
Tailored synthesis of molecularly thin platinum nanosheets using designed 2D surfactant solids†
Eisuke
Yamamoto
 *ab,
Akiko
Suzuki
a,
Makoto
Kobayashi
a and
Minoru
Osada
*ac
*ab,
Akiko
Suzuki
a,
Makoto
Kobayashi
a and
Minoru
Osada
*ac
aDepartment of Materials Chemistry & Institute of Materials and Systems for Sustainability (IMaSS), Nagoya University, Nagoya 464-8601, Japan
bPrecursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST), Saitama 332-0012, Japan
cInternational Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), Tsukuba 305-0044, Japan
First published on 29th June 2022
Abstract
The assembly of the surfactants has been utilized as unique templates for the controlled synthesis of metal nanosheets. However, current strategies for metal nanosheets have mainly focused on the liquid-phase surfactant assembly. Herein, we found the solid-state surfactants as designable crystals suitable for nanostructural control and proposed a novel synthetic route for molecularly thin Pt metal nanosheets using solid surfactant crystals as a precursor. The 2D surfactant crystals containing planarly arranged Pt complexes were prepared, and the subsequent UV-ozone treatment and reduction process allowed us to obtain Pt metal nanosheets. Pt metal nanosheets had a distinct morphology with various thicknesses (from 1.5 nm to 3.0 nm), characteristic of 2D surfactant crystals.
Introduction
The controlled synthesis of nanomaterials into the desired architectures is at the heart of nanomaterials research.1–4 Nanostructured metals are one of the essential targets in this regard; nanostructured metals with different shapes and sizes often exhibit unique functionalities unlike their bulk counterparts. The surfactants play a key role in the design of the morphology and structure of nanostructured metals owing to their usefulness in versatile conditions, such as growth controls and increasing dispersibility.5 In particular, the surfactants can be assembled into various superstructures, including micelles,6 liquid crystals,7,8 and self-assembled monolayer,9 offering nanoscale design by templating the assembled structures. Therefore, the surfactant templating methods are fundamental techniques for nanostructured metals despite the demanding control on the superstructure. The further development of the surfactant assembly depends on finding a novel approach that enables the precise design of nanostructures with desired functionalities.10Such an issue is particularly important for designing 2D metal nanosheets.11,12 2D metal nanosheets have attracted great attention because they exhibit a drastic change in their physicochemical properties owing to 2D confinement effects.13 In particular, due to their unique 2D morphology with exceptionally large exposed surface area, 2D nanosheets can significantly enhance the surface reactions, redox processes, and charge separations. The synthetic approach using the surfactant assembly is one of the solutions for generating 2D metal nanosheets.14–17 Typically, the concentrated surfactant micelle solutions form lamellar liquid crystals possessing the 2D nanospace; the use of interlayer nanospace of the lamellar liquid crystals has been well documented as an ideal template for the confined growth of 2D metal nanosheets. However, the control of the thickness and morphology of the metal nanosheets still remains a real challenge. Due to their fragile nature, lamellar liquid crystals usually lack a strong confinement force to regulate the 2D anisotropic growth; metal atoms tend to form 3D structures. In addition, the structure of the lamellar liquid crystals was easily changed by various factors, including temperature, concentration, and precursor amount.18–20 Therefore, the synthesis of the metal nanosheets using the surfactant assembly faces difficulty in the systematic design.
The previous strategies using the surfactant assembly have only focused on the liquid-phase surfactants; nevertheless, it is known that the surfactants form unique solid phases with a crystalline state under the Krafft point.21,22 The crystalline solid phase (Lc phase) surfactants have a lamellar structure with densely arranged counter ions in the interlayer space and the distinct morphology reflecting the crystal habit. Interestingly, the structure and morphology of surfactant crystals have the potential for design depending on the component's structure and composition, unlike liquid crystals.21,23–25 Therefore, templating the solid surfactant crystals with distinct thickness and morphology will allow the precise design of metal nanosheets, offering the potential for a novel technique for the nanoarchitectonics of metal nanosheets. However, to the best of our knowledge, the surfactant assembly with the solid crystalline phase has never been used for the designed synthesis of nanostructured materials.
Herein, we report a new concept for the designed synthesis of molecularly thin metal nanosheets using solid-state surfactant crystals. As typical metal nanosheets, Pt metal nanosheets were synthesized because they are potential candidates for future nanosheets devices and catalysts.26 We synthesized the surfactant crystals with planarly arranged Pt complexes as a precursor and designed the morphology and thickness of the 2D surfactant crystals via the recrystallization process. Then, the 2D surfactant crystals were treated with UV-ozone and reduced by H2(5%)/Ar. These processes lead to the synthesis of molecularly thin Pt metal nanosheets, which is inherent from the thickness and morphology of 2D surfactant crystals (Fig. 1). We have also succeeded in the structural analysis of surfactant crystals and found that there is a clear correlation between the thickness and morphology of 2D surfactant crystals and the resulting metal nanosheets. This method is potentially useful for the precise design of molecularly thin metal nanosheets with distinct morphology and thickness.
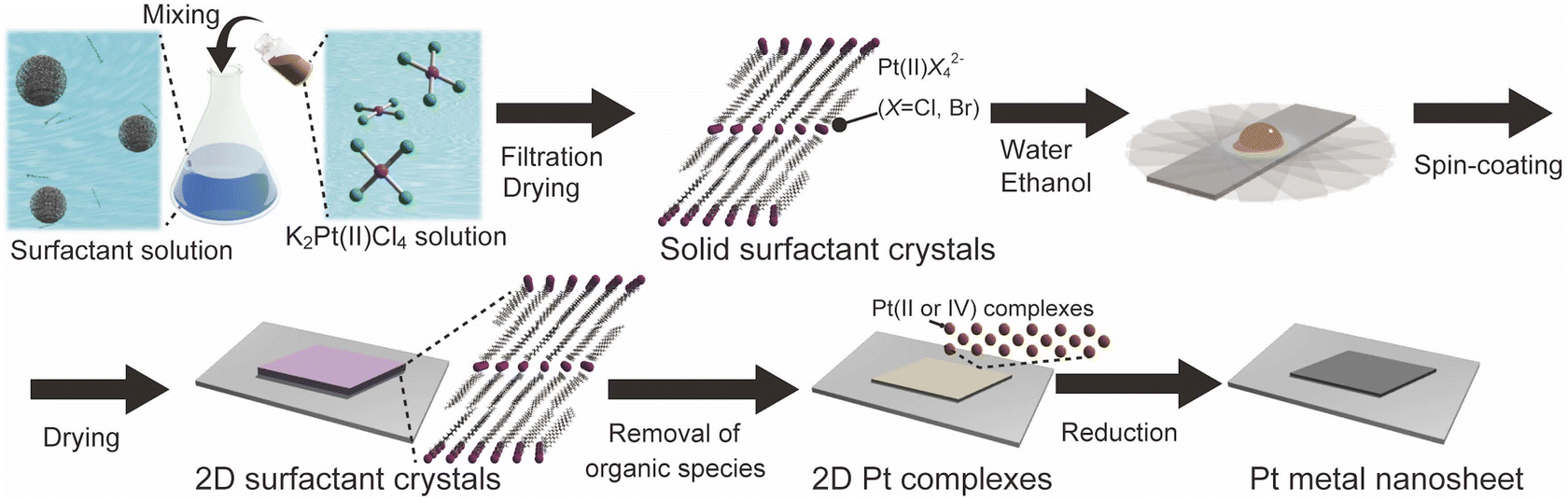 | ||
| Fig. 1 Schematic process of the molecularly thin Pt metal nanosheets using surfactant crystals. | ||
Results and discussion
Preparation of solid surfactant crystals containing Pt complexes
The surfactant crystals were prepared as powders by mixing the aqueous solution of cetyltrimethylammonium bromide (CTAB) and K2Pt(II)Cl4. For the evaluation of the crystal structure, the single crystal X-ray diffraction measurement was conducted. The single crystal of the surfactant with a distinct rhombus morphology was prepared by adding water as a poor solvent to the mixed solvent of N-methylformamide and 2-propanol dissolving the powder surfactant crystal at 298 K (Fig. 2(a)). The structure was determined to have a triclinic system (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) with a composition of [(C16H33)N(CH3)3]2[PtBr0.4Cl3.6] and lattice constants of a = 7.59250(10) Å, b = 8.14060(10) Å, c = 20.0035(2) Å, α = 88.8910(10)°, β = 80.8020(10)°, and γ = 66.2290(10)°. Pt ions are coordinated by Br and Cl ions, which occupy two sites disorderly, and have a square-planar geometry with a C2h symmetry. The alkylammonium cation are aligned orderly in the ab-plane, resulting in the formation of the layered structure, which indicates the formation of the solid-state crystalline phase. In addition, the Pt complexes are arranged planarly as the interlayer counter ions. The distance between two Pt complexes at different interlayer spaces is approximately 2.0 nm (Fig. 2(b)). The condition for crystal structure refinement is shown in the ESI.†
) with a composition of [(C16H33)N(CH3)3]2[PtBr0.4Cl3.6] and lattice constants of a = 7.59250(10) Å, b = 8.14060(10) Å, c = 20.0035(2) Å, α = 88.8910(10)°, β = 80.8020(10)°, and γ = 66.2290(10)°. Pt ions are coordinated by Br and Cl ions, which occupy two sites disorderly, and have a square-planar geometry with a C2h symmetry. The alkylammonium cation are aligned orderly in the ab-plane, resulting in the formation of the layered structure, which indicates the formation of the solid-state crystalline phase. In addition, the Pt complexes are arranged planarly as the interlayer counter ions. The distance between two Pt complexes at different interlayer spaces is approximately 2.0 nm (Fig. 2(b)). The condition for crystal structure refinement is shown in the ESI.†
 | ||
| Fig. 2 (a) Optical image of the single crystalline surfactants with Pt complexes, and (b) the refined crystal structure.27 | ||
Synthesis of Pt metal nanosheets
2D surfactant crystals were prepared on an Si substrate by evaporating the mixed solution of water and ethanol dissolving the surfactant crystals. For the formation of the 2D surfactant crystals, the control of the evaporation process was essential. The simple drying of the liquid droplet is not practical for the 2D growth of the surfactant crystals because it led to the formation of bulk state crystals (Fig. S1(a) of the ESI†). We found that thin 2D surfactant crystals could be obtained under the centrifugal force via spin coating. With the continuous loss of the surfactant solutions via spin coating, the fluid layer becomes thinner and flat; 2D surfactant crystals can form in such a thin fluid layer. Finding the proper conditions, the rotation speed of the spin coating was varied from 1500 rpm to 5000 rpm. At the slow rotation speed of 1500 rpm, the large-sized bulk crystals were mainly obtained (Fig. S1(b) of the ESI†), while a homogeneous non-crystalline film was formed by applying over 5000 rpm (Fig. S1(d) of the ESI†). The optimal condition was found to be 2000–4000 rpm, where the controlled synthesis of the 2D surfactant crystals was achieved (Fig. S1(c), (d), and (e) of the ESI†). Almost all the 2D surfactant crystals showed a rhombus morphology, reflecting the crystal structure. The obtained 2D surfactant crystals have wide thickness distribution under any condition. For example, 2D surfactant crystals with approximately 60 nm mean thickness (in the range from 20 nm to 90 nm) were formed at the 2000 rpm condition. Although they have a wide distribution in the thickness range, the mean thickness of the 2D nanosheets tends to decrease slightly on increasing the rotation speed of the spin coating.As a typical example, 2D surfactant crystals were prepared via 2000 rpm coating, and the thickness of the sheet was monitored at each step. As seen from the AFM image, the thickness of the 2D surfactant crystal was evaluated to be 49 nm, which corresponds to 24 layers of the surfactant bilayer considering the crystal structure (Fig. 3(a) and (d)). XRD and XPS measurements and Raman spectroscopy analyses were conducted to confirm the composition and crystal structure. Fig. S2(a) of the ESI† shows the XRD pattern of the 2D surfactant crystals. The XRD pattern showed clear peaks at 4.5, 9.0, and 13.5°, which is assigned to 001, 002, and 003 reflections of the surfactant crystals, respectively. The Raman spectrum showed the retention of the surfactant, as shown by broad peaks derived from the stretching of C–H groups in the alkyl chain (CH2 and CH3) at 2900 cm−1 (Fig. S2(c) of the ESI†). In addition, the XPS spectrum of the 2D surfactant crystals showed clear peaks at 75.2 eV and 72.0 eV derived from Pt2+4f5/2 and Pt2+4f7/2, respectively (Fig. 1(g)). Although the peaks derived from Br 3d (68 eV) are unclear, a peak derived from Cl 2p3/2 was clearly observed at 198 eV, indicating the retention as Pt halides (Fig. S2(e) of the ESI†). These results indicate the formation of 2D surfactant crystals with the same structure as that of the single crystal bulk.
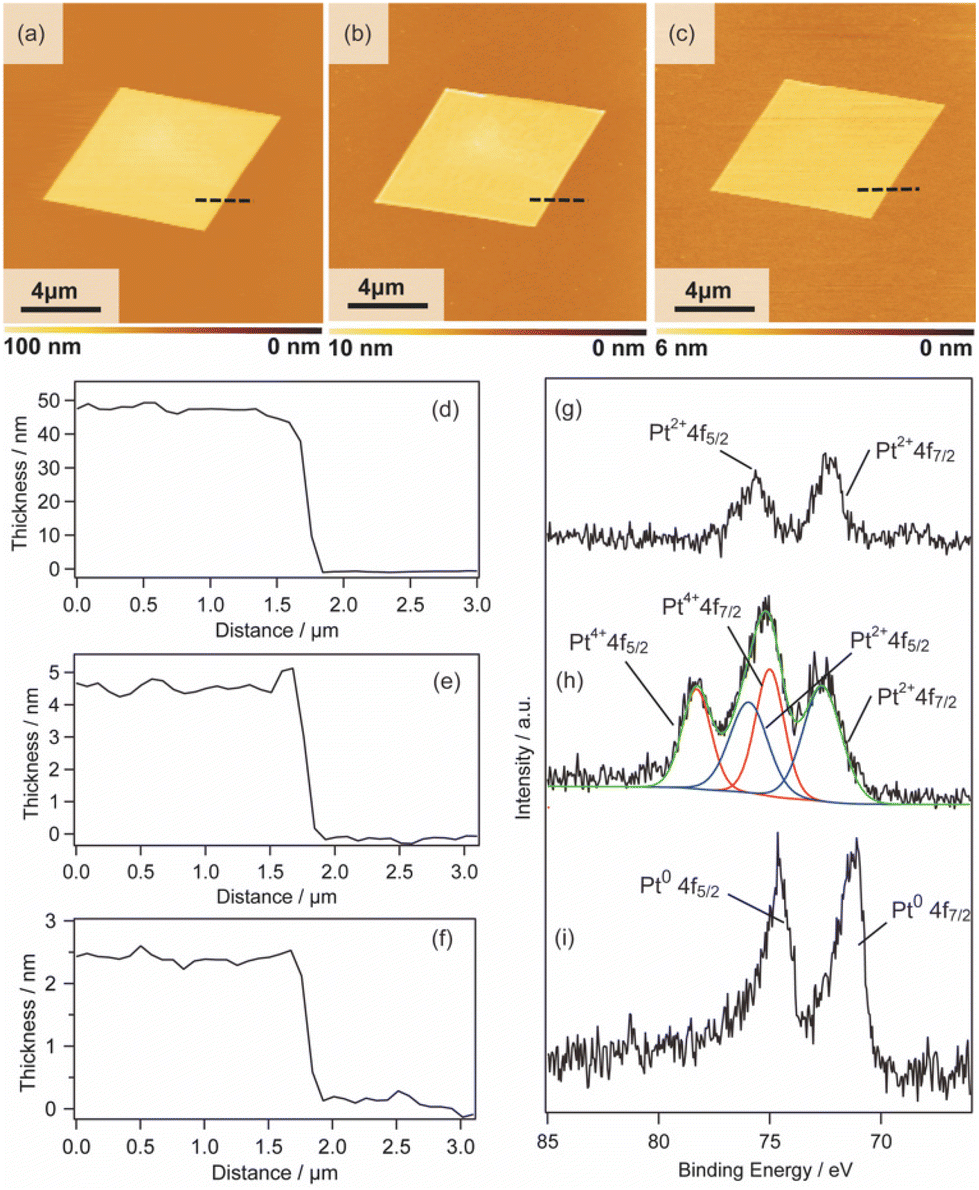 | ||
| Fig. 3 AFM images, height profiles, and XPS spectra of the 2D sheets: (a), (d), and (g) 2D surfactant crystals, (b), (e), and (h) 2D Pt complexes, and (c), (f), and (i) Pt metal nanosheets. | ||
Pt metal nanosheets were synthesized via UV-ozone treatment of 2D surfactant crystals and a subsequent reduction process under H2 (5%)/Ar flow. We characterized the samples at each step for clarifying the effects of UV-ozone treatment and reduction process under H2 (5%)/Ar flow. Firstly, the 2D surfactant crystals were treated with UV-ozone to selectively remove the organic species. The other processes, such as calcination at mild conditions, lead to the collapse of the 2D structure (Fig. S3 of the ESI†). A key for maintaining the 2D morphology is the intermediate UV-ozone treatment; the direct reduction of the 2D surfactant crystals leads to the formation of discrete large-sized Pt metal nanoparticles (Fig. S4 of the ESI†). The morphological change by UV-ozone treatment on the 2D Pt complexes was observed by AFM (Fig. 3(b)). The thickness of the 2D Pt complexes was drastically decreased from 49 nm to 4.7 nm with the retention of the surface roughness and lateral size. In addition, the composition and structure of the 2D Pt complexes were evaluated by Raman spectroscopy analysis and XRD measurement. No clear peak attributed to the stretching of C–H groups (2900 cm−1) was observed in the Raman spectrum (Fig. S2(d) in the ESI†), indicating the removal of CTA+. The removal of the surfactant corresponds to the fact that the XRD pattern did not show the peaks derived from the ordered structure (Fig. S2(b) in the ESI†). Notably, the Pt species were partially oxidized via UV-ozone treatment, as detected by the XPS measurement. The peaks derived from Pt2+4f5/2, Pt2+4f7/2, Pt4+4f7/2, and Pt4+4f5/2 were observed, and the ratio of Pt2+/Pt4+ was roughly calculated to be 1.2 (Fig. 3(h)). The mixture of Pt oxide and halide should be formed after UV-ozone treatment considering the broad peaks of Cl 2p3/2. From these results, it was confirmed that the 2D nanosheets composed of the Pt complex without organic species were formed after UV-ozone treatment.
The molecularly thin Pt metal nanosheets were synthesized by reducing the 2D Pt complexes under H2 (5%)/Ar flow. For optimizing the conditions, the reductions were carried out at 120, 200, and 300 °C. The reduction condition of the Pt complex is an important factor, and discrete nanoparticles assemblies were formed at 200 and 300 °C (Fig. S5(a) and (b) of the ESI†). We succeeded in maintaining a nanosheet structure through reduction at 120 °C (Fig. 3(c)). Note that the complete reduction of the Pt species required a long-time treatment because Pt2+ species partially remained when the nanosheets were reduced for 5 h (Fig. S5(c) and (d) of the ESI†). After reduction treatment for 20 h, the asymmetrical peaks attributed to Pt04f5/2 and Pt04f7/2 were mainly obtained, indicating the formation of Pt metal nanosheets (Fig. 3(i)). The AFM image of the sample after hydrogen reduction at 120 °C for 20 h showed a significant decrease in the thickness of the nanosheet from 4.7 to 2.3 nm (Fig. 3(e)). The surface roughness of the nanosheets is almost the same as that of the substrate. In addition, the high-resolution AFM image showed that the surface of the nanosheet was very smooth, although nanoparticles were observed (Fig. S6(a) and (b) of the ESI†). The theoretical thickness calculated by the amount of Pt species in the 2D surfactant crystals with 49 nm thickness should be approximately 0.7 nm considering 21.45 g cm−3 as the density. It is known that the AFM observation of 2D materials often overestimates the thickness due to adsorbed molecules on the nanosheet surface and water adlayers on the substrate. For example, the thickness of the monolayer graphene was evaluated to be 0.7 nm in spite of 0.4 nm being the real thickness because of the water adlayers.28 However, the measured 2.3 nm thickness is too different from the theoretical value (0.7 nm) even if there were adsorbates. Therefore, there should be some voids in the nanosheet structure, which may be due to the interparticle space of polycrystalline continuous Pt metal nanosheets. Although the obtained nanosheets should be composed of polycrystalline Pt metal nanoparticles, such polycrystalline nanosheets also tend to exhibit unique properties unlike nanoparticles and bulks.29 The obtained molecularly thin Pt metal nanosheets with large lateral size and distinct rhombus morphology should be useful for application and the fundamental investigation of their properties.
The synthesis of Pt metal nanosheets with varied thicknesses and morphologies
We also investigated the versatility of this method using different substrates, such as MgO and Al2O3. The 2D surfactant crystals were formed successfully regardless of the kinds of substrates, and metal nanosheets should be formed via reduction after UV-ozone treatment. All the obtained nanosheets were appropriately 2 nm in thickness, as shown by the AFM images (Fig. S7(a), (b), (c), and (d) in the ESI†). Therefore, this method has potential for the synthesis of Pt metal nanosheets on various substrates as long as the substrate is stable against UV-ozone treatment and hydrogen reduction at 120 °C.In addition, the morphologies of the Pt metal nanosheets synthesized using different precursors were investigated. As typical examples, Pt metal nanosheets were also synthesized using H2Pt(IV)Cl6 as a metal complex precursor (Fig. 4(a) and (b)) or CTAC as a surfactant precursor (Fig. S7(e) and (f) in the ESI†). When the metal nanosheets were synthesized using CTAC, the morphology of the nanosheets was not changed from the rhombus type (Fig. S7(g) and (h) in the ESI†). In contrast, interestingly, the use of H2Pt(IV)Cl6 leads to the morphological change of the nanosheet into a hexagon, probably reflecting the crystal habit of the surfactant crystals (Fig. 4). Although the crystal structure of the surfactant is under investigation, there should be a possibility of the control of the morphology through the choice of the precursors. Considering that the previous molecularly thin Pt metal nanosheets have undefined morphology, this method may open a platform for designing the morphology of molecularly thin Pt metal nanosheets.
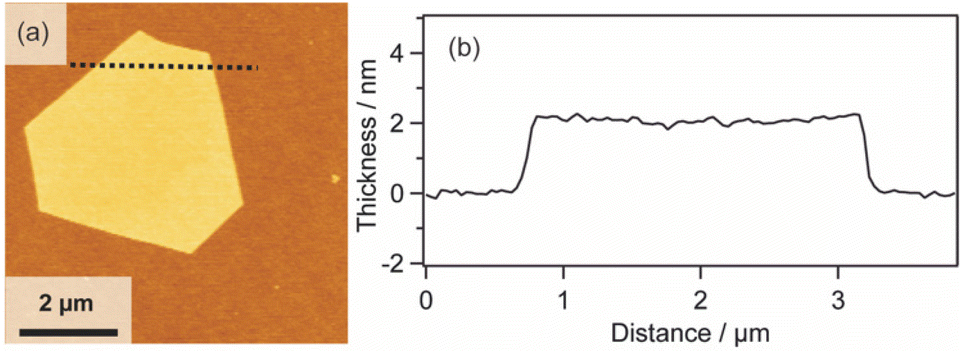 | ||
| Fig. 4 (a) AFM image and (b) height profile of the Pt metal nanosheet synthesized using H2Pt(IV)Cl6 as a metal complex precursor. | ||
Furthermore, it is known that the thickness of metal nanosheets has a significant impact on their structures and properties, and the sub-nm level thickness control is an important issue.13 Although many methods have been proposed to synthesize Pt metal nanosheets,15,29–39 the thickness and morphology control still remain challenging issues. In order to control the thickness of the molecularly thin nanosheets, we investigated the relationship of thickness between the 2D surfactant crystals and Pt metal nanosheets. The thickness of the nanosheets was monitored at each step using 4 samples with different thicknesses (Fig. S8 in the ESI†). Table 1 shows the thickness variation for each of the four nanosheets. In all cases, the thicknesses were decreased to 10% when the 2D surfactant crystals were changed into 2D Pt complexes. Subsequently, the thickness was decreased to 7–8% when the 2D complexes were reduced to Pt metal nanosheets. Considering the relationship, the thickness of the Pt metal nanosheets could be varied quite finely via the thickness control of the 2D surfactant crystals. This method should have the potential for sub-nm thickness control of Pt metal nanosheets. In addition, this method may also have the potential for increasing the lateral size of nanosheets via the optimization of the 2D surfactant synthesis condition. Pt metal nanosheets with a large lateral size (15 μm) were also synthesized using a coincidently formed large 2D surfactant as a precursor (Fig. S9 in the ESI†). Although the synthetic condition for the 2D surfactant with a large lateral size is not optimized, such large-sized nanosheets have been preferred to investigate the fundamental property of the metal nanosheets.
| No. | 2D surfactant crystal | 2D Pt complex | Pt metal nanosheet |
|---|---|---|---|
| 1 | 48 nm | 4.5 nm | 3.0 nm |
| 2 | 30 nm | 3.0 nm | 2.3 nm |
| 3 | 29 nm | 2.8 nm | 2.2 nm |
| 4 | 20 nm | 2.0 nm | 1.5 nm |
Stability of the Pt metal nanosheets
We investigated their stability against different solvents, which is essential for their practical use. The obtained nanosheets were immersed for 1 day in water or ethanol and their structural changes were investigated. However, the obtained Pt metal nanosheets were unstable in both the solvents. For example, when the nanosheet with a thickness of 2.0 nm was immersed in water for 1 day, the thickness increased to 3 nm (Fig. 5(a) and (c)). The increase in the thickness was caused via the morphological change of the nanosheet component, which means the formation of large-sized nanoparticles, as shown by the high-resolution AFM image (Fig. 5(b)). The surface roughness (Ra) of the nanosheets was increased from 140 pm to 490 pm, also supporting the formation of the nanoparticles.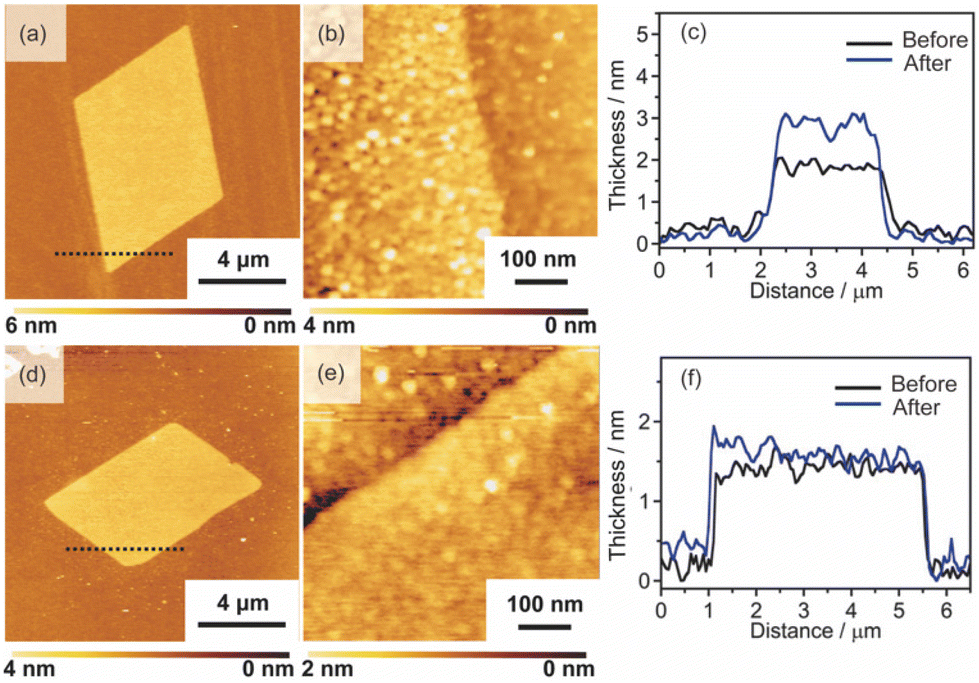 | ||
| Fig. 5 AFM images and height profiles of the Pt metal nanosheets after immersing those in water for 1 day: (a), (b), and (c) the Pt metal nanosheet before calcination, and (d), (e), and (f) the Pt metal nanosheet after calcination. | ||
We found that the calcination of Pt metal nanosheets at 800 °C suppresses the formation of the large-sized nanoparticles. The calcined nanosheets exhibited high stability against solvents, and no morphological change was observed by immersing the nanosheets into water for 1 day (Fig. 5(a), (b), and (c)). Therefore, we evaluated the structure of the Pt metal nanosheets after calcination. In the calcination process, the thickness of the nanosheets slightly changed from 2.3 nm to 2.0 nm, while the morphology was retained (Fig. S10(a) in the ESI†). The decrease in the thickness should be derived from the removal of adsorbed molecules or the shrinkage of the void volume. The surface of the nanosheets was very flat, and no particle was observed (Fig. S11(b) in the ESI†). The chemical states of the nanosheets were not changed, as shown by the XPS spectrum, which showed only peaks derived from metallic Pt species (Fig. S10(c) in the ESI†). The in-plane XRD measurement probably showed very weak peaks, which are assigned to Pt with an fcc structure (Fig. S10(d) in the ESI†). Although a complete understanding of the structure of the obtained nanosheets awaits further studies, this process may be effective for obtaining stable metal nanosheets with precisely controlled thickness.
Conclusions
We have demonstrated a synthetic approach of molecularly thin 2D Pt metal nanosheets using solid surfactant crystals as a precursor. This method requires three steps: (i) Preparation of surfactant crystals containing planarly arranged Pt complex and their 2D anisotropic growth on the target substrate. (ii) Formation of 2D Pt complex through the selective removal of alkylammonium cation via UV-ozone treatment. (iii) The reduction of the 2D Pt complex at mild conditions under H2 (5%)/Ar flow. This method has potential for various substrates, which have stability against UV-ozone treatment and hydrogen gas treatment under 120 °C. In addition, the thickness of the nanosheets can be controlled from 1.5 nm to 3.0 nm, depending on the thickness of the 2D surfactant crystals, offering the potential for a novel method for precisely-controlled metal nanosheets. Although the obtained Pt metal nanosheets were unstable against the solvents, calcination led to a drastic improvement of the stability. This method is potentially useful for precisely designing molecularly thin metal nanosheets with distinct morphology and thickness.Experimental
Materials
All the materials were used without further purification. Hexadecyltrimethylammonium bromide (CTAB), hexadecyltrimethylammonium chrolide (CTAC), ethanol, and 2-propanol (>99.7%) were purchased from Kishida Chemical Co., Ltd. Potassium tetrachloroplatinate(II) (99.9%), hexachloroplatinate(IV) hexahydrate (98.5%), and N-methylformamide (NMF) (>99.0%) were purchased from FUJIFILM Wako Pure Chemical Co., Ltd.Preparation of solid surfactant crystals containing Pt complexes
K2Pt(II)Cl4 (0.5 g) was dissolved in 10 mL water, and 10 mL of water-containing CTAB (0.878 g) was added into the mixture at 60 °C. The precipitates were filtered after standing the solution for 24 h, and the obtained precipitates were washed with water. The collected slurry was dried at room temperature to form surfactant crystals. The surfactant crystals were also prepared using H2Pt(IV)Cl6 or CTAC. The formation of the surfactant crystals prepared with Pt(IV)Cl6 was confirmed by XANES and XRD measurements, Raman spectroscopic analysis, and SEM observation (Fig. S11 in the ESI†).Synthesis of molecularly thin Pt metal nanosheets
The obtained solid surfactant crystals (0.5 mg) were dissolved in a mixture of ethanol (2 mL) and water (1 mL). Then, the obtained dispersion was dropped on different substrates including Si, MgO, and Al2O3. Subsequently, spin-coating was conducted twice at 1500, 2000, 3000, 4000, or 5000 rpm. The 2D surfactant crystals were formed after drying the substrates. Then, the 2D surfactant crystals were treated by UV-Ozone for 4 days to remove the organic species selectively. Finally, the sample was reduced under H2 gas flow condition (H2(5%)/Ar, 50 mL min−1) at 120 °C for 20 h.X-ray crystallography
Single crystals of surfactants were prepared by adding water as a poor solvent to the mixed solvent of NMF and 2-propanol dissolving the surfactant with Pt complex at 298 K. A colorless platelet single crystal with a dimension of 0.10 × 0.07 × 0.02 mm3 was picked up and was affixed to a Mitegen mount with a drop of Paratone-N oil. It was set to a goniometer of a single-crystal X-ray diffractometer (Rigaku, XtaLAB). The X-ray diffraction data of the single crystal were collected using MoKα radiation at 151 K with multi-layer mirrors and a hybrid pixel array detector (Rigaku, HyPix-6000 area detector). Unit cell refinement and absorption collection were performed by the program CrysAlisPro 1.171.40.82a (Rigaku Oxford Diffraction). The crystal structure was solved by direct methods using the SHELXT program,40 and the structural parameters were refined by full-matrix least-squares on F2 using the SHELXL2018 program.41 All calculations were conducted using the WinGX software package.42 The details of crystallographic data, data collection, and structure refinement are summarized in Table S1,† and a moiety of the compound is presented in Fig. S12 in the ESI.† Crystallographic data reported in this manuscript have been deposited with Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 2161128.†Characterization
AFM images were obtained on an MFP3D-origin (Oxford Instruments). Scanning electron microscopy (SEM) images were observed by a JSM-7610FPlus (JEOL) operated at 5 kV. Powder X-ray diffraction (XRD) patterns were observed by a SmartLab (RIGAKU) operating at 45 kV and 200 mA. The crystal structure was illustrated with the VESTA software package.27 Confocal laser microscopy images were obtained by an OLS4000 (Olympus). CHN elemental analyses were conducted by a micro corder JM10 (J-Science Lab). Thermogravimetry (TG) curves were obtained on a STA7200 (Hitachi High-Tech) under air flow at a heating rate of 10 °C min−1 up to 900 °C. Raman spectra were obtained by a LabRAM HR800 (HORIBA Jobin Yvon). XPS spectra were obtained by a ESCALAB 250Xi (Thermo Fisher Scientific). The X-ray absorption find structure (XAFS) data were collected at BL01B1 beamline in SPring-8.Author contributions
The manuscript was written through the contributions of all authors. E. Y. contributed to make the concept of the synthesis and preparation of the manuscript. E. Y. and A. S. experimented with the synthesis of nanosheets. M. K. performed the single crystalline X-ray analysis, evaluation on the composition of surfactant crystals and discussed the data. M. O. discussed the data and revised essential content in the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Grant-in-Aid for Scientific Research for young scientists (19K15656), JSPS, PRESTO (JPMJPR19TA), JST, the Grant-in-Aid for Scientific Research (21H05015, 21H04617, 19K22104), JSPS, and A-STEP, JST, DEJI2MA, MEXT, and the joint usage/research program of IMaSS, Nagoya University, Japan. In addition, we thank Prof. H. Kato (Tohoku University), Dr I. Hayashi (Nagoya University), Dr Y. Taniguchi (Oxford instruments) and Mr D. Kurimoto (Nagoya University) for the experimental assistance.Notes and references
- C. Zhu, D. Du, A. Eychmüller and Y. Lin, Chem. Rev., 2015, 115, 8896–8943 CrossRef CAS PubMed.
- G. V. Hartland, Chem. Rev., 2011, 111, 3858–3887 CrossRef CAS PubMed.
- Q. Shi, C. Zhu, D. Du and Y. Lin, Chem. Soc. Rev., 2019, 48, 3181–3192 RSC.
- M. Zahmakırana and S. Özkar, Nanoscale, 2011, 3, 3462–3481 RSC.
- B. Wu and N. Zheng, Nano Today, 2013, 8, 168–197 CrossRef CAS.
- T. K. Sau and A. L. Rogach, Adv. Mater., 2010, 22, 1781–1804 CrossRef CAS PubMed.
- K. Ariga, A. Vinu, Y. Yamauchi, Q. Ji and J. P. Hill, Bull. Chem. Soc. Jpn., 2012, 85, 1–32 CrossRef CAS.
- Y. Yusuke, T. Azusa, K. Masaki, S. Makoto, O. Tetsu and K. Kazuyuki, Chem. Mater., 2008, 20, 1004–1011 CrossRef.
- N. K. Chaki and K. Vijayamohanan, Biosens. Bioelectron., 2002, 17, 1–12 CrossRef CAS PubMed.
- K. Ariga, X. Hu, S. Mandal and J. P. Hill, Nanoscale, 2010, 2, 198–214 RSC.
- R. Dong, T. Zhang and X. Feng, Chem. Rev., 2018, 118, 6189–6235 CrossRef CAS PubMed.
- F. Nosheen, N. Wasfi, S. Aslam, T. Anwar, S. Hussain, N. Hussain, S. N. Shah, N. Shaheen, A. Ashraf, Y. Zhu, H. Wang, J. Ma, Z. Zhang and W. Hu, Nanoscale, 2020, 12, 4219–4237 RSC.
- L. Wang, Z. Zeng, W. Gao, T. Maxson, D. Raciti, M. Giroux, X. Pan, C. Wang and J. Greeley, Science, 2019, 363, 870–874 CrossRef CAS PubMed.
- J. Niu, D. Wang, H. Qin, X. Xiong, P. Tan, Y. Li, R. Liu, X. Lu, J. Wu, T. Zhang, W. Ni and J. Jin, Nat. Commun., 2014, 5, 3313 CrossRef PubMed.
- H. Kawasaki, M. Uota, T. Yoshimura, D. Fujikawa, G. Sakai, M. Annaka and T. Kijima, Langmuir, 2005, 21, 11468–11473 CrossRef CAS PubMed.
- H. L. Qin, D. Wang, Z. L. Huang, D. M. Wu, Z. C. Zeng, B. Ren, K. Xu and J. Jin, J. Am. Chem. Soc., 2013, 135, 12544–12547 CrossRef CAS PubMed.
- B. Jiang, Y. Guo, J. Kim, A. E. Whitten, K. Wood, K. Kani, A. E. Rowan, J. Henzie and Y. Yamauchi, J. Am. Chem. Soc., 2018, 140, 12434–12441 CrossRef CAS PubMed.
- P. B. Warren and M. Buchanan, Curr. Opin. Colloid Interface Sci., 2001, 6, 287–293 CrossRef CAS.
- M. G. Berni, C. J. Lawrence and D. Machin, Adv. Colloid Interface Sci., 2002, 98, 217–243 CrossRef CAS PubMed.
- A. Khan, Curr. Opin. Colloid Interface Sci., 1996, 1, 614–623 CrossRef CAS.
- R. M. Miller, J. T. Cabral, E. S. J. Robles, N. J. Brooks and O. Ces, CrystEngComm, 2018, 20, 6834–6843 RSC.
- T. Lee, K. Yeh, J. You, Y. Fan, Y. Cheng and D. Pratama, ACS Omega, 2020, 5, 1068–1079 CrossRef CAS PubMed.
- V. M. Coiro and F. Mazza, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1989, 45, 1132–1136 CrossRef.
- V. M. Coiro and F. Mazza, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 1169–1173 CrossRef.
- L. A. Smith, R. B. Hammond, K. J. Roberts, D. Machin and G. McLeod, J. Mol. Struct., 2000, 554, 173–182 CrossRef CAS.
- W. Sugimoto and D. Takimoto, Chem. Lett., 2021, 50, 1304–1312 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- X. Ke, C. Peigen and H. R. James, Science, 2010, 329, 1188–1191 CrossRef PubMed.
- A. Funatsu, H. Tateishi, K. Hatakeyama, Y. Fukunaga, T. Taniguchi, M. Koinuma, H. Matsuura and Y. Matsumoto, Chem. Commun., 2014, 50, 8503–8506 RSC.
- M. Sodeno, S. Kato, H. Nanao and M. Shirai, Catal. Today, 2021, 375, 48–55 CrossRef CAS.
- M. Shirai, Chem. Rec., 2019, 19, 1263–1271 CrossRef CAS PubMed.
- M. Chhetri, M. Rana, B. Loukya, P. K. Patil, R. Datta and U. K. Gautam, Adv. Mater., 2015, 27, 4430–4437 CrossRef CAS PubMed.
- M. Shirai, K. Igeta and M. Arai, Chem. Commun., 2000, 623–624 RSC.
- Z. Fan, Y. Zhu, X. Huang, Y. Han, Q. Wang, Q. Liu, Y. Huang, C. L. Gan and H. Zhang, Angew. Chem., Int. Ed., 2015, 54, 5672–5676 CrossRef CAS PubMed.
- F. Saleem, Z. Zhang, B. Xu, X. Xu, P. He and X. Wang, J. Am. Chem. Soc., 2013, 135, 18304–18307 CrossRef CAS PubMed.
- X. Bao, Y. Gong, X. Zheng, J. Chen, S. Mao and Y. Wang, J. Energy Chem., 2020, 51, 272–279 CrossRef.
- S. Takenaka, S. Miyake, S. Uwai, H. Matsune and M. Kishida, J. Phys. Chem. C, 2015, 119, 12445–12454 CrossRef CAS.
- S. Takenaka, H. Arita, K. Sugiyama and K. Nakatani, Chem. Lett., 2018, 47, 975–978 CrossRef CAS.
- F. Saleem, B. Xu, B. Ni, H. Liu, F. Nosheen, H. Li and X. Wang, Adv. Mater., 2015, 27, 2013–2018 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2161128. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2nr01807a |
| This journal is © The Royal Society of Chemistry 2022 |