
Platinum(II) metallacycles as highly affinitive hosts for dendritic amino acids with tunable circularly polarized luminescence†
Ning
Wang‡
a,
Jianjian
Zhao‡
a,
Qian
Xu
a,
Pengyao
Xing
 *a,
Shengyu
Feng
a,
Xing-Dong
Xu
*a and
Hai-Bo
Yang
b
*a,
Shengyu
Feng
a,
Xing-Dong
Xu
*a and
Hai-Bo
Yang
b
aKey Laboratory of Special Functional Aggregated Materials of Ministry of Education, Shandong Key Laboratory of Advanced Organosilicon Materials and Technologies, School of Chemistry and Chemical Engineering, National Engineering Research Center for Colloidal Materials, Shandong University, Jinan 250100, People's Republic of China. E-mail: xingpengyao@sdu.edu.cn; xuxd@sdu.edu.cn
bShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
First published on 17th March 2022
Abstract
The incorporation of platinum(II) metallacycles into supramolecular chiral arrays has potential in introducing intriguing chiroptical activities and applications; however, it remains a considerable challenge due to their shape-persistent feature. Herein, we present the noncovalent modulation of supramolecular chirality in platinum(II) metallacycle-involved host–guest complexation. Tetraphenylethylene-based platinum(II) metallacycles were designed and synthesized, which exhibited aggregation-induced emission with fine-tailored luminescence colors. A hex-armed tyrosine derivative showed size matching towards the metallacycles. Driven by multiple electrostatic attractions, highly affinitive host–guest complexation occurred with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometry and a constant with a 108 order of magnitude. Host–guest complexation turned on the fluorescence of the metallacycles, which also appended chirality to the photoexcited state. Circularly polarized luminescence at different wavelengths was observed, benefiting from the robust complexation, realizing the noncovalent modulation of chirality to the platinum(II) metallacycles. This work establishes a strategy to transfer chirality via host–guest interaction to shape-persistent metallacycles with potential chiroptical applications.
:1 binding stoichiometry and a constant with a 108 order of magnitude. Host–guest complexation turned on the fluorescence of the metallacycles, which also appended chirality to the photoexcited state. Circularly polarized luminescence at different wavelengths was observed, benefiting from the robust complexation, realizing the noncovalent modulation of chirality to the platinum(II) metallacycles. This work establishes a strategy to transfer chirality via host–guest interaction to shape-persistent metallacycles with potential chiroptical applications.
 Pengyao Xing | Pengyao Xing is currently a full professor at the School of Chemistry and Chemical Engineering, Shandong University. He received his BSc and PhD degrees from Shandong University under the supervision of Prof. Aiyou Hao. He was a postdoctoral scholar with Prof. Yanli Zhao at Nanyang Technological University. Since Dec 2018, he has been a full professor of the School of Chemistry and Chemical Engineering, Shandong University. His research interests mainly focus on the emergence and control over chirality at hierarchical levels. Prof. Xing has published more than 40 scientific papers as a corresponding author and has been awarded with the High-Level Young Talents of China award in 2021. |
Introduction
The last several decades have witnessed the prosperity of supramolecular chemistry based on host–guest interactions. The development of novel host compounds allows for the emergence of advanced functions and applications. Macrocyclic hosts are able to provide microenvironments to accommodate diverse guest molecules depending on the intrinsic structures and properties of the hosts. Compared to pure organic hosts, the use of supramolecular coordination complexes is a favorable route to synthesize discrete metallacycles with high fidelity and yields.1–3 By utilizing metal–ligand bonding and synthetic approaches, supramolecular complexes with various topologies, sizes and dimensions (like 2D polygons and 3D polyhedra) have been designed and fabricated.4 Among them, platinum(II)-based coordinated metallacycles are discrete hosts constituting the conjugation of Lewis-acid acceptors and Lewis-base donors. Pioneered by Stang and Fujita et al.,5–7 Pt(II)-based metallacycles have been appended with tremendous functionalities. Host–guest complexation between Pt(II)-metallacycles and guest molecules has afforded extended supramolecular polymers.8 Due to the existence of cationic pyridinium, electrostatic attraction occurs between the metallacycles and anionic molecules such as in 1,3,6,8-tetrasulfonate pyrene,9 tobacco mosaic virus,10 phosphopeptides,11 sulfated glycosaminoglycan polymer12 and heparin,13 showing promising applications in crystal engineering, biosensing, protein recovery and stimulus-responsive materials.14,15 The presence of cationic pyridinium and the corresponding photo-induced electron transfer suppress the luminescence of Pt(II) metallacycles. This issue has been extensively addressed by Stang et al. by introducing aggregation-induced emission (AIE) entities such as tetraphenylethylene (TPE), whereby rational control over the luminescence properties has been realized,16–19 and luminescent liquid crystals20 and nanoparticles for theranostics21,22 have been developed by Stang and Yang.The combination of host–guest complexation and supramolecular chirality endows intriguing properties and functions. To this end, either hosts or guests with intrinsic molecular chirality should be employed. Cyclodextrins comprising 6–8 glucoses conjugated by a 1,4-glucosidic bond possess chiral cavity and periphery due to the D-configuration of glucose. Thus, the complexation and cyclodextrins transfer chirality to the included compounds.23 Additionally, the metal–organic frameworks built by cyclodextrins provide chiral environments to allow for chirality transfer to the entrapped guests to afford chiroptical properties.23–25 Recently, the intrinsic chirality of pillararenes has attracted considerable attention.26–28 The orientation of substituents (methoxyl group) on the 1,4-position of benzenes forms propeller chirality after separation by high-performance liquid chromatography. Chiral pillararenes facilitate the fabrication of chiral switches and rotaxanes by host–guest interaction. Alternatively, chirality may be transferred from chiral guests to hosts after complexation, whereby achiral guests are appended with supramolecular chirality. For instance, Jiang utilized naphthotubes as an achiral host to recognize and sense chiral amines and alcohols with precise ee% resolution.29,30 Chirality can be transferred from chiral guests to the hosts with chromophores to enable chiroptical responses such as Cotton effects and circularly polarized luminescence (CPL).
Utilization of Pt(II)-metallacycles as hosts to include chiral guests with high affinity can endow novel chiroptical properties to the supramolecular complex; however, this remains a major challenge and limitation.31,32 In this regard, in this work, we illustrate the highly affinitive host–guest complexation between Pt(II)-metallacycles and amino acid-based guests (Scheme 1). TPE ligands were conjugated to di-Pt(II) acceptors to afford hexagonal macrocycles, as evidenced by their mass and nuclear magnetic resonance spectra. To obtain high affinity of the host–guest complexation, a hex-armed tyrosine derivative was designed and synthesized. Structural matching enabled a 1:1 binding stoichiometry with a constant of up to 108 orders of magnitude, which is close to the behavior of cucurbiturils. Complexation induced the emergence of luminescence ascribed to the AIE property of the metallacycles. Chirality was transferred from the amino acid-based guest to the rigid metallacycles, allowing for the emergence of CPL. By varying the electron-donating groups, yellow to red emission and CPL were obtained. This work, emphasizing on the delicate design of guests to afford giant host–guest complexes with supramolecular chirality and chiroptical properties, establishes an alternative route to the function of supramolecular coordinated macrocycles.
 | ||
| Scheme 1 (a) Chemical structures of the metallacycles and guests. (b) Highly affinitive host–guest complexation. | ||
Results and discussion
In order to synthesize the hexagonal metallacycles, “V”-shaped TPE-based ligands with either methoxyl or N,N-dimethyl amino groups were conjugated to di-Pt(II) acceptors. The ligands and acceptors with a 1:1 molar ratio were added to a mixture of acetone and deionized water (4:1 by volume), followed by overnight incubation at 55 °C. The starting materials were almost quantitatively reacted to afford high-yield products, which were characterized by nuclear magnetic resonance (NMR) and mass spectra (MS). In the 1H NMR spectra (Fig. 1a), H1 to H3 adjacent to the coordination sites of Ligand1 shifted downfield after coordination because the coordination of pyridine to Pt(II) led to decreased electron density. The remote H4 shows a slight upfield shift possibly due to the deshielding effect. In addition, the upfield shifts of H5 and H6 from the Pt(II) acceptor suggest the successful coordination to the pyridine segment with high affinity. Then, 31P{1H} NMR spectroscopy was used (Fig. S26, ESI†). The singlet at 20.14 ppm for the pristine acceptor shifted to 15.80 ppm after the coordination-driven self-assembly, supporting the formation of discrete coordination metallacycles. Ligand2 exhibits similar coordination behavior in the 1H and 31P{1H} NMR spectra (Fig. 1b and Fig. S26, ESI†). Electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS), which has been recognized as a powerful technique to characterize discrete metallacycles, was further employed to confirm the formation of the metallacycles (Fig. 1c and d). As shown in Fig. 1e and f, several peaks corresponding to the [M − xNO3]x+ (x = 4–5) segments, which can be assigned to the [3+3] coordination assembly with lost counter nitrate counterions, were observed. The peaks at m/z = 1240.3128 and 979.8518 are designated to the [M − 4NO3]4+ and [M − 5NO3]5+ complexes, respectively, which are in good agreement with the corresponding calculated values of 1240.3438 and 979.8774. A similar scenario was found in the Ligand2-coordinated complexes. For instance, m/z = 1259.8551 corresponds to the [M – 4NO3]4+ charged species. The isotopically resolved peaks with good consistency with the calculated values suggest the successful synthesis of hexagonal Cycle1 and Cycle2 with high yield and fidelity.
 | ||
| Fig. 1 (a and b) Comparison of 1H NMR spectra between components in the solvent of acetone-d6. (c and d) Electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS) of Cycle1 with different peaks, where blue and red stand for the calculated and measured spectra, respectively. (e and f) Calculated and measured ESI-TOF-MS peaks of Cycle2. | ||
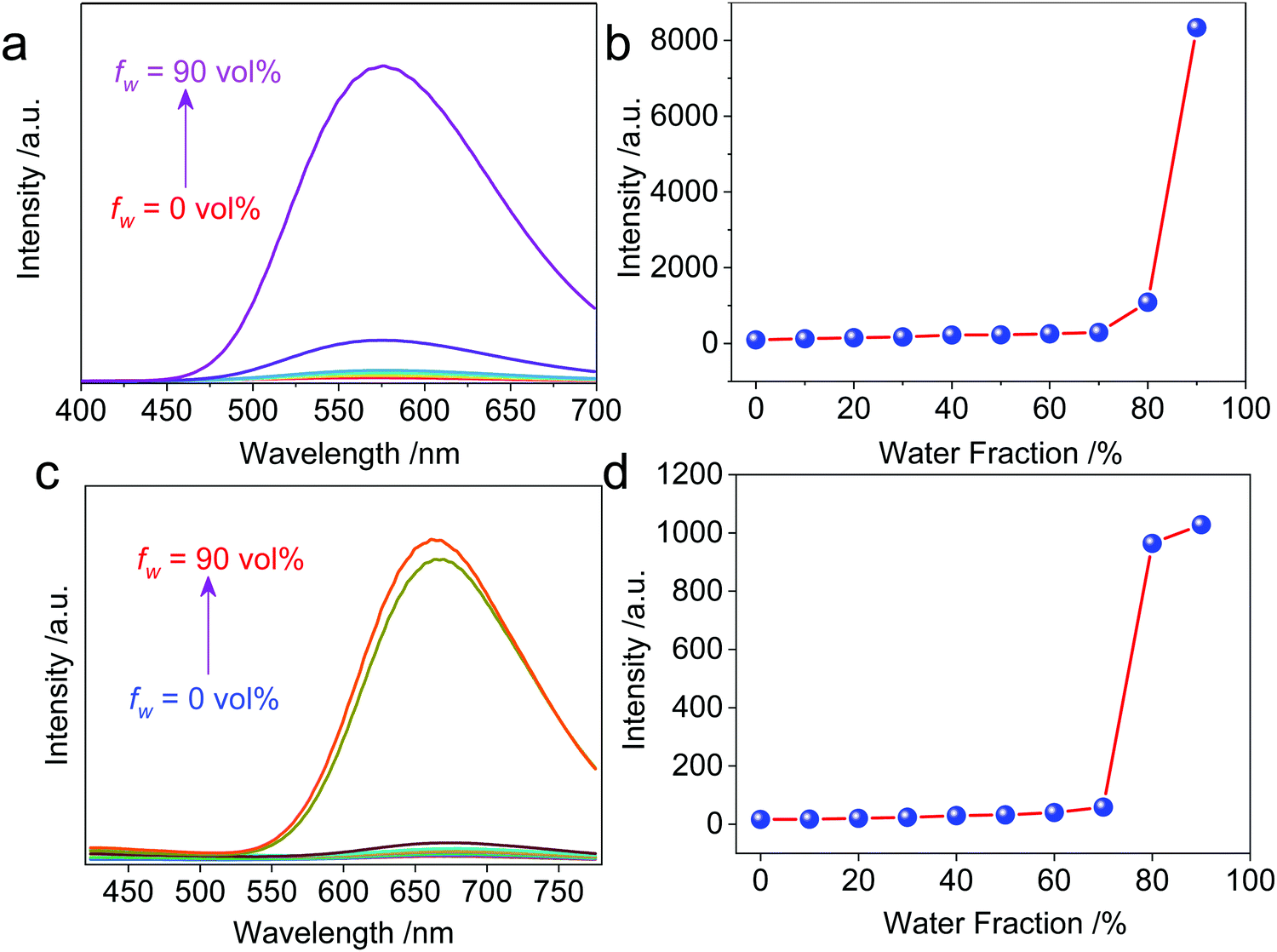 | ||
| Fig. 2 (a) Water fraction-dependent fluorescent spectra of Cycle1versus acetone (Ex. = 365 nm, c = 0.2 mM). (b) Fluorescent intensity at 576 nm as a function of the water fraction. (c and d) The corresponding fluorescence variation of Cycle2 (Ex. = 365 nm, c = 0.2 mM, collected at 662 nm). | ||
The resultant cationic metallacycles are barely emissive in good solvents (acetone). Considering the presence of TPE segments, their AIE properties were evaluated. By varying the good/poor solvent fraction (acetone versus water), typical AIE behavior was observed for both Cycle1 and Cycle2. The increase of the water fraction up to 80 vol% led to the emergence of fluorescence intensity with an enhanced factor up to 86 and 64-fold for Cycle1 and Cycle2, respectively (Fig. 2), which could be attributed to the spontaneous aggregation that restricted the molecular motion and low structural relaxation at the photoexcited state to reduce the non-irradiative emission. Cycle1 and Cycle2, with methoxyl and N,N-dimethyl amino substituents, exhibit distinct yellow and red emission centered at 576 and 662 nm, respectively. The emission of Cycle2 in the aggregation state is in the red emission region, which possesses potential applications in bioimaging. We also measured the absolute fluorescence quantum yield of Cycle1 and Cycle2 in solution and in the solid state. Cycle1 and Cycle2 showed weak fluorescence, and the fluorescence quantum yields (Φ) were 0.28% and 0.21%, respectively. In the solid state, the fluorescence emission of Cycle1 and Cycle2 was significantly enhanced and showed higher quantum yields of 20.55% and 13.16%, respectively.
Then, we moved on to the supramolecular complexation of metallacycles with the hex-armed amino acids. To verify the binding stoichiometric ratio, Job's plots were prepared based on fluorescence (Fig. 3a and b). The extremum values at R (Tyr/Cycle) = 0.5 indicated a 1:1 binding ratio for both Cycle1 and Cycle2, which is consistent with our assumption. Geometry and size compatibility contribute majorly to the 1:1 supramolecular complexation. The host–guest behaviors were further confirmed using fluorescent titration by adding Tyr into the metallacycle solutions. In the diluted solution (5 μM) of Cycle1, increasing the concentration of Tyr from 0 to 15 μM enhanced the emission intensity by up to 16-fold (Fig. 3c). The fluorescence enhancement was initiated by the host–guest interaction that restricts the structural relaxation of the metallacycles, similar to the AIE process. The intensity at 537 nm as a function of Tyr concentration in Fig. 3d shows a plateau propensity at 5 μM, corresponding to the 1:1 binding stoichiometry, and the curve was fitted to the non-linear fit with the n = 1 mode, affording the binding constant Ka = 1.38 × 108 M−1. Such a binding constant of up to 108 orders of magnitude reflects the high affinity between Cycle1 and the Tyr guest, which is higher than most hosts, such as cyclodextrin and pillararenes. Similarly, the emission intensity of Cycle2 was enhanced 14-fold after binding with Tyr, and the non-linear fit gave a binding constant Ka = 2.12 × 106 M−1, which is much lower than that of Cycle1. The hosts comprise TPE segments, which belong to the AIEgen family. Due to the cyclic topology, the TPE segments were restricted to some extent. However, the emission of these two hosts is rather weak because of the cationic Pt, which undergoes photoinduced electron transfer (PET) to quench the luminescence. The addition of the Tyr guest reduces the PET effect by appending anionic ions. In addition, further aggregation was realized after co-assembly to enhance the emission. Such a different binding affinity may be due to the substituent electronic effect. The –N(CH3)2 group possesses higher electron-donating capability than the –OCH3 group due to the lower electronegativity, which reduces the electropositivity of pyridinium as well as the electrostatic affinity between the guest and host molecules. In the extinction spectra, host–guest complexation enabled a slight bathochromic shift of Cycle1 with increased absorbance in the visible region (Fig. S27, ESI†), indicating the emergence of aggregates. By cooling down the Cycle1/Tyr inclusion complex from 363 K to 303 K, decreasing absorbance was observed, and the temperature-dependent absorbance was not in accordance with the cooperative or isodesmic aggregation modality. Such a temperature-directed aggregation–disassociation process showed inconformity with the typical supramolecular aggregation behavior. Generally speaking, the formation of aggregates decreases the absorbance or extinction spectra due to the lowered absorbance coefficient of the colloidal aggregates. In the present case, decreasing temperature induces the formation of aggregates accompanied with reduced absorbance.
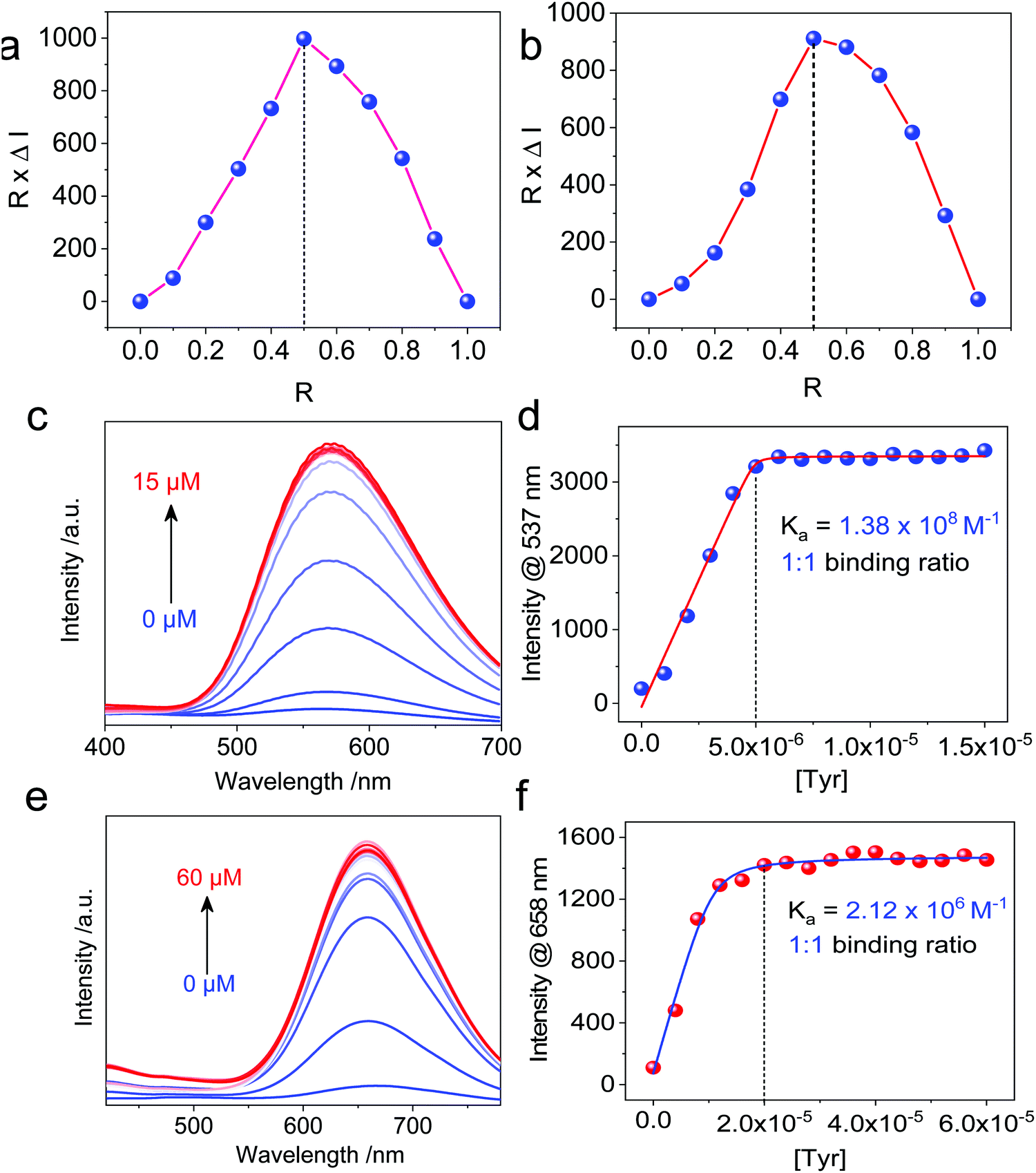 | ||
| Fig. 3 (a and b) Job's plot curves of Cycle1 and Cycle2, respectively, with Tyr (total concentration = 5 μM in the mixture of acetone and water). R stands for the molar fraction of Tyr. (c and d) Tyr concentration-dependent emission spectra of Cycle1 (5 μM), Ex. = 365 nm. (e and f) Tyr concentration-dependent emission spectra of Cycle2 (20 μM), Ex. = 400 nm. | ||
Molecular geometries of the cyclic hosts and guest were optimized to evaluate the structural factors in the supramolecular complexation behavior. As shown in Fig. 4, both Cycle1 and Cycle2 adopt a hexagonal geometry, where six Pt(II)-coordinates behave as linkers to conjugate the ligands. Such structure-persistent geometries show barely any distortion, ascribed to the rigid ligand and coordinated motifs. The lateral length and width of the hexagon were determined to be 37.7 and 25.2 Å, respectively, for both Cycle1 and Cycle2. This indicates that the substitution on the tetraphenylethylene segments did not interfere with the size of the metallacycles. The Tyr guest features a hexagonal geometry as well. Due to steric hindrance, six arms are distributed on two sides. The up-down side arm orientation enables a 12 Å distance under the side view, and the lateral length under the top view affords 24 Å. Therefore, the size of the guest is well suited to loading by the hosts of Cycle1 and Cycle2. Due to the size matching and potential electrostatic attraction, high binding affinity with 1:1 binding stoichiometry is expected. The optimized complex structure of Cycle1/l-Tyr indicates a distorted motif induced by host–guest interaction, which verifies the size matching effect. The distorted motif might be attributed to the chirality transfer.
 | ||
| Fig. 4 Geometry-optimized structures of Cycle1, Cycle2, guest l-Tyr and the Cycle1/l-Tyr complex using the PM6 method of the Gaussian 16 program. | ||
In order to probe the inclusion scenario in detail, the 1H NMR spectra were recorded. The inclusion complexes of Tyr@Cycle1 and Tyr@Cycle2 as well as their proton assignments are shown in Fig. 5a–d. Compared to the pristine Cycle1, the supramolecular complex with the 1:1 ratio exhibited humped peaks without apparent splitting, suggesting that the host–guest complexation triggered aggregation with lower resolution. Particularly, the resolution of protons from Tyr (H12–H15) is profoundly declined compared to that of Cycle1, which indicates the difference in the conformation of Tyr in the host–guest complex. According to NMR, the metallacycles rather than the complexes of Tyr@Cycle1 are characterized by discrete and rigid features. Moreover, the dynamic behavior demonstrated by NMR occurs between the metallacycles and Tyr. Partial protons including H1, H2 and H3 slightly shifted to high fields with Δ = 0.014, 0.021 and 0.024 ppm, respectively. In comparison, H4, H5, H6 and H8 were barely shifted. This suggests that the binding regions are located around the pyridinium domains, and the approach of electron-rich carbonate contributes to the shifts towards high fields. Supramolecular complexation also enabled the dramatic shift of protons from the guest molecule, whereby the shifts of H12–H15 were up to 0.12 ppm. In contrast, the inclusion of Tyr caused general shifts of protons H1 to H9 at the aromatic rings of Cycle2 towards the high field with Δ ∼ 0.06 ppm. Additionally, significant shifts were observed for Tyr in spite of the binding constant of Cycle2 being much lower than that of Cycle1 based on fluorescent titration. Then, 2D nuclear Overhauser enhancement spectroscopy (NOESY) was used to obtain more information regarding the binding areas (Fig. 5d and e). The NOESY spectra show the correlation between H16 (the adjacent proton of carbonate) and H8 (the interior proton near pyridinium) from Tyr and the Cycles, respectively, for both the Cycle1 and Cycle2 complexation systems, which further supports the electrostatic attraction-driven complexation. Direct evidence for host–guest complexation was further provided by ESI-TOF-MS characterization (Fig. 5f and g). Experimentally, for the Tyr@Cycle1 complex, three major m/z distributions at 2107.6370, 1581.2300 and 1264.9852 were found, which were designated to [MCycle1/Tyr + 3H]3+, [MCycle1/Tyr + 4H]4+ and [MCycle1/Tyr + Na + 4H]5+, respectively. The isotopically resolved spectra are in good consistency with the calculated m/z values of 2107.6441, 1581.2352 and 1264.9894, respectively. We also found the corresponding complexation of Tyr@Cycle2 by ESI-TOF-MS, where 2133.7074, 1600.5324 and 1280.6274 were designated to [MCycle2/Tyr + 3H]3+, [MCycle2/Tyr + 4H]4+ and [MCycle2/Tyr + 5H]5+, respectively. These results support the existence of host–guest complexes with 1:1 stoichiometry. The structural rigidity and high binding affinity afford the integrity seen in the ESI-TOF-MS characterization.
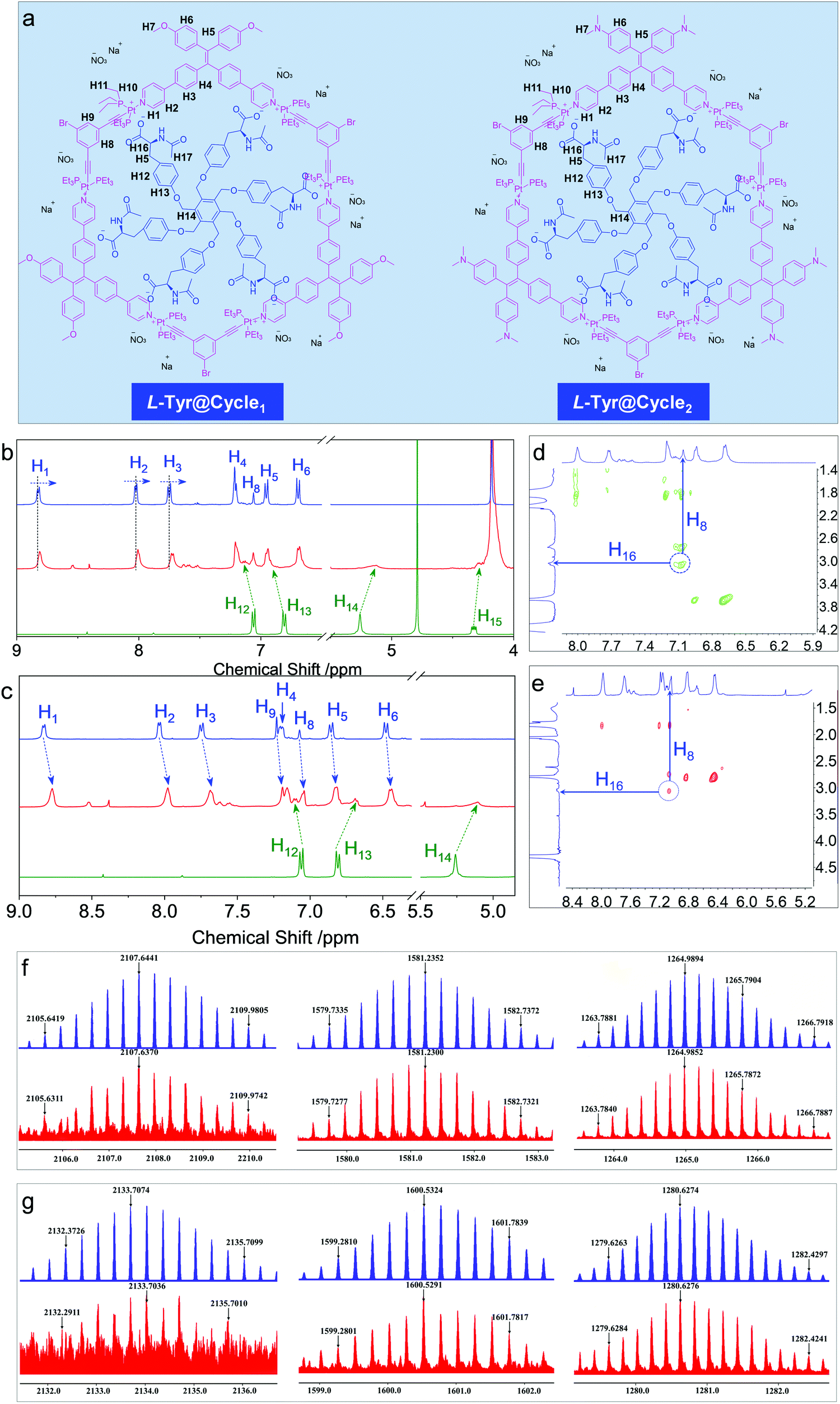 | ||
| Fig. 5 (a) Proton assignment in the inclusion complexes. (b and c) 1H NMR spectra of the Cycles and guest molecule in a mixture of acetone-d6 and deuterated water (fw = 60 vol%). (d and e) The corresponding 2D NOESY spectra. (f and g) ESI-TOF-MS profiles of the Tyr@Cycle1 and Try@Cycle2 inclusion complexes. | ||
Host–guest complexation and co-assembly enabled chirality transfer from the chiral guests to the host molecules. Chiroptical activity could reflect the chirality behavior that occurred with chromophores. Circular dichroism (CD) and circularly polarized luminescence (CPL) represent the chirality at the ground and photoexcited states, respectively. In aqueous media, supramolecular complexation induces the emergence of weak Cotton effects at the extremum wavelength of 365 nm. Mirror spectra were obtained for the L- and D-configurations. The Cotton effect reflects the chirality transfer from the amino acids to the rigid metallacycles. The appended supramolecular chirality was also observed in the CPL spectra (Fig. 6). In order to overcome the relatively weak chirality transfer to the rigid metallacycles, the molar ratio of Tyr was in excess to afford a sufficient chirality environment. Left- and right-CPL with dissymmetry g-factors of 1.9 × 10−3 and −1.7 × 10−3 at the extremum wavelength of Cycle1 were observed for L- and D-Tyr, respectively. The yellow CPL signals at around 550 nm suggested chirality behavior at the photoexcited state. By changing the metallacycle from Cycle1 to Cycle2 with a different emission, red-CPL at around 650 nm was observed (Fig. 6b) with slightly increased corresponding g-factors of 2.1 × 10−3 and −2.3 × 10−3. Compared to Cycle1, the Cycle2 complexes with Tyr exhibited inverse CPL handedness. Such a deviation implies that the electron-donating group may interfere with the energy levels at the excited state.
 | ||
| Fig. 6 (a and b) CPL spectra of Tyr@Cycle1 (0.3 mM:0.1 mM) and Tyr@Cycle2 (1.75 mM:0.25 mM). Ex. = 365 nm and 400 nm, respectively. | ||
Based on the fitted fluorescent titration, Cycle1 shows a higher binding constant compared to Cycle2. Cycle1 possesses methoxy groups, while Cycle2 has dimethyl amino groups. Both the groups are electron-donating, yet the dimethyl amino group has better electron-donating capability ascribed to its lower electronegativity. Therefore, the positive charge on the Pt atom is compensated for to a larger extent by the dimethyl amino group, which lowers the electrostatic attraction between Cycle2 and Tyr to afford a lower binding constant. Normally, the binding constant is associated with the structure-persistency of the complexes, which may contribute to the chirality at the ground state. On top of that, only the Cycle1/Tyr complex gave small CD signals. However, the CPL spectra reflect chirality at the photoexcited state, which has less correlation with the structure–persistency or the binding constant. Both the complexes show active CPL spectra. To exclude the artifacts in the measurement of CPL, repeated tests were carried out. As shown in Fig. S30 (ESI†), the results display similar opposite signals, suggesting that the CPL inversion is reliable. The substituents are electron-donating rather than electron-withdrawing. Oxygen and nitrogen groups have two effects when linked to aryl groups, i.e. electron-inducing and electron-conjugating effects. Both of the groups show overall electron-donating effects. The handedness of CPL is associated with many effects, which sometimes are not correlated to the CD spectra since the ground state to the photoexcited state experiences structural relaxation. In the present case, Cycle1 and Cycle2 show different binding constants towards the Tyr guest. We attribute the CPL signal inversion behavior to the possibly different conformations of the complexes. The values of gabs and glum are associated with the rotational strength, which can be calculated by the dot product of the electric and magnetic dipole moments. In the supramolecular self-assemblies, the chiral packing distance and screw angles determine the values and magnitudes of the asymmetric g-factors.33 The emergence and magnitudes of gabs and glum have no absolute correlation, established through a report that summarized the rough correlation between glum and gabs by Mori et al.34 They also explained that the correlation between glum and gabs is unpredictable in luminophores without rigid and structure-persistent features. In the present case, the geometry optimization indicates that the structure of the cycles might be distorted by the host molecules. Moreover, the Stokes shifts are higher than 300 nm in the emission spectra. These facts indicate a pronounced structural relation that might occur after photoexcitation, in contrast to the ground state. Therefore, the CD and CPL spectra could not match each other, with distinct gabs and glum values.
The metallacycles possess hydrophilic cationic domains as well as hydrophobic segments, which endow amphiphilicity to the hosts. We investigated the spontaneous aggregation and self-assembly behaviors corresponding to the host–guest complexation. In diluted solution, both Cycle1 and Cycle2 afforded nanoparticles without apparent interior and exterior boundaries (Fig. 7a and b) observed under transmission electron microscopy (TEM). The atomic force microscopy (AFM) images were consistent with the TEM images. The statistical size distribution (Fig. 7e) shows that most particles range from 20 to 30 nm in diameter with a median of 25 nm. The micelle size distribution was verified by the DLS profiles shown in Fig. 7g, which exhibit a wider distribution with a major distribution at around 20 nm–30 nm. Cycle1 and Cycle2 share a similar aggregation behavior in both morphology and size. The variation of the electron-donating group has negligible effect on the aggregation behavior. The participation of the guest Tyr resulted in the agglomeration of discrete micelles into 2D aggregates under TEM observation (Fig. 7c and d). The lateral lengths are up to hundreds of nanometers to micrometers in scale. Then, AFM was utilized to inspect the 2D morphology. Cycle1/Tyr complexation produced 2D planar aggregates (Fig. S29, ESI†) with lateral lengths up to the micrometer scale, in agreement with the TEM observation. The cross-sectional height profile suggested ultra-thin thickness at 6 nm. Considering the super-large host–guest complex, only a few molecular layers are expected within the nanofilms (Fig. 7f). Co-assembly behavior for the Cycle2/Tyr complexation displayed almost identical morphology to that of Cycle1, exhibiting a 2D thin film assembly as well (Fig. 7h). Although Cycle1 and Cycle2 possessed different binding constants and CPL properties, their self-assembly behaviors at the nanoscale are identical, illustrating a strong structure–property correlation. Incorporating the specific guests in the cavity of the metallacycles altered the hydrophobic balance and changed the cationic species into zwitterionic complexes. Additionally, the participation of Tyr with abundant amides and carboxylates introduced multiple hydrogen bonds with high directionality, which facilitates the transformation from amorphous nanoparticles to 2D thin films with enhanced and ordered molecular packing.
 | ||
| Fig. 7 (a and b) TEM images of Cycle1 and Cycle2 (0.1 mM) individual assemblies in water. (c and d) TEM images of the Cycle1/Tyr and Cycle2/Tyr co-assemblies (0.1:0.1 mM), respectively. (e) AFM (1 × 1 μm) images and particle size distribution of the Cycle2 individual assembly (0.1 mM). (f) AFM images (5 × 5 μm and 1 × 1 μm) and cross-sectional height profile of the Cycle2/Tyr co-assembly (0.1:0.1 mM). (g) Size distribution of the Cycle1 and Cycle2 (0.1 mM) individual assemblies in water using dynamic light scattering. (h) Schematic representation of the morphological transformation after host–guest complexation. | ||
Conclusions
In summary, two Pt(II) metallacycles were designed and synthesized, which could bind specific guest molecules via host–guest complexation. Supramolecular complexation enhanced the fluorescence of hosts and was ascribed to the AIE behavior. The high binding constant and 1:1 binding stoichiometry were characterized by mass spectroscopy, geometry optimization, NMR and fluorescence titration. The size matching and multiple binding sites were attributed to the high affinity between the cycles and Tyr. Finally, we realized chirality transfer from the chiral guest Tyr to the shape-persistent metallacycles, achieving CPL activity, which showed dependence on the absolute chirality of tyrosine. This work builds an alternative protocol to design highly affinitive guests to modulate the chirality of Pt(II)-based metallacycles.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Qilu Young Scholarship Funding and the Young Scholars Program (2018WLJH40) of Shandong University. This works is also supported by the National Natural Science Foundation of China (Nos 21872087, 22171165, 21602124) and the Natural Science Foundation of Jiangsu Province (BK20190209). We also acknowledge the financial support from the Youth cross-scientific innovation group of Shandong University (2020QNQT003). The authors acknowledge the assistance of Shandong University Structural Constituent and Physical Property Research Facilities.References
- K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC.
- S. Chakraborty and G. R. Newkome, Chem. Soc. Rev., 2018, 47, 3991–4016 RSC.
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed.
- D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- Y. Sun, C. Chen, J. Liu and P. J. Stang, Chem. Soc. Rev., 2020, 49, 3889–3919 RSC.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- B. Shi, Z. Zhou, R. T. Vanderlinden, J.-H. Tang, G. Yu, K. Acharyya, H. Sepehrpour and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 11837–11841 CrossRef CAS PubMed.
- A. Kaloudi-Chantzea, E. Martinou, K. Seintis, N. Karakostas, P. Giastas, F. Pitterl, H. Oberacher, M. Fakis and G. Pistolis, Chem. Commun., 2016, 52, 3388–3391 RSC.
- Y. Tian, X. Yan, M. L. Saha, Z. Niu and P. J. Stang, J. Am. Chem. Soc., 2016, 138, 12033–12036 CrossRef CAS PubMed.
- L.-J. Chen, S. J. Humphrey, J.-L. Zhu, F.-F. Zhu, X.-Q. Wang, X. Wang, J. Wen, H.-B. Yang and P. A. Gale, J. Am. Chem. Soc., 2021, 143, 8295–8304 CrossRef CAS PubMed.
- L.-J. Chen, Y.-Y. Ren, N.-W. Wu, B. Sun, J.-Q. Ma, L. Zhang, H. Tan, M. Liu, X. Li and H.-B. Yang, J. Am. Chem. Soc., 2015, 137, 11725–11735 CrossRef CAS PubMed.
- Z.-T. Shi, Y.-X. Hu, Z. Hu, Q. Zhang, S.-Y. Chen, M. Chen, J.-J. Yu, G.-Q. Yin, H. Sun, L. Xu, X. Li, B. L. Feringa, H.-B. Yang, H. Tian and D.-H. Qu, J. Am. Chem. Soc., 2021, 143, 442–452 CrossRef CAS PubMed.
- K. Acharyya, S. Bhattacharyya, H. Sepehrpour, S. Chakraborty, S. Lu, B. Shi, X. Li, P. S. Mukherjee and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14565–14569 CrossRef CAS PubMed.
- Z.-Y. Li, Y. Zhang, C.-W. Zhang, L.-J. Chen, C. Wang, H. Tan, Y. Yu, X. Li and H.-B. Yang, J. Am. Chem. Soc., 2014, 136, 8577–8589 CrossRef CAS PubMed.
- Z. Guo, G. Li, H. Wang, J. Zhao, Y. Liu, H. Tan, X. Li, P. J. Stang and X. Yan, J. Am. Chem. Soc., 2021, 143, 9215–9221 CrossRef CAS PubMed.
- C. Mu, Z. Zhang, Y. Hou, H. Liu, L. Ma, X. Li, S. Ling, G. He and M. Zhang, Angew. Chem., Int. Ed., 2021, 133, 12401–12405 CrossRef.
- Y. Li, S. S. Rajasree, G. Y. Lee, J. Yu, J.-H. Tang, R. Ni, G. Li, K. N. Houk, P. Deria and P. J. Stang, J. Am. Chem. Soc., 2021, 143, 2908–2919 CrossRef CAS PubMed.
- X. Yan, T. R. Cook, P. Wang, F. Huang and P. J. Stang, Nat. Chem., 2015, 7, 342–348 CrossRef CAS PubMed.
- Y.-X. Hu, X. Hao, L. Xu, X. Xie, B. Xiong, Z. Hu, H. Sun, G.-Q. Yin, X. Li, H. Peng and H.-B. Yang, J. Am. Chem. Soc., 2020, 142, 6285–6294 CrossRef CAS PubMed.
- G. Yu, M. Zhang, M. L. Saha, Z. Mao, J. Chen, Y. Yao, Z. Zhou, Y. Liu, C. Gao, F. Huang, X. Chen and P. J. Stang, J. Am. Chem. Soc., 2017, 139, 15940–15949 CrossRef CAS PubMed.
- X. Yan, H. Wang, C. E. Hauke, T. R. Cook, M. Wang, M. L. Saha, Z. Zhou, M. Zhang, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2015, 137, 15276–15286 CrossRef CAS PubMed.
- I. Roy and J. F. Stoddart, Acc. Chem. Res., 2021, 54, 1440–1453 CrossRef CAS PubMed.
- L. Hu, K. Li, W. Shang, X. Zhu and M. Liu, Angew. Chem., Int. Ed., 2020, 59, 4953–4958 CrossRef CAS PubMed.
- C. Yang, D. Yang, X. Zhu, Y. Meng and M. Liu, Langmuir, 2020, 36, 12366–12374 CrossRef CAS PubMed.
- W.-J. Li, Q. Gu, X.-Q. Wang, D.-Y. Zhang, Y.-T. Wang, X. He, W. Wang and H.-B. Yang, Angew. Chem., Int. Ed., 2021, 60, 9507–9515 CrossRef CAS PubMed.
- S. Fa, K. Adachi, Y. Nagata, K. Egami, K. Kato and T. Ogoshi, Chem. Sci., 2021, 12, 3483–3488 RSC.
- H. Zhu, Q. Li, Z. Gao, H. Wang, B. Shi, Y. Wu, L. Shangguan, X. Hong, F. Wang and F. Huang, Angew. Chem., Int. Ed., 2020, 59, 10868–10872 CrossRef CAS PubMed.
- L.-L. Wang, Z. Chen, W.-E. Liu, H. Ke, S.-H. Wang and W. Jiang, J. Am. Chem. Soc., 2017, 139, 8436–8439 CrossRef CAS PubMed.
- L.-L. Wang, M. Quan, T.-L. Yang, Z. Chen and W. Jiang, Angew. Chem., Int. Ed., 2020, 59, 24025–24032 CrossRef.
- L.-J. Chen and H.-B. Yang, Acc. Chem. Res., 2018, 51, 2699–2710 CrossRef CAS PubMed.
- H. Zhu, Q. Li, B. Shi, H. Xing, Y. Sun, S. Lu, L. Shangguan, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 17340–17345 CrossRef CAS PubMed.
- G. Albano, G. Pescitelli and L. Di Bari, Chem. Rev., 2020, 120, 10145–10243 CrossRef CAS PubMed.
- H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2tc00032f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |