
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of polymer carriers on the occurrence and development of autophagy in drug delivery
Changduo
Wang
a,
Yang
Li
b,
Yu
Tian
a,
Wenyuan
Ma
a and
Yong
Sun
 *a
*a
aDepartment of Pharmaceutics, School of Pharmacy, Qingdao University, Qingdao 266000, China. E-mail: sunyong@qdu.edu.cn; Tel: +86-532-82991203
bDepartment of Pharmacy, Qingdao Municipal Hospital, Qingdao 266000, China
First published on 20th July 2022
Abstract
Autophagy is an evolutionarily conserved catabolic process that can degrade cytoplasmic materials and recycle energy to maintain metabolite homeostasis in cells. Autophagy is closely related to various physiological or pathological processes. Macromolecular materials are widely used in drug delivery systems and disease treatments due to their intrinsic effects, such as altered pharmacokinetics and biodistribution. Interaction of autophagic flux or the signal pathway with macromolecules may cause autophagy inhibition or autophagy cell death. This review covers autophagy regulation pathways and macromolecular materials (including functional micelles, biodegradable and pH-sensitive polymers, biomacromolecules, dendrimers, coordination polymers, and hybrid nanoparticles) mediated autophagy modulation.
Introduction
Autophagy is a highly conserved process activated in limited growth conditions (such as hypoxia, nutrient starvation, and chemo-radiotherapy), which can recycle energy to maintain homeostasis in cells.1 Autophagy plays a vital role in various physiological processes. Metal and inorganic nanomaterials have been reported to induce autophagy for cancer treatment. For example, nano-gold (Au NPs) alkalizes and impairs lysosomes to block the fusion between autophagosome and lysosome.2 Quantum dots induce autophagic cell death via producing excessive reactive oxygen species (ROS).3 It has been demonstrated that oxidative stress is closely related to inducing autophagy.4 Rare-earth oxide nanocrystals (including Sm2O3, Eu2O3, Gd2O3, and Tb2O3) have autophagy-inducing activity, which is dose and time dependent.5 Compared with inorganic nanomaterials, organic polymers have the advantage of higher modifiability, functional diversity, and better biodegradability and have been widely applied in drug delivery systems. Polymeric drugs are employed in various diseases due to their intrinsic therapeutic effects, such as altered pharmacokinetics and biodistribution.6 Polymeric drugs could covalently or non-covalently interact with small molecule drugs to improve the therapeutic effects, as a result of polymers enhancing cell internalization or regulating signal processes.7 However, the studies on organic polymers inducing autophagy are fewer than those on metal and inorganic nanomaterials. Thus, the research on organic macromolecules and autophagy can help us solve many problems clinically, which shows enormous potential in drug delivery and disease treatment.The mechanistic pathway of autophagic flux
Autophagy is an evolutionarily conserved catabolic process, which was originally proposed by Deer R L and Deduve C in 1967.8 Autophagy can degrade cytoplasmic materials (such as damaged organelles, obsolete proteins, and invading pathogens) and recycle energy to maintain metabolite homeostasis in cells.9 According to the pathways of cytoplasmic materials into lysosomes, autophagy can be classified as macroautophagy, microautophagy, and chaperone-mediated autophagy.10 The main mechanism in macroautophagy is transferring and degrading damaged proteins and organelles in eukaryocyte.11 Macroautophagy is initiated by the formation of a phagophore, and phagophores can package cytoplasmic materials to form autophagosomes, which can degrade substances through fusion with lysosomes.12 The process of macroautophagy is generally divided into three parts: phagophore assembly, autophagosome formation and maturation, and autophagolysosome degradation.13 The term “autophagy” generally refers to macroautophagy.The initiation of autophagy
The activation of autophagy is closely related to the mammalian target of rapamycin (mTOR).14 mTOR is a nuclear serine/threonine protein kinase implicated in many signaling pathways in animals, which could form two different complexes with several proteins, called mTORC1 and mTORC2, respectively.15 mTOR is positively regulated by growth factors or amino acids, while it is negatively regulated under the conditions of low energy, hypoxia, and DNA damage.16 Many studies have shown that mTORC1 is an important modulator of autophagy, which takes part in all autophagy processes. mTORC1 is composed of mTOR, Raptor (regulatory-associated protein of mTOR), PRAS40 (a substrate of protein kinase Akt), mLST8 (mammalian lethal with SEC13 protein 8), and DEPTOR (DEP-domain containing mTOR-interacting protein).17,18 The mTORC1 can inhibit ULK1/Atg1 kinase complex via phosphorylation of ATG13, which controls the autophagy initiation.19 In addition, mTOR could be inhibited by AMP-activated protein kinase (AMPK). Meanwhile, AMPK also indirectly activates ULK1 to promote autophagy.20The phagophore assembly
Autophagy is initiated by the cytoplasmic emergence of cup-shaped structures called phagophores and activated in limited growth conditions such as hypoxia, nutrient starvation, and chemo-radiotherapy.21 Beclin 1 (BCN1) is a tumor suppressor gene and is the first gene that proved to be an autophagy modulator.22 The Class III phosphatidylinositol-3-kinase (PI3KC3) complex is a multiprotein assembly composed of Beclin1, lipid kinase Vps34, and serine/threonine kinase Vps15.23 PI3KC3, as a phosphatidylinositol-3-phosphates (PI3Ps) producer, is essential for the autophagy process that involves PI3P-enriched membrane vesicles.24 The positive regulators Atg14L and UV irradiation resistance-associated gene (UVRAG) can interact with Beclin1 in a mutually exclusive manner to form PI3KC3/Atg14L and PI3KC3/UVRAG complexes, which can enhance lipid kinase activity of PI3KC3.25 ULK1/Atg1 can also activate PI3KC3 via the phosphorylation of Beclin 1.26 However, the autophagic activity of the PI3KC3 complex is suppressed by anti-apoptosis Bcl-2.27 Atg9 complex can translocate phospholipids (raised by Atg2) from cytoplasmic to phagophore by Atg9, thereby driving autophagosomal membrane expansion.28The autophagosome formation and maturation
The phagophore continually elongates into a closed double-membrane structure and encapsulates cytoplasmic contents such as damaged organelles. During autophagosome formation, the ATG12/Atg12-conjugation ubiquitin-like system is closely associated with autophagosomal membrane expansion. Atg12, activated by Atg7, is conjugated to Atg5 via the E2 enzyme Atg10. Then, the Atg12-Atg5 conjugate is stabilized by Atg16L proteins to form the Atg12-Atg5-Atg16L complex, which can activate the LC3 -conjugation system. LC3, microtubule-associated protein light chain 3, has been widely investigated as autophagy-induction marker in disease research. Cytosolic LC3-I is activated by the E1 enzyme ATG7 and transferred to the E2 enzyme ATG3, and then it will conjugate to the amino group of the lipid phosphatidylethanolamine (PE) (LC3-I/PE, known as LC3-II).29,30 Catalyzed by the E3 enzyme Atg12-Atg5-Atg16L complex, the phagophore membrane continues to expand and finally forms autophagosomes when the Atg12-Atg5-Atg16L complex falls from the autophagosome outer-membrane. LC3-II is located at the autophagosome membrane and interacts with p62 to mediate substrate degradation. Noteworthily, the total level of LC3 is not necessary to change for the generation of autophagic flux but the conversion of LC3-I to LC3-II is.The autophagolysosome degradation
Autophagosome is a vacuole with a double-membrane structure but does not have hydrolase for degradation. The autophagolysosomes are formed by fusion with lysosomes to degrade the contents to maintain energetic homeostasis and viability. SQSTM1/p62 is able to participate in the transduction of various signaling pathways, including autophagy flux, which mediates the degradation of ubiquitinated protein in autophagy. The accumulation of p62, generally, is closely related to autophagy inhibition. The detection of autophagic flux cannot simply be evaluated by the level of LC3 or p62, but autophagy is detected via the combination of LC3-I/II and p62 (Fig. 1).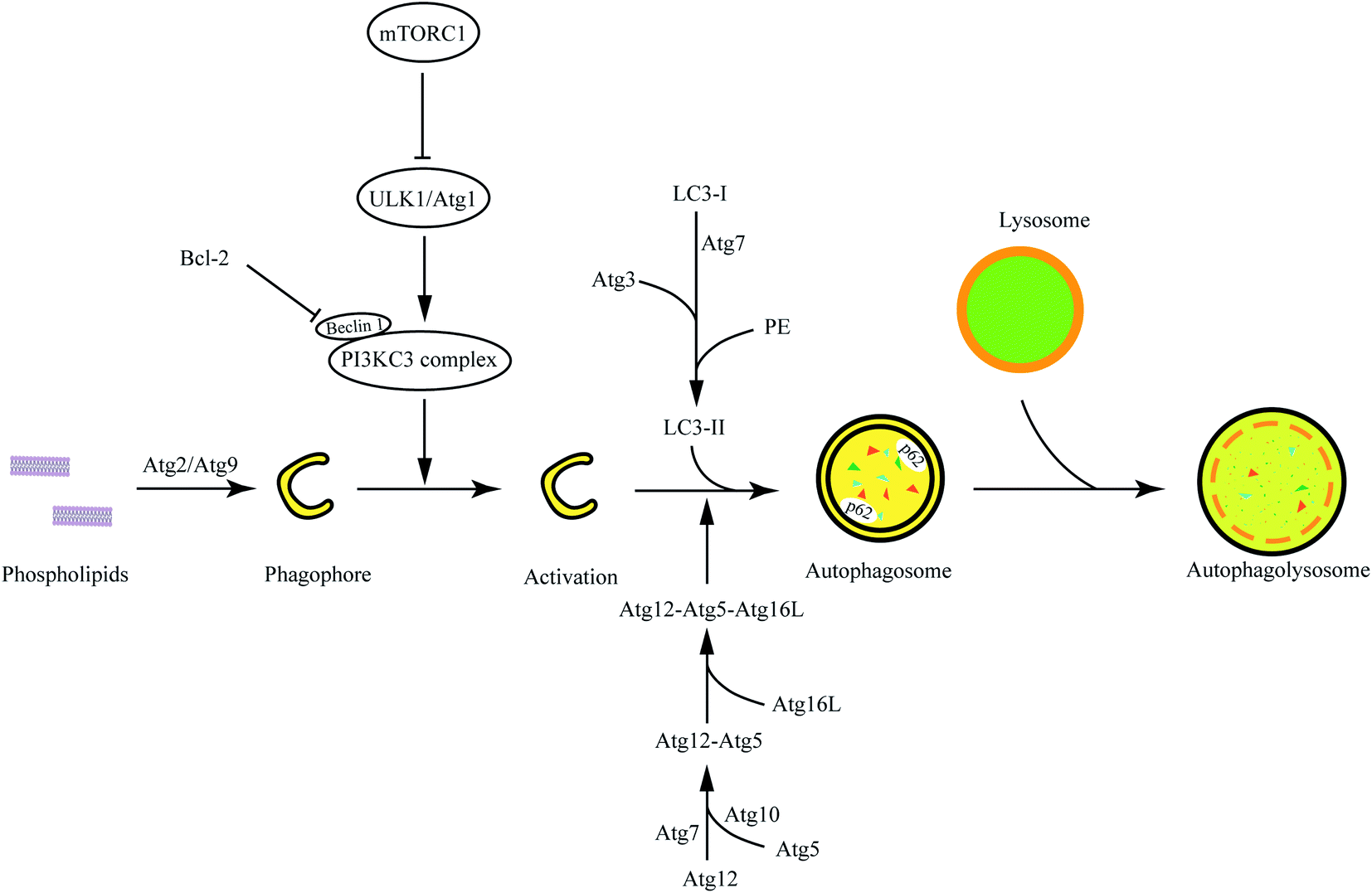 | ||
| Fig. 1 The overall process and mechanism of autophagy. The process of macroautophagy is generally divided into three parts: phagophore assembly, autophagosome formation and maturation, and autophagolysosome degradation. | ||
Besides macroautophagy, microautophagy and chaperone-mediated autophagy also are important metabolic pathways. Microautophagy refers to a process in which lysosomes directly devour cytoplasmic contents via membrane invagination. Macroautophagy and microautophagy are non-specific autophagy processes, but chaperone-mediated autophagy can specifically identify protein substrates mediated by molecular chaperone.13 Molecular chaperone (Heatshockcongnate70, Hsc70) and related factors can recognize proteins (containing KFERQ pentapeptide) and bind with transmembrane protein Lamp-2A on the membrane of lysosomes to unfold protein substrates.31 Then, the unfolded proteins can be delivered into lysosomes via metathesis polymerization. In this review, autophagy is referred to as macroautophagy. We will introduce the advances in macromolecular materials in autophagy modulation in the following text.
The regulation of autophagy
Autophagy can degrade abnormal proteins or organelles to maintain intracellular homeostasis.32–34 The pathological proteins or lipids can be “self-eaten” by inducing autophagy.35,36 There are many autophagy modulators, such as rapamycin, lithium chloride, Brefeldin A, 3-methyladenine, bafilomycin A1, and hydroxychloroquine, which have been widely used in autophagy research.35 Autophagy can be activated by inducing a stress environment; the induction of endoplasmic reticulum (ER) stress can be mediated by Brefeldin A (blocking the protein transport from endoplasmic reticulum to Golgi apparatus), Thapsigargin (microsomal Ca2+-ATPase inhibitor), and Tunicamycin (N-linked glycosylation inhibitor). Establishing hunger conditions via Earle's balanced salt solution (EBSS) is also a common method to induce autophagy. Rapamycin is a potent and specific mTOR inhibitor, which is widely applied as an autophagy activator. Besides, Xestospongin C (an inositol 1,4,5-trisphosphate receptor (IP3R) inhibitor) and Ceramide 2 (protein phosphatase 1 (PP1) activator) can also be used to induce autophagy. On the other hand, autophagy can protect cancer cells in tumor treatment.1 The inhibition of autophagy would sensitize cytotoxic drugs and improve the effectiveness of chemotherapy.37 3-Methyladenine (a Class III PI3K inhibitor), Bafilomycin A1 (a vacuolar H+-ATPase inhibitor), and chloroquine/hydroxychloroquine (alkalizing lysosome) are used to inhibit or block autophagy. Besides, autophagy-related genes can be knocked down by RNA interference, which also is a common means of the regular autophagy process.38,39 Recently, more and more small compounds with bioactivities have been discovered for regulating autophagy, but biomaterials have greater potential in autophagy and drug delivery.Macromolecular materials mediating autophagy modulation
Functional micelles
Many cationic polymers have been reported to have the ability to induce autophagy. Polyaspartimide was covalently linked with ethylene diamine to form cationic surface nanoparticles. Polyaspartimide can promote the conversion from LC3-I into LC3-II compared with the control without any primary amine, suggesting the significant role of primary amines in autophagy induction.40 The up-regulated autophagy process by polyaspartimide can clear protein aggregates from cells against neurodegenerative diseases. Polyethylenimine (PEI) is a well-accepted polycation in gene therapy, which has been found to induce autophagy.41,42 Through detecting the Rab5 (located on early endosomes) and Rab7 (located on later endosomes) on PEI-induced autophagy, it was found that PEI-induced autophagy was mainly correlated with lysosome damage at the early stage but was mainly related with mitochondrial injury at the later phase.41 Furthermore, the lactosylation of PEI can significantly decrease PEI-induced autophagy and cytotoxicity.43 The lactosylated N-alkyl polyethylenimine-coated nanoparticles could increase the protein level of LC3 and p62, suggesting that it might have activated the formation of autophagosome-like structure but blocked autophagic flux via alkalizing lysosomes.44 Hyperbranched polymers, driven by spontaneous phenylboronate linking between OEI600-PBA and HBPO, could promote the activation of autophagy caused by chemotherapeutic drugs.45 The cytotoxicity caused by gene carriers can be efficiently decreased by suppressing autophagy.In order to understand the mechanism of cationic polymers inducing autophagy, PEI and poly(L-lysine) (PLL) were studied. Both PEI and PLL can induce autophagosome formation with increasing LC3 and decreasing p62. The proton sponge effect of PEI could disrupt the enzymatic function of lysosomes in cells and protect LC3-II from degradation but the LC3-II detection in PLL-treated cells would be inhibited in the presence of protease inhibitors because PLL does not have the proton sponge effect (Fig. 2).42
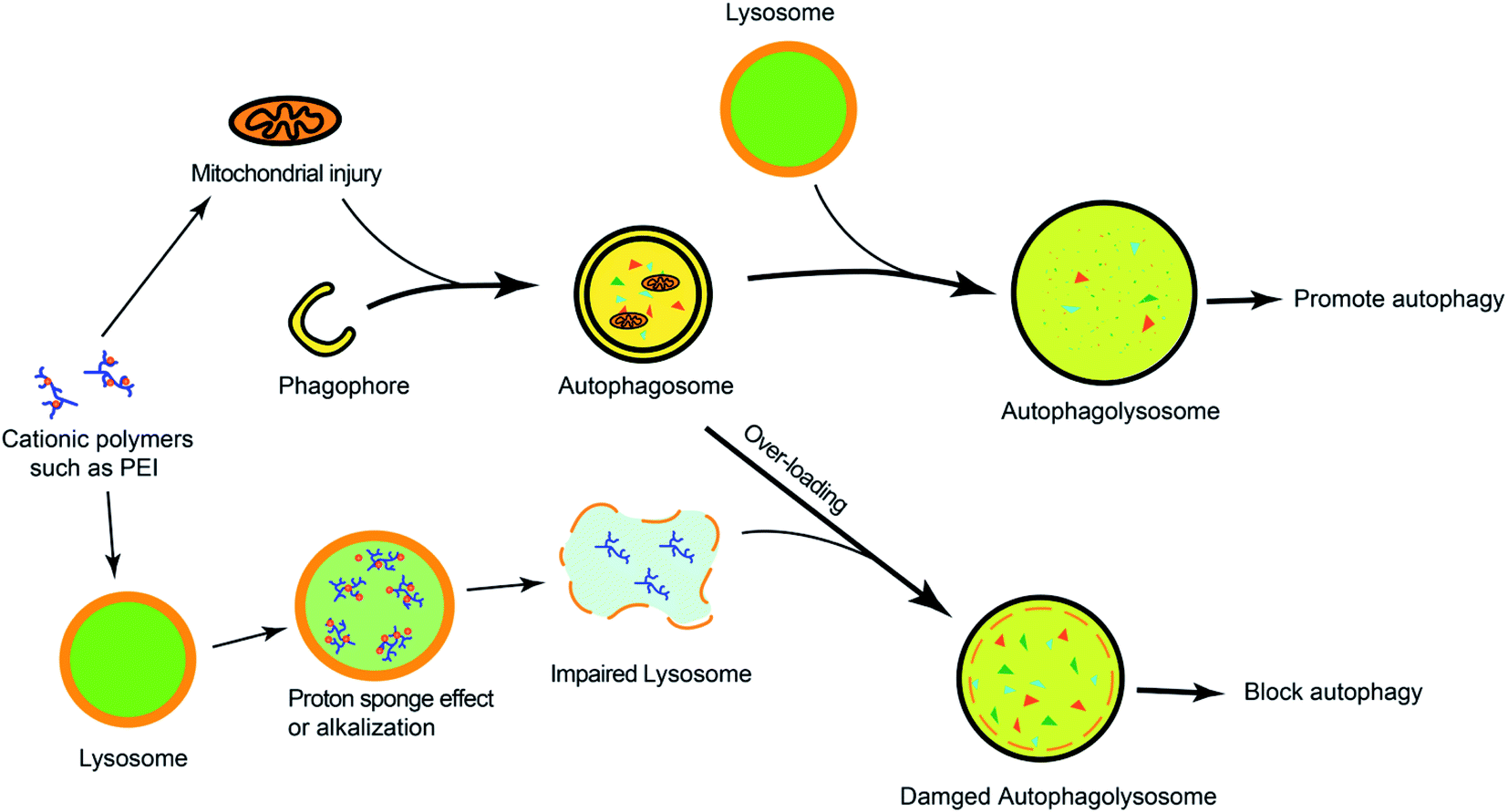 | ||
| Fig. 2 The overview of cationic polymers modulating autophagy. Cationic polymers (such as PEI) could block autophagic flux via lysosome impairment and mitochondrial injury. | ||
Recent research shows chloroquine (CQ) and hydroxychloroquine (HCQ), antimalarial drugs, can block autophagic flux via alkalifying lysosomes. Methacryloyl chloroquine (MACQ) was grafted within 2-(dimetheylamino)ethyl methacrylate (DMAEMA) to synthesise CQ-containing copolymers PDmCQn, which have the ability to inhibit autophagy. Compared to the control group (without chlorine atom in the quinolone ring), CQ-containing copolymers PDmCQn remarkably could significantly increase LC3-II and p62, which also suggests that the 7-Cl of the quinolone ring is necessary for keeping the autophagy-blockage of CQ.46 The presence of CQ component not only largely improved the transfection efficacy, but also sensitized cancer cell apoptosis via autophagy inhibition. In addition, through copolymerizing HCQ and N-(2-hydroxypropyl)methacrylamide (HPMA), the pCQ polymers showed more efficient translocation to the cytoplasm with lower cytotoxicity than HCQ. However, the incorporation of HCQ into HPMA resulted in a significant loss of autophagy–inhibition activity.6 These results suggested that the cytotoxicity of HCQ is related to autophagic cell death. HPMA has a similar structure as DMAEMA, but their CQ-containing copolymers have different effects on autophagy, which may be because DMAEMA is a positively charged polymer and easily degrades than HPMA. Thus, less cytotoxic pCQ may be a promising polymeric drug platform in anticancer therapies. Poly(methacrylic acid) (PMASH) is used to prepare multilayered redox-active microcapsules (μCs) via disulfide-crosslinking, which could induce strong autophagy and cytotoxicity. However, the conversion of the carboxyl groups of PMASH into the neutral amide of poly(hydroxypropylmetacylamide) (pHPMASH) could effectively prevent the induction of autophagy, further suggesting the ζ-potential of NPs may play an important role in autophagy induction.47
In addition, the strategy can trigger autophagy in which mitochondria-targeting molecules impair mitochondria and lead to a lower ATP concentration. The impaired mitochondria can be encapsulated and degraded by autophagosomes, but the overloading mitochondria in lysosomes would suppress the degradative capacity, which results in the dysfunction of lysosomes and the blockage of autophagic flux (Fig. 3).48
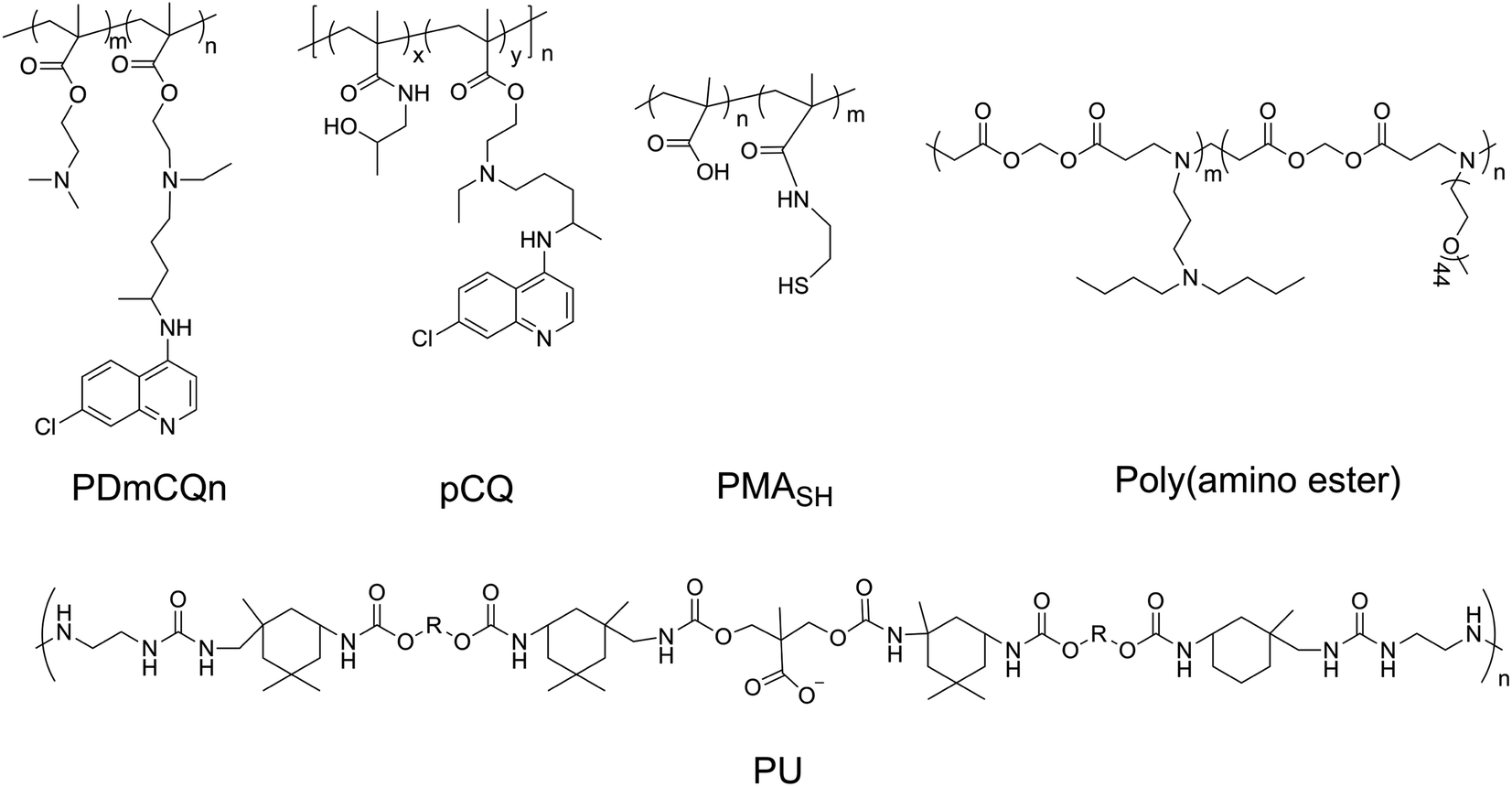 | ||
| Fig. 3 The chemical structures of PDmCQn, pCQ, PMASH, poly(amino ester), and PU. | ||
Biodegradable and pH-sensitive polymers
Poloxamer 188 (P188) is an excipient commonly used, which can protect cells and restore lysosomal membrane integrity to impair autophagy flux.49 Bioresorbable poly-D/L-lactide polymers could promote oxidative stress and induce excessive autophagy with up-regulating LC3-I/II.50 Poly(lactic-co-glycolic acid) (PLGA) is a biodegradable material that has been widely used for drug delivery.51 PLGA itself could induce weak autophagy, but the PEG and TPGS modified PLGA-based NPs revealed a stronger ability to induce autophagy via regulating ER located Class III PI3K complex,52 which may be due to the NPs enhancing internalization and mitochondria-targeting TPGS impairing mitochondria. PLGA NPs are co-delivered with autophagy inhibitors such as 3 MA and CQ, which can inhibit autophagy and improve the advantages of nanomedicines. Polyurethane (PU) has gained great attention in biomedical-related fields; the carboxyl-functionalized PU NPs can induce autophagy activation by increasing intracellular calcium and limiting the NF-kB activities of macrophages. A higher number of carboxyl groups on the PU may have a stronger ability to induce autophagy due to increased Ca2+ adsorption by the COO− groups.53 Meanwhile, PU NPs demonstrate COO− dependent immunosuppressive properties without carrying any anti-inflammatory agents. Likewise, three similar poly(alkylcyanoacrylate) (PACA) particles with different alkyl side chains, ethylbutyl (PEBCA), butyl (PBCA), and octyl (POCA), affected the unfolded protein response and antioxidant response. The ζ-potential of PACA is weakly negative (approximately −3 mV) rather than positive, but PACA nanoparticles can also induce autophagy. This research found that the highly similar NPs have different impacts on autophagy but do not induce lysosome dysfunction: PEBCA up-regulates LC3-II expression via the ISR-ATF4 stress signaling but do not affect autophagy degradation; PBCA can inhibit LC3-I conversion and autophagy degradation; POCA can block the autophagosome-lysosome fusion.54 Notably, it is found that the autophagy-stimulating effect is not only related to different side chains but also to different concentrations. A high concentration of PBCA could inhibit LC3-I conversion and block autophagy cargo flux induced by mTOR inhibition, but a low concentration would enhance autophagic flux without an increase in LC3-II (Fig. 4).55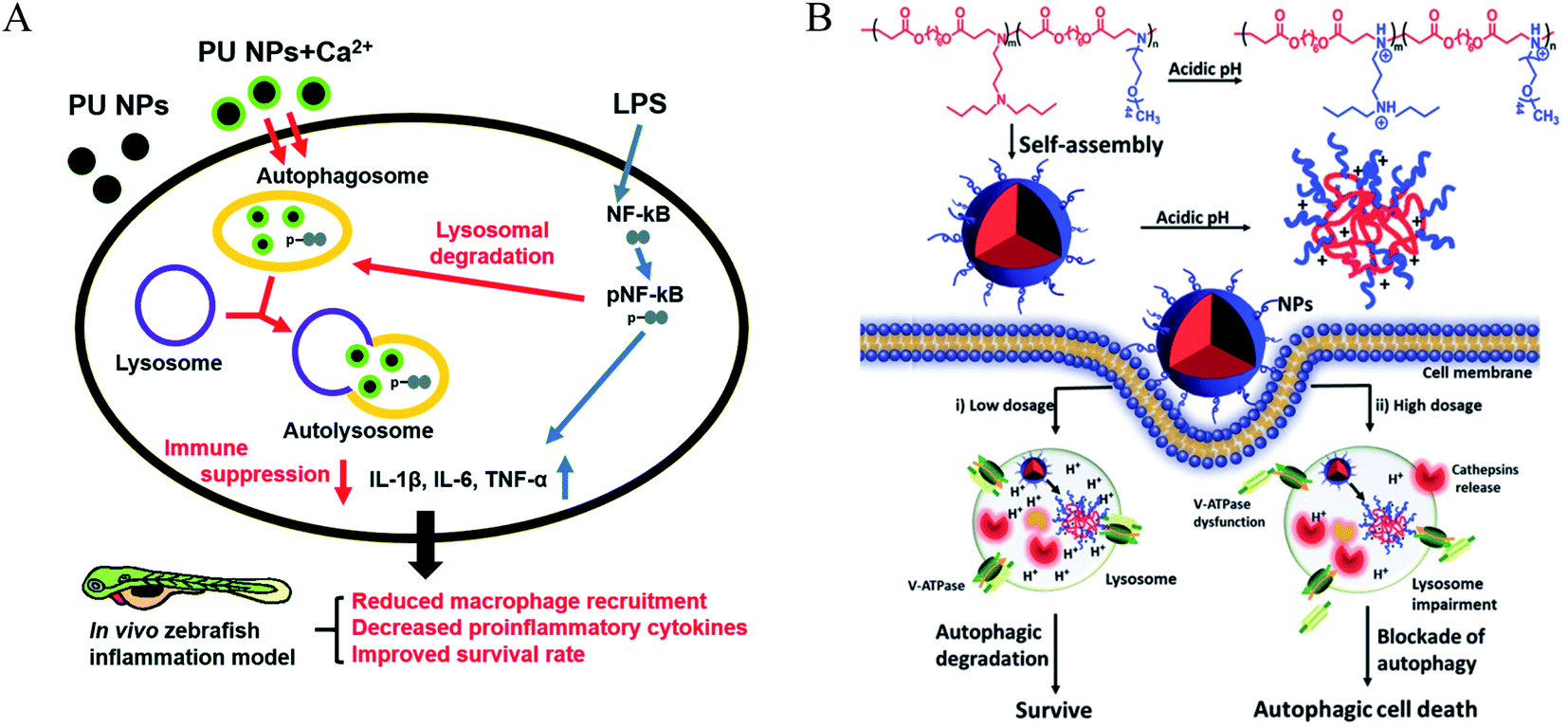 | ||
| Fig. 4 (A) Scheme of immune suppressive activities of PU NPs in macrophages. PU NPs adsorbed calcium through surface carboxyl groups and transported the calcium into macrophages. The increased intracellular calcium may enhance autophagy and reduce NF-kB pathway via autophagy-lysosomal degradation. Copyright 2015, Royal Society of Chemistry. (B) Schematic illustration displays the autophagic effects of pH-sensitive poly(β-amino ester)s. Copyright 2016, Wiley Online Library. | ||
Polydopamine (PDA), as a pH-sensitive material, could control the drug release under pH-stimulus.56 PDA-coated mesoporous silica nanoparticles could increase LC3-II and p62 levels and decrease beclin1 levels via the inhibition of the AKT-mTOR-p70S6K signaling pathway.57 The protonation of poly(β-amino ester)s, as pH-sensitive materials, could trigger the dissociation of micelles under acidic conditions. Furthermore, the poly(β-amino ester)s can impair lysosomes and suppress the degradation of autophagosomes, resulting in the blockage of autophagic flux.58 The pH-sensitive nanomaterials can induce cell death through the regulation of autophagy, which may open an avenue for cancer therapy. Noteworthily, pH sensitivity is one of the most important factors in inducing autophagy, and a low concentration of micelles induced autophagy but a high concentration would block autophagy.59 Silica nanoparticles with a pH-sensitive poly(diethylaminoethyl methacrylate) (PDEAEM) shell demonstrated a significantly higher ratio of autophagy than without the PDEAEM shell.60 This suggested that the endosomal escape caused by the pH-sensitive polymers may promote the induction of autophagy. The pH-sensitive polymers can also be used to detect the activity of lysosomes and further autophagy.61 However, the relationship between pH sensitivity and autophagy induction is still unclear.
Biomacromolecules
Ceramic is a bioactive sphingolipid that plays a role in regulating various bioactivities such as cell division, cell differentiation, apoptosis, and so on.62 Ceramic can induce autophagy through deactivating mTOR pathways and dissociating the Beclin1: Bcl-2 complex.63 PEG-ceramide is composed of hydrophobic ceramide and hydrophilic PEG-2000, which could be widely used in the preparation of lipidosome or micelles. Moreover, PEG-ceramide could increase the LC3-II/LC3-I ratio and increase p62 levels, demonstrating that it enhances the autophagic flux.64 PEG-ceramide nanomicelles can induce autophagy and degrade tau proteins, which show great promise as agents in the treatment of AD.Beclin1, encoded by the BECN1 gene, is a mammalian homolog of Atg6 and takes part in the initiation of autophagosome formation through binding to PI3K.65 It could be a successful strategy to induce autophagy in which Beclin1 or its derived peptide are delivered into cells by nano-carriers, which can promote the LC3-I conversion and p62 degradation.66 Polymer-peptide has longer blood circulation for tumor-targeted delivery and enhances therapeutic effects. Beclin1 was covalently grafted onto pH-sensitive polymers (poly(β-amino ester)), which self-assemble into nanoparticles (P-Bec1) with a PEG shell. P-Bec1 can alkalize and impair lysosomes as a result of polymer dissociation under the lysosomal environment. Compared with Beclin1, P-Bec1 can escape from lysosomes and block autophagy degradation.67 Moreover, the P-Bec1 nanoparticles efficiently induced autophagy and inhibited MCF-7 cell growth. In addition, the 14-amino-acid polycationic peptide (KLAKLAK)2 (abbr. KLAK) can disrupt anionic prokaryotic to antibacterial. Peptide KLAK was conjugated with PEG to form P-KLAK, which exhibits excellent antitumor effects by disrupting mitochondrial membranes. Meanwhile, P-KLAK can cause lysosome impairment and result in autophagosome accumulation.68 In addition, P-KLAK might induce U87 cell death through autophagy blockage (Fig. 5).
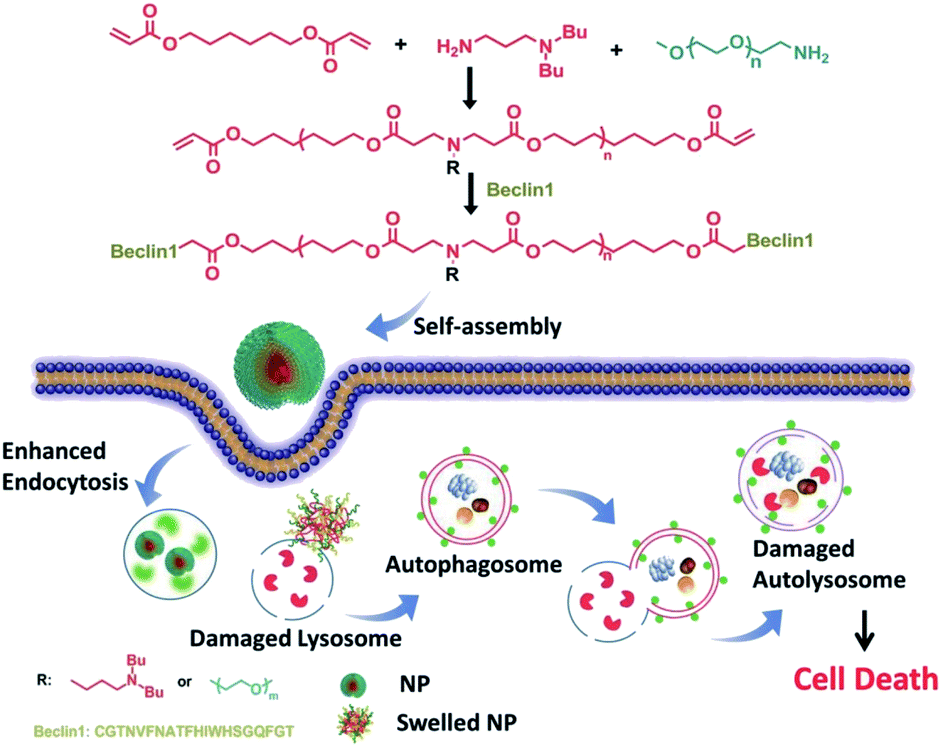 | ||
| Fig. 5 Schematic illustration of P-Bec1 nanoparticles for a highly efficient autophagy induction process. Copyright 2015, Wiley Online Library. | ||
Polysaccharides are suitable as carriers for different drug preparations due to theirs biological and chemical properties.69 Recently, polysaccharides extracted from herbal medicines have been reported as having the ability to modulate autophagy. Polysaccharides can inhibit PRV replication and cope with virus infection by relieving virus-induced autophagy.70Astragalus polysaccharide can up-regulate the gene and protein expressions of Beclin1 and LC3-II while down-regulating p62, which is used to sensitize cisplatin.71 Besides, a water-soluble polysaccharide extract from Inonotus taiwanensis could suppress LC3 expression that is dose-dependent and time-dependent, which shows anti-cancerous efficacy.72Pseudomonas aeruginosa lipopolysaccharide can lead to increased p62 expression via activating SIRT1, decreased p53 acetylation, and Bcl-2 protein expression, suggesting the blockage of autophagy degradation.73 In addition, lipopolysaccharide, the major constituent of the Gram negative bacterial outer wall, could cause deposition of autophagy flow via PARP-1 to promote pyroptosis in cardiomyocytes.74 Thus, promoting autophagy may inhibit LPS-induced pyroptosis in cardiomyocytes. Low-degree polymerization chitooligosaccharide (COS) is the major degradation product of chitosan via chemical or enzymatic hydrolysis.75 Moreover, COS can increase sensitivity to chemotherapy of cisplatin and demonstrates anti-tumor effects on osteosarcoma.
Fatty acids are substantial components of lipids and cell membranes in the form of phospholipids.76 By feeding a high-fat diet to the livers of C57BL/6 mice for up to 16 weeks, LC3-II expression of hepatocytes was markedly increased in HFD-fed mice. In SMMC-7721 and HepG2 cells, the palmitic acid (a saturated fatty acid) increases LC3-II expression in a time- and dose-dependent manner via JNK2 activation but the unsaturated fatty acid oleic acid does not.77 However, a study demonstrated that oleic acid (an unsaturated fatty acid) could induce autophagy yet have a weaker autophagy-induced effect than palmitic acid.78 Omega-3 polyunsaturated fatty acids were also proved to induce autophagy as well.79 Autophagy modulation has therapeutic benefits for obesity-induced steatosis and liver injury.
Coordination polymers
Coordination polymers (CPs) are applied in many research fields because of their unique topological structures.80 The Mn(II) coordination polymer [Mn2(L)(hfpd)(H2O)]·1.75H2O}n (L = 4′4-bis(imidazole-1-yl)-biphenyl and H4hfpd = 4,4′-(hexafluoroisopropylidene)diphthalic acid) could down-regulate the Beclin1 and Bcl-2 expression effectively in the macrophages in a dose-dependent manner.81 Besides, the Cu2+ polymer complexes of 5-(2,3-dimethyl-1-phenylpyrazol-5-one azo)-8-hydroxyquinoline (HL) ligand were prepared and investigated for their anti-microbial activity. The results of bio-TEM showed the Cu(II) polymer complex can deactivate autophagy flux by which the number of autophagosomes decreased as the Cu(II) increased.82 The different metal ions of coordination polymer may cause different effects of autophagy. For instance, the flexible benzoimidazole-type ligand 1-(2-(1H-benzoimidazol-1-yl)ethyl)-1H-benzoimidazole (beb) coordination polymers of Zn(II) and Co(II) had different influences on Bcl-2 and Beclin1, with the Co(II) polymer obviously up-regulated the autophagy level as detected by RT-PCR but the Zn(II) polymer could not.83 However, another Zn(II) coordination polymer, [Zn(LOMe)(μ1-dca)(μ1.5-dca)]n, could induce the initiation of autophagic flux and block the degradation of autophagolysosome (Fig. 6).84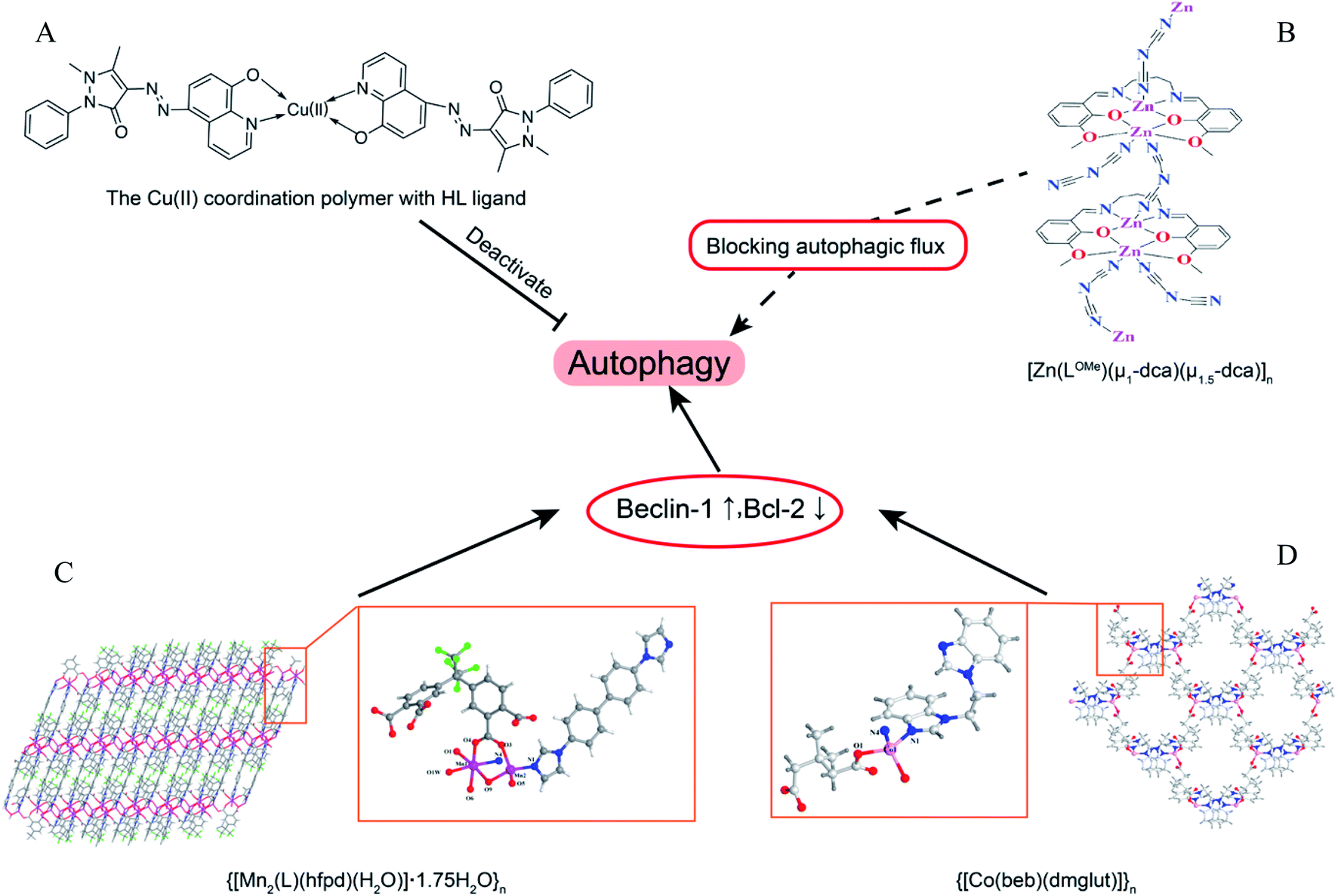 | ||
| Fig. 6 The overview of coordination polymers affecting autophagy. (A) Chemical structure of Cu(II) coordination polymer with HL ligand.82 (B) Zn(II) coordination polymer, [Zn(LOMe)(μ1-dca)(μ1.5-dca)]n, blocked autophagic flux. The asymmetry unit views and skeleton structures for (C) {[Mn2(L)(hfpd)(H2O)]·1.75H2O}n (ref. 81) and (D) {[Co(beb)(dmglut)]}n,83 which induce autophagy via regulating Beclin1 and BCL-2. | ||
Dendrimers
Dendrimer, a polymerized molecule, is widely used in drug delivery due to its highly symmetrical, monodisperse and outstanding load capacity.85 Cationic polyaminoamide (PAMAM) has many amino groups on its terminal surface, which shows a strong positive charge. PAMAM and the coated NPs could enhance the level of autophagy via inhibiting Akt/mTOR and activating Erk1/2 signaling pathways.86,87 However, anionic PAMAM G5.5 does not induce the accumulation of autophagosomes under TME compared with cationic PAMAM G3, suggesting that the surface potentials of polymers or nanoparticles play key roles in autophagy modulation.88 Besides, polystyrene (PS) dendrimers can be easily prepared to various NPs sizes and be widely applied in biomaterials. Cationic NH2-PS can induce autophagy via generating ROS and endoplasmic reticulum (ER) stress caused by misfolded protein aggregation.89 In addition, the PDA-coated zeolitic imidazolate framework compound could induce autophagosome-lysosome formation but not impair autophagy degradation through detecting autophagy by mRFP-GFP-LC3 adenovirus transfection.90Hybrid nanoparticles
Polymers can be combined with metals and inorganic nanoparticles to form hybrid nanoparticles, which extend their applications to fluorescence imaging.91,92 Fluorescent polymers (CN-PPV polymer and NIR775 dye) were used to prepare magneto-gold-fluorescent nanoparticles (MGFs), and then tumor-homing peptide (LyP-1) was conjugated on the surface of MGFs to form MGFs-LyP-1, which can induce the accumulation of LC3-II and be sensitive to chemotherapy.93 Poly(L-lactic acid)/graphene microfibers (PLLA/Gr) with the nanoporous surface can upregulate the protein expression of LC3-I/II and p62 via the induction of the production of mROS and ATP. In addition, the Gr in/on the PLLA fibers could offer additional physicochemical stimuli to induce the damage of mitochondria, and excessively damaged organelles would block the degradation of autophagosomes.94 Laminarin polysaccharides decorated selenium nanoparticles (LP-SeNPs) induced the up-regulation of LC3-II and p62 and the down-regulation of Bcl-2, suggesting the activation of early autophagy but blocking the late phase of autophagy.95 LP-SeNPs exerted their cytotoxicity in HepG2 cells by inhibiting autophagy and inducing apoptosis. In these hybrid nanoparticles, polymers are simply used to improve the effects of metal nanoparticles or microfibers as supplementary. The metal nanoparticles or microfibers may be the main inducer of autophagy rather than polymers, although the polymeric effects on autophagy were not further studied.Chiral polymer
Recently, some studies have shown that chiral polymers can selectively induce autophagy of cancer cells. Chiral poly(acryloyl-L/D-valine) (L/D-PAV) were used to coat AuNPs (PAV-AuNPs) for chiral selectivity induction of autophagy. Compared with L-PAV-AuNPs, D-PAV-AuNPs can induce autophagy more efficiently, which might be due to excellent internalization, higher ROS production, and more serious lysosome function.96 Furthermore, D-PAV-AuNPs could largely suppress tumor growth and were less toxic. D-Protein nanomimics (D-PNs) would activate efficient and continuous autophagy mimicking acute turnover of endogenous redundant proteins compared with L-PNs.97D-Cysteine-modified nanocrystals caused more extensive autophagy than L-cysteine-modified, and the D-homochirality definitively affected their interactions with biologically active substances.98 The chirality-dependent autophagy-inducing ability may be due to the enhanced accumulation in living cells.99 The L-phospholipid molecular layer on cells is easier to combine with D-polymers. Thus, D-NPs show better absorption than L-NPs (Fig. 7).100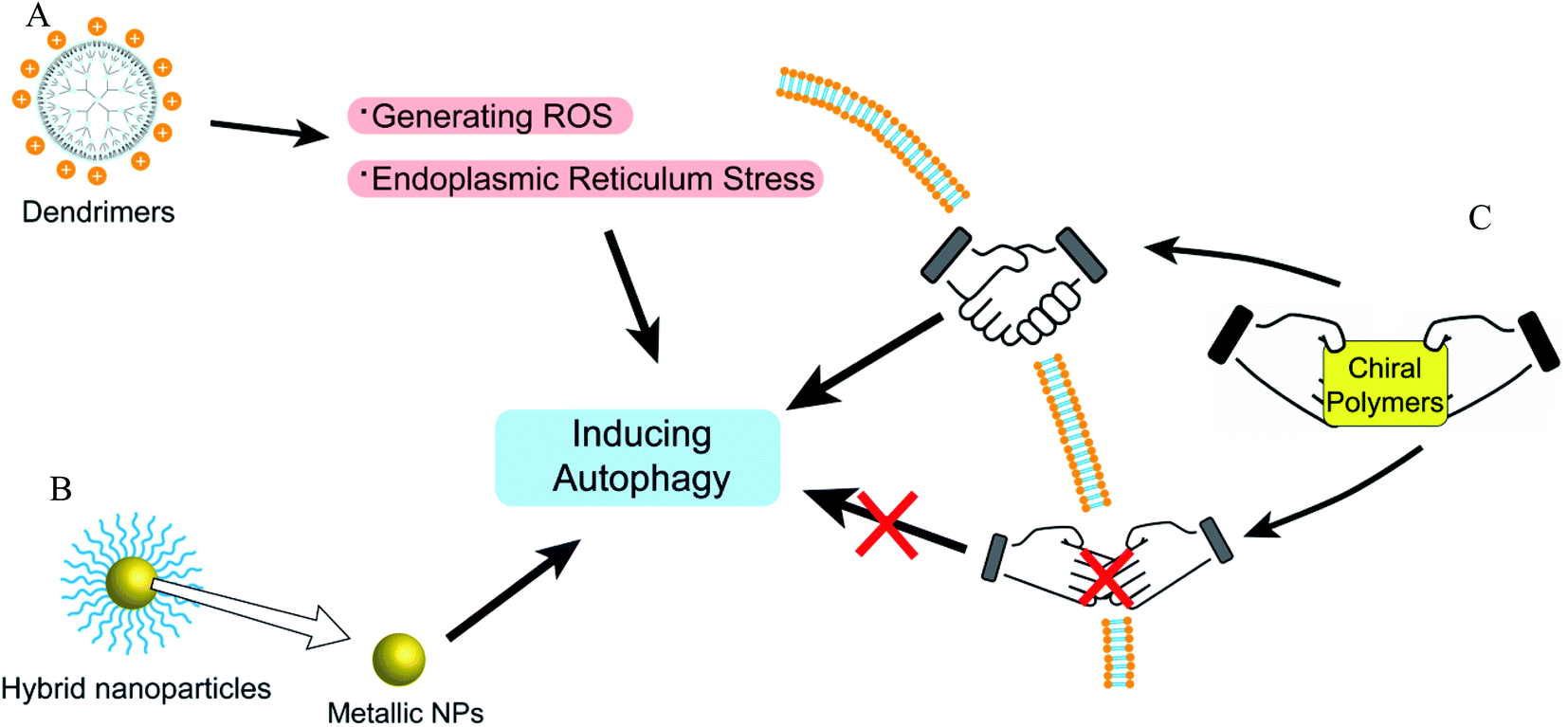 | ||
| Fig. 7 The overview of autophagy-modulating dendrimers, coordination polymers, and hybrid nanoparticles. (A) Dendrimers could induce autophagy via generating ROS and ER stress. (B) The metal nanoparticles in hybrid nanoparticles play important roles in autophagy modulation. (C) The autophagy modulation of chiral polymers is related to whether the chiral polymers are easy to be absorbed. | ||
Future prospect
Autophagy is closely related to various physiological processes, and the research on autophagy can help us solve problems in clinical treatments. Polymers have many advantages, such as high stability, easy modification, and drug loading. Through modulation by polymers, chemotherapeutic drugs can be sensitized for overcoming drug-resistant tumors.75,101 Excessive autophagy can promote apoptosis and kill MDR cancer cells.48 On the other hand, autophagy could protect cancer cells from chemotherapy or phototherapy.102 Autophagy is able to clear abnormal proteins in cells, and thus neurodegenerative diseases, such as Alzheimer's disease64 and Parkinson's disease,49 could be controlled by inducing autophagy.40,65 It is also applied in posterior capsular opacification in which abnormal organelles or proteins are degraded by autophagy.86 There have been advances in endometriosis,66 mitochondrial responses,94 anti-inflammatory platform,44,53,74 and antimicrobial activities.82Macromolecular materials are generally used as carriers to deliver drugs into target cells.103–105 The cytotoxicity caused by polymer carriers is an undesired effect for the treatment of diseases, which may be closely related to autophagic cell death. Thus, better drug carriers (with efficient delivery and less cytotoxicity) could be prepared and modified by inhibiting cell autophagy.43 In addition, autophagy is strongly associated with drug-resistance and phototherapy-resistance. The down regulation of autophagy could re-sensitize chemotherapeutic drugs and enhance the treatment effect of photothermal (or photodynamic) therapy.37,106 Compared to co-delivering autophagy inhibitors, it may be safer and more efficacious if the polymer carriers are modified with the ability to modulate autophagy. Furthermore, autophagy can degrade abnormal proteins and damaged organelles in cells. We could utilize this phenomenon to design materials that promote autophagolysosomal degradation to clear lesions such as precancerous lesions, fibrotic lesions, amyloid lesions, and atherosclerosis lesions. Meanwhile, excessive autophagy induced by macromolecular materials would promote the cell apoptosis of tumor cells (Fig. 8).
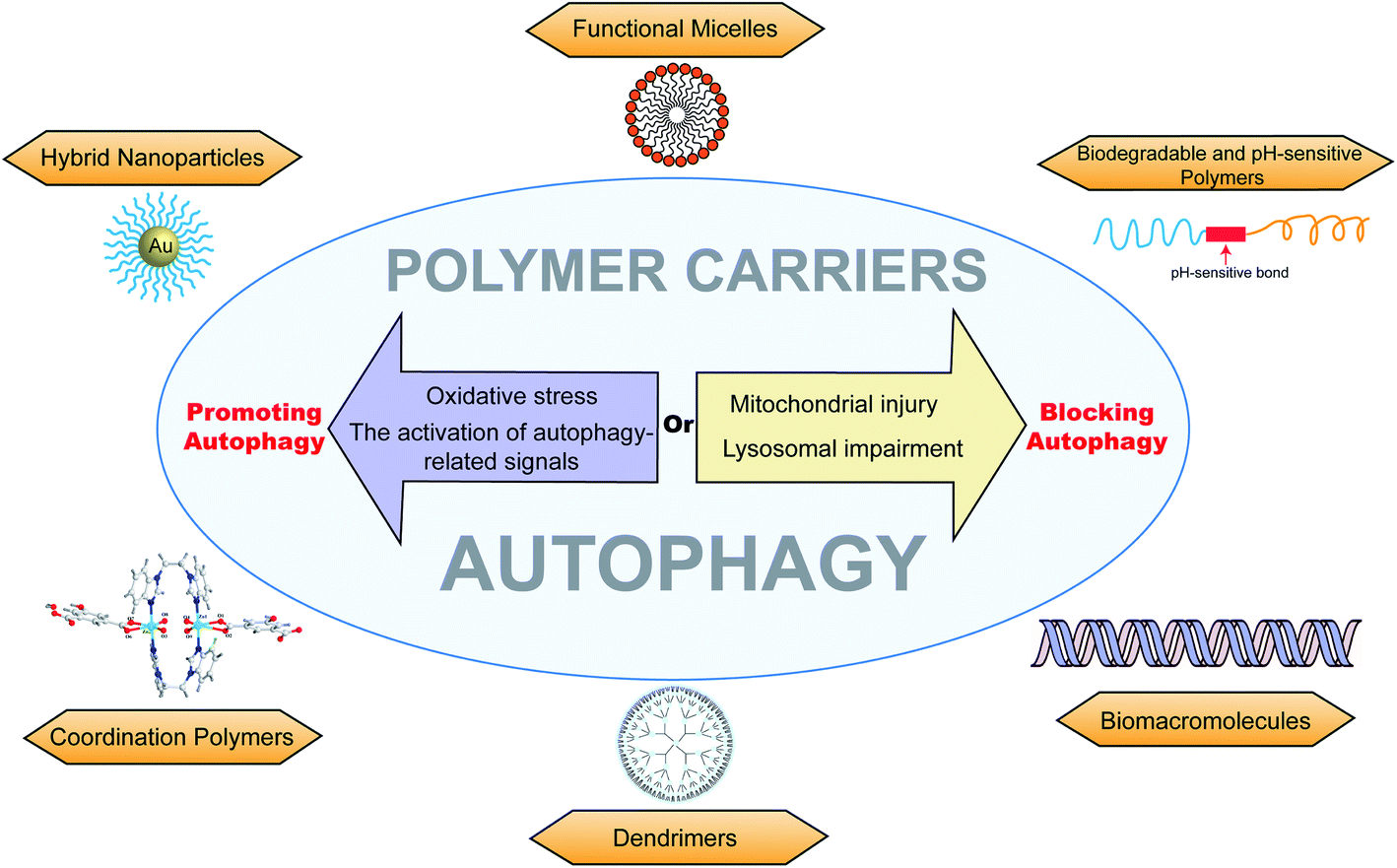 | ||
| Fig. 8 The overview of different autophagy-modulating polymer carriers, including functional micelles, biodegradable and pH-sensitive polymers, biomacromolecules, dendrimers, coordination polymers, and hybrid nanoparticles. The polymer carriers can promote autophagy via oxidative stress or the activation of autophagy-related signals and can block autophagy via lysosomal impairment or mitochondrial injury. | ||
Autophagy provides possible explanations and directions for the mechanism of biomacromolecules and the development of active substances. It may open up a new research area where activated macromolecules maintain general homeostasis and treat diseases through modulating autophagy (Table 1).
| Sample | Autophagy-related level | Mechanism or key groups | Refer. | |
|---|---|---|---|---|
| Functional micelles | ||||
| Polyaspartimide | LC3-II/LC3-I ↑ | Primary anime | 40 | |
| Polyethylenimine (PEI) | LC3-II/LC3-I ↑, p62 ↓ | Lysosome damage at the early stage Mitochondrial injury at the later phase | 41 and 42 | |
| Lactosylated N-alkyl polyethylenimine | LC3-II/LC3-I ↑, p62 ↑ | Alkalizing lysosomes | 43 | |
| Poly(L-lysine) (PLL) | LC3-II/LC3-I ↑, p62 ↓ | — | 42 | |
| PDmCQn copolymers | LC3-II/LC3-I ↑, p62 ↑ | The autophagy-blockage of CQ | 46 | |
| pCQ polymers | LC3-II/LC3-I ↑, p62 ↑ | The autophagy-blockage of CQ | 6 | |
| Poly(methacrylic acid) (PMASH) | LC3-II/LC3-I ↑ | Impaired mitochondria | 47 | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Biodegradable and pH-sensitive polymer | ||||
| Poloxamer 188 (P188) | Inhibit LC3-I conversion | Recruit the lysosome membrane | 49 | |
| Poly-D/L-lactide polymers | LC3-II/LC3-I ↑ | Promote oxidative stress | 50 | |
| TPGS modified PLGA-based NPs | LC3-II/LC3-I ↑ | Regulating ER located class III PI3K complex | 52 | |
| Polyurethane (PU) | LC3-II/LC3-I ↑ | Increasing intracellular calciumLimit the NF-kB activities of macrophages | 53 | |
| Poly(alkylcyanoacrylate) (PACA) | PEBCA | LC3-II/LC3-I ↑, p62 ↓ | ISR-ATF4 stress signaling | 54 and 55 |
| PBCA | Inhibit LC3-I conversion | Block autophagy cargo flux induced by mTOR inhibition | ||
| POCA | LC3-II/LC3-I ↑, p62 ↑ | Block autophagosome-lysosome fusion | ||
| Polydopamine (PDA) | LC3-II/LC3-I ↑, p62 ↑, Beclin 1 ↓ | Inhibition of the AKT-mTOR-p70S6K signaling pathway | 56 | |
| Poly(β-amino ester)s | LC3-II/LC3-I ↑, p62 ↑ | Impair lysosome | 58 | |
| Poly(diethylaminoethyl methacrylate) (PDEAEM) | LC3-II/LC3-I ↑ | — | 60 | |
|
||||
| Biomacromolecule | ||||
| Lipopolysaccharide | LC3-II/LC3-I ↑ | LPS-induced pyroptosis | 74 | |
| Chitooligosaccharides (COS) | LC3-II/LC3-I ↑ | Activation of p53 and deactivation of mTOR pathways | 75 | |
| P-Bec1 polymers | LC3-II/LC3-I ↑, p62 ↑ | Alkalize and impairment of lysosomes | 67 | |
| PEG-ceramide | LC3-II/LC3-I ↑, p62 ↑ | Deactivating mTOR pathways | 64 | |
| Polycationic peptide (KLAKLAK)2 | LC3-II/LC3-I ↑ | Disrupting mitochondrial membranes | 68 | |
| Astragalus polysaccharide | LC3-II/LC3-I ↑, p62 ↓, Beclin 1 ↑ | — | 71 | |
| Inonotus taiwanensis polysaccharide | LC3-II/LC3-I ↓ | — | 72 | |
| Pseudomonas aeruginosa lipopolysaccharide | p62 ↑ | Activating SIRT1 decreased p53 acetylation and Bcl-2 protein expression | 73 | |
| Palmitic acid | LC3-II/LC3-I ↑ | JNK2 activation | 77 | |
| Oleic acid | LC3-II/LC3-I ↑ | — | 78 | |
| Omega-3 polyunsaturated fatty acids | LC3-II/LC3-I ↑ | — | 79 | |
|
||||
| Dendrimers | ||||
| Polyaminoamide (PAMAM) | LC3-II/LC3-I ↑ | Inhibiting Akt/mTOR and activating Erk1/2 signaling pathways | 86 and 87 | |
| Polystyrene (PS) dendrimer | LC3-II/LC3-I ↑, p62 ↑, Beclin 1 ↑ | Generating ROS and endoplasmic reticulum (ER) stress | 89 | |
|
||||
| Coordination polymers | ||||
| Mn(II) coordination polymer [Mn2(L)(hfpd)(H2O)]·1.75H2O}n | BECN1 ↑, BCL-2 ↓ | — | 81 | |
| Cu(II) polymer of 5-(2,3-dimethyl-1-phenylpyrazol-5-one azo)-8-hydroxyquinoline (HL) ligand | Accumulating of autophagosomes | — | 82 | |
| Co(II) polymers of benzoimidazole-type ligand 1-(2-(1H-benzoimidazol-1-yl)ethyl)-1H-benzoimidazole (beb) | BECN1 gene ↑, BCL-2 gene ↓ | — | 83 | |
| [Zn (LOMe)(μ1-dca)(μ1.5-dca)]n | p-mTOR ↓, LC3-ii/LC3-I ↑, p62 ↑ | — | 84 | |
|
||||
| Hybrid nanoparticles | ||||
| CN-PPV polymer | LC3-II/LC3-I ↑ | — | 93 | |
| Poly(L-lactic acid)/graphene microfibers (PLLA/Gr) | LC3-II/LC3-I ↑, p62 ↑ | Inducing the production of mROS and ATP | 94 | |
| Laminarin polysaccharides decorated selenium nanoparticles (LP-SeNPs) | LC3-II/LC3-I ↑, p62 ↑, Bcl-2 ↓ | — | 95 | |
|
||||
| Chiral polymer | ||||
| D-PAV-AuNPs | LC3-II/LC3-I ↑ | Higher ROS production | 96 | |
| D-Protein nanomimics (D-PNs) | LC3-II/LC3-I ↑ | Higher ROS production | 97 | |
| D-Cysteine-modified nanocrystals | LC3-II/LC3-I ↑ | — | 100 | |
Conclusion
In this review, several macromolecules that could modulate autophagy are presented. These macromolecules affect the autophagic flux through different mechanisms. Macromolecular materials could induce or block autophagy via various pathways. Polymers mainly induce autophagy via mTOR-ULK1-PI3KC3, accompanied by upregulation of LC3-I/II and degradation of p62. Blocking autophagic flux will induce the accumulation of p62 and autophagolysosomes via impairing lysosomes and dysfunctioning mitochondria. However, there still are some questions that are unknown and undefined. Firstly, the relationship between lysosome escape and autophagy is not clear; research has shown that the lysosome escape may block autophagic flux via dysfunctional lysosomes. Some pH-sensitive polymers can escape lysosomes and block autophagy via damaging and alkalizing lysosomes. However, blocking autophagic flux would cause the accumulation of damaged autophagolysosomes, which seem to be contradictory to that it could escape from lysosomes to the cytoplasm. If the drug in autophagolysosomes could produce a medicinal effect by affecting the target located in the cytoplasm, then how is it done? Moreover, distinguishing between simply inducing autophagy and inducing excessive autophagy would encourage us to further develop new macromolecular materials. Regulating autophagy moderately can be used to clear lesions, but excessive autophagy induces cell apoptosis. Most materials affect autophagy in a time-dependent and dose-dependent manner; meanwhile, high concentrations and low concentrations may result in contrasting effects. Thus, it is important for materials to control the degree of autophagy.Considering the significance of autophagy in various diseases and the rapid development of drug carriers in recent years, we summarize the main mechanism of autophagy, outline the occurrence of various nano-carriers based on autophagy, and give an outlook on the future development of polymer-mediated autophagy. We believe that this review will have great guiding significance in drug delivery and disease treatment.
Author contributions
Each named author has substantially contributed to drafting and revising this manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the Qingdao Science and Technology Demonstration and Guidance Project (21-1-4-rkjk-10-nsh), and Key technology research and industrialization demonstration projects of Qingdao (22-3-3-hygg-25-hy).References
- Y. J. Li, Y. H. Lei, N. Yao, C. R. Wang, N. Hu, W. C. Ye, D. M. Zhang and Z. S. Chen, Chin. J. Cancer, 2017, 36, 52 CrossRef PubMed.
- X. Ma, Y. Wu, S. Jin, Y. Tian, X. Zhang, Y. Zhao, L. Yu and X. J. Liang, ACS Nano, 2011, 5, 8629–8639 CrossRef CAS PubMed.
- O. Zabirnyk, M. Yezhelyev and O. Seleverstov, Autophagy, 2007, 3, 278–281 CrossRef CAS PubMed.
- M. I. Khan, A. Mohammad, G. Patil, S. A. Naqvi, L. K. Chauhan and I. Ahmad, Biomaterials, 2012, 33, 1477–1488 CrossRef CAS PubMed.
- L. Yu, Y. Lu, N. Man, S. H. Yu and L. P. Wen, Small, 2009, 5, 2784–2787 CrossRef CAS PubMed.
- F. Yu, J. Li, Y. Xie, R. L. Sleightholm and D. Oupický, J. Controlled Release, 2016, 244, 347–356 CrossRef CAS PubMed.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E. W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- R. L. Deter and C. De Duve, J. Cell Biol., 1967, 33, 437–449 CrossRef CAS PubMed.
- H. Maes, N. Rubio, A. D. Garg and P. Agostinis, Trends Mol. Med., 2013, 19, 428–446 CrossRef CAS PubMed.
- W. W. Li, J. Li and J. K. Bao, Cell. Mol. Life Sci., 2012, 69, 1125–1136 CrossRef CAS PubMed.
- Y. Feng, D. He, Z. Yao and D. J. Klionsky, Cell Res., 2014, 24, 24–41 CrossRef CAS PubMed.
- X. Wen and D. J. Klionsky, J. Mol. Biol., 2016, 428, 1681–1699 CrossRef CAS PubMed.
- R. Sahu, S. Kaushik, C. C. Clement, E. S. Cannizzo, B. Scharf, A. Follenzi, I. Potolicchio, E. Nieves, A. M. Cuervo and L. Santambrogio, Dev. Cell, 2011, 20, 131–139 CrossRef CAS PubMed.
- C. Porta, C. Paglino and A. Mosca, Front. Oncol., 2014, 4, 64 Search PubMed.
- W. Fu and M. N. Hall, Genes, 2020, 11 Search PubMed.
- R. A. Saxton and D. M. Sabatini, Cell, 2017, 169, 361–371 CrossRef CAS PubMed.
- Y. Hu, H. Su, C. Liu, Z. Wang, L. Huang, Q. Wang, S. Liu, S. Chen, J. Zhou, P. Li, Z. Chen, H. Liu and G. Qing, Oncogene, 2017, 36, 1038–1047 CrossRef CAS PubMed.
- C. C. Dibble and L. C. Cantley, Trends Cell Biol., 2015, 25, 545–555 CrossRef CAS PubMed.
- C. Puente, R. C. Hendrickson and X. Jiang, J. Biol. Chem., 2016, 291, 6026–6035 CrossRef CAS PubMed.
- D. G. Hardie, F. A. Ross and S. A. Hawley, Nat. Rev. Mol. Cell Biol., 2012, 13, 251–262 CrossRef CAS PubMed.
- B. Levine and G. Kroemer, Cell, 2019, 176, 11–42 CrossRef CAS PubMed.
- A. Salminen, K. Kaarniranta and A. Kauppinen, Ageing Res. Rev., 2013, 12, 520–534 CrossRef CAS PubMed.
- Q. Yang, X. Qiu, X. Zhang, Y. Yu, N. Li, X. Wei, G. Feng, Y. Li, Y. Zhao and R. Wang, J. Med. Chem., 2021, 64, 13475–13486 CrossRef CAS PubMed.
- C. He and B. Levine, Curr. Opin. Cell Biol., 2010, 22, 140–149 CrossRef CAS PubMed.
- R. C. Russell, Y. Tian, H. Yuan, H. W. Park, Y. Y. Chang, J. Kim, H. Kim, T. P. Neufeld, A. Dillin and K. L. Guan, Nat. Cell Biol., 2013, 15, 741–750 CrossRef CAS PubMed.
- D. F. Egan, M. G. Chun, M. Vamos, H. Zou, J. Rong, C. J. Miller, H. J. Lou, D. Raveendra-Panickar, C. C. Yang, D. J. Sheffler, P. Teriete, J. M. Asara, B. E. Turk, N. D. Cosford and R. J. Shaw, Mol. Cell, 2015, 59, 285–297 CrossRef CAS PubMed.
- C. Liang, P. Feng, B. Ku, I. Dotan, D. Canaani, B. H. Oh and J. U. Jung, Nat. Cell Biol., 2006, 8, 688–699 CrossRef CAS PubMed.
- K. Matoba, T. Kotani, A. Tsutsumi, T. Tsuji, T. Mori, D. Noshiro, Y. Sugita, N. Nomura, S. Iwata, Y. Ohsumi, T. Fujimoto, H. Nakatogawa, M. Kikkawa and N. N. Noda, Nat. Struct. Mol. Biol., 2020, 27, 1185–1193 CrossRef CAS PubMed.
- N. Mizushima, T. Yoshimori and B. Levine, Cell, 2010, 140, 313–326 CrossRef CAS PubMed.
- E. Karanasios, S. A. Walker, H. Okkenhaug, M. Manifava, E. Hummel, H. Zimmermann, Q. Ahmed, M. C. Domart, L. Collinson and N. T. Ktistakis, Nat. Commun., 2016, 7, 12420 CrossRef CAS PubMed.
- A. E. Majeski and J. F. Dice, Int. J. Biochem. Cell Biol., 2004, 36, 2435–2444 CrossRef CAS PubMed.
- A. V. Onorati, M. Dyczynski, R. Ojha and R. K. Amaravadi, Cancer, 2018, 124, 3307–3318 CrossRef PubMed.
- Q. Li, Y. Liu and M. Sun, Cell. Mol. Neurobiol., 2017, 37, 377–388 CrossRef CAS PubMed.
- R. Singh, S. Kaushik, Y. Wang, Y. Xiang, I. Novak, M. Komatsu, K. Tanaka, A. M. Cuervo and M. J. Czaja, Nature, 2009, 458, 1131–1135 CrossRef CAS PubMed.
- T. Hamano, S. Enomoto, N. Shirafuji, M. Ikawa, O. Yamamura, S. H. Yen and Y. Nakamoto, Int. J. Mol. Sci., 2021, 22 Search PubMed.
- M. B. Khawar, H. Gao and W. Li, Adv. Exp. Med. Biol., 2019, 1206, 359–374 CrossRef CAS PubMed.
- S. Ruan, R. Xie, L. Qin, M. Yu, W. Xiao, C. Hu, W. Yu, Z. Qian, L. Ouyang, Q. He and H. Gao, Nano Lett., 2019, 19, 8318–8332 CrossRef CAS PubMed.
- X. Zhang, L. L. Wang, B. Wang, H. L. Liu, J. Zhang, Y. H. Li and L. H. Wang, Am. J. Transl. Res., 2020, 12, 2052–2061 CAS.
- B. Mathew, M. Chennakesavalu, M. Sharma, L. A. Torres, C. R. Stelman, S. Tran, R. Patel, N. Burg, M. Salkovski, K. Kadzielawa, F. Seiler, L. N. Aldrich and S. Roth, Autophagy, 2021, 17, 1479–1499 CrossRef CAS PubMed.
- K. Debnath, N. R. Jana and N. R. Jana, ACS Biomater. Sci. Eng., 2019, 5, 390–401 CrossRef CAS PubMed.
- X. Gao, L. Yao, Q. Song, L. Zhu, Z. Xia, H. Xia, X. Jiang, J. Chen and H. Chen, Biomaterials, 2011, 32, 8613–8625 CrossRef CAS PubMed.
- C. W. Lin, M. S. Jan, J. H. Kuo, L. J. Hsu and Y. S. Lin, Eur. J. Pharm. Sci., 2012, 47, 865–874 CrossRef CAS PubMed.
- J. Du, W. Zhu, L. Yang, C. Wu, B. Lin, J. Wu, R. Jin, T. Shen and H. Ai, Regener. Biomater., 2016, 3, 223–229 CrossRef CAS PubMed.
- T. Shen, W. Zhu, L. Yang, L. Liu, R. Jin, J. Duan, J. M. Anderson and H. Ai, Regener. Biomater., 2018, 5, 141–149 CrossRef CAS PubMed.
- H. Z. Jia, W. Zhang, J. Y. Zhu, B. Yang, S. Chen, G. Chen, Y. F. Zhao, J. Feng and X. Z. Zhang, J. Controlled Release, 2015, 216, 9–17 CrossRef CAS PubMed.
- J. F. Wang, X. F. Zhou, H. F. Wang, Q. Xiao, K. F. Ding, X. Dong, S. F. Xu, B. Shen, J. H. Sun, Z. X. Zhou, J. B. Tang, X. R. Liu and Y. Q. Shen, Biomater, 2020, 255 Search PubMed.
- G. L. Beretta, M. Folini, F. Cavalieri, Y. Yan, E. Fresch, S. Kaliappan, C. Hasenöhrl, J. J. Richardson, S. Tinelli, A. Fery, F. Caruso and N. Zaffaroni, Nanoscale, 2015, 7, 6261–6270 RSC.
- Y. X. Zhu, H. R. Jia, G. Gao, G. Y. Pan, Y. W. Jiang, P. Li, N. Zhou, C. Li, C. She, N. W. Ulrich, Z. Chen and F. G. Wu, Biomaterials, 2020, 232, 119668 CrossRef CAS PubMed.
- H. Dong, Y. Qin, Y. Huang, D. Ji and F. Wu, Neurochem. Int., 2019, 126, 178–186 CrossRef CAS PubMed.
- G. H. Steven Wood and B. Tesfamariam, J. Am. Coll. Cardiol., 2015, 66, B209 CrossRef.
- D. N. Kapoor, A. Bhatia, R. Kaur, R. Sharma, G. Kaur and S. Dhawan, Ther. Delivery, 2015, 6, 41–58 CrossRef CAS PubMed.
- X. Zhang, Y. Dong, X. Zeng, X. Liang, X. Li, W. Tao, H. Chen, Y. Jiang, L. Mei and S. S. Feng, Biomaterials, 2014, 35, 1932–1943 CrossRef CAS PubMed.
- Y. J. Huang, K. C. Hung, F. Y. Hsieh and S. H. Hsu, Nanoscale, 2015, 7, 20352–20364 RSC.
- T. Sønstevold, N. Engedal, Ý. Mørch, T. G. Iversen, T. Skotland, K. Sandvig and M. L. Torgersen, J. Biomed. Nanotechnol., 2020, 16, 432–445 CrossRef PubMed.
- T. Sonstevold, N. Engedal and M. L. Torgersen, Cells, 2021, 10 Search PubMed.
- J. Lu, L. Cai, Y. Dai, Y. Liu, F. Zuo, C. Ni, M. Shi and J. Li, Chem. Rec., 2021, 21, 781–796 CrossRef CAS PubMed.
- Y. Duo, Y. Li, C. Chen, B. Liu, X. Wang, X. Zeng and H. Chen, RSC Adv., 2017, 7, 39641–39650 RSC.
- Y. X. Lin, Y. J. Gao, Y. Wang, Z. Y. Qiao, G. Fan, S. L. Qiao, R. X. Zhang, L. Wang and H. Wang, Mol. Pharmaceutics, 2015, 12, 2869–2878 CrossRef CAS PubMed.
- Y. X. Lin, Y. Wang, S. L. Qiao, H. W. An, R. X. Zhang, Z. Y. Qiao, R. P. Rajapaksha, L. Wang and H. Wang, Small, 2016, 12, 2921–2931 CrossRef CAS PubMed.
- A. Parodi, M. Evangelopoulos, N. Arrighetti, A. Cevenini, M. Livingston, S. Z. Khaled, B. S. Brown, I. K. Yazdi, F. Paradiso, J. N. Campa-Carranza, A. De Vita, F. Taraballi and E. Tasciotti, Small, 2020, 16, e1907693 CrossRef PubMed.
- T. Zhang, F. Huo, W. Zhang, J. Chao and C. J. S. Yin, Chemical, 2021, 345, 130393 CAS.
- M. Dziadek, E. Stodolak-Zych and K. Cholewa-Kowalska, Mater. Sci. Eng., C, 2017, 71, 1175–1191 CrossRef CAS PubMed.
- S. Pattingre, C. Bauvy, T. Levade, B. Levine and P. Codogno, Autophagy, 2009, 5, 558–560 CrossRef CAS PubMed.
- J. Gao, X. Chen, T. Ma, B. He, P. Li, Y. Zhao, Y. Ma, J. Zhuang and Y. Yin, Int. J. Nanomed., 2020, 15, 6779–6789 CrossRef CAS PubMed.
- A. Ashkenazi, C. F. Bento, T. Ricketts, M. Vicinanza, F. Siddiqi, M. Pavel, F. Squitieri, M. C. Hardenberg, S. Imarisio, F. M. Menzies and D. C. Rubinsztein, Nature, 2017, 545, 108–111 CrossRef CAS PubMed.
- L. Ren, Y. Gao and Y. Cheng, Bioact. Mater., 2022, 9, 44–53 CrossRef CAS PubMed.
- Y. Wang, Y. X. Lin, Z. Y. Qiao, H. W. An, S. L. Qiao, L. Wang, R. P. Rajapaksha and H. Wang, Adv. Mater., 2015, 27, 2627–2634 CrossRef CAS PubMed.
- Z. Y. Qiao, W. J. Lai, Y. X. Lin, D. Li, X. H. Nan, Y. Wang, H. Wang and Q. J. Fang, Bioconjugate Chem., 2017, 28, 1709–1721 CrossRef CAS PubMed.
- X. Y. Wang, D. D. Zhang, J. Y. Yin, S. P. Nie and M. Y. Xie, Crit. Rev. Food Sci. Nutr., 2019, 59, S96–s115 CrossRef CAS PubMed.
- Y. Xing, L. Wang, G. Xu, S. Guo, M. Zhang, G. Cheng, Y. Liu and J. Liu, Res. Vet. Sci., 2021, 140, 18–25 CrossRef CAS PubMed.
- Q. L. Zhai, X. D. Hu, J. Xiao and D. Q. Yu, China J. Chin. Mater. Med., 2018, 43, 805–812 Search PubMed.
- T. L. Chao, T. Y. Wang, C. H. Lee, S. J. Yiin, C. T. Ho, S. H. Wu, H. L. You and C. L. Chern, Int. J. Mol. Sci., 2018, 19, 393 CrossRef PubMed.
- X. Shi, W. Wei and N. Wang, Oncol. Lett., 2018, 15, 9609–9616 Search PubMed.
- Y. F. He, J. Huang, Y. Qian, D. B. Liu and Q. F. Liu, J. Cardiovasc. Dis. Diagn., 2021, 11, 1025–1035 Search PubMed.
- Z. Pan, D. D. Cheng, X. J. Wei, S. J. Li, H. Guo and Q. C. Yang, Carbohydr. Polym., 2021, 258, 117596 CrossRef CAS PubMed.
- E. Tvrzicka, L. S. Kremmyda, B. Stankova and A. Zak, Biomed. Pap., 2011, 155, 117–130 CrossRef CAS PubMed.
- Q. Q. Tu, R. Y. Zheng, J. Li, L. Hu, Y. X. Chang, L. Li, M. H. Li, R. Y. Wang, D. D. Huang, M. C. Wu, H. P. Hu, L. Chen and H. Y. Wang, Acta Pharmacol. Sin., 2014, 35, 504–512 CrossRef CAS PubMed.
- S. Mei, H. M. Ni, S. Manley, A. Bockus, K. M. Kassel, J. P. Luyendyk, B. L. Copple and W. X. Ding, J. Pharmacol. Exp. Ther., 2011, 339, 487–498 CrossRef CAS PubMed.
- Y. Li, Y. Tang, S. Wang, J. Zhou, J. Zhou, X. Lu, X. Bai, X. Y. Wang, Z. Chen and D. Zuo, Front. Immunol., 2016, 7, 350 Search PubMed.
- Y. Liu, S. Lv, D. Liu and F. Song, Acta Biomater., 2020, 116, 16–31 CrossRef CAS PubMed.
- Y.-X. Xue and C.-X. Lou, J. Cluster Sci., 2022, 33, 1353–1360 CrossRef CAS.
- M. I. Abou-Dobara, N. F. Omar, M. A. Diab, A. Z. El-Sonbati, S. M. Morgan, O. L. Salem and A. M. Eldesoky, Mater. Sci. Eng., C, 2019, 103, 109727 CrossRef CAS PubMed.
- D. Zhao, X. Lin, M. Lv, Y. Duan and J. Chen, J. Cluster Sci., 2020, 1–9 Search PubMed.
- D. Majumdar, Y. Agrawal, R. Thomas, Z. Ullah, M. K. Santra, S. Das, T. K. Pal, K. Bankura and D. Mishra, Appl. Organomet. Chem., 2020, 34, e5269 CrossRef CAS.
- S. da Silva Santos, E. Igne Ferreira and J. Giarolla, Molecules, 2016, 21 Search PubMed.
- C. Qin, S. Liu, S. Wen, Y. Han, S. Chen, J. Qie, H. Chen and Q. Lin, J. Mater. Chem. B, 2021, 9, 793–800 RSC.
- Y. Li, S. Wang, Z. Wang, X. Qian, J. Fan, X. Zeng, Y. Sun, P. Song, M. Feng and D. Ju, Nanotechnology, 2014, 25, 365101 CrossRef PubMed.
- C. Li, H. Liu, Y. Sun, H. Wang, F. Guo, S. Rao, J. Deng, Y. Zhang, Y. Miao, C. Guo, J. Meng, X. Chen, L. Li, D. Li, H. Xu, H. Wang, B. Li and C. Jiang, J. Mol. Cell Biol., 2009, 1, 37–45 CrossRef CAS PubMed.
- H. W. Chiu, T. Xia, Y. H. Lee, C. W. Chen, J. C. Tsai and Y. J. Wang, Nanoscale, 2015, 7, 736–746 RSC.
- X. Chen, R. Tong, B. Liu, H. Liu, X. Feng, S. Ding, Q. Lei, G. Tang, J. Wu and W. Fang, Biomater. Sci., 2020, 8, 1380–1393 RSC.
- R. Xie, S. Ruan, J. Liu, L. Qin, C. Yang, F. Tong, T. Lei, M. Shevtsov, H. Gao and Y. Qin, Biomaterials, 2021, 275, 120891 CrossRef CAS PubMed.
- Y. Zhao, L. Meng, K. Zhang, Y. Sun, Y. Zhao, Z. Yang, Y. Lin, X. Liu, H. Sun, B. Yang and Q. Lin, Chin. Chem. Lett., 2021, 32, 266–270 CrossRef CAS.
- G. Wang, K. Qian and X. Mei, Nanoscale, 2018, 10, 10467–10478 RSC.
- M. Li, W. S. Deng, J. Zhang, W. P. Zheng, T. Yu and Q. H. Zhou, Adv. Mater. Interfaces, 2021, 8, 2100229 CrossRef CAS.
- D. Cui, J. Ma, T. Liang, L. Sun, L. Meng, T. Liang and Q. Li, Int. J. Biol. Macromol., 2019, 137, 829–835 CrossRef CAS PubMed.
- L. Yuan, F. Zhang, X. Qi, Y. Yang, C. Yan, J. Jiang and J. Deng, J. Nanobiotechnol., 2018, 16, 55 CrossRef PubMed.
- C. Hu, X. Xu, X. Zhang, Y. Li, Y. Li and Z. Gu, Chem. Mater., 2017, 29, 7658–7662 CrossRef CAS.
- Y. Wang and Y. Xia, J. Mater. Chem. B, 2020, 8, 7921–7930 RSC.
- M. Sun, T. Hao, X. Li, A. Qu, L. Xu, C. Hao, C. Xu and H. Kuang, Nat. Commun., 2018, 9, 4494 CrossRef PubMed.
- Z. Peng, L. Yuan, J. XuHong, H. Tian, Y. Zhang, J. Deng and X. Qi, J. Nanobiotechnol., 2021, 19, 220 CrossRef PubMed.
- P. Chen, C. Xia, S. Mei, J. Wang, Z. Shan, X. Lin and S. Fan, Biomaterials, 2016, 81, 1–13 CrossRef CAS PubMed.
- S. A. Umar and S. A. Tasduq, RSC Adv., 2020, 10, 36317–36336 RSC.
- Y. Wang, M. J. van Steenbergen, N. Beztsinna, Y. Shi, T. Lammers, C. F. van Nostrum and W. E. Hennink, J. Controlled Release, 2020, 328, 970–984 CrossRef CAS PubMed.
- H. Xia, Y. Liang, K. Chen, C. Guo, M. Wang, J. Cao, S. Han, Q. Ma, Y. Sun and B. He, Colloids Surf., B, 2021, 203, 111733 CrossRef CAS PubMed.
- W. Fan, L. Zhang, Y. Li and H. Wu, Curr. Med. Chem., 2019, 26, 2356–2376 CrossRef CAS PubMed.
- W. K. Martins, R. Belotto, M. N. Silva, D. Grasso, M. D. Suriani, T. S. Lavor, R. Itri, M. S. Baptista and T. M. Tsubone, Front. Oncol., 2020, 10, 610472 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |