
Dual agonist immunostimulatory nanoparticles combine with PD1 blockade for curative neoadjuvant immunotherapy of aggressive cancers‡
Prabhani U.
Atukorale
*abc,
Taylor J.
Moon†
a,
Alexandr R.
Bokatch‡
a,
Christina F.
Lusi
c,
Jackson T.
Routhier
a,
Victoria J.
Deng
a and
Efstathios
Karathanasis
 *ab
*ab
aDepartment of Biomedical Engineering, School of Medicine, Case Western Reserve University, Cleveland, OH 44106, USA. E-mail: stathis@case.edu; patukorale@umass.edu
bCase Comprehensive Cancer Center, Cleveland, OH 44106, USA
cDepartment of Biomedical Engineering, University of Massachusetts, Amherst, MA 01003, USA
First published on 7th January 2022
Abstract
Lethal cancer is characterized by drug-resistant relapse and metastasis. Here, we evaluate the efficacy of a neoadjuvant therapeutic strategy prior to surgery that combines the immune checkpoint inhibitor anti-PD1 with a powerful immunostimulatory nanoparticle (immuno-NP). Lipid-based immuno-NPs are uniquely designed to co-encapsulate a STING and TLR4 agonist that are functionally synergistic. Efficacy of neoadjuvant combination immunotherapy was assessed in three aggressive murine tumor models, including B16F10 melanoma and 4T1 and D2.A1 breast cancer. Primary splenocytes treated with dual-agonist immuno-NPs produced a 75-fold increased production of interferon β compared to single-agonist treatments. Systemic delivery facilitated the widespread deposition of immuno-NPs in the perivascular space throughout the tumor mass and their preferential uptake by tumor-resident antigen-presenting cells. Our findings strongly suggested that immuno-NPs, when administered in combination with anti-PD1, harnessed and activated the otherwise “exhausted” CD8+ T cells as key mediators of tumor clearance. Neoadjuvant combination immunotherapy resulted in significant efficacy, curative responses, and protective immunological memory in 71% of good-responding mice bearing B16F10 melanoma tumors and showed similar trends in the two breast cancer models. Finally, this neoadjuvant combination immunotherapy drove the generation of B and T cell de novo epitopes for a comprehensive memory response.
1. Introduction
Due to significant advances in surgical precision, primary tumor resection is typically the first and most effective treatment option in the majority of cancers.1–3 However, the aggressive forms of cancer display a high risk of metastatic disease and recurrence, which exhibit pro-survival and chemo/radio-resistant phenotypes, making current standard-of-care therapies ineffective. Further, in aggressive solid tumors that are initially unresectable or high-risk resectable, there is significant interest in the development of new neoadjuvant treatments to reduce tumor size and viability prior to surgery.4–6 Due to increased resistance of aggressive cancers to conventional treatments, immunotherapies are increasingly exploited as neoadjuvant pre-surgical treatments.An effective neoadjuvant immune intervention can exploit the primary tumor and its diverse microenvironment and tumor antigens, increasing the likelihood of the entire tumor being turned into a vaccine. From an immunotherapy design perspective, the tumor microenvironment (TME) itself offers a substantial local immune cell infiltrate that, if modulated effectively, can be harnessed for tumor clearance. In advanced tumors, this immune infiltrate can make up approximately 50% of the cells of the tumor mass and includes both phagocytic innate immune cells, such as neutrophils, macrophages, dendritic cells (DCs), and natural killer (NK) cells, and adaptive immune cells, such as T and B cells.1,7–9 Notably, while early tumors are inflamed and “hot” with significant numbers of proinflammatory anti-tumor immune cells such as DCs, M1 macrophages, and CD8+ T cells, aggressive tumors advance to become noninflamed, immune-excluded, and “cold” by upregulating the immunosuppressive function of tumor-promoting cell subsets such as neutrophils, M2 macrophages, and regulatory T cells (Tregs) to render proinflammatory cells dysfunctional and “exhausted”.10,11 Tumor immunity, however, can be adeptly modulated if accessed effectively. Neoadjuvant immunotherapies exploit this very malleability of tumor immune cells for therapeutic purposes. Despite some modest efficacy, neoadjuvant therapies using the immune checkpoint inhibitor anti-PD1 have been limited in their narrow, singular focus solely on CD8+ T cells, which relies on these “exhausted” cells in “cold” tumors to mediate tumor clearance entirely by themselves. Given that the majority of tumor-resident CD8+ T cells are dysfunctional and “exhausted” due to an overwhelming immunosuppressive “cold” TME milieu, “cold”-to-“hot” immunostimulatory strategies to harness and activate the proinflammatory innate immune cells of the tumor to reduce TME immunosuppression have garnered significant attention as a necessary component of CD8+ T cell therapies.11–17
Towards this goal of developing effective “cold”-to-“hot” strategies, a nascent but fast-growing class of immunotherapies involving the direct intratumoral administration of immunostimulatory innate agonists as neoadjuvant treatments prior to surgery has gained notable traction in recent years.12,18–20 Such neoadjuvant immunostimulatory therapies seek to reprogram and activate the antigen-presenting cells (APCs) in the TME by delivering agonists of powerful pattern recognition receptor (PRR) pathways, chief classes of which are the families of the stimulator of interferon genes pathway (STING)21–26 and Toll-like receptor (TLR) pathway.27–29
Here we report on the development of a novel immunostimulatory nanoparticle-based neoadjuvant therapeutic strategy to achieve both curative results and protective immunological memory (Fig. 1). Notably, we combine these immuno-nanoparticles (immuno-NPs) with anti-PD1 to powerfully augment the pivotal tumor-killing function of local CD8+ T cells. The neoadjuvant immuno-NP treatment integrates three key features. First, we designed a biocompatible lipid-based nanoparticle system that co-encapsulates two synergistic immune agonists, the STING pathway agonist cyclic di-guanylate monophosphate (cdGMP)30,31 and the TLR4 pathway agonist monophosphoryl lipid A (MPLA)32–36 on the same particle.37,38 In recent work, we have shown that and co-delivery of the two agonists to the same target APC is an essential driver of synergistic IFNβ production.23,37,39 Second, this dual-agonist immuno-NP system can be systemically delivered to achieve widespread access to the tumor perivascular region that is rich in target APCs (Fig. 1, step 1). Systemic administration of immuno-NPs resulted in preferential and significant uptake by tumor APCs and promoted significant IFNβ production by these cells in multiple aggressive murine tumor models including melanoma, triple-negative breast cancer, and pancreatic cancer. Systemic administration offers distinct benefits over the standard intratumoral administration methods, which not only rely on accurate prior knowledge of tumor locations that is often clinically infeasible, but also have limitations in achieving the necessary widespread delivery of agonists throughout the tumor mass.40 Compared to delivery of free agonists, the co-encapsulation of the two synergistic cargoes in immuno-NP enables high potency at the tumor target site at a reduced dose resulting in a high safety profile where any toxicity is only minimal and transient. Third, we combined the immuno-NP treatment with tumor PD1 blockade in efforts to augment and facilitate a tumor-clearing CD8+ T cell response (Fig. 1, step 2). Activation of local tumor-resident CD8+ T cells that are otherwise “exhausted” allows us to harness their tumor-killing potential for a significant therapeutic advantage.41–43 Immuno-NP uptake by and activation of target APCs that continue to sample antigens from dying tumor cells promotes their production of a powerful local Type I IFN cytokine gradient that serves to activate other innate and adaptive immune cells. Notably, these APCs can prime and activate local tumor-specific CD8+ T cells that are otherwise “exhausted”, leading to T cell-mediated tumor killing and clearance (Fig. 1, step 3). The neoadjuvant combination immunotherapy can generate potent antitumor immunity leading to a substantial reduction of the tumor size within just a few days, which can increase the likelihood of success of the subsequent surgical resection. Finally, immunological memory is generated to protect against tumor recurrence (Fig. 1, step 4). We further demonstrate the major role local tumor-resident CD8+ T cells play in tumor clearance. We establish that immuno-NPs drive de novo CD8+ T and B cell epitope recognition,44–47 enabling more comprehensive defense and protection against tumor recurrence.
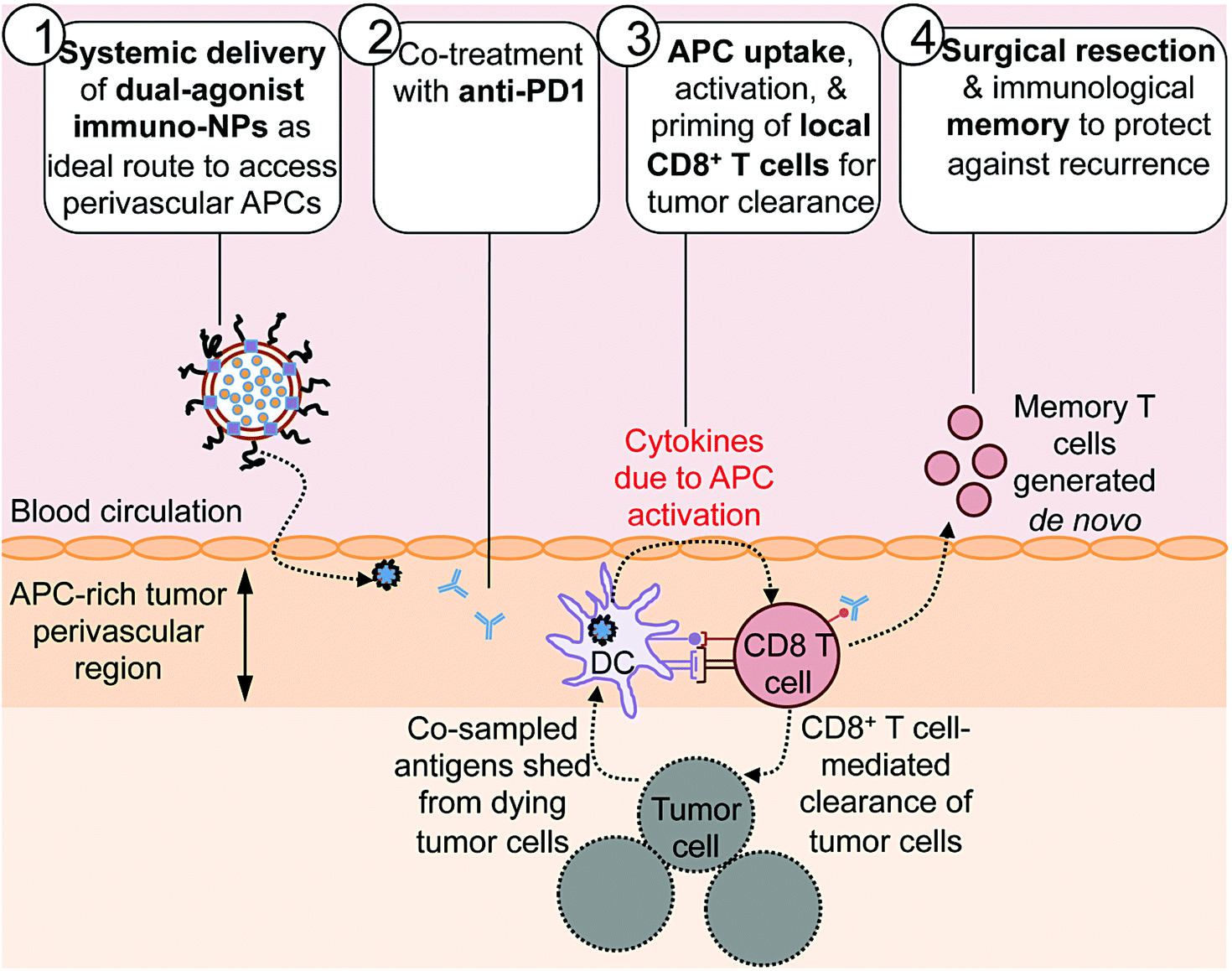 | ||
| Fig. 1 Combining systemic dual agonist immunostimulatory NPs with anti-PD1 as a powerful neoadjuvant treatment prior to surgical resection of aggressive tumors. Our treatment relied on systemic delivery of dual-agonist immuno-NPs as an ideal route to access perivascular APCs (step 1) and combination with PD1 blockade as a co-treatment (step 2). Immuno-NP uptake by local APCs and their simultaneous sampling of antigens shed by tumor cells drives the priming and activation of local CD8+ T cells that become key drivers of tumor clearance (step 3). Surgical resection of the small residual tumor mass ensures complete elimination, while immunological memory generated from the immunotherapy protects against cancer recurrence (step 4). | ||
2. Experimental section
2.1. Experimental design
The purpose of this study was to develop an immunostimulatory nanoparticle-based combination therapy with PD1 checkpoint blockade as a neoadjuvant immunotherapy for aggressive cancers. Institutional, local, state, and federal guidelines under a Case Western Reserve University School of Medicine IACUC-approved protocol were followed for all in vivo animal studies using syngeneic murine tumor models. In vitro cell culture analysis was performed using both established cell lines and primary cells. Results from rigorous prior studies were used as a guideline to establishing the optimal number of animals and samples used in each study. Sample size was assessed to ensure adequate power and selected based on historical data. All mice were females to eliminate any confounding gender differences and also because this gender was most relevant for the breast cancer models used in this study. Treatment groups were formed by random assigning of mice, and all analyses were blinded. No animals or samples were excluded from analysis, neither were any outliers. The specific number of independent biological replicates, sample size, and details of statistical analyses are included in the figure legends.2.2. Immuno-nanoparticle synthesis & characterization
Dual-agonist immunostimulatory nanoparticles (immuno-NPs) were synthesized by ultrasonication. Equimolar amounts of DOPC (48.5 mol% 1,2-dioleoyl-sn-glycero-3-phosphocholine, Avanti) and DPPC (48.5 mol% 1,2-dipalmitoyl-sn-glycero-3-phosphocholine, Avanti) were prepared in chloroform, along with 3 mol% mPEG2000-DSPE [methoxy-poly(ethyleneglycol)-2000 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N, Laysan Bio]. 100 μg of MPLA (Sigma-Aldrich) was added per 42 μmol of lipid. Lipid films were formed by evaporation of chloroform. These films were hydrated in phosphate-buffered saline (PBS) containing 200 μg of cdGMP (Invivogen), heated to 56 °C for 40 min, with vortexing every 5 min for 30 s intervals. Samples were then ultrasonicated on ice using 30 s cycles, with pulsing at 20% amplitude for 20 s followed by a 10 s pause for a total of 5 min. Immuno-NPs were dialyzed for 2–4 h against sterile PBS and stored immediately at 4 °C. Dynamic light scattering (DLS) and zeta potential measurements were used to measure immuno-NP hydrodynamic size and surface charge, respectively (Beckman Coulter). High permeation liquid chromatography (HPLC, Shimadzu) was used to measure encapsulated cdGMP. Stability studies were performed in independent triplicate at both 22 °C and 37 °C over short (1–4 h) and long (24 h) time points. 0.2 mol% of DiR (Thermo Fisher Scientific) was incorporated into lipid films during synthesis for fluorescent immuno-NP studies. Each dose of immuno-NPs carried 7 μg each of cdGMP and MPLA. The MTT assay (Abcam) was used for viability/cytotoxicity in vitro studies in B16F10 melanoma cells using immuno-NPs carrying doses of 7 μg each of cdGMP and MPLA and an equivalent concentration of empty NPs as a control.2.3. Cell lines & animal models
Murine B16F10 melanoma cells, 4T1 triple-negative breast cancer cells, D2.A1 breast cancer cells, and RAW 264.7 macrophages were cultured in DMEM (Gibco) supplemented with 10% FBS (HyClone). 4T1 and D2.A1 cells were transfected to express GFP and luciferase and a gift from the Schiemann Laboratory at the Case Comprehensive Cancer Center. RAW macrophages were purchased from ATCC. Quality control of cell lines was routinely performed using short tandem repeat authentication and testing for Mycoplasma contamination.All animal procedures were conducted under a protocol (number: 2015-0116) approved by the Institutional Animal Care and Use Committee (IACUC) of Case Western Reserve University (CWRU). CWRU follows the Guide for the Care and Use of Laboratory Animals, which is required by the United States Public Health Service Policy (PHS) on humane care and use of laboratory animals.
For B16F10 melanoma studies, C57/BL6 mice (Jackson Laboratories) were inoculated by orthotopic subcutaneous injection of 1 × 106 B16F10 cells on the dorsal flank. Tumor burden was monitored by measurement of physical tumor dimensions by digital calipers and mouse weight every 1–2 days post-inoculation. Tumor-bearing animals were treated on day 7 after inoculation. Immuno-NPs were administered by intravenous (i.v.) tail vein injection. Anti-PD1 (250 μg, clone RMP1-14; BioXCell) was administered by subcutaneous injection adjacent to the primary tumor mass. On day 14 post-inoculation, tumors were surgically removed under 1–2% isoflurane anesthesia with 5.0 mg kg−1 carprofen as analgesic for 2–3 days post-surgery.
For D2.A1 and 4T1 breast cancer studies, Balb/C mice (Jackson Laboratories) were inoculated by orthotopic subcutaneous injection of 5 × 105 tumor cells in mammary fat pad #9. Tumor burden was monitored by bioluminescence imaging (IVIS Spectrum, PerkinElmer), measurement of physical tumor dimensions by digital calipers, and mouse weight every 1–2 days post-inoculation. Tumor-bearing animals were treated on day 7 after inoculation, with immuno-NPs/anti-PD1 treatment administered and surgical resection performed as for B16F10 tumor-bearing mice as detailed above.
2.4. Flow cytometry
Anti-mouse CD45 (clone 30-F11), Ly6G (clone 1A8), Ly-6C (clone AL-21), CD11b (clone M1/70), CD11c (clone HL3), F4/80 (clone BM8), CD49b (clone DX5), CD3ε (clone 145-2C11), CD4 (clone GK1.5), CD8α (clone Ly2, 53-6.7), and CD19 (clone 1D3) flow cytometry antibodies were purchased from BD Biosciences. Anti-mouse CD80 (clone 16-10A1), CD86 (clone GL1), CD206 (clone C068C2), CD25 (clone PC61), and CD127 (clone SB/199) were also purchased from BD Biosciences and BioLegend. Flow cytometry analysis was performed on blood, tumor, and spleen samples 4 days post-treatment and on a weekly basis as indicated. Blood was collected by retro-orbital bleeding and tumors were excised via surgical resection or immediately following euthanization along with spleens. Blood samples were treated with ACK lysing buffer to lyse erthyrocytes and the remaining blood leukocytes were washed in FACS buffer (0.5% FBS in PBS-EDTA) prior to staining. Organs were gently disrupted by progressive mincing/straining into single-cell suspensions. Cells were blocked with anti-mouse CD18/CD32 and stained in FACS buffer to identify immune cell populations. Samples were analyzed using a BD FACS LSR II Flow Cytometer (Becton Dickinson) and data analysis was performed using FlowJo software.For IgG antibody coating studies for investigation of de novo B cell epitope recognition, treated mice that were initially inoculated with B16F10 or D2.A1 tumor cells that were good responders were bled and serum was isolated at 4 °C. Tumor cells were cultured in sterile PBS containing 5% of serum from these good responders for 30 min also at 4 °C. Samples were stained with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200 of anti-mouse IgG Alexa Fluor 647-conjugated secondary antibody for 1 h at 25 °C. Samples were washed in FACS buffer and analyzed for IgG staining using a BD FACS LSR II Flow Cytometer (Becton Dickinson).
:200 of anti-mouse IgG Alexa Fluor 647-conjugated secondary antibody for 1 h at 25 °C. Samples were washed in FACS buffer and analyzed for IgG staining using a BD FACS LSR II Flow Cytometer (Becton Dickinson).
2.5. Immunostaining and confocal microscopy
B16F10 tumor-bearing mice were treated with combination immuno-NPs/anti-PD1, immuno-NPs only, anti-PD1 only, or left untreated on day 7 following inoculation. Mice were euthanized on day 11, 4 days following the start of therapy. Immediately following, organs were harvested, fixed in 4% PFA/PBS, dehydrated in 30% sucrose/PBS, and embedded and frozen in optimum cutting temperature medium (OCT medium, Thermo Fisher Scientific). Primary antibodies to mouse anti-PD1 and anti-PDL1 were purchased from BioXCell, while secondary antibodies were purchased from Thermo Fisher Scientific. 7 μm thick frozen sections were sectioned, blocked with goat serum, and stained with 1:50–1:100 primary antibodies overnight at 4 °C, followed by staining with 1:50–1:100 secondary antibodies for 1 h at 25 °C. Following staining, sections were mounted with no. 1.5 glass coverslips using Vectashield DAPI aqueous mounting medium (Vector Laboratories). Samples were imaged using a Leica TCS SP8 gated STED confocal microscope (Leica Microsystems).
2.6. Immunosequencing
For T cell receptor repertoire expansion studies, PCR amplification bias-controlled immunosequencing technology, ImmunoSEQ, by Adaptive Biotechnologies was used. Genomic DNA was analyzed from whole blood of mice cured from B16F10 melanoma, 7 days following re-challenge with B16F10 tumor cells. Data was analyzed using ImmunoSEQ Analyzer software.2.7. Transmission electron microscopy with negative staining
For TEM imaging, carbon film grids were first treated by glow discharge for 3 min. NPs were diluted 10-fold from treatment concentrations and applied to treated grids. Following 30 s of sedimentation time, grids were washed with 5 mM Tris buffer and stained immediately with 2% uranyl acetate. Samples were imaged using a Tecnai G2 SpiritBT Electron Microscope (FEI) operated at 80 kV.2.8. ELISAs
C57/BL6 splenocytes were harvested as for flow cytometry and 6 million cells were plated per well of a 24-well plate in triplicate and treated with dual-agonist immuno-NPs containing 20 μg mL−1 each of cdGMP and MPLA, single-agonist NPs containing equivalent amounts of cdGMP or MPLA, empty NPs that served as the vehicle control, or left untreated. Cell culture supernatants were harvested 16 and 40 h after seeding and treatment, clarified by centrifugation at 4 °C, and analyzed per manufacturers’ protocols for the presence of IFNβ (LumiKine Xpress Bioluminescent Cytokine ELISA Kit, Invivogen) and TNFα (ELISA MAX Deluxe Kit, BioLegend). For RAW 264.7 macrophage ELISAs, identical methods were used with cell culture supernatants harvested at 2, 6, and 24 h after seeding and treatment.2.9. Statistical analysis
All statistical analyses are detailed in figure legends. Prism 8 (GraphPad Software) was used to analyze data by one- or two-way ANOVA with Tukey or Sidak's post-test. To be considered statistically significant, P values were less than 0.05. All values are reported as the mean ± standard error (SEM) of at least 3 independent biological replicates, unless otherwise noted. For animal studies, at least 3–5 mice were included in each group. In representative studies, study groups consisted of up to 25 mice when pooled from up to 3 independent study repetitions.3. Results and discussion
3.1. Synergistic dual agonist immuno-NPs drive activation of primary DCs
The effective design of the immuno-NP itself is fundamental to the premise of the novel nanoparticle-based neoadjuvant combination immunotherapy we report here (Fig. 2). Since our goal was to generate a robust proinflammatory Type I interferon response driven specifically by APCs, we elected to co-deliver agonists of two powerful PRR pathways that overlapped in downstream effectors, STING and TLR4 (Fig. 2A). Given our very recent findings that co-delivery of STING agonist cdGMP and TLR4 agonist MPLA on the same liposomal NP promoted the synergistic production of Type I IFNβ,37 we elected to use the same lipid-based design (Fig. 2B). We constructed immuno-liposomes by ultrasonication of an equimolar lipid matrix of the low- and high-Tm lipids DOPC and DPPC, respectively. This lipid-based design was ideally suited for the co-encapsulation of the hydrophilic cdGMP within the aqueous internal core of the NP and the hydrophobic lipidic MPLA by insertion within the lipid bilayer shell. Driven by design criteria that dictated both optimal tumor draining from the systemic blood circulation and internalization within a local target APC, we elected to coat the immuno-NP with 3 mol% poly(ethylene glycol) (PEG) for an overall diameter of ∼50 nm (Fig. 2B). Characterization by transmission electron microscopy (TEM) with negative staining indicated stable lipid ultrastructure (Fig. 2C). Dynamic light scattering (DLS) and zeta potential measurements indicated a mean ∼50 nm hydrodynamic size with neutral surface charge (Fig. 2D, E, ESI Table 1 and ESI Fig. 1A‡). DLS measurements over short (1–4 h) and long (24 h) time points indicated that NPs were stable in physiologically relevant solvents, including PBS, water, and DMEM medium supplemented with 10% serum (ESI Fig. 1B‡). Further, in vitro cytotoxicity measurements indicated that there were no changes to cell viability following treatment with dual-agonist NPs compared to controls (Fig. 2F).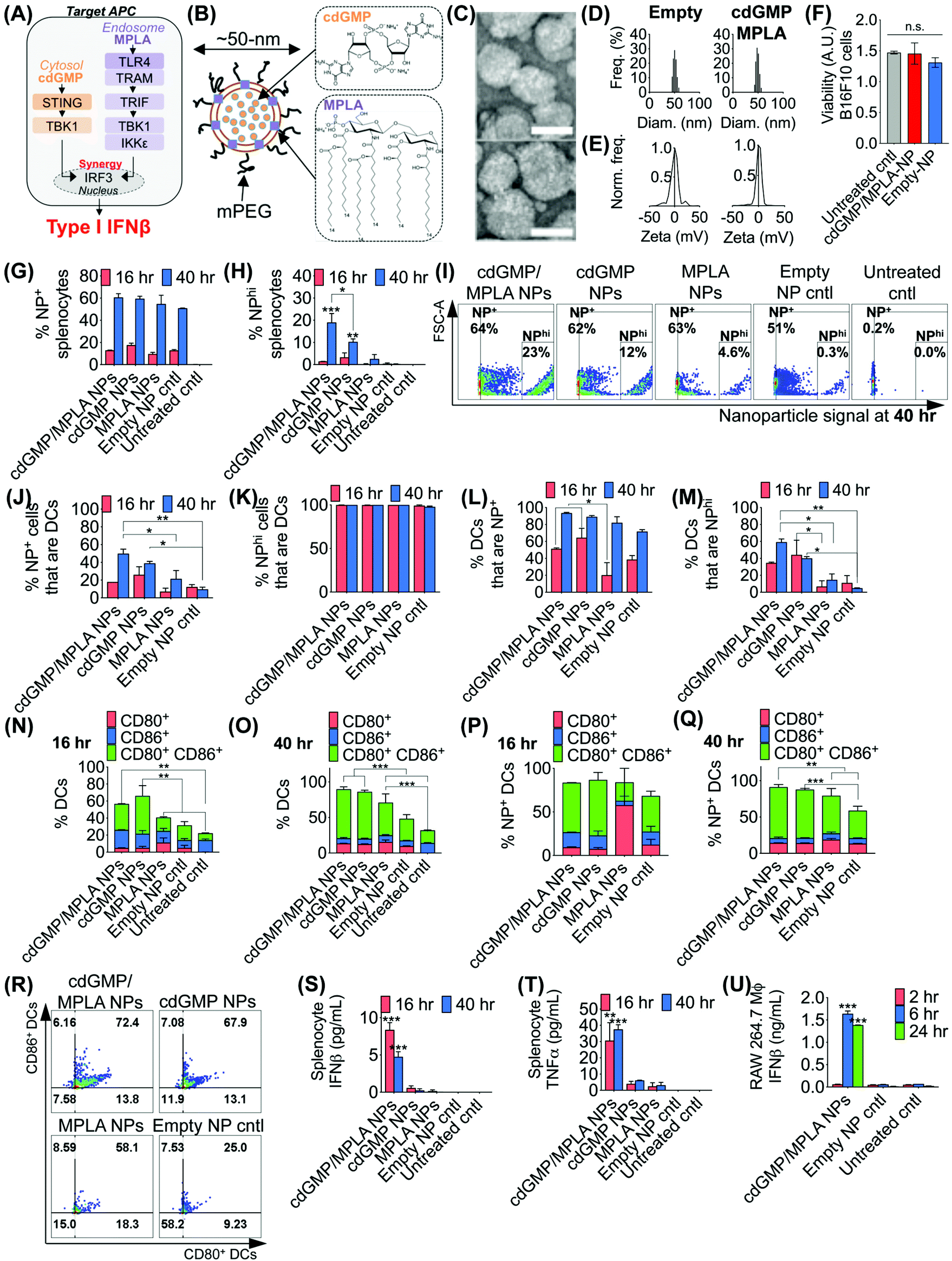 | ||
| Fig. 2 Co-encapsulation of cdGMP and MPLA on the same immunostimulatory NP promotes synergistic production of IFNβ and significant uptake in primary DCs. Schematic depiction of synergistic STING and TLR4 pathways (A) and dual-agonist immuno-NP co-encapsulating cdGMP and MPLA (B). Ultrastructure of immuno-NPs by transmission electron microscopy (TEM) with negative staining (C), scalebar 50 nm. DLS (D) and zeta potential (E) measurements of empty control NPs and cdGMP/MPLA dual-agonist NPs. (F) Viability studies in B16F10 melanoma cells. Analysis by flow cytometry of NP+ splenocytes (G) and NPhi splenocytes (H) with representative dot plots (I). Analysis by flow cytometry of NP+ (J) and NPhi (K) cells that are primary DCs, and primary DCs that are NP+ (L) and NPhi (M). Expression of activation markers CD80 and CD86 in primary DCs at 16 h (N) and 40 h (O), and in NP+ DCs at 16 h (P) and 40 h (Q). Flow cytometry dot plot plots for activation markers CD80 and CD86 in primary DCs (R). ELISA analysis for production of IFNβ (S) and TNFα (T) in primary splenocytes, and of IFNβ in RAW 264.7 macrophages (U). All experiments were performed in independent biological triplicate. Statistics were analyzed by Student's t test, 1-/2-way ANOVA with Tukey's post-test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). | ||
To assess the functionality of immuno-NPs in vitro, we used primary splenic immune cell cultures from C57BL6 mouse strains to quantify immuno-NP uptake, DC activation, and promotion of synergistic production of proinflammatory cytokines. Dual-agonist immuno-NPs, single-agonist NPs, and empty NPs were prepared for these studies (ESI Fig. 2‡). Uptake of fluorescent NPs by splenocytes was continuously increasing from 16 to 40 h of treatment (Fig. 2G), but a distinct population of NP+ splenocytes appeared to endocytose particles with significantly higher affinity at 40 h, with cells that were more than 10000-times higher in NP fluorescence that other splenocytes. Within this NPhi population, at 40 h of treatment, dual-agonist immuno-NPs and cdGMP-only NPs had significantly more NPhi splenocytes, with dual-agonist immuno-NPs having the highest, a significant 1.8-fold greater than cdGMP-only NPs, 7.5-fold greater than MPLA-only NPs, and 61.3-fold greater than empty NP vehicle controls (Fig. 2H and I). Further investigation indicated that at 40 h, 50% of NP+ splenocytes were DCs in the case of treatment with dual-agonist immuno-NPs, which was significantly higher than the single-agonist and control comparisons (Fig. 2J). Significantly, nearly 100% of NPhi splenocytes across all treatment conditions were DCs, which is expected given their proficient phagocytic activity (Fig. 2K). In splenocytes treated with dual-agonist immuno-NPs and cdGMP-only NPs, 58% of DCs were NP+ at 16 h of treatment, which was a significant 2.9-fold higher than with MPLA-only NPs (Fig. 2L). At 40 h, 91% of DCs were NP+ in splenocytes treated with dual-agonist immuno-NPs and cdGMP-only NPs (Fig. 2L). In splenocytes treated with dual-agonist immuno-NPs, a significant 60% of DCs are NPhi at 40 h compared to cells treated with single-agonist and control formulations (Fig. 2M). Taken together, these data suggest a significant role for APCs in general and DCs in particular in driving dual-agonist immuno-NP efficacy. Next, we investigated immuno-NP-mediated DC activation. At both 16 and 40 h, DCs in splenocyte cultures treated with dual-agonist immuno-NPs and cdGMP-only NPs were significantly expressing both surface activation markers, CD80 and CD86, compared to MPLA-only NPs, empty NPs, and untreated controls (Fig. 2N and O). In dual-agonist immuno-NP-treated splenocyte cultures, 57% of DCs were expressing CD80, CD86, or both activation markers at 16 h of treatment, while 90% of DCs were expressing either or both activation markers at 40 h. At 40 h of treatment, dual-agonist immuno-NP- and cdGMP-only NP-treated splenocyte cultures had at least 2.1-fold more strongly activated CD80+ CD86+ double-positive DCs compared to cultures treated with MPLA-only NPs and control cultures (Fig. 2O). Similar trends were observed for NP+ DCs, where at 40 h, there were 1.6-fold more CD80+ CD86+ DCs compared to MPLA-only NPs and empty NPs (Fig. 2P–R). Having quantitatively assessed NP uptake and NP-mediated activation, we finally sought to assess biological function by measuring their promotion of proinflammatory IFNβ and TNFα in treated splenocytes. Strikingly, splenocytes treated with dual-agonist immuno-NPs significantly produced the highest IFNβ and TNFα compared to single-agonist NPs, empty NPs, and untreated controls (Fig. 2S and T). Notably, dual-agonist immuno-NPs promoted 45- and 75-fold increased production of IFNβ at 16 and 40 h of treatment, respectively, and 19- and 16-fold increased production of TNFα at 16 and 40 h of treatment (Fig. 2S and T). Production of both cytokines is strongly synergistic and does not correlate solely to any differences in uptake (i.e., a 1.8-fold increase in uptake of dual-agonist immuno-NPs compared to cdGMP-only NPs at 40 h cannot alone account for the 75-fold increase in IFNβ production and 16-fold increase in TNFα production). To assess kinetics of this response, we treated RAW 264.7 macrophages with dual-agonist immuno-NPs and measured a significant IFNβ production after 6 h of treatment, but not as early as 2 h, compared to controls (Fig. 2U).
3.2. Combination with PD1 blockade augments reduction in tumor burden
Initially, we sought to assess whether an initial combination treatment of immuno-NP and anti-PD1 can provide therapeutic benefits in terms of activation of the tumor-resident immune cells and control of tumor growth for at least a week prior to surgery (next study). In previous work,37 evaluation of the immuno-NP's biodistribution indicated that 6% of the dose accumulated in the tumor 28 hours after injection with the rest of the nanoparticles found primarily in the liver and spleen. Notably, flow cytometry analysis of the primary tumor showed that about 53% of immuno-NPs were taken up by CD45+ immune cells, including DCs and macrophages, but less than 1% of immuno-NPs were found in tumor cells.37 These findings point out to the benefits of systemic delivery of immuno-NPs and their passive and preferential accumulation in the perivascular TME for gaining access to the majority of tumor-resident APCs. We tested the efficacy of the immuno-NP/anti-PD1 combination in vivo using the B16F10 melanoma model. When tumor were well-established with sizes of 100–150 mm3 (day 7 post-inoculation), animals were treated with 3 consecutive daily doses each of immuno-NPs and anti-PD1 with anti-PD1 treatment staggered ahead to start 24 h prior to the start of immuno-NP treatment (Fig. 3A). On days where mice received both immuno-NPs and anti-PD1, treatments were staggered to be administered 12 h apart. While immuno-NPs were administered systemically via intravenous (i.v.) injection in the tail vein, anti-PD1 was administered subcutaneously (s.c.) directly adjacent to the tumor mass to minimize any transport and delivery challenges that might arise due to the large globular size of the antibody. We recognize that a lymph node-activated CD8+ T cell response would likely not occur for 4–7 days following tumor-resident DC activation by immuno-NPs and DC trafficking to lymph nodes. However, we hypothesized that the striking and near-immediate tumor clearance driven by immuno-NP treatment alone that occurs within 24 h of initial treatment may not be due simply to the activity of phagocytic innate immune cells but also due to the activation of local otherwise “exhausted” tumor-specific CD8+ T cells. As such, to significantly augment this particular local CD8+ T cell response, we elected to administer anti-PD1 at the very start of the combination therapy adjacent to the tumor, allowing for direct draining to the tumor mass (Fig. 3A). Tumor burden was monitored by caliper measurements of tumor volume (Fig. 3B) and overall mouse weight (ESI Fig. 3‡). Just 24 h following the first immuno-NP treatment, tumor volumes in both the combination and immuno-NP groups reduced by up to 25%, indicating near-immediate tumor clearance. While tumor sizes remained significantly small in mice that received combination therapy for at least 8 days following (45% reduced), this clearance was sustained for only 5 days in mice that received immuno-NPs alone (39% reduced). Throughout the study, mice in the combination group significantly had the smallest tumors, 57% smaller than mice receiving immuno-NPs alone, and 75% smaller than mice receiving anti-PD1 alone or untreated mice. Notably, in specific treatment groups where anti-PD1 was given continually every 2 days following the first 3 consecutive doses, there appeared to be no added benefit (Fig. 3B).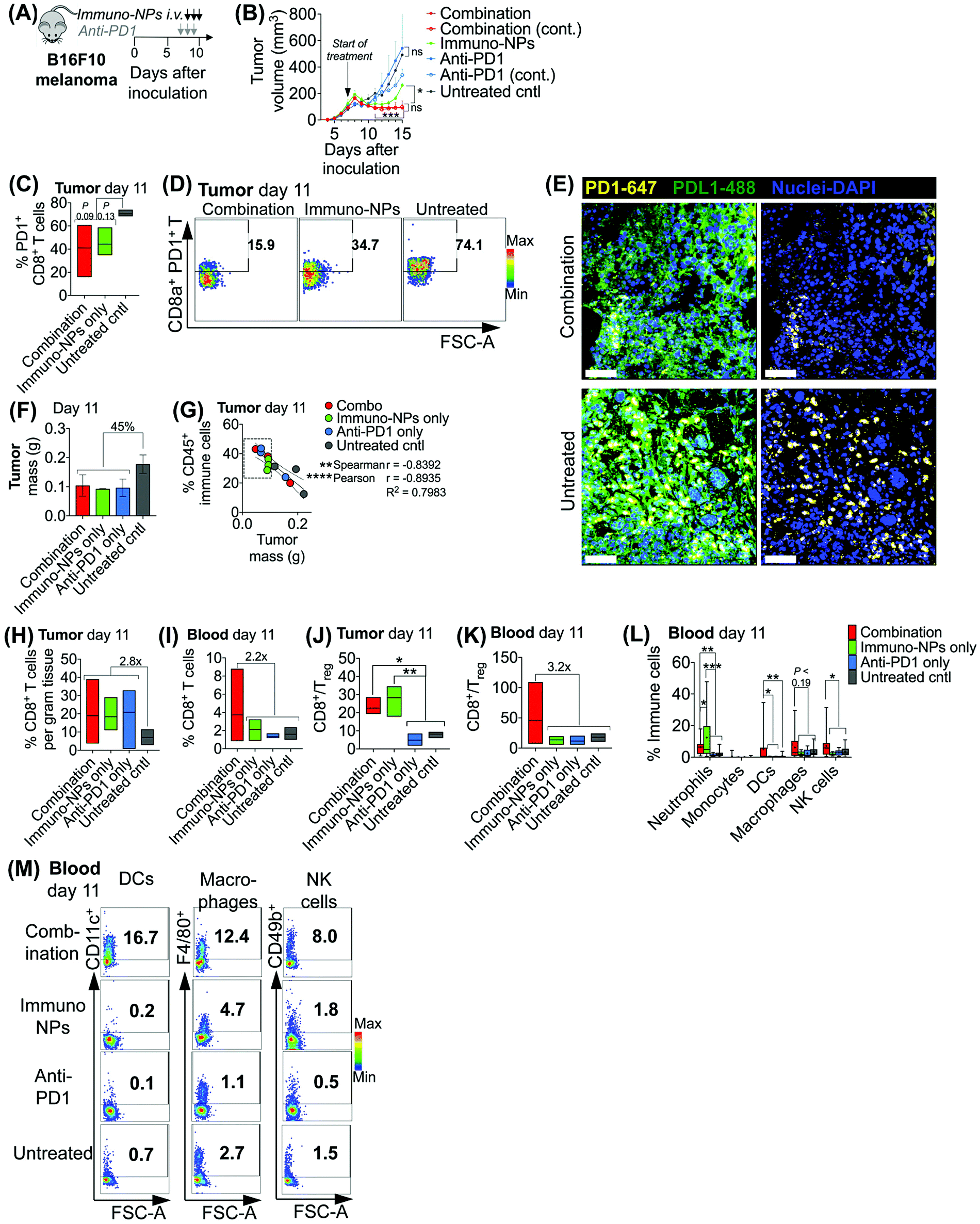 | ||
| Fig. 3 Combination therapy of immuno-NPs with anti-PD1 significantly improves overall survival in vivo. Combination treatment schematic of B16F10 orthotopic melanoma-bearing mice (A). Average physical tumor volume measurements (B). Flow cytometry analysis for PD1+ CD8+ T cells in the tumor (C) and representative dot plots (D). Confocal micrographs for expression of PD1 (yellow, stained with AlexaFluor 647 secondary antibody), PDL1 (green, stained with AlexaFluor 488 antibody), and nuclei (blue, stained with DAPI) in B16F10 tumor tissue (E), scalebar 50 μm. B16F10 tumor mass measurements upon excision on day 11 (F). Flow cytometry analysis for CD45+ immune cells in the tumor in correlation to tumor mass upon excision (G). Flow cytometry analysis of CD8+ T cells in the tumor (H) and blood (I), CD8+/Treg ratios in the tumor (J) and blood (K), and neutrophils, monocytes, DCs, macrophages, and NK cells in the blood (L) on day 11. Flow cytometry dot plots of DCs, macrophages, and NK cells in the blood on day 11 (M). All experiments were performed with at least 3–5 mice per study group. Statistics were analyzed by Student's t test, 1-/2-way ANOVA with Tukey's post-test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). | ||
To investigate the mechanistic basis underlying these functional results, we treated mice bearing B16F10 melanoma tumors with combination therapy or individual monotherapies and analyzed the immune cell makeup of the tumor and blood on day 11 post-inoculation, 4 days following the start of the treatment. We found 37–42% reduced PD1 on the surface of tumor-resident CD8+ T cells in mice that received combination therapy or immuno-NPs compared to untreated mice (Fig. 3C and D). Confocal microscopy showed that both PD1 and PDL1 were upregulated in untreated tumors compared to their treated counterparts (Fig. 3E). On day 11, we found that excised tumor masses of all treated groups were comparable and 45% smaller than those of the untreated group (Fig. 3F). Further, splenic masses across all groups, treated and untreated, were comparable, suggesting that there was no significant systemic inflammation (ESI Fig. 4‡). Tumor size had a strong significant inverse correlation with the percentage of CD45+ immune cells in the tumor on day 11, with all tumors having more than 28% of their cells CD45+ and a mass less than 100 mg being from mice that received a treatment (Fig. 3G, dashed box). Specifically with respect to CD8+ T cells, all treatment groups had 2.8-fold greater tumor-resident CD8+ T cells compared to untreated groups on day 11 (Fig. 3H). Mice that received the combination treatment had 2.2-fold more CD8+ T cells in the blood compared to mice from other groups on day 11 (Fig. 3I). We noted a 3.9-fold increased CD8+/Treg ratio in the tumor in the combination and immuno-NP treatment groups compared to the anti-PD1 and untreated controls (Fig. 3J). Similarly in the blood, we found a 3.2-fold increased CD8+/Treg ratio in the combination treatment group compared to the other groups (Fig. 3K). Further, we measured significantly increased levels of innate immune cells, specifically neutrophils, DCs, macrophages, and NK cells, in the blood compartment of mice receiving combination therapy on day 11 compared to all other treatment groups and untreated controls (Fig. 3L and M).
3.3. Surgical resection following neoadjuvant combination immunotherapy enables curative responses with memory
Given the efficacy of the immuno-NP/anti-PD1 combination therapy for the necessary 7-day window before surgery, we next investigated the efficacy of a complete neoadjuvant scenario with the addition of surgery. Since the treated tumor itself serves as an essential source of information for the immune system to generate a robust protective memory response, we hypothesized that sufficient time was necessary following the start of therapy to allow for local tumor clearance, DC trafficking to lymph nodes for priming of tumor-specific T and B cells, and the generation of adaptive memory clones. In mice bearing B16F10 melanoma tumors, we therefore constructed an identical treatment regimen with surgical resection of tumors 7 days following the start of therapy, on day 14 (Fig. 4A), where we found in our previous studies that tumor size remained at its smallest (Fig. 3B). On day 31 following original inoculation and approximately 2 weeks following surgery, we prepared to re-challenge surviving mice or good responders with tumor cells to assess for protective immunological memory. Mice bearing orthotopic B16F10 tumors were either treated with neoadjuvant combination immunotherapy followed by surgery or treated by surgery alone (Fig. 4B). Compared to mice that received neoadjuvant combination immunotherapy, mice that received surgery alone succumbed significantly more to primary tumor regrowth post-surgery, to sudden weight loss, and to tumor re-challenge (Fig. 4C). Overall survival was significantly prolonged in mice that received combination immunotherapy followed by surgery with 71% of the treated group being cancer-free (compared to just 20% for mice treated by surgery alone). These mice were classified as good responders, all of which were cured with immunological memory that protected against recurrence (Fig. 4D–G). Following surgery, the resected tumor masses of mice that received combination immunotherapy were 52% smaller than those of mice that received surgery alone (Fig. 4H). Further, within each treatment group, the resected tumor masses of good responders were 3–5 times smaller than those of poor responders (Fig. 4I). Resected tumors in mice that received combination therapy prior to surgery had 2.7-fold more CD45+ immune cells compared to mice that received surgery alone (Fig. 4J). Notably, excised tumor masses of mice that received combination therapy had significantly more tumor-infiltrating B cells (Fig. 4K and L), which suggested a key role for humoral immunity in the memory response (investigated in studies reported later in Fig. 7). Resected tumors of mice that received combination immunotherapy also had greater cross-presenting DCs (3.6-fold), significantly higher M1/M2 ratios (5.3-fold), and higher CD8+/Treg ratios (6.7-fold) (Fig. 4M–O, respectively). Four days following surgery, on day 18 post-inoculation, mice that received combination immunotherapy had significantly higher levels of blood neutrophils, DCs, macrophages, and NK cells compared to mice that received surgery alone (Fig. 4P), as well as significantly elevated blood CD3e+ T cells (Fig. 4Q). One and 7 days following re-challenge, mice that received combination therapy had notably 3.2-fold increased CD4+ T cells, 2.8-fold increased CD8+ T cells, and 2.1-fold increased B cells in the blood (Fig. 4R and S).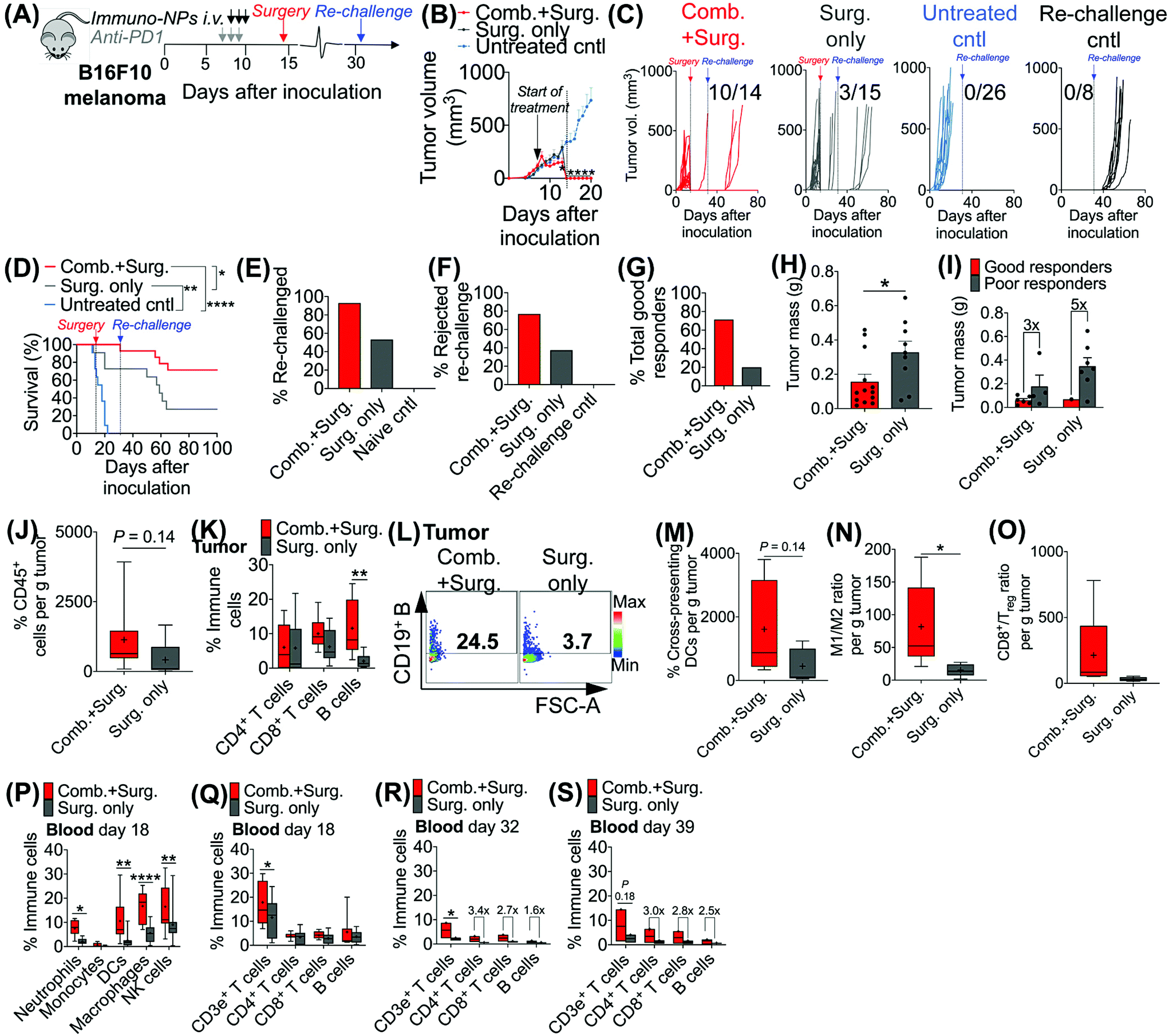 | ||
| Fig. 4 Addition of surgery following combination immunotherapy drives curative outcomes with protective immunological memory. Schematic of combination treatment followed by surgery of B16F10 orthotopic melanoma-bearing mice (A). Average (B) and individual curves (C) for physical tumor volume measurements with corresponding Kaplan–Meier survival (D). Percentage of mice that was re-challenged (E), percentage of mice that rejected re-challenge (F), and percentage of total good responders (G). Tumor masses following surgical resection of all mice (H) and tumor masses of good versus poor responders (I). Flow cytometry analysis for CD45+ immune cells in the tumor (J) and T and B cells in the tumor (K), with a representative dot plot for tumor B cells (L). Flow cytometry analysis for cross-presenting DCs (M), M1/M2 ratio (N), and CD8+/Treg ratio (O), all in the tumor. Flow cytometry analysis for neutrophils, monocytes, DCs, macrophages, and NK cells in the blood on day 18 (P), and T and B cells in the blood on day 18 (Q), day 32 (R), and day 39 (S). All experiments were performed with at least 5–10 mice per study group. Statistics were analyzed by Student's t test, 1-/2-way ANOVA with Tukey's post-test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). | ||
3.4. Local tumor-resident CD8+ T cells are central mediators of tumor clearance
Following these findings of rapid tumor clearance, curative treatment, and subsequent long-term protective immunological memory, we sought to elucidate immune cell players with pivotal roles in these processes (Fig. 5). Towards this goal, we followed an identical neoadjuvant combination immunotherapy treatment regimen with re-challenge in mice bearing orthotopic B16F10 melanoma tumors and selectively depleted macrophages, NK cells, CD8+ T cells, or B cells. Depletions were initiated 24 h prior to the start of therapy and continued 2–3 times per week until tumors were surgically removed (Fig. 5A). Mice that were depleted of macrophages, NK cells, and B cells all responded to immuno-NP/anti-PD1 combination therapy similarly to mice where no cells were depleted, where tumor sizes across all of these groups were 51% smaller than tumors of untreated mice (Fig. 5B). Strikingly, however, mice that were depleted of CD8+ T cells did not appear to respond to combination therapy at all (mice in other groups had 61% smaller tumors) and tumor sizes in these mice with depleted CD8+ T cells were statistically similar to the large sizes of untreated controls (Fig. 5B). These results strongly suggested that local tumor-resident CD8+ T cells that are otherwise “exhausted” likely play a major role in the near-immediate clearance of tumor cells following combination immunotherapy. In this particular cohort of mice, with no depletions, 100% of mice had curative responses with protective immunological memory against tumor re-challenge (Fig. 5C). With CD8+ T cells depleted, 50% of mice succumbed to large tumors prior to surgery, 25% of mice had primary tumors that regrew, and the remaining 25% of mice that were re-challenged did not control the re-challenge, indicating that they did not have a significant adaptive memory subset (Fig. 5D). In groups that were depleted of macrophages, NK cells, and B cells, 100% of mice were tumor-free at re-challenge, but while none of the mice with depleted NK cells and B cells controlled the re-challenge, 50% of mice with depleted macrophages survived with curative outcomes (Fig. 5E). Taken together, these data suggested that while primarily CD8+ T cells play a major role in primary tumor clearance, CD8+ T, NK, and B cells all play critical roles in the immunological memory response (Fig. 5F and G). They also indicated that macrophages may be important APCs at the start of the therapy, but unlike DCs, they may not be critical for efficacy (Fig. 5F and G). Flow cytometry analysis of surgically excised tumor masses indicated a significantly increased tumor mass in mice depleted of CD8+ T cells (at least 2-fold, Fig. 5H) and significantly reduced levels of CD45+ immune cells (Fig. 5I), DCs (Fig. 5J), macrophages (Fig. 5K), and CD8+ T cells as expected (Fig. 5L).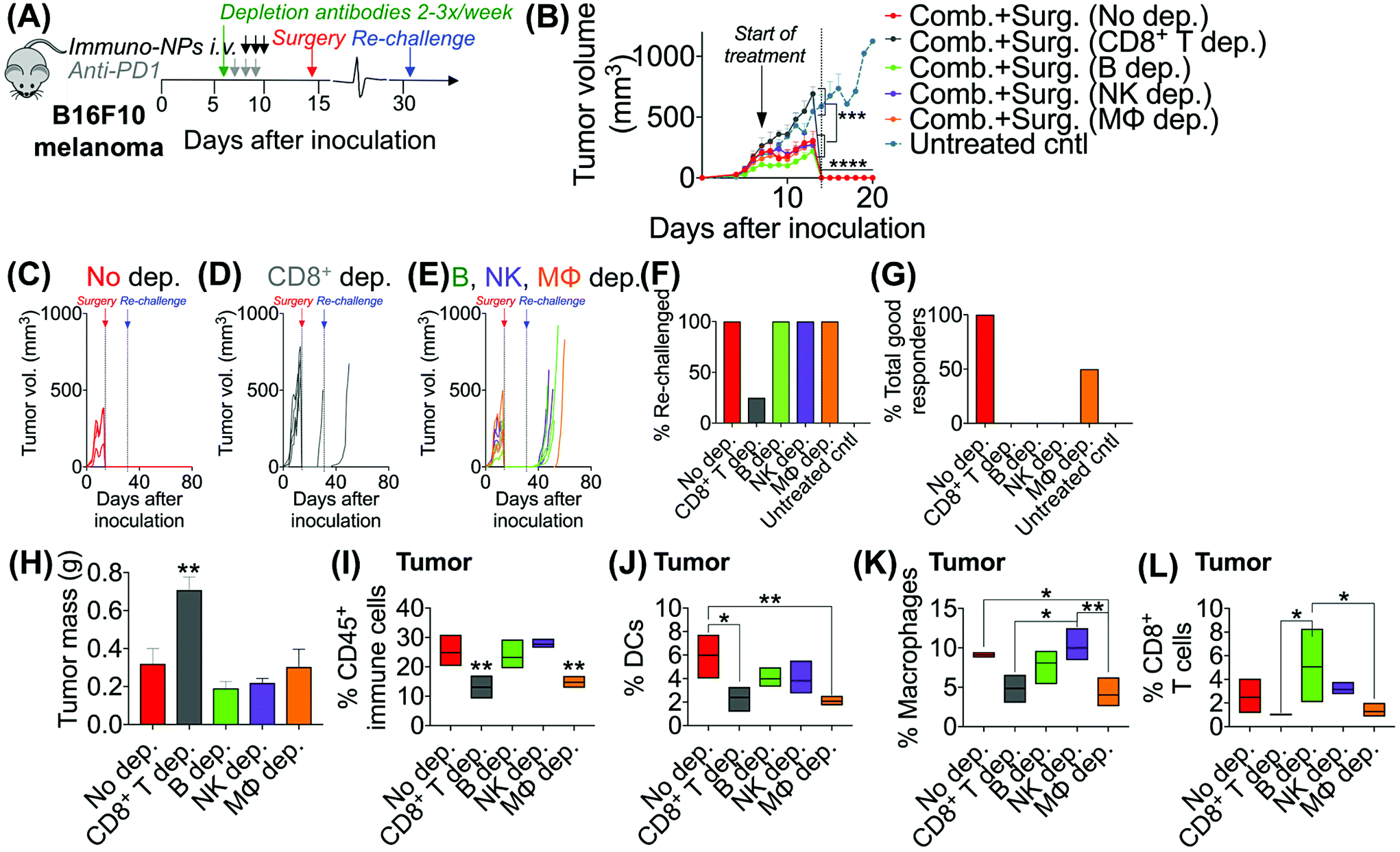 | ||
| Fig. 5 Immune cell depletions suggest key tumor clearance role for CD8+ T cells. Schematic of neoadjuvant combination treatment of B16F10 orthotopic melanoma-bearing mice with depletion antibodies administered 2–3 times per week from days 6–14 (A). Average (B) and individual (C–E) tumor volume measurements. Percentage of mice that was re-challenged (F) and percentage of total good responders (G). Tumor masses following surgical resection (H). Flow cytometry analysis for CD45+ immune cells (I), DCs (J), macrophages (K), and CD8+ T cells (L) in the tumor. All experiments were performed with at least 4–6 mice per study group. Statistics were analyzed by Student's t test, 1-/2-way ANOVA with Tukey's post-test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). | ||
Given these results, we sought to investigate the efficacy of neoadjuvant combination immunotherapy in two mouse models of metastatic breast cancer (D2.A1 and 4T1 models). We treated mice bearing either D2.A1 or 4T1 tumors using the same neoadjuvant treatment regimen we established for B16F10 tumor-bearing mice (Fig. 6A). For both models, primary orthotopic breast cancer cells were inoculated in the mammary fat pad and allowed to grow until well established at the start of treatment on day 7. On day 14, primary tumors were surgically resected and by day 21, metastasis was detected predominantly in the lungs (Fig. 6B, trajectory of tumor development shown for representative D2.A1 tumor-bearing mice and is similar for 4T1 tumor-bearing mice). In mice bearing D2.A1 tumors, primary tumor volume was 1.7-fold decreased significantly in mice treated with combination therapy by the time of surgical resection (Fig. 6C). Primary tumor cell viability as measured by luciferase luminescence decreased significantly 4.2-fold within just 2 days of the start of treatment (Fig. 6D). Strikingly, early metastasis signal also decreased 2.8-fold in mice bearing D2.A1 tumors given immuno-NP/anti-PD1 combination treatment within just 2 days. Metastatic signal remained low in these mice even 11 days post-surgery, 25 days post-inoculation, in contrast to mice that were treated by surgery alone that developed significant advanced metastases with 20-fold higher signal (Fig. 6E). We re-challenged the good responders 31 days post-inoculation and 100% of these mice rejected the re-challenge, which was significant compared to naïve controls (Fig. 6F). Similarly in mice bearing 4T1 tumors, mice that received combination treatment had 1.5–2.0-fold smaller tumors consistently up to 3 days prior to surgery (Fig. 6G). Further, 7 days post-surgery and 21 days post-inoculation, mice that received only surgery had advanced metastasis with a signal that was 32 times that of 4T1 tumor-bearing mice that received neoadjuvant combination treatment prior to surgery (Fig. 6H). 4T1 tumor-bearing mice that received combination therapy had resected tumors that were 25% smaller by mass compared to mice that received surgery alone (Fig. 6I). Tumors of these treated mice had 1.9-fold increased CD45+ immune cells (Fig. 6J), 1.6-fold and 1.2-fold increased DCs and NK cells, respectively (Fig. 6K), and 1.4-fold increased cross-presenting DCs (Fig. 6L). Further, these tumors had 1.2-fold elevated CD3e+ T cells, 1.4-fold elevated CD8+ T cells, and 2-fold elevated B cells (Fig. 6M). Notably, DCs were significantly elevated in the blood of 4T1 tumor-bearing mice on day 18, 4 days post-surgery (Fig. 6N). B cells were also significantly elevated on days 11 and 18, 3 days prior to surgery and 4 days after surgery, respectively, again indicating that an immunological memory response may have a strong humoral component (Fig. 6O and P).
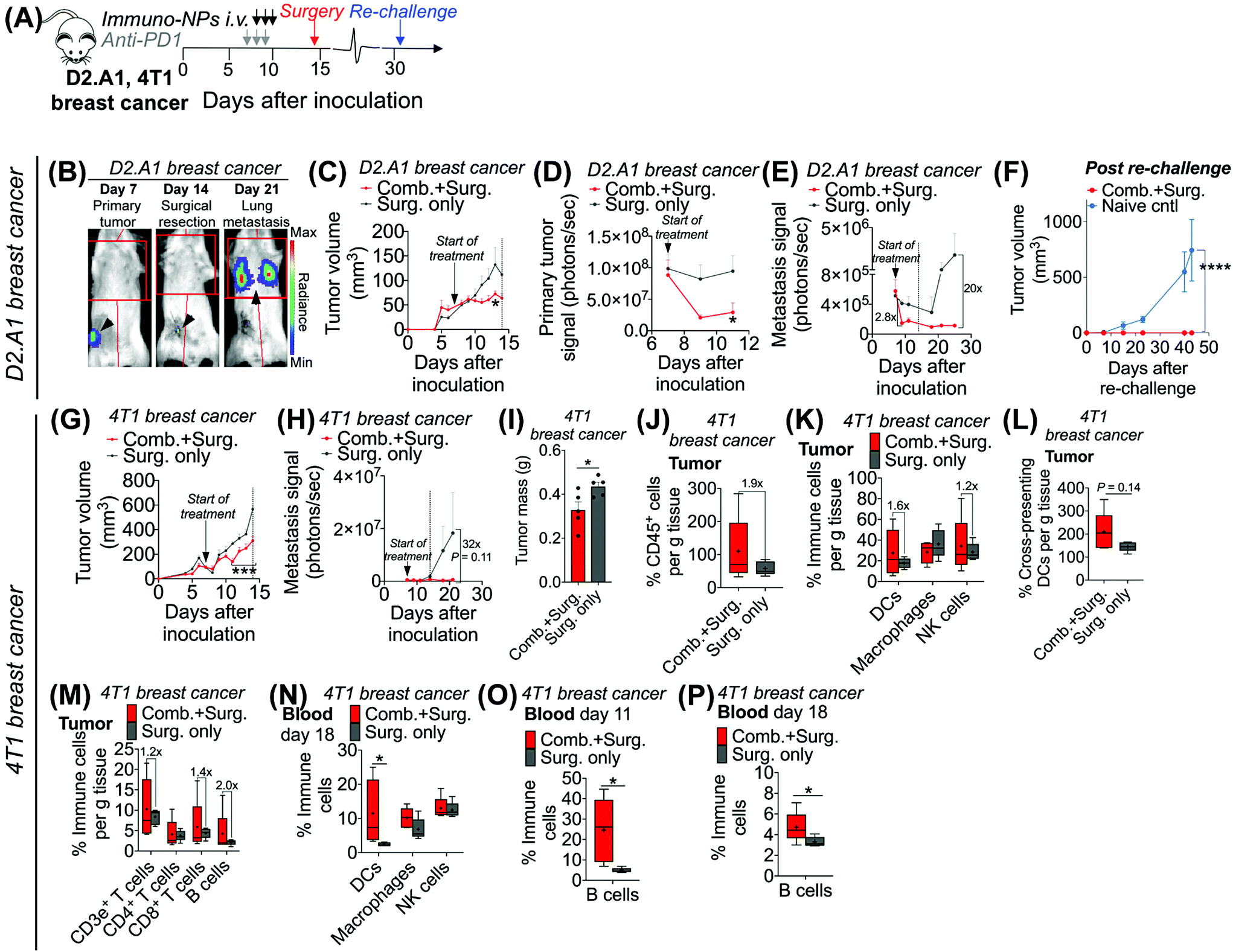 | ||
| Fig. 6 Analysis in two models of metastatic breast cancer corroborate efficacy of neoadjuvant combination immunotherapy. Schematic of neoadjuvant combination treatment of D2.A1 and 4T1 orthotopic breast tumor-bearing mice (A). In vivo bioluminescence imaging in D2.A1 tumor-bearing mice for orthotopic primary tumor signal on day 7, surgical resection on day 14, and lung metastasis on day 21 (B). 4T1 tumor-bearing mice had a similar tumor growth trajectory. Average tumor volume (C), primary tumor bioluminescence signal (D), and metastatic bioluminescence signal (E) for D2.A1 tumor-bearing mice. Average tumor volume in D2.A1 re-challenged mice (F). Average tumor volume (G) and metastatic signal (H) in 4T1 tumor-bearing mice. Surgically resected 4T1 tumor masses (I). Flow cytometry analysis for tumor-infiltrating CD45+ immune cells (J), DCs, macrophages, and NK cells (K), cross-presenting DCs (L), and T and B cells (M) in 4T1 tumor-bearing mice. Flow cytometry analysis for DCs, macrophages, and NK cells in the blood on day 18 (N) and for B cells in the blood on days 11 (O) and 18 (P) in 4T1 tumor-bearing mice. All experiments were performed with at least 5–7 mice per study group. Statistics were analyzed by Student's t test, 1-/2-way ANOVA with Tukey's post-test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). | ||
3.5. Dual agonist immuno-NPs drive increased recognition of de novo T and B cell epitopes
Taken together, these data in multiple models of aggressive cancer provided evidence of a protective immunological memory. Given that the immuno-NPs can by themselves drive an active APC-mediated sampling and surveillance of the tumor microenvironment without co-delivery of a specific tumor antigen, we sought to establish whether this technology enhances de novo epitope recognition. Using the established neoadjuvant combination immunotherapy treatment scheme in mice bearing B16F10 melanoma tumors (Fig. 7A), we assayed blood of re-challenged mice early during the course of treatment, 5 and 7 days following re-challenge particularly for tumor-specific memory CD8+ T cells (Fig. 7B). Notably, these samples were taken 3–5 days before the appearance of even minimally palpable tumors in the naïve controls and 9–11 days before any differences in tumor size between the treated and control groups were statistically significant (Fig. 7B). To induce tumor-specific memory CD8+ T cells, peripheral blood mononuclear cells (PBMCs) were co-cultured with increasing numbers of B16F10 tumor cells for 30 h prior to Golgi transport inhibition and for another 18 h following for a total of 48 h incubation. While there was no significant CD8+ T cell proliferation when day-5 PBMCs were co-cultured with 5 × 103 B16F10 tumor cells, there was a significant 2.5-fold increase in CD8+ T cells in co-cultures with 10 × 103 B16F10 tumor cells that remained as high in co-cultures with 20 × 103 B16F10 tumor cells (Fig. 7B). However, CD8+ T cells co-cultured with all three concentrations of B16F10 tumor cells were producing significantly increased amounts of functional cytokines TNFα and IFNγ when compared to CD8+ T cells that were not co-cultured with tumor cells (Fig. 7D). Notably, levels of these cytokines and CD8+ T cells that were producing both TNFα and IFNγ and therefore fully functional increased significantly on day 7 compared to day 5 (Fig. 7C–E). To assess directly for increased peripheral CD8+ T cell clones in the blood of mice that received combination immunotherapy prior to surgery versus mice that received surgery alone, we selected good responders from each group (71% of mice who received neoadjuvant therapy were good responders, while 20% of mice who received surgery alone were good responders). ImmunoSEQ (Adaptive Biotechnologies) genetic sequencing analysis on day 7 following re-challenge in mice formerly bearing B16F10 tumors indicated that mice that received neoadjuvant combination immunotherapy had 2.5-fold increased CD8+ T cells in peripheral blood and a 2.6-fold increased number of unique clones (Fig. 7F). Further, mice that received neoadjuvant combination immunotherapy shared 22 times as many unique clones between them compared to mice that received surgery alone (Fig. 7G and H). Following this analysis of CD8+ T cells, we also assayed for increased de novo antibody responses, especially considering the significant levels of tumor-infiltrating and circulating B cells observed in multiple tumor models. Selecting representative good responders among mice originally bearing either B16F10 or D2.A1 tumors, we obtained blood serum samples ∼300 days post-inoculation. Incubation of good responder serum with B16F10 or D2.A1 tumor cells and analysis for surface IgG antibody coating as a measurement of tumor antigen recognition indicated a clear increased antibody response in mice that received neoadjuvant combination immunotherapy in both tumor models, 8.8-fold for mice cured of B16F10 melanoma and 6.1-fold for mice cured of D2.A1 breast cancer compared to surgery-only and naïve controls (Fig. 7I–K).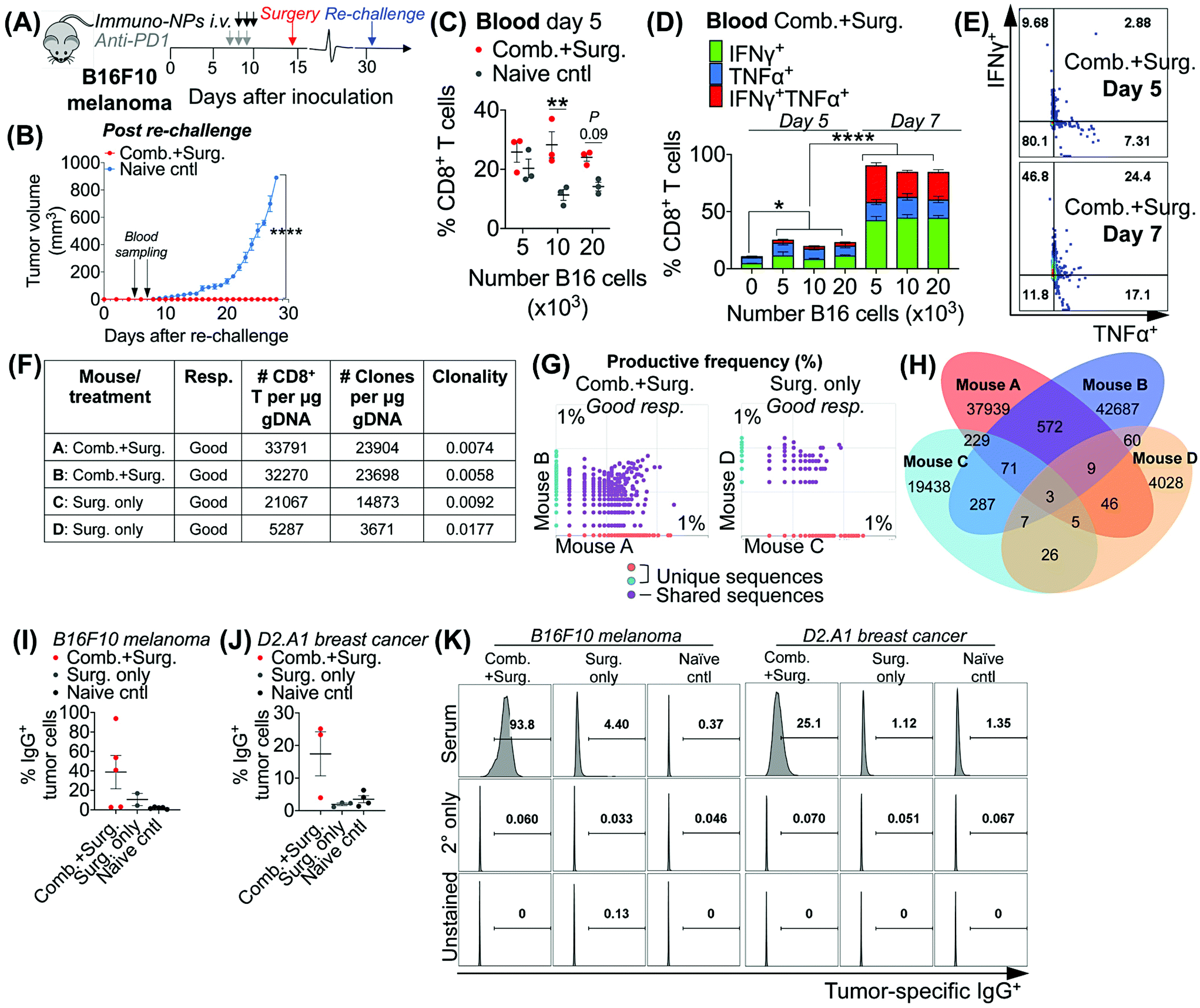 | ||
| Fig. 7 Combination therapy drives CD8+ T cell and humoral memory with de novo epitope recognition. Schematic of neoadjuvant combination treatment of B16F10 melanoma-bearing mice (A). Average tumor volumes following re-challenge (B). Following in vitro co-culture of blood cells with B16F10 tumor cells, flow cytometry analysis for proliferating CD8+ T cells on day 5 post re-challenge (C) and activated CD8+ T cells on day 7 expressing IFNγ, TNFα, or both cytokines (D) with representative dot plots (E). ImmunoSEQ analysis for CD8+ T cell clones in blood of mice on day 7 post re-challenge with data summary (F), pairwise plots for productive frequency (G), and Venn diagram for shared versus individual clones (H). Following in vitro incubation of serum antibodies with tumor cells, flow cytometry analysis for IgG antibody-coated B16F10 melanoma (I) and D2.A1 breast cancer (J) tumor cells. Representative flow cytometry histogram plots for IgG-coated tumor cells (K). All experiments were performed with at least 3–5 mice per study group. In vitro studies were performed in independent biological triplicate. Statistics were analyzed by Student's t test, 1-/2-way ANOVA with Tukey's post-test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). | ||
4. Conclusions
These results highlight the efficacy of this novel neoadjuvant nanoparticle-based combination immunotherapy. Two central components of this therapeutic approach are at the crux of its efficacy. First, the design of the immuno-NPs that enables co-encapsulation of two synergistic immune agonists, cdGMP a STING agonist and MPLA a TLR4 agonist, on the same NP is critical in promoting the robust IFNβ production that potentiates an APC-driven immune response. Notably, in recent results, we have also shown that NP-encapsulated agonists have significant immune-promoting efficacy compared to both free agonists administered i.v. or even directly by i.t. injection.37 We have also shown the functional synergy of the two agonists can be altered by adjusting the ratio of the two agonists loaded in the immuno-NP.38 Further, due to the high potency of the synergistic cytokine-promoting immuno-NP treatment, the required doses for efficacy can be reduced allowing for high systemic safety profiles. A single dose of immuno-NPs carries 7 μg each of cdGMP and MPLA, intentionally chosen to be 7–21 times lower than published doses for free agonists.24,48,49 Further as previously reported, our assessment of systemic hepatotoxicity in terms of release of liver enzymes alanine transaminase (ALT) and aspartate aminotransferase (AST) indicated only minimal and transient elevation in these enzymes that was not statistically significant.37 Similarly, quantification of weight changes in treated mice also indicated only a transient weight loss within 2 days of the start of treatment that is recovered fully and rapidly within 2–4 days. Within 7 days and onwards following the start of treatment, mice that received the combination treatment gain weight. Second, the addition of anti-PD1 as a combination companion therapeutic is central in augmenting a tumor-resident CD8+ T cell-driven tumor clearance response. Since our depletion studies indicated that these T cells were key drivers of immediate clearance (i.e., within 1–2 days of the start of immuno-NP treatment), these results reinforce the powerful efficacy of our tumor-draining immuno-NPs since they are able to initiate and drive an immune response that can move well beyond the innate myeloid compartment and harness an effective adaptive response. These results further support the essential role that nanotechnology and delivery systems can play in cancer immunotherapy.50,51In recent publications,37,38 we reported on the preferential and significant uptake of immuno-NPs by tumor-resident APCs, specifically DCs and macrophages, in vivo in both the primary tumor and sites of disseminated metastasis. We also noted that NP+ APCs were producing significantly increased levels of IFNβ. Nanoparticles loaded with anticancer drugs have traditionally relied on the so-called EPR effect. However, systemic administration often results in heterogeneous and perivascular deposition of nanoparticles in tumors leading to failure to deliver their anticancer drug to the majority of cancer cells. In the case of immunostimulatory NPs, systemic administration allows NPs to passively deposit into the tumor's perivascular space, which coincides with the APC-rich tumor areas, resulting in predominant uptake by the desirable innate immune cells. Overall, while near-perivascular deposition of nanoparticles may be considered a limitation in the context of delivery of cytotoxic cancer drugs, NPs can seamlessly access the APC-rich perivascular region of tumors.37,38
In the current work, the results suggest a pivotal tumor clearance role for CD8+ T cells. This is significant, since this cell subset is otherwise “exhausted” and dysfunctional in aggressive, advanced, and highly immunosuppressive cancers.41,52 These findings emphasize the powerful efficacy of immuno-NPs and their intratumoral perivascular deposition in harnessing an APC subset that is strong enough to activate otherwise dysfunctional and “exhausted” CD8+ T cells. Notably, rescuing “exhausted” CD8+ T cells is a significant area of investigation. The key role for CD8+ T cells established here in these studies suggests that DC activation by immuno-NPs may be more pronounced and significant, since it is only DCs that can prime and activate CD8+ T cells. This significant role for activated DCs also has implications for long-term therapeutic efficacy since these DCs have the potential to traffic to lymph nodes to prime, activate, and recruit other tumor-specific T cells for clearance.
Finally, the neoadjuvant application of the combination immunotherapy (immuno-NP and anti-PD1) provides an essential immune intervention, while the primary tumor and its diverse microenvironment and tumor antigens are still present, allowing the prospect of the entire tumor to be turned into a vaccine. Our findings also indicate that immuno-NPs drive de novo T and B cell epitope recognition.10,44,45,47 We expect that uptake of immuno-NPs and activation of these target cells drives a self-amplifying response that, along with co-sampling of tumor antigens shed by dying tumor cells (e.g., by APCs), leads to immune recognition of a large and increasing number of diverse tumor-specific T and B cell clones. We performed these studies in good responders with an immunological memory response that protected against a tumor re-challenge. These studies indicated that the memory cells also contained tumor-specific clones that were generated de novo. Notably, this response of de novo epitope recognition can result in a more comprehensive protective immunological memory response. To further validate the potency of the antitumor immunity, we will perform detailed investigation of the tumor antigen-specific responses in the future using mixed lymphocyte reaction assays.
Taken together, here we report on the development of an effective combination neoadjuvant therapeutic strategy that combines a dual-agonist immunostimulatory nanoparticle with anti-PD1 checkpoint blockade. Our findings suggest that this therapeutic strategy is effective in multiple models of aggressive cancer. In particular, the significance of this approach fundamentally relies on the co-encapsulation of a STING agonist and TLR4 agonist on the same immuno-NP that together promote the synergistic production of Type I IFNβ upon uptake by tumor-resident APCs. Further, the combination with anti-PD1 highlighted the role of otherwise “exhausted” tumor-infiltrating CD8+ T cells in immediate tumor clearance. As such, this unique nanoparticle-based strategy has significant implications for the development of effective clinical neoadjuvant immunotherapies.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by grants from the National Cancer Institute (R01CA253627, U01CA198892, E. K.), the Case Comprehensive Cancer Center Support Grant (P30CA043703) and the Shiverick Family Fund (E. K.), and VeloSano Foundation (P. U. A.). We acknowledge the Case Center for Imaging Research, Case Comprehensive Cancer Center Flow Cytometry Core, and the Case School of Medicine Light Microscopy Core (NIH grant S10-RR031845).References
- D. Hanahan and R. A. Weinberg, Hallmarks of cancer: the next generation, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- D. S. Park, et al., The Goldilocks Window of Personalized Chemotherapy: Getting the Immune Response Just Right, Cancer Res., 2019, 79, 5302–5315 CrossRef CAS PubMed.
- C. Guo, et al., Therapeutic Cancer Vaccines: Past, Present and Future, Adv. Cancer Res., 2013, 119, 421–475 CrossRef CAS PubMed.
- L. A. Korde, et al., Neoadjuvant Chemotherapy, Endocrine Therapy, and Targeted Therapy for Breast Cancer: ASCO Guideline, J. Clin. Oncol., 2021, 39, 1485–1505 CrossRef PubMed.
- M. Kaufmann, et al., Recommendations From an International Expert Panel on the Use of Neoadjuvant (Primary) Systemic Treatment of Operable Breast Cancer: An Update, J. Clin. Oncol., 2006, 24, 1940–1949 CrossRef PubMed.
- J. Collignon, L. Lousberg, H. Schroeder and G. Jerusalem, Triple-negative breast cancer: treatment challenges and solutions, Breast Cancer, 2016, 8, 93–107 CAS.
- H. Yu, M. Kortylewski and D. Pardoll, Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment, Nat. Rev. Immunol., 2007, 7, 41–51 CrossRef CAS PubMed.
- H. Tang, J. Qiao and Y.-X. Fu, Immunotherapy and tumor microenvironment, Cancer Lett., 2016, 370, 85–90 CrossRef CAS PubMed.
- S. Ostrand-Rosenberg, Immune surveillance: a balance between protumor and antitumor immunity, Curr. Opin. Genet. Dev., 2008, 18, 11–18 CrossRef CAS PubMed.
- V. Thorsson, et al., The Immune Landscape of Cancer, Immunity, 2018, 48, 812–830.e14 CrossRef CAS PubMed.
- P. S. Hegde, V. Karanikas and S. Evers, The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition, Clin. Cancer Res., 2016, 22, 1865–1874 CrossRef CAS PubMed.
- T. F. Cloughesy, et al., Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma, Nat. Med., 2019, 25, 477–486 CrossRef CAS PubMed.
- K. A. Schalper, et al., Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma, Nat. Med., 2019, 25, 470–476 CrossRef CAS PubMed.
- D. M. Pardoll, The blockade of immune checkpoints in cancer immunotherapy, Nat. Rev. Cancer, 2012, 12, 252–264 CrossRef CAS PubMed.
- K. Esfahani, et al., A review of cancer immunotherapy: from the past, to the present, to the future, Curr. Oncol., 2020, 27, S87–S97 CrossRef CAS PubMed.
- T. Nakamura, et al., STING agonist loaded lipid nanoparticles overcome anti-PD-1 resistance in melanoma lung metastasis via NK cell activation, J. Immunother. Cancer, 2021, 9, e002852 CrossRef PubMed.
- S. Yan, et al., Improving Cancer Immunotherapy Outcomes Using Biomaterials, Angew. Chem., 2020, 132, 17484–17495 CrossRef.
- E. A. Rozeman, et al., Survival and biomarker analyses from the OpACIN-neo and OpACIN neoadjuvant immunotherapy trials in stage III melanoma, Nat. Med., 2021, 27, 256–263 CrossRef CAS PubMed.
- J. S. O'Donnell, E. P. Hoefsmit, M. J. Smyth, C. U. Blank and M. W. L. Teng, The Promise of Neoadjuvant Immunotherapy and Surgery for Cancer Treatment, Clin. Cancer Res., 2019, 25, 5743–5751 CrossRef PubMed.
- J. Liu, et al., Timing of neoadjuvant immunotherapy in relation to surgery is crucial for outcome, OncoImmunology, 2019, 8, e1581530 CrossRef PubMed.
- O. Demaria, et al., Harnessing innate immunity in cancer therapy, Nature, 2019, 574, 45–56 CrossRef CAS PubMed.
- D. B. Stetson and R. Medzhitov, Recognition of Cytosolic DNA Activates an IRF3-Dependent Innate Immune Response, Immunity, 2006, 24, 93–103 CrossRef CAS PubMed.
- D. B. Stetson and R. Medzhitov, Type I Interferons in Host Defense, Immunity, 2006, 25, 373–381 CrossRef CAS PubMed.
- L. Corrales, et al., Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity, Cell Rep., 2015, 11, 1018–1030 CrossRef CAS PubMed.
- L. Corrales, S. M. McWhirter, T. W. Dubensky and T. F. Gajewski, The host STING pathway at the interface of cancer and immunity, J. Clin. Invest., 2016, 126, 2404–2411 CrossRef PubMed.
- L. Corrales and T. F. Gajewski, Molecular Pathways: Targeting the Stimulator of Interferon Genes (STING) in the Immunotherapy of Cancer, Clin. Cancer Res., 2015, 21, 4774–4779 CrossRef CAS PubMed.
- Y.-C. Lu, W.-C. Yeh and P. S. Ohashi, LPS/TLR4 signal transduction pathway, Cytokine, 2008, 42, 145–151 CrossRef CAS PubMed.
- J. Garaude, A. Kent, N. van Rooijen and J. M. Blander, Simultaneous Targeting of Toll- and Nod-Like Receptors Induces Effective Tumor-Specific Immune Responses, Sci. Transl. Med., 2012, 4, 120ra16–120ra16 Search PubMed.
- T. Kawai and S. Akira, TLR signaling, Cell Death Differ., 2006, 13, 816–825 CrossRef CAS PubMed.
- M. C. Hanson, et al., Nanoparticulate STING agonists are potent lymph node–targeted vaccine adjuvants, J. Clin. Invest., 2015, 125, 2532–2546 CrossRef PubMed.
- G. N. Barber, STING: infection, inflammation and cancer, Nat. Rev. Immunol., 2015, 15, 760–770 CrossRef CAS PubMed.
- C. W. Cluff, Monophosphoryl Lipid A (MPL) as an Adjuvant for Anti-Cancer Vaccines: Clinical Results, Landes Bioscience, 2013 Search PubMed.
- C. R. Casella and T. C. Mitchell, Putting endotoxin to work for us: monophosphoryl lipid A as a safe and effective vaccine adjuvant, Cell. Mol. Life Sci., 2008, 65, 3231–3240 CrossRef CAS PubMed.
- A. Hernandez, et al., The role of MyD88- and TRIF-dependent signaling in monophosphoryl lipid A-induced expansion and recruitment of innate immunocytes, J. Leukocyte Biol., 2016, 100, 1311–1322 CrossRef CAS PubMed.
- W. Cui, et al., TLR4 ligands lipopolysaccharide and monophosphoryl lipid a differentially regulate effector and memory CD8+ T Cell differentiation, J. Immunol., 2014, 192, 4221–4232 CrossRef CAS PubMed.
- P. Pradhan, et al., TRAF6-IRF5 kinetics, TRIF, and biophysical factors drive synergistic innate responses to particle-mediated MPLA-CpG co-presentation, Sci. Adv., 2021, 7, eabd4235 CrossRef CAS PubMed.
- P. U. Atukorale, et al., Nanoparticle encapsulation of synergistic immune agonists enables systemic co-delivery to tumor sites and interferon β-driven anti-tumor immunity, Cancer Res., 2019, 79, 5394–5406 CrossRef CAS PubMed.
- M. E. Lorkowski, et al., Immunostimulatory nanoparticle incorporating two immune agonists for the treatment of pancreatic tumors, J. Controlled Release, 2021, 330, 1095–1105 CrossRef CAS PubMed.
- L. Zitvogel, L. Galluzzi, O. Kepp, M. J. Smyth and G. Kroemer, Type I interferons in anticancer immunity, Nat. Rev. Immunol., 2015, 15, 405–414 CrossRef CAS PubMed.
- P. U. Atukorale, G. Covarrubias, L. Bauer and E. Karathanasis, Vascular targeting of nanoparticles for molecular imaging of diseased endothelium, Adv. Drug Delivery Rev., 2017, 113, 141–156 CrossRef CAS PubMed.
- S. P. Arlauckas, et al., In vivo imaging reveals a tumor-associated macrophage–mediated resistance pathway in anti–PD-1 therapy, Sci. Transl. Med., 2017, 9, eaal3604 CrossRef PubMed.
- F. Balkwill, Cancer and the chemokine network, Nat. Rev. Cancer, 2004, 4, 540–550 CrossRef CAS PubMed.
- M. Fankhauser, et al., Tumor lymphangiogenesis promotes T cell infiltration and potentiates immunotherapy in melanoma, Sci. Transl. Med., 2017, 9, eaal4712 CrossRef PubMed.
- A. Ito, et al., Cancer Neoantigens: A Promising Source of Immunogens for Cancer Immunotherapy, J. Clin. Cell. Immunol., 2015, 1–7, DOI:10.4172/2155-9899.1000322.
- T. N. Schumacher and R. D. Schreiber, Neoantigens in cancer immunotherapy, Science, 2015, 348, 69–74 CrossRef CAS PubMed.
- Z. Hu, et al., Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma, Nat. Med., 2021, 27, 515–525 CrossRef CAS PubMed.
- Z. Hu, P. A. Ott and C. J. Wu, Towards personalized, tumour-specific, therapeutic vaccines for cancer, Nat. Rev. Immunol., 2018, 18, 168–182 CrossRef CAS PubMed.
- M. C. Hanson, et al., Liposomal vaccines incorporating molecular adjuvants and intrastructural T-cell help promote the immunogenicity of HIV membrane-proximal external region peptides, Vaccine, 2015, 33, 861–868 CrossRef CAS PubMed.
- D. J. Irvine, M. C. Hanson, K. Rakhra and T. Tokatlian, Synthetic Nanoparticles for Vaccines and Immunotherapy, Chem. Rev., 2015, 115, 11109–11146 CrossRef CAS PubMed.
- R. S. Riley, C. H. June, R. Langer and M. J. Mitchell, Delivery technologies for cancer immunotherapy, Nat. Rev. Drug Discovery, 2019, 18, 175–196 CrossRef CAS PubMed.
- W. Nie, et al., Non-viral vector mediated CKb11 with folic acid modification regulates macrophage polarization and DC maturation to elicit immune response against cancer, Bioact. Mater., 2021, 6, 3678–3691 CrossRef CAS PubMed.
- E. Ahn, et al., Role of PD-1 during effector CD8 T cell differentiation, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4749–4754 CrossRef CAS PubMed.
Footnotes |
| † These authors contributed equally. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr06577g |
| This journal is © The Royal Society of Chemistry 2022 |