
Synergistic Lewis acid and Pd active sites of metal–organic frameworks for highly efficient carbonylation of methyl nitrite to dimethyl carbonate†
Han-Ying
Wu
abc,
Ye-Yan
Qin
a,
Yi-Hong
Xiao
bc,
Jian-Shan
Chen
a,
Rong
Guo
a,
Si-Qi
Wu
abc,
Lei
Zhang
 bc,
Jian
Zhang
bc and
Yuan-Gen
Yao
*abc
bc,
Jian
Zhang
bc and
Yuan-Gen
Yao
*abc
aKey Laboratory of Coal to Ethylene Glycol and Its Related Technology, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, 350002, P. R. China
bState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, P. R. China
cSchool of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, P.R. China. E-mail: yyg@fjirsm.ac.cn
First published on 6th April 2022
Abstract
Metal–organic frameworks (MOFs) have recently been applied as versatile platforms for constructing efficient catalysts with improved performance in many fields. Herein, for the first time, we prepare a MOF UiO-66 based catalyst showing the synergistic effect of Lewis acid and Pd(II) active sites, and it shows highly efficient methyl nitrite (MN) carbonylation to dimethyl carbonate (DMC). Lewis acid sites were first introduced to UiO-66 by adding different amounts of trifluoroacetic acid (TFA) and activating at different temperatures. Pd(II) sites were then supported on UiO-66x-X to obtain the resulting Pd-UiO-66x-X (x, the molar percentage of TFA; X, activation temperature). The Lewis acid sites not only act as the center of CO adsorption, but affect the electron density of Pd(II) for the generation of more COOCH3* intermediates, resulting in a higher catalytic performance. As a result, the best catalytic performance based on the DMC selectivity based on CO (SDMC/CO) near 100%, SDMC/MN up to 67.4%, CO conversion to DMC (CCO) as high as 68.4%, and the weight time yield (WTY) of DMC up to 2056 g kgcat−1 h−1 was achieved by the composite material Pd-UiO-66TFA-0.25-290. This work provides efficient catalysts for carbonylation of MN to DMC and presents a significant synergistic methodology for improving the catalytic performance of MOF materials.
Introduction
Dimethyl carbonate (DMC) is one of the promising materials used to substitute for phosgene and other poisonous reagents, according to the basic concept of green chemistry,1,2 and has been widely used in polycarbonate production3 and the lithium battery industry,4 and as a methylating agent.5 In the past decade, several technical routes have been applied to synthesize DMC involving transesterification,6–8 methanol oxidative carbonylation,9–11 methyl nitrite (MN) carbonylation,12–14etc. Particularly, the carbonylation of MN to DMC has attracted widespread attention due to its more economical and environmentally friendly characteristics, and catalysts composed of active sites and supports have been used to improve the catalytic performance.13,15,16 Accordingly, Pd(II) species in catalysts are widely acknowledged as active sites, and supports are usually porous and large surface area materials including zeolites,14 activated carbon,17 metal oxides,18etc. To date, the states of Pd active sites (e.g. morphology, dispersion), the effects of various supports, and the interactions between promoters (e.g. Cu2+) and active sites have been extensively studied to achieve highly efficient performance.12,13,15 However, the effect of active sites and supports on the catalytic performance is still unclear.Metal–organic frameworks (MOFs) as a class of crystalline porous materials, due to their large surface area, high porosity, and tunable structures,19–26 have become excellent supports for accommodating highly dispersed metal centers and allow the reactants to diffuse effectively on the catalyst to enhance the catalytic performance.27–30 As for the carbonylation of MN to DMC, Lewis acid sites of MOFs have been considered as an effective way to improve the catalytic performance, and could be designed using coordinatively unsaturated metal nodes.31 Particularly, Pd-based Lewis acid MOFs possessing both metal active sites and Lewis acid sites have been known as representative multisite catalysts.32–36 Although the interaction between active sites and supports has been reported in the literature,37 the synergistic role of Lewis acid sites and Pd(II) in MOF-based materials has not been systematically investigated for the carbonylation of MN to DMC. Therefore, designing such catalysts is of great significance and challenge, which promotes the understanding of the whole catalytic reaction and offers a new route to expand functional MOF-based materials for DMC catalysts.
The classical MOF UiO-66 is one of the most promising supports for MN carbonylation to DMC owing to its outstanding thermal stability, high tolerance for regulation, and simple synthesis.38–40 Herein, we first prepared Pd(II)-supported UiO-66 (Pd-UiO-66x-X) with Lewis acid sites by adding different amounts of TFA (x, the molar percentage of TFA: 0, 0.25, 0.50, and 0.75%) and adjusting the activation temperature (X: 250, 270, 290, and 320 °C) for the highly efficient carbonylation of MN to DMC. As illustrated in Fig. 1, as a modifier, different amounts of TFA were added, where terephthalic acid (BDC) was partly substituted to obtain UiO-66x, and UiO-66x-X with different Lewis acid sites were obtained by activating UiO-66x to remove TFA. Afterward, Pd(II) was incorporated into UiO-66x-X, resulting in Pd-UiO-66x-X. The NH3 temperature-programmed desorption (TPD) profiles and pyridine FT-IR spectra proved that the concentration of Lewis acid sites increased with the addition of TFA and the activation temperature. The results of MN carbonylation to DMC showed that Pd-UiO-66TFA-0.25-290 showed the best catalytic performance (DMC selectivity based on CO (SDMC/CO) near 100%, SDMC/MN up to 67.4%, CO conversion to DMC (CCO) as high as 68.4%, and the weight time yield (WTY) of DMC of 2056 g kgcat−1 h−1) and that it is a record performance among MOF based materials, suggesting that the appropriate amount of Lewis acid sites played a significant role in the performance. The CO adsorption diffuse reflectance infrared Fourier-transform spectra (DRIFTS) and CO-TPD profiles indicated that besides serving as the adsorption center of CO, Lewis acid sites also act as the electron acceptor to Pd(II) that affected the formation of COOCH3* intermediates, revealing the synergistic effect of Lewis acid sites and Pd(II) in Pd-UiO-66x-X. Furthermore, the in situ DRIFTS spectra demonstrated DMC generated on the catalysts and COOCH3* intermediates was easily formed on Pd-UiO-66TFA-0.25-290, which was in good agreement with the excellent catalytic performance.
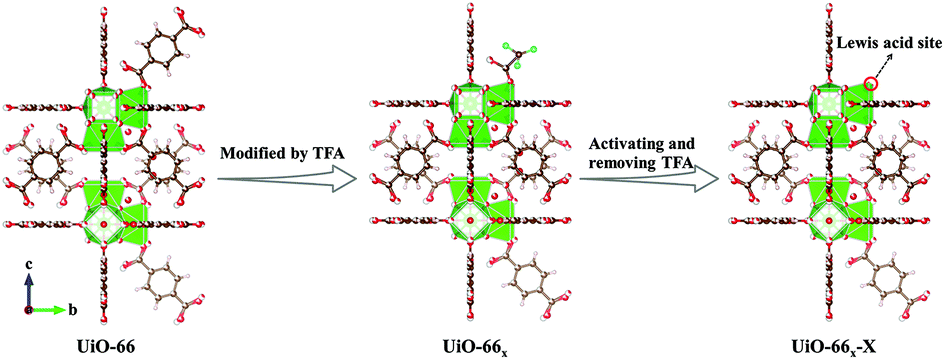 | ||
| Fig. 1 Schematic illustration of the synthesis of UiO-66x-X. | ||
Experimental
Synthesis of UiO-66x-X supports
ZrCl4 (3.5 g, 15 mmol) and terephthalic acid (2.5 g, 15 mmol) were dissolved in DMF (155 ml, 2 mol) and stirred for 30 min to obtain a transparent solution. In the modulated synthesis, TFA (molar percentage of TFA: 0, 0.25% (2.9 ml) 0.5% (5.8 ml) and 0.75% (8.7 ml)) was added to the mixture and further stirred for 15 min. Then the mixture was transferred into a Teflon-lined autoclave (250 ml), which was sealed and heated at 120 °C for 24 hours. After that, the Teflon-lined autoclave was cooled down to room temperature naturally, and the product was collected by centrifugation, thoroughly washed with DMF (3 times) and methanol (3 times), and finally dried in a vacuum oven. The powders were activated by heating at 250 °C for 12 h to obtain the resulting UiO-66x (x: molar percentage of TFA). The activation temperature was changed to 270 °C, 290 °C and 320 °C to obtain a series of UiO-66TFA-0.25 with different activated temperatures, which were denoted as UiO-66TFA-0.25-270, UiO-66TFA-0.25-290, and UiO-66TFA-0.25-320, respectively.Synthesis of Pd-UiO-66x-X catalysts
Pd-UiO-66x-X (x: the molar percentage of TFA, X: activation temperature) catalysts were obtained by the traditional wet impregnation method. Typically, 5 g of UiO-66x-X was dispersed in 50 ml of acetone and the mixture was stirred for 30 min. After that, 0.105 g of palladium acetate (the mass ratio of Pd to UiO-66x-X was 1%) dissolved in 10 ml of acetone solution was added to the mixture while being stirred. Then the compound was further stirred for 20 h at room temperature. The products were collected by centrifugation, washed with acetone several times, and dried in a vacuum oven. Finally, the products were calcined at 200 °C for 4 h to enhance the interaction between UiO-66x-X and Pd(II) to obtain the resulting Pd-UiO-66x-X (Pd-UiO-66-250, Pd-UiO-66TFA-0.25-250, Pd-UiO-66TFA-0.5-250, Pd-UiO-66TFA-0.75-250, Pd-UiO-66TFA-0.25-270, Pd-UiO-66TFA-0.25-290, and Pd-UiO-66TFA-0.25-320, respectively).In situ FT-IR measurement
In situ diffuse reflectance FT-IR spectra were recorded using a Nicolet 6700 Fourier-transform spectrometer equipped with a specially designed reaction cell. Each spectrum was recorded by averaging 128 scans at a resolution of 4 cm−1. For CO adsorption, 200 mg of the sample without diluents was placed into the cell and pretreated under N2 (10 ml min−1) at 120 °C for 60 min to remove water. After cooling down to 20 °C, the IR spectrum was recorded as a reference spectrum. Then the sample was exposed to CO–N2 (25% CO, 5 ml min−1) for 50 min, and the spectra were collected every 5 min. Subsequently, flushing with N2 (10 ml min−1) was done for 20 min to remove CO in the gas phase. For the experiment of monitoring the reaction process between CO and MN, 200 mg of the sample without diluents was placed into the cell and pretreated under N2 at 120 °C for 60 min to remove water. Then the sample was exposed to MN–N2 (10% MN, 5 ml min−1) for 50 min and purged with N2 (10 ml min−1) for 15 min to remove MN in the gas phase and the IR spectrum was recorded as a reference spectrum. After that, CO (CO–N2 (25% CO), 5 ml min−1) was introduced into the chamber and the DRIFTS spectra were collected every 1 min.Catalyst evaluation
The catalytic performances for carbonylation of methyl nitrite (MN) to dimethyl carbonate (DMC) were evaluated in a continuous fixed-bed reactor under atmospheric pressure using a mixture gas of 50% N2, 10% CO, and 40% MN at 120 °C, and a gas hourly space velocity (GHSV) of 3000 h−1. The reaction products were collected using an online gas chromatograph (GC) (7900, Thanghai Tianmei) with an FID detector.The weight time yield (WTY) of DMC was calculated as follows:
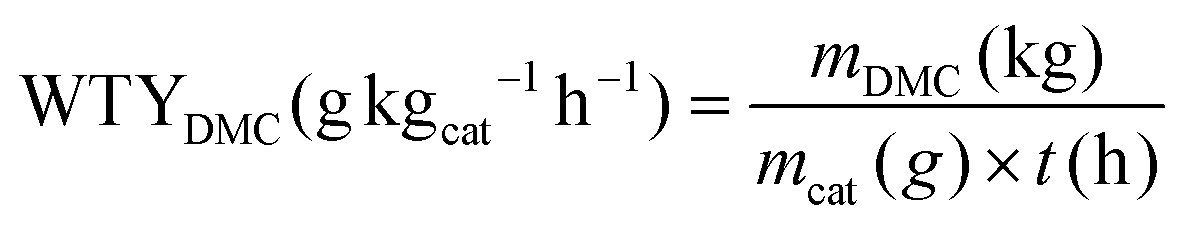
The selectivity of DMC based on CO or MN was calculated as follows:
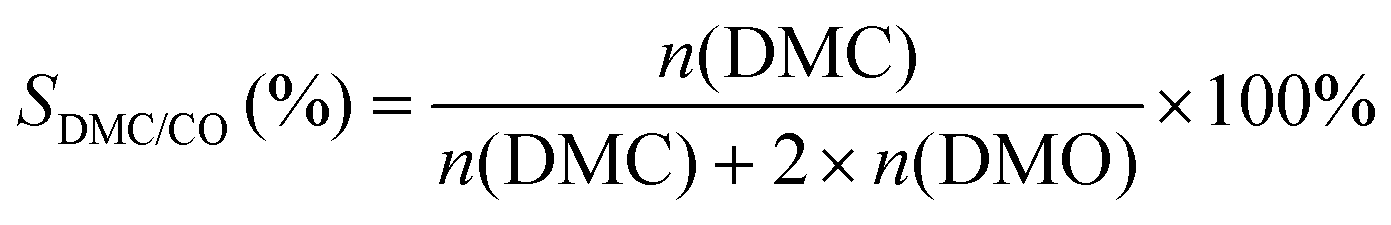
where mDMC represents the mass of DMC (kg), mcat is the mass of the catalyst, t is the reaction time (given in hours), and n(MeOH), n(MF), n(DMM), n(DMC), and n(DMO) represent the molar number of MeOH, MF, DMM, DMC and DMO species, respectively.
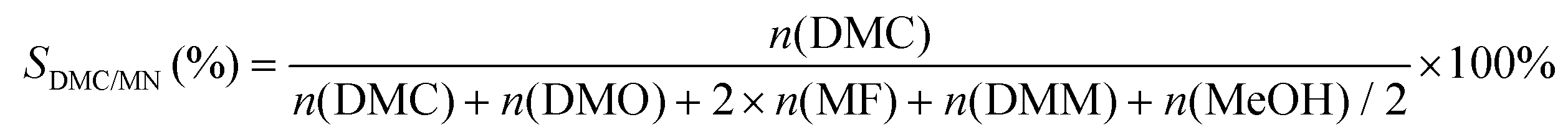
Results and discussion
Firstly, UiO-66x were synthesized by a solvothermal method, with partial substitution of BDC by different amounts of TFA (x from 0, 0.25, 0.50 to 0.75%, see the details in the ESI†). The XRD patterns showed that all the peaks of UiO-66x were indexed to simulated peaks (Fig. 2a), indicating that the addition of TFA would not change the phase of UiO-66. As shown in Fig. 2b, thermogravimetric analysis (TGA) data showed that UiO-66 modified with TFA loses more weight than pristine UiO-66 in the range 100–320 °C, indicating that TFA could be removed at these temperatures.41 Besides, the structure of UiO-66 would be maintained below 450 °C. Furthermore, the activation temperature was selected as 250 °C, and the structure of UiO-66x remained after activation (UiO-66x-250, Fig. S1 and S2†). Then Pd species were supported on UiO-66x-250 by the traditional wet impregnation method. The unchanged XRD patterns indicated the high stability of UiO-66 even when Pd was loaded (Fig. 2c). Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) images demonstrated that there were no distinctly aggregated Pd in UiO-66TFA-0.25-250 (Fig. S3–S5† and Fig. 2d). The Pd loading was about 0.8 wt% determined by ICP-OES (Table S1†). High angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and elemental mapping (Fig. 2e and f) showed the higher dispersion of Pd species on the supports, although a small amount of Pd aggregated that may be Pd(II) species did not fully enter the pores inevitably or some defects in UiO-66TFA-0.25-250 during the calcination process. The N2 adsorption isotherms of UiO-66TFA-0.25-250 and Pd-UiO-66TFA-0.25-250 (Fig. S6 and Table S2†) demonstrated that the specific surface area of UiO-66TFA-0.25-250 was significantly reduced after loading Pd(II) species, indicating that a part of the pores of the MOF was occupied by Pd(II) species, which could be corroborated with a homogeneous distribution of the Pd element in UiO-66TFA-0.25-250 in STEM mapping images (Fig. 2f) despite a small amount of Pd aggregated.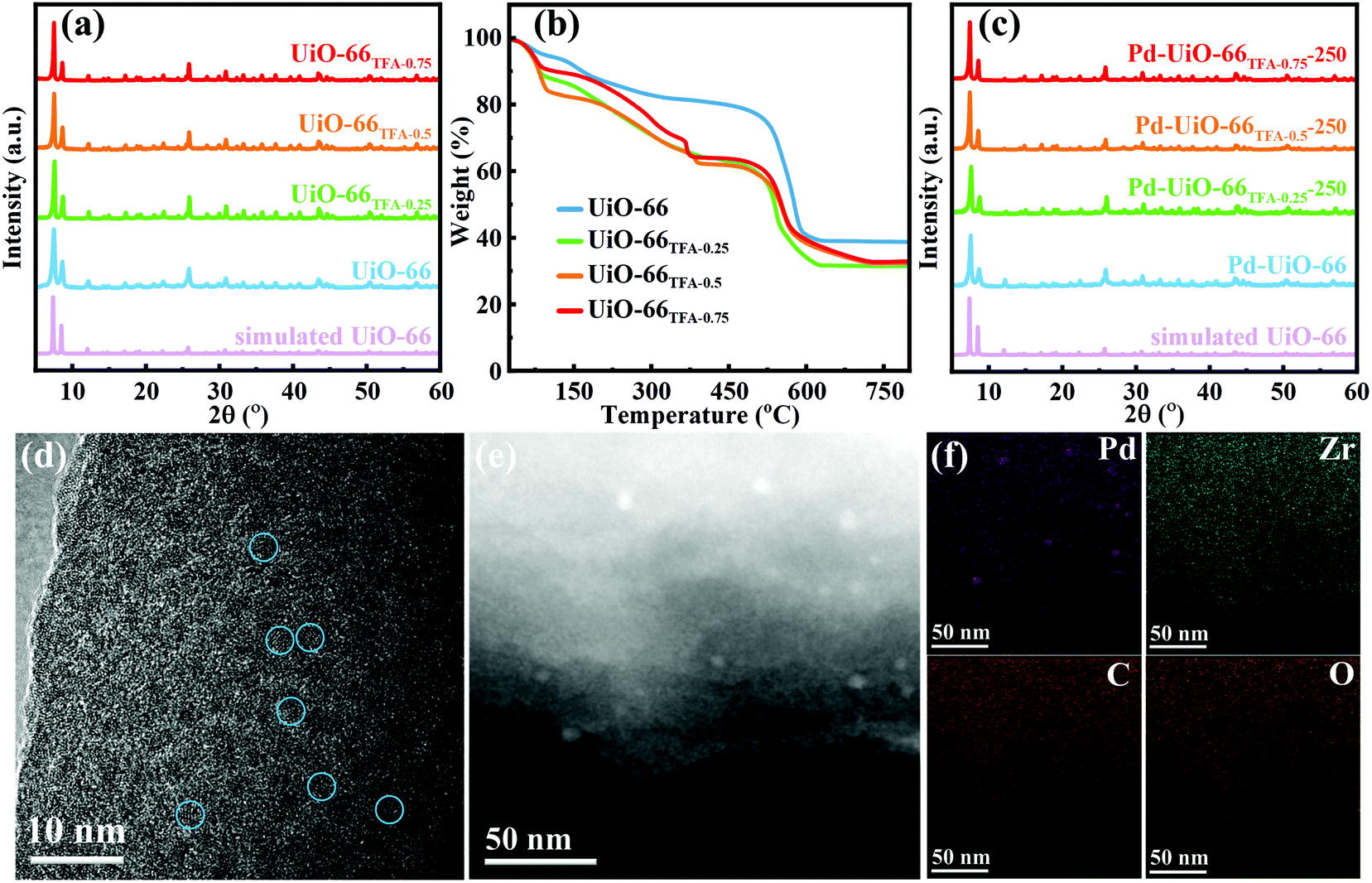 | ||
| Fig. 2 (a) XRD patterns of UiO-66x (UiO-66, UiO-66TFA-0.25, UiO-66TFA-0.5 and UiO-66TFA-0.75); (b) TGA curves of UiO-66x; (c) XRD patterns of Pd-UiO-66x-250; (d) HTEM image; (e) HAADF-STEM image; and (f) the corresponding Pd, Zr, C, and O elemental mapping of Pd-UiO-66TFA-0.25-250. | ||
Since TFA in UiO-66 could be removed by activating at a higher temperature to form unsaturated Zr6,42,43 NH3-TPD was employed to investigate the Lewis acid sites in UiO-66x-250. As shown in Fig. 3a, there were two desorption peaks in the range 50–320 °C for all supports, the first peak below 150 °C is attributed to NH3 adsorbed at weak Lewis acid sites,31,44 and the intensity increased with the addition of TFA. The other desorption peak located between 150 and 300 °C corresponds to NH3 adsorbed at medium Lewis acid sites,31 and it shifted to higher temperature with the addition of TFA, indicating that the Lewis acidity of UiO-66x-250 strengthened with the amount of TFA. To further study the Lewis acid characteristics of UiO-66x-250, pyridine FT-IR spectroscopy was performed. Obviously, the pyridine FT-IR spectra (Fig. 3b) exhibited two signals at ∼1033 cm−1 and ∼1068 cm−1 for UiO-66x-250, corresponding to the pyridine absorbed on Lewis acid sites.31,45,46 As expected, the amount of Lewis acid sites increased with the addition of TFA, which was identical to the analysis of NH3-TPD.
 | ||
| Fig. 3 (a) NH3-TPD of UiO-66x-250. (b) FTIR-spectra of pyridine chemisorption on UiO-66x-250; the catalytic performance of MN carbonylation to DMC on (1) Pd-UiO-66-250, (2) Pd-UiO-66TFA-0.25-250, (3) Pd-UiO-66TFA-0.5-250 and (4) Pd-UiO-66TFA-0.75-250. (c) DMC selectivity based on CO (SDMC/CO), DMC selectivity based on MN (SDMC/MN) and CO conversion (CCO), and (d) WTY of DMC. | ||
To disclose the influence of Pd-UiO-66x-250 with different additions of TFA on MN carbonylation to DMC, the catalytic activity measurement was evaluated under 120 °C, where reactant gases composed of 50% N2, 40% MN, and 10% CO were used. Fig. 3c shows that the DMC selectivity and CO conversion could be effectively modulated by modifying the catalyst supports via changing the addition of TFA, and the Pd-UiO-66TFA-0.25-250 catalyst showed the best performance in terms of selectivity and conversion (SDMC/MN up to 63.4% and CCO as high as 59.3%). The trend of the WTY of DMC (Fig. 3d) was consistent with the DMC selectivity and CO conversion, and Pd-UiO-66TFA-0.25-250 showed a maximum value of 1590 g kgcat−1 h−1. This indicated that there was an optimal concentration of Lewis acid sites for achieving the best catalytic performance.
The TFA in UiO-66x could be eliminated with activation as observed in 19F NMR spectra. In Fig. 4, UiO-66TFA-0.25 showed the typical signal of the –CF3 group in TFA located at −79 ppm, which is up-field shifted relative to the physisorbed TFA (−75 ppm) revealing that TFA coordinated with Zr.42,47 The signal of UiO-66TFA-0.25-250 showed a lower intensity, a sharper peak shape, and a slight up-field shift to −82 ppm, which may be due to the partial removal of TFA from the structure of UiO-66TFA-0.25 through activating at 250 °C. When the activation temperature was up to 270 °C, the signals of 19F almost disappeared, indicating that TFA had been removed from UiO-66TFA-0.25 and made Zr unsaturated resulting in Lewis acid sites. Therefore, the 19F NMR spectra also indicated that the Lewis acid sites could be regulated via increasing the activation temperature. Additionally, the phase of UiO-66 was maintained when the activation temperature was below 320 °C (Fig. S7†). Moreover, the loaded Pd could not change the phase of UiO-66TFA-0.25-X (Fig. S8 and Table S3†), and the distribution states of Pd in UiO-66TFA-0.25-X are shown in Fig. S9–S11.†
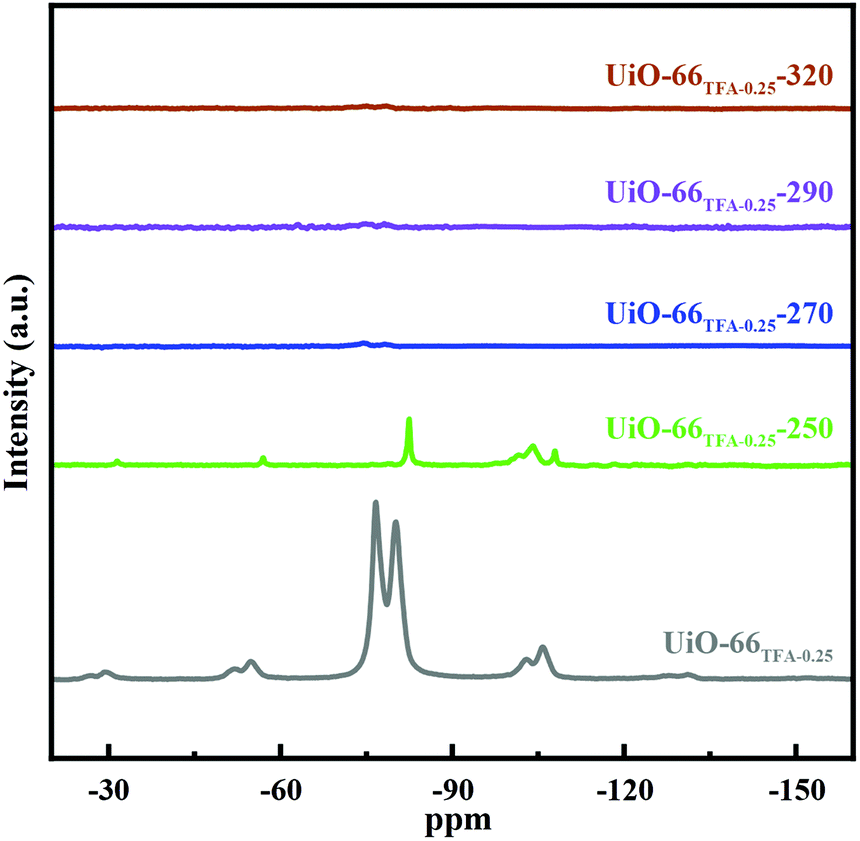 | ||
| Fig. 4 Mass-normalized 19F NMR spectra of UiO-66TFA-0.25-X (UiO-66TFA-0.25, UiO-66TFA-0.25-250, UiO-66TFA-0.25-270, UiO-66TFA-0.25-290 and UiO-66TFA-0.25-320). | ||
The Lewis acid characteristics of UiO-66TFA-0.25-X were further investigated using NH3-TPD and pyridine FT-IR. As expected, the NH3-TPD profiles and pyridine FT-IR spectra of UiO-66TFA-0.25-X (Fig. 5a and b) showed that the amount of Lewis acid sites increased with activation temperature. However, the amount of Lewis acid sites over UiO-66TFA-0.25-X only slightly increased from 270 to 320 °C because TFA had been removed substantially as the activation temperature rose to 270 °C. Fig. 5c and d show that when the activation temperature rose from 250 to 290 °C, SDMC/MN increased from 63.4 to 67.4%, CCO increased from 59.3 to 68.4%, and the WTY of DMC increased from 1590 to 2056 g kgcat−1 h−1 for the UiO-66TFA-0.25-X supported Pd. However, when the temperature continued to rise to 320 °C, SDMC/MN, CCO, and the WTY of DMC decreased. This demonstrated that the catalytic performance of MN carbonylation to DMC could be regulated by adjusting the activation temperature, and there was an optimal activation temperature for achieving the best catalytic performance. A long-term stability test was carried out for the Pd-UiO-66TFA-0.25-290 catalyst. As shown in Fig. S12,† it was found that CCO, SDMC/MN, SDMC/CO, and the WTY of DMC were not significantly decreased with the increase of reaction time, indicating that the catalyst had higher stability. The unchanged XRD pattern of the catalyst after the reaction (Fig. S13†) indicated that the structure of the catalyst was stable during the process.
 | ||
| Fig. 5 (a) NH3-TPD of UiO-66TFA-0.25-X. (b) FTIR-spectra of pyridine chemisorption on UiO-66TFA-0.25-X; the catalytic performance of MN carbonylation to DMC on (2) Pd-UiO-66TFA-0.25-250, (5) Pd-UiO-66TFA-0.25-270, (6) Pd-UiO-66TFA-0.25-290 and (7) Pd-UiO-66TFA-0.25-320. (c) SDMC/CO (i), SDMC/MN (ii), and CCO (iii), and (d) WTY of DMC. | ||
Moreover, the microstructure of the catalysts was analyzed in detail. N2 adsorption experiments of UiO-66x-X were carried out at 77 K, which showed that the BET surface areas increased with the amount of Lewis acid sites in UiO-66x-250 (Fig. S14 and Table S4†). Besides, UiO-66TFA-0.25-X also showed a similar change in BET surface areas (Fig. S15 and Table S5†). The Lewis acid site was generated by removing TFA from the structure of UiO-66x-X, and more open sites were formed to absorb more N2. In addition, the pore size distributions of UiO-66x-X (Fig. S16 and S17†) revealed that the pore sizes did not change significantly with the amount of Lewis acid sites. On the other hand, the IR spectra (Fig. S18–S24†) showed that the peak at ∼1654 cm−1 disappeared after activating at 250 °C, proving that the DMF solvent had been completely removed.48,49 Interestingly, as the amount of Lewis acid sites increased, the signal of u3-OH (480 cm−1) decreased while the signal of u3-O (553 cm−1) increased,50,51 suggesting that the concentration of Lewis acid sites affected the removal of hydroxyl groups, which resulted in the decomposition of MN. The UV-vis spectra of UiO-66x-X showed that the absorption edge had a slightly blue-shift with the increase of the amount of Lewis acid sites (Fig. S25 and S26†), indicating the presence of weak Zr–O, which enhanced the interaction between O and Pd.52 Inevitably, the UV edge showed a red-shift and broadening after Pd species were supported on UiO-66x-X, indicating the interaction between Pd(II) and Zr–O (Fig. S27 and S28†).52,53 As expected, the edge exhibited a red-shift with the concentration of Lewis acid sites, suggesting that Lewis acid sites were beneficial for strengthening the interaction of Zr–O–Pd.
To further investigate the effect of Pd(II) on supports, X-ray photoelectron spectroscopy (XPS) was carried out. As shown in Fig. S29 and S30,† there were four peaks of Pd 3d + Zr 3p in all samples, belonging to Zr 3p1/2 (346.7 eV), Zr 3p3/2 (333.1 eV), Pd2+ 3d3/2 (∼343.2 eV) and Pd2+ 3d5/2 (∼337.2 eV), respectively.54–56 In the XPS spectra of Pd-UiO-66x-X, the binding energies of Pd 3d and Zr 3p had partial overlap. To clearly compare the binding energies of Pd 3d in different catalysts, the peaks were fitted and the signal of Pd 3d was extracted as shown in Fig. 6a and b. Fig. 6a shows that the peaks of Pd2+ 3d moved to higher binding energy with the addition of TFA, revealing that the electron density of Pd2+ decreased with the amount of Lewis acid sites. Similarly, for the Pd-UiO-66TFA-0.25-X catalysts (Fig. 6b), with the increase of activation temperature from 250 to 320 °C, the binding energy of Pd5/22+ increased from 336.97 to 337.42 eV, and the binding energy of Pd3/22+ slightly increased from 342.9 eV to 343.15 eV, indicating that there exists monotonicity between the electron density of Pd(II) and the amount of Lewis acid sites. This suggested that the Lewis acid sites could serve as electron acceptors to Pd species and form Pd(II) electron-deficient, promoting the catalytic reaction to generate DMC. Electrochemical impedance spectra (EIS) were recorded to further study the electron transfer between the active sites of Pd(II) and supports. Fig. 6c and d show that the Nyquist plot semicircle became smaller with the amount of TFA increasing from 0 to 0.75%, and the Nyquist plot semicircle was also smaller with the activation temperature, indicating that the amount of Lewis acid sites enhanced the electron transport between Pd(II) and supports. Therefore, the more Lewis acid sites, the stronger ability to accept electrons, ensuring that Pd(II) possesses a lower electron density for enhancing DMC product generation.
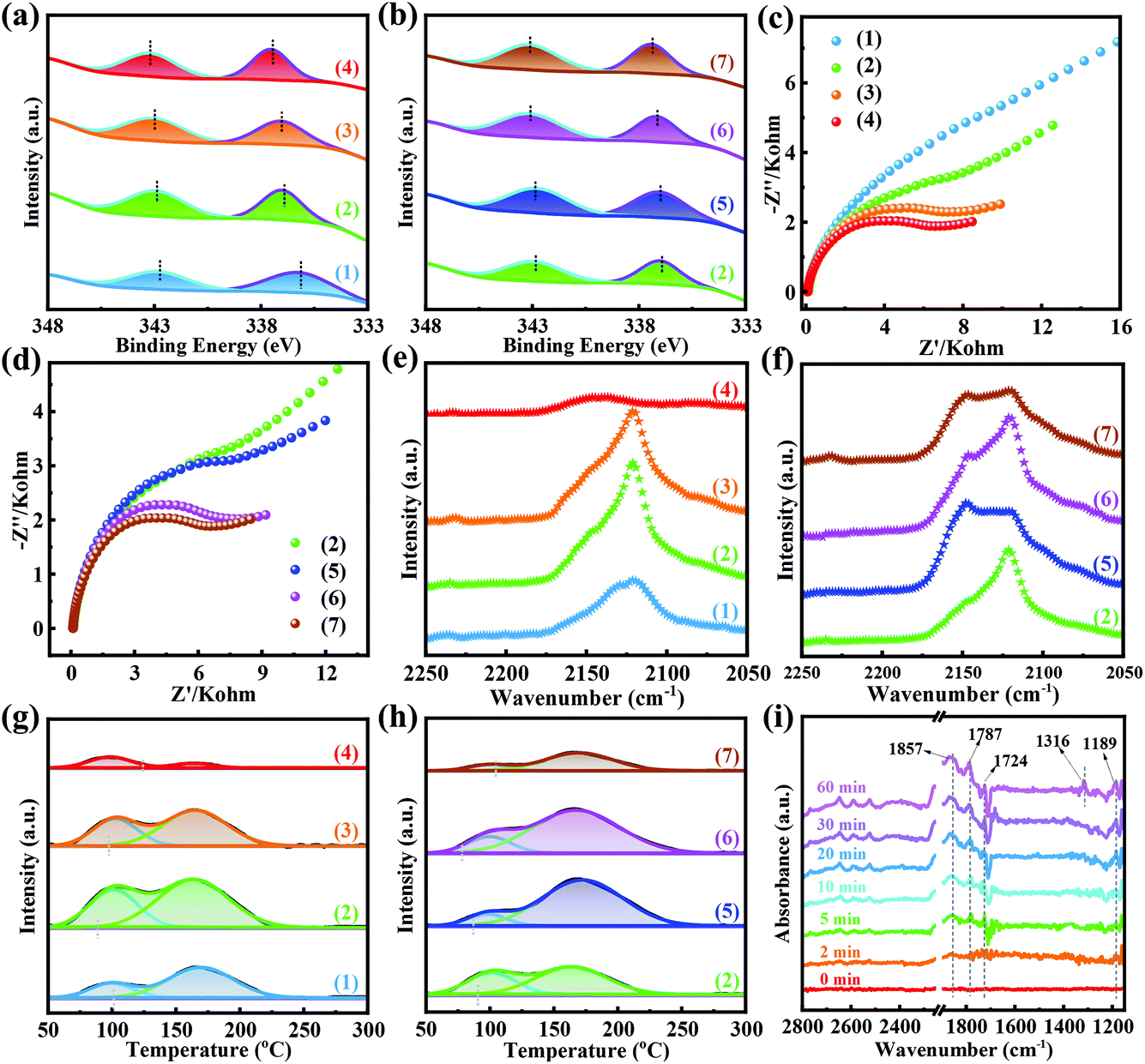 | ||
| Fig. 6 Pd 3d XPS spectra of (a) Pd-UiO-66x-250 and (b) Pd-UiO-66TFA-0.25-X. EIS spectra of (c) Pd-UiO-66x-250 and (d) Pd-UiO-66TFA-0.25-X at an overpotential of −1 V. DRIFTS spectra of CO adsorbed on (e) Pd-UiO-66x-250 and (f) Pd-UiOTFA-0.25-X after exposing to CO at 20 °C for 50 min and the cell was purged with N2 for 20 min. CO-TPD profiles of (g) Pd-UiO-66x-250 and (h) Pd-UiO-66TFA-0.25-X. (i) In situ DRIFTS spectra of Pd-UiO-66TFA-0.25-250 pre-adsorbed with MN at 120 °C as a function of CO. | ||
As the Pd(II) electron density and Lewis acid sites will influence CO adsorption, the DRIFTS measurement was performed. The CO adsorption spectra (Fig. 6e and f) of the Pd-UiO-66x-X catalysts were recorded at 20 °C after exposing to CO (5 mL min−1) for 50 min and purging with N2 (10 mL min−1) for 20 min to remove CO in the gas phase, and a single absorption peak was observed at 2120 cm−1 corresponding to linear CO adsorbed on the surface of catalysts.57–59 Accordingly, the linear CO adsorbed on Pd-UiO-66x-250 increased with the addition of TFA (from 0 to 0.25%) reaching the maximum value for Pd-UiO-66TFA-0.25-250 (Fig. 6e). However, on further increasing the amount of TFA (from 0.25 to 0.75%), the linear CO adsorption was weakened. Similarly, the linear CO adsorbed on Pd-UiO-66TFA-0.25-X catalysts (Fig. 6f) increased with the activation temperature until 290 °C. As CO could be absorbed on Lewis acid sites, CO adsorption increased with the amount of Lewis acid sites. Interestingly, in this system, in addition to serving as sites to adsorb CO, the Lewis acid sites also affect the electron density of Pd(II). Pd species are recognized as the adsorption sites of CO, whose electron density plays a vital role in CO adsorption. Combined analysis of XPS and EIS proved that with the amount of Lewis acid sites, the electron density of Pd(II) decreased, which decreased the back-donation of electrons from Pd(II) to the 2π* antibonding orbitals of CO, leading to the reduction of CO adsorption and the weakening of the activation of the C–O bond. Hence, it showed the synergistic effect between Lewis acid sites and Pd(II) active sites, and there exists an optimal amount of Lewis acid sites for achieving the best CO chemisorption. It is widely accepted that CO chemisorption is beneficial for generating COOCH3* intermediates in the catalytic reaction of MN carbonylation to DMC. The MN carbonylation to DMC could be divided into two processes: non-catalytic MN decomposition to CH3O* and NO (eqn (1)), and the catalytic reaction of CH3O* and CO* on the catalyst surface to produce DMC (eqn (2) and (3)). However, there existed side reactions in which CH3O* species reacted with proton-bearing substances on the catalyst surface (eqn (2)) or CH3O* undergoes a self-disproportionation reaction (eqn (3)) to form methanol by-products. Moreover, when CH3O* lost protons, it formed CH2O* and CHO*, and continued to react with CH3O* to form two other by-products, dimethoxymethane (DMM) and methyl formate (MF). Therefore, the reaction between CH3O* species and CO* species to generate COOCH3* intermediates plays a vital role in the selectivity of DMC based on MN. Due to the CO chemisorption being conducive to generating the COOCH3* intermediates, there should be a positive correlation between the SDMC/MN and the amount of CO chemisorption on catalyst surfaces, which is well consistent with the result of SDMC/MN in the tests of catalytic performance.
| CH3ONO(g) → CH3O* + NO | (1) |
| CH3O* + CO* → COOCH3* | (2) |
| COOCH3* + CH3O* → (CH3O)2CO (DMC) | (3) |
| CH3O* + H* → CH3OH | (4) |
| CH3O* + CH3O* → CH3OH + CH2O | (5) |
| CH3O* − H* → CH2O* | (6) |
| CH2O* + CH3OH → CH3OCH2CH3O (DMM) | (7) |
| CH2O* − H* → CHO* | (8) |
| CH3O* + CHO* → HCOOOCH3 (MF) | (9) |
To further analyze the influence of Pd(II) electron density on CO adsorption and desorption on the catalysts, CO-TPD was performed. The CO-TPD profiles (Fig. 6g and h) showed that there were two CO desorption peaks for Pd-UiO-66x-X catalysts. The CO desorption peak at a lower temperature (located at ∼100 °C) could be assigned to the weak interaction of linear Pd–CO, while the signal at ∼167 °C was related to the stronger interaction of linear Pd–CO.60 The CO desorption peak areas were computed to compare the amount of linear CO, and the results are listed in Table S6.† It revealed that there was no monotonicity between the desorption peak areas of CO chemisorption and the amount of Lewis acid sites, and Pd-UiO-66TFA-0.25-290 had the best CO chemisorption, which was well consistent with the results of the CO adsorption DRIFTS spectra. Besides, the starting temperature of the stronger interaction of the Pd–CO signal had a significant shift with different amounts of Lewis acid sites on the Pd-UiO-66x-X catalysts. As for Pd-UiO-66x-250, the Pd-UiO-66TFA-0.25-250 catalyst possessed the lowest starting temperature. With the activation temperature, the starting temperature continued to decrease until the activation temperature was up to 290 °C. It likely contributed to the synergistic effect between Lewis acid sites and Pd(II), which accounted for the best catalytic performance in MN carbonylation to DMC over Pd-UiO-66TFA-0.25-290.
Combined with the in situ DRIFTS CO adsorption spectra and the CO-TPD profiles, it was confirmed that the synergistic effect between Lewis acid sites and Pd(II) played a dominant role in linear CO adsorption. There is an optimal synergistic effect between Lewis acid sites and Pd(II) for achieving the best adsorption of CO, which is beneficial for the generation of COOCH3* intermediates, affecting the catalytic performance in terms of SDMC/MN, CCO, and WTY of DMC. Moreover, the synergistic effect of Lewis acid sites and Pd(II) affected the CO desorption and ultimately affected the stability of Pd species in the reaction. It demonstrated that Pd-UiO-66TFA-0.25-290 had the largest CO adsorption areas and the lowest starting temperature of linear CO desorption for achieving the best catalytic performance in MN carbonylation to DMC.
Furthermore, the reaction process between CO and MN on the Pd-UiO-66x-X catalysts was monitored by in situ DRIFTS. The baseline was obtained after exposing to MN at 120 °C for 50 min and purging with N2 for 15 min. The in situ DRIFTS spectra of Pd-UiO-66-250, Pd-UiO-66TFA-0.25-250, and Pd-UiO-66TFA-0.25-290 showed the increased signals from 0 to 60 min under exposure to CO at 120 °C (Fig. S31 and S32† and Fig. 6i). The bands at 1189 cm−1 and 1724 cm−1 were assigned to the C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode, respectively, of COOCH3* intermediates.14,61 The signal (1780 cm−1) could be attributed to the stretching vibrations of DMC, indicating that the DMC product increased with the passing time.14 Besides, the absorption band at 1857 cm−1 was ascribed to the gaseous NO that could be generated during this process.14,61 Significantly, a new peak (1316 cm−1) appeared assigned to the stretching vibration of DMO, indicating that a part of Pd(II) had been reduced to Pd (0) after exposing to CO for 60 min at 120 °C.61 Besides, the signals of COOCH3* intermediates had a remarkable difference in these three catalysts for different exposure times (2 min, 30 min, and 60 min) (Fig. S33–S35†). Accordingly, the stronger signals of COOCH3* appeared for the Pd-UiO-66TFA-0.25-290 catalyst, indicating that the COOCH3* species could be easily generated on Pd-UiO-66TFA-0.25-290. Meanwhile, the Pd-UiO-66TFA-0.25-290 catalyst showed the strongest absorption of DMC, further demonstrating that the catalyst had the best catalytic performance for DMC.
O stretching mode, respectively, of COOCH3* intermediates.14,61 The signal (1780 cm−1) could be attributed to the stretching vibrations of DMC, indicating that the DMC product increased with the passing time.14 Besides, the absorption band at 1857 cm−1 was ascribed to the gaseous NO that could be generated during this process.14,61 Significantly, a new peak (1316 cm−1) appeared assigned to the stretching vibration of DMO, indicating that a part of Pd(II) had been reduced to Pd (0) after exposing to CO for 60 min at 120 °C.61 Besides, the signals of COOCH3* intermediates had a remarkable difference in these three catalysts for different exposure times (2 min, 30 min, and 60 min) (Fig. S33–S35†). Accordingly, the stronger signals of COOCH3* appeared for the Pd-UiO-66TFA-0.25-290 catalyst, indicating that the COOCH3* species could be easily generated on Pd-UiO-66TFA-0.25-290. Meanwhile, the Pd-UiO-66TFA-0.25-290 catalyst showed the strongest absorption of DMC, further demonstrating that the catalyst had the best catalytic performance for DMC.
Conclusions
In summary, we successfully prepared UiO-66 supported Pd(II) with different amounts of Lewis acid sites as catalysts for MN carbonylation to DMC. The concentration of Lewis acid sites had a significant influence on the catalytic performance. The optimal concentration of Lewis acid sites appeared on the Pd-UiO-66TFA-0.25-290 catalyst, which showed excellent catalytic activity: SDMC/CO was near 100%, SDMC/MN was up to 67.4%, CCO was as high as 68.4%, and the WTY was 2056 g kgcat−1 h−1. The CO adsorption DRIFTS and CO-TPD profiles demonstrated that there existed a synergistic effect of Lewis acid sites and Pd(II) in Pd-UiO-66x-X. In addition to being CO adsorption sites, Lewis acid sites also affected the electron density of Pd(II) for the generation of COOCH3* intermediates and finally contributed to the catalytic performance. Besides, the in situ DRIFTS spectra proved that the easiest formation of COOCH3* intermediates and the strongest signal of DMC appeared on the Pd-UiO-66TFA-0.25-290 catalyst. This work provides an in-depth understanding of the mechanism of reaction, and offers a new route for designing highly efficient MOF-based catalysts to synthesize DMC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2017YFB0307301, 2017YFA0206802, 2018YFA0704502), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA21020800), and the Science Foundation of Fujian Province (2018J05029, 2019J05156, 2019H0053).Notes and references
- D. Delledonne, F. Rivetti and U. Romano, Developments in the Production and Application of Dimethylcarbonate, Appl. Catal., A, 2001, 221, 241–251 CrossRef CAS.
- Y. Ono, Catalysis in the Production and Reactions of Dimethyl Carbonate, an Environmentally benign Building Block, Appl. Catal., A, 1997, 155, 133–166 CrossRef CAS.
- T. Tabanelli, S. Cailotto, J. Strachan, A. F. Masters, T. Maschmeyer, A. Perosa and F. Cavani, Process Systems for the Carbonate Interchange Reactions of DMC and Alcohols: Efficient Synthesis of Catechol Carbonate, Catal. Sci. Technol., 2018, 8, 1971–1980 RSC.
- Q. Zhou, S. Dong, Z. Lv, G. Xu, L. Huang, Q. Wang, Z. Cui and G. Cui, A Temperature - Responsive Electrolyte Endowing Superior Safety Characteristic of Lithium Metal Batteries, Adv. Energy Mater., 2020, 10, 1903441 CrossRef CAS.
- A. Duval and L. Avérous, Mild and Controlled Lignin Methylation with Trimethyl Phosphate: Towards a Precise Control of Lignin Functionality, Green Chem., 2020, 22, 1671–1680 RSC.
- J.-Q. Wang, J. Sun, W.-G. Cheng, C.-Y. Shi, K. Dong, X.-P. Zhang and S.-J. Zhang, Synthesis of Dimethyl Carbonate Catalyzed by Carboxylic Functionalized Imidazolium Salt via Transesterification Reaction, Catal. Sci. Technol., 2012, 2, 600–605 RSC.
- T. Wei, M. Wang, W. Wei, Y. Sun and B. Zhong, Synthesis of Dimethyl Carbonate by Transesterification over CaO/Carbon Composites, Green Chem., 2003, 5, 343–346 RSC.
- Y.-L. Gan, X.-Q. Hu, L.-Z. Wen, J. Xu and B. Xue, Metal-Free Synthesis of Dimethyl Carbonate via Transesterification of Ethylene Carbonate Catalyzed by Graphitic Carbon Nitride Materials, New J. Chem., 2020, 44, 3215–3223 RSC.
- Y. Z. Yuan, W. Cao and W. Z. Weng, CuCl2 Immobilized on Amino-Functionalized MCM-41 and MCM-48 and Their Catalytic Performance toward the Vapor-Phase Oxy-Carbonylation of Methanol to Dimethylcarbonate, J. Catal., 2004, 228, 311–320 CrossRef CAS.
- Y. Zhang, D. N. Briggs, E. De Smit and A. T. Bell, Effects of Zeolite Structure and Composition on the Synthesis of Dimethyl Carbonate by Oxidative Carbonylation of Methanol on Cu-Exchanged Y, ZSM-5, And Mordenite, J. Catal., 2007, 251, 443–452 CrossRef CAS.
- M. C. Figueiredo, V. Trieu, S. Eiden and M. T. Koper, Spectro-Electrochemical Examination of the Formation of Dimethyl Carbonate from CO and Methanol at Different Electrode Materials, J. Am. Chem. Soc., 2017, 139, 14693–14698 CrossRef CAS PubMed.
- R. Guo, Y. Qin, L. Qiao, J. Chen, X. Wu and Y. Yao, Enhancement of the Catalytic Performance in Pd-Cu/NaY Catalyst for Carbonylation of Methyl Nitrite to Dimethyl Carbonate: Effects of Copper Doping, Catal. Commun., 2017, 88, 94–98 CrossRef CAS.
- Y. Yamamoto, T. Matsuzaki, S. Tanaka, K. Nishihira, K. Ohdan, A. Nakamura and Y. Okamoto, Catalysis and Characterization of Pd/NaY for Dimethyl Carbonate Synthesis from Methyl Nitrite and CO, J. Chem. Soc., Faraday Trans., 1997, 93, 3721–3727 RSC.
- H.-Z. Tan, Z.-N. Chen, Z.-N. Xu, J. Sun, Z.-Q. Wang, R. Si, W. Zhuang and G.-C. Guo, Synthesis of High-Performance and High-Stability Pd(II)/NaY Catalyst for CO Direct Selective Conversion to Dimethyl Carbonate by Rational Design, ACS Catal., 2019, 9, 3595–3603 CrossRef CAS.
- Z. Q. Wang, J. Sun, Z. N. Xu and G. C. Guo, CO Direct Esterification to Dimethyl Oxalate and Dimethyl Carbonate: the Key Functional Motifs for Catalytic Selectivity, Nanoscale, 2020, 12, 20131–20140 RSC.
- Y. Ge, Y. Dong, S. Wang, Y. Zhao, J. Lv and X. Ma, Influence of Crystalline Phase of Li-Al-O Oxides on the Activity of Wacker-Type Catalysts in Dimethyl Carbonate Synthesis, Front. Chem. Sci. Eng., 2012, 6, 415–422 CrossRef CAS.
- S. Wang, L. Wei, Y. Dong, Y. Zhao and X. Ma, Dimethyl Carbonate Synthesis from Methyl Nitrite and CO over Activated Carbon Supported Wacker-type Catalysts: the Surface Chemistry of Activated Carbon, Catal. Commun., 2015, 72, 43–48 CrossRef CAS.
- G. L. Zhuo and X. Z. Jiang, Catalytic Decompostiton of Methyl Nitrite over Supported Palladium Catalysts in Vapor Phase, React. Kinet. Catal. Lett., 2002, 77, 219–226 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The Chemistry and Applications of Metal-Organic Frameworks, Science, 2013, 341, 6149 CrossRef PubMed.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Zr-Based Metal-Organic Frameworks: Design, Synthesis, Structure, and Applications, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- Y. H. Xiao, W. M. Tian, S. Y. Jin, Z. G. Gu and J. Zhang, Host-Guest Thin Films by Confining Ultrafine Pt/C QDs into Metal-Organic Frameworks for Highly Efficient Hydrogen Evolution, Small, 2020, 16, 2005111 CrossRef CAS PubMed.
- Z. H. Gao, B. Y. Xu, T. J. Zhang, Z. Liu, W. G. Zhang, X. Sun, Y. Liu, X. Wang, Z. F. Wang, Y. L. Yan, F. Q. Hu, X. G. Meng and Y. S. Zhao, Spatially Responsive Multicolor Lanthanide-MOF Heterostructures for Covert Photonic Barcodes, Angew. Chem., Int. Ed., 2020, 59, 19060–19064 CrossRef CAS PubMed.
- J.-D. Yi, R. Xie, Z.-L. Xie, G.-L. Chai, T.-F. Liu, R.-P. Chen, Y.-B. Huang and R. Cao, Highly Selective CO2 Electroreduction to CH4 by In situ Generated Cu2O Single-Type Sites on a Conductive MOF: Stabilizing Key Intermediates with Hydrogen Bonding, Angew. Chem., Int. Ed., 2020, 59, 23641–23648 CrossRef CAS PubMed.
- S. Sharma, A. V. Desai, B. Joarder and S. K. Ghosh, A Water-Stable Ionic MOF for the Selective Capture of Toxic Oxoanions of SeVI and AsV and Crystallographic Insight into the Ion-Exchange Mechanism, Angew. Chem., 2020, 132, 7862–7866 CrossRef.
- D. Buzek, S. Adamec, K. Lang and J. Demel, Metal-Organic Frameworks vs. Buffers: Case Study of UiO-66 Stability, Inorg. Chem. Front., 2021, 8, 720–734 RSC.
- M. Ding and H.-L. Jiang, Improving Water Stability of Metal–Organic Frameworks by a General Surface Hydrophobic Polymerization, CCS Chem., 2021, 3, 2740–2748 CrossRef CAS.
- G. Huang, Q. H. Yang, Q. Xu, S. H. Yu and H. L. Jiang, Polydimethylsiloxane Coating for a Palladium/MOF Composite: Highly Improved Catalytic Performance by Surface Hydrophobization, Angew. Chem., Int. Ed., 2016, 55, 7379–7383 CrossRef CAS PubMed.
- B. Li, H. M. Wen, Y. J. Cui, W. Zhou, G. D. Qian and B. L. Chen, Emerging Multifunctional Metal-Organic Framework Materials, Adv. Mater., 2016, 28, 8819–8860 CrossRef CAS PubMed.
- L. L. Gong, W. T. Yao, Z. Q. Liu, A. M. Zheng, J. Q. Li, X. F. Feng, L. F. Ma, C. S. Yan, M. B. Luo and F. Luo, Photoswitching Storage of Guest Molecules in Metal-Organic Framework for Photoswitchable Catalysis: Exceptional Product, Ultrahigh Photocontrol, and Photomodulated Size Selectivity, J. Mater. Chem. A, 2017, 5, 7961–7967 RSC.
- G. R. Cai, M. L. Ding, Q. Y. Wu and H. L. Jiang, Encapsulating Soluble Active Species into Hollow Crystalline Porous Capsules beyond Integration of Homogeneous and Heterogeneous Catalysis, Natl. Sci. Rev., 2020, 7, 37–45 CrossRef CAS PubMed.
- Y.-P. Xu, Z.-Q. Wang, H.-Z. Tan, K.-Q. Jing, Z.-N. Xu and G.-C. Guo, Lewis Acid Sites in MOFs Supports Promoting the Catalytic Activity and Selectivity for CO Esterification to Dimethyl Carbonate, Catal. Sci. Technol., 2020, 10, 1699–1707 RSC.
- G. Li, H. Abroshan, Y. Chen, R. Jin and H. J. Kim, Experimental and Mechanistic Understanding of Aldehyde Hydrogenation Using Au25 Nanoclusters with Lewis Acids: Unique Sites for Catalytic Reactions, J. Am. Chem. Soc., 2015, 137, 14295–14304 CrossRef PubMed.
- M. Shamzhy, M. Opanasenko, P. Concepcion and A. Martinez, New Trends in Tailoring Active Sites in Zeolite-Based Catalysts, Chem. Soc. Rev., 2019, 48, 1095–1149 RSC.
- S. Zhang, Z. Xia, Y. Zou, M. Zhang and Y. Qu, Spatial intimacy of Binary Active-Sites for Selective Sequential Hydrogenation-Condensation of Nitriles into Secondary Imines, Nat. Commun., 2021, 12, 3382 CrossRef CAS PubMed.
- L. Jiao, J. Wang and H.-L. Jiang, Microenvironment Modulation in Metal–Organic Framework-Based Catalysis, Acc. Mater. Res., 2021, 2, 327–339 CrossRef CAS.
- D. Li, H.-Q. Xu, L. Jiao and H.-L. Jiang, Metal–Organic Frameworks for Catalysis: State of the Art, Challenges, and Opportunities, EnergyChem, 2019, 1, 100005 CrossRef.
- Y. Dong, S. Huang, S. Wang, Y. Zhao, J. Gong and X. Ma, Synthesis of Dimethyl Carbonate through Vapor-Phase Carbonylation Catalyzed by Pd-Doped Zeolites: Interaction of Lewis Acidic Sites and Pd Species, ChemCatChem, 2013, 5, 2174–2177 CrossRef CAS.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Synthesis and Stability of Tagged UiO-66 Zr-MOFs, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Disclosing the Complex Structure of UiO-66 Metal Organic Framework: A Synergic Combination of Experiment and Theory, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, Unusual and Highly Tunable Missing-Linker Defects in Zirconium Metal–Organic Framework UiO-66 and Their Important Effects on Gas Adsorption, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- J. Wang, L. Liu, C. Chen, X. Dong, Q. Wang, L. Alfilfil, M. R. AlAlouni, K. Yao, J. Huang, D. Zhang and Y. Han, Engineering Effective Structural Defects of Metal–Organic Frameworks to Enhance Their Catalytic Performances, J. Mater. Chem. A, 2020, 8, 4464–4472 RSC.
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, Synthesis Modulation as a Tool To Increase the Catalytic Activity of Metal–Organic Frameworks: The Unique Case of UiO-66(Zr), J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- N. Ronaghi, D. Shade, H. J. Moon, S. Najmi, J. W. Cleveland, K. S. Walton, S. France and C. W. Jones, Modulation and Tuning of UiO-66 for Lewis Acid Catalyzed Carbohydrate Conversion: Conversion of Unprotected Aldose Sugars to Polyhydroxyalkyl and C-Glycosyl Furans, ACS Sustainable Chem. Eng., 2021, 9, 11581–11595 CrossRef CAS.
- F. Martinovic, F. A. Deorsola, M. Armandi, B. Bonelli, R. Palkovits, S. Bensaid and R. Pirone, Composite Cu-SSZ-13 and CeO2-SnO2 for Enhanced NH3-SCR Resistance towards Hydrocarbon Deactivation, Appl. Catal., B, 2021, 282, 119536 CrossRef CAS.
- H. Leclerc, A. Vimont, J.-C. Lavalley, M. Daturi, A. D. Wiersum, P. L. Llwellyn, P. Horcajada, G. Férey and C. Serre, Infrared Study of the Influence of Reducible Iron(III) Metal Sites on the Adsorption of CO, CO2, Propane, Propene and Propyne in the Mesoporous Metal-Organic Framework MIL-100, Phys. Chem. Chem. Phys., 2011, 13, 11748–11756 RSC.
- C. Volkringer, H. Leclerc, J.-C. Lavalley, T. Loiseau, G. Ferey, M. Daturi and A. Vimont, Infrared Spectroscopy Investigation of The Acid Sites in the Metal–Organic Framework Aluminum Trimesate MIL-100 (Al), J. Phys. Chem. C, 2012, 116, 5710–5719 CrossRef CAS.
- A. Lingel, A. Vulpetti, T. Reinsperger, A. Proudfoot, R. Denay, A. Frommlet, C. Henry, U. Hommel, A. D. Gossert and B. Luy, Comprehensive and High–Throughput Exploration of Chemical Space Using Broadband 19F NMR–Based Screening, Angew. Chem., Int. Ed., 2020, 59, 14809–14817 CrossRef CAS PubMed.
- J. T. Xu, M. D. Sun, Y. Kuang, H. T. Bi, B. Liu, D. Yang, R. C. Lv, S. L. Gai, F. He and P. P. Yang, Markedly Enhanced Up-Conversion Luminescence by Combining IR-808 Dye Sensitization and Core-Shell-Shell Structures, Dalton Trans., 2017, 46, 1495–1501 RSC.
- M. Nasrabadi, M. A. Ghasemzadeh and M. R. Z. Monfared, The Preparation and Characterization of UiO-66 Metal-Organic Frameworks for the Delivery of the Drug Ciprofloxacin and an Evaluation of Their Antibacterial Activities, New J. Chem., 2019, 43, 16033–16040 RSC.
- X. Chen, Q. Li, J. J. Li, J. Chen and H. P. Jia, Modulating Charge Separation via In Situ Hydrothermal Assembly of Low Content Bi2S3 into UiO-66 for Efficient Photothermocatalytic CO2 Reduction, Appl. Catal., B, 2020, 270, 118915 CrossRef CAS.
- R. M. Xu, Q. H. Ji, P. Zhao, M. P. Jian, C. Xiang, C. Z. Hu, G. Zhang, C. C. Tang, R. P. Liu, X. W. Zhang and J. H. Qu, Hierarchically Porous UiO-66 with Tunable Mesopores and Oxygen Vacancies for Enhanced Arsenic Removal, J. Mater. Chem. A, 2020, 8, 7870–7879 RSC.
- C. Y. Xu, Y. T. Pan, G. Wan, H. Liu, L. Wang, H. Zhou, S. H. Yu and H. L. Jiang, Turning on Visible-Light Photocatalytic C-H Oxidation over Metal-Organic Frameworks by Introducing Metal-to-Cluster Charge Transfer, J. Am. Chem. Soc., 2019, 141, 19110–19117 CrossRef CAS PubMed.
- K. Hendrickx, J. J. Joos, A. De Vos, D. Poelman, P. F. Smet, V. Van Speybroeck, P. Van Der Voort and K. Lejaeghere, Exploring Lanthanide Doping in UiO-66: A Combined Experimental and Computational Study of the Electronic Structure, Inorg. Chem., 2018, 57, 5463–5474 CrossRef CAS PubMed.
- F. K. Bi, X. D. Zhang, J. F. Chen, Y. Yang and Y. X. Wang, Excellent Catalytic Activity and Water Resistance of UiO-66-Supported Highly Dispersed Pd Nanoparticles for Toluene Catalytic Oxidation, Appl. Catal., B, 2020, 269, 118767 CrossRef CAS.
- Y. Yao, K. X. Huang, Y. Liu, T. T. Luo, G. Tian, J. X. Li, S. Zhang, G. G. Chang and X. Y. Yang, A Hierarchically Multifunctional Integrated Catalyst with Intimate and Synergistic Active Sites for One-Pot Tandem Catalysis, Inorg. Chem. Front., 2021, 8, 3463–3472 RSC.
- F. Morales-Lara, V. K. Abdelkader-Fernández, M. Melguizo, A. Turco, E. Mazzotta, M. Domingo-García, F. J. López-Garzón and M. Pérez-Mendoza, Ultra-Small Metal Nanoparticles Supported on Carbon Nanotubes Through Surface Chelation and Hydrogen Plasma Reduction for Methanol Electro-Oxidation, J. Mater. Chem. A, 2019, 7, 24502–24514 RSC.
- M. Skotak, Z. Karpinski, W. Juszczyk, J. Pielaszek, L. Kepinski, D. V. Kazachkin, V. I. Kovalchuk and J. L. d'Itri, Characterization and Catalytic Activity of Differently Pretreated Pd/Al2O3 Catalysts: the Role of Acid Sites and of Palladium-Alumina Interactions, J. Catal., 2004, 227, 11–25 CrossRef CAS.
- J. F. Weaver, J. Choi, V. Mehar and C. J. Wu, Kinetic Coupling among Metal and Oxide Phases during CO Oxidation on Partially Reduced PdO(101): Influence of Gas-Phase Composition, ACS Catal., 2017, 7, 7319–7331 CrossRef CAS.
- A. W. Aylor, L. J. Lobree, J. A. Reimer and A. T. Bell, Investigations of the Dispersion of Pd in H-ZSM-5, J. Catal., 1997, 172, 453–462 CrossRef CAS.
- F. H. Meng, P. Z. Zhong, Z. Li, X. X. Cui and H. Y. Zheng, Surface Structure and Catalytic Performance of Ni-Fe Catalyst for Low-Temperature CO Hydrogenation, J. Chem., 2014, 2014, 534842 Search PubMed.
- C. Z. Wang, L. P. Han, P. J. Chen, G. F. Zhao, Y. Liu and Y. Lu, High-performance, Low Pd-Loading Microfibrous-Structured Al-Fiber@ns-AlOOH@Pd Catalyst for CO Coupling to Dimethyl Oxalate, J. Catal., 2016, 337, 145–156 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qi00302c |
| This journal is © the Partner Organisations 2022 |