DOI:
10.1039/D2BM00650B
(Paper)
Biomater. Sci., 2022,
10, 3892-3900
Nanodrug regulates lactic acid metabolism to reprogram the immunosuppressive tumor microenvironment for enhanced cancer immunotherapy†
Received
27th April 2022
, Accepted 24th May 2022
First published on 10th June 2022
Abstract
A majority of cancers fail to respond to immunotherapy due to the immunosuppressive tumor microenvironment (TME), and metabolic regulation of the TME has been a promising strategy to improve immunotherapy. Lactate is a key metabolic player in tumor immune response since its excess secretion aggravates tumor immune escape by favoring the polarization of tumor-associated macrophages (TAMs) to an immunosuppressive phenotype meanwhile impeding the tumor infiltration of the cytotoxic T lymphocyte. Here, we proposed a metabolic reprogramming mechanism to ameliorate tumor immunosuppression by using lonidamine and syrosingopine incorporated liposomes (L@S/L) to regulate lactate production and efflux. Concretely, lonidamine reduced lactate production by affecting the glycolytic metabolic pathway while syrosingopine decreased lactate efflux by inhibiting the key protein expression of the lactate transporter MCT-4. Consequently, both the drugs synergistically normalize the pH of the TME to overcome the tumor immunosuppressive microenvironment. In vivo studies demonstrated that the decreased extracellular lactate preferentially polarized TAMs to the M1 phenotype, simultaneously increased the proportion of NK cells and reduced the number of Treg cells. These results validated an efficient tumor immunotherapy in the breast cancer model. This new strategy of lactic acid metabolism regulation is proposed to operate in concert with immune modulation in the TME, which shows great potential for immunotherapy of immunologically “cold” tumors.
Introduction
Triple-negative breast cancer (TNBC) features an immunologically “cold” tumor microenvironment (TME) with abundant immunosuppressive cell infiltration.1 Abnormal tumor cell metabolism is an important factor contributing to the formation of the immunosuppressive TME.2–4 Most solid cancer cells rely on aerobic glycolysis for energy production, known as the famous “Warburg effect” which was firstly reported by Otto Warburg.5 The high level of aerobic glycolysis in cancer cells leads to the secretion of a large amount of lactate with the concentration reaching 40 mM in the tumor cytoplasm and TME,6 which in parallel, promotes an acidic TME to result in tumor immunosuppression and metastasis.7,8 Concretely, the lactate in TME inhibits tumor immunity function via affecting several types of immune cells, e.g., directly suppressing the infiltration of natural killer (NK) cells, recruiting regulatory T cells (Treg), and promoting the arginase activity and HIF-1α stability to induce M2 polarization of macrophages.6,8,9 Therefore, the lactate in the TME plays a crucial role in cancer progression and treatment, the changes in lactate concentrations are the current standard for detecting the sensitivity to antitumor drugs to some extent.10–13 Accordingly, the regulation of lactic acid metabolism seems to be a promising target for cancer immunotherapy.
It has been reported that 3-bromotyrosine, a hexokinase (HK) II inhibitor, could mediate mitochondrial dysfunction to induce cancer cell death via adenosine 5-(gamma-thio) triphosphate (ATP) depletion-dependent apoptosis, necrosis and ROS generation.14 In addition, lonidamine, a derivative of 3-carboxy-indole, has been found to act on the glycolytic activity inhibition associated with HK in the mitochondria15 to reduce the production of intracellular ATP and lactate in cancer cells and inhibit Ehrlich ascites tumors.16 Owing to the flexibility of metabolic pathways in cells, bypass metabolisms can reverse the effect of single pathway suppression.17 For example, HK inhibitors appear to be sufficient for suppressing lactate production in cancer cells,18 whereas the intrinsic and adaptive antioxidant capacity of cancer cells will still weaken the anti-tumor ability of lonidamine to maintain redox homeostasis, which limits the potential anti-tumor strategies of regulating lactate secretion.19 Notably, the lactate efflux inhibitor of syrosingopine silences the expression of monocarboxylate transporter 4 (MCT-4), resulting in tumor cell autophagy to decrease lactate metabolism and cancer cell proliferation.20,21 Taking the above mechanisms together, we speculate that synergistic mitochondrial damage and lactate efflux suppression may synergistically reduce the lactate content to reverse the acidic immunosuppressive TME, which provides an effective way for tumor immunotherapy.
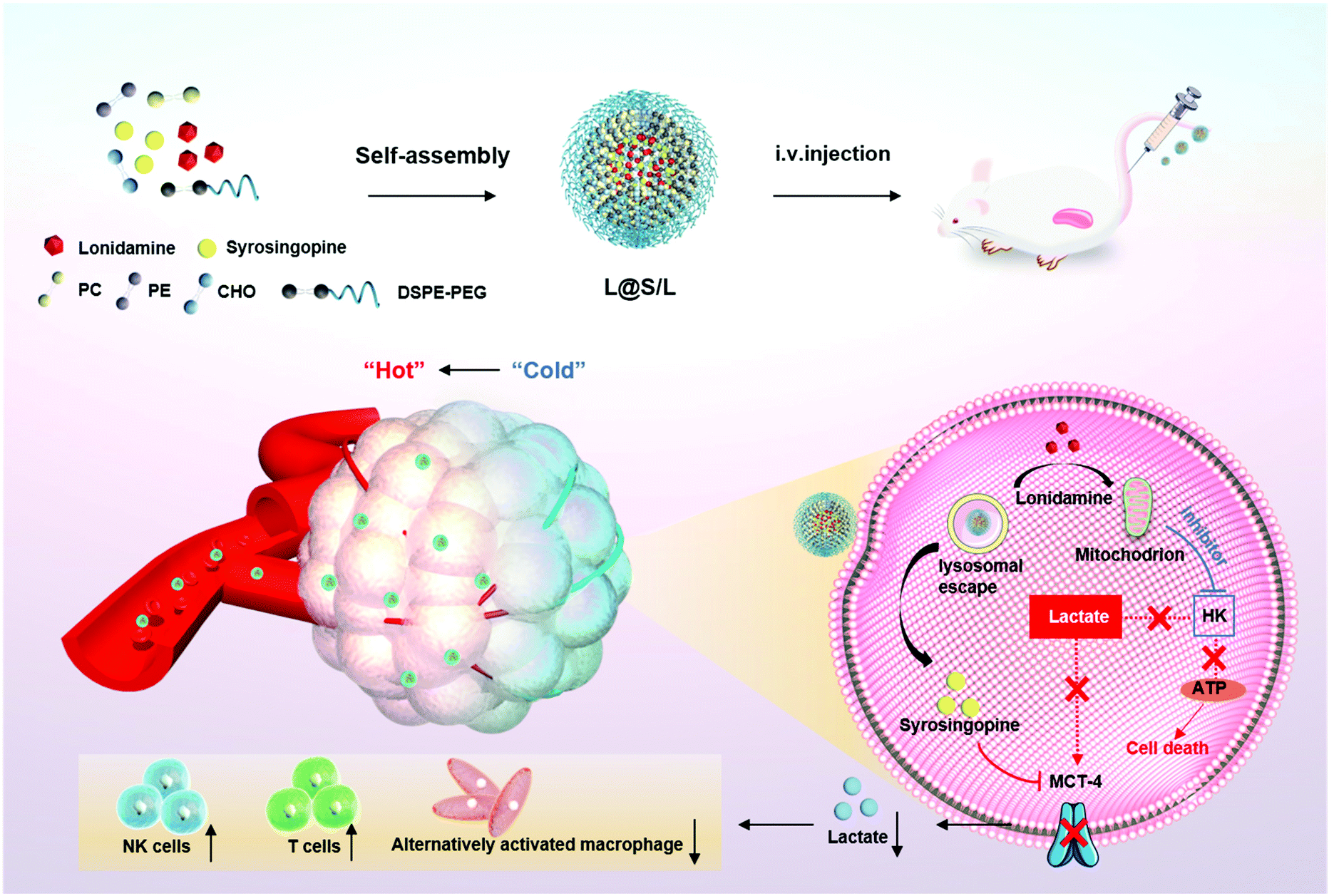
Here, we present an innovative intra/extracellular lactic acid exhaustion strategy for synergistic antitumor metabolism administration and immunotherapy. Lonidamine (LND) to affect the glycolytic process and syrosingopine (Sy) to inhibit the expression of MCT-4 are simultaneously encapsulated with PEG-modified liposomes to form the metabolic and immune regulation nanodrug (denoted as L@S/L) as shown in Fig. 1. PEG shielding enhances the accumulation of the nanodrug at the tumor site via the enhanced permeability and retention (EPR) effect.22,23 After being internalized into tumor cells, LND impairs glycolysis and lactate production by inhibiting mitochondrial hexokinase.24 Meanwhile, Sy inhibits the key proteins (MCT-4) of lactate efflux on tumor cell membranes,25 which synergistically regulate the pH of the TME. Specifically, the extracellular lactate depletion for metabolism interference is expected to promote NK cell activity and induce the M1 phenotype of macrophages, which eventually reverses the immunosuppressive TME and further inhibits tumor growth.26
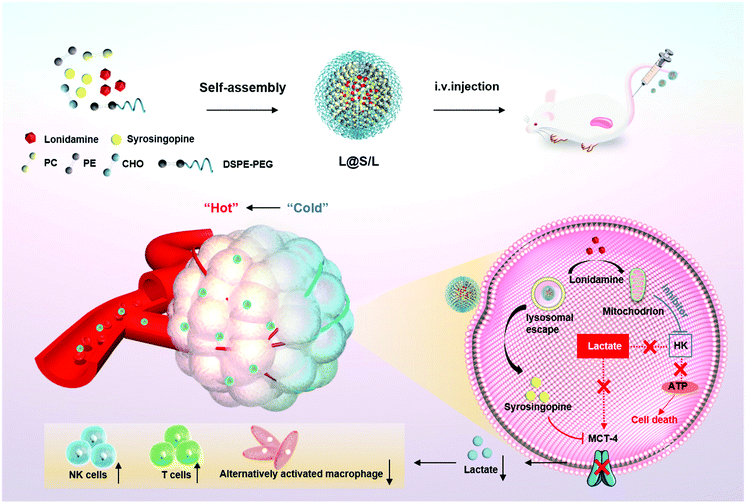 |
| | Fig. 1 Schematic illustration to show the preparation of lonidamine and syrosingopine incorporated liposomes (L@S/L) nanodrugs and the mechanism of combined metabolic-immune cancer therapy. | |
Materials and experiments
Materials
Lecithin (PC), cholesterol (Chol), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy] (polyethylene glycol)-2000 (DSPE-PEG2000) were bought from Advanced Vehicle Technology (Shanghai, China). Phosphatidylethanolamine (PE) was obtained from Macklin (Shanghai, China). Lonidamine and syrosingopine were purchased from Acmec and TargetMol respectively. Coumarin 6 (C6) was purchased from Aladdin. Anhydrous chloroform, dimethyl sulfoxide (DMSO) and other reagents used in this experiment were purchased from Guangzhou Chemical Reagent Factory, and they were all analytical grade.
Nanodrug preparation and characterization
Nanodrugs were prepared by the thin film hydration method. Briefly, 30 mg lipids with a molar ratio of PC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PE:Chol:DSPE-PEG2000 of 50:25:25:1 were dissolved in 10 mL of chloroform and 5 mg lonidamine and 2 mg syrosingopine were dissolved in 200 μL of DMSO, respectively. The solutions were added into a 250 mL eggplant-shaped bottle and rotary evaporated (40 °C, 120 rpm, 30 min) to remove the solvent for film formation using a rotary evaporator. Then 5 mL of deionized water was added under sonication (SONICS VCX130, power 20 W, 20 kHz, 30 min). Subsequently, the solution was dialyzed (MWCO: 14 kDa) against deionized water for 24 h to remove DMSO. Finally, the solution in the dialysis bag was filtered through a syringe filter (pore size: 450 nm) to eliminate large aggregates. The same method was used to synthesize the blank control (BL), single drug-loaded nanodrugs (e.g., L@L and L@S) and fluorescently-labeled nanodrugs (e.g., L@C6 and L@DiR).
:PE:Chol:DSPE-PEG2000 of 50:25:25:1 were dissolved in 10 mL of chloroform and 5 mg lonidamine and 2 mg syrosingopine were dissolved in 200 μL of DMSO, respectively. The solutions were added into a 250 mL eggplant-shaped bottle and rotary evaporated (40 °C, 120 rpm, 30 min) to remove the solvent for film formation using a rotary evaporator. Then 5 mL of deionized water was added under sonication (SONICS VCX130, power 20 W, 20 kHz, 30 min). Subsequently, the solution was dialyzed (MWCO: 14 kDa) against deionized water for 24 h to remove DMSO. Finally, the solution in the dialysis bag was filtered through a syringe filter (pore size: 450 nm) to eliminate large aggregates. The same method was used to synthesize the blank control (BL), single drug-loaded nanodrugs (e.g., L@L and L@S) and fluorescently-labeled nanodrugs (e.g., L@C6 and L@DiR).
The particle size of the nanodrugs was determined by dynamic light scattering (DLS, Brookhaven Instruments Corp Particle Solutions), and the morphology of nanodrugs was observed using transmission electron microscopy (TEM, JEM-1400 Plus). To determine the stability of the nanodrug, 0.1 mL of L@S/L was suspended in 0.9 mL of PBS solution and 0.9 mL of PBS solution containing 10% fetal bovine serum at room temperature, respectively, with DLS examination at different times to measure its particle size. In order to measure the drug loading and encapsulation efficiency of L@S/L, the sample was freeze-dried into powder using a freeze dryer (SP Industries, 2KBTES), which was convenient for subsequent testing.
In vitro drug release
To explore the cumulative release of LND and Sy in vitro, 10 mL of L@S/L solution was divided into two aliquots, placed into two dialysis bags (14 kDa) and then transferred to 10 mL of release medium (PBS containing 0.5% Tween 80; pH 5.5 and 7.4 respectively). 1 mL solution outside the dialysis bag was collected (supplemented with fresh PBS) at different time points. The concentrations of LND and Sy were measured by UV-Vis absorbance (PerkinElmer UV750) and high performance liquid chromatography (Waters Breeze GP system) respectively, and cumulative drug release was calculated and plotted against time.
Cell culture
4T1 mouse breast cancer cells were cultured in a DMEM high glucose medium (Gibco) containing 10% fetal bovine serum and 1% penicillin/streptomycin, and placed in a constant temperature (37 °C) cell incubator (Froma Scientific) containing 5% CO2. Mouse bone marrow-derived macrophages (BMDMs) were extracted from the bone marrow of mice, and differentiated into M0-type macrophages as reported previously.27
Cell uptake of the nanodrug
The 4T1 cells were seeded at a density of 1 × 104 cells per well in 35 mm confocal culture dishes overnight and then incubated with L@C6 for various times (0, 0.5, 1, 2, and 4 h). Subsequently, the cells were washed with PBS and fixed with 4% paraformaldehyde (Jie Tewei, Guangzhou). Finally, after 4′,6-diamino-2-phenylindole (DAPI) was added to stain the nuclei for 5 min, the cells were observed under a laser confocal microscope (CLSM) (Nikon C2+, Japan).
4T1 cells were seeded in a 12-well plate at a density of 1 × 106 cells per well, and cultured for 24 h. L@C6 was added and incubated for 0, 0.5, 1, 2, and 4 h. Then, the cells were collected and resuspended in PBS and analyzed using a flow cytometer (NovoCyte Quanteon, USA). Datas were analyzed using FlowJo 10.0 (Treestar, USA).
MTT assay
The 4T1 cells were seeded overnight at a density of 5000 cells per well in 96-well plate, and samples (PBS, BL, L, and L@L) with different liposome drug concentrations were added after 24 h. After another 24 h of co-incubation, the MTT kit (Beyotime) was used to detect cell viability.
Lactic acid test and macrophage differentiation
After 4T1 cells were treated with different nanodrugs (PBS, L@L, L@S and L@S/L) for 24 h, the supernatant was obtained for subsequent experiments. The lactate content in the supernatant (extracellular lactic) and the lactate level in the 4T1 cell lysate (intracellular lactic) were measured using a lactate kit purchased from Nanjing Jiancheng Bioengineering Institute; the final value of extra/intracellular lactic acid concentration was calculated according to the manufacturer's protocol. In addition, the supernatant was co-cultured with M0 macrophages for 48 h. Afterward, the cells were collected and macrophage polarization was measured by flow cytometry.
Western blot assay
The total protein from 4T1 cells or tumor tissues was extracted by radioimmunoprecipitation with protease inhibitors. Antibodies used in this experiment were purchased from Abcam. Protein equivalents were separated using SDS-PAGE gel and then transferred to polyvinylidene fluoride (PVDF, Bio-Rad, USA) membranes. Next, PVDF membranes were blocked with 5% nonfat milk for 1 h and then incubated with primary antibodies MCT-4 and GAPDH overnight at 4 °C. Then, the membrane was washed 5 times with Tris-buffer physiological saline (TBST) containing Tween 20, incubated with secondary antibody IgG-horseradish peroxidase (HRP) for 1 h, and then washed with TBST. Finally, immunoreactive protein bands were visualized on a chemiluminescence detection system (Imagequant LAS 500, USA).
In vivo biodistribution
4T1 (1 × 106 cells) were subcutaneously implanted into female Balb/c mice (6 weeks old) and tumors were allowed to grow for a week to reach a size of ∼100 mm3. L@DiR was injected into tumor-bearing mice via the tail vein at a DiR dose of 0.75 mg per kg (body weight). Then, fluorescence images were captured at specific time points using a fluorescence imaging system (In Vivo FX, Carestream, USA). 24 h after injection, the ex vivo fluorescence images of major organs and tumors were also obtained from the same animal.
Therapeutic effect of nanodrugs in vivo
All animal experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals of Sun Yat-sen University and approved by the Animal Ethics Committee of Sun Yat-sen University. All experiments involving animals strictly abide by the “China Animal Management Regulations” (1988, revised in 2017) and “China's Guidelines for Humane Treatment of Laboratory Animals” (MOST 2006). After cleaning the hair, 1 × 106 4T1 cells were injected subcutaneously into the right leg of Balb/c mice at a dose of 100 μL to establish an animal tumor model. When the tumor volumes reached approximately 50 mm3,28 the mice were randomly divided into four groups and treated with PBS, L@L, L@S, and L@S/L, respectively. Nanodrugs were intravenously injected once every two days. The Sy and LND doses per injection were 1 mg per kg and 2.5 mg per kg (body weight), respectively. The tumor volume was measured with calipers every two days and calculated as follows: tumor volume = 0.5 × length × width2.
Immune response
After the treatment, the mice were sacrificed to collect their tumors, and the single-cell suspension of tumor tissues was extracted by the grinding method. To determine Treg cell subsets, cell suspensions were stained with antibodies (e.g., anti-mouse CD3-APC, anti-mouse CD4-FITC, anti-mouse CD25-BV785 and anti-mouse FOXP3-PE) for 30 min. To identify tumor-associated macrophages (TAMs), cell suspensions were incubated with anti-mouse CD45-FITC, anti-mouse CD11b-Alexa700, anti-mouse F4/80-PE/Cyanine7, anti-mouse CD86-PE, and anti-mouse CD206-APC for 30 min. To identify NK cells, cell suspensions were labeled with antibodies against mouse CD3-APC, anti-mouse CD11b-PE, and anti-mouse NK1.1-FITC. Finally, the number of immune cell suspensions was assessed using flow cytometry. All fluorescently labeled flow cytometry antibodies in this section were purchased from BioLegend (San Diego, CA, USA).
Histological and immunohistochemical assays
Tumors were collected to prepare paraffin-embedded sections for hematoxylin/eosin (HE) and TUNEL immunofluorescence staining and analysis. HE staining of the main organs (heart, liver, spleen, lung, kidney) was performed with a HE staining kit. Then, the sections were scanned with a fully automated slide scanning system (AxioScan.Z1). TUNEL immunofluorescence staining was performed using the kit from KeyGEN BioTECH following the manufacturer's instructions and analyzed by confocal microscopy.
Statistics
The statistics were analyzed by one-way ANOVA (Graphpad Prism 6.0, USA). The data are expressed as means ± standard deviation (SD). p < 0.05 was considered statistically significant. All statistical tests were two-tailed.
Results and discussion
Preparation and characterization of nanodrugs
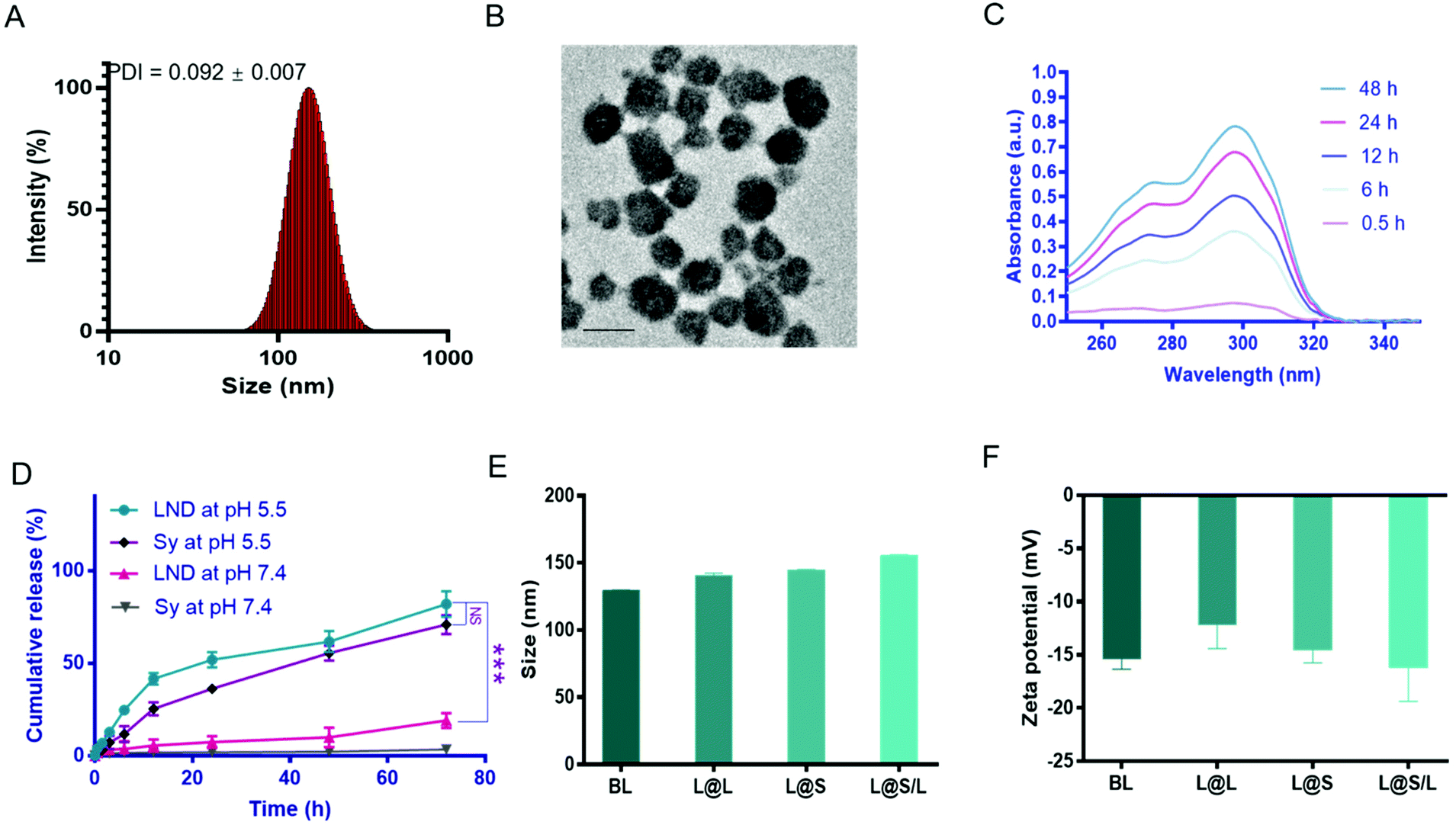
The liposome for drug encapsulation is composed of lecithin (PC), phosphatidylethanolamine (PE), cholesterol (CHO), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy] (polyethylene glycol)-2000 (DSPE-PEG2000). Then the drug LND and the small molecule inhibitor Sy were successfully encapsulated into liposomes by the thin film hydration method with the loading contents of LND and Sy of 6.92 ± 0.27% and 3.62 ± 0.43%, respectively. The L@S/L exhibited a particle size of around 150 nm, a good polydispersity index (PDI) of 0.101 ± 0.018, and a negatively charged zeta potential of around −16.2 mV as measured by dynamic light scattering (DLS) (Fig. 2A and Table S1†), which was suitable for long time circulation in vivo.29 Observation by transmission electron microscopy (TEM) confirmed that L@S/L appeared as small spheres of about 100 nm with uniform size distribution (Fig. 2B). The release behaviors of LND and Sy from L@S/L at pH 5.5 and 7.4 were respectively measured, as shown in Fig. 2C and D. The loaded drug released slowly and the liposomes remained basically stable at pH 7.4. In 72 hours, only 19.02 ± 4.04% of LND, and 3.50 ± 2.32% of Sy were released. However, at pH 5.5, the loaded drug release was accelerated, with 41.53 ± 3.19% of LND and 25.39 ± 3.49% of Sy released at 12 h, and 81.99 ± 6.92% of LND and 70.90 ± 5.14% of Sy released at 72 h. The particle sizes and zeta potentials of blank liposome (BL), LND-loaded liposome (L@L), and Sy-loaded liposome (L@S) were similar to L@S/L (Fig. 2E and F). Additionally, there was no significant change in the particle size of L@S/L in phosphate buffered saline (PBS) and PBS containing 10% fetal bovine serum for 72 h at 37 °C, indicating that nanodrugs were stable during blood circulation (Fig. S1 and S3†). And the PDI was low for all samples, indicating a very narrow size distribution (Tables S2 and S3†).
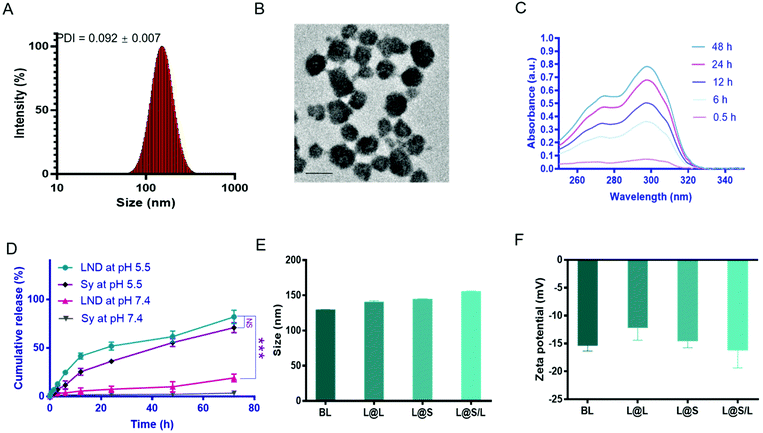 |
| | Fig. 2 Characterization of the nanodrugs. (A) The size distribution of L@S/L. (B) Transmission electron microscopy (TEM) image of L@S/L. Scale bar, 100 nm. (C) UV–vis absorption spectra of cumulative release of LND from L@S/L at pH 5.5. (D) Cumulative release of LND and Sy from L@S/L at pH 5.5 and 7.4, respectively. (E) The particle size of BL, L@L, L@S and L@S/L. (F) The zeta potential of BL, L@L, L@S and L@S/L. (mean ± SD, n = 3). ***p < 0.001, NS: no significant difference. | |
In vitro cellular uptake of L@S/L
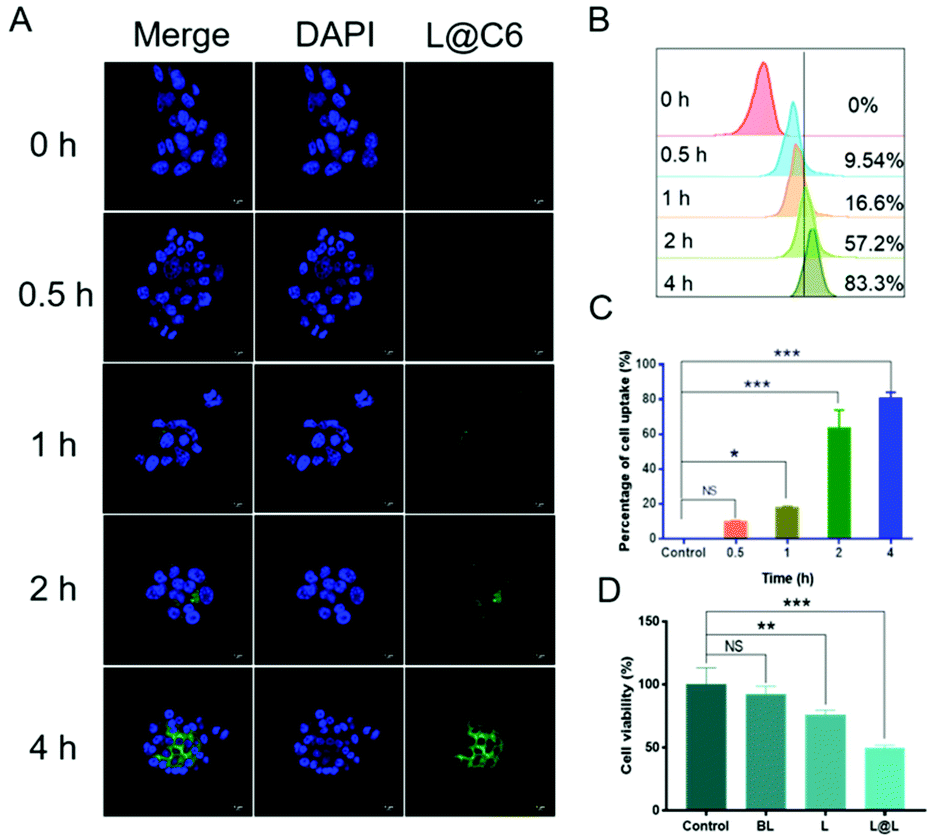
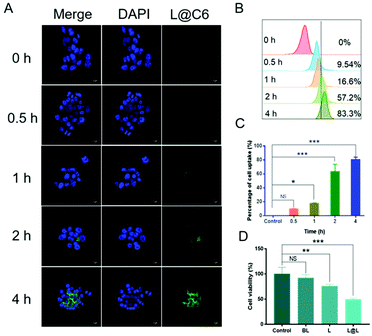
Confocal laser scanning microscopy (CLSM) and flow cytometry were used to analyze the ability of nanoparticles to be phagocytosed by 4T1 breast cancer cells. The fluorescent C6 was encapsulated instead of LND and Sy into the nanoparticles to visualize the cellular uptake and intracellular distribution of the nanodrug. CLSM observations showed that the green fluorescence representing C6 became gradually strengthened over time, indicating that cell incubation significantly increased the internalization of nanoparticles in 4T1 cells (Fig. 3A). Consistent with the confocal data, quantitative analysis by flow cytometry showed that the cellular uptake rate reached 83.3% after 4 h of incubation (Fig. 3B and C). The above results implied that the nanosystem could be effectively taken up into cancer cells. Meanwhile, the survival rate of the cells receiving blank liposome (BL) treatment reached 93.58 ± 5.15%, indicating good compatibility (Fig. 3D). In contrast, the survival rate of the cells treated with free lonidamine (L) and liposome encapsulated lonidamine (L@L) was decreased to 75.93 ± 2.82% and 49.33 ± 2.13%, respectively.
 |
| | Fig. 3
In vitro uptake of nanodrugs in 4T1 cells (concentration of nanodrug, 0.1 mg mL−1). (A) Cellular uptake of L@C6 determined by CLSM imaging. Scale bar, 5 μm. (B) Cellular uptake of L@C6 determined by flow cytometry. (C) Statistical analysis of cellular uptake of L@C6 determined by flow cytometry. (D) The survival rate of the cells receiving different treatments (mean ± SD, n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, NS: no significant difference. | |
Effect of lactate on macrophage polarization in vitro
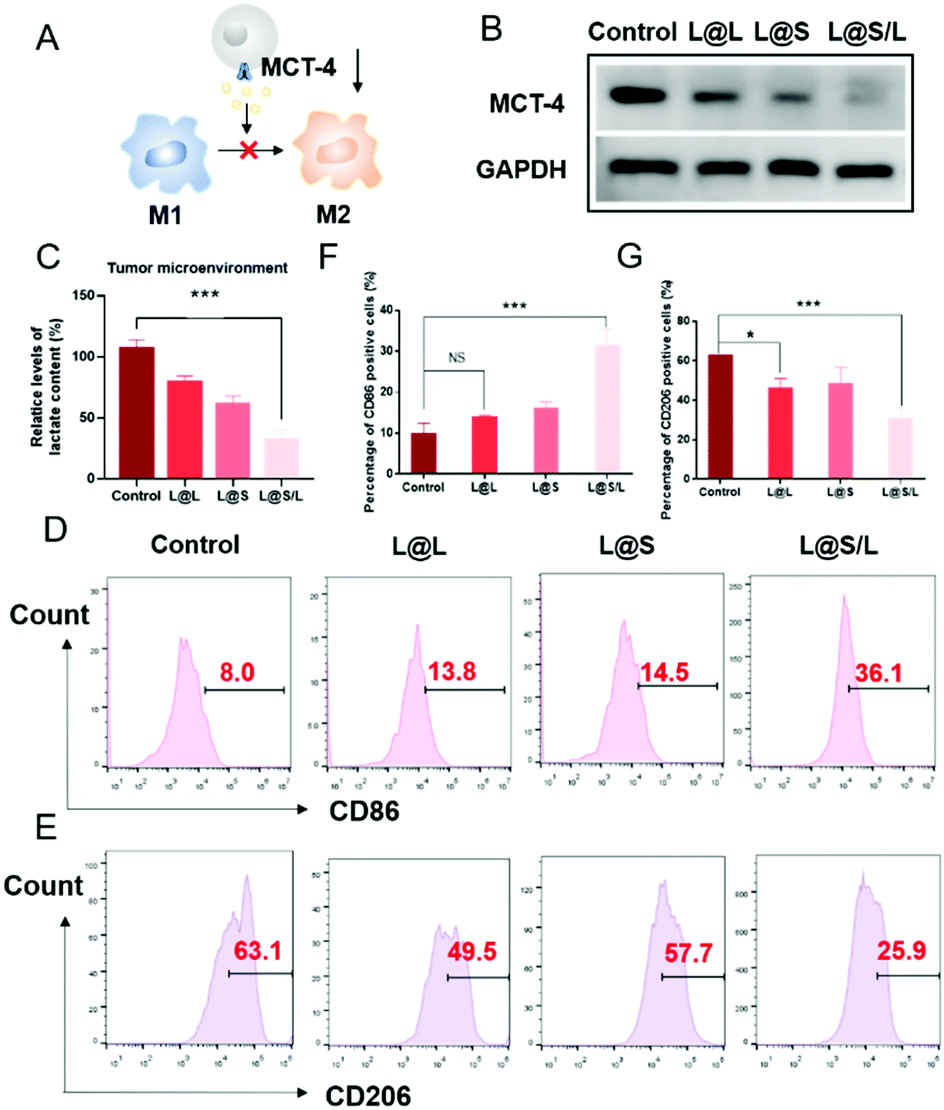
Lonidamine suppresses tumor cell glycolysis which reduces the lactic acid production and syrosingopine inhibits the expression of MCT-4 on the cell membrane.30,31 As such, we have mechanistically investigated the effects of LND in combination with Sy on lactate production, and explored the effect of the combination treatment regimen on macrophage cells (Fig. 4A). As shown in Fig. 4B, the extracellular lactic acid level was decreased in the LND-loaded nanodrug (L@L) treatment group. The Sy loaded nanodrug (L@S) group also showed inhibition of extracellular lactate. Synthetically, the L@S/L group showed extremely low levels of extracellular lactate (Fig. 4B). Meanwhile, intracellular lactate levels in the L@L group were not different from those in the control group, while L@S treatment increased the intracellular lactic acid level, probably because Sy inhibited the expression of MCT-4 to reduce lactate efflux (Fig. 4C). Notably, the cells receiving L@S/L treatment showed the lowest extracellular lactate content. Thus, we speculate that the combined effect of LND inhibiting glycolysis to induce less lactate production and Sy silencing MCT-4 to cause low lactate efflux (Fig. 4B). To verify this, the cell membrane transporter MCT-4 was further measured by western blotting assay. As shown in Fig. 4D, L@S/L obviously down-regulated the expression of MCT-4, potently suggesting that Sy could inhibit MCT-4 expression in 4T1 cells and LND cooperatively inhibits MCT-4 expression.32
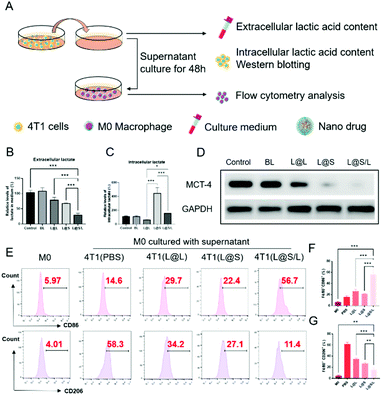 |
| | Fig. 4 Effect of lactate on macrophage polarization in vitro (concentration of L@L, L@S, L@S/L: 0.2 mg mL−1). (A) Coculture model. (B) Normalized extracellular lactate content of 4T1 cells. (C) Normalized intracellular lactate content of 4T1 cells. (D) Expression of MCT-4 in 4T1 cells after different treatments. (E) Flow cytometry analysis of the expression of CD86 and CD206 in macrophages after coculturing with the supernatant. (F) and (G) Statistical analysis of the expression of CD86 and CD206 in macrophages after coculturing with the supernatant (mean ± SD, n = 3). **p < 0.01, ***p < 0.001, NS: no significant difference. | |
Furthermore, we assessed the effects of L@S/L on macrophage polarization. After co-cultivation for 48 h, the cells were harvested, and the percentages of macrophages with M1 or M2 phenotype were analyzed by flow cytometry (Fig. 4E). Compared with the control group or other treatment groups, the proportion of M1 macrophages significantly increased and the proportion of M2 macrophages was the lowest in the L@S/L group (Fig. 4F and G). Thus, the L@S/L nanodrug system inhibited lactate production and efflux in the TME to promote the polarization of the M1 phenotype.33
In vivo distribution and tumor accumulation of L@S/L
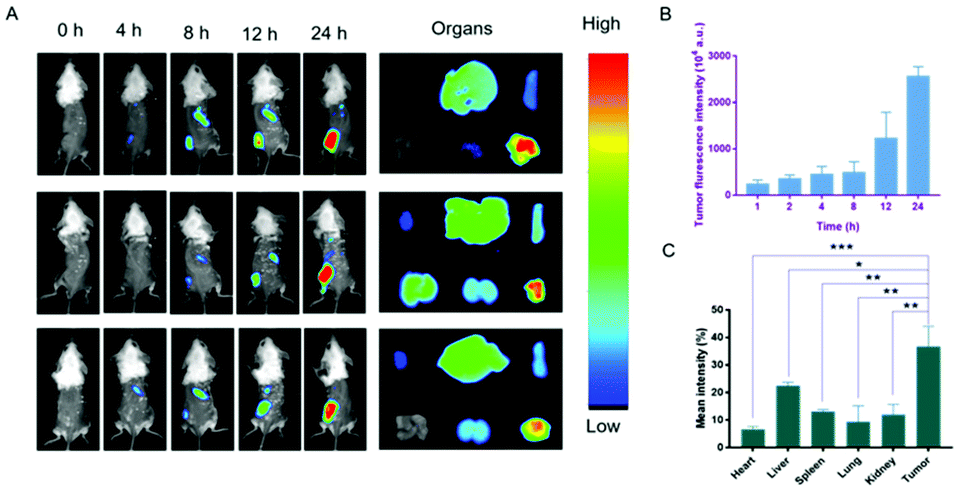
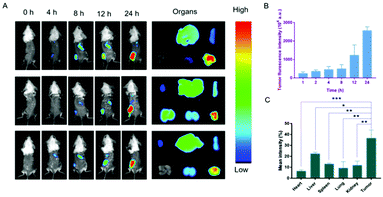
The biodistribution of the nanodrug in 4T1 tumor-bearing mice was analyzed using a small animal imaging system in vivo and fluorescent dye 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide (DiR) instead of Sy or LND into the liposome (L@DiR) for imaging. After an intravenous injection of L@DiR via the tail vein, DiR fluorescence intensity accumulated at the tumor site at 8 h post-injection, gradually increased at around 24 h, indicating the highly efficient tumor accumulation of the nanodrug (Fig. 5A and B). Additionally, the distribution of the nanodrug was also investigated by the ex vivo fluorescence imaging of the tumors and main organs after 24 h post-injection. The result revealed that the nanodrug mainly accumulated in the tumor except for the liver (Fig. 5A and C).34 These results suggested that L@S/L has a strong potential for tumor accumulation via the EPR effect.
 |
| | Fig. 5
In vivo fluorescence imaging of the nanodrug. (A) In vivo fluorescence imaging of 4T1 tumor-bearing Balb/c mice at different time points after tail vein injection of L@DiR and ex vivo fluorescence imaging of main organs and tumor from sacrificed mice after intravenous injection at 24 h. (B) Statistical analysis of fluorescence intensity of tumors in vivo at different time points after injection. (C) Statistical analysis of ex vivo fluorescence intensity of organs (mean ± SD, n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, NS: no significant difference. | |
In vivo therapeutic effect of L@S/L
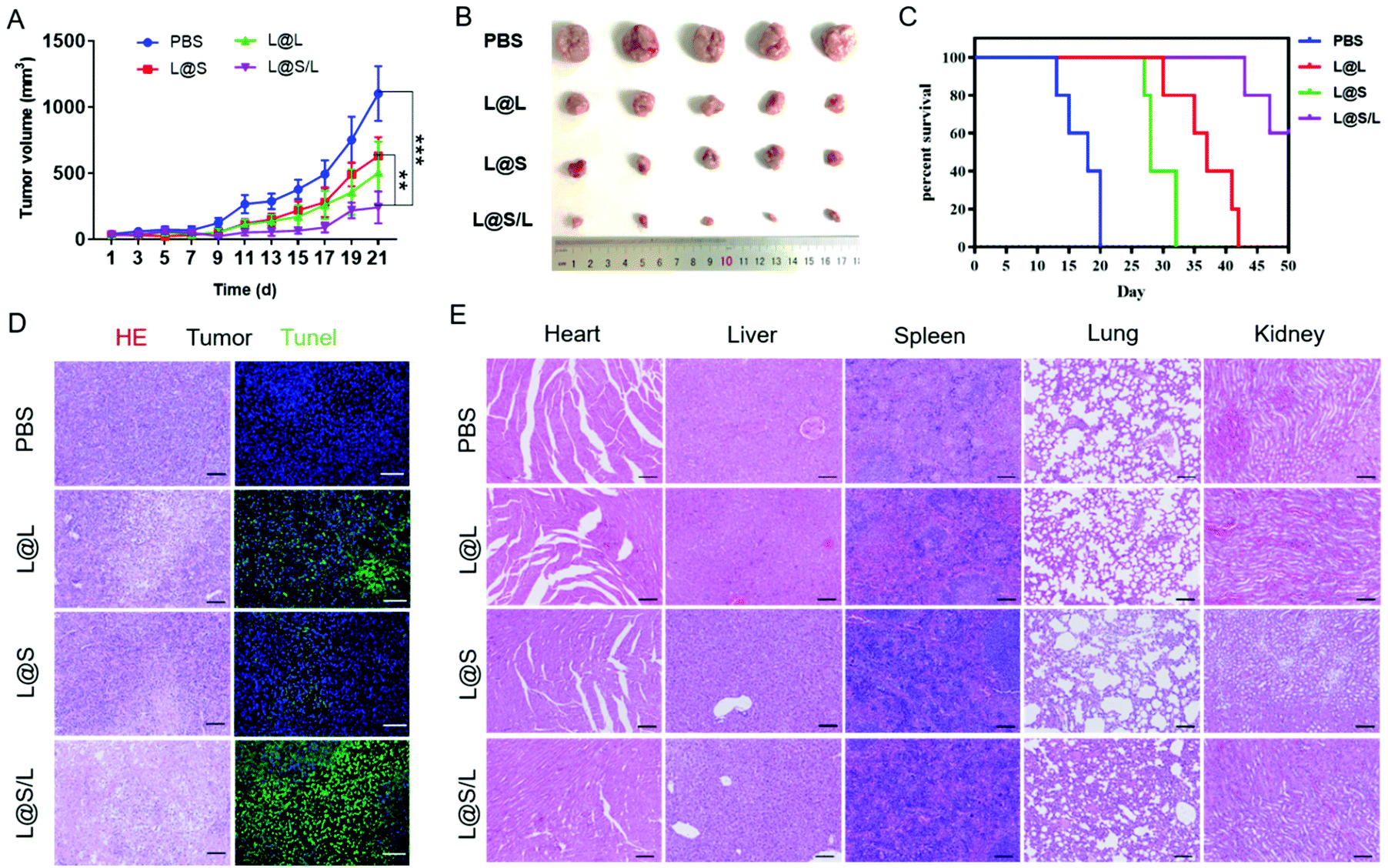
Encouraged by the highly efficient tumor accumulation of the nanodrug, the synergistic antitumor effects of LND and Sy in vivo were further explored. All nanodrug formulations were separately injected into 4T1 tumor-bearing Balb/c mice via the tail vein every 3 days. As shown in Fig. 6A and B, the tumor rapidly grew in the PBS-treated group, and the LND-alone (L@L) or Sy-alone (L@S) group showed moderate inhibition of tumor growth. At the same dosage, the combination therapy of LND and Sy (L@S/L) exhibited the most optimal tumor growth inhibition. In addition, the recorded survival rates of mice were highly dependent on the therapeutic formulations to show a consistent trend with the tumor growth inhibition data. As shown in Fig. 6C, no mouse survived in the PBS group at day 20 after treatment, whereas 60% of mice survived at day 50 when receiving the treatment of L@S/L. These results indicated that the combination of LND and Sy showed a synergistic effect in inhibiting tumor growth and prolonging the survival time of mice. More evidence about the anti-tumor effect and toxicity of nanodrugs could be found by the histological analysis of tumors. As shown in Fig. 6D, the tumor tissues of the L@S/L-treated group showed the highest tumor apoptosis and necrosis, and the lowest cell proliferation, which was in line with the results of tumor growth inhibition. The body weights of mice were measured to assess the safety of the nanodrug. All the nanodrug treated mice showed no significant difference in body weight compared with the PBS group during treatment (Fig. S3†). Moreover, compared with the PBS group, no obvious histological lesion or abnormality was observed in the major organs, i.e. the heart, liver, spleen, and lung, of mice receiving the nanodrug treatment, indicating the safety of the liposome system (Fig. 6E).
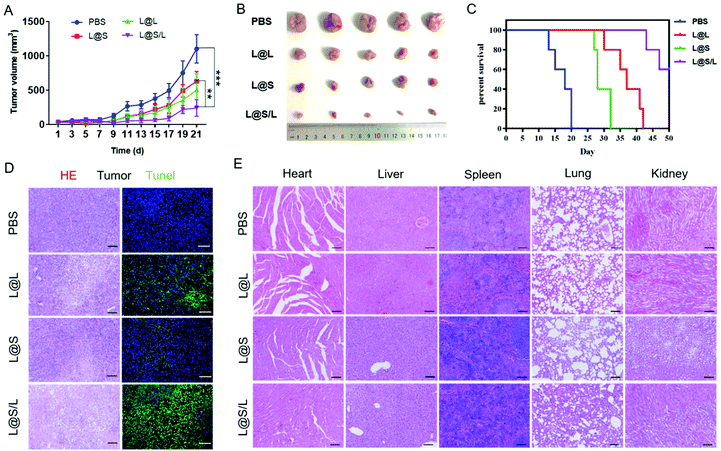 |
| | Fig. 6
In vivo synergistic antitumor therapy of nanodrugs. (A) Tumor growth curves for mice receiving different treatments (PBS, L@L, L@S and L@S/L, n = 5). (B) Images of tumor in 4T1 tumor-bearing Balb/c mice. (C) Cumulative survival of 4T1 tumor-bearing mice after treatments with PBS, L@L, L@S and L@S/L (n = 5). (D) Photograph of tumors excised from mice receiving various treatments. Scale bar: 100 μm. (E) Photographs of excised organs (heart, liver, spleen, lung, and kidney) from mice that received various treatments. Scale bar, 100 μm. **p < 0.01, ***p < 0.001, NS: no significant difference. | |
In vivo immune response regulated by nanodrugs
As such results suggest, we speculated that the efficient anti-cancer function of L@S/L was probably attributed to the inhibition of lactate efflux to reduce the production of M2-type macrophages, leading to enhanced anti-tumor immunity (Fig. 7A). Hence, we monitored the lactate levels and examined the infiltration of various immune cells, including TAMs, Treg cells and NK cells in tumor tissues, as well as measured cytokine levels in the TME at the end of the treatment. As shown in Fig. 7B, L@S/L reduced the expression of the cell membrane transporter MCT-4 in tumor tissues. Moreover, compared with the L@L group and L@S group, the L@S/L group significantly reduced the lactate content in the TME (Fig. 7C). During macrophage phenotype detection in tumor tissues (Fig. 7D), the proportion of M1-like macrophages in L@S/L treated mice showed a dramatic increase. Compared with the control group, the M2-like macrophages treated with L@L decreased slightly, but the M2-like macrophages decreased drastically after L@S/L treatment, from 63.1% in the control group to 25.9% in the L@S/L group (Fig. 7E). The corresponding statistical analysis is shown in Fig. 7F and G. These results were in line with in vitro studies that nanodrug-mediated lactate metabolism regulation promotes M1-like polarization of macrophages.
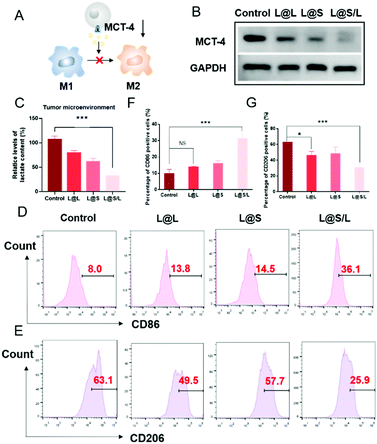 |
| | Fig. 7 L@S/L reduced the production of M2-type macrophages in 4T1 tumors. (A) Inhibition of lactate efflux to reduce the production of M2-type macrophages. (B) MCT-4 expression after different treatments. (C) Statistical analysis of the lactate content in the TME. (D) Quantification of M1-type macrophages (gated on CD45+CD11b+F4/80+CD86+) in 4T1 tumors analyzed by flow cytometry. (E) Quantification of M2-type macrophages (gated on CD45+CD11b+F4/80+CD206+) in 4T1 tumors analyzed by flow cytometry. (F) Statistical analysis of M1-type macrophages after different treatments. (G) Statistical analysis of M2-type macrophages after different treatments (mean ± SD, n = 3). *p < 0.05, ***p < 0.001, NS: no significant difference. | |
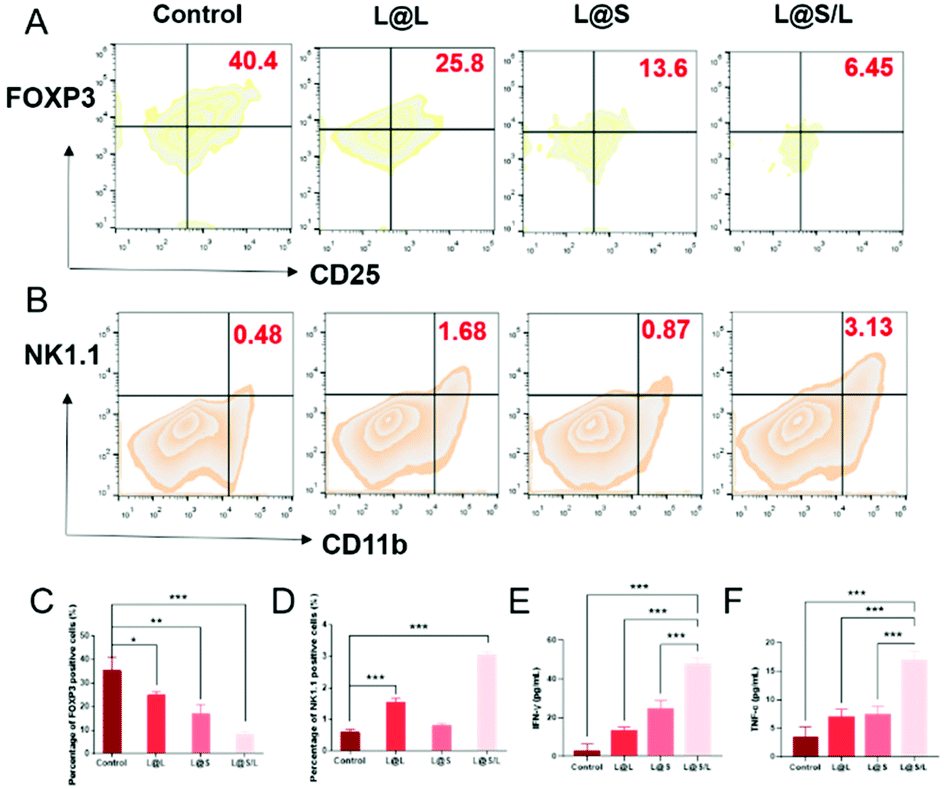
Treg cells have strong suppression effects against effector T cells and can utilize lactate as a carbon fule source to maintain their survival and immunosuppressive function in the TME.35 As shown in Fig. 8A, as a result of decreased lactate efflux, the number of Treg cells in the tumor tissues from L@S/L treated mice decreased significantly. Then, the impact of the L@S/L treatment on NK cell infiltration was also examined. The density of NK cells in the tumor tissue of L@S/L-treated mice was significantly higher than those in other treatment groups (Fig. 8B). The same trends were also observed in the quantitative data (Fig. 8C and D). The effective infiltration of NK cells into tumors is one of the possible intrinsic mechanisms of L@S/L responsible for the suppression of tumor growth.36 The levels of tumor-killing cytokines (e.g., IFN-γ and TNF-α) secreted by M1-like macrophage and NK cells were determined using ELISA kits. As shown in Fig. 8E and F, the results of cytokine detection showed that L@S/L treatment significantly enhanced IFN-γ and TNF-α in tumor tissues. Our results indicate that L@S/L, which inhibits glycolysis and reduces lactate accumulation, can regulate the acidified tumor microenvironment reducing the population of Treg cells.37,38 Furthermore, the inhibition of lactate efflux reduces M2 macrophages in the TME. Therefore, the scheme of reprogramming the TME by interfering with lactate metabolism and enhancing the sensitivity of anti-tumor immunity is highly feasible.39,40
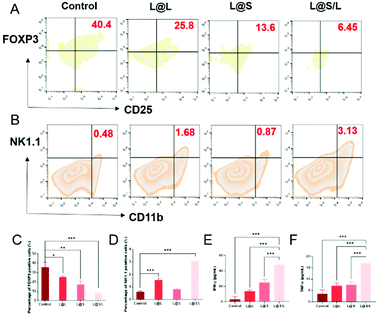 |
| | Fig. 8 L@S/L promoted infiltration of immune cells into tumor. (A) Quantification of Treg cells (gated on CD4+CD25+FOXP3+) in 4T1 tumors analyzed by flow cytometry. (B) Quantification of NK cell (gated on CD3−CD11b+NK1.1+) activation of different nanodrugs determined by flow cytometry. (C) Statistical analysis of Treg cells (gated on CD4+CD25+FOXP3+) inhibition of different treatments in 4T1 tumors analyzed by flow cytometry. (D) Statistical analysis of NK cell activation of different nanodrugs determined by flow cytometry. Statistical analysis of IFN-γ (E) and TNF-α (F) in 4T1 tumors analyzed using ELISA kits (mean ± SD, n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, NS: no significant difference. | |
Conclusions
In conclusion, we prepared a nanodrug using liposome co-encapsulated LND and Sy (L@S/L) for immune “cold” tumor treatment. Firstly, the nanodrug could effectively accumulate in the tumor site. Then the inhibition of the acidified TME and the reduced lactate accumulation could be regarded as a concrete embodiment of the L@S/L function to enhance immune response. As demonstrated in vivo, L@S/L increased the recruitment of NK cells and meanwhile decreased the population of immune-suppressing Treg cells, which enhanced the anti-tumor immune effects. Additionally, this anti-tumor strategy could reduce the amount of M2-like TAMs and enhance tumor-killing cytokines such as IFN-γ and TNF-α in the TME. Consequently, the present study indicated the L@S/L-based lactate metabolism regulation to be a great potential approach for cancer therapy.
Author contributions
L. T. and M. L. conceived and designed the nanodrug and experimental program. H. Z., Y. C. and B. L. performed the experiments and data analysis. X. S. wrote the manuscript with the assistance of Z. X. All authors provided feedback on the final manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by the Key Technologies Research and Development Program of Guangzhou (202007020006), the National Natural Science Foundation of China (51933011, 31971296, and 82102194), the Natural Science Foundation of the Guangdong Province (2020A1515111206 and 2021A1515111006), and the China Postdoctoral Science Foundation (2020M680119 and 2021M703763).
Notes and references
- Y. R. Murciano-Goroff, A. B. Warner and J. D. Wolchok, Cell Res., 2020, 30, 507–519 CrossRef PubMed
 .
.
- G. Kroemer and J. Pouyssegur, Cancer Cell, 2008, 13, 472–482 CrossRef CAS PubMed .
- Z. Xiao, Y. Cai, X. Wang, L. Hu, M. Lin, K. Zhu, Y. Wang and X. Shuai, Nano Today, 2022, 44, 101490 CrossRef .
- Y. Ding, Y. Wang and Q. Hu, Exploration, 2022, 20210106 CrossRef .
- P. Icard, S. Shulman, D. Farhat, J. M. Steyaert, M. Alifano and H. Lincet, Drug Resistance Updates, 2018, 38, 1–11 CrossRef PubMed .
- R. Perez-Tomas and I. Perez-Guillen, Cancers, 2020, 12, 3244–3272 CrossRef CAS .
- K. G. de la Cruz-Lopez, L. J. Castro-Munoz, D. O. Reyes-Hernandez, A. Garcia-Carranca and J. Manzo-Merino, Front. Oncol., 2019, 9, 1143–1163 CrossRef .
- C. Roma-Rodrigues, R. Mendes, P. V. Baptista and A. R. Fernandes, Int. J. Mol. Sci., 2019, 20, 840–870 CrossRef CAS PubMed .
- M. J. Ernsting, B. Hoang, I. Lohse, E. Undzys, P. Cao, T. Do, B. Gill, M. Pintilie, D. Hedley and S. D. Li, J. Controlled Release, 2015, 206, 122–130 CrossRef CAS PubMed .
- N. Santos, A. Pereira-Nunes, F. Baltazar and S. Granja, Immunometabolism, 2019, 1, e190015 Search PubMed .
- E. Saulle, I. Spinello, M. T. Quaranta, L. Pasquini, E. Pelosi, E. Iorio, G. Castelli, M. Chirico, M. E. Pisanu, T. Ottone, M. T. Voso, U. Testa and C. Labbaye, Front. Oncol., 2020, 10, 621458 CrossRef PubMed .
- C. Chen, Z. Wu, P. Ding, N. Sun, H. Liu, Y. Chen, Z. Wang and R. Pei, Adv. Fiber Mater., 2020, 2, 186–193 CrossRef CAS .
- M. Wang, Y. Tan, D. Li, G. Xu, D. Yin, Y. Xiao, T. Xu, X. Chen, X. Zhu and X. Shi, Adv. Fiber Mater., 2021, 3, 192–202 CrossRef CAS .
- J. S. Kim, K. J. Ahn, J. A. Kim, H. M. Kim, J. D. Lee, J. M. Lee, S. J. Kim and J. H. Park, J. Bioenerg. Biomembr., 2008, 40, 607–618 CrossRef CAS .
- S. Fakhri, S. Z. Moradi, M. H. Farzaei and A. Bishayee, Semin. Cancer Biol., 2022, 80, 276–305 CrossRef CAS .
- N. Braidy, J. Berg, J. Clement, F. Khorshidi, A. Poljak, T. Jayasena, R. Grant and P. Sachdev, Antioxid. Redox Signal., 2019, 30, 251–294 CrossRef CAS PubMed .
- C. M. Metallo and M. G. Vander Heiden, Mol. Cell, 2013, 49, 388–398 CrossRef CAS PubMed .
- S. P. Mathupala, Y. H. Ko and P. L. Pedersen, Oncogene, 2006, 25, 4777–4786 CrossRef CAS .
- H. Q. Ju, J. F. Lin, T. Tian, D. Xie and R. H. Xu, Signal Transduction Targeted Ther., 2020, 5, 231–242 CrossRef CAS PubMed .
- C. Anfray, A. Ummarino, F. T. Andon and P. Allavena, Cells, 2019, 9, 46–70 CrossRef PubMed .
- P. Wu, W. Gao, M. Su, E. C. Nice, W. Zhang, J. Lin and N. Xie, Front. Cell Dev. Biol., 2021, 9, 641469 CrossRef .
- J. Park, S. R. Hwang, J. U. Choi, F. Alam and Y. Byun, Int. J. Pharm., 2018, 535, 38–46 CrossRef CAS PubMed .
- S. Guo, K. Li, B. Hu, C. Li, M. Zhang, A. Hussain, X. Wang, Q. Cheng, F. Yang, K. Ge, J. Zhang, J. Chang, X. J. Liang, Y. Weng and Y. Huang, Exploration, 2021, 1, 35–49 CrossRef .
- J. W. Chambers, M. L. Fowler, M. T. Morris and J. C. Morris, Mol. Biochem. Parasitol., 2008, 158, 202–207 CrossRef CAS .
- K. M. Kennedy and M. W. Dewhirst, Future Oncol., 2010, 6, 127–148 CrossRef CAS PubMed .
- F. Gao, Y. Tang, W. L. Liu, M. Z. Zou, C. Huang, C. J. Liu and X. Z. Zhang, Adv. Mater., 2019, 31, e1904639 CrossRef PubMed .
- Q. Zhang, H. Li, Y. Mao, X. Wang, X. Zhang, X. Yu, J. Tian, Z. Lei, C. Li, Q. Han, L. Suo, Y. Gao, H. Guo, D. M. Irwin, G. Niu and H. Tan, Int. J. Mol. Med., 2020, 45, 10–22 CAS .
- X. Duan, C. Chan, N. Guo, W. Han, R. R. Weichselbaum and W. Lin, J. Am. Chem. Soc., 2016, 138, 16686–16695 CrossRef CAS PubMed .
- P. Gadgil, J. Shah and D. S. Chow, Int. J. Pharm., 2018, 544, 265–277 CrossRef CAS PubMed .
- G. Cheng, Q. Zhang, J. Pan, Y. Lee, O. Ouari, M. Hardy, M. Zielonka, C. R. Myers, J. Zielonka, K. Weh, A. C. Chang, G. Chen, L. Kresty, B. Kalyanaraman and M. You, Nat. Commun., 2019, 10, 2205 CrossRef PubMed .
- D. Kolb, N. Kolishetti, B. Surnar, S. Sarkar, S. Guin, A. S. Shah and S. Dhar, ACS Nano, 2020, 14, 11055–11066 CrossRef CAS .
- B. Nancolas, L. Guo, R. Zhou, K. Nath, D. S. Nelson, D. B. Leeper, I. A. Blair, J. D. Glickson and A. P. Halestrap, Biochem. J., 2016, 473, 929–936 CrossRef CAS PubMed .
- L. Cassetta and J. W. Pollard, Nat. Rev. Drug Discovery, 2018, 17, 887–904 CrossRef CAS PubMed .
- W. Jiang, L. Su, M. Ao, X. Guo, C. Cheng, Y. Luo, Z. Xie, X. Wang, J. Wang, S. Liu, Y. Cao, P. Li, Z. Wang, H. Ran, Z. Zhou and J. Ren, J. Nanobiotechnol., 2021, 19, 200–222 CrossRef CAS PubMed .
- Y. Ohue and H. Nishikawa, Cancer Sci., 2019, 110, 2080–2089 CrossRef CAS PubMed .
- X. Li, T. Gruosso, D. Zuo, A. Omeroglu, S. Meterissian, M. C. Guiot, A. Salazar, M. Park and H. Levine, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 3678–3687 CrossRef CAS PubMed .
- D. Chen, X. Zhang, Z. Li and B. Zhu, Theranostics, 2021, 11, 1016–1030 CrossRef CAS PubMed .
- A. Morrot, L. M. da Fonseca, E. J. Salustiano, L. B. Gentile, L. Conde, A. A. Filardy, T. N. Franklim, K. M. da Costa, C. G. Freire-de-Lima and L. Freire-de-Lima, Front. Oncol., 2018, 8, 81 CrossRef PubMed .
- K. E. Allison, B. L. Coomber and B. W. Bridle, Immunology, 2017, 152, 175–184 CrossRef CAS PubMed .
- E. Yang, X. Wang, Z. Gong, M. Yu, H. Wu and D. Zhang, Signal Transduction Targeted Ther., 2020, 5, 242 CrossRef PubMed .
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Bo
Li
b,
Ze-Cong
Xiao
a,
Bo
Li
b,
Ze-Cong
Xiao