
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe structures of ring-expanded NHC supported copper(I) triphenylstannyls and their phenyl transfer reactivity towards heterocumulenes†
Rex S. C.
Charman
 ,
Mary F.
Mahon
,
John P.
Lowe
and
David J.
Liptrot
*
,
Mary F.
Mahon
,
John P.
Lowe
and
David J.
Liptrot
*
Department of Chemistry, University of Bath. Claverton Down, Bath, BA2 7AY UK. E-mail: dl260@bath.ac.uk
First published on 22nd December 2021
Abstract
Three ring-expanded N-heterocyclic carbene-supported copper(I) triphenylstannyls have been synthesised by the reaction of (RE-NHC)CuOtBu with triphenylstannane (RE-NHC = 6-Mes, 6-Dipp, 7-Dipp). The compounds were characterised by NMR spectroscopy and X-ray crystallography. Reaction of (6-Mes)CuSnPh3 with di-p-tolyl carbodiimide, phenyl isocyanate and phenylisothiocyanate gives access to a copper(I) benzamidinate, benzamide and benzothiamide respectively via phenyl transfer from the triphenylstannyl anion with concomitant formation of (Ph2Sn)n. Attempts to exploit this reactivity under a catalytic regime were hindered by rapid copper(I)-catalysed dismutation of Ph3SnH to Ph4Sn, various perphenylated tin oligomers, H2 and a metallic material thought to be Sn(0). Mechanistic insight was provided by reaction monitoring via NMR spectroscopy and mass spectrometry.
Organostannanes have found widespread use in a range of organic synthetic procedures, most notably Stille-type cross-couplings where carbon–tin bonds are useful C-nucleophilic synthons.1,2 To facilitate the synthesis of such species, a plethora of stannyl transfer reagents have been developed. Of these, organostannyl anions constitute a particularly attractive class of reagents owing to the net umpolung elaboration represented by the C–X to C–Sn transformation when combined with transition-metal catalysed cross-coupling. Group 1 organostannyl anions have received extensive attention,3–8 and reports have begun to elucidate the organostannyl-transfer potential of alkaline earth–tin bonds.9–12 For example, Hill and co-workers reported the formation of a structurally unprecedented stannaamidinate formed by the insertion of diisopropyl carbodiimide into a Ca–Sn bond.10
Homodehydrocoupling of stannanes is a useful route to di- and oligotin compounds. Alongside methods applying electrochemistry13 and organic hydrogen acceptors,14 a number of d-block catalysed couplings have been described,15–18 and some key metal–stannyl intermediates have been characterised.16 Structural characterisation of organostannyl-coinage metal complexes, however, is limited to a very small number of N-heterocyclic carbene (NHC) supported copper(I) complexes19,20 alongside a single phosphine supported gold(I) system.21 Sadighi and co-workers reported the first structurally authenticated copper stannyl, synthesising (IPr)CuSnPh3 (IPr = 1,3-bis-(2,6-diisopropylphenyl)-imidazol-2-ylidene) by the deprotonation of Ph3SnH by (IPr)CuX (X = H, OtBu).19 Kleeberg and co-workers built on this work, describing the closely related (IPr)CuSnMe3 as well as [IMe4CuSnMe3]3 (IMe4 = 1,3,4,5-tetramethyl-imidazol-2-ylidene), synthesised through a Cu–O/Sn–B σ-bond metathesis between (NHC)CuOtBu and Me3SnB((NiPr)2(C2H4)).20 (IPr)CuSnR3 (R = Me, Ph) each contained a two-coordinate near linear copper(I) atom, whilst [IMe4CuSnMe3]3 formed a stannyl-bridged trimer with extremely short Cu⋯Cu distances (Scheme 1).
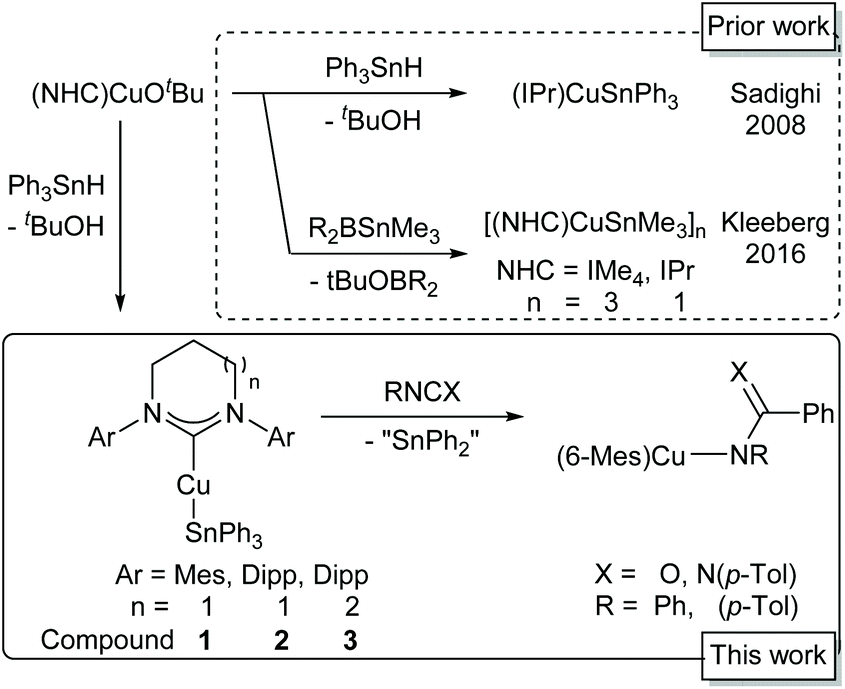 | ||
| Scheme 1 Synthesis and reactivity of (NHC)CuSnR3 complexes. | ||
These sparse examples are matched by a correspondingly limited, although intriguing, set of reactivity studies. Sadighi and co-workers showed that (IPr)CuSnPh3 acts not as a stannyl transfer agent, but as a source of the phenyl anion alongside diphenylstannylene, generating benzoates on reaction with CO2 and benzene when reacted with proton sources.19 These authors proposed two possible mechanisms for this reactivity, postulating that either concerted phenyl transfer from the intact (IPr)CuSnPh3 was operant, or that (IPr)CuSnPh3 exists in equilibrium with (IPr)CuPh and SnPh2.
We and others have recently reported on the effects of ring-expanded NHCs on their copper(I) complexes.22–27 Notably, RE-NHC copper(I) complexes often show reduced molecularity and/or enhanced stability with respect to their 5-membered cousins. For example, we recently reported the synthesis and structures of a number of NHC-copper(I) phosphides including [(NHC)CuPPh2]2 (NHC = SIMes, 6-Mes) which were dimeric in the solid state but where the 6-Mes supported system was proposed to monomerise in solution.27 We also recently described the formation of (6-Dipp)CuBpin which showed profoundly greater stability than (IPr)CuBpin.28
In order to explore the effect of RE-NHC ligands on copper(I) systems more thoroughly, and with the hope of gaining additional insight into the reactivity of copper(I)-triphenylstannyl systems, we set out to synthesise a range of systems of the form (RE-NHC)CuSnPh3.
Herein we report the results of these explorations; the synthesis and characterisation of (RE-NHC)CuSnPh3 (RE-NHC = 6-Mes, 1; 6-Dipp, 2; 7-Dipp, 3), and examples of their reactivity. (6-Mes)CuSnPh3 was found to transfer a phenyl group to both phenyl isocyanate and di-p-tolyl carbodiimide to generate an benzamidate and benzamidinate respectively.
Compounds 1–3 were initially synthesised by the reaction of an equimolar mixture of triphenyltin hydride and (RE-NHC)CuOtBu in C6D6, and the reaction mixture interrogated by NMR spectroscopy (see Fig. S1–S6†). In each case, 1H NMR spectroscopy reflected the formation of tBuOH and 119Sn NMR spectroscopy suggested the formation of a copper(I)-triphenylstannyl by resonances at −79.1 (1), −83.1 (2) and −85.4 ppm (3) respectively. Repetition of these reactions on a preparative scale in toluene, followed by removal of all solvent in vacuo gave yellow powders in good yields (1, 84%; 2, 79%; 3, 77%) which provided NMR spectroscopy data consistent with the formation of the desired (RE-NHC)CuSnPh3 systems (see Fig. S24–S32†). Recrystallisation of 1 by diffusion of hexane into its saturated toluene solution, and of 2 and 3 by cooling their saturated toluene solutions to ca. −18 °C provided material suitable for single crystal X-ray crystallography which confirmed the structures of 1–3 as RE-NHC supported copper(I) triphenylstannyl complexes (Fig. 1, Table 1).
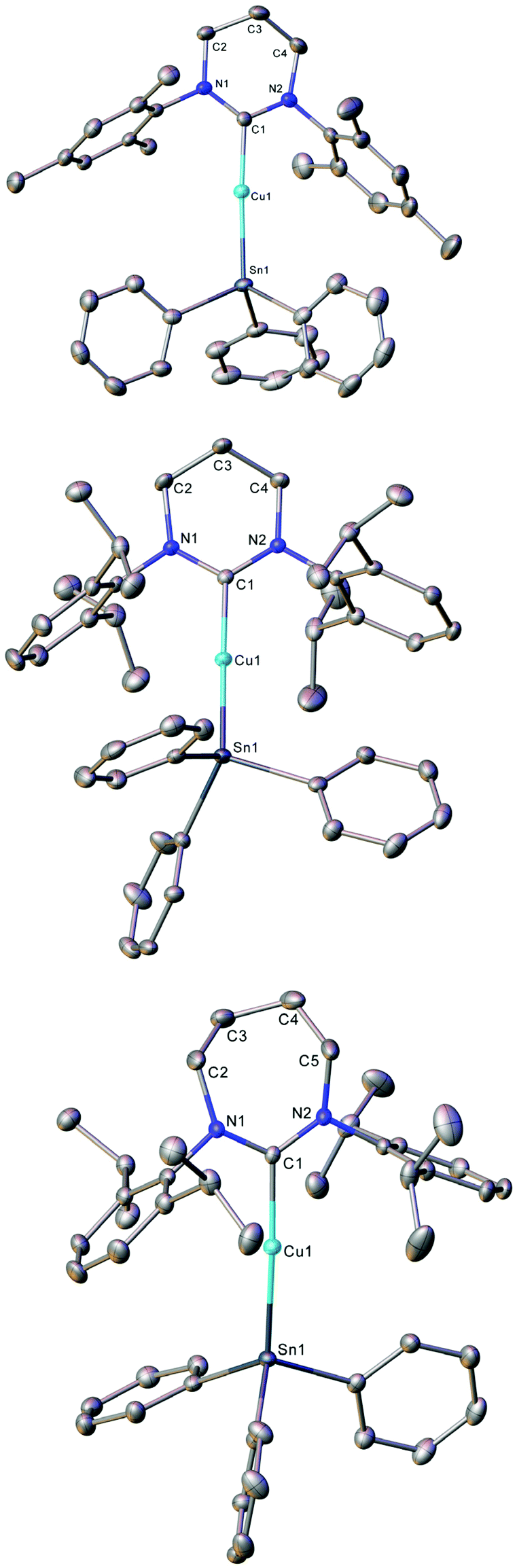 | ||
| Fig. 1 Molecular structure (30% probability ellipsoids) of compounds 1–3. Hydrogen atoms are omitted for clarity. | ||
(IPr)CuSnPh3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 19 |
1 | 2 | 3 | |
|---|---|---|---|---|
| N–Ccarbene (Å) | 1.353(2) | 1.331(4) | 1.333(4) | 1.331(3) |
| 1.358(2) | 1.334(4) | 1.343(4) | 1.347(3) | |
| Ccarbene–Cu (Å) | 1.914(2) | 1.927(3) | 1.934(3) | 1.938(2) |
| Cu–Sn (Å) | 2.469(5) | 2.4567(4) | 2.4742(4) | 2.4686(3) |
| Sn–Cphenyl (Å) | 2.168(2) | 2.171(3) | 2.176(3) | 2.175(3) |
| 2.176(2) | 2.173(3) | 2.188(4) | 2.176(2) | |
| 2.1852(19) | 2.184(3) | 2.190(3) | 2.182(2) | |
| N–Ccarbene–N (°) | 103.84(15) | 118.5(3) | 117.9(3) | 120.0(2) |
| Ccarbene–Cu–Sn (°) | 169.6(8) | 172.87(10) | 171.27(9) | 172.02(7) |
Compounds 1–3 all crystallise as monomers with 2-coordinate near linear copper(I) atoms. The Cu–Sn parameters for 1–3 (1, 2.4567(4) Å; 2, 2.4742(4) Å; 3, 2.4686(3) Å) are similar to previously reported terminal copper(I) triorganostannyl complexes, which show Cu–Sn distances in the range of 2.47–2.50 Å.19,20
Furthermore, the variation in ligand identity seems to have negligible effects on the C–Cu–Sn core, with other metric parameters being essentially unchanged between IPr (C–Cu, 1.914(2) Å; C–Cu–Sn, 169.6(8)°),19 6-Mes (C–Cu, 1.927(3) Å; C–Cu–Sn, 172.87(10)°), 6-Dipp (C–Cu, 1.934(3) Å; C–Cu–Sn, 171.27(9)°) and 7-Dipp (C–Cu, 1.938(2) Å; C–Cu–Sn, 172.02(7)°).
These consistent parameters are in contrast to a significant variation in N–C–N angle for each of these species (IPr, 103.84(15)°;19 6-Mes, 118.5(3)°; 6-Dipp, 117.9(3)°; 7-Dipp 120.0(2)°), which is itself unsurprising in light of the varying ring size of these species.
With these complexes characterised, we were anxious to explore their reactivities. We have recently reported on the reaction of a number of (RE-NHC)CuPPh2 systems with heterocumulenes, often noting distinct structural features and reactivity during reactions with CO2, CS2, isocyanates, and isothiocyanates.27 In light of the thorough exploration of the reaction of (IPr)CuSnPh3 with CO2 reported by Sadighi and co-workers,19 we turned our attention to the reactivity of 1 towards carbodiimides, isocyanates and isothiocyanates. Compound 1 was reacted with one equivalent of di-p-tolyl carbodiimide in C6D6 (see Fig. S7 and S8†). Within minutes, the 119Sn NMR spectrum of this reaction showed consumption of 1 and formation of a peak at −43.6 ppm. Sadighi and co-workers noted a corresponding resonance in the 119Sn NMR spectrum of the reaction between (IPr)CuSnPh3 and CO2 attributing it to an unknown (Ph2Sn) derived product. In the 1H NMR spectrum of 1, resonances ascribed to a new 6-Mes supported copper species were observed, as well as complete consumption of the carbodiimide. From these data, we inferred the formation of an amidinate in an analogous fashion, yielding (6-Mes)Cu(p-Tol-N)C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N-p-Tol)Ph. This interpretation was corroborated by the presence of the [M + H]+ ion peak with an m/z of 301.1703, attributed to HN(p-Tol)C(N-p-Tol)Ph, in the mass spectrum.
N-p-Tol)Ph. This interpretation was corroborated by the presence of the [M + H]+ ion peak with an m/z of 301.1703, attributed to HN(p-Tol)C(N-p-Tol)Ph, in the mass spectrum.
The purported formation of a copper amidate, and the synthetic route used to access 1–3 raised an intriguing possibility; that under catalytic conditions the amidinate, (6-Mes)CuN(p-Tol)C(N-p-Tol)Ph, could deprotonate Ph3SnH to regenerate 1. In the presence of excess p-TolNCN-p-Tol and stannane, the net hydroarylation of a carbodiimide might thus be accessible with concomitant formation of (Ph2Sn)6. Attempts to extend this transformation to a catalytic regime (triphenylstannane, di-p-tolyl carbodiimide and 1 in a 10:10:1 ratio in C6D6) proved unsuccessful, with significant competition from stannane dismutation (see Fig. S9 and S10†).9,29 This result is unsurprising in light of the competence of (6-Mes)CuOtBu in mediating this same reaction (see Fig. S11 and S12†).
Addition of one equivalent of phenyl isocyanate to 1 in C6D6 resulted in a rapid attenuation of the peaks associated with 1 in the 119Sn NMR spectrum, and the growth of a resonance at −209.4 ppm, which was assigned to (Ph2Sn)6 based on literature precedent (see Fig. S13 and S14†).29 These data were interpreted as an implication that phenyl transfer occurs to the isocyanate to generate (6-Mes)CuN(Ph)C(O)Ph in a manner analogous to the formation of (IPr)CuOC(O)Ph from (IPr)CuSnPh3 and CO2. Mass spectrometry of the reaction mixture between 1 and PhNCO provided further evidence for this thesis, by the presence of the [M + H]+ ion peak with an m/z of 198.0914. This corresponds to N-phenylbenzamide, PhN(H)C(O)Ph, which was, most likely, protonated away from the copper centre due to the sample preparation in wet acetonitrile. Reactions attempted with (IPr)CuSnPh3 showed analogous reactivity (see Fig. S15 and S16†).
Interrogation of a preliminary reaction between Ph3SnH and PhNCO in the presence of 10 mol% of 1 in C6D6, by 1H NMR spectroscopy, revealed the complete consumption of both starting materials by the loss of their resonances in the spectrum (see Fig. S17 and S18†). Examination of the corresponding 119Sn NMR spectrum was somewhat less promising, however, indicating the presence of Ph3SnSnPh3 and a number of other phenylstannane derivatives, once again thought to originate from stannane dismutation.9,29 These data compelled a reexamination of the 1H NMR spectrum where a resonance at ca. 4.5 ppm was attributed to the presence of H2.30 Thus, the ultimate fate of the PhNCO was found to be trimerisation to the corresponding isocyanurate, based on the presence of this species in the mass and 1H NMR spectra of the reaction mixtures.31
Addition of 1 equivalent of PhNCS to 1 gave somewhat divergent results (see Fig. S19–S21†). After 30 minutes, the 119Sn NMR spectrum contained resonances at −43.6 and −202.5 ppm. The latter resonance reduced in intensity overnight, and the resultant 1H NMR spectrum contained features consistent with (6-Mes)CuN(Ph)C(S)Ph. The presence of the corresponding thioimidate anion was once again verified by appropriate mass spectral data. More notably, however, was the mass spectrum after 30 minutes which contained an [M + H]+ peak with an m/z of 488.0491. This aligns to the expected mass of HN(Ph)C(S)SnPh3, and allows the tentative attribution of the peak at −202.5 ppm in the 119Sn NMR spectrum to (6-Mes)CuN(Ph)C(S)SnPh3 which converts to (6-Mes)CuN(Ph)C(S)Ph and “Ph2Sn” overnight. Overall, these observations indicate a third mechanistic possibility which may operate in the phenylation of heterocumulenes as shown in Scheme 2.
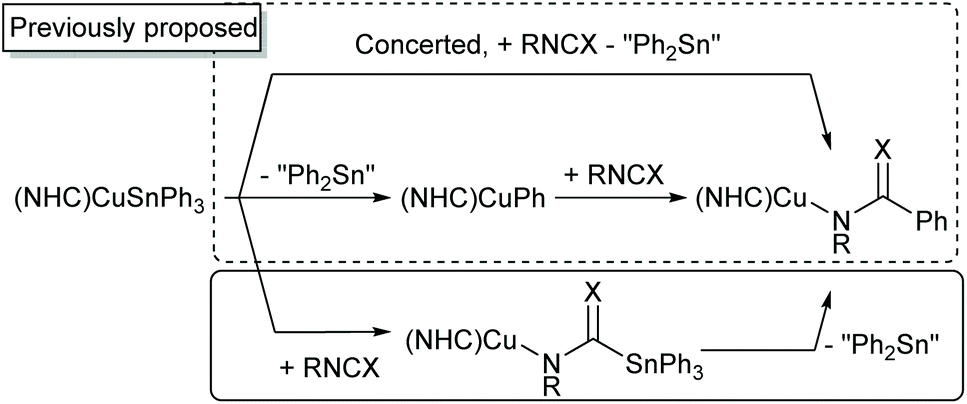 | ||
| Scheme 2 Possible mechanistic pathways in the phenylation of heterocumulenes by copper(I) triphenylstannyls. The binding mode of the amidinate, amidate and thioamidate ligands was not ascertained from current data. | ||
These results raised the possibility that this mechanism may also contribute to the reactivity of 1 towards di-p-tolyl carbodiimide and phenyl isocyanate. Whilst these reactions appeared to be quantitative within the timeframe of the initial NMR experiment, instantaneous quenching of the reaction mixture after the addition of the heterocumulene to 1 in wet acetonitrile followed by mass spectrometry was informative. In the case of di-p-tolyl carbodiimide, the mass spectrum contained a [M + H]+ peak with an m/z of 575.1494, whilst in the case of phenyl isocyanate a [M + Na]+ value of 494.0531 was obtained. These correspond to the masses of HN(p-Tol)C(N-p-Tol)SnPh3 and HN(Ph)C(O)SnPh3 respectively.
The rapid consumption of 1 observed in these reactions prompted us to attempt to observe relevant intermediates using VT-NMR spectroscopy. An equimolar reaction of 1 and di-p-tolyl carbodiimide was made up in d8-toluene and quickly frozen in liquid nitrogen. Thawing in the NMR spectrometer at −50 °C followed by interrogation by 119Sn NMR spectroscopy indicated the previously observed resonance of “(Ph2Sn)” at −43.6 ppm and a hitherto unobserved peak at −245.3 ppm, attributed to (6-Mes)CuN(p-Tol)C(N-p-Tol)SnPh3, which disappeared entirely within 45 minutes (see Fig. S22†). Reaction of 1 with phenyl isocyanate under equivalent conditions provided a new resonance at −216.7 ppm, attributed to (6-Mes)CuN(Ph)C(O)SnPh3, but which persisted at −50 °C. Slow warming of the reaction mixture to room temperature resulted in attenuation and eventually disappearance of this peak, favouring its assignment as an intermediate.
At this stage it is impossible to definitively conclude which pathway (or pathways) are operant during this class of reactions. Nevertheless, these preliminary results raise an intriguing mechanistic possibility and demand further exploration to elucidate the molecular origin of this unusual reactivity. We note the extensive computational work performed on the reaction of copper tetryls with CO232 and hope this experimental study will, in time, allow a more conclusive theoretical understanding of these intriguing C–C bond forming reactions.
Conclusions
In conclusion, we have described three new (NHC)CuSnPh3 species, supported by the ring-expanded NHCs; 6-Mes (1), 6-Dipp (2) and 7-Dipp (3). In addition to characterisation of these compounds by NMR spectroscopy and X-ray crystallography we have also investigated the reactivity of 1 towards carbodiimides, isocyanates and isothiocyanates broadening the scope of the unusual phenyl transfer ability of copper(I) triphenylstannyls to generate benzamidinates, benzamides, and benzothiamides respectively. Furthermore, in situ reaction monitoring has provided evidence for the possibility of an additional mechanistic pathway in the phenylation of heterocumulenes by copper(I) triphenylstannyls.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DJL thanks the Royal Society for the support of a University Research Fellowship.Notes and references
- P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704–4734 CAS.
- M. M. Heravi and L. Mohammadkhani, J. Organomet. Chem., 2018, 869, 106–200 CrossRef CAS.
- D. Reed, D. Stalke and D. S. Wright, Angew. Chem., Int. Ed. Engl., 1991, 30, 1459–1460 CrossRef.
- P. B. Hitchcock, M. F. Lappert, G. A. Lawless and B. Royo, J. Chem. Soc., Chem. Commun., 1993, 554–555 RSC.
- B. E. Eichler, A. D. Phillips and P. P. Power, Organometallics, 2003, 22, 5423–5426 CrossRef CAS.
- T. Schollmeier, U. Englich, R. Fischer, I. Prass, K. Ruhlandt, M. Schürmann and F. Uhlig, Z. Naturforsch., B: J. Chem. Sci., 2004, 59, 1462–1470 CrossRef CAS.
- C. Kleeberg, J. Grunenberg and X. Xie, Inorg. Chem., 2014, 53, 4400–4410 CrossRef CAS PubMed.
- J. T. B. H. Jastrzebski, M. van Klaveren and G. van Koten, Organometallics, 2015, 34, 2600–2607 CrossRef CAS.
- U. Englich, K. Ruhlandt-Senge and F. Uhlig, J. Organomet. Chem., 2000, 613, 139–147 CrossRef CAS.
- L. J. Morris, M. S. Hill, I. Manners, C. L. McMullin, M. F. Mahon and N. A. Rajabi, Chem. Commun., 2019, 55, 12964–12967 RSC.
- L. J. Morris, N. A. Rajabi, M. F. Mahon, I. Manners, C. L. McMullin and M. S. Hill, Dalton Trans., 2020, 49, 10523–10534 RSC.
- P. M. Chapple, J. Cartron, G. Hamdoun, S. Kahlal, M. Cordier, H. Oulyadi, J.-F. Carpentier, J.-Y. Saillard and Y. Sarazin, Chem. Sci., 2021, 12, 7098–7114 RSC.
- J. Nokami, H. Nose and R. Okawara, J. Organomet. Chem., 1981, 212, 325–328 CrossRef CAS.
- H. Schneider, M. J. Krahfuß and U. Radius, Z. Anorg. Allg. Chem., 2016, 642, 1282–1286 CrossRef CAS.
- P. Braunstein, J. Durand, X. Morise, A. Tiripicchio and F. Ugozzoli, Organometallics, 2000, 19, 444–450 CrossRef CAS.
- N. R. Neale and T. D. Tilley, J. Am. Chem. Soc., 2002, 124, 3802–3803 CrossRef CAS PubMed.
- S. M. Thompson and U. Schubert, Inorg. Chim. Acta, 2003, 350, 329–338 CrossRef CAS.
- S. M. Thompson and U. Schubert, Inorg. Chim. Acta, 2004, 357, 1959–1964 CrossRef CAS.
- K. X. Bhattacharyya, J. A. Akana, D. S. Laitar, J. M. Berlin and J. P. Sadighi, Organometallics, 2008, 27, 2682–2684 CrossRef CAS.
- J. Plotzitzka and C. Kleeberg, Inorg. Chem., 2016, 55, 4813–4823 CrossRef CAS PubMed.
- R. D. Adams, Y. O. Wong and Q. Zhang, J. Organomet. Chem., 2015, 795, 40–44 CrossRef CAS.
- J. K. Park, H. H. Lackey, B. A. Ondrusek and D. T. McQuade, J. Am. Chem. Soc., 2011, 133, 2410–2413 CrossRef CAS PubMed.
- J. Lie, W.-X. Shen and X.-R. Li, Curr. Org. Chem., 2012, 16, 2879–2891 CrossRef.
- A. J. Jordan, C. M. Wyss, J. Bacsa and J. P. Sadighi, Organometallics, 2016, 35, 613–616 CrossRef CAS.
- F. Sebest, J. J. Dunsford, M. Adams, J. Pivot, P. D. Newman and S. Díez-González, ChemCatChem, 2018, 10, 2041–2045 CrossRef CAS PubMed.
- A. J. Jordan, R. K. Walde, K. M. Schultz, J. Bacsa and J. P. Sadighi, Inorg. Chem., 2019, 58, 9592–9596 CrossRef CAS PubMed.
- T. M. Horsley Downie, J. W. Hall, T. P. Collier Finn, D. J. Liptrot, J. P. Lowe, M. F. Mahon, C. L. McMullin and M. K. Whittlesey, Chem. Commun., 2020, 56, 13359–13362 RSC.
- T. M. Horsley Downie, R. S. C. Charman, J. W. Hall, M. F. Mahon, J. P. Lowe and D. J. Liptrot, Dalton Trans., 2021, 50, 16336–16342 RSC.
- B. Wrackmeyer, in Annual Reports on NMR Spectroscopy, ed. G. A. Webb, Academic Press, 1999, vol. 38, pp. 203–264 Search PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- S. R. Foley, G. P. A. Yap and D. S. Richeson, Organometallics, 1999, 18, 4700–4705 CrossRef CAS.
- A. Ariafard, N. J. Brookes, R. Stranger and B. F. Yates, Organometallics, 2011, 30, 1340–1349 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General synthetic experimental details, NMR spectra, X-ray analysis of compounds 1–3. CCDC 2109091–2109093. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03109k |
| This journal is © The Royal Society of Chemistry 2022 |