DOI:
10.1039/D2QI01443B
(Research Article)
Inorg. Chem. Front., 2022,
9, 5735-5744
A Ce-MOF as an alkaline phosphatase mimic: Ce-OH2 sites in catalytic dephosphorylation†
Received
5th July 2022
, Accepted 1st September 2022
First published on 1st September 2022
Abstract
The field of biomimetic catalysis poses several challenges, including selective dephosphorylation of enzymes. Lanthanide-based metal–organic frameworks (Ln-MOFs) are widely used as hydrolytic enzymes due to their well-controlled structural motifs and having comparable enzyme cofactors, making them ideal for biomimetic catalysis. In this work, we have synthesized and structurally characterized a Ce–OH2–Ce motif-containing MOF (Ce-MOF) to mimic the active sites of alkaline phosphatase. Single crystal X-ray diffraction (SXRD) analysis illustrates that the Ce-MOF has a robust ladder-like supramolecular network that is stable in a wide range of solvents and basic aqueous solutions, which is confirmed through independent powder XRD (PXRD) analysis. The catalytic activity of the Ce-MOF was investigated by UV-visible spectroscopy by hydrolyzing the model substrate, the disodium salt of 4-nitrophenyl phosphate (4-NPP), taken in an N-methylmorpholine buffer aqueous solution. Experimental studies reveal that the Ce-MOF has the highest catalytic activity for the hydrolysis of the P–O bond of 4-NPP at pH 9.0. To the best of our knowledge, this is the first time a cerium-MOF has been used as a catalyst in an alkaline medium to mimic phosphatase enzymes. Importantly, the high catalytic activity of the Ce-MOF towards 4-NPP hydrolysis was found owing to the synergistic effect of the Ce(III) ion, which reinforces the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond with the metal, and metal hydroxide activation under basic circumstances. The calculated turnover number (kcat) for 4-nitrophenyl phosphate (4-NPP) hydrolysis was 7.42 × 10−3 min−1. The formation of phosphate ions during the hydrolytic reaction has been monitored through time-dependent 31P-NMR spectroscopy and this provides very crucial information on the possible mechanistic pathway.
O bond with the metal, and metal hydroxide activation under basic circumstances. The calculated turnover number (kcat) for 4-nitrophenyl phosphate (4-NPP) hydrolysis was 7.42 × 10−3 min−1. The formation of phosphate ions during the hydrolytic reaction has been monitored through time-dependent 31P-NMR spectroscopy and this provides very crucial information on the possible mechanistic pathway.
Introduction
Phosphatases are a class of hydrolytic enzymes composed of bivalent Zn(II) ions that are linked by a hydroxide bridge in their active sites and catalyze the hydrolysis of the phosphate ester (P–O) bonds of mono-, di-, and tri-esters, fluorophosphate, fluorophosphonate, phosphoric anhydrides, and many other similar enzymes.1–8 These enzymes not only play an active role in the hydrolysis of organophosphorus compounds in several biochemical processes, such as DNA fragmentation,9,10 RNA replication, and bone metabolism,11 in agriculture (removal of phosphate-containing pesticides)12,13 and in nerve agents14,15 but also selectively govern the phosphorylation of biomolecules in vivo and abnormal hyperphosphorylation, which is strongly linked to Alzheimer's disease, lung cancer, and other human diseases.16–18 Alkaline phosphatase (Scheme 1a) from E. coli (ALP, EC 3.1.3.1), particularly the mammalian enzyme, aids in the hydrolysis of phosphate ester (P–O) bonds and the release of inorganic phosphate in a basic environment. ALP is a non-specific phosphomonoesterase enzyme that is homodimeric and contains 449 amino acids per monomer.19 It is a thermostable enzyme that works best at high pH values. Alkaline phosphatase is widely employed in biology to remove phosphates from DNA and RNA, inhibiting self-ligation.20 As a result, alkaline phosphatase is a critical component of tool enzymes in molecular biology laboratories.21 Natural biological enzymes, fortunately, can rapidly hydrolyze the phosphate ester bond with high catalytic efficiency and specificity, despite the fact that this bond is extremely stable, having a half-life of thousands of years.22 However, numerous inherent flaws in enzymes, such as limited chemical stability, high separation, and purification costs, and recycling issues, limit the range of uses of natural phosphatases. To address the deficiencies of natural phosphatases, it is essential to develop a simulated phosphatase based on a thorough understanding of its structure and mechanism.23,24 Phosphatase biomimetic research is widely used as one of the most effective strategies for overcoming the technological limitations and inherent problems of natural enzymes.25
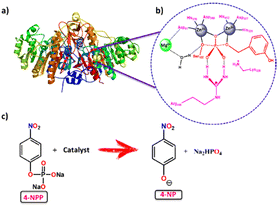 |
| | Scheme 1 Overview of MOF catalysts for the biomimetic hydrolysis of alkaline phosphatase: (a) the structure of alkaline phosphatase (ALP),48 (b) the neighbouring environment of the active site in ALP, and (c) hydrolysis of the phosphate ester bond of 4-NPP using a catalyst. | |
Although there are some reports on imitating phosphatase as hydrolytic metalloenzymatic models using certain homogeneous catalysts such as Ni2+, Cu2+, Mn2+/Mn3+, Fe3+, and Zn2+ complexes as well as metallic nanoclusters,26–31 the most challenging issue is the homogeneous nature of the catalyst. For these homogeneous catalytic systems, the recovery and reuse of the catalysts for prolonged use is a major obstacle to sustainability. To overcome this problem, many heterogeneous catalysts, based on metal oxides,32,33 metal nanoparticles,34,35 and metal–organic frameworks (MOFs),36–39 have been developed. MOFs are extremely promising among such heterogeneous catalysts,40,41 based on the following considerations: (1) MOFs are the naturally right choice for biomimetic studies because they can provide fine-tunable structures for thoughtfully introducing catalytic sites,42 (2) the Lewis acidity and low toxicity of MOFs with metal centers help to achieve the hydrolysis of phosphate ester bonds, which can be useful in biology, and (3) MOFs’ exceptional thermal and chemical stability allows the catalysts to be reused several times in reactions due to their ordered structure. The phosphatase reaction can be carried out efficiently over MOF-based catalysts. Among them, lanthanum-based MOFs demonstrate high catalytic activity towards this hydrolytic reaction because of their high Lewis acidity, oxidation state, charge density, coordination number, unique binding sites, and remarkable stability in diverse solvents and acid/base solutions.43 Zr6 cluster-containing MOFs such as Uio-66, NU-1000, and MOF-808 are very much familiar with higher rates for organophosphate-based nerve agents.44,45 Farah's group investigated a study comparing the activity of Ce(IV) and Zr(IV) based MOFs in the hydrolysis process of the phosphate ester bond and found that Ce(IV) had superior activity than Zr(IV).46 This higher activity of Ce(IV) towards this reaction is owing to the mixing of 4f orbitals of Ce(IV) with the orbitals of the PO bond.47 From the standpoint of catalytic design, the active site of alkaline phosphatase has a ZnII–OH–ZnII motif with a MgII ion (Scheme 1b), which works together to cleave P–O bonds; one ZnII center binds and activates the P–O bond, while the other transfers an OH− to promote the cleavage of an –OR group from the substrate. Metal ions influence the following factors in phosphate ester bond cleavage: (i) metal ion coordination stabilizes the transition state, (ii) formation of a nucleophilic metal coordinated hydroxide, (iii) hydrolyzed product's stabilization, and (iv) product construction as well as catalyst regeneration.28 With these ideas in mind, we propose developing a CeIII–OH2–CeIII motif-containing metal–organic framework (MOF) to mimic phosphatase, which is most similar to natural enzyme's ZnII–OH–ZnII active site.
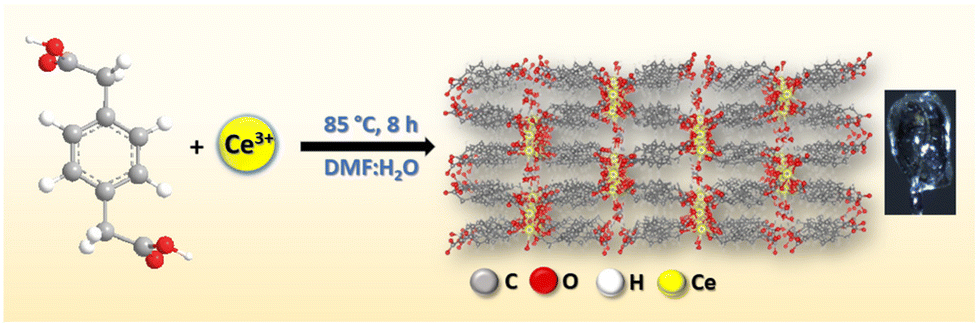
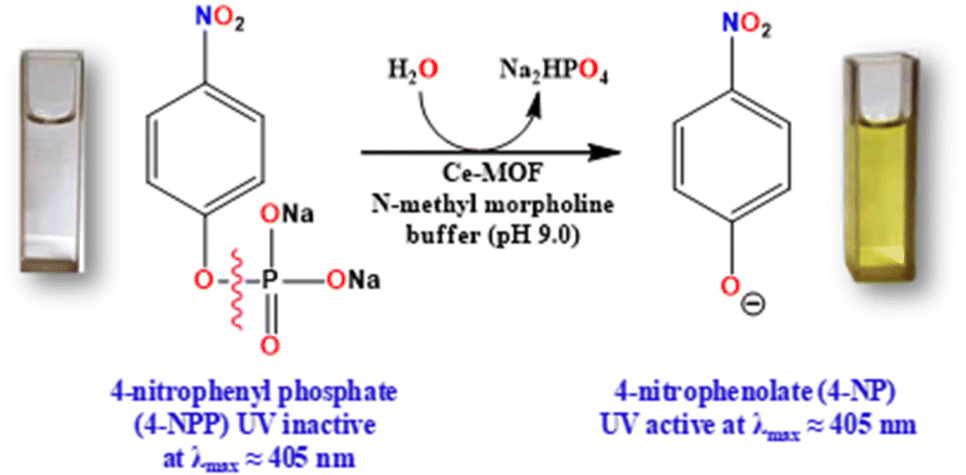
Therefore, in this context, we have designed and synthesized Ce-MOF [{Ce2(PDA)3(H2O)}·2H2O] (PDA = 1,4-phenylendiacetate dianion) through hydrothermolysis employing the ligand 1,4-phenyldiacetic acid and Ce(NO3)3·6H2O as the salt in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. Single-crystal X-ray diffraction was used to characterize its structure. The structure has a three-dimensional supramolecular network that looks like a ladder. The Ce-MOF has excellent stability in a variety of solvents and basic aqueous solutions. The catalytic activity of the Ce-MOF was investigated by hydrolyzing the model substrate, the disodium salt of 4-nitrophenyl phosphate (4-NPP), in N-methylmorpholine buffering aqueous solution (pH 9.0) and determining the generation rate of the product, 4-nitrophenol (4-NP) (Scheme 1c), using a UV-vis experimental technique to monitor the increment of the absorbance peak at 405 nm. For the hydrolysis of 4-nitrophenyl phosphate (4-NPP), the calculated turnover number (kcat) is 7.42 × 10−3 min−1. The existence of distinct active sites (Ce–H2O) in the Ce-MOF allows it to successfully mimic alkaline phosphatase. Control experiments show that the Ce metal in the Ce-MOF is important in the unique binding site and that water molecules can help to break the phosphate ester bond of 4-NPP. A probable mechanistic pathway has been explored with the formation of phosphate ions during the hydrolytic reaction and this is monitored by time-dependent 31P-NMR spectroscopic studies.
:2. Single-crystal X-ray diffraction was used to characterize its structure. The structure has a three-dimensional supramolecular network that looks like a ladder. The Ce-MOF has excellent stability in a variety of solvents and basic aqueous solutions. The catalytic activity of the Ce-MOF was investigated by hydrolyzing the model substrate, the disodium salt of 4-nitrophenyl phosphate (4-NPP), in N-methylmorpholine buffering aqueous solution (pH 9.0) and determining the generation rate of the product, 4-nitrophenol (4-NP) (Scheme 1c), using a UV-vis experimental technique to monitor the increment of the absorbance peak at 405 nm. For the hydrolysis of 4-nitrophenyl phosphate (4-NPP), the calculated turnover number (kcat) is 7.42 × 10−3 min−1. The existence of distinct active sites (Ce–H2O) in the Ce-MOF allows it to successfully mimic alkaline phosphatase. Control experiments show that the Ce metal in the Ce-MOF is important in the unique binding site and that water molecules can help to break the phosphate ester bond of 4-NPP. A probable mechanistic pathway has been explored with the formation of phosphate ions during the hydrolytic reaction and this is monitored by time-dependent 31P-NMR spectroscopic studies.
Experimental section
Materials and instrumentation
For both synthesis and applications, required chemicals such as 1,4-phenyldiacetic acid (Sigma Aldrich), ceric nitrate nonahydrate (Spectrochem, India), and DMF (Rankem, India) were used without any further purification. The powder X-ray diffraction (PXRD) patterns of the as-synthesized bulk sample were investigated using a Bruker AXS D8 Advanced SWAX diffractometer (Cu Kα, λ = 0.15406 nm). The percentage of carbon, hydrogen, and nitrogen was analysed using a PerkinElmer 240C CHN analyser. X-ray photoelectron spectroscopic analysis was performed using an Omicron Nanotechnology XPS 0571 spectrometer. A PerkinElmer spectrophotometer was used for the recording of the FT-IR spectra in the range of 400–4000 cm−1 with solid KBr pellets. The solid-state UV-vis DRS spectroscopic studies of the ligand and Ce-MOF, and the solution-state UV-vis spectral analysis of the Ce-MOF for alkaline phosphatase activity were performed using a Shimadzu UV-2401PC (Japan) instrument. The scanning electron microscopy (SEM) analysis of the Ce-MOF was performed with a JEOL JSM-6700F field emission microscope. The thermal stability (TGA) profile of the Ce-MOF was investigated using a TA Instruments TA-SDQ Q-600 thermal analyzer under continuous airflow. 31P-NMR spectra were recorded on a Bruker AVANCE REO-600 MHz spectrometer.
Synthesis of [{Ce2(PDA)3(H2O)}.2H2O]∞ (Ce-MOF)
In a typical synthesis of the Ce-MOF (Scheme 2), the ligand 1,4-phenyldiacetic acid (PDA) (0.1 mmol, 19.42 mg) and Ce(NO3)3·6H2O (0.2 mmol, 86.84 mg) were placed in a Teflon lined glass vial, and 4 mL of DMF and 2 mL of H2O solvent were added to this reaction mixture with continuous stirring for 30 minutes. Under static conditions, this reaction mixture was heated to 85 °C for 8 hours. Colourless diffractive needle-shaped crystals were found at the bottom of the glass vial after it was slowly cooled to room temperature.
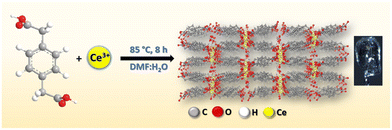 |
| | Scheme 2 Synthetic route to the Ce-MOF. | |
Yield: ∼72% based on PDA. Anal. calcd for C30H30Ce2O15: C, 39.56; H, 3.32. Found: C, 39.31; H, 3.12. FT-IR (4000–400 cm−1; KBr pellet, ν/cm−1) 3459 (br s), 2905 (w), 1585 (S), 1525 (W), 1375(W), 1376 (S), 1091 (W), 769 (W), 703 (W).
X-ray crystallographic data collection and refinement
A single crystal of Ce-MOF was mounted on the tip of the goniometer head in a Bruker Smart Apex diffractometer equipped with a CCD detector. X-ray data were collected using graphite monochromated Mo-K radiation (λ = 0.71073 Å). For unit cell refinement,49 integration, indexing, scaling, and other tasks, Bruker's APEX-3 software was used. The structure was analysed using direct methods and refined using full-matrix least-squares methods on F2 with SHELXTL version 6.1.50 The WinGX System, Ver2013.3.45, was used to refine the structure.51 Crystal data, as well as the details of refinements, are given in Table 1.
Table 1 Crystal parameters of the Ce-MOF
| CCDC no. |
2163330† |
| Empirical formula |
C30H30Ce2O15 |
| Formula weight |
910.78 |
| Temperature/K |
145(2) |
| Crystal system |
Monoclinic |
| Space group |
P21/c |
|
a/Å |
22.0700(13) |
|
b/Å |
10.1231(6) |
|
c/Å |
14.1332(8) |
|
α/° |
90 |
|
β/° |
91.600(2) |
|
γ/° |
90 |
| Volume/Å3 |
3156.4(3) |
|
Z
|
4 |
|
ρ
calc(g cm−3) |
1.917 |
|
μ/mm−1 |
2.921 |
|
F(000) |
2960.0 |
| Crystal size/mm3 |
0.6 × 0.4 × 0.2 |
| Radiation |
Mo Kα (λ = 0.71073) |
| 2θ range for data collection/° |
4.95 to 51.444 |
| Index ranges |
−22 ≤ h ≤ 26, −12 ≤ k ≤ 12, −17 ≤ l ≤ 17 |
| Reflections collected |
30528 |
| Independent reflections |
6014 [Rint = 0.1184, Rsigma = 0.0630] |
| Data/restraints/parameters |
6014/0/432 |
| Goodness-of-fit on F2 |
1.053 |
| Final R indexes [I ≥ 2σ (I)] |
R
1 = 0.0362, wR2 = 0.0876 |
| Final R indexes [all data] |
R
1 = 0.0459, wR2 = 0.0975 |
| Largest diff. peak/hole/e Å−3 |
1.28/−1.35 |
Preparation of the SEM sample
The Ce-MOF was dispersed in a water medium to create a homogeneous solution by sonication for 1 h. On a glass disk, the dispersed solution was added dropwise. The disk was then dried overnight at room temperature in air before being subjected to a SEM experiment to capture the images.
Pphosphatase-like activity of 4-NPP
The catalytic hydrolysis reaction (dephosphorylation reaction) of 4-NPP was monitored by UV-vis spectroscopy using disodium (4-nitrophenyl) phosphate (4-NPP) hexahydrate as the model substrate. The formation of the 4-nitrophenolate ion from the cleavage of the phosphoester (P–O) bond in 4-NPP at 25 °C in the presence of the catalyst (Ce-MOF) was monitored by an increase in the absorption band at λmax ∼ 405 nm. The catalyst Ce-MOF (5 × 10−3 M) was dispersed in an N-methylmorpholine buffer medium. In a 3 mL cuvette, 30 μL of catalyst solution was added directly to the buffer solution, followed by the addition of (150 μL) 1 × 10−3 M 4-NPP at 25 °C (substrate:catalyst = 5:1). After appropriate shaking, the reactive substance was examined using a UV-vis spectrophotometer as quickly as possible. Following that, we obtained the data collected with the help of a kinetic model, as well as the absorbance–time plots. Then, using the steady-state kinetics method, we studied the kinetics of the Ce-MOF by looking at the rate of increase in the absorption band at λmax ∼ 405 nm with an increasing order of 4-nitrophenolate concentration. The substrate concentrations ranged from 0.25 to 1.8 mM, and the catalyst concentration was maintained constant at 5 × 10−5 (M). Saturation kinetics with Michaelis–Menten-like behaviour was revealed by determining the initial rates as a function of the 4-NPP concentration. Michaelis–Menten's approach for enzyme kinetics was used to determine the kinetic parameters (kcat, Vmax, kM) for the hydrolysis of the phosphate ester bond of 4-NPP.
Results and discussion
X-ray-quality single crystals of Ce-MOF were synthesized in DMF:H2O medium under solvothermal conditions, using a molar ratio of 1:2 of 1,4-phenyldiacetic acid (PDA) and Ce(NO3)3·6H2O (where PDA acts as a linker and the Ce(NO3)3·6H2O salt acts as a holder). Jiang's group previously reported Ln-based MOFs made with 1,4-phenyldicarboxylic acid in water:dimethylacetamide medium at 105 °C for 48 h, which are structurally identical to ours.52 However, the synthetic process took a long time and required a high temperature. We changed the reaction conditions in this study to make it take less time and have a lower temperature. A colorless single crystal of Ce-MOF was finally characterized using several physicochemical methods such as SCXRD, FT-IR, PXRD, solid-state UV-vis, TGA, and SEM.
Crystal structure description of [{Ce2(PDA)3(H2O)}.2H2O]∞ (Ce-MOF)
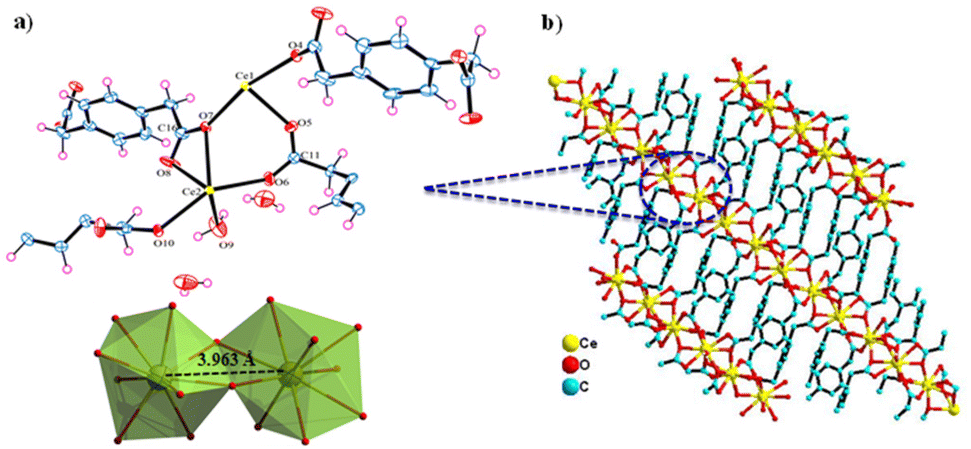
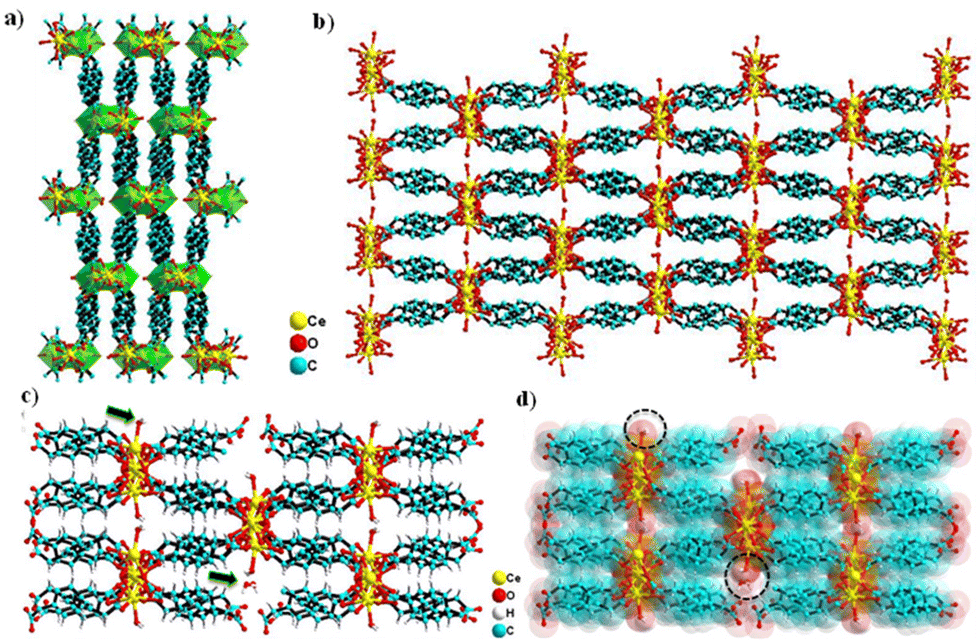
The Ce-MOF crystallizes in the monoclinic P21/c space group. The asymmetric unit comprises two Ce3+ cations that are crystallographically independent, four PDA anions (two full and two half molecules of PDA), one coordinated water molecule, and two uncoordinated water molecules outside the coordination sphere as the solvent of crystallization (Fig. 1a and b). Both cerium(III) ions have a nine-coordinated distorted-capped-square-antiprismatic geometry, with Ce1 being coordinated by nine carboxylate oxygen atoms from six distinct PDA ligands, of which O2, O4, O7, and O13 are situated at the equatorial sites, and O1, O5, O10, O11, and O12 are situated at the vertexes. Ce2 is coordinated by eight carboxylate oxygen atoms and a water molecule where O3, O7, and O8 are filled in the equatorial sites and O2, O4, O6, O10, O13, and OW9 are capped in the vertexes. The Ce–O (carboxylate) bond distance varies between 2.391 and 2.704 Å, while the Ce–O (aqua) bond distance is 2.458 Å. The trans bond angles of O–Ce1–O and O–Ce2–O are 158.26° and 145.69°, respectively, whereas the equatorial bond angles of O–Ce1–O are 103.31–119.64° and that of O–Ce2–O is just 85.21°. In conclusion, the O–Ce2–O equatorial bond angle is much smaller than other O–Ce1–O equatorial bond angles. The metal–metal bond distance of Ce(1) …Ce(2) is 3.963 Å based on the asymmetric unit (Fig. 1a). Selected bond distances and angles are presented in Tables S1 and S2.† Two carboxylate groups (–COO−) of a PDA ligand attach to the Ce3+ center in the η1 and η2 binding modes, resulting in a 1D network with an extended zigzag orientation. Such a 1D network involving the Ce3+ and carboxylate groups of PDA are further cross-linked via the –CH2C6H4CH2– spacers coming from the PDA ligand, creating a 3D ladder-like supramolecular network (Fig. 2a and b).
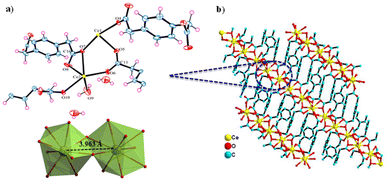 |
| | Fig. 1 Crystal structure of Ce-MOF: (a) asymmetric unit with a distorted-capped-square-antiprismatic geometry around the metal (Ce1 and Ce2) core and (b) crystal packing comprised of Ce3+ and PDA anions viewed along the crystallographic b axis. | |
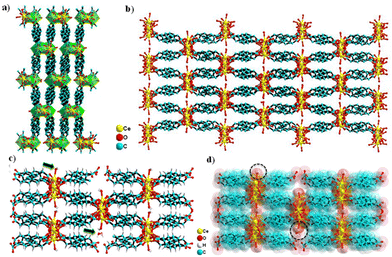 |
| | Fig. 2 Crystal structure illustration of the Ce-MOF: (a) 3D ladder-like structure (with a polyhedral view) viewed along the crystallographic c axis, (b) 3D network viewed along the crystallographic c axis and the cross-section of the open channel in the Ce-MOF, (c) ball and stick model, and (d) superimposed space-filling model. | |
The lattice water molecules hang in the channels, while the coordinate water molecules drift away from the Ce-MOF frame and into the channels. (Fig. 2c and d). The TOPOS study has provided a deeper understanding of the crystal structure by revealing that the Ce-MOF exhibits a 4-nodal network 4,4,6,6T3 topology with the point symbol {42.62.82}{43.63}2{46.67.82}{48.67} (Fig. S2†). Selected crystallographic data and refinement details are given in Table 1.
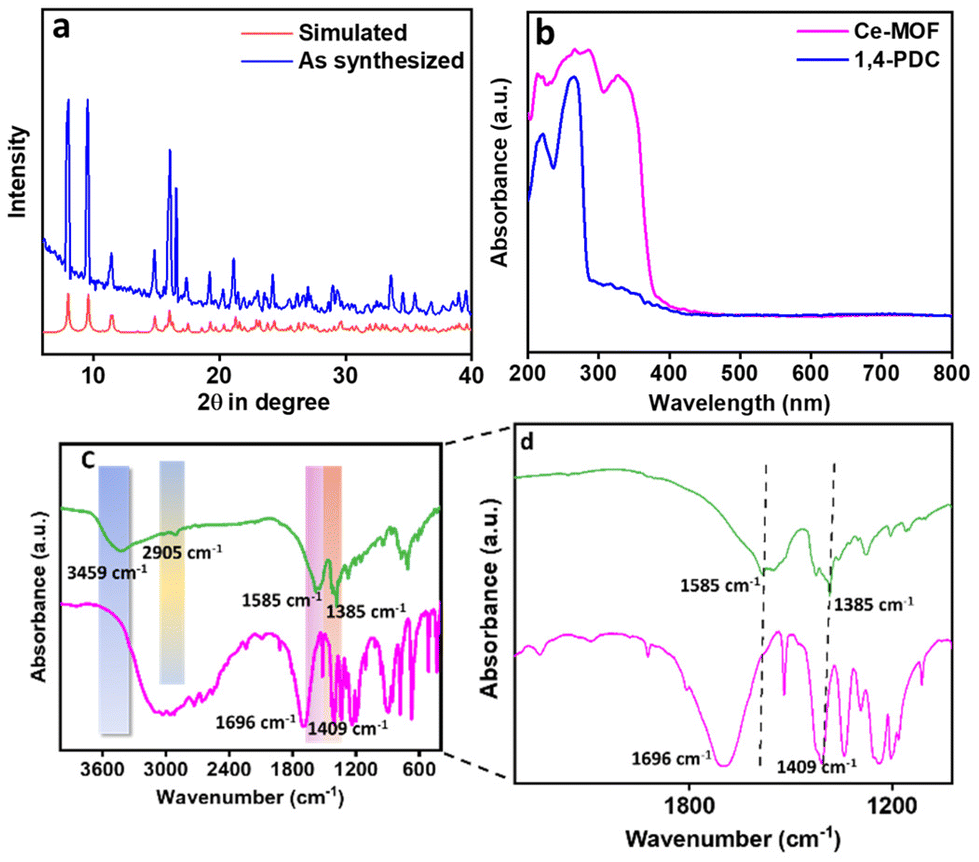
The bulk phase purity of the Ce-MOF was determined by examining the PXRD patterns (see Fig. 3a). The figure shows that the bulk Ce-MOF PXRD profile and the simulated one are very similar. The micropores in the Ce-MOF are represented by two peaks at 8.03 and 9.54°. We evaluated the sample stability in various solvent media once more (Fig. S3†). The UV-vis DRS spectra of both the Ce-MOF and ligand were recorded separately and are shown in Fig. 3b. In 1,4-PDC, peaks were near 220–260 nm for π–π* transition. These peaks shifted towards 287 nm in the Ce-MOF. A new peak was observed at 327 nm due to the electronic interaction between the metal and the ligand moiety. Furthermore, the FT-IR spectra, as shown in Fig. 3c and d, corroborated the bonding connectivity between the metal and the ligand. Because of the asymmetric and symmetric stretching of the –COOH group in 1,4-PDC, the ligand showed two distinct peaks at 1696 and 1409 cm−1. The absence of these two peaks in the Ce-MOF confirms the coordination of the metal and ligand. Two peaks in the Ce-MOF were found at 1585 and 1385 cm−1, indicating that the metal and –COOH of the ligand were connected.53 The observed peak at 3459 cm−1 could be attributed to the presence of surface water. The coordinated water molecules exhibit a peak at 769 cm−1. The peak near 2905 cm−1 is due to the presence of the –C–H bond of the methylene moiety present in the linker. The sample was soaked in N-methylmorpholine buffer (pH 9.0) before collecting the FT-IR data, which are shown in Fig. S4,† indicating that the Ce-MOF has good stability in N-methylmorpholine buffer solution.
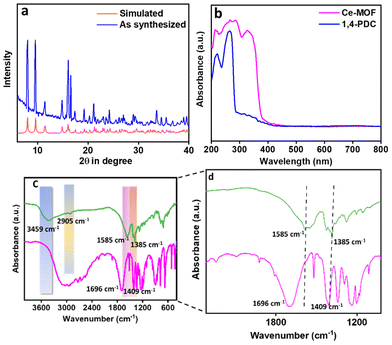 |
| | Fig. 3 (a) Simulated (red) and experimental (blue) PXRD patterns of the as-synthesized Ce-MOF (b) UV-vis DRS spectra of both the ligand and the Ce-MOF, and FT-IR spectra of both the ligand (green) and the Ce-MOF (pink): (c) zoomed out and (d) zoomed in images. | |
The chemical oxidation states of C, O and Ce related to the Ce-MOF were assessed by means of XPS analysis and this is shown in Fig. S5.† The C1s analysis showed three peaks at 284.4, 285.5 and 288.4 eV representing C(sp2), C(sp3) and C–O, respectively. The oxidation state of Ce was investigated by analysing the spectrum of the Ce 3d core level. Four peaks are produced by the Ce(III) state at energies of 880.6, 885.6, 900.0, and 904.8 eV.54,55 The spectrum shows the splitting of the peaks into two, which are the 3d5/2 and 3d3/2 peaks of Ce, respectively, at 885.6 and 904.8 eV. The two peaks observed at 531.6 and 532.8 eV could be attributed to the CO and C–O, respectively. The TGA profile of the Ce-MOF is shown in Fig. S6.† The steep weight loss of 5.93% at 315 °C is due to the loss of coordinated water and lattice water molecules from the Ce-MOF. The overall TGA profile suggests that the Ce-MOF is highly stable up to 394 °C and beyond this temperature the structure collapsed. We used scanning electron microscopy techniques to assess the morphological view of the Ce-MOF, as shown in Fig. 4. This study demonstrated the hexagonal-shaped prism-like structure of the Ce-MOF with a uniform particle distribution throughout the region. We also performed the elemental mapping of the Ce-MOF (Fig. 4c) to ensure atom distribution, which validates the finely distributed atoms. The chemical composition of the nano prisms is believed to be the same as that of the matching as-synthesized bulk crystals, according to energy dispersive X-ray (EDX) spectroscopy (Fig. S7 in the ESI†).
 |
| | Fig. 4 Scanning electron microscopy (SEM) analysis of the Ce-MOF: (a) low magnification image; (b) high magnification image; (c) elemental mapping of the Ce-MOF. | |
Phosphatase activity
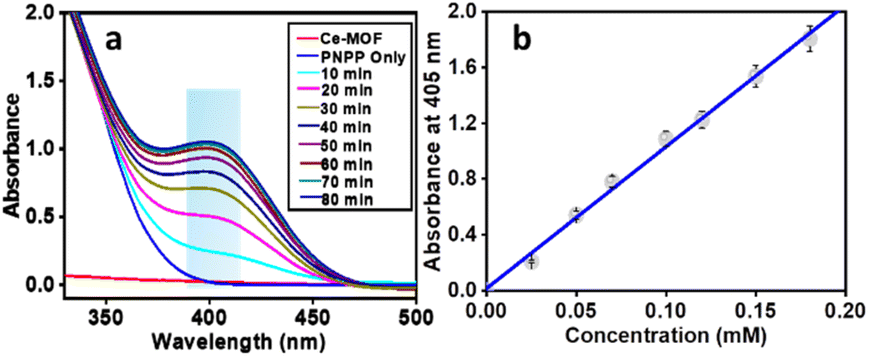
The phosphatase-like activity of the Ce-MOF was studied using the disodium salt of (4-nitrophenyl)-phosphate hexahydrate (4-NPP) as the model substrate. The catalytic hydrolysis of the phosphate ester (P–O) bond in 4-NPP generates free phosphate anions and 4-nitrophenolate (4-NP) in an aqueous N-methylmorpholine buffer solution (Scheme 3). The substrate 4-NPP exhibits an absorption peak at 310 nm and its hydrolysis product 4-NP displays a characteristic absorption peak at 405 nm in the UV-vis spectra, which makes it very easy to monitor the catalytic hydrolysis reaction of 4-NPP and the production of 4-NP. Thus, the hydrolytic behavior of the Ce-MOF was observed spectrophotometrically by tracking the time evolution of 4-nitrophenolate (λmax ≈ 405 nm) over a wavelength range of 200 to 600 nm and the color change from colorless to pale yellow to deep yellow with time (Fig. S8†) in an aqueous N-methylmorpholine buffer medium. The spectral change behaviour of the Ce-MOF and the standard curve of 4-NPP are shown in Fig. 5a and b, respectively. Firstly, we optimized the catalytic conditions using five buffer systems (HEPES buffer, Tris-HCl buffer, carbonate buffer, N-methylmorpholine buffer, and water).
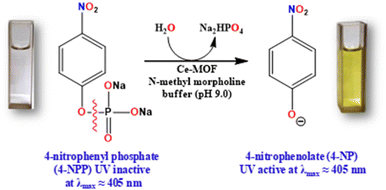 |
| | Scheme 3 Overview of Ce-MOF catalysts for the biomimetic hydrolysis of the phosphate ester (P–O) bond of (4-nitrophenyl)-phosphate hexahydrate (4-NPP). | |
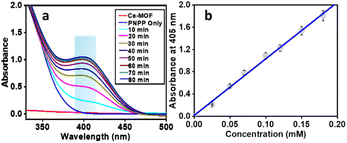 |
| | Fig. 5 Wavelength scan for the hydrolysis of 4-NPP in the presence of the Ce-MOF in N-methylmorpholine buffer medium recorded at 25 °C at intervals of 10 min (a) and a standard curve of 4-NPP for determining the phosphatase activity (b). | |
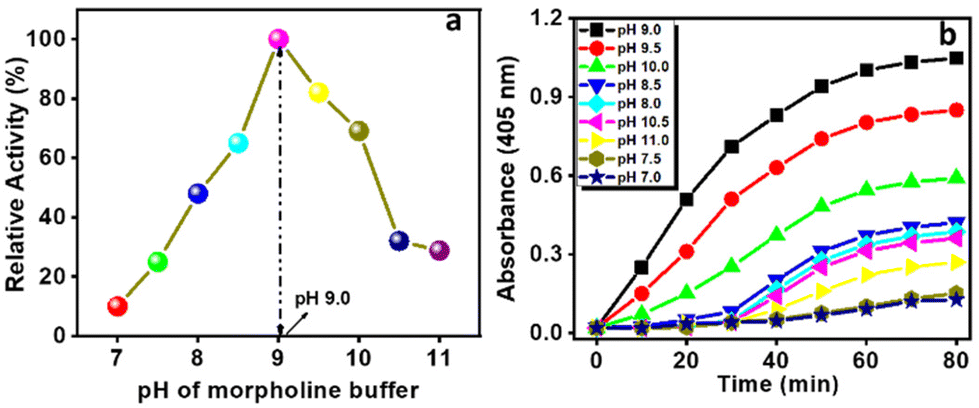
Here, we also employed the pH-dependent hydrolytic behaviour of the Ce-MOF to estimate the optimum activity of the catalyst at 25 °C as shown in Fig. 6a and b.
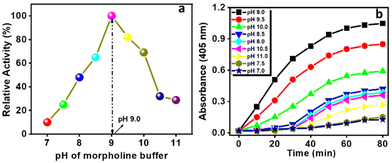 |
| | Fig. 6 The catalytic activity (a) and absorbance (at 405 nm) vs. time plot (b) of the Ce-MOF at different pH values of N-methylmorpholine buffer. | |
Among them, N-methylmorpholine buffer exhibited paramount performance at room temperature for this type of catalytic hydrolysis reaction (Fig. S9†) and was employed as a default buffer in subsequent experiments.
The activity of the catalytic hydrolysis reaction increased with the increase of the pH value from 7 and optimum activity was observed at pH 9.0. In the neutral solution (pH 7.0) (Fig. 6a), the catalytic activity of the Ce-MOF was too low (10% conversion). As the pH of the solution was increased from 7.0 to 8.5, the conversion of 4-NP considerably increased to 64% and at pH 9.0 the activity reached 100%, signifying pH-dependent catalytic performance.
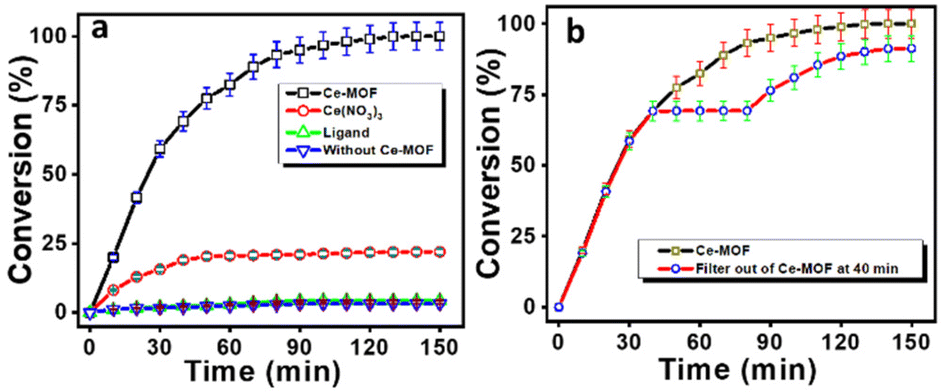
Thereafter, further increasing the pH values of the reaction solutions resulted in slightly diminishing catalytic performance as the increase in pH causes loss of crystallinity of the Ce-MOF and its significant degradation. The stability of the Ce-MOF towards the base can be obtained from pH-dependent PXRD (Fig. S10†) and FT-IR (Fig. S11†) patterns. The conversion profiles of the Ce-MOF, Ce(NO3)3, ligand 1,4 PDC, and blank experiment in N-methylmorpholine buffer solution (pH 9.0) are shown in Fig. 7a. In the conversion profiles (Fig. 7a), the Ce-MOF exhibited excellent catalytic performance towards 4-NPP hydrolysis after 120 min, whereas the raw material of the as-synthesized Ce-MOF, such as ligand 1,4 PDC, had no effect on the hydrolysis of 4-NPP but Ce(NO3)3 showed slight activity (<25%); but it was far less than that of the catalytic activity of the Ce-MOF (Table S3†).
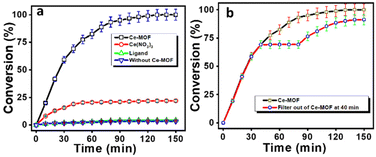 |
| | Fig. 7 Hydrolysis profiles of 4-NPP with the Ce-MOF, Ce(NO3)3, ligand 1,4 PDC, and blank experiment (a). The catalytic activity of the Ce-MOF, where the catalyst was filtered out at 40 min for the hydrolysis of the phosphate ester bond of 4-NPP (b). | |
We also investigated the nature of the hydrolytic reaction. After the removal of the Ce-MOF catalyst from the reaction mixture by centrifugation, no reaction was noticed at 40 min (Fig. 7b). Afterward, the reaction again started after adding the Ce-MOF catalyst into the reaction system at 80 min.
Kinetic studies of the hydrolysis of 4-NPP
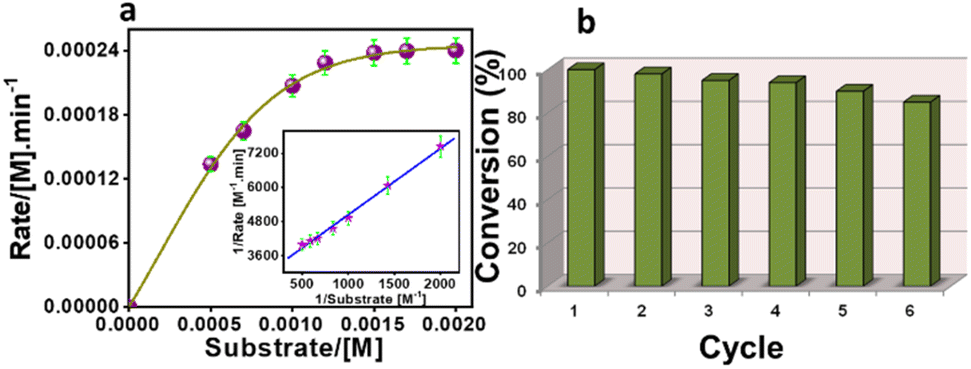
The kinetic study of the Ce-MOF was performed by the steady-state kinetics method following the rate of increase in the absorption band at λmax ∼ 405 nm, corresponding to an increase in the concentration of 4-nitrophenolate in aqueous solution. To determine the dependence of rate constants on the substrate concentration, the Ce-MOF was treated with seven different concentrations of the substrate. The binding constant (KM), maximum velocity (Vmax), and rate constant for the hydrolysis of substrates (i.e., turnover number, kcat) were calculated for the Ce-MOF using a Lineweaver–Burk plot of 1/V vs. 1/[S], using the equation 1/V = {KM/Vmax} × {1/[S]} + 1/Vmax as per the Michaelis–Menten treatment of enzymatic kinetics.56 The Lineweaver–Burk plot and enzymatic kinetics plot of the rate vs. substrate concentrations of the Ce-MOF are shown in Fig. 8a. All the kinetic parameters are summarized in Table 2. The calculated turnover number (kcat in min−1) of the Ce-MOF was 7.42 × 10−3 min−1.
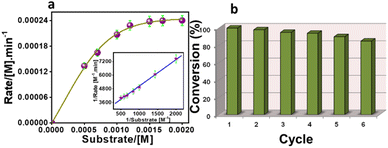 |
| | Fig. 8 Steady-state kinetic assays of the Ce-MOF (a), where the Lineweaver–Burk plot is shown in the inset. Recycling results in the hydrolysis of 4-NPP in the presence of the Ce-MOF (b). | |
Table 2 First-order kinetic parameters for phosphatase activity obtained by the Michaelis–Menten treatment of the Ce-MOF
| |
k
cat (min−1) |
V
max
|
K
M
|
| Ce-MOF |
7.42 × 10−3 |
3.7 × 10−4 |
7.2 × 10−4 |
A comparison table is provided in Table S4† to compare the kcat values with those of previously reported heterogeneous catalysts. The Ce-MOF catalyst was reused for up to six reaction cycles under the same conditions, where only a very little reduction in the catalytic performance was observed (Fig. 8b). The regenerated catalyst was further characterized by SEM, PXRD analysis, and FT-IR spectroscopy (Fig. S12, S13, and S14†). These characterization results indicated that the catalyst still maintained its integrity after the sixth catalytic cycle.
Mechanistic insight
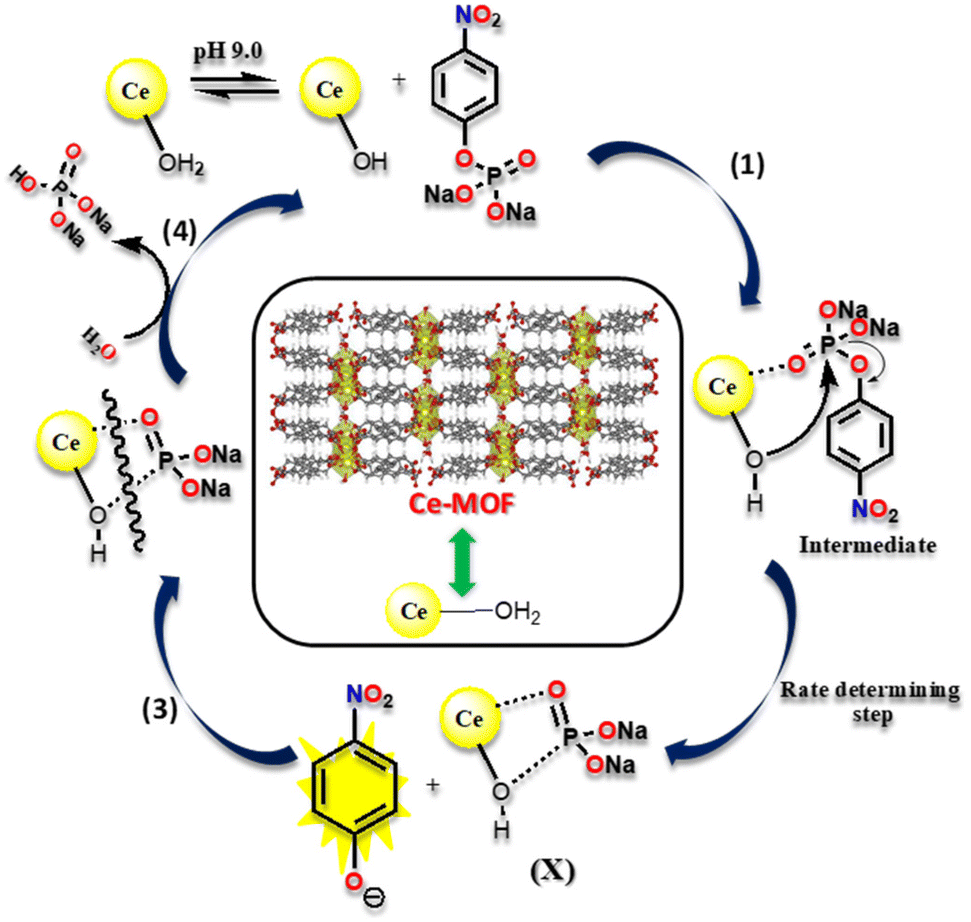
Fig. 9 depicts a possible reaction mechanism for the catalytic hydrolysis of 4-NPP over the Ce-MOF. The catalytic reaction occurs at the surface of the Ce-MOF catalyst because the water molecules obstruct its inner pores. In the rate-determining step, the surface Ce-bound water molecules and 4-NPP react with each other and 4-NP is released from the surface of the Ce-MOF catalyst (Fig. S15†). To begin with, N-methylmorpholine abstracts protons from Ce-bound water (Ce-H2O) molecules in an alkaline (N-methylmorpholine buffer solution) environment, resulting in the formation of nucleophilic cerium hydroxide (Ce–OH) species.57,58
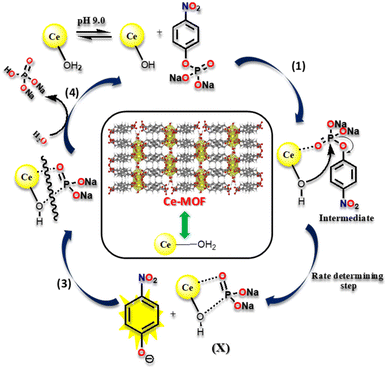 |
| | Fig. 9 A pictorial illustration of the catalytic hydrolysis cleavage mechanism of 4-NPP by the Ce-MOF. | |
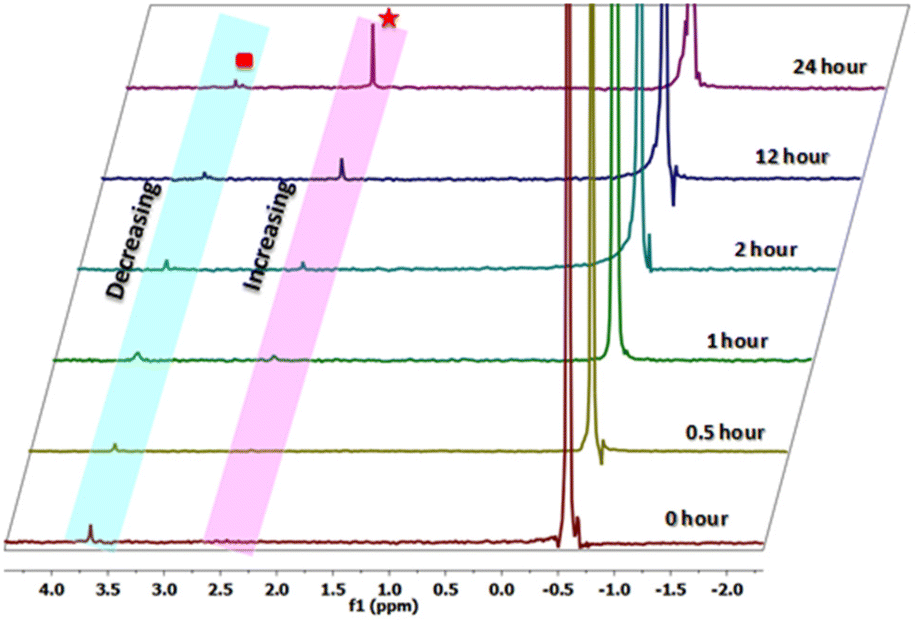
In step 1, the oxygen atom (OP–) of 4-NPP coordinates with the aqua-hydroxyl active species Ce metal centre as it contains vacant 4f orbitals to form an intermediate species, which is useful for converting an intermolecular reaction to an intramolecular reaction. Step 2 involves the attack of the phosphorous atom of the 4-NPP molecule by an intramolecular metal hydroxide, which aids the release of the 4-nitrophenolate ion from the surface of the Ce-MOF catalyst, which was monitored using UV-vis spectroscopy. Furthermore, step 3 involves the breaking of the X species with water and the removal of the phosphate ion from the active species (Ce–OH). This has been observed from 31P-NMR analysis (Fig. 10). In step 4, the regenerated active Ce-hydroxide species coordinates to 4-NPP and enters into the next cycle.
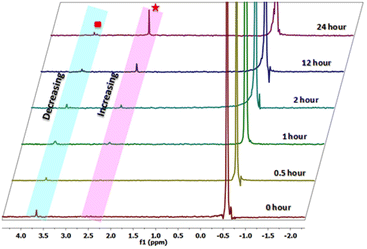 |
| | Fig. 10
31P-NMR spectra showing the species Na2HPO4 generated during the hydrolysis of 4-NPP in an H2O:D2O (90:10) medium at 25 °C. Assignments:  4-NPP + N-methyl morpholine adduct, 4-NPP + N-methyl morpholine adduct,  Na2HPO4 (inorganic phosphate). Data were collected at 0 h, 0.5 h, 1 h, 2 h, 12 h and 24 h after the addition of the Ce-MOF. Na2HPO4 (inorganic phosphate). Data were collected at 0 h, 0.5 h, 1 h, 2 h, 12 h and 24 h after the addition of the Ce-MOF. | |
Conclusion
Designing MOF-based biomimetic catalysts that mimic enzyme activity has emerged as one of the most promising areas of biomimetic research. In this regard, we have developed a Ce-MOF with a 1,4-phenylendiacetate linker and its Ce(III) node closely resembles the active sites of alkaline phosphate (ALP). The superior stability of the Ce-MOF has been explored in a variety of solvents and basic aqueous buffering solutions. In an N-methylmorpholine buffered environment (pH 9.0), the Ce-MOF possesses good catalytic activity for the hydrolysis of the phosphate ester bond of 4-NPP. To our knowledge, this is the first time that a cerium-MOF has been exploited as a catalyst in an alkaline medium to mimic phosphatase enzymes. Importantly, the high catalytic activity of the Ce-MOF towards 4-NPP hydrolysis was found owing to the synergistic effect of the Ce(III) ion, which reinforces the PO bond with the metal, and metal hydroxide activation under basic circumstances. The calculated turnover number (kcat) for the hydrolysis of 4-nitrophenylphosphate (4-NPP) is 7.42 × 10−3 min−1. Control experiments suggest that the Ce metal of the Ce-MOF plays an important role in the unique binding site, and that water molecules can favor the breaking of the phosphate ester bond. The generation of phosphate ions during the hydrolytic reaction, which is monitored by time-dependent 31P-NMR spectra, has been explored as a possible mechanistic approach and this will open up new opportunities in biomimetic catalysis.
Author contributions
Sudip Bhattacharjee: methodology, data curation, formal analysis, and writing draft manuscript. Tonmoy Chakraborty: methodology, data curation, formal analysis, and writing draft manuscript. Asim Bhaumik: supervision, funding acquisition, formal analysis, and writing–reviewing and editing.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
SB thanks the IGSTC, New Delhi, for a Junior Research Fellowship, and TC thanks the IACS, Kolkata, for a Research Associate Fellowship. TC and AB would like to thank Greenitio Pte. Ltd, Singapore for project research grant (grant number: Appt./Greenitio-Proj/SMS/RA-1/AB/2022-02). AB thanks the IGSTC, New Delhi, for an Indo-German project research grant (Project IGSTC/Call2018/CO2BioFeed/15/2019-20).
References
- S. H. Gellman, R. Petter and R. Breslow, Catalytic hydrolysis of a phosphate triester by tetracoordinated zinc compounds, J. Am. Chem. Soc., 1986, 108, 2388–2394 CrossRef CAS PubMed.
- D. R. Jones, L. F. Lindoy and A. M. Sargeson, Enhanced base hydrolysis of coordinated phosphate esters: the reactivity of an unusual cobalt(III) amine dimer, J. Am. Chem. Soc., 1984, 106, 7807–7819 CrossRef CAS.
- B. Anderson, R. M. Milburn, J. M. Harrowfield, G. B. Robertson and A. M. Sargeson, Cobalt(III)-promoted hydrolysis of a phosphate ester, J. Am. Chem. Soc., 1977, 99, 2652–2661 CrossRef CAS.
- F. J. Farrell, W. A. Kjellstrom and T. G. Spiro, Metal ion activation of phosphate transfer by bidentate coordination, Science, 1969, 164, 320–321 CrossRef CAS.
- H. Sigel, F. Hofstetter, R. B. Martin, R. M. Milburn, V. Scheller-Krattiger and K. H. Scheller, Hydrolysis of Nucleoside Phosphates. 8. General Considerations of Transphosphorylations: Mechanism of the Metal Ion Facilitated Dephosphorylation of Nucleoside 5′-triphosphates Including Promotion of ATP Dephosphorylation by Addition of Adenosine 5′-monophosphate, J. Am. Chem. Soc., 1984, 106, 7935–7946 CrossRef CAS.
- R. N. Bose, R. D. Cornelius and R. E. Violo, Kinetics and Mechanisms of Platinum(II)-Promoted Hydrolysis of Inorganic Poly Phosphates, Inorg. Chem., 1985, 24, 3989–3996 CrossRef CAS.
- M. J. Jedrjezas and P. Setlow, Comparison of the Binuclear Metalloenzymes Diphosphoglycerate-Independent Phosphoglycerate Mutase and Alkaline Phosphatase: Their Mechanism of Catalysis via a Phosphoserine Intermediate, Chem. Rev., 2001, 101, 607–618 CrossRef.
- Y. Y. Guo, X. Q. Li, Y. M. Dong and G. L. Wang, Acid Phosphatase Invoked Exquisite Enzyme Cascade for Amplified Colorimetric Bioassay, ACS Sustainable Chem. Eng., 2019, 7, 7572–7579 CrossRef CAS.
- P. U. Maheswari, S. Roy, H. Dendulk, S. Barends, G. V. Wezel and J. Reedijk, The square-planar cytotoxic [CuII(pyrimol)Cl] complex acts as an efficient DNA cleaver without reductant, J. Am. Chem. Soc., 2006, 128, 710–711 CrossRef CAS.
- M. E. Branum, A. K. Tipton, S. Zhu and L. Que, Double-strand hydrolysis of plasmid dna by dicerium complexes at 37 °C, J. Am. Chem. Soc., 2001, 123, 1898–1904 CrossRef CAS PubMed.
- F. Mancin, P. Scrimin, P. Tecilla and U. Tonellato, Artificial metallonucleases, Chem. Commun., 2005, 2540–2548 RSC.
- L. Gonzalez, F. J. Carmona, N. M. Padial, J. A. R. Navarro, E. Barea and C. R. Maldonado, Dual removal and selective recovery of phosphate and an organophosphorus pesticide from water by a zr-based metal-organic framework, Mater. Today Chem., 2021, 22, 100596–100603 CrossRef CAS.
- B. K. Singh and A. Walker, Microbial Degradation of Organophosphorus Compounds, FEMS Microbiol. Rev., 2006, 30, 428–471 CrossRef CAS PubMed.
- I. Lyagin and E. Efremenko, Enzymes, reacting with organophosphorus compounds as detoxifiers: diversity and functions, Int. J. Mol. Sci., 2021, 22, 1761–1807 CrossRef CAS PubMed.
- L. Jiang, Y. Sun and P. Chen, Nan. From DNA to Nerve Agents–The Biomimetic Catalysts for the Hydrolysis of Phosphate Esters, ChemistrySelect, 2020, 5, 9492–9516 CrossRef CAS.
- F. Ardito, M. Giuliani, D. Perrone, G. Troiano and L. L. Muzio, The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy, Int. J. Mol. Med., 2017, 40, 271–280 CrossRef CAS PubMed.
- K. Iqbal, C. Del, A. Alonso, S. Chen, M. O. Chohan, E. El-Akkad, C.-X. Gong, S. Khatoon, B. Li, F. Liu, A. Rahman, H. Tanimukai and I. Grundke-Iqbal, Tau Pathology in Alzheimer Disease and other Tauopathies, Biochim. Biophys. Acta, Mol. Basis Dis., 2005, 1739, 198–210 CrossRef CAS.
- S. Zhao, D. Sedwick and Z. Wang, Genetic
Alterations of Protein Tyrosine Phosphatases in Human Cancers, Oncogene, 2015, 34, 3885–3894 CrossRef CAS PubMed.
- T. W. Reid and I. B. Wilson, E. coli Alkaline Phosphatase, The enzymes, 1971, 4, 373–415 CAS . Academic Press, New York.
-
A. Maxam and W. Gilbert, Sequencing end-labeled DNA with Base-Specific Chemical Cleavages, Academic Press, New York, 1980, 65, 499–560 Search PubMed.
- Y. J. Zhan, S. T. Yang, L. F. Chen, Y. B. Zeng, L. Li, Z. Y. Lin, L. H. Guo and W. Xu, Ultrahigh efficient FRET ratiometric fluorescence biosensor for visual detection of alkaline phosphatase activity and its inhibitor, ACS Sustainable Chem. Eng., 2021, 9, 12922–12929 CrossRef CAS.
- M. M. C. Wang, W. X. Li, L. Guo, Z. J. Cai, B. R. Wang, J. Chen and R. F. Shen, Changes of acid and alkaline phosphatase activities in long-term chemical fertilization are driven by the similar soil properties and associated microbial community composition in acidic soil, Eur. J. Soil Biol., 2021, 104, 103312 CrossRef.
- J. E. Coleman, Structure and Mechanism of Alkaline Phosphatase, Annu. Rev. Biophys. Biomol. Struct., 1992, 21, 441–483 CrossRef CAS PubMed.
- K. M. Holtz and E. R. Kantrowitz, The mechanism of the Alkaline Phosphatase Reaction: Insights from NMR, Crystallography and Site-Specific Mutagenesis, FEBS Lett., 1999, 462, 7–11 CrossRef CAS PubMed.
- B. Stec, K. M. Holtz and E. R. Kantrowitz, A Revised Mechanism for the Alkaline Phosphatase Reaction involving three metal ions, J. Mol. Biol., 2000, 299, 1303–1311 CrossRef CAS PubMed.
- T. Chakraborty, S. Mukherjee, S. Dasgupta, B. Biswas and D. Das, Anion-Mediated Bio-Relevant Catalytic Activity of Dinuclear Nickel(II) Compounds Derived from end-off Compartmental Ligand, Dalton Trans., 2019, 48, 2772–2784 RSC.
- T. Chakraborty, A. Sarkar, A. Adhikary, N. Chakiroy and D. Das, Synthesis of structurally diverse ferrimagnetically and antiferromagnetically coupled MII−MnII (M=Cu, Ni) heterometallic Schiff base compounds with a dicyanamide spacer and study of biomimetic catalytic activity, Cryst. Growth Des., 2019, 19, 7336–7348 CrossRef CAS.
- T. Chakraborty, S. Dasgupta, E. Zangrando and D. Das, Portraying the Role of Halo Ligands and the Auxiliary Part of Ligands of Mononuclear Manganese(III)-Schiff Base Compounds in Catalyzing Phosphor-Ester Bond Hydrolysis, New J. Chem., 2018, 42, 14933–14942 RSC.
- T. Chowdhury, A. Adhikary, M. Roy, E. Zangrando, D. Samanta and D. Das, Mapping of Solvent-Mediated Molecular Self-Assembly of Iron(III) Discrete Compounds: Exploring Their Magnetic Behavior and Phosphatase-Like Activity, Cryst. Growth Des., 2020, 20, 1254–1265 CrossRef CAS.
- T. Chakraborty, S. Mukherjee, R. Parveen, A. Chandra, D. Samanta and D. Das, A combined experimental and theoretical rationalization of an unusual zinc(II)-mediated conversion of 18-membered schiff-base macrocycles to 18-membered imine-amine macrocycles with imidazolidineside rings: an investigation of their bio-relevant catalytic activities, New J. Chem., 2021, 45, 2550–2562 RSC.
- T. Chowdhury, S. Dasgupta, S. Khatua, K. Acharya and D. Das, Executing a series of zinc(II) complexes of homologous schiff base ligands for a comparative analysis on hydrolytic, antioxidant, and antibacterial activities, ACS Appl. Bio Mater., 2020, 3, 4348–4357 CrossRef CAS PubMed.
- X. Hu, T. Huang, H. Liao, L. Hu and M. Wang, The phosphatase-like activity of zirconium oxide nanoparticles and their application in near-infrared intracellular imaging, J. Mater. Chem. B, 2020, 8, 4428–4433 RSC.
- K. Kato, S. Lee and F. Nagata, Catalytic performance of ceria fibers with phosphatase-like activity and their application as protein carriers, Adv. Powder Technol., 2020, 31, 2880–2889 CrossRef CAS.
- T. Li, W. Zhong, C. Jing, X. Li, T. Zhang, C. Jiang and W. Chen, Enhanced hydrolysis of p-nitrophenyl phosphate by iron (hydr) oxide nanoparticles: roles of exposed facets, Environ. Sci. Technol., 2020, 54, 8658–8667 CrossRef CAS PubMed.
- H. Liu and J. Liu, Self–limited Phosphatase–Mimicking CeO2 Nanozymes, ChemNanoMat, 2020, 6, 947–952 CrossRef CAS.
- S. K. Das, S. Chatterjee, S. Bhunia, A. Mondal, P. Mitra, V. Kumari, A. Pradhan and A. Bhaumik, A new strongly paramagnetic cerium-containing microporous MOF for co2 fixation under ambient conditions, Dalton Trans., 2017, 46, 13783–13792 RSC.
- C. Q. Meng, Y. Cao, Y. L. Luo, F. Zhang, Q. Q. Kong, A. A. Alshehri, K. A. Alzahrani, T. S. Li, Q. Liu and X. P. Sun, A Ni-MOF nanosheet array for efficient oxygen evolution electrocatalysis in alkaline media, Inorg. Chem. Front., 2021, 8, 3007–3011 RSC.
- D. Chakraborty, A. Chowdhury, M. Chandra, R. Jana, S. Shyamal, M. K. Bhunia, D. Chandra, M. Hara, D. Pradhan, A. Datta and A. Bhaumik, Novel Tetradentate Phosphonate Ligand Based Bioinspired Co-Metal–Organic Frameworks: Robust Electrocatalyst for the Hydrogen Evolution Reaction in Different Mediums, Cryst. Growth Des., 2021, 21, 2614–2626 CrossRef CAS.
- N. Ogiwara, H. Kobayashi, M. Inuka, Y. Nishiyama, P. Concepcion, F. Rey and H. Kitagawa, Ligand-Functionalization-Controlled Activity of Metal-Organic Framework-Encapsulated Pt Nanocatalyst toward Activation of Water, Nano Lett., 2020, 20, 426–432 CrossRef CAS.
- D. Chakraborty, S. Bej, S. Sahoo, S. Chongdar, A. Ghosh, P. Banerjee and A. Bhaumik, Novel Nanoporous Ti-Phosphonate Metal-Organic Framework for Selective Sensing of 2,4,6-Trinitrophenol and a Promising Electrode in an Energy Storage Device, ACS Sustainable Chem. Eng., 2021, 9, 14224–14237 CrossRef CAS.
- J. W. Lan, Y. Qu, Z. J. Wang, P. Xu and J. M. Sun, A facile fabrication of a multi-functional and hierarchical Zn-based MOF as an efficient catalyst for CO2 fixation at room-temperature, Inorg. Chem. Front., 2021, 8, 3085–3095 RSC.
- R. A. Molla, K. Ghosh, B. Banerjee, M. A. Iqubal, S. K. Kundu, S. M. Islam and A. Bhaumik, Silver Nanoparticles Embedded Over Porous Metal Organic Frameworks for Carbon Dioxide Fixation via Carboxylation of Terminal Alkynes at Ambient Pressure, J. Colloid Interface Sci., 2016, 477, 220–229 CrossRef CAS PubMed.
- M. Xu, L. Feng, L. N. Yan, S. S. Meng, S. Yuan, M. J. He and H. C. Zhou, Discovery of Precise pH-Controlled Biomimetic Catalysts: Defective Zirconium Metal–Organic Frameworks as Alkaline Phosphatase Mimics, Nanoscale, 2019, 11, 11270–11278 RSC.
- M. J. Katz, J. E. Mondloch, R. K. Totten, J. K. Park, S. T. Nguyen, O. K. Farha and J. T. Hupp, Simple and Compelling Biomimetic Metal-organic Framework Catalyst for the Degradation of Nerve Agent Simulants, Angew. Chem., Int. Ed., 2014, 53, 507–511 CrossRef.
- S. Y. Moon, Y. Liu, J. T. Hupp and O. K. Farha, Instantaneous Hydrolysis of Nerve–Agent Simulants with a Six–Connected Zirconium–based Metal–Organic Framework, Angew. Chem., Int. Ed., 2015, 54, 6899–6903 CrossRef.
- T. Islamoglu, A. Atilgan, S.-Y. Moon, G. W. Peterson, J. B. DeCoste, M. Hall, J. T. Hupp and O. K. Farha, Cerium(IV) vs Zirconium(IV) Based Metal-Organic Frameworks for Detoxification of a Nerve Agent, Chem. Mater., 2017, 29, 2672–2675 CrossRef CAS.
- H. Shigekawa, M. Ishida, K. Miyake, R. Shioda, Y. Iijima, T. Imai, H. Takahashi, J. Sumaoka and M. Komiyama, Extended X-Ray Absorption Fine Structure Study on the Cerium(IV)-Induced DNA Hydrolysis: Implication to the Roles of 4f Orbitals in the Catalysis, Appl. Phys. Lett., 1999, 74, 460–462 CrossRef CAS.
-
https://en.wikipedia.org/wiki/File:1ALK.png
.
- G. M. Sheldrick, Crystal Structure Refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3–8 Search PubMed.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia and G. X. Win, ORTEP for Windows: an Update, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- Y. W. Ren, J. X. Liang, J. X. Lu, B. W. Cai, D. B. Shi, C. R. Qi, H. F. Jiang, J. Chen and D. Zheng, 1,4-Phenylenediacetate-Based Ln MOFs-Synthesis, Structures, Luminescence, and Catalytic Activity, Eur. J. Inorg. Chem., 2011, 4369–4376 CrossRef CAS.
- P. Thangasamy, S. Shanmuganathan and V. Subramanian, A NiCo-MOF Nanosheet Array Based Electrocatalyst for the Oxygen Evolution Reaction, Nanoscale Adv., 2020, 2, 2073–2079 RSC.
- J. Ruchi, A. Dubey, M. K. Ghosalya and C. S. Gopinath, Gas-solid interaction of H2-Ce0.95Zr0.05O2: new insights into surface participation in heterogeneous catalysis, Catal. Sci. Technol., 2016, 6, 1746–1756 RSC.
- D. Rana, B. Sakthivel, M. K. Ghosalya, A. Dhakshinamoorthy and S. Biswas, A cerium-based metal-organic framework having inherent oxidase-like activity applicable for colorimetric sensing of biothiols and aerobic oxidation of thiols, CrystEngComm, 2017, 19, 5915–5925 RSC.
- Q. Wang, Q. T. Li, Y. W. Lu, X. D. Zhang and Y. M. Huang, Rational Design of N-Doped Carbon Nanocage-Equipped Co-N-x Active Sites for Oxidase Mimicking and Sensing Applications, ACS Sustainable Chem. Eng., 2021, 9, 7668–7677 CrossRef CAS.
- J. Du, S. Qi, J. Chen, Y. Yang, T. Fan, P. Zhang and C. Zhu, Fabrication of Highly Active Phosphatase-Like Fluorescent Cerium-Doped Carbon Dots for in Situ Monitoring the Hydrolysis of Phosphate Diesters, RSC Adv., 2020, 10, 41551–41559 RSC.
- A. A. Vernekar, T. Das and G. Mugesh, Vacancy–Engineered Nanoceria: Enzyme Mimetic Hotspots for the Degradation of Nerve Agents, Angew. Chem., Int. Ed., 2016, 55, 1412–1416 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2163330. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qi01443b |
| ‡ These authors contributed equally to this paper. |
|
| This journal is © the Partner Organisations 2022 |

 ,
Tonmoy
Chakraborty‡
,
Tonmoy
Chakraborty‡


 4-NPP + N-methyl morpholine adduct,
4-NPP + N-methyl morpholine adduct,  Na2HPO4 (inorganic phosphate). Data were collected at 0 h, 0.5 h, 1 h, 2 h, 12 h and 24 h after the addition of the Ce-MOF.
Na2HPO4 (inorganic phosphate). Data were collected at 0 h, 0.5 h, 1 h, 2 h, 12 h and 24 h after the addition of the Ce-MOF.