodes composed of the 60 wt% polymer.95 Reprinted with permission from ref. 95 Copyright 2021, Wiley-VCH. | ||
Despite the fact that nitroxide radical conjugated polymers (NCPs) have long been investigated in the field of electrochemical energy storage, their practical application in electrodes is still limited due to their challenging synthesis and complex charge transfer and storage mechanism.55,103 Xie et al.94 found that the coulombic efficiency can be significantly affected by the different redox kinetics between the nitrioxide radicals and the polyacetylene backbone due to the side reaction between the free radical and the backbone (Fig. 3c). For example, when such polymers are charged at low current densities, the nitrogen oxide radical and the conjugate backbone are oxidized to ammonium oxide ion and polaron, respectively. Nevertheless, when the electrode was exposed to a rapid charge and discharge (i.e., a high C-rate) within a small potential window, the internal electron transfer was reduced and a high Coulombic efficiency was obtained.
:1 v/v DOL/DME as an electrolyte. When the polymer electrode thickness is approximately 20 microns, it has a load capacity of 1.28 mg cm−2 and is capable of achieving a theoretical specific capacity of 66% at an ultrahigh test rate of 100 C. In addition, when the polymer content was increased to 80%wt, the theoretical specific capacity was 68% at 10 C and 55% at 12 C (Fig. 3d–g). However, the effect of the EG side chain content on the electrochemical properties of NDI polymer materials still needs further study.
However, the formal potential of the quinone group needs to be improved due to its low potential when combined with lithium anode materials since it is required to be compatible with lithium-based electrolytes. Fortunately, the electron-withdrawing substitutions on the quinone ring had been found to be effective in achieving this.110 Wang et al.111 created a series of quinone-based conducting redox polymers by substituting quinone pendant groups (PGs) with electron-withdrawing substituents (Fig. 4a). As predicted, the addition of electron-withdrawing substituents resulted in an increase in the quinone formal potential. Nevertheless, in situ measurements revealed that the onset potential for the polymer backbone conductance was affected by the PG redox chemistry, where a reduced and lithiated pendant caused structural distortion. An assembly of this material was used in Li-ion batteries as cathode materials, and the reversible voltage output was between 2.8 and 3.2 V with a 67% capacity retention after 200 cycles.
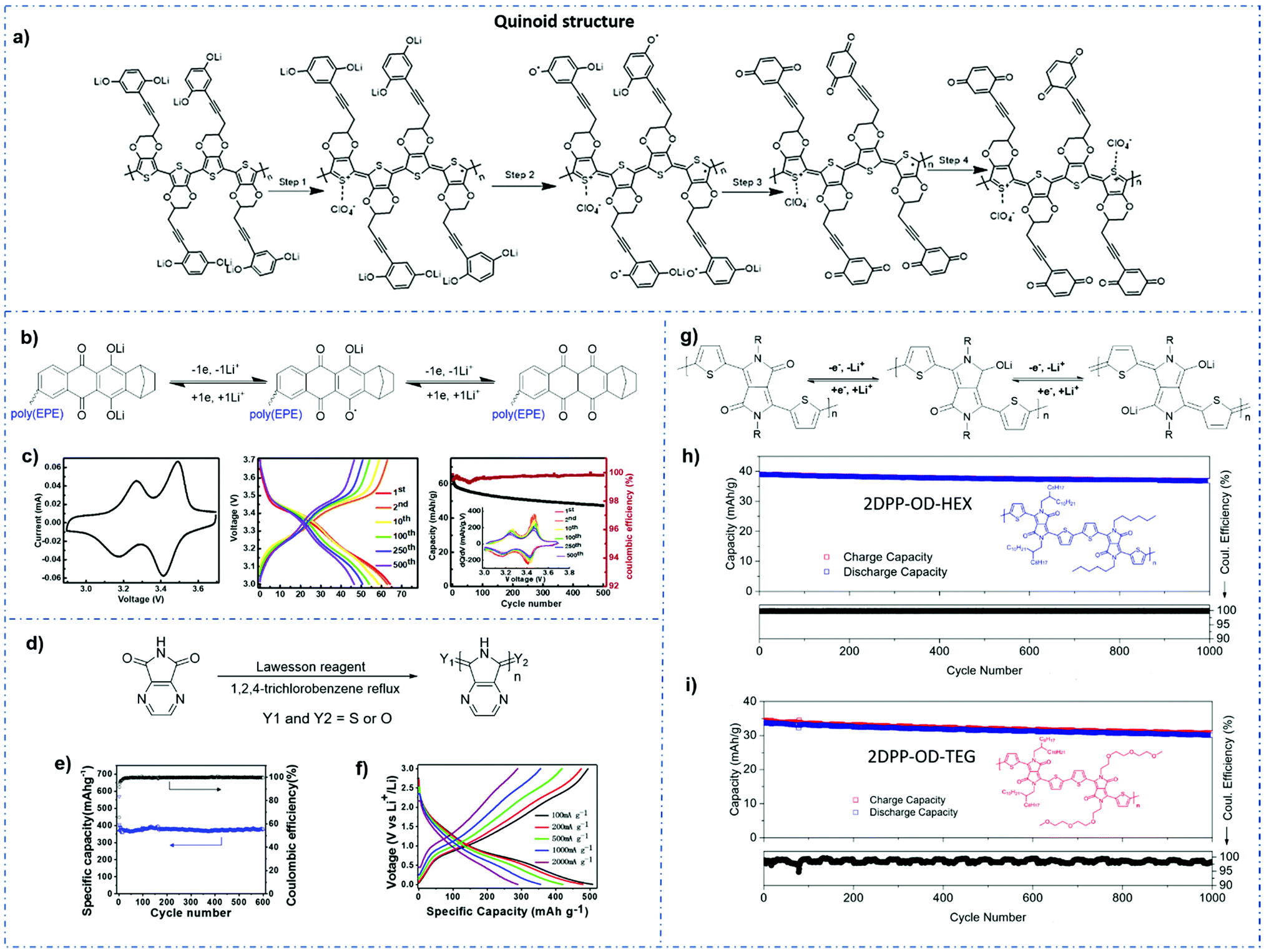 | ||
| Fig. 4 (a) The redox process occurring in Q-PEDOT in the LiClO4/MeCN electrolyte.111 Reprinted with permission from ref. 111 Copyright 2019, the American Chemical Society. (b) Redox process of poly (Qz-EPE) in Li+-based electrolytes. Battery evaluation in the 1 M LiPF6/EC/DEC electrolyte with poly (Qz-EPE) as the cathode. (c) Eelctrochemical performances of the batteries.115 Reprinted with permission from ref. 115 Copyright 2021, Elsevier. (d) Structure of PDAPI. (e and f) Electrochemical performances of the batteries.116 Reprinted with permission from ref. 116 Copyright 2020, the American Chemical Society. (g) The proposed electrochemical redox reactions of 2DPP-OD-HEX. (h and i) Charge–discharge capacities of 2DPP-OD-HEX and 2DPP-OD-TEG over 1000 cycles.117 Reprinted with permission from ref. 117 Copyright 2021, the American Chemical Society. | ||
It is worth noting that the polycyclic aromatic substitution was found to produce considerably greater alterations in the formal potential.112–114 In their further study,115 quinizarin (Qz) was functionalized onto a thiophene-based trimer backbone, and a norbornane group was added to the quinone ring to protect the susceptible locations near the redox-active carbonyl groups on Qz. The results showed an average discharge voltage of 3.3 V, a discharge capacity of 65 mA h g−1 at 1.5 C and a capacity retention of 74% after 500 cycles (Fig. 4b and c).
Duan et al.116 synthesized a conducting polymer polydiazaphthalimide (PDAPI) with a quinoline structure by a simple one-step process containing three different end groups, i.e., two carbonyls, one carbonyl and one thiocarbonyl, or two thiocarbonyls. Li foil was applied as the anode and 1 M LiPF6 in EC/DEC (1/1, w/w) was used as the electrolyte. The presence of these end groups and unsaturated nitrogen groups promotes efficient lithiation/delithiation processes (Fig. 4d–f). The quinoline structure also provides excellent electrical conductivity to the polymer and preserves the polymer from being dissolved by the electrolyte.
Samuel et al.117 developed two donor–acceptor polymers, each with a conjugated backbone made up of repeating units containing diketopyrrolopyrrole (DPP) and thiophene (T) groups. The Li anode and 1 M LiClO4 in 1:1 (v/v) DOL/DME as the electrolyte were applied in this work. They revealed that, during a two-step redox process, the oxygen atoms of the carbonyl group on DPP as redox-active sites coordinate with the Li ion (Fig. 4g). Furthermore, the bonded ring structure of DPP provided the polymer with backbone coplanarity, which aided in increased interchain packing and stronger contacts, hence facilitating the intermolecular charge transfer with high electron mobilities.118 Notably, triethylene glycol (TEG) side chains increase the polymer backbone's packing and crystallinity, resulting in greater carrier mobilities than hexyl (HEX).119 Li-ion batteries based on DPP-based polymers demonstrated a reliable cycling up to 1000 cycles, strong rate performance with 70% capacity retention at a C-rate of 500 C, and relatively high potentials of 2.2 V versus Li+/Li (Fig. 4h and i). Moreover, these polymers were shown to be used as cathode materials in other types of ion insertion batteries, such as Na-ion batteries.
Additionally, the design and synthesis of porous materials has been a new strength to address the natural challenge of these materials. For example, as a mixed ion–electron conductor, poly(3,4-ethylenedioxythiophene)-poly(styrenesulfonate)(PEDOT:PSS) is considered to be a conducting material candidate due to its aqueous-based processability, good conductivity, and excellent hybridization ability with other materials.126–128 However, most researchers have only focused on improving the electrical conductivity of the PEDOT:PSS film.129
Youngsang Ko et al.130 fabricated the PEDOT:PSS/cellulose nanofiber (CNF) nanocomposite paper with excellent flexibility through post-treatment with an organic solvent. The CV results indicated a faradaic redox reaction due to the redox activity of PEDOT:PSS. In addition, excellent capacitance retention after 500 charge–discharge cycles showed that the nanocomposite paper after post-treatment may be used in applications requiring electrochemical energy storage ability.
The exploration of bifunctional redox polymers as cathode and anode materials has become a practical strategy to develop symmetric polymer batteries, which has been pursued as a promising sustainable energy storage technology.60 Mecerreyes et al. developed dual redox-active polyimides based on phenothiazine and naphthalene tetracarboxylic dianhydride units for symmetric all-organic batteries.131 The importance of phenothiazine for faster charge–discharge processes has been revealed. When applied as the anode and cathode, phenothiazine functional polyimide shows two redox active voltages at around 2.5 and 3.7 V (vs. Li/Li+) with a high discharge capacity of 160 mA h g−1. This all-polymer battery delivered an outstanding power density (1542 W kg−1) and an excellent cycling stability (94% of the capacity retention after 1000 cycles at 800 mA g−1).
Xue et al.132 combined theory and experiment to argue the effect of the structural design of multiple pyrene-functionalized radical copolymers (2,2,6,6-tetramethyl-1-piperidinyloxy, TEMPO) to improve the battery performance. The coin cell (CR2032) was fabricated by stacking the polymer-based cathode, the lithium metal anode and a separator film with the LP30 electrolyte. Compared to methylene-ester linkages, amide-bonded pyrene containing polymers indicated relatively table redox peaks at around 3.55 V vs. Li/Li+. Additionally, the latter copolymer has a higher π–π interaction with CNTs than the pyrene–TEMPO copolymers with methylene-ester linkages, which improve the battery performance.
4.2 Li–S batteries
Lithium–sulfur batteries (LSBs) have become one of the most attractive new generation of energy storage devices with their theoretical specific capacity and energy density far exceeding those of traditional lithium batteries. However, the insulation of active substances, the shuttle effect of lithium sulfide in the charging and discharging process and the volume expansion of sulfur have always affected the practical use of lithium–sulfur batteries.To overcome the above obstacles, a common strategy is to design suitable cathode materials. Organic polymers with strong designability, diverse molecular structures and rich resources have been widely concerned.
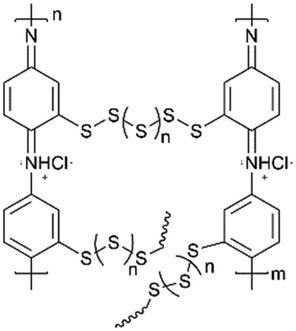
Li et al.137 researched an aniline-based polyorganodisulfide (PDTAn) containing an S–S bond on the side chain as cathode materials (Fig. 5a). The polymer backbone was similar to conducting polyaniline (PANI), which possessed high electronic conductivity with fast redox kinetics. As expected, the redox reaction of the S–S bond did not cause the depolymerization of the polyaniline chain, and showed a discharge capacity of 225 mA h g−1 at 10 mA g−1 and a Coulombic efficiency of more than 80% in the following cycles. However, there might be a bulky condensed aliphatic heterocyclic substituent or the existence of an oligomer in the polymers, leading to the fact that the electronic conductivity was poorer than their expectation.
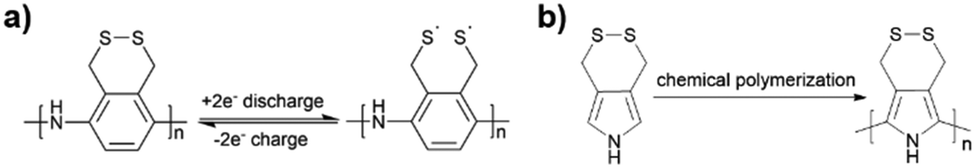 | ||
| Fig. 5 (a) The mechanism of the discharge–charge process of the polymer PDTAn.137 Reprinted with permission from ref. 137 Copyright 2004, Elsevier. (b) The chemical polymerization of the PPy derivative with the disulphide bond.138 Reprinted with permission from ref. 138 Copyright 2006, Elsevier. | ||
In previous studies, Amaike et al.138 developed a novel polypyrrole (PPy) derivative in which the cyclic disulfide bond is covalently attached to the polymer backbone (Fig. 5b), accompanied by a redox process for reversible open-loop cracking–recombination. The good electrical conductivity of this structure and the polypyrrole skeleton itself allows the polymer to exhibit better electrochemical activity and conductivity, with a theoretical capacity of 398 mA h g−1.
Oschmann et al.140 first reported on the copolymertization of sulfur and allyl-terminated poly (3-hexylthiophene-2,5-diyl) (P3HT) derived by the conversion of sulfur radicals formed by the thermolytic cleavage of S8 rings with an allyl end-group. The rapid chemical interaction between polythiophene and lithium polysulfides (LIPSs) from the covalent linking resulted in the rapid charge transfer and smaller loss of active materials (Fig. 6a).
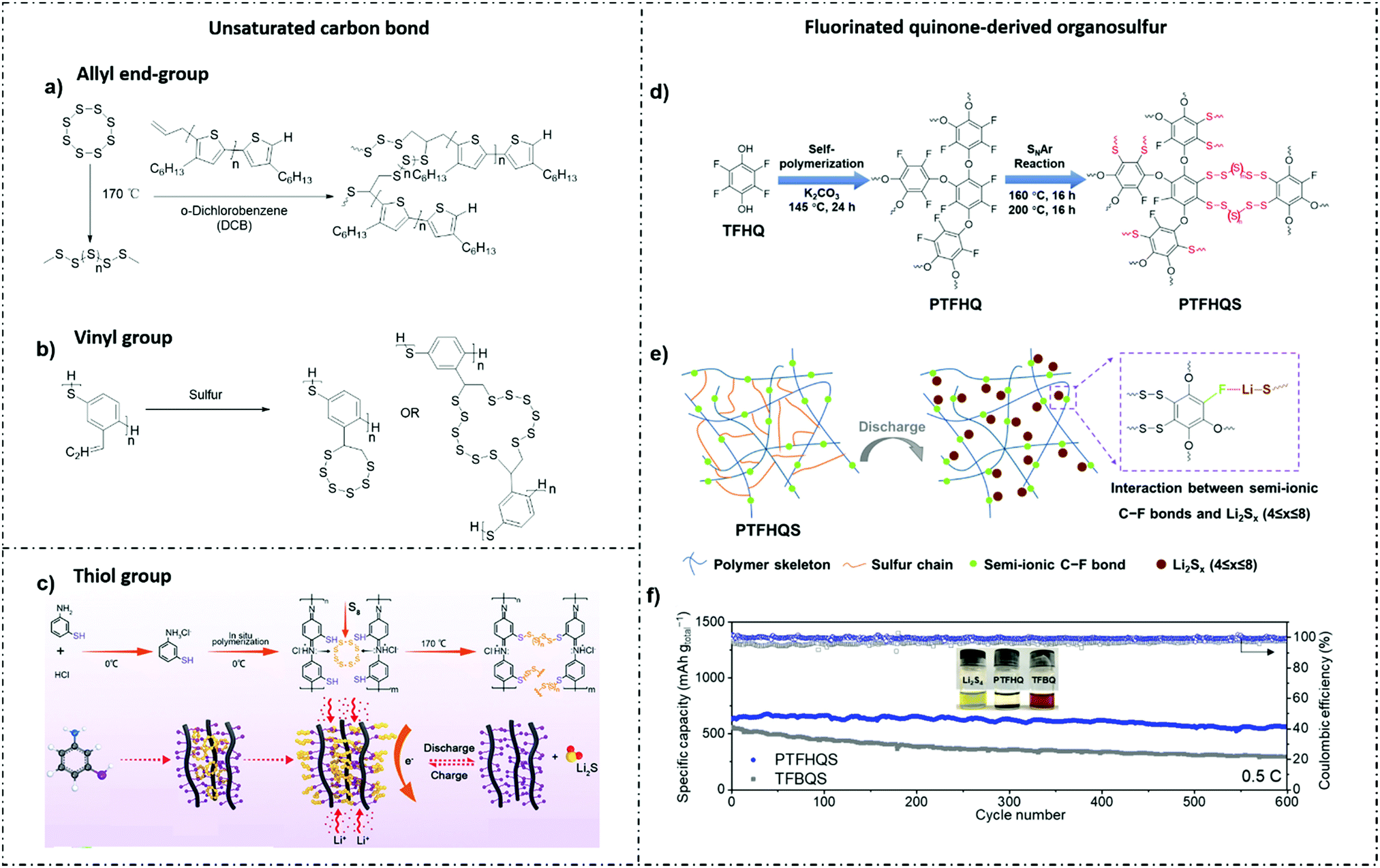 | ||
| Fig. 6 (a) Synthetic approach for the copolymerization of allyl-terminated P3HT and sulfur.140 Reprinted with permission from ref. 140 Copyright 2015, the American Chemical Society. (b) Schematic of the inverse vulcanization of S into the poly(1,4-phenylene sulfide) polymer via addition reactions.141 Reprinted with permission from ref. 141 Copyright 2020, The Royal Society of Chemistry. (c) The synthesis of the cp(S-PMAT) copolymer and the structural evolution in the synthesis of cp(S-PMAT) and the discharge–charge process.142 Reprinted with permission from ref. 142 Copyright 2020, European Chemical Societies Publishing. (d) TFHQ is first self-polymerized into PTFHQ and then covalently linked to sulfur via the SNAr reaction. The as-obtained copolymer is referred to as PTFHQS. (e) Li⋯F interaction between Li2Sx intermediates and the PTFHQS polymer skeleton with abundant semi-ionic C–F bonds. (f) Long-term cycling performance of the PTFHQS and TFBQS cathodes at 0.5 C. The inset shows the comparison test of the visual Li2S4 adsorption of PTFHQ and TFBQ monomers.143 Reprinted with permission from ref. 143 Copyright 2021, Elsevier. | ||
Gao et al.141 modified the conducting poly(1,4-phenylene sulfide) via the vinyl group to improve the ability to vulcanize with element S. The unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds were the most favorable sites for vulcanization via crosslinking. Compared with the condensation polymer, poly(2-vinyl-1,4-phenylene sulfide) via crosslinking showed better conductivity, higher specific capacity and better kinetic stability as LSB cathodes. The maximum specific capacity and gravimetric energy density for crosslinking polymers were 413 mA h g−1 and 626 Wh kg−1, respectively (for the fully vulcanized crosslinking configuration) (Fig. 6b).
C bonds were the most favorable sites for vulcanization via crosslinking. Compared with the condensation polymer, poly(2-vinyl-1,4-phenylene sulfide) via crosslinking showed better conductivity, higher specific capacity and better kinetic stability as LSB cathodes. The maximum specific capacity and gravimetric energy density for crosslinking polymers were 413 mA h g−1 and 626 Wh kg−1, respectively (for the fully vulcanized crosslinking configuration) (Fig. 6b).
Although many studies have found that sulfur-rich copolymers can be formed by thermally activated free radical polymerization of sulfur with polymerizable monomers or polymers with reactive groups, most of these copolymers are insulating or less conducting, which is not desired to improve the rate capacity.140,146–151 Zeng et al.121–142 uniformly polymerized the thiol functional group with m-aminophenol, and then by reverse sulfurization copolymerization with elemental sulfur to form the conducting functional polymer CP S-poly(m-aminothiophenol) (S-PMAT), which was used as the cathode material of LSBs, and tested under an active load condition of 1.5 mg cm−2. The results show that the reversible capacities at 0.1 C, 1 C and 5 C are 1240 mA h g−1, 880 mA h g−1 and 600 mA h g−1, respectively. Even after charging and discharging 1000 times at 2 C, the capacity retention rate is 66.9%, and the Coulombic efficiency is reduced only 0.04%. The results provide a feasible example for designing conducting polymers with reactive functional groups as effective electrode materials for high-performance LSBs (Fig. 6c).
:1 (v/v) DOL/DME as the electrolyte), the sulfur–polyaniline nanotubes (SPANI-NT/S) had a high sulfur content of 62 wt%. Benefiting from the flexible framework of SPANI and the electropositive amine and imine groups, the sulfur was both physically confined in the 3D network and chemically anchored to molecular chains. Besides the obviously inhibited shuttle effect, the SPANI-NT/S performed a stable capacity of 837 mA h g−1 at 0.1 C after 100 cycles due to the high electronic conductivity of the polyaniline skeleton.
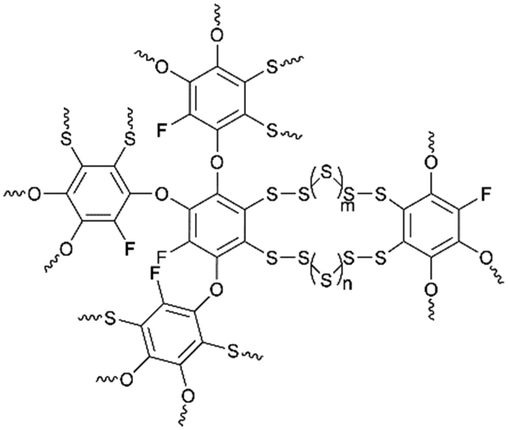
Unexpectedly, some conventional organosulfur copolymers are subjected to poor electronic/ionic conductivity and low sulfur content, which resulted in lower rate capability and energy density. To this end, exploring new organosulfur copolymers with rational molecular structures and functional groups becomes rapidly desirable. Yan et al.143 reported fluorinated quinone-derived organosulfur copolymers poly(tetrafluorohydroquinone)–sulfur (PTFHQ) with high sulfur content as cathode materials for LSBs. Benefiting from the homogeneously distributed sulfur in the copolymers at the molecular level and the abundant semi-ionic C–F bonds in the TFHQS skeleton, the shuttle effect of polysulfides was effectively suppressed. Meanwhile, the PTFHQ backbone showed higher electronic conductivity than the monomer, and increased the Li ion transportation, which was helpful in ensuring higher rate capability and sulfur utilization. As a result, the PTFHQS cathode with the 71 wt% sulfur content delivered a high discharge capacity of 906 mA h g−1 at 0.5 C, a stable Coulombic efficiency of over 99%, a good rate performance of 432 mA h g−1 at 2.0 C and an excellent capacity retention of 87% at 0.5 C after 600 cycles (Fig. 6d–f).
According to the previous work, conventional conducting polymers can hardly produce long-term stable cycling due to the lack of a polar functional group to provide strong chemical absorbing with polar LiPSs.152,153 Moreover, the undesired porous structure (i.e. irregular pore size distribution) and low specific surface area made weak physical confinement. Furthermore, the strict and changeable conditions in the electrochemical system required higher stability of conducting polymers.154–157 Combining the microporous structure with the conducing polymer is a theoretically ideal strategy to address these challenges.123,158,159 Chen et al.160 developed a covalent organic framework (COF)-linked conjugated microporous polyaniline(CMPA) serving for fixing sulfur in a carbon nanofiber (CNF) current collector to suppress the shuttle effect for Li–S batteries. Benefiting from the N-rich systems provide an excellent polar group, CMPA showed stronger affinity towards LiPSs. To date, bulk COFs with functional groups (e.g., N and B atoms, vinyl groups, and fluorinated groups) have been explored as the sulfur host for Li–S batteries. Such COF-based cathodes not only can physically confine LiPSs by well-defined pores, but also their pore surface can interact with LiPSs through sulfiphilic and/or lithiophilic interactions. Therefore, the soluble LiPSs can be effectively restrained in the cathode region, providing the high utilization of sulfur and superior cycling stability. In spite of the abovementioned advantages, bulk COFs usually show limited surface utilization, which hinders the great potential of COFs as the efficient sulfur host for practical applications. Recently, Ning et al.161 developed polyimide COFs (PI-COFs) with a well-defined lamellar structure, which can be exfoliated into ultrathin (∼1.2 nm) 2D polyimide nanosheets (PI-CONs) with a large size (∼6 μm) and large quantity (40 mg per batch). Being explored as new sulfur host materials for LSBs, PI-COF and PI-CONs deliver high capacities (1330 and 1205 mA h g–1 at 0.1 C, respectively), excellent rate capabilities (620 and 503 mA h g−1 at 4 C, respectively), and a superior cycling stability (96% capacity retention at 0.2 C for PI-CONs) by virtue of the synergy of robust conjugated porous frameworks and strong oxygen–lithium interactions.
4.3 Aqueous batteries
Low-cost and sustainable aqueous batteries are believed to overcome the limitations of the traditional organic electrolyte in virtue of the safety and environmentally friendly nature of aqueous electrolyte. Significant efforts have been made to achieve high energy density, high power density, and a long cycle life. Compared to inorganic materials (e.g. vanadium-based compounds), organic material-based electrodes have attracted increasing interest due to their rich redox chemistry and relatively low cost. In this section, aqueous organic batteries including but not limited to monovalent-ion batteries have been summarized. A series of the recently reported redox-active organic materials, mainly conducting or conjugated polymers are included combing the material design with the explanation of different working mechanisms.4.3.1.1 Imide groups. As discussed in the above sections, carbonyl-containing polymers are intensively investigated as redox-active n-type organic electrode materials for Li-ion batteries. Among the various structures, polyimide (PI) has more suitable working voltage, higher capacity and better structure stability, therefore attracting additional attention beyond organic lithium batteries.162–166 Qin et al.162 successfully demonstrated aqueous Li- and Na-ion batteries using 1,4,5,8-naphthalenetetracarboxylic dianhydride (NTCDA)- and perylene-3,4,9,10-tetracarboxylic dianhydride (PTCDA)-derived PI anodes based on their good redox activity and structural diversity on the insertion of different ions. LiCoO2 and NaVPO4F were applied as the cathodes for lithium and sodium batteries, respectively. A full aqueous Li-ion battery with an average operating voltage of 1.12 V and a specific capacity of 71 mA h g−1 was developed with a cycle life of 200 cycles. The aqueous Na-ion battery employing the same PI-based anode and NaVPO4F cathode delivered a moderate specific capacity of ∼40 mA h g−1.
On the basis that aqueous batteries rely on water as the electrolyte solvent, additional requirements are proposed to organic electrode materials beyond the redox activity and conductivity, which is the hydrophilic properties when compared to those used for organic batteries. Ethylene glycol groups have been thus applied to improve the ion diffusion of the aqueous electrolyte. Davide Moia et al.163 synthesized a 3,30-dialkoxybithiophene homo-polymer (p-type polymer) with ethylene glycol side chains and attached the polar zwitterion side chains to the naphthalene-1,4,5,8-tetracarboxylic-diimide-dialkoxybithiophene (NDI-gT2) copolymers. The n-type polymer was applied as the anode and the p-type polymer as the cathode with a 0.1 M NaCl aqueous solution as the supporting electrolyte. The polymer films demonstrate the high reversibility redox reaction in aqueous electrolytes. Furthermore, the thin films of these polymers combined in two-electrode batteries showed a reversible charging up to 1.4 V. (Fig. 7a)
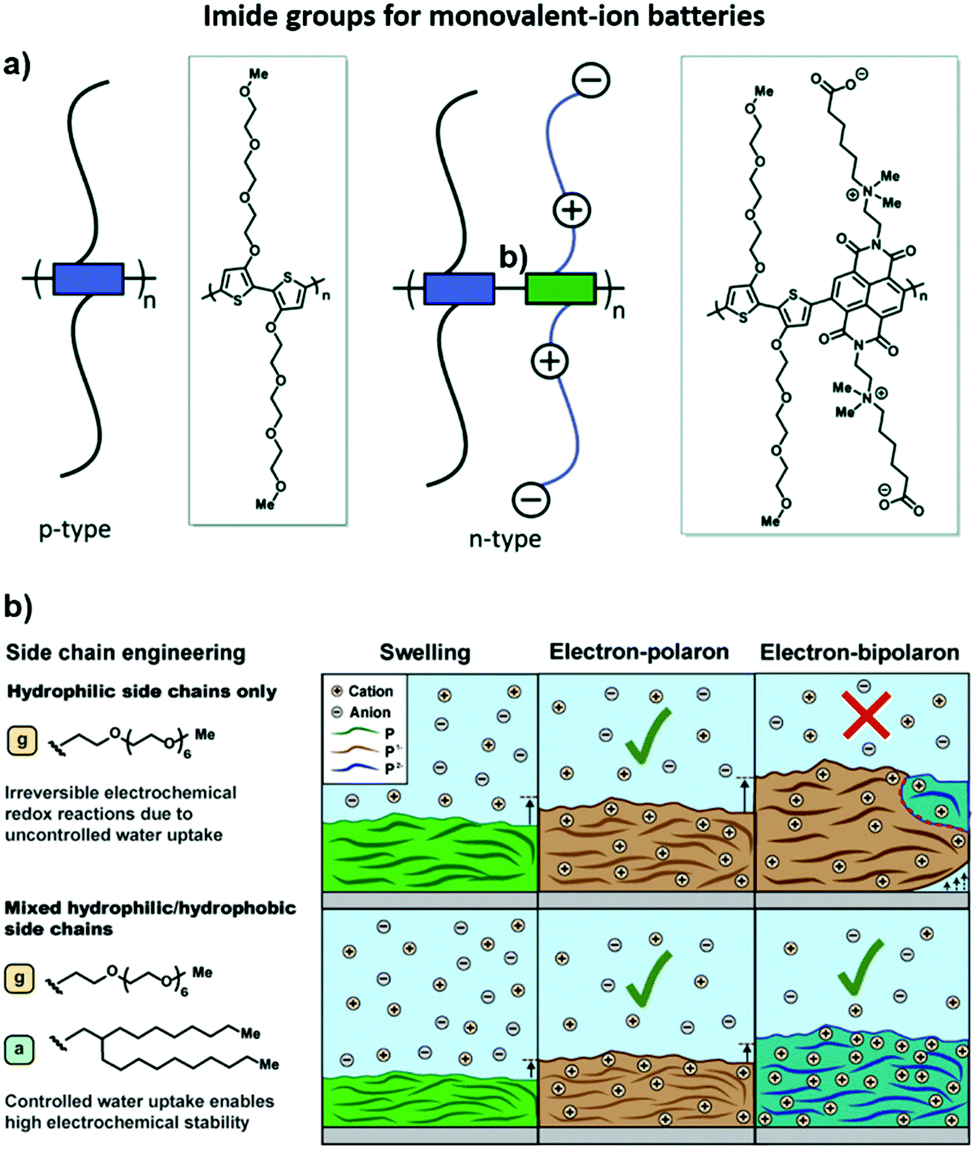 | ||
| Fig. 7 (a) p-Type homo-polymer with TEG side chains p(gT2) and n-type donor–acceptor copolymer with zwitterion side chains on the acceptor units (marked as green blocks in the schematic) and TEG side chains on the donor units (blue blocks) p(ZI-NDI-gT2).163 Reprinted with permission from ref. 163 Copyright 2019, The Royal Society of Chemistry. (b) Illustration of the charging mechanism of redox-active polymers with controlled water uptake (e.g., polymers with mixed hydrophilic/hydrophobic side chains) and uncontrolled water uptake (e.g., polymers with hydrophilic side chains) to the electron–polaron and electron bipolaronic states. The swelling of the polymer film is illustrated by the thickness of the polymer chain, where the color change represents the charging of the neutral polymer (green) to the electron polaronic (brown) and electron bipolaronic (blue) states. Solid-lined arrows represent the swelling of the polymer electrode, and dashed-line arrows indicate the detachment of the swollen or charged polymer from the current collector. The red dashed line illustrates mechanical stress that results in the detachment of the polymer.164 Reprinted with permission from ref. 164 Copyright 2021, the American Chemical Society. | ||
Noteworthily, the excellent hydrophilicity of the side chain easily forms hydrogen bonds with aqueous molecules, which may cause excessive aqueous absorption and damage the microstructure of the polymer, resulting in the decline of the electrochemical performance. Then, Anna A. Szumska et al.164 attempted to replace some hydrophilic (glycol) side chains with hydrophobic(alkyl) groups. When excessive aqueous absorption was inhibited, the swelling of the polymer disappeared, and the reversible double reduction and dipolarity sate appeared during electrochemical charging, which increased the actual capacity to more than 70% of the theoretical capacity. Therefore, the introduction of functional groups can affect the electrochemical performance of the polymer under the condition of affecting the local environment (Fig. 7b).
Based on the previous studies, Tan et al.165 researched the recyclable energy storage device with redox-active conjugated polymers as the electrode. A conjugated polymer based on NDI and T2 units was employed as the anode, while, for the cathode, a conjugated homopolymer based on g3T2 was used. The hydrophilic side chains based on ethylene glycol not only can promote fast ion transport from the aqueous electrolyte into the electrode, but also can enable high solubility in organic solvents for reclaiming electrode materials from solution. Furthermore, to increase the electrochemical stability of redox-active polymers, 25% of hydrophobic side chains were linked to the polymer backbone due to the lengthy hydrophilic side chains in the anode.
4.3.1.2 Quinone pedant group. Quinone pendant groups providing high potentials, for example, catechol or dihydroxyanthraquinones, would make better use of the stability window of water. The capacity could be improved by optimizing the linkage unit and increasing the number of redox groups per repeat unit in the polymer. For aqueous systems, such a quinone group recently has been developed for all-organic proton batteries without any carbon additive and binder by Strietzel et al.167 In this study, conjugated polymers pEP(NQ)E and pEP(QH2)E with both the conducting thiophene backbone and redox-active quinone pedant group were designed as the anode and cathode, respectively. (E = 3,4-ethylenedioxythiophene; NQ = naphthoquinone; NQH2 = naphthohydroquinone; P = 3,4-propylenedioxythiophene; p = polymerized; Q = benzoquinone; QH2 = hydroquinone). HSO4− anions were predoped into the polymers to ensure the satisfactory conductivity of both electrodes. The battery added with 0.5 M aqueous sulfuric acid (H2SO4) delivered a higher capacity retention of 85% after 500 cycles. The battery could even function at a sub-zero temperature of −24 °C due to the electron transport process within the conjugated backbone was not affected during the electrochemical reaction. This work demonstrated how thiophene-based trimeric structures with naphthoquinone or hydroquinone redox-active pendent groups can be processed in solution, deposited, dried and subsequently polymerized in the solid state to form conducting (redox) polymer layers without any additives. Thus, the construction of additive-free all-organic aqueous batteries of the rocking-chair type using protons as cycling ions has been demonstrated as a proof-of-concept.
4.3.1.3 Carboxylic dianhydride. Another typical carbonyl containing polymer is β-perylene-3,4,9,10-tetracarboxylic dianhydride (β-PTCDA), which has been commonly used as an anode material. Besides Li and Na metals, potassium (K) metal exhibits similar activity vs. such carbonyl-containing polymers. For example, Chen et al.168 demonstrated a K aqueous battery using as the cathode material. An initial specific capacity of 145 mA h g−1 was achieved and 82% of the capacity could be maintained at 2 A g−1 after 500 cycles. A full K-ion battery composed of the β-PTCDA anode and the potassium iron(II) hexacyanoferrate (KFHCF) cathode was fabricated and delivered a higher capacity retention of 89% after 1000 cycles at a high current rate of 12.5 C.
4.3.2.1 Quinone group. Polymers with embedded quinones in backbones are intensively applied not only for monovalent-ion batteries, but also for multivalent-ion batteries such as Zn+ ion batteries. In such studies, quinone groups can get more nucleophilic sites for Zn+ hosting, which can improve the energy density.169 Previous studies have demonstrated that faster electron transport pathways can be provided by the grafting of quinone groups to long conjugated polymer chains.167,170 Wang et al.171 were inspired to graft twisting para-benzoquinone (pBQ) to the ladder-type dithieno[3,2-b
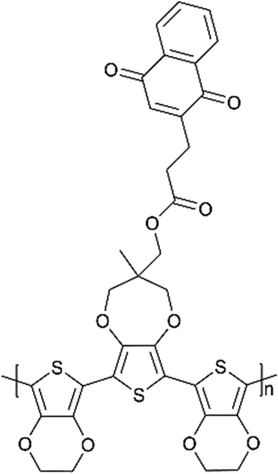
:2′3′-d]pyrrole core as the redox conducting polymer of ZIB cathodes. The hydroxyl group of quinone groups was oxidized to form oxygen radicals, providing a large number of active sites that bind only to Zn2+, and showing highly reversible Zn2+ insertion/release during the charge and discharge of the battery. Moreover, the cathodes demonstrated 120 mA h g−1 and 52.5% of capacity retention at 5 A g−1, while the high Coulombic efficiency upon 99% (Fig. 8a–c). Owing to the dendrite formation on the zinc anode, Oka et al.21 designed polymer–manganese secondary batteries based on a naphthoquinone (NQ) pendant group onto the trimer of EPE (E = 3,4-ethylenedioxythiophene; P = 3,4-propylenedioxythiophene) used as the anode. A discharge voltage of 1.0 V was yielded, which was close to the potential window of water (Fig. 8d–f). Besides, the battery displayed a discharging capacity of 76 mA h g−1 and a highly rate capability up to 10 C.
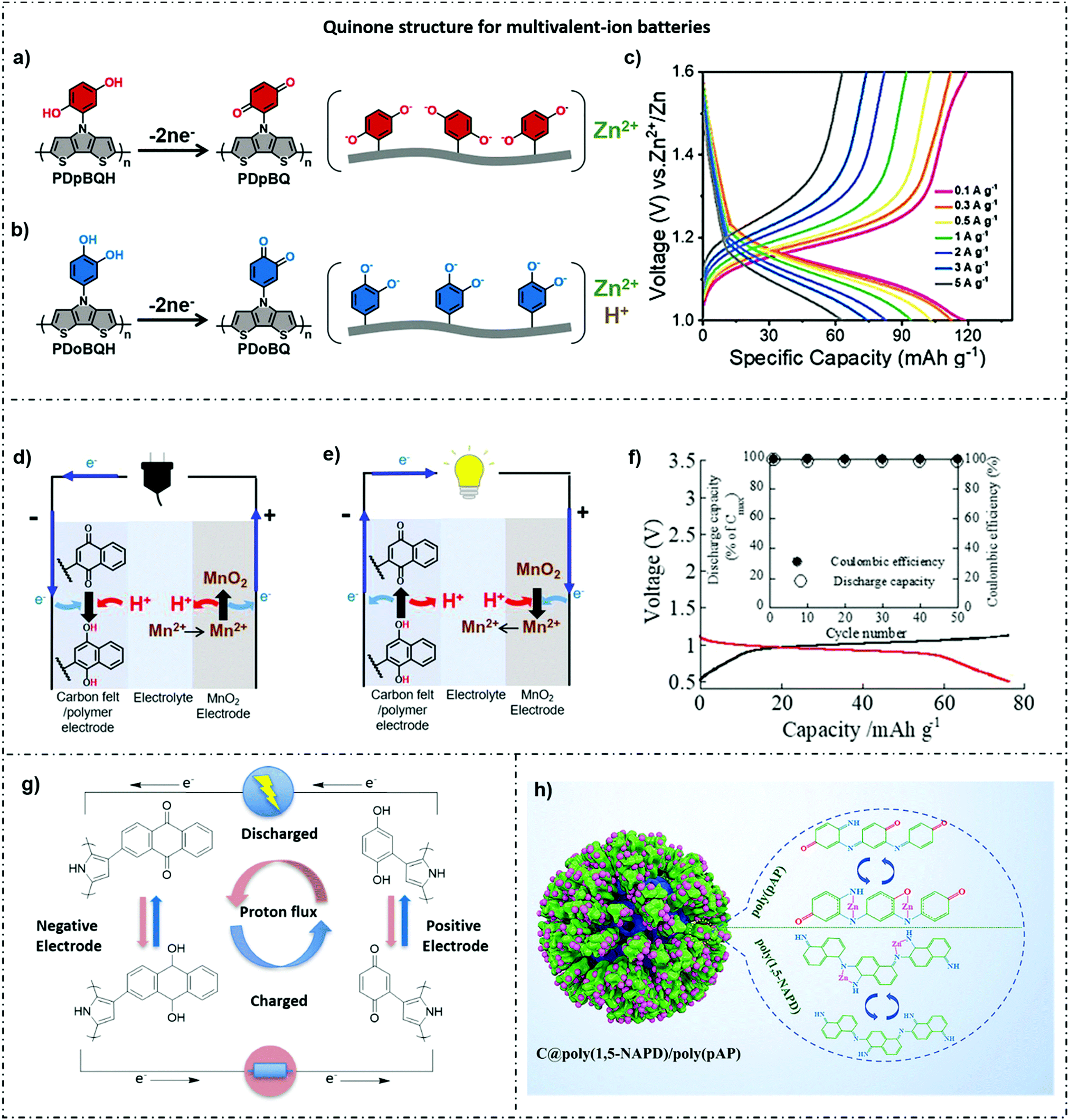 | ||
| Fig. 8 (a and b) Schematic of our polymers PDpBQH/PDoBQH were oxidized to work in ZIBs. PDpBQ shows only the Zn2+ involved insertion/extraction reaction with excellent reversibility, while PDoBQ undergoes both Zn2+ and hydrion involved redox processes which concurrently exhibit the degenerative electrochemical performance. (c) Electrochemical performances of PDpBQ at different current densities.171 Reprinted with permission from ref. 171 Copyright 2022, Wiley-VCH. (d) Schematic image of the rechargeable conducting redox polymer–manganese battery (charging). (e) Schematic image of the battery (discharging). (f) The charging (black) and discharging (red) curves of the battery at 1 C. Inset: Capacity retention for 50 cycles upon the galvanostatic charge and discharge of the battery at 1 C.21 Reprinted with permission from ref. 21 Copyright 2020, Wiley-VCH. (g) Schematic representation of the processes occurring during the charge (red arrows) and discharge (blue arrows) of the battery with P1 as the negative electrode and P10 as the positive electrode.172 Reprinted with permission from ref. 172 Copyright 2021, Elsevier. (h) Illustration of the fabrication process and Zn2+ storage mechanism of C@poly(1,5-NAPD)/poly(pAP) cathodes for Zn–organic batteries.173 Reprinted with permission from ref. 173 Copyright 2021, Wiley-VCH. | ||
Through changing the quinone structures to tune the working potential was also applied in aqueous batteries. Huang et al.172 synthesized a series of quinone–pyrrole dyads with various substituents on the quinone, and they had been successfully copolymerized with pyrrole, resulting in a series of polypyrrole (PPy) based CRPs with different quinone formal potentials. The cell potentials resulted in the galvanostatic charge–discharge of the cell showed a positive coincide with the difference in the redox potential between the quinone substituents used in the anode and cathode (Fig. 8g).
Zhao et al.173 attempted to introduce multiple redox centres into organic composite materials to solve the problem of low capacity due to over-reliance on cations reacting only with a single functional group in zinc batteries. They first electrodeposited poly(1,5-naphthalenediamine, 1,5-NAPD) in situ on nanoporous carbon as the stable intermediate layer and then daubed poly(para-aminophenol, pAP) skin on top of 1,5-NAPD as an external conducting layer. The C–N/C = N groups in poly(1,5-NAPD) clearly increase the nucleation sites and the stability of the organic–organic cathode, while the C–O and C–N groups in the poly(pAP) skin can improve the Zn2+ reaction kinetics and redox potential. The polymer cathode has a high capacity (348 mA h g−1), an excellent rate capability (132 mA h g−1 at 40 A g−1), a longer lifetime of over 5000 cycles, and a superior areal capacity of 2.8 mA h cm−2 even at the practical (10.2 mg cm−2), demonstrating the advantage of the 3D organic/organic heterostructure (Fig. 8h).
4.4 Metal–iodine batteries
Metal–iodine batteries (MIBs) hold practical promise for large-scale energy storage owing to the advantages of natural abundance, low cost, and high gravimetric/volumetric energy densities. In spite of the considerable advancements in MIBs, the inferior kinetics and high iodine solubility severely restrict their practical applications. To date, scientists have made enormous efforts to the design of advanced electrode materials to address the key scientific issues of inhibiting iodine dissolution under reversible reaction conditions. A series of effective strategies using polymer-based materials to promote the comprehensive characteristics of the iodine electrode have been developed. For instance, polyvinylpyrrolidone (PVP) and its nanocomposite PVP@C were developed by Han et al. and Deng et al.,174,175 respectively. These PVP based polymers can immobilize polyiodide by the chemical interaction between the host material and iodine species. Consequently, the suppressed polyiodide shuttle for Li–I and Na–I batteries has been demonstrated. However, the poor conductivity of the PVP retards the electrochemical redox kinetics of the iodine cathode. Using conducing polymers as organic electrode materials becomes a practical strategy. As an example, polyaniline (PANI) was selected as a model by Liang et al.176 They revealed that polyiodide can be strongly confined in the polymer backbone during the redox reaction and demonstrated a proof-of-concept about polyiodide doped conducting polymers for aqueous Zn–I batteries. In addition to conventional polymers,177 conjugated MOFs show great potential as advanced electrode materials, which is beneficial for a high I2 loading, high tolerance to sever volume change, and fast electrolyte penetration. Significantly, the modulation of metal–ligand orbital hybridization in the conjugated MOFs allows for profound investigations on the charge storage mechanisms at the molecular level. Similarly, the design and synthesis of well-defined redox pendant functional groups play a similar role in exploring highly efficient electrode materials for MIBs, which can provide an additional platform for the MIB study and has not been fully studied to date.4.5 Beyond alkali metal batteries
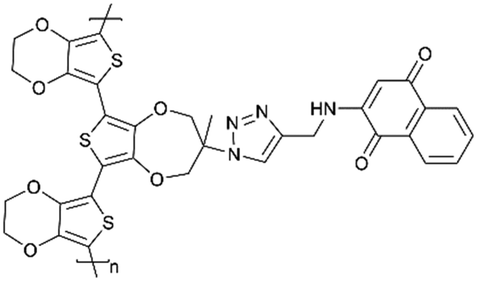
Oka et al.178 designed the naphthoquinone pendant group onto the conducting redox polymer based on the poly(3,4-ethylenedioxythiophene) backbone, which is formed from a stable suspension of a trimeric precursor and an oxoammonium cation as an oxidant. It has been demonstrated a promising candidate as the anode-active material for a polymer–air secondary battery combining a conventional Pt/C catalyst as the cathode, and sulfuric acid aqueous solution as the electrolyte. A discharge voltage of 0.50 V and good cycling stability with 97% capacity retention after 100 cycles have been delivered. In addition, this type of battery shows rapid cycling capacities, keeping the full discharge capacity up to 20 C. Similarly, using 1,4-dihydroxyanthraquinone (Qz) and naphthoquinone (NQ) as side chains, the conducting polymer backbone was covalently attached, respectively, as the cathode and anode of an all-organic proton rocking chair battery with a proton ionic liquid electrolyte. During the rapid potentiostatic charging and galvanostatic discharge at 4.5 C, the battery showed a specific capacity of 62 mA h g−1 and had 80% capacity retention after 500 cycles.179
5. Summary
The current review surveys the progress in the application of CRPs as efficient organic electrode materials and discusses the state-of-the-art understanding in this field. In addition to the intrinsic low cost and density, earth-abundance, and tunable chemical properties, the combination of CPs with redox active groups mainly induces a synergism that helps in solving the major problems of organic electrode materials, which include but are not limited to the improvement of conductivity, specific capacity, and the suppression of the dissolution of active materials. In this regard, we mainly focus on those CRPs with well-designed redox pendant groups. We first introduce the fundamentals of conducting polymers and electrochemically active redox groups with respect to their underlying chemistry in electrochemical energy storage. Afterwards, the structure- and property-dominated applications of CRPs as organic materials (both the cathode and anode) for different rechargeable batteries are discussed in detail according to the classification of different active pendant groups (nitroxide radical, quinone structure, quinoline structure, thiol group, imide groups, etc.). Emerging research studies including monovalent-ion batteries (e.g., Li/Na/K-ion) and multivalent-ion batteries with either an organic or aqueous electrolyte have been summarized. We particularly focus on the function of the well-designed pendant groups in each battery for addressing the specific issues in their applications.Although an impressively large number of studies have accumulated and the applications related with CRPs are highly expected to reach the practical batteries, the strategy of combining organic conducting polymer skeletons with redox-active functional groups has proven to be an initial success. In addition to what has been achieved, challenges covering the rational design of CRPs and the development of synthetic techniques still exist. Regarding the requirement of electrode materials for electrochemical energy storage, we have summarized and anticipated several of the abovementioned challenges based on the reported studies, which may be helpful in guiding the development of practical organic electrode materials.
(1) The first on-going task would be the design and synthesis of CRPs with sufficient redox-active groups that are capable of providing more energy storage sites to achieve high specific capacities (i.e. > 200 mA h g−1) and long-term cycling stabilities (i.e. > 85% capacity retention for 1 K cycles). Despite great efforts, the up-to-date design of CRPs has been reported to limited redox-active groups both in quantity and types as examples shown in the review.
(2) Preservation of the intrinsic conductivity of the polymer backbone is another challenge during functionalization with redox-active groups and the process of charge–discharge, where repeated cracking and recombination of the covalent bonds usually destroy the conjugated structure of the polymer backbone. An in-depth study into the working mechanism is required for the rational design of CRPs with stable conducting backbones.
(3) The steps for employing CRPs with ultralow redox potential as anode materials to realize all-polymer batteries are challenging due to the fact that polymers with low redox potential are less abundant and versatile than the cathode candidates.
(4) From the sustainability perspective, the green synthesis of CRPs from renewable resources such as biomass is attractive for the development of future energy storage devices.
Other obstacles blocking the transfer from a fundamental study to practical applications may be the high cost, the harsh conditions, and the complexity in the synthesis route. Therefore, we deem that low-cost and sustainable energy storage systems with high energy density and long working life are expected to be realized by addressing the abovementioned issues existing in the exploration of new CRPs. Regarding the technical difficulties in the molecular design and synthesis of CRPs, the non-covalent combination of polymers with different functions offers an additional opportunity to create multifunctional organic electrodes. In practice, the construction of a non-covalently linked composite containing conducting and redox polymers could achieve comparable functionality to CRPs, which show great potential to develop advanced electrode materials in a distinct manner. As an example, interpenetrating networks consisting of conducting and redox-active polymers (e.g., polypyrrole and lignosulfonate) have been demonstrated by Inganas et al. as efficient charge and energy storage materials, which have been developed as an attractive route to combine the advantages of conducting and redox active polymers.180,181 This non-covalent combination strategy raises a new platform for the construction of a multifunctional electrode and allows the application of renewable biopolymers to produce low cost electrodes with improved safety.
We hope that this review will not only provide readers with a panoramic view of CRP-based electrode materials for battery applications, but also inspire researchers to develop more strategies for the design and synthesis of CRPs or polymer composites with diverse properties and functions, further expanding their application in the electrochemical energy storage field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (No. 22075027 and 52003030), the starting Grant from the Beijing Institute of Technology (3020012222019) and financial support from the State Key Laboratory of Explosion Science and Technology (YBKT21-06).References
- G. He, Y. Liu, D. E. Gray and J. Othon, Compos. Commun., 2021, 27, 100882 CrossRef
.
- C. P. Grey and J. M. Tarascon, Nat. Mater., 2017, 16, 45–56 CrossRef PubMed
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed
- M. V. Reddy, A. Mauger, C. M. Julien, A. Paolella and K. Zaghib, Materials, 2020, 13, 1884 CrossRef CAS PubMed
- X.-Q. Zhang, C.-Z. Zhao, J.-Q. Huang and Q. Zhang, Engineering, 2018, 4, 831–847 CrossRef CAS
- H. Zhao, N. Deng, J. Yan, W. Kang, J. Ju, Y. Ruan, X. Wang, X. Zhuang, Q. Li and B. Cheng, Chem. Eng. J., 2018, 347, 343–365 CrossRef CAS
- H.-J. Peng, J.-Q. Huang, X.-B. Cheng and Q. Zhang, Adv. Energy Mater., 2017, 7, 1600168 Search PubMed
- H. Zhang, X. Liu, H. Li, I. Hasa and S. Passerini, Angew. Chem., Int. Ed., 2021, 60, 598–616 CrossRef CAS PubMed
- V. Verma, S. Kumar, W. Manalastas Jr, R. Satish and M. Srinivasan, Adv. Sustainable Syst., 2019, 3, 1800111 CrossRef
- C. Hou, W. Yang, X. Xie, X. Sun, J. Wang, N. Naik, D. Pan, X. Mai, Z. Guo, F. Dang and W. Du, J. Colloid Interface Sci., 2021, 596, 396–407 CrossRef CAS PubMed
- Y. Yuan, Z. Chen, H. Yu, X. Zhang, T. Liu, M. Xia, R. Zheng, M. Shui and J. Shu, Energy Stor. Mater., 2020, 32, 65–90 CrossRef
- Y. Wang, R. Zhang, J. Chen, H. Wu, S. Lu, K. Wang, H. Li, C. J. Harris, K. Xi, R. V. Kumar and S. Ding, Adv. Energy Mater., 2019, 9, 1900953 CrossRef
- A. Ghosh, P. Cherepanov, C. Nguyen, A. Ghosh, A. Kumar, A. Ahuja, M. Kar, D. R. MacFarlane and S. Mitra, Appl. Mater. Today, 2021, 23, 101062 CrossRef
- Y. Zou, D. Guo, B. Yang, L. Zhou, P. Lin, J. Wang, X. A. Chen and S. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 50093–50100 CrossRef CAS PubMed
- Y. Zhou, Y. Zhang and X. Li, Mater. Today Energy, 2021, 19, 100591 CrossRef CAS
- T. Sun, Z.-J. Li, Y.-F. Zhi, Y.-J. Huang, H. J. Fan and Q. Zhang, Adv. Funct. Mater., 2021, 31, 2010049 CrossRef CAS
- F. Wan and Z. Niu, Angew. Chem., Int. Ed., 2019, 58, 16358–16367 CrossRef CAS PubMed
- D. Chen, M. Lu, D. Cai, H. Yang and W. Han, J. Energy Chem., 2021, 54, 712–726 CrossRef
- Y. Zhang, L. Tao, C. Xie, D. Wang, Y. Zou, R. Chen, Y. Wang, C. Jia and S. Wang, Adv. Mater., 2020, 32, 1905923 CrossRef CAS PubMed
- J. J. Shea and C. Luo, ACS Appl. Mater. Interfaces, 2020, 12, 5361–5380 CrossRef CAS PubMed
- K. Oka, R. Löfgren, R. Emanuelsson, H. Nishide, K. Oyaizu, M. Strømme and M. Sjödin, ChemElectroChem, 2020, 7, 3336–3340 CrossRef CAS
- L. M. Zhu, A. W. Lei, Y. L. Cao, X. P. Ai and H. X. Yang, Chem. Commun., 2013, 49, 567–569 RSC
- M. Armand, S. Grugeon, H. Vezin, S. Laruelle, P. Ribière, P. Poizot and J. M. Tarascon, Nat. Mater., 2009, 8, 120–125 CrossRef CAS PubMed
- C.-X. Zhang, S.-L. Mei, X.-H. Chen, E.-T. Liu and C.-J. Yao, J. Mater. Chem. C, 2021, 9, 17182–17200 RSC
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, Á. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed
- V. Ramezankhani, I. K. Yakuschenko, A. V. Mumyatov, S. G. Vasil'ev, I. S. Zhidkov, E. Z. Kurmaev, A. F. Shestakov and P. A. Troshin, J. Power Sources, 2022, 517, 230711 CrossRef CAS
- M. D. Hager, B. Esser, X. Feng, W. Schuhmann, P. Theato and U. S. Schubert, Adv. Mater., 2020, 32, 2000587 CrossRef CAS PubMed
- Z. Song, Y. Qian, T. Zhang, M. Otani and H. Zhou, Adv. Energy Mater., 2015, 2, 1500124 Search PubMed
- J. Xie, P. Gu and Q. Zhang, ACS Energy Lett., 2017, 2, 1985–1996 CrossRef CAS
- Y. Shi, L. L. Peng, Y. Ding, Y. Zhao and G. H. Yu, Chem. Soc. Rev., 2015, 44, 6684–6696 RSC
- C.-J. Yao, J. Xie, Z. Wu, Z. J. Xu, S. Zhang and Q. Zhang, Chem. – Asian J., 2019, 14, 2210–2214 CrossRef CAS PubMed
- P. Novák, K. Müller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207–282 CrossRef PubMed
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, Chem. Soc., Chem. Commun., 1977, 578–580 RSC
- C. K. Chiang, Polymer, 1981, 22, 1454–1456 CrossRef CAS
- D. MacInnes, M. A. Druy, P. J. Nigrey, D. P. Nairns, A. G. MacDiarmid and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1981, 317–319 RSC
- G. Guzmán-González, S. Vauthier, M. Alvarez-Tirado, S. Cotte, L. Castro, A. Guéguen, N. Casado and D. Mecerreyes, Angew. Chem., Int. Ed., 2022, 61, e202114024 CrossRef PubMed
- C. Wei, L. Tan, Y. Zhang, K. Zhang, B. Xi, S. Xiong, J. Feng and Y. Qian, ACS Nano, 2021, 15, 12741–12767 CrossRef CAS PubMed
- C.-J. Yao, Z. Wu, J. Xie, F. Yu, W. Guo, Z. J. Xu, D.-S. Li, S. Zhang and Q. Zhang, ChemSusChem, 2020, 13, 2457–2463 CrossRef CAS PubMed
- D. Zhu, G. Xu, M. Barnes, Y. Li, C.-P. Tseng, Z. Zhang, J.-J. Zhang, Y. Zhu, S. Khalil, M. M. Rahman, R. Verduzco and P. M. Ajayan, Adv. Funct. Mater., 2021, 31, 2100505 CrossRef CAS
- Y. Hu, L. J. Wayment, C. Haslam, X. Yang, S.-H. Lee, Y. Jin and W. Zhang, EnergyChem, 2021, 3, 100048 CrossRef CAS
- Y. Lu, Y. Cai, Q. Zhang and J. Chen, J. Phys. Chem. Lett., 2021, 12, 8061–8071 CrossRef CAS PubMed
- T. Janoschka, M. D. Hager and U. S. Schubert, Adv. Mater., 2012, 24, 6397–6409 CrossRef CAS PubMed
- N. Casado, G. Hernandez, H. Sardon and D. Mecerreyes, Prog. Polym. Sci., 2016, 52, 107–135 CrossRef CAS
- N. Goujon, N. Casado, N. Patil, R. Marcilla and D. Mecerreyes, Prog. Polym. Sci., 2021, 122, 101449 CrossRef CAS
- T. Nokami, T. Matsuo, Y. Inatomi, N. Hojo, T. Tsukagoshi, H. Yoshizawa, A. Shimizu, H. Kuramoto, K. Komae, H. Tsuyama and J.-I. Yoshida, J. Am. Chem. Soc., 2012, 134, 19694–19700 CrossRef CAS PubMed
- B. Häupler, R. Burges, C. Friebe, T. Janoschka, D. Schmidt, A. Wild and U. S. Schubert, Macromol. Rapid Commun., 2014, 35, 1367–1371 CrossRef PubMed
- B. Häupler, T. Hagemann, C. Friebe, A. Wild and U. S. Schubert, ACS Appl. Mater. Interfaces, 2015, 7, 3473–3479 CrossRef PubMed
- A. Kassam, D. J. Burnell and J. R. Dahn, Electrochem. Solid-State Lett., 2011, 14, A22–A23 CrossRef CAS
- P. Sharma, D. Damien, K. Nagarajan, M. M. Shaijumon and M. Hariharan, J. Phys. Chem. Lett., 2013, 4, 3192–3197 CrossRef CAS
- J. Geng, J.-P. Bonnet, S. Renault, F. Dolhem and P. Poizot, Energy Environ. Sci., 2010, 3, 1929–1933 RSC
- F. Rosciano, M. M. Salamone, R. Ruffo, M. Sassi and L. Beverina, J. Electrochem. Soc., 2013, 160, A1094–A1098 CrossRef CAS
- C. Karlsson, H. Huang, M. Strømme, A. Gogoll and M. Sjödin, RSC Adv., 2015, 5, 11309–11316 RSC
- H. Wang, C.-J. Yao, H.-J. Nie, K.-Z. Wang, Y.-W. Zhong, P. Chen, S. Mei and Q. Zhang, J. Mater. Chem. A, 2020, 8, 11906–11922 RSC
- M. Aydın, B. Esat, Ç. Kılıç, M. E. Köse, A. Ata and F. Yılmaz, Eur. Polym. J., 2011, 47, 2283–2294 CrossRef
- N. Casado, G. Hernández, A. Veloso, S. Devaraj, D. Mecerreyes and M. Armand, ACS Macro Lett., 2016, 5, 59–64 CrossRef CAS PubMed
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS
- B. Häupler, A. Wild and U. S. Schubert, Adv. Energy Mater., 2015, 5, 1402034 CrossRef
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020, 120, 6490–6557 CrossRef CAS PubMed
- J. Xie, Z. Wang, Z. J. Xu and Q. Zhang, Adv. Energy Mater., 2018, 8, 1703509 CrossRef
- R. Emanuelsson, M. Sterby, M. Strømme and M. Sjödin, J. Am. Chem. Soc., 2017, 139, 4828–4834 CrossRef CAS PubMed
- G. Hernández, N. Casado, A. M. Zamarayeva, J. K. Duey, M. Armand, A. C. Arias and D. Mecerreyes, ACS Appl. Energy Mater., 2018, 1, 7199–7205 CrossRef
- J. Qin, Q. Lan, N. Liu, F. Men, X. Wang, Z. Song and H. Zhan, iScience, 2019, 15, 16–27 CrossRef CAS PubMed
- N. Patil, A. Mavrandonakis, C. Jérôme, C. Detrembleur, N. Casado, D. Mecerreyes, J. Palma and R. Marcilla, J. Mater. Chem. A, 2021, 9, 505–514 RSC
- R. Emanuelsson, C. Karlsson, H. Huang, C. Kosgei, M. Strømme and M. Sjödin, Russ. J. Electrochem., 2017, 53, 8–15 CrossRef CAS
- Y. Wu, R. Zeng, J. Nan, D. Shu, Y. Qiu and S.-L. Chou, Adv. Energy Mater., 2017, 7, 1700278 CrossRef
- B. Genorio, K. Pirnat, R. Cerc-Korosec, R. Dominko and M. Gaberscek, Angew. Chem., Int. Ed., 2010, 49, 7222–7224 CrossRef CAS PubMed
- D. W. Leedy and D. L. Muck, J. Am. Chem. Soc., 1971, 93, 4264–4270 CrossRef CAS
- Y. Xu, T. Jiang, J. Wen, H. Gao, J. Wang and X. Xue, Hydrometallurgy, 2018, 179, 60–72 CrossRef CAS
- E. P. Tomlinson, M. E. Hay and B. W. Boudouris, Macromolecules, 2014, 47, 6145–6158 CrossRef CAS
- T. Sukegawa, A. Kai, K. Oyaizu and H. Nishide, Macromolecules, 2013, 46, 1361–1367 CrossRef CAS
- K. Nakahara, S. Iwasa, M. Satoh, Y. Morioka, J. Iriyama, M. Suguro and E. Hasegawa, Chem. Phys. Lett., 2002, 359, 351–354 CrossRef CAS
- T. Suga, H. Konishi and H. Nishide, Chem. Commun., 2007, 1730–1732 RSC
- T. Suga, Y.-J. Pu, K. Oyaizu and H. Nishide, Bull. Chem. Soc. Jpn., 2004, 77, 2203–2204 CrossRef CAS
- T. Suga, K. Yoshimura and H. Nishide, Macromol. Symp., 2006, 245-246, 416–422 CrossRef
- J. Arias-Pardilla, T. F. Otero, R. Blanco and J. L. Segura, Electrochim. Acta, 2010, 55, 1535–1542 CrossRef CAS
- K. Oyaizu, H. Tatsuhira and H. Nishide, Polym. J., 2015, 47, 212–219 CrossRef CAS
- S. Conte, G. G. Rodríguez-Calero, S. E. Burkhardt, M. A. Lowe and H. D. Abruña, RSC Adv., 2013, 3, 1957–1964 RSC
- C. Su, F. Yang, L. Xu, X. Zhu, H. He and C. Zhang, ChemPlusChem, 2015, 80, 606–611 CrossRef CAS PubMed
- T. F. Otero, J. Arias-Pardilla, H. Herrera, J. L. Segura and C. Seoane, Phys. Chem. Chem. Phys., 2011, 13, 16513–16515 RSC
- Y. Liang, Z. Chen, Y. Jing, Y. Rong, A. Facchetti and Y. Yao, J. Am. Chem. Soc., 2015, 137, 4956–4959 CrossRef CAS PubMed
- R. Fang, J. Xu and D.-W. Wang, Energy Environ. Sci., 2020, 13, 432–471 RSC
- L. Ma, H. L. Zhuang, S. Wei, K. E. Hendrickson, M. S. Kim, G. Cohn, R. G. Hennig and L. A. Archer, ACS Nano, 2016, 10, 1050–1059 CrossRef CAS PubMed
- C. Chae, J. Kim, J. Y. Kim, S. Ji, S. S. Lee, Y. Kang, Y. Choi, J. Suk and S. Jeong, ACS Appl. Mater. Interfaces, 2018, 10, 4767–4775 CrossRef CAS PubMed
- J. Liang, Z.-H. Sun, F. Li and H.-M. Cheng, Energy Stor. Mater., 2016, 2, 76–106 CrossRef
- G. Zhou, E. Paek, G. S. Hwang and A. Manthiram, Nat. Commun., 2015, 6, 7760 CrossRef CAS PubMed
- T.-Z. Hou, W.-T. Xu, X. Chen, H.-J. Peng, J.-Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 8178–8182 CrossRef CAS PubMed
- L. Borchardt, M. Oschatz and S. Kaskel, Chem. – Eur. J., 2016, 22, 7324–7351 CrossRef CAS PubMed
- Z. Chen, J. Droste, G. Zhai, J. Zhu, J. Yang, M. R. Hansen and X. Zhuang, Chem. Commun., 2019, 55, 9047–9050 RSC
- M. Liu, S. J. Visco and L. C. De Jonghe, J. Electrochem. Soc., 1989, 136, 2570 CrossRef CAS
- S. Visco, M. Liu, M. Armand and L. De Jonghe, Mol. Cryst. Liq. Cryst., 1990, 190, 185–195 CrossRef CAS
- H. Tsutsumi, Y. Oyari, K. Onimura and T. Oishi, J. Power Sources, 2001, 92, 228–233 CrossRef CAS
- F. Li, S. Wang, Y. Zhang and J. L. Lutkenhaus, Chem. Mater., 2018, 30, 5169–5174 CrossRef CAS
- Y. Xie, K. Zhang, M. J. Monteiro and Z. Jia, ACS Appl. Mater. Interfaces, 2019, 11, 7096–7103 CrossRef CAS PubMed
- X. Li, Y. Li, K. Sarang, J. Lutkenhaus and R. Verduzco, Adv. Funct. Mater., 2021, 31, 2009263 CrossRef CAS
- J.-K. Kim, G. Cheruvally, J.-H. Ahn, Y.-G. Seo, D. S. Choi, S.-H. Lee and C. E. Song, J. Ind. Eng. Chem., 2008, 14, 371–376 CrossRef CAS
- K. Nakahara, J. Iriyama, S. Iwasa, M. Suguro, M. Satoh and E. J. Cairns, J. Power Sources, 2007, 165, 398–402 CrossRef CAS
- F. Li, Y. Zhang, S. R. Kwon and J. L. Lutkenhaus, ACS Macro Lett., 2016, 5, 337–341 CrossRef CAS PubMed
- M. Aydin and B. Esat, J. Solid State Electrochem., 2015, 19, 2275–2281 CrossRef CAS
- Y. Zhang, A. M. Park, S. R. McMillan, N. J. Harmon, M. E. Flatté, G. D. Fuchs and C. K. Ober, Chem. Mater., 2018, 30, 4799–4807 CrossRef CAS
- L. Xu, F. Yang, C. Su, L. Ji and C. Zhang, Electrochim. Acta, 2014, 130, 148–155 CrossRef CAS
- J.-j Lu, J.-q Ma, J.-m Yi, Z.-l Shen, Y.-j Zhong, C.-a Ma and M.-c Li, Electrochim. Acta, 2014, 130, 412–417 CrossRef CAS
- S. Bahceci and B. Esat, J. Power Sources, 2013, 242, 33–40 CrossRef CAS
- B. Bilgiç, Ç. Kılıç and B. Esat, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 115207 CrossRef
- F. Li, D. N. Gore, S. Wang and J. L. Lutkenhaus, Angew. Chem., 2017, 129, 9988–9991 CrossRef
- M. A. Ratner and D. F. Shriver, Chem. Rev., 1988, 88, 109–124 CrossRef CAS
- P. V. Wright, J. Appl. Polym. Sci., 1976, 14, 955–957 CAS
- Y. Liang, Y. Jing, S. Gheytani, K.-Y. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841–848 CrossRef CAS PubMed
- Z. Luo, L. Liu, Q. Zhao, F. Li and J. Chen, Angew. Chem., Int. Ed., 2017, 56, 12561–12565 CrossRef CAS PubMed
- C. Karlsson, A. Gogoll, M. Strømme and M. Sjödin, J. Phys. Chem. C, 2013, 117, 894–901 CrossRef CAS
- H. Wang, R. Emanuelsson, H. Liu, K. Edström, F. Mamedov, M. Strømme and M. Sjödin, ACS Appl. Energy Mater., 2019, 2, 7162–7170 CrossRef CAS
- S. Er, C. Suh, M. P. Marshak and A. Aspuru-Guzik, Chem. Eng. Sci., 2015, 6, 885–893 CAS
- M. Namazian, S. Siahrostami and M. L. Coote, J. Fluorine Chem., 2008, 129, 222–225 CrossRef CAS
- K. C. Kim, T. Liu, K. H. Jung, S. W. Lee and S. S. Jang, Energy Stor. Mater., 2019, 19, 242–250 CrossRef
- H. Wang, R. Emanuelsson, H. Liu, F. Mamedov, M. Strømme and M. Sjödin, Electrochim. Acta, 2021, 391, 138901 CrossRef CAS
- R. Duan, Z. Liu, Z. Wu, M. Baumgarten and C. Li, ACS Appl. Energy Mater., 2020, 3, 8179–8183 CrossRef CAS
- J. J. Samuel, V. K. Karrothu, R. K. Canjeevaram Balasubramanyam, A. A. Mohapatra, C. Gangadharappa, V. R. Kankanallu, S. Patil and N. P. B. Aetukuri, J. Phys. Chem. C, 2021, 125, 4449–4457 CrossRef CAS
- R. S. Ashraf, I. Meager, M. Nikolka, M. Kirkus, M. Planells, B. C. Schroeder, S. Holliday, M. Hurhangee, C. B. Nielsen, H. Sirringhaus and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 1314–1321 CrossRef CAS PubMed
- C. Kanimozhi, N. Yaacobi-Gross, E. K. Burnett, A. L. Briseno, T. D. Anthopoulos, U. Salzner and S. Patil, Phys. Chem. Chem. Phys., 2014, 16, 17253–17265 RSC
- M. Miroshnikov, K. P. Divya, G. Babu, A. Meiyazhagan, L. M. Reddy Arava, P. M. Ajayan and G. John, J. Mater. Chem. A, 2016, 4, 12370–12386 RSC
- M. Tang, H. Li, E. Wang and C. Wang, Chin. Chem. Lett., 2018, 29, 232–244 CrossRef CAS
- J. Wu, X. Rui, G. Long, W. Chen, Q. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 7354–7358 CrossRef CAS PubMed
- Z. Luo, L. Liu, J. Ning, K. Lei, Y. Lu, F. Li and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 9443–9446 CrossRef CAS PubMed
- M. Tang, S. Zhu, Z. Liu, C. Jiang, Y. Wu, H. Li, B. Wang, E. Wang, J. Ma and C. Wang, Chem, 2018, 4, 2600–2614 CAS
- J. Yang, Y. Shi, P. Sun, P. Xiong and Y. Xu, ACS Appl. Mater. Interfaces, 2019, 11, 42305–42312 CrossRef CAS PubMed
- Y. Ko, J. Kim, H. Y. Jeong, G. Kwon, D. Kim, M. Ku, J. Yang, Y. Yamauchi, H.-Y. Kim, C. Lee and J. You, Carbohydr. Polym., 2019, 203, 26–34 CrossRef CAS PubMed
- S. Stříteský, A. Marková, J. Víteček, E. Šafaříková, M. Hrabal, L. Kubáč, L. Kubala, M. Weiter and M. Vala, J. Biomed. Mater. Res., Part A, 2018, 106, 1121–1128 CrossRef PubMed
- Z. Li, Y. Guo, X. Wang, W. Ying, D. Chen, X. Ma, X. Zhao and X. Peng, Chem. Commun., 2018, 54, 13865–13868 RSC
- X. Liu, Z. Xu, A. Iqbal, M. Chen, N. Ali, C. Low, R. Qi, J. Zai and X. Qian, Nanomicro Lett., 2021, 13, 54 Search PubMed
- Y. Ko, J. Kim, D. Kim, G. Kwon, Y. Yamauchi and J. You, Nanomaterials, 2019, 9, 612 CrossRef CAS PubMed
- N. Casado, D. Mantione, D. Shanmukaraj and D. Mecerreyes, ChemSusChem, 2020, 13, 2464–2470 CrossRef CAS PubMed
- W. Xue, H. Mutlu, H. Li, W. Wenzel and P. Theato, Polym. Chem., 2021, 12, 2643–2650 RSC
- M. M. Doeff, S. J. Visco and L. C. De Jonghe, J. Electrochem. Soc., 1992, 139, 1808–1812 CrossRef CAS
- M. M. Doeff, M. M. Lerner, S. J. Visco and L. C. De Jonghe, J. Electrochem. Soc., 1992, 139, 2077–2081 CrossRef CAS
- A. Bhargav, M. E. Bell, Y. Cui and Y. Fu, ACS Appl. Energy Mater., 2018, 1, 5859–5864 CrossRef
- H. Tsutsumi, K. Okada, K. Fujita and T. Oishi, J. Power Sources, 1997, 68, 735–738 CrossRef CAS
- J. Li, H. Zhan, L. Zhou, S. Deng, Z. Li and Y. Zhou, Electrochem. Commun., 2004, 6, 515–519 CrossRef CAS
- M. Amaike and T. Iihama, Synth. Met., 2006, 156, 239–243 CrossRef CAS
- B. Duan, W. Wang, A. Wang, K. Yuan, Z. Yu, H. Zhao, J. Qiu and Y. Yang, J. Mater. Chem. A, 2013, 1, 13261–13267 RSC
- B. Oschmann, J. Park, C. Kim, K. Char, Y.-E. Sung and R. Zentel, Chem. Mater., 2015, 27, 7011–7017 CrossRef CAS
- G. Gao, X. Sun and L.-W. Wang, J. Mater. Chem. A, 2020, 8, 21711–21720 RSC
- S. Zeng, L. Li, L. Xie, D. Zhao, N. Wang and S. Chen, ChemSusChem, 2017, 10, 3378–3386 CrossRef CAS PubMed
- W. Yan, K.-Y. Yan, G.-C. Kuang and Z. Jin, Chem. Eng. J., 2021, 424, 130316 CrossRef CAS
- L. Xiao, Y. Cao, J. Xiao, B. Schwenzer, M. H. Engelhard, L. V. Saraf, Z. Nie, G. J. Exarhos and J. Liu, Adv. Mater., 2012, 24, 1176–1181 CrossRef CAS PubMed
- P. Geng, S. Cao, X. Guo, J. Ding, S. Zhang, M. Zheng and H. Pang, J. Mater. Chem. A, 2019, 7, 19465–19470 RSC
- H. Kim, J. Lee, H. Ahn, O. Kim and M. J. Park, Nat. Commun., 2015, 6, 7278 CrossRef CAS PubMed
- H. Cheng and S. Wang, J. Mater. Chem. A, 2014, 2, 13783–13794 RSC
- S. Diez, A. Hoefling, P. Theato and W. Pauer, Polymers, 2017, 9, 59 CrossRef PubMed
- A. Chang, Q. Wu, X. Du, S. Chen, J. Shen, Q. Song, J. Xie and W. Wu, Chem. Commun., 2016, 52, 4525–4528 RSC
- Y. Cui and Y. Fu, ACS Appl. Mater. Interfaces, 2015, 7, 20369–20376 CrossRef CAS PubMed
- S. Zeng, L. Li, D. Zhao, J. Liu, W. Niu, N. Wang and S. Chen, J. Phys. Chem. C, 2017, 121, 2495–2503 CrossRef CAS
- J. Zhang, G. Li, Y. Zhang, W. Zhang, X. Wang, Y. Zhao, J. Li and Z. Chen, Nano Energy, 2019, 64, 103905 CrossRef CAS
- X. Fan, S. Chen, W. Gong, X. Meng, Y. Jia, Y. Wang, S. Hong, L. Zheng, L. Zheng, C. W. Bielawski and J. Geng, Energy Stor. Mater., 2021, 41, 14–23 CrossRef
- A. Bhargav and A. Manthiram, Adv. Energy Mater., 2020, 10, 2001658 CrossRef CAS
- J. Xu, W. Tang, F. Yu, S. Zhao, D. Niu, X. Zhang, Z. Xin and R. Chen, J. Mater. Chem. A, 2020, 8, 19001–19010 RSC
- J. Xu, F. Yu, J. Hua, W. Tang, C. Yang, S. Hu, S. Zhao, X. Zhang, Z. Xin and D. Niu, Chem. Eng. J., 2020, 392, 123694 CrossRef CAS
- J. Wang, S. Yi, J. Liu, S. Sun, Y. Liu, D. Yang, K. Xi, G. Gao, A. Abdelkader, W. Yan, S. Ding and R. V. Kumar, ACS Nano, 2020, 14, 9819–9831 CrossRef CAS PubMed
- M. A. Weret, C.-F. Jeffrey Kuo, T. S. Zeleke, T. T. Beyene, M.-C. Tsai, C.-J. Huang, G. B. Berhe, W.-N. Su and B.-J. Hwang, Energy Stor. Mater., 2020, 26, 483–493 CrossRef
- Z. Lei, Q. Yang, Y. Xu, S. Guo, W. Sun, H. Liu, L.-P. Lv, Y. Zhang and Y. Wang, Nat. Commun., 2018, 9, 576 CrossRef PubMed
- X. Chen, Y. Wang, J. Wang, J. Liu, S. Sun, L. Zhu, Q. Ma, N. Zhu, X. Wang, J. Chen and W. Yan, J. Mater. Chem. A, 2022, 10, 1359–1368 RSC
- H. Duan, K. Li, M. Xie, J.-M. Chen, H.-G. Zhou, X. Wu, G.-H. Ning, A. I. Cooper and D. Li, J. Am. Chem. Soc., 2021, 143, 19446–19453 CrossRef CAS PubMed
- H. Qin, Z. P. Song, H. Zhan and Y. H. Zhou, J. Power Sources, 2014, 249, 367–372 CrossRef CAS
- D. Moia, A. Giovannitti, A. A. Szumska, I. P. Maria, E. Rezasoltani, M. Sachs, M. Schnurr, P. R. F. Barnes, I. McCulloch and J. Nelson, Energy Environ. Sci., 2019, 12, 1349–1357 RSC
- A. A. Szumska, I. P. Maria, L. Q. Flagg, A. Savva, J. Surgailis, B. D. Paulsen, D. Moia, X. Chen, S. Griggs, J. T. Mefford, R. B. Rashid, A. Marks, S. Inal, D. S. Ginger, A. Giovannitti and J. Nelson, J. Am. Chem. Soc., 2021, 143, 14795–14805 CrossRef CAS PubMed
- S. T. M. Tan, T. J. Quill, M. Moser, G. LeCroy, X. Chen, Y. Wu, C. J. Takacs, A. Salleo and A. Giovannitti, ACS Energy Lett., 2021, 6, 3450–3457 CrossRef CAS
- A. Giovannitti, I. P. Maria, D. Hanifi, M. J. Donahue, D. Bryant, K. J. Barth, B. E. Makdah, A. Savva, D. Moia, M. Zetek, P. R. F. Barnes, O. G. Reid, S. Inal, G. Rumbles, G. G. Malliaras, J. Nelson, J. Rivnay and I. McCulloch, Chem. Mater., 2018, 30, 2945–2953 CrossRef CAS PubMed
- C. Strietzel, M. Sterby, H. Huang, M. Strømme, R. Emanuelsson and M. Sjödin, Angew. Chem., Int. Ed., 2020, 59, 9631–9638 CrossRef CAS PubMed
- H. Chen, Z. Zhang, Z. Wei, G. Chen, X. Yang, C. Wang and F. Du, Sustain. Energy Fuels, 2020, 4, 128–131 RSC
- G. Dawut, Y. Lu, L. Miao and J. Chen, Inorg. Chem. Front., 2018, 5, 1391–1396 RSC
- J. Cao, F. Ding, H. Chen, H. Wang, W. Wang, Z. Chen and J. Xu, J. Power Sources, 2019, 423, 316–322 CrossRef CAS
- X. Wang, J. Xiao and W. Tang, Adv. Funct. Mater., 2022, 32, 2108225 CrossRef CAS
- H. Huang, M. Strømme, A. Gogoll and M. Sjödin, Electrochim. Acta, 2021, 389, 138758 CrossRef CAS
- Y. Zhao, Y. Huang, F. Wu, R. Chen and L. Li, Adv. Mater., 2021, 33, 2106469 CrossRef CAS PubMed
- Z. Meng, H. Tian, S. Zhang, X. Yan, H. Ying, W. He, C. Liang, W. Zhang, X. Hou and W.-Q. Han, ACS Appl. Mater. Interfaces, 2018, 10, 17933–17941 CrossRef CAS PubMed
- G. Zhang, H. Wang, S. Zhang and C. Deng, J. Mater. Chem. A, 2018, 6, 9019–9031 RSC
- X. Zeng, X. Meng, W. Jiang, J. Liu, M. Ling, L. Yan and C. Liang, ACS Sustainable Chem. Eng., 2020, 8, 14280–14285 CrossRef CAS
- F. Wang, Z. Liu, C. Yang, H. Zhong, G. Nam, P. Zhang, R. Dong, Y. Wu, J. Cho, J. Zhang and X. Feng, Adv. Mater., 2020, 32, 1905361 CrossRef CAS PubMed
- K. Oka, C. Strietzel, R. Emanuelsson, H. Nishide, K. Oyaizu, M. Strømme and M. Sjödin, ChemSusChem, 2020, 13, 2280–2285 CrossRef CAS PubMed
- H. Wang, R. Emanuelsson, C. Karlsson, P. Jannasch, M. Strømme and M. Sjödin, ACS Appl. Mater. Interfaces, 2021, 13, 19099–19108 CrossRef CAS PubMed
- G. Milczarek and O. Inganäs, Science, 2012, 335, 1468–1471 CrossRef CAS PubMed
- J. Ruhl, L. M. Riegger, M. Ghidiu and W. G. Zeier, Adv. Energy Sustainability Res., 2021, 2, 2000077 CrossRef
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |