
Urea-engineering mediated hydrogen-bond donating Friedel–Crafts alkylation of indoles and nitroalkenes in a dual-functionalized microporous metal–organic framework with high recyclability and pore-fitting-induced size-selectivity†
Manpreet
Singh
 ab and
Subhadip
Neogi
*ab
ab and
Subhadip
Neogi
*ab
aInorganic Materials & Catalysis Division, CSIR-Central Salt and Marine Chemicals Research Institute (CSIR-CSMCRI), Bhavnagar, Gujarat 364002, India. E-mail: sneogi@csmcri.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
First published on 4th March 2022
Abstract
As an effective alternative to Lewis-acid activation, hydrogen-bond donating (HBD) organo-catalysis represents a powerful construction tool for important classes of carbon–carbon bonds, wherein metal–organic frameworks (MOFs) alleviate issues like self-quenching, solubility and reactivity. However, size-selectivity is rather challenging in such catalysis, while the status quo is still unexplored when both H-bonding and open-metal sites (OMS) are present together in a single system. The pillar-bilayer Cd(II) MOF with a rare (3,8)-connected 2-nodal network upholds uni-directional microporous channels integrated with free –NH groups from the urea-moiety of the N,N-donor linker, and aqua-molecule bound [Cd3(COO)6] cluster. The activated framework allows the highly efficient Friedel–Crafts alkylation of indole and β-nitrostyrene under relatively mild conditions with low catalyst loading and no leaching. The strategically designed MOF exhibits unaltered activity over multiple catalytic cycles, and corroborates its effectivity towards a wide range of substituted electrophiles and nucleophiles. Importantly, suitably sized pores generated by two-fold interpenetration restrict the entry of a sterically encumbered substrate and result in poor conversion, demonstrating the rarest pore-fitting-induced size-selectivity. Given that this pore-engineered MOF contains both coordination unsaturated Cd(II) centres and unbound –NH groups as active interaction sites, explicit proof of the interaction of the MOF functionality with the –NO2 group of the reactant is elaborated for the first time in light of the change in emission intensity of the framework in the presence of an electrophile, a judicious choice of substrate, and an in-depth comparison of the catalytic activity of an isostructural framework without a urea-moiety. These control experiments unprecedentedly authenticate urea-moiety-mediated two-point hydrogen bonding in the proposed catalytic route, and simultaneously exclude any major role for OMS at the SBU. Apart from pore-induced size-exclusive reactions, this MOF exemplifies site-specific Friedel–Crafts alkylation, and paves the way to tailor-made engineering of advanced functionalities in contemporary materials for unconventional HBD reactions at the interface of structure–property synergies.
Introduction
Recent advances in developing a practical approach for the construction of important classes of carbon–carbon bonds will substantially dictate the overall synthetic efficacy of technologically relevant building blocks. For example, the Friedel–Crafts alkylation reaction of nitroalkene with indole is one of the fundamental C–C bond forming reactions, catalyzed by the presence of suitable Lewis acids.1–4 Among diverse substrates, β-nitrostyrene is considered the most preferred Michael acceptor in this reaction because of the strong electron-withdrawing nature of the nitro group.5,6 However, the lower nucleophilicity of non-N-substituted indoles compared to their N-alkylated (viz. 1-methylindole) analogues7,8 leads to much more sluggish Friedel–Crafts reactions. Clearly, there is an urgent need for an advanced methodology to overcome the barriers in this crucial transformation, which often assists in synthesizing tryptamine derivatives as precursors to important biologically active alkaloids.9–14 In this milieu, hydrogen bond donor (HBD) catalysts have emerged as potential pseudo-Lewis-acidic compounds, capable of activating electrophiles towards nucleophilic addition via cooperative hydrogen bonding, and have been shown to be an alternative approach to Lewis-acid activation because of their rigid conformation, tunable reactivity and anchored functional sites.15–18 Generally, thiourea or urea groups are used as key functional parts in such catalysts that make use of two-point hydrogen bonding through acidic N–H bonds. In principle, such hydrogen bonding diminishes the lowest unoccupied molecular orbital (LUMO) of the electrophile, thereby lowering the activation barrier for subsequent nucleophilic attack.15 Thus far, homogeneous HBD catalysts have proven capable of plentiful bond-forming reactions, including Claisen rearrangements,19 Mannich reactions,20 Diels–Alder reactions,21 Strecker reactions,22 and Friedel–Crafts reactions23 with excellent selectivity and yield. However, these functional groups are prone to self-quenching that involves unproductive self-assemblies formed through dimerization or oligomerization of the catalyst. Alternatively, the presence of intermolecular hydrogen bonding further engenders a noteworthy reduction in the catalytic activity of these assemblies.24,25 Keeping these drawbacks in mind, the best possible approach for efficient HBD catalysis could be to amalgamate metal-free (organo-catalytic systems) and heterogeneous catalysts over a single platform.26 One promising strategy consists of incorporating homogeneous catalytic sites into a porous structure so as to nullify the possibility of self-quenching by the way of uniform spatial regulation of the responsive H-bonding centres without compromising the specific surface area.27,28Metal–organic frameworks (MOFs) are well-defined porous crystalline materials with intriguing properties suitable for a plethora of applications, viz. gas adsorption and separation, heterogeneous catalysis, luminescent sensing, proton conduction, water purification, drug delivery, magnetism and so forth.29–36 Due to the definite structural organization, tunable porosity, and strategic incorporation of guest-responsive functional sites, these hybrids are extensively used in different avenues of multifaceted attributes.37–39 In view of their efficient HBD catalysis, their key capabilities lie in the careful incorporation of functional hydrogen-bonding sites, and their uniform distribution throughout the porous structure in MOFs. However, this feat is often tricky as functional organic groups (FOGs) either coordinate with metal ions or are not precisely oriented along the pore structure to exhibit desired catalytic activity. Then again, a suitable choice of hydrogen-bond donor system is critical, considering the fact that thioureas generally reveal thermal stability issues during MOF synthesis, whereas the corresponding ureas have no such effects, yet exhibit high tendency to act as hydrogen-bond donors.21,40 Importantly, the periodic arrangement of building units aids in developing a reliable structure–performance correlation, leading to an unambiguous elucidation of the role of appropriate functionality in the underlying catalytic mechanism, which is otherwise unavailable with traditional porous solid systems.41–43 A number of reports have thus far overlaid the mechanistic conduit in MOF-based HBD catalysis, using FOGs as the sole active sites. However, the status quo is still unprecedented when both an open-metal node and an H-bonding site are simultaneously present in a single system. In contrast to surface catalysis, these materials can also provide a micro-reactor type environment due to the pore confinement effect, which assists in increasing the effective number of collisions between substrates and active centers, and promises pore-fitting-mediated size selectivity.26
Considering the above-mentioned facts and challenges, coupled with our ongoing quest to develop functionalized porous crystalline materials for environmentally benign and size-exclusive catalysis,44–50 we constructed a Cd(II)-based framework, CSMCRI-12 (CSMCRI = Central Salt & Marine Chemicals Research Institute) via the self-assembly of a tripodal carboxylate ligand and a urea functionality grafted di-pyridyl linker. Structural elucidation divulged a number of fascinating attributes like (i) activation-induced generation of high-density unsaturated Cd(II) centers, (ii) N-functionality-actuated guest interaction sites in the carboxylate ligand, (iii) perfect positioning of urea functional groups within the cavity, and (iv) the two-fold-interpenetration-mediated formation of suitably sized unidirectional pores. The catalytic performance of the desolvated framework in the Friedel–Crafts alkylation reaction of indole and β-nitrostyrene under mild reaction conditions is studied with a detailed understanding of substrate scope and recyclability. For the best HBD coupling reaction, the narrow pore channel assists the diffusion of substrates with compatible molecular dimensions and unveils the rarest size-selectivity. The major role of the hydrogen-bonding interaction between the functionalized linker and the nitro group of electrophiles over an open metal site in the dual-functionalized MOF is supported by a battery of experimental evidence, including comparison with a similar framework without a urea-moiety and in view of the change in luminescence intensity of the MOF upon addition of β-nitrostyrene.
Results and discussion
Crystal structure of CSMCRI-12
Block-shaped colourless crystals of CSMCRI-12 were solvothermally grown by the reaction of Cd(NO3)2·4H2O (0.1 mmol), C3-symmetric 4,4′,4′′-tricarboxytriphenylamine (H3TCA) ligand (0.05 mmol) and functionalized 1,3-di(pyridin-4-yl)urea (L) linker (0.1 mmol) in a mixed-solvent system of N,N-dimethylacetamide/water (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 85 °C. Single-crystal X-ray diffraction (SCXRD) analysis showed triclinic space group P
:1) at 85 °C. Single-crystal X-ray diffraction (SCXRD) analysis showed triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with three crystallographically independent Cd(II) ions, two deprotonated TCA3− ligands, one linker, and a metal-bound water molecule in the asymmetric unit. Three Cd(II) ions are bridged together by six carboxylate groups from diverse TCA3− ligands to form a tri-nuclear [Cd3(COO)6] cluster (Fig. S2, ESI†) with average Cd–Cd distance of 3.78 Å, which is regarded as the secondary building unit (SBU). The coordination environment around each Cd(II) center in the SBU reveals six coordination with distorted octahedral geometries from dissimilar atom-connections. While the middle Cd(II) ion is entirely ligated to six carboxylate oxygen atoms, each terminal metal node is attached to one pyridyl ring of the linker. These SBUs are joined through long aromatic arms of carboxylate ligands and propagate along the crystallographic ac plane to create a two-dimensional (2D) bilayer framework, containing large hexagonal rings (Fig. 1a) of dimension 18.77 × 15.98 Å2 (considering atom-to-atom connection).
with three crystallographically independent Cd(II) ions, two deprotonated TCA3− ligands, one linker, and a metal-bound water molecule in the asymmetric unit. Three Cd(II) ions are bridged together by six carboxylate groups from diverse TCA3− ligands to form a tri-nuclear [Cd3(COO)6] cluster (Fig. S2, ESI†) with average Cd–Cd distance of 3.78 Å, which is regarded as the secondary building unit (SBU). The coordination environment around each Cd(II) center in the SBU reveals six coordination with distorted octahedral geometries from dissimilar atom-connections. While the middle Cd(II) ion is entirely ligated to six carboxylate oxygen atoms, each terminal metal node is attached to one pyridyl ring of the linker. These SBUs are joined through long aromatic arms of carboxylate ligands and propagate along the crystallographic ac plane to create a two-dimensional (2D) bilayer framework, containing large hexagonal rings (Fig. 1a) of dimension 18.77 × 15.98 Å2 (considering atom-to-atom connection).
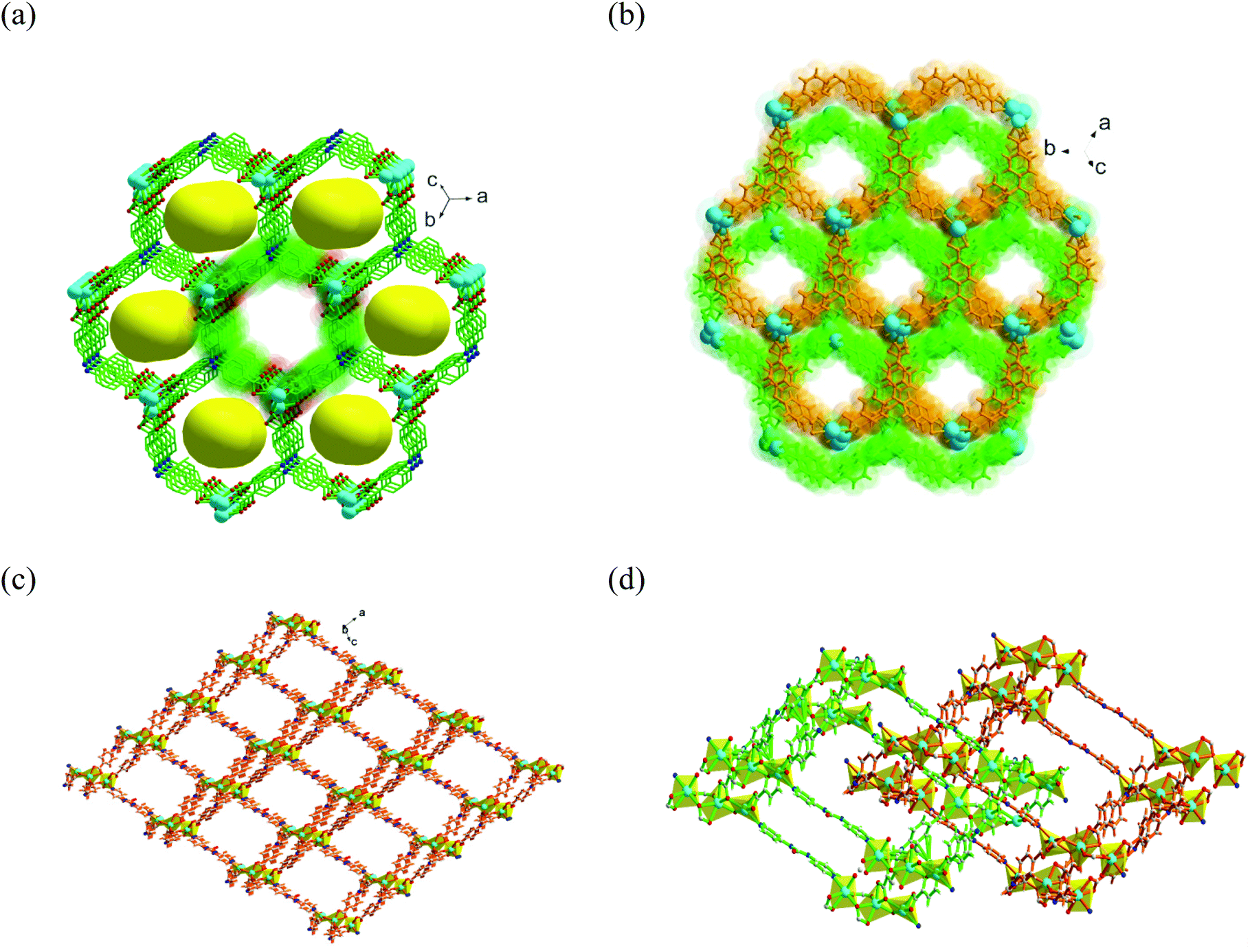 | ||
| Fig. 1 (a) View of the nearly hexagonal pores in a single net of CSMCRI-12 along the ac plane. (b) Representation of the doubly interpenetrated nets along the ac plane (for clarity, each single net is shown in a different color). (c) Rhombohedral channels in a single net along the b axis. (d) Two-fold interpenetration in the framework. | ||
The 2D layers are further connected through the di-pyridyl linker that results in the formation of a bilayer-pillar framework.45 A perspective view of the single-net 3D structure is depicted in Fig. 1c, which shows large rhombohedral channels of size 25.11 × 22.33 Å2 along the b axis. Importantly, the urea functionalities in the linker remain uncoordinated and are projected inside these porous channels, conferring an appropriate environment for the activation of the substrate through their accessible (N–H) hydrogen-bonding sites. The formation of big pores by the presence of a long tripodal ligand as well as a urea-grafted linker (10.29 Å) prompts two-fold interpenetration into the overall framework (Fig. 1d). Topological analysis revealed a rare (3,8)-connected 2-nodal 3D network (Fig. S2c and S2d, ESI†) with the corresponding Schläfli symbol (436248)(43)2.51 Though voids are somewhat reduced, nearly rectangular cavities of dimensions 6.67 × 10.61 Å2 still exist along the b axis (Scheme 1) with a sufficient solvent-accessible void volume (44%) per unit cell.52 Nevertheless, interpenetration instigates a couple of positive effects for CSMCRI-12 as (i) the number of suitably oriented, free urea-groups virtually doubles per cavity, and (ii) the generation of smaller-sized channels (Fig. 1b) can result in size-selective catalysis by virtue of the pore-confinement effect. The guest solvent molecules inside the pores could not be crystallographically mapped because of high disorder. However, their presence was otherwise confirmed from the combined inputs of Fourier-transform infrared (FT-IR) spectral data, elemental analysis, thermogravimetric weight loss, and PLATON calculations that correspond to the molecular formula [Cd3(TCA)2L(H2O)]·2DMA·10H2O (see ESI† for calculation details).
 | ||
| Scheme 1 Illustration of the salient features of multiple functionalities in the ligand, linker and SBU in CSMCRI-12 along with its microporous structure and an image of single-crystals. | ||
Structural characterizations and porosity measurement of the framework
The phase purity of CSMCRI-12 was endorsed by the exact correlation of powder X-ray diffraction (PXRD) peaks of the bulk material to those of simulated peaks (Fig. 2a), obtained from the X-ray structure. Thermogravimetric analysis (TGA) under an N2 atmosphere (Fig. S3, ESI†) revealed a two-step curve without a plateau, where 11.93% initial weight loss (calcd 11.97%) occurs around 120 °C. A second loss of 9.34% (cal. 9.77%) ensues up to 300 °C, and further heating results in framework decomposition. The as-synthesized MOF was soaked in various common organic solvents for 24 h and the PXRD patterns were recorded. As depicted in Fig. 2b, the PXRD profiles in all these media display fair accordance to the pristine material and substantiate its structural robustness. Subsequently, guest solvents in CSMCRI-12 were exchanged upon soaking in acetone for 3 days, and the solvent-free framework (12a) was acquired by heating the solvent-exchanged crystals at 120 °C under vacuum for 6 h. The PXRD pattern of the desolvated MOF showed insignificant changes in the peak positions (Fig. 2a), confirming the unaltered structural integrity and crystallinity. The FTIR spectrum of CSMCRI-12 (Fig. S4, ESI†) showed strong peaks at 1596 cm−1 and 1384 cm−1, corresponding to antisymmetric stretching of the coordinated carboxylate groups. On the other hand, broad peaks in the range ∼3200–3400 cm−1 resemble the presence of water molecules while the carbonyl stretching frequency for the free DMA molecule could be observed at 1755 cm−1. In contrast, the FT-IR spectrum of 12a reveals the absence of these peaks and confirms the removal of the solvent molecules, which was further endorsed by there being no weight loss in the TGA curve up to 300 °C (Fig. S3, ESI†). The microstructure of CSMCRI-12 was studied through field emission-scanning electron microscopy (FE-SEM), which revealed a nearly rectangular block-shaped morphology (Fig. 2d), consistent with the transmission electron microscopy (TEM) image (Fig. S5, ESI†) of the same crystal. Further, SEM energy dispersive X-ray (EDX) analysis showed uniformly distributed elements: C, N, O and Cd (Fig. 2d) throughout the MOF surface. To obtain insight into the electronic structure and surface composition of the as-synthesized framework, X-ray photoelectron spectroscopy (XPS) was employed. The survey spectra (Fig. S6, ESI†) showed the presence of the expected elements. Further deconvolution of the spectrum was carried out to understand the bonding of elements (Fig. 3a–d). The high-resolution C 1s spectrum shows binding energy (BE) values at 284.5, 285.63, and 287.7 eV, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–N, and C–O bonding, respectively.53,54 The high-resolution XPS of N 1s was fitted to two nearly symmetrical peaks because of the presence of the bridge-head tertiary N-atom of tripodal ligand, and N–H binding in the N-donor linker at 399.59 and 400.62 eV, respectively.55,56 Two spin–orbit orientations in the high-resolution XPS spectrum of Cd 3d correspond to Cd 3d5/2, and Cd 3d3/2 that appeared at 412.27 eV and 405.54 eV, respectively, with a spin–orbit splitting value ∼7 eV.57,58 The distinguishing peaks of O 1s were further deconvoluted into two peaks that relate to the metal-bound ligand moiety (O–Cd), and O–C bonding in the organic struts at BE 530.3 and 531.9 eV, respectively.59
C, C–N, and C–O bonding, respectively.53,54 The high-resolution XPS of N 1s was fitted to two nearly symmetrical peaks because of the presence of the bridge-head tertiary N-atom of tripodal ligand, and N–H binding in the N-donor linker at 399.59 and 400.62 eV, respectively.55,56 Two spin–orbit orientations in the high-resolution XPS spectrum of Cd 3d correspond to Cd 3d5/2, and Cd 3d3/2 that appeared at 412.27 eV and 405.54 eV, respectively, with a spin–orbit splitting value ∼7 eV.57,58 The distinguishing peaks of O 1s were further deconvoluted into two peaks that relate to the metal-bound ligand moiety (O–Cd), and O–C bonding in the organic struts at BE 530.3 and 531.9 eV, respectively.59
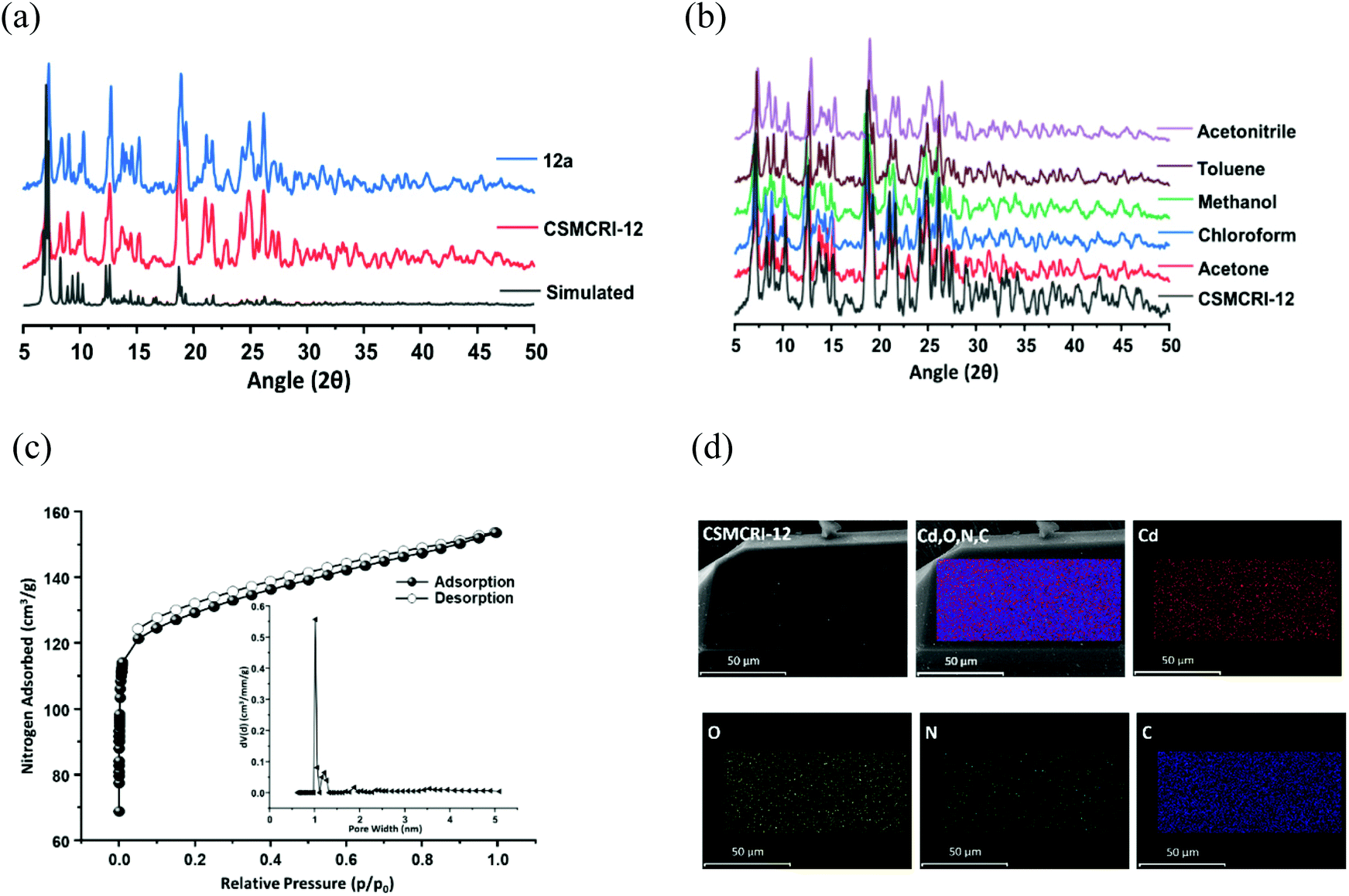 | ||
| Fig. 2 (a) Simulated (black), as-synthesized (red) and activated (blue) PXRD patterns of the framework. (b) PXRD profiles of CSMCRI-12 after exposure to common organic solvents. (c) N2 adsorption isotherm of 12a up to a relative pressure (p/p0) of 1.0 at 77 K (the inset shows the pore-size distribution plot). (d) SEM images of the as-synthesised MOF, and elemental mapping from SEM-EDX showing the uniform distribution of elements (mixed elements, Cd, O, N, C) in the selected area of the crystal. | ||
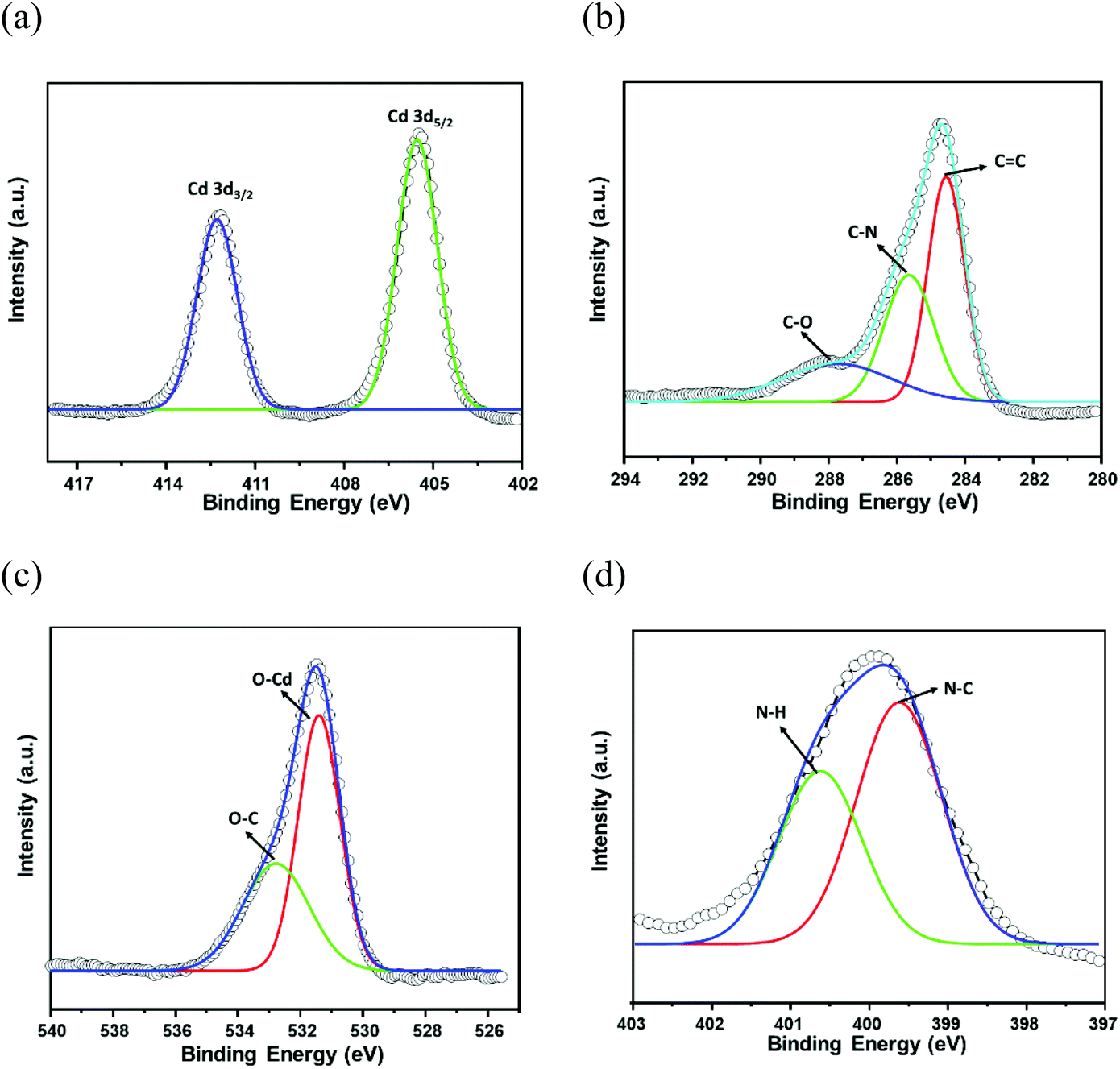 | ||
| Fig. 3 High-resolution XPS spectra of the framework: (a) Cd 3d, (b) C 1s, (c) O 1s, and (d) N 1s. | ||
The porosity of 12a was measured from the N2 adsorption isotherm at 77 K, which displayed a nearly type-I isotherm with a steep rise in the low-pressure region (Fig. 2c), archetypal of a microporous material. The saturated N2 uptake of 149.77 cm3 g−1 corresponds to a Brunauer–Emmett–Teller (BET) surface area of 503 m2 g−1 and the pore volume was calculated to be 0.239 cm3 g−1 from this experimental adsorption isotherm. The non-local density functional theory (NLDFT) model-derived pore size distribution (PSD) curve (Fig. 2c, inset) shows an average pore diameter of 1.022 nm that stems from the presence of smaller-sized pores in the two-fold interpenetrated framework. In spite of the moderate BET surface area, the porosity of this framework exceeds that of many recent MOFs, including TMOF-1 (Table S3, entry 1, ESI†), NKMOF-1-Ni (Table S3, entry 3, ESI†), CoMOF-2 (Table S3, entry17, ESI†), CSMCRI-7 (Table S3, entry 22, ESI†), and CSMCRI-8 (Table S3, entry 22, ESI†). Moreover, the PXRD pattern of the framework after surface area analysis was checked and found to comply with that of the as-made one, substantiating the maintenance of structural integrity (Fig. S7, ESI†).
Friedel–Crafts alkylation of indole and β-nitrostyrene with recyclability
On the basis of strategically positioned urea functionality in CSMCRI-12, together with its robust nature and suitably sized pores generated by two-fold interpenetration, we set out to determine the heterogeneous catalytic activity of the activated MOF in the alkylation of nitroalkenes with indoles. The coupling between 1H-indole (0.12 mmol) and (E)-(2-nitrovinyl)benzene (β-nitrostyrene) (0.1 mmol) was considered as a model reaction (Scheme 2) and formation of the product was analyzed by 1H NMR spectral data (Fig. S8–S22, ESI†).41 The influences of different parameters, including reaction medium, catalyst amount, temperature, and duration were thoroughly checked (Table 1). From the knowledge that low-polar solvents reduce unfavourable interactions with the active sites in an HBD catalyst,60 the reaction was performed in solvents like dichloromethane, chloroform, acetonitrile, and toluene, which showed toluene to be the most suitable (Table 1, entries 1–4). The coupling between indole and β-nitrostyrene hardly afforded any product at room temperature (Table 1, entry 6), and the alkylation was therefore targeted at higher temperature. Maximum yield of the desired product 3-(1-nitro-2-phenylethyl)-1H-indole was perceived upon increasing the temperature to 60 °C (Fig. 4). It may be noted that the present reaction temperature is still below that of contemporary reports.23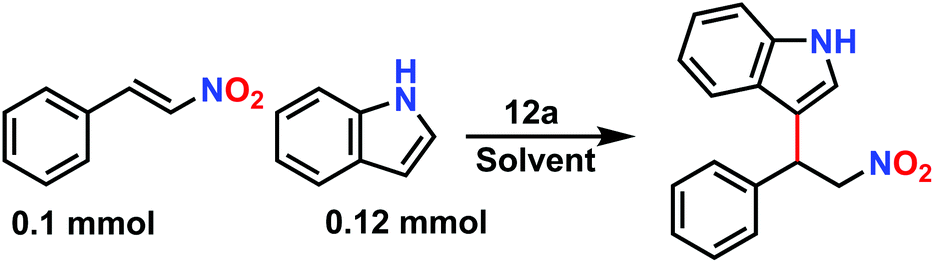 | ||
| Scheme 2 Model reaction between β-nitrostyrene and indole, catalysed by 12a. | ||
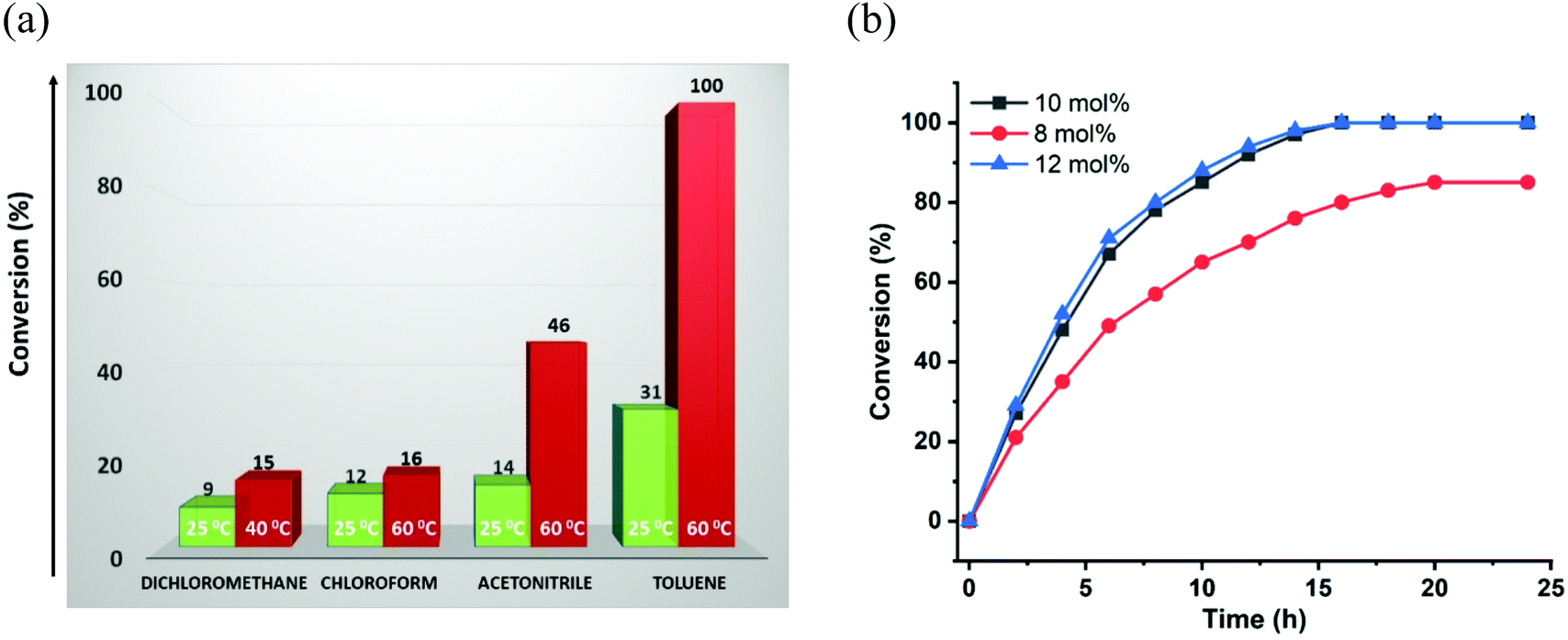 | ||
| Fig. 4 (a) Effect of solvent and temperature on conversion (%) of HBD reaction. (b) Kinetic study of the model reaction using various concentrations of the catalyst (12a) in toluene. | ||
| S. no. | Catalyst | Catalyst mol (%) | Time | Solvent | Temp (°C) | Conversiona (%) |
|---|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy. | ||||||
| 1. | 12a | 10 | 16 | DCM | 40 | 15 |
| 2. | 12a | 10 | 16 | Chloroform | 60 | 16 |
| 3. | 12a | 10 | 16 | Acetonitrile | 60 | 46 |
| 4. | 12a | 10 | 16 | Toluene | 60 | 100 |
| 5. | 12a | 10 | 24 | Toluene | 60 | 100 |
| 6. | 12a | 10 | 24 | Toluene | 25 | 31 |
| 7. | 12a | 5 | 16 | Toluene | 60 | 61 |
| 8. | 12a | 8 | 16 | Toluene | 60 | 85 |
| 9. | 12a | 12 | 16 | Toluene | 60 | 100 |
| 10. | Cd(NO3)2·4H2O | 10 | 16 | Toluene | 60 | 39 |
| 11. | L | 10 | 16 | Toluene | 60 | 65 |
| 12. | L /Cd(NO3)2·4H2O | 10 | 16 | Toluene | 60 | 74 |
| 13. | H3TCA | 10 | 16 | Toluene | 60 | 7 |
| 14. | CSMCRI-12 | 10 | 16 | Toluene | 60 | 29 |
| 15. | No catalyst | 10 | 24 | Toluene | 60 | 5 |
| 16. | Activated CSMCRI-945 |
12 | 24 | Toluene | 60 | 34 |
Subsequently, Friedel–Crafts alkylation was monitored at different loadings of 12a, which showed that an increase in catalyst amount from 5 mol% (conversion: 61%) to 8 mol% (conversion: 85%) has a positive effect on conversion (Table 1, entries 7 and 8). Maximum product was achieved with 10 mol% of catalyst in toluene at 60 °C (Table 1, entry 5). A comparison of available data of MOF-catalysed HBD reactions showed that this catalyst loading is comparable to others.8,61 Then again, the reaction was found to be very dependent on time in the sense that a noteworthy decline in conversion occurred with a shorter time span. This trend is depicted in a plot of conversion (%) vs. time presented in Fig. 4b, which elaborates a 46% conversion up to a period of 4 h that increased to 68% in 6 h. The reaction showed maximum conversion (100%) at 16 h, and the activity remained unchanged even up to 24 h. In addition to this, we performed another set of experiments (Table 1, entry 9) considering a slightly higher amount of 12a (12 mol%), and the kinetics profiles exhibited maximum conversion in the case of the 10 mol% catalyst. Given that an excess amount of 12a did not make any difference to the conversion (%), all the additional experiments and substrate scopes were performed by using the 10 mol% catalyst.
Therefore, the optimized conditions for alkylation reaction were established as 10 mol% of 12a, at 60 °C for 16 h in toluene, and all additional experiments were performed under these conditions. Though there are a handful examples of HBD catalysis by MOFs (Table S4, ESI†), some reports suffer seriously from long reaction time, the need for additives, and multistep catalyst synthesis through post-synthetic exchange (PSE) and/or post-synthetic modification (PSM), which limit their practical applicability.61–64 In view of these limitations, the present system is advantageous as the MOF can be easily synthesized, and used for the C–C coupling reaction under moderate conditions after mild activation. The heterogeneous nature of 12a in the reaction between indole and nitroalkene was confirmed from leaching as well as hot filtration tests. First, the catalyst was separated after completion of the reaction, and the remaining mixture was evaporated to dryness. Inductively coupled plasma-optical emission spectroscopy (ICP-OES) of the resulting solid mass revealed insignificant cadmium (0.002 ppm), ruling out any possibility of leaching of metal ions into the product. Next, the progress of the reaction was monitored after 6 h, and the dispersed catalyst was then removed via filtration. No further conversion to the product could be observed upon continuing the reaction for an additional 18 h (Fig. S50, ESI†), indicating that heterogeneous catalyst 12a is essential for the progress of this reaction. To crosscheck the catalyst recyclability, the material was first recovered from the reaction mixture after the initial run, washed with acetone and heated at 120 °C for 4 h before further use. To make sure that the catalyst was free from reactants or products, we heated the recovered 12a in toluene at 60 °C for 6 h, and the solution did not reveal the presence of either reactant or product in 1H NMR. We were delighted to find that the catalyst reusability shows insignificant loss of activity up to 5 cycles (Fig. 6a). Nevertheless, a slight decrease in the product yield is ascribed to a minor loss of catalyst during the recovery process of each cycle.
The XPS studies of the recovered catalyst (Fig. 6b) show it is identical to the pristine MOF without any reduction in cadmium content and/or additional peaks associated with ligand/linker moieties (Fig. S45, ESI†), indicating no degradation of the material. The symmetric and asymmetric stretching peaks of the carboxylate group and peak for the urea moiety in FT-IR are consistent with the pristine framework (Fig. S44, ESI†). Moreover, every peak position in the PXRD profile of recovered 12a matches that of CSMCRI-12 (Fig. S43, ESI†), while the selected area electron diffraction (SAED) pattern after catalysis showed (Fig. S47, ESI†) maintenance of the crystalline nature. Alongside, we measured the BET surface area of the filtered catalyst after the first and fifth catalytic cycles, which revealed the similar nature of the N2 adsorption isotherm (Fig. S51, ESI†) to that of 12a. Further, SEM images of the recovered MOF showed no alteration in the surface morphology with a uniform distribution of elements (Fig. 6c). These observations mutually confirm that the structural integrity, crystallinity and phase purity of the microporous architecture are well maintained after multicyclic Friedel–Crafts alkylation.
With knowledge of the optimum reaction conditions, we subsequently strove to figure out the impact of the strategic design of the present framework in Friedel–Crafts alkylation. As described in entry 15 of Table 1, no reaction ensues in the absence of 12a, whereas very low conversion occurred in the presence of a tripodal carboxylate ligand (Table 1, entry 13) due to the lack of potential active sites. While Cd(NO3)2·4H2O as a catalyst showed only moderate conversion to the product over a period of 24 h (Table 1, entry 10), the use of a functionalized linker afforded 65% conversion under identical conditions (Table 1, entry 11). Unsurprisingly, this reduction in the catalytic activity of L in comparison to 12a is attributed to the self-quenching of the free N–H bonds through di/oligomerization as a means of generating non-constructive self-assembly.61,65,66 To support this verdict more explicitly, and to investigate how functional-group-assisted pore-environment modulation can substantially mitigate HBD catalysis via alleviating unfavourable self-aggregation of the urea-based linker, a control experiment with a urea-linker-free MOF is a worthy alternative.27,28 In fact, such a study should be also beneficial as this dual-functionalized framework contains both frustrated Cd(II) centres and a urea-moiety as interaction sites, where a convincing conclusion on an exact active site can be drawn from direct comparison of activity between the MOFs with identical structure and topology, differing in the nature of functionalization. Though reports of well-known systems (MIL or UiO) are available in the literature (Table S4, ESI†), such an approach is still cumbersome with new systems because of the unfavourable binding between additional functionality and transition metal ions.8,61 Fortunately, one of our previous systems, CSMCRI-9 has an identical 2-fold interpenetrated bilayer-pillar framework and (3,8)-connected 2-nodal 3D network (Fig. S2, ESI†).45 Similar to CSMCRI-12, this robust MOF is also composed of a TCA3− ligand, and aqua molecule bound [Cd3(COO)6] SBUs, except for the fact that the pyridyl linker lacks a urea-moiety. A control experiment with higher loading (12 mol%) of this new catalyst (9a) provided only 34% conversion to the product under a prolonged reaction time (Table 1, entry 16). This observation undoubtedly signifies that the noteworthy catalytic efficacy in dual-functionalized 12a arises mainly from the presence of urea groups as two-point hydrogen-bonding donor moieties. Moreover, substrate variation studies also reflected this corroboration (vide infra). Nevertheless, the observed catalytic activity of framework 9a arises due to Lewis acidity around the central metal ions in the SBU, which is not unprecedented and is in compliance with prior reports.23,45,64 These results collectively endorse that engineering of the well-organized, porous 3D architecture of CSMCRI-12 with a urea-unit is highly beneficial as it prevents catalytically detrimental self-association of active sites via a spatially controlled arrangement, and “turns-on” the performance of the hydrogen-bonding motif. To the best of our knowledge, such a systematic study focussing on the exact active site in HBD catalysis using a dual-functionalized MOF has hitherto been unexplored, and promises the opening up of a new avenue to tailor-made engineering of task-specific functionalities for advanced attributes at the interface of structure–property synergies.
Substrate variation and size-selectivity in HBD catalysis
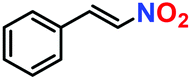
The generosity of C–C bond formation reaction with various substrates was targeted under optimized conditions to understand the functional group tolerance in targeted transformation. The conversion (%) was confirmed by the method described in the Experimental section (vide infra) and observed catalytic data are provided in Table 1. At the outset, the effect on β-nitrostyrene was studied by varying substituents at different positions. A maximum conversion of 100% was witnessed upon the inclusion of a methoxy group (Table 2, entry 2) at the para position, which leaves no residual peak of unreacted (E)-1-methoxy-4-(2-nitrovinyl)benzene (Fig. S24, ESI†). Fortunately, we were able to grow quality single-crystals of 3-(1-(4-methoxyphenyl)-2-nitroethyl)-1H-indole (Table 2, entry 2), and SCXRD analysis furnished the crystal structure of the product (Fig. 8). While substitution of the methyl group (Table 2, entry 3) somewhat reduced the conversion to 56% (Fig. S25, ESI†), chloro substitution provided a satisfactory transformation with a decent yield (Table 2, entries 4 and 5). These variations can be ascribed to diverse electronic effects of substituents on the aromatic ring of β-nitrostyrene, wherein inclusion of an electron-withdrawing group increases the electrophilicity at the attacking centre, while incorporation of a –CH3 group reduces the same through electron donation and causes lower conversion.23,41| Entry | β-Nitrostyrene | Indole | Product | Conversiona | Yieldb | TOFc (h−1) |
|---|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy (refer to ESI†). b Isolated yield after purification of the products. c Number of moles of product per mole of the catalyst per hour. | ||||||
| 1. |
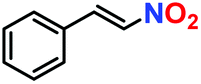
|
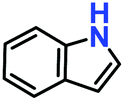
|
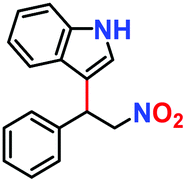
|
100% | 96% | 53 |
| 2. |
|
|
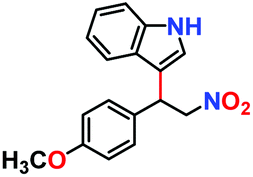
|
100% | 97% | 53 |
| 3. |
|
|
|
56% | 52% | 29 |
| 4. |
|
|
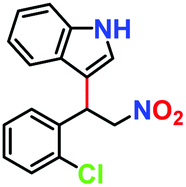
|
87% | 84% | 46 |
| 5. |
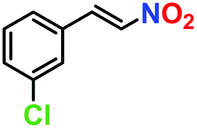
|
|
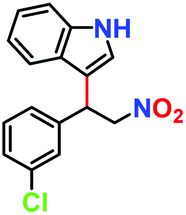
|
81% | 77% | 42 |
| 6. |
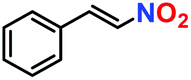
|
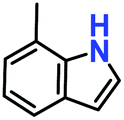
|
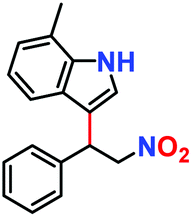
|
100% | 94% | 52 |
| 7. |
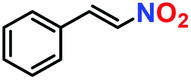
|
|
|
57% | 52% | 29 |
| 8. |
|
|
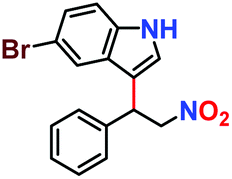
|
65% | 58% | 32 |
| 9. |
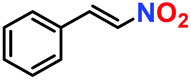
|
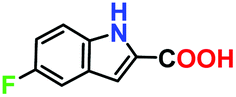
|
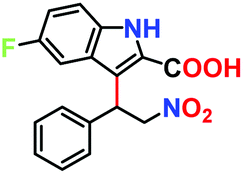
|
No product | — | — |
| 10. |
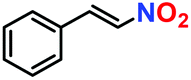
|
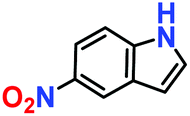
|
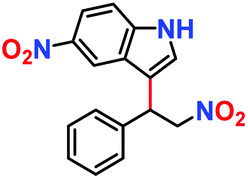
|
10% | — | — |
| 11. |
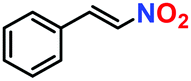
|
|
|
19% | — | — |
| 12. |
|
|
|
5% | — | — |
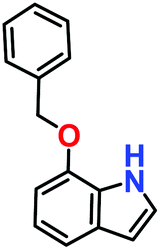
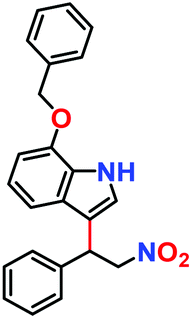
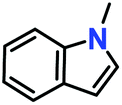
Likewise, substituents on the nucleophile were varied to understand the impact on the C-3 centre of the indole moiety. The results revealed considerable alteration from the catalytic efficacy of 12a. For example, electron-rich 7-methyl-indole (Table 2, entry 6) assists activation of the indole ring toward nucleophilic attack, resulting in 100% conversion to the respective product. In contrast, electron-withdrawing groups containing 6-chloro-indole (Table 2, entry 7) or 5-bromo-indole (Table 2, entry 8) furnished 52% and 58% yields of the products, respectively. A drastic reduction in yield for 5-nitro-indole (Table 2, entry 10) further confirms this point. In particular, N-alkylated indole gave a dramatically decreased yield (Table 2, entry 12) compared to that of free indole, as also observed previously.23 Surprisingly, 7-(benzyloxy)-1H-indole (Table 2, entry 11) produced only 19% conversion based on NMR data (Fig. S33, ESI†). This phenomenon points to the fact that this particular indole derivative may have difficulty in diffusing through the relatively narrow pore aperture in the interpenetrated framework to interact with the H-bonding sites of the MOF.
A pore-size distribution (PSD) plot using NLDFT (inset of Fig. 2c) shows the micropores of 12a are centred around 1.022 nm. Given that the model substrate (Fig. 5b) has molecular dimensions of 2.84 × 4.47 Å2, it can quickly diffuse inside the cavity to form the desired product. However, the reaction of bulkier 7-benzyloxyindole, with molecular dimensions of 11.36 × 4.47 Å2 (Fig. 5c), might occur over the catalyst surface, resulting in lower conversion to the product. To explicitly prove this hypothesis, we did an additional controlled experiment, wherein recovered 12a was used as it was for the model reaction without any proper washing. The 1H NMR (Fig. S49, ESI†) showed only 28% conversion that can be attributed to the non-availability of adequate H-bonding sites for the HBD reaction.67,68 In contrast, thorough washing of the same material followed by the usual activation furnished the expected conversion (99%) in the subsequent cycle (Fig. 7b) as a result of the regeneration of the porous structure, which allows the reaction to take place inside the MOF channel. These experimental outcomes clearly support the optimal pore-aperture in 12a aiding pore-fitting-induced catalysis, and exemplifies the second-ever size-selective Friedel–Crafts alkylation through the HBD reaction in MOFs.23
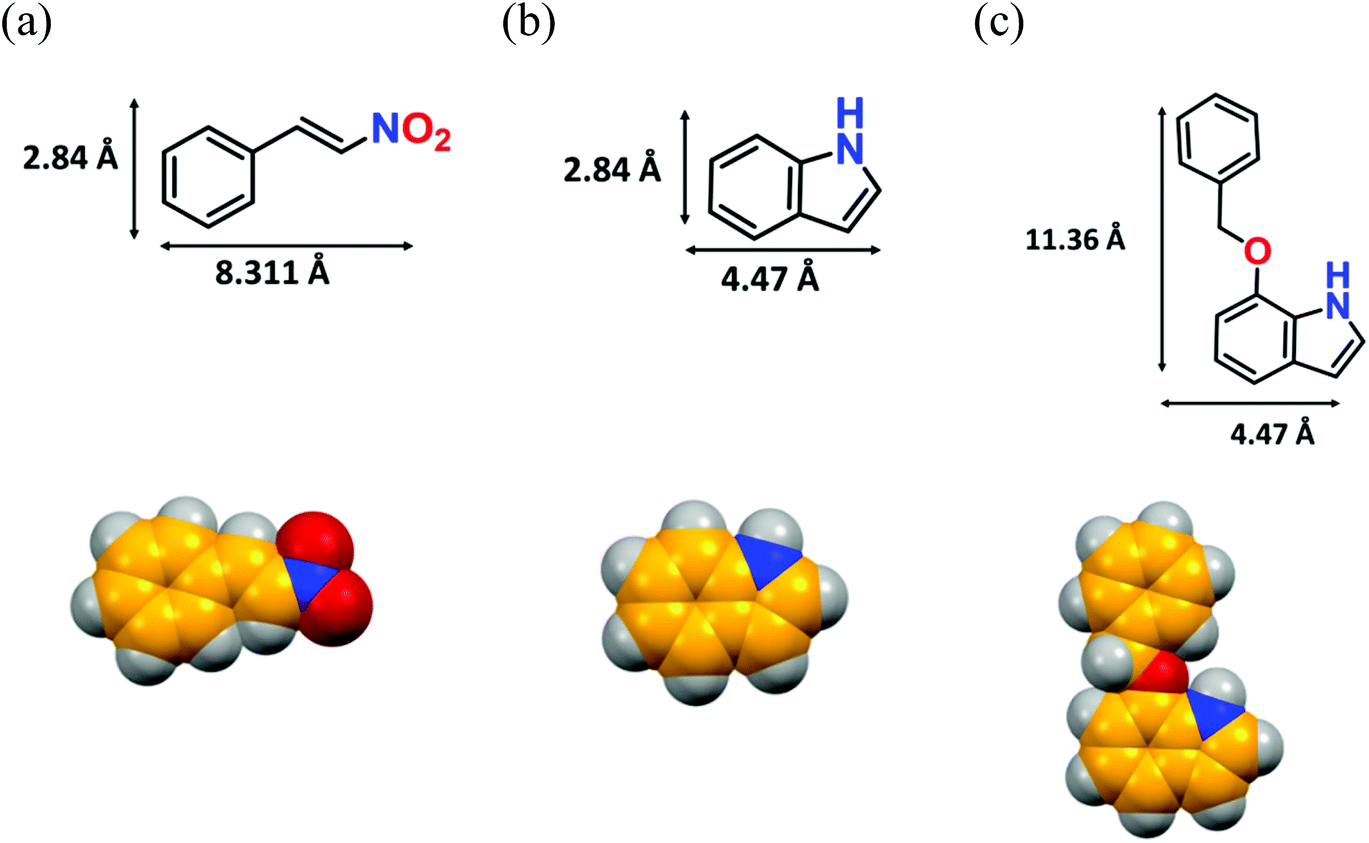 | ||
| Fig. 5 Optimised structures of (a) β-nitrostyrene, (b) indole, and (c) 7-(benzyloxy)-1H-indole with their molecular dimensions (considering atom-to-atom connection). | ||
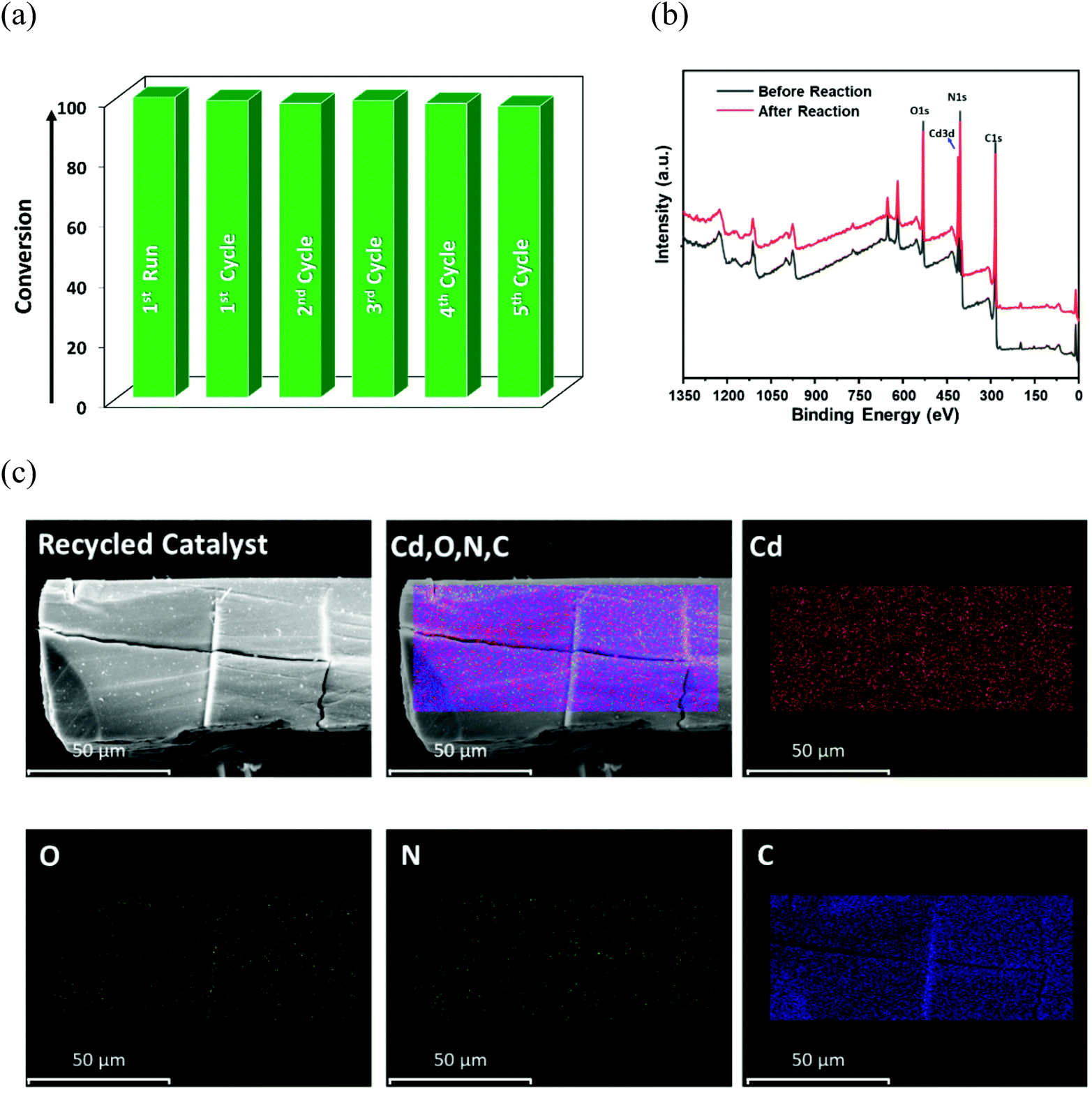 | ||
| Fig. 6 (a) Multicyclic catalytic performance of 12a. (b) Intact XPS spectra before and after catalysis. (c) Elemental mapping (SEM-EDX) showing the uniform distribution of elements (mixed elements Cd, O, N, C) in the selected area of the material after catalysis. | ||
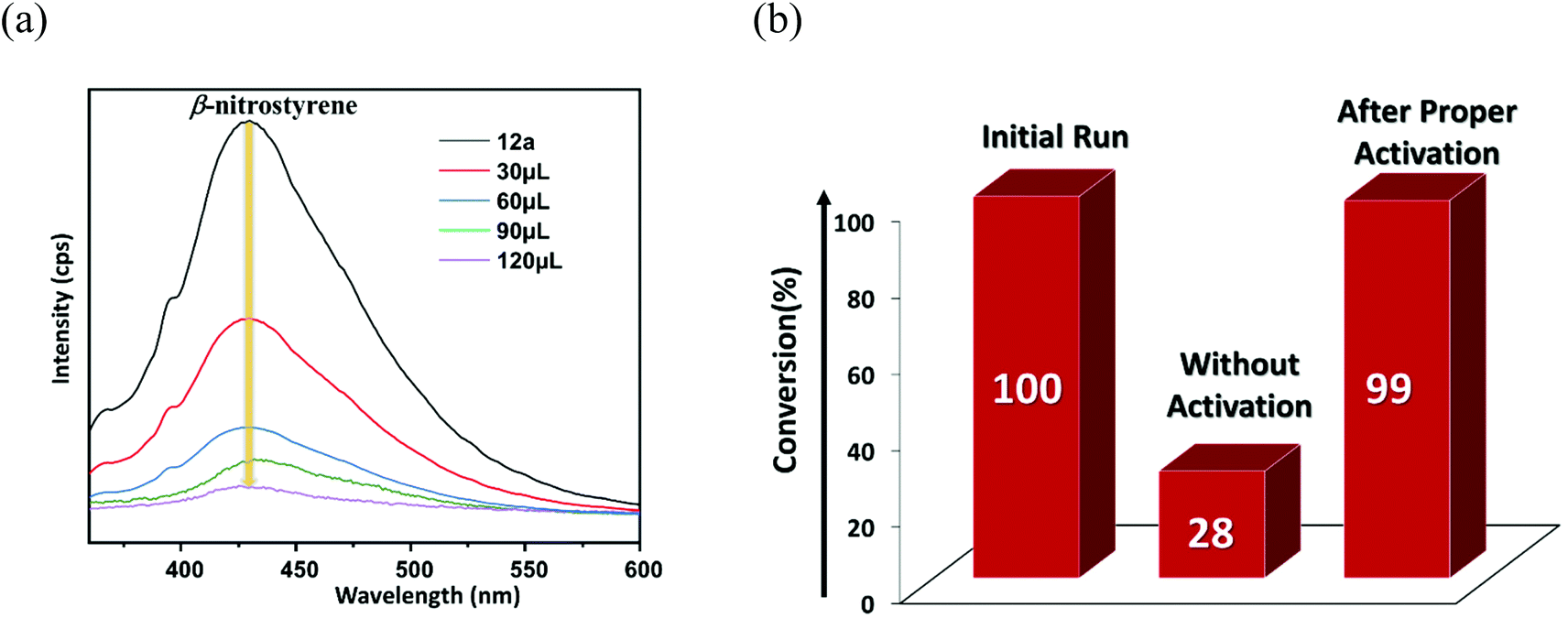 | ||
| Fig. 7 (a) Change in the luminescence intensity of 12a upon gradual addition of β-nitrostyrene. (b) Conversion (%) to the product without and with proper activation of the catalyst. | ||
Mechanistic validation of H-bonding driven Friedel–Crafts alkylation
Building on the knowledge that Friedel–Crafts alkylation in this MOF occurs inside the urea-moiety engineered pores, we subsequently strove to find a tentative mechanism from experimental observations and previous reports in the literature (Table S4, ESI†). Fig. 8 details the route to HBD catalysis, wherein a part of the framework containing the urea functionality is considered for clarity. The cyclic hydrogen-bonded motif between the urea-group of the framework and the β-nitrostyrene molecule (step-1) leads to electron deficiency at the CC position that consecutively enhances the electrophilic character of the carbon atom to facilitate attack from the C-3 position of the indole.26,69,70
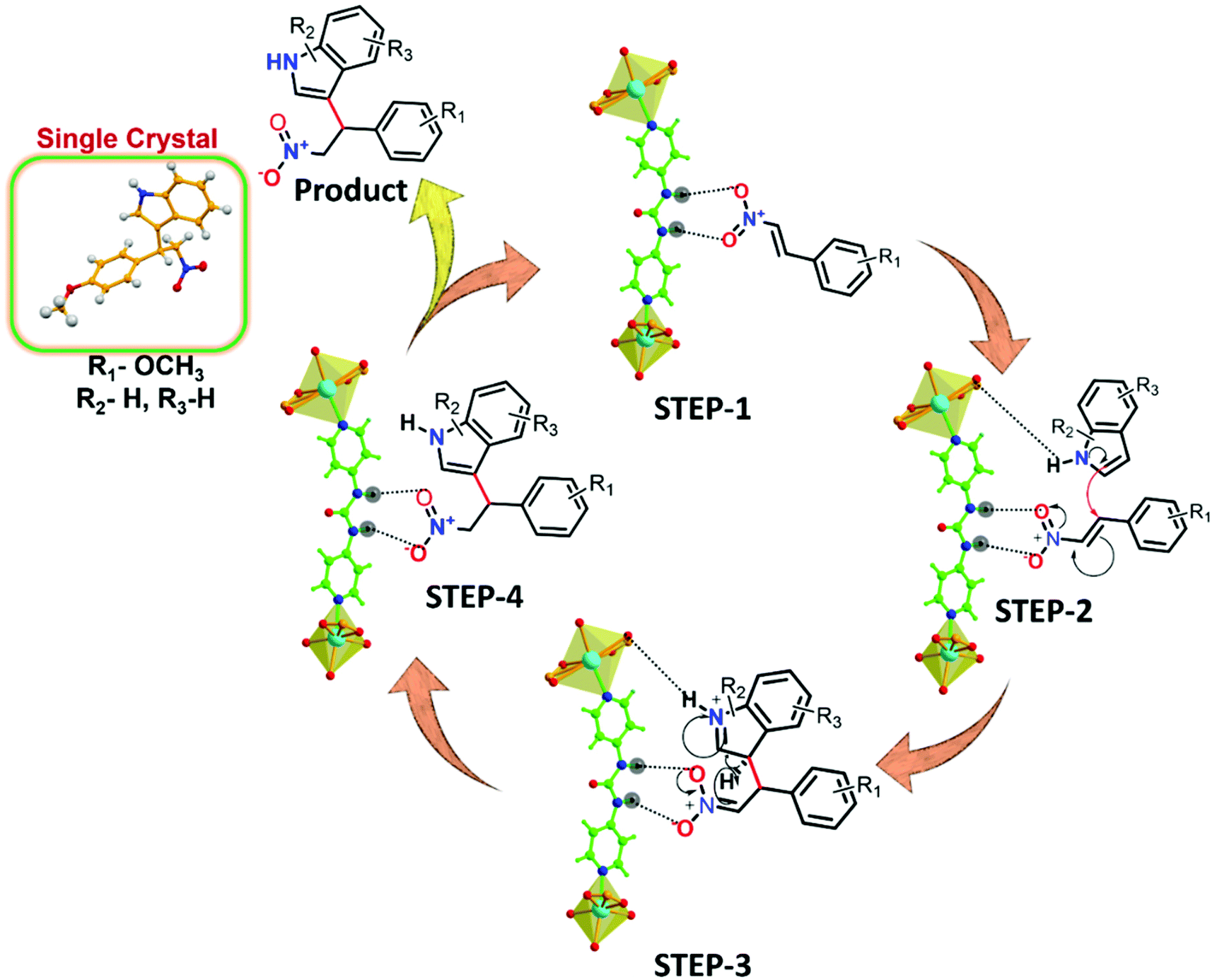 | ||
| Fig. 8 Plausible H-bonding-mediated reaction mechanism for the Friedel–Crafts alkylation of indole with β-nitrostyrene by 12a (a partial view of the framework is presented for clarity). | ||
Fluorescence spectroscopy was used as a measure to support41 the interaction of β-nitrostyrene with functional organic sites in 12a. 2 mg of finely grounded MOF powder was well-dispersed (in 2 mL of toluene) in a cuvette, and freshly prepared β-nitrostyrene solution (1 mM in toluene) was incrementally added under stirring. As displayed in Fig. 7a, a significant decrease in the luminescence intensity of the MOF is evidenced, signifying host–guest interactions.45,46 Nevertheless, a similar addition of indole did not greatly influence the spectral intensity of MOF (Fig. S48, ESI†).41 In a separate experiment, 5-fluoro-1H-indole-2-carboxylic acid (Table 2, entry 9) did not afford any product with the nucleophile under optimised reaction conditions. This phenomenon is ascribed to the strong interaction between the carboxylic acid group in the substrate and the urea-moiety, which leaves no place for the –NO2 group to interact, and alternatively confirms the two-point H-bonding in HBD catalysis. Both these investigations clearly support the interaction between the –NO2 group of β-nitrostyrene and the H-bonding sites in the framework. In step-2, indole attacks the α-carbon atom of β-nitrostyrene through its C-3 carbon centre to provide the expected product through delocalization of electron density (step-3), and finally releases the MOF catalyst for participation in the next cycle (step-4).
Conclusions
In conclusion, we have demonstrated the construction of a rare (3,8)-connected 2-nodal 3D network CSMCRI-12, from an astute combination of a C3-symmetric tricarboxylate ligand, a functionalized N,N-donor linker and a [Cd3(COO)6] cluster containing secondary building units (SBUs). The microporous metal–organic framework (MOF) upholds a number of impressive structural attributes, including (i) the two-fold-interpenetration-mediated formation of suitably sized pores, allowing the entrance of guests with an appropriate molecular dimension, (ii) the provision of activation-induced generation of coordination-frustrated Cd(II) ions along the pore-wall in the SBUs, and (iii) free –NH groups of the urea-moiety in the pillaring strut as active hydrogen-bonding sites for substrate molecules. The guest-free structure (12a) reveals effective Friedel–Crafts alkylation of indole with β-nitrostyrene under relatively mild conditions with broad substrate scope, including a variety of substituted indoles and nitro-alkenes, wherein single-crystal X-ray diffraction analysis is used for the first time as a characterization tool for the product. Remarkably, 12a displays multicyclic usability towards the C–C coupling reaction with low catalyst loading and no leaching, signifying the potential of the catalyst system for long-term usage. For the best Friedel–Crafts alkylation, sterically encumbered substrates with an incompatible molecular dimension to that of two-fold-interpenetration-generated pores reveal poor conversion and corroborate pore-fitting-induced size-selectivity. Since this dual-functionalized MOF contains both unsaturated Cd(II) centres and a urea-moiety as active interaction sites, a series of controlled experiments were employed to confirm the exact mechanistic route to catalysis, including consideration of an isostructural framework without a urea-moiety, change in emission intensity of the framework in the presence of both an electrophile and a nucleophile, and a judicious choice of substrate molecules. The results unequivocally validate hydrogen-bond donating (HBD) catalysis through two-point H-bonding interaction of free –NH groups from a urea-engineered MOF with the nitro group of an electrophile, and simultaneously excludes any major role for the open metal site at the SBU. This report not only exemplifies how functional-group-assisted pore-environment modulation in the MOF can mitigate unfavourable self-aggregation-induced quenching and recyclability issues in homogeneous HBD catalysis through uniform spatial regulation of the responsive H-bonding centres, but also sheds light on the tremendous potential of optimised pores in these crystalline hybrids for size-exclusive reactions.Experimental section
Synthesis of ligands
Synthetic details of the ligand and linker are provided in the ESI.†71
Synthesis of CSMCRI-12
A mixture of Cd(NO3)2·4H2O (30 mg, 0.1 mmol), linker (L) (21.4 mg, 0.1 mmol) and H3TCA (19 mg, 0.05 mmol) was dissolved in a mixed solvent system (N,N-dimethylacetamide:water = 4 mL:2 mL) in a 15 mL glass vial. The mixture was homogenised under ultrasonic treatment for 15 min, followed by heating at 85 °C for 4 days, and then slowly cooled down to room temperature. Shiny, colourless, and rectangular block-shaped X-ray quality crystals were obtained, which were then filtered, thoroughly washed with fresh DMA and air dried (yield: 75%). Anal. Calcd for [Cd3(TCA)2L(H2O)]·2DMA·10H2O: Calculated: C, 43.39; H, 4.42; N, 6.64; Found: C, 44.15, H, 5.39; N, 5.94.
General procedure for HBD catalysis
β-Nitrostyrene (15 mg, 0.1 mmol), indole (14 mg, 0.12 mmol), and 10 mol% catalyst were mixed in 5 mL round-bottom screw cap vials. To these, 500 μL of toluene was added and the reaction mixture was stirred at 60 °C for varying times as per Table 1. After the reaction mixture had cooled to room temperature, the catalyst was centrifuged and separated. The solvent was evaporated under a vacuum to get the final product. The product was analysed by 1H NMR (CDCl3). Selected products were purified using a 25% ethyl acetate/hexane mixture and reported as isolated yield.Author contributions
Manpreet Singh: methodology, formal analysis, data curation, writing-original draft. Subhadip Neogi: conceptualization, funding acquisition, supervision, writing–review and editing.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
M. S. thanks UGC, Delhi, for providing a senior research fellowship. S. N. acknowledges the DST-SERB (grant no. CRG/2021/002529) and the CSIR (grant no. MLP-0028). The analytical support from AESD & CIF (CSMCRI) and crystallographic help from Dr E. Suresh are gratefully acknowledged. CSMCRI communication no. 220/2021.References
-
G. A. Olah, R. Khrisnamurti and G. K. S. Prakash, Comprehensive Organic Synthesis, 1991 Search PubMed
.
- A. Z. Halimehjani, M. V. Farvardin, H. P. Zanussi, M. A. Ranjbari and M. Fattahi, Friedel–Crafts Alkylation of N,N-dialkylanilines with Nitroalkenes Catalyzed by Heteropolyphosphotungstic Acid in Water, J. Mol. Catal. A: Chem., 2014, 381, 21–25 CrossRef CAS
- S. L. You, Q. Cai and M. Zeng, Chiral Brønsted Acid Catalyzed Friedel–Crafts Alkylation Reactions, Chem. Soc. Rev., 2009, 38, 2190–2201 RSC
- Z. H. Shi, H. Sheng, K. F. Yang, J. X. Jiang, G. Q. Lai, Y. Lu and L. W. Xu, Diarylprolinol Silyl Ether Catalyzed Asymmetric Friedel–Crafts Alkylation of Indoles with α,β-unsaturated Aldehydes: Enhanced Enantioselectivity and Mechanistic Investigations, Eur. J. Org. Chem., 2011, 66–70 CrossRef
- R. Kastl and H. Wennemers, Peptide-Catalyzed Stereoselective Conjugate Addition Reactions Generating All-Carbon Quaternary Stereogenic Centers, Angew. Chem., Int. Ed., 2013, 52, 7228–7232 CrossRef CAS PubMed
- O. M. Berner, L. Tedeschi and D. Enders, Asymmetric Michael Additions to Nitroalkenes, Eur. J. Org. Chem., 2002, 1877–1894 CrossRef CAS
- Y.-X. Jia, S.-F. Zhu, Y. Yang and Q.-L. Zhou, Asymmetric Friedel–Crafts alkylations of Indoles with Nitroalkenes Catalyzed by Zn(II)–bisoxazoline Complexes, J. Org. Chem., 2006, 71, 75–80 CrossRef CAS PubMed
- C. M. McGuirk, M. J. Katz, C. L. Stern, A. A. Sarjeant, J. T. Hupp, O. K. Farha and C. A. Mirkin, Turning on Catalysis: Incorporation of a Hydrogen-Bond-Donating Squaramide Moiety into a Zr Metal–Organic Framework, J. Am. Chem. Soc., 2015, 137, 919–925 CrossRef CAS PubMed
- J. J. Lalonde, D. E. Bergbreiter and C. H. Wong, Enzymic Kinetic Resolution of .Alpha.-Nitro .Alpha.-Methyl Carboxylic Acids, J. Org. Chem., 1988, 53, 2323–2327 CrossRef CAS
- M. W. Majchrzak, J. N. Zobel and D. J. Obradovich, A Facile Synthesis of Racemic Nα-FMOC-2-phenyltryptophan, Synth. Commun., 1997, 27, 3201–3211 CrossRef CAS
- R. A. Glennon, M. Lee, J. B. Rangisetty, M. Dukat, B. L. Roth, J. E. Savage, A. McBride, L. Rauser, S. Hufeisen and D. K. H. Lee, 2-Substituted Tryptamines: Agents with Selectivity for 5-HT6 Serotonin Receptors, J. Med. Chem., 2000, 43, 1011–1018 CrossRef CAS PubMed
- C. C. J. Loh, G. Raabe and D. Enders, Enantioselective Synthesis of Tetrahydrocarbazoles through a Michael Addition/Ciamician–Plancher Rearrangement Sequence: Asymmetric Synthesis of a Potent Constrained Analogue of MS-245, Chem. – Eur. J., 2012, 18, 13250–13254 CrossRef CAS PubMed
- W. Cai, M. Hassani, R. Karki, E. D. Walter, K. H. Koelsch, H. Seradj, J. P. Lineswala, H. Mirzaei, J. S. York, F. Olang, M. Sedighi, J. S. Lucas, T. J. Eads, A. S. Rose, S. Charkhzarrin, N. G. Hermann, H. D. Beall and M. Behforouz, Synthesis, Metabolism and In Vitro Cytotoxicity Studies on Novel Lavendamycin Antitumor Agents, Bioorg. Med. Chem., 2010, 18, 1899–1909 CrossRef CAS PubMed
- G. J. Marek and G. K. Aghajanian, Indoleamine and the Phenethylamine Hallucinogens: Mechanisms of Psychotomimetic Action, Drug Alcohol Depend., 1998, 51, 189–198 CrossRef CAS PubMed
- P. R. Schreiner, Metal-Free Organocatalysis through Explicit Hydrogen Bonding Interactions, Chem. Soc. Rev., 2003, 32, 289–296 RSC
- M. S. Taylor and E. N. Jacobsen, Asymmetric Catalysis by Chiral Hydrogen-Bond Donors, Angew. Chem., Int. Ed., 2006, 45, 1520–1543 CrossRef CAS PubMed
- A. G. Doyle and E. N. Jacobsen, Small-Molecule H-bond Donors in Asymmetric Catalysis, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS PubMed
- S. Ghosh, N. Nagarjun, M. Alam, A. Dhakshinamoorthy and S. Biswas, Friedel-Crafts Alkylation Reaction Efficiently Catalyzed by A Di-Amide Functionalized Zr(IV) Metal-Organic Framework, Mol. Catal., 2021, 112007 Search PubMed
- M. C. Etter and S. M. Reutzel, Hydrogen Bond Directed Cocrystallization and Molecular Recognition Properties of Acyclic Imides, J. Am. Chem. Soc., 1991, 113, 2586–2598 CrossRef CAS
- A. G. Wenzel and E. N. Jacobsen, Asymmetric Catalytic Mannich Reactions Catalyzed by Urea Derivatives: Enantioselective Synthesis of β-aryl-β-amino acids, J. Am. Chem. Soc., 2002, 124, 12964–12965 CrossRef CAS PubMed
- P. R. Schreiner and A. Wittkopp, H-bonding Additives Act Like Lewis Acid Catalysts, Org. Lett., 2002, 4, 217–220 CrossRef CAS PubMed
- M. S. Sigman and E. N. Jacobsen, Schiff Base Catalysts for the Asymmetric Strecker Reaction Identified and Optimized from Parallel Synthetic Libraries, J. Am. Chem. Soc., 1998, 120, 4901–4902 CrossRef CAS
- A. Das, N. Anbu, M. Sk, A. Dhakshinamoorthy and S. Biswas, Highly Active Urea-Functionalized Zr(IV)-UiO-67 Metal–Organic Framework as Hydrogen Bonding Heterogeneous Catalyst for Friedel–Crafts Alkylation, Inorg. Chem., 2019, 58, 5163–5172 CrossRef CAS PubMed
- S. V. Deshapande, C. C. Meredith and R. A. Pasternak, Crystallographic Data on Disubstituted Symmetric Ureas, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1968, 24, 1396–1397 CrossRef CAS
- S. Stankovic and G. D. Andreetti, N,N′-Bis(3,4-dichlorophenyl)urea, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, B34, 3787–3790 CrossRef CAS
- J. M. Roberts, B. M. Fini, A. A. Sarjeant, O. K. Farha, J. T. Hupp and K. A. Scheidt, Urea Metal–Organic Frameworks as Effective and Size-Selective Hydrogen-Bond Catalysts, J. Am. Chem. Soc., 2012, 134, 3334–3337 CrossRef CAS PubMed
- C. K. Brozek, J. T. Miller, S. A. Stoian and M. Dincă, NO Disproportionation at a Mononuclear Site-Isolated Fe2+ Center in Fe2+-MOF-5, J. Am. Chem. Soc., 2015, 137, 7495–7501 CrossRef CAS PubMed
- M. J. Ingleson, J. P. Barrio, J. Bacsa, C. Dickinson, H. Park and M. J. Rosseinsky, Generation of a Solid Brønsted Acid Site in a Chiral Framework, Chem. Commun., 2008, 1287–1289 RSC
- B. Parmar, K. K. Bisht, Y. Rachuri and E. Suresh, Zn(II)/Cd(II) Based Mixed Ligand Coordination Polymers as Fluorosensors for Aqueous Phase Detection of Hazardous Pollutants, Inorg. Chem. Front., 2020, 7, 1082–1107 RSC
- P. Patel, B. Parmar, R. S. Pillai, A. Ansari, N.-u. H. Khan and E. Suresh, CO2 Fixation by Cycloaddition of Mono/Disubstituted Epoxides Using Acyl Amide Decorated Co(II) MOF as a Synergistic Heterogeneous Catalyst, Appl. Catal., A, 2020, 590, 117375 CrossRef CAS
- A. Pal, S. Chand, J. C. Boquera, F. Lloret, J.-B. Lin, S. C. Pal and M. C. Das, Three Co(II) Metal–Organic Frameworks with Diverse Architectures for Selective Gas Sorption and Magnetic Studies, Inorg. Chem., 2019, 58, 6246–6256 CrossRef CAS PubMed
- S. C. Pal and M. C. Das, Superprotonic Conductivity of MOFs and Other Crystalline Platforms beyond 10–1 S cm−1, Adv. Funct. Mater., 2021, 31, 2101584 CrossRef CAS
- S. Chand, S. C. Pal, D.-W. Lim, K. Otsubo, A. Pal, H. Kitagawa and M. C. Das, A 2D Mg(II)-MOF with High Density of Coordinated Waters as Sole Intrinsic Proton Sources for Ultrahigh Superprotonic Conduction, ACS Mater. Lett., 2020, 2, 1343–1350 CrossRef CAS
- M. Singh, A. S. Palakkal, R. S. Pillai and S. Neogi, N-Functionality Actuated Improved CO2 Adsorption and Turn-on Detection of Organo-toxins with Guest-induced Fluorescence Modulation in Isostructural Diamondoid MOFs, J. Mater. Chem. C, 2021, 9, 7142–7153 RSC
- M. Singh, G. Kumar and S. Neogi, Devising Mixed-Ligand Based Robust Cd(II)-Framework From Bi-Functional Ligand for Fast Responsive Luminescent Detection of Fe3+ and Cr(VI) Oxo-Anions in Water With High Selectivity and Recyclability, Front. Chem., 2021, 9, 651866 CrossRef CAS PubMed
- R. Goswami, S. Das, N. Seal, B. Pathak and S. Neogi, High-Performance Water Harvester Framework for Triphasic and Synchronous Detection of Assorted Organotoxins with Site-Memory-Reliant Security Encryption via pH-Triggered Fluoroswitching, ACS Appl. Mater. Interfaces, 2021, 13, 34012–34026 CrossRef CAS PubMed
- G. Chakraborty, P. Das and S. K. Mandal, Strategic Construction of Highly Stable Metal–Organic Frameworks Combining both Semi-Rigid Tetrapodal and Rigid Ditopic Linkers: Selective and Ultrafast Sensing of 4-Nitroaniline in Water, ACS Appl. Mater. Interfaces, 2018, 10, 42406–42416 CrossRef CAS PubMed
- G. Chakraborty, P. Das and S. K. Mandal, Polar Sulfone-Functionalized Oxygen-Rich Metal–Organic Frameworks for Highly Selective CO2 capture and Sensitive Detection of Acetylacetone at ppb Level, ACS Appl. Mater. Interfaces, 2020, 12, 11724–11736 CrossRef CAS PubMed
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H.-C. Zhou, Stable Metal–Organic Frameworks: Design, Synthesis, and Applications, Adv. Mater., 2018, 30, 1704303 CrossRef PubMed
- E. Fan, S. A. Van Arman, S. Kincaid and A. D. Hamilton, Molecular Recognition: Hydrogen-Bonding Receptors that Function in Highly Competitive Solvents, J. Am. Chem. Soc., 1993, 115, 369–370 CrossRef CAS
- D. Markad and S. K. Mandal, Design of a Primary-Amide-Functionalized Highly Efficient and Recyclable Hydrogen-Bond-Donating Heterogeneous Catalyst for the Friedel–Crafts Alkylation of Indoles with β-Nitrostyrenes, ACS Catal., 2019, 9, 3165–3173 CrossRef CAS
- C. Zhu, Q. Mao, D. Li, C. Li, Y. Zhou, X. Wu, Y. Luo and Y. Li, A Readily available Urea Based MOF that Act as A Highly Active Heterogeneous Catalyst for Friedel-Crafts Reaction of Indoles and Nitrostryenes, Catal. Commun., 2018, 104, 123–127 CrossRef CAS
- Q. Sun, Z. Dai, X. Meng and F.-S. Xiao, Porous Polymer Catalysts with Hierarchical Structures, Chem. Soc. Rev., 2015, 44, 6018–6034 RSC
- G. Kumar, M. Singh, R. Goswami and S. Neogi, Structural Dynamism-Actuated Reversible CO2 Adsorption Switch and Postmetalation-Induced Visible Light Cα–H Photocyanation with Rare Size Selectivity in N-Functionalized 3D Covalent Organic Framework, ACS Appl. Mater. Interfaces, 2020, 12, 48642–48653 CrossRef CAS PubMed
- N. Seal, M. Singh, S. Das, R. Goswami, B. Pathak and S. Neogi, Dual-Functionalization Actuated Trimodal Attribute in an Ultra-Robust MOF: Exceptionally Selective Capture and Effectual Fixation of CO2 with Fast-Responsive, Nanomolar Detection of Assorted Organo-Contaminants in Water, Mater. Chem. Front., 2021, 5, 979–994 RSC
- N. Seal, A. S. Palakkal, M. Singh, R. Goswami, R. S. Pillai and S. Neogi, Chemically Robust and Bifunctional Co(II)-Framework for Trace Detection of Assorted Organo-Toxins and Highly Cooperative Deacetalization–Knoevenagel Condensation with Pore-Fitting-Induced Size-Selectivity, ACS Appl. Mater. Interfaces, 2021, 13, 28378–28389 CrossRef CAS PubMed
- M. Singh, P. Solanki, P. Patel, A. Mondal and S. Neogi, Highly Active Ultrasmall Ni Nanoparticle Embedded inside A Robust Metal–Organic Framework: Remarkably Improved Adsorption, Selectivity, and Solvent-Free Efficient Fixation of CO2, Inorg. Chem., 2019, 58, 8100–8110 CrossRef PubMed
- N. Seal, K. Karthick, M. Singh, S. Kundu and S. Neogi, Mixed-Ligand-Devised Anionic MOF with Divergent Open Co(II)-Nodes as Chemo-Resistant, Bi-Functional Material for Electrochemical Water Oxidation and Mild-Condition Tandem CO2 Fixation, Chem. Eng. J., 2022, 429, 132301 CrossRef CAS
- N. Seal and S. Neogi, Intrinsic-Unsaturation-Enriched Biporous and Chemorobust Cu(II) Framework for Efficient Catalytic CO2 Fixation and Pore-Fitting Actuated Size-Exclusive Hantzsch Condensation with Mechanistic Validation, ACS Appl. Mater. Interfaces, 2021, 13, 55123–55135 CrossRef CAS PubMed
- M. Singh, S. Senthilkumar, S. Rajput and S. Neogi, Pore-Functionalized and Hydrolytically Robust Cd(II)-Metal–Organic
Framework for Highly Selective, Multicyclic CO2 Adsorption and Fast-Responsive Luminescent Monitoring of Fe(III) And Cr(VI) Ions with Notable Sensitivity and Reusability, Inorg. Chem., 2020, 59, 3012–3025 CrossRef CAS
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Applied Topological Analysis of Crystal Structures with the Program Package TOPOSPRO, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS
- A. Spek, Single-Crystal Structure Validation with the Program Platon, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS
- C.-a. Tao, J. Wang, S. Qin, Y. Lv, Y. Long, H. Zhu and Z. Jiang, Fabrication of pH-Sensitive Graphene Oxide–Drug Supramolecular Hydrogels as Controlled Release Systems, J. Mater. Chem., 2012, 22, 24856–24861 RSC
- S. Bag, K. Roy, C. S. Gopinath and C. R. Raj, Facile Single-Step Synthesis of Nitrogen-Doped Reduced Graphene Oxide-Mn3O4 Hybrid Functional Material for the Electrocatalytic Reduction of Oxygen, ACS Appl. Mater. Interfaces, 2014, 6, 2692–2699 CrossRef CAS PubMed
- H. B. Aiyappa, J. Thote, D. B. Shinde, R. Banerjee and S. Kurungot, Cobalt-Modified Covalent Organic Framework as a Robust Water Oxidation Electrocatalyst, Chem. Mater., 2016, 28, 4375–4379 CrossRef CAS
- K. Nath, K. Bhunia, D. Pradhan and K. Biradha, MOF-Templated Cobalt Nanoparticles Embedded in Nitrogen-Doped Porous Carbon: A Bifunctional Electrocatalyst for Overall Water Splitting, Nanoscale Adv., 2019, 1, 2293–2302 RSC
- H.-L. Cao, F.-Y. Cai, K. Yu, Y.-Q. Zhang, J. Lü and R. Cao, Photocatalytic Degradation of Tetracycline Antibiotics over Cds/Nitrogen-Doped–Carbon Composites Derived from In Situ Carbonization of Metal–Organic Frameworks, ACS Sustainable Chem. Eng., 2019, 7, 10847–10854 CrossRef CAS
- H.-B. Huang, Y. Wang, W.-B. Jiao, F.-Y. Cai, M. Shen, S.-G. Zhou, H.-L. Cao, J. Lü and R. Cao, Lotus-Leaf-Derived Activated-Carbon-Supported Nano-CdS as Energy-Efficient Photocatalysts under Visible Irradiation, ACS Sustainable Chem. Eng., 2018, 6, 7871–7879 CrossRef CAS
- C. Fan, B. Zhu, X. Zhang, C. Bi, D. Zhang, Z. Zong and Y. Fan, Highly Stable Acid-induced Emission-enhancing Cd-MOFs: Synthesis, Characterization, and Detection of Glutamic Acid in Water and Fe ions in Acid, Inorg. Chem., 2021, 60, 6339–6348 CrossRef CAS
- H. Zhang, Y.-H. Liao, W.-C. Yuan and X.-M. Zhang, Organocatalytic Enantioselective Friedel–Crafts Alkylation of Sesamol with Nitro Olefins, Eur. J. Org. Chem., 2010, 3215–3218 CrossRef CAS
- X. W. Dong, T. Liu, Y. Z. Hu, X. Y. Liu and C. M. Che, Urea Postmodified in A Metal–Organic Framework as A Catalytically Active Hydrogen-Bond-Donating Heterogeneous Catalyst, Chem. Commun., 2013, 49, 7681–7683 RSC
- E. A. Hall, L. R. Redfern, M. H. Wang and K. A. Scheidt, Lewis Acid Activation of a Hydrogen Bond Donor Metal–Organic Framework for Catalysis, ACS Catal., 2016, 6, 3248–3252 CrossRef CAS
- X. Zhang, Z. Zhang, J. Boissonnault and S. M. Cohen, Design and Synthesis of Squaramide-based MOFs as Efficient MOF-Supported Hydrogen-Bonding Organocatalysts, Chem. Commun., 2016, 52, 8585–8588 RSC
- P. C. Rao and S. Mandal, Friedel-Crafts Alkylation of Indoles with Nitroalkenes through Hydrogen-Bond-Donating Metal-Organic Framework, ChemCatChem, 2017, 9, 1172–1176 CrossRef CAS
- C. M. McGuirk, C. L. Stern and C. A. Mirkin, Small Molecule Regulation of Self-Association and Catalytic Activity in A Supramolecular Coordination Complex, J. Am. Chem. Soc., 2014, 136, 4689–4696 CrossRef CAS PubMed
- L. Fischer and G. Guichard, Folding and Self-Assembly of Aromatic and Aliphatic Urea Oligomers: Towards Connecting Structure and Function, Org. Biomol. Chem., 2010, 8, 3101–3117 RSC
- J. V. Alegre-Requena, E. Marqués-López, R. P. Herrera and D. D. Díaz, Metal–Organic Frameworks
(MOFs) Bring New Life to Hydrogen-Bonding Organocatalysts In Confined Spaces, CrystEngComm, 2016, 18, 3985–3995 RSC
- S. Lancianesi, A. Palmieri and M. Petrini, Synthetic Approaches to 3-(2-Nitroalkyl) Indoles and their use to Access Tryptamines and Related Bioactive Compounds, Chem. Rev., 2014, 114, 7108–7149 CrossRef CAS PubMed
- G. Dessole, R. P. Herrera and A. Ricci, H-Bonding Organocatalysed Friedel-Crafts Alkylation of Aromatic and Heteroaromatic Systems with Nitroolefins, Synlett, 2004, 2374–2378 CAS
- D. P. Curran and L. H. Kuo, Altering the Stereochemistry of Allylation Reactions of Cyclic.Alpha.-Sulfinyl Radicals with Diarylureas, J. Org. Chem., 1994, 59, 3259–3261 CrossRef CAS
- N. Seal, R. Goswami, M. Singh, R. S. Pillai and S. Neogi, An Ultralight Charged MOF as Fluoro-Switchable Monitor for Assorted Organo-Toxins: Size-Exclusive Dye Scrubbing and Anticounterfeiting Applications via Tb3+ Sensitization, Inorg. Chem. Front., 2021, 8, 296–310 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Materials and physical measurements; experimental details; single-crystal X-ray crystallography; asymmetric unit and topology of CSMCRI-12; TGA curves; FT-IR spectra; nitrogen adsorption–desorption isotherms; PXRD patterns; FE-SEM images; NMR spectra; crystal data; and refinement parameters. CCDC 2125430 and 2125432. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2qi00206j |
| This journal is © the Partner Organisations 2022 |