
Restraining polysulfide shuttling by designing a dual adsorption structure of bismuth encapsulated into carbon nanotube cavity†
Xingyan
Zeng
,
Yakun
Tang
,
Lang
Liu
 *,
Qingtao
Ma
,
Yang
Gao
,
Mao
Qian
and
Dianzeng
Jia
*,
Qingtao
Ma
,
Yang
Gao
,
Mao
Qian
and
Dianzeng
Jia
State Key Laboratory of Chemistry and Utilization of Carbon Based Energy Resources; Key Laboratory of Advanced Functional Materials, Autonomous Region; College of Chemistry; Institute of Applied Chemistry, Xinjiang University, China. E-mail: liulang@xju.edu.cn
First published on 7th May 2021
Abstract
The shuttle effect derived from the dissolution of lithium polysulfides (LIPs) seriously hinders commercialization of lithium–sulfur (Li–S) batteries. Hence, we skillfully designed 1D cowpea-like CNTs@Bi composites with a double adsorption structure, where the bismuth nanoparticles/nanorods are encapsulated in the cavities of CNTs, avoiding the aggregation of bismuth nanoparticles during cycling and improving the conductivity of the electrode. Meanwhile, the sulfur was evenly distributed on the surface of bismuth nanoparticles/nanorods, ensuring effective catalytic activity and displaying high sulfur loading. Under the synergetic effects of the physical detention of abundant pores and chemical adsorption of bismuth, LIPs can be minimised, effectively curbing the shuttle effect. Benefiting from the above advantages, the CNTs@Bi/S cathodes exhibit a high capacity of 1352 mA h g−1, long cycling lifespan (708 mA h g−1 after 200 cycles at 1 C) and excellent coulombic efficiency. As the anodes of lithium-ion batteries (LIBs), the CNTs@Bi composites also show excellent performance due to the encapsulated structure to accommodate the serious volume change. This work offers an innovative strategy for improving the performances of the Li–S batteries and LIBs.
Lithium-ion batteries (LIBs) have been widely used in emergency illuminations and mobile electronic devices due to their excellent electrochemical performance.1,2 Recently, with the application of more large-scale electronic equipment and the popularization of electronic vehicles, main works have been focused on improving the performance of LIBs and developing alternative energy-storage technology to replenish LIBs for meeting the demand of energy storage equipment.3,4 Li–S batteries are drawing a good deal of attention as next-generation storage, because of their high theoretical capacity (1675 mA h g−1) and energy density (2600 W h kg−1), abundant sulfur resource, low cost, and environmental friendliness.5,6 Regrettably, the shortcomings of inherent insulation,7 unfavorable shuttle effect and severe volume expansion have prevented the commercialization of Li–S batteries.8,9
Tremendous effort has been devoted to solving the above detrimental effects. Porous carbon materials, which can detain lithium polysulfides (LIPs) and reduce the shuttle effect to a certain extent, have been the first choice as the sulfur host.10,11 But the dissolution of LIPs is inescapable as a result of the weak binding force between the nonpolar carbon and polar LIPs.12 In addition, chemical adsorbents such as metal sulfide,13 oxide and nitride, can firmly grasp LIPs,14,15 but amounts of LIPs accumulate on cathodes, resulting in inferior conductivity and few available active materials.16 Recently, metal nanoparticles with excellent electrocatalysis have been used as hosts, which is another effective strategy that can accelerate the conversion of LIPs into Li2S, thereby ensuring high availability of active materials.17,18 However, on account of the high energy surface, the metal nanoparticles agglomerate easily to form nanoclusters, leading to low ion/electron diffusion rate and few active sites for LIPs conversion.19,20 Although the performance of Li–S batteries have been largely enhanced to some extent, the results are still unsatisfactory.
To further improve the efficient application of metal nanoparticles, many methods have been used to design various structures such as nanotubes,21 nanospheres,22,23 nanosheets,24 double-shelled hollow carbon spheres,25,26 and 3D nanostructures; in which, the metal nanoparticles are coated or dispersed in a carbon matrix, thereby it is difficult to aggregate to some extent. But after many cycles of charge/discharge, the structure of electrodes is usually destroyed, and lots of the metal nanoparticles are exposed to the electrolyte, resulting in undesirable side reactions.27 Therefore, a more effective method is to introduce a physical barrier that the metal nanoparticles can be completely segregated by a carbon matrix, which can successfully hinder metal nanoparticles contacting with the electrolyte and avoid side reactions. In addition, such a strategy combines the conductivity and porosity of carbon materials with the catalytic activity of the metal nanoparticles, which can accelerate the ion transmission rate, enlarge active sites and promote the kinetic conversion of LiPs.28,29 Moreover, under the dual effects of the physical barrier and the chemical sorbents, LIPs can be firmly held during cycling, reducing the impact of the shuttle effect.30,31 Therefore, integrating the metal into carbon materials for a sulfur host, provides a vast prospect for advancing the performance of Li–S batteries.
In recent years, bismuth (Bi), with excellent electrocatalytic ability and strong adsorption capacity, was thought to have great potential to integrate with sulfur as the cathode in Li–S batteries.32–34 Moreover, on account of the high theoretical gravimetric capacity (385 mA h g−1) and volumetric capacity (3800 mA h cm−3),35 and low cost, Bi has also attracted considerable interest as the anode of LIBs.36,37 Specially, the large lattice layer spacing can accommodate Li-ion insertion/extraction and alleviate volume variation during cycling.38 Unfortunately, it is easy to aggregate owing to the high surface free energy, which is unfavorable to promote the electrochemical property of LIBs and Li–S batteries. Inspired by the above depiction, dispersing the Bi nanoparticles in the carbon matrix can not only effectively solve the problem of agglomeration, but also enhance the conductivity, avoid side reactions,39 and shorten the ion diffusion pathway.40 Such a structure is beneficial to increase the property of LIBs and Li–S batteries.
Here, we synthesized 1D cowpea-like carbon nanotubes@bismuth (CNTs@Bi) composites via the sol–gel method and subsequent heat treatment, in which the Bi nanoparticles/nanorods are immobilized in the cavities of CNTs, so it is difficult to pulverize and aggregate during cycling. Meanwhile, the sulfur is uniformly distributed on the surface of Bi which can enlarge the contacting area between the electrolyte and the active materials, and the utilization rate of sulfur is effectively improved owing to the high catalytic activity of Bi. More importantly, the composite can be an effective LIP fixator under the constraint of the carbon layer and the strong adsorption of Bi. As a result, it delivers a high capacity, long lifetime and excellent Coulomb efficiency as the cathode of Li–S batteries. Additionally, when the CNTs@Bi composites are applied in LIBs, the electrode also shows high capacity, excellent rate performance and a long lifespan due to sufficient void space that can release the vast volume change during cycling and provide a lot of active sites. The work in this article puts forward a novel idea for LIBs and Li–S batteries with super performance.
The CNTs@Bi composites were synthesized by the sol–gel and calcination method as schematically shown in Scheme 1. Firstly, the precursors for bismuth neodecanoate@sulfonated polymer nanotubes (SPNTs) are obtained through the sol–gel method. Then, through calcination, Bi3+ is reduced to Bi, and SPNTs are turned into CNTs, leading to the formation of the 1D cowpea-like CNTs@Bi, and the composites obtained at the calcination temperature of 550, 650, 750 and 850 °C are marked as BNT1, BNT2, BNT3 and BNT4, respectively. Finally, the cathode materials (CNTs@Bi/S) are attained by in situ growing, fusing and infiltrating of the sulfur on CNTs@Bi.
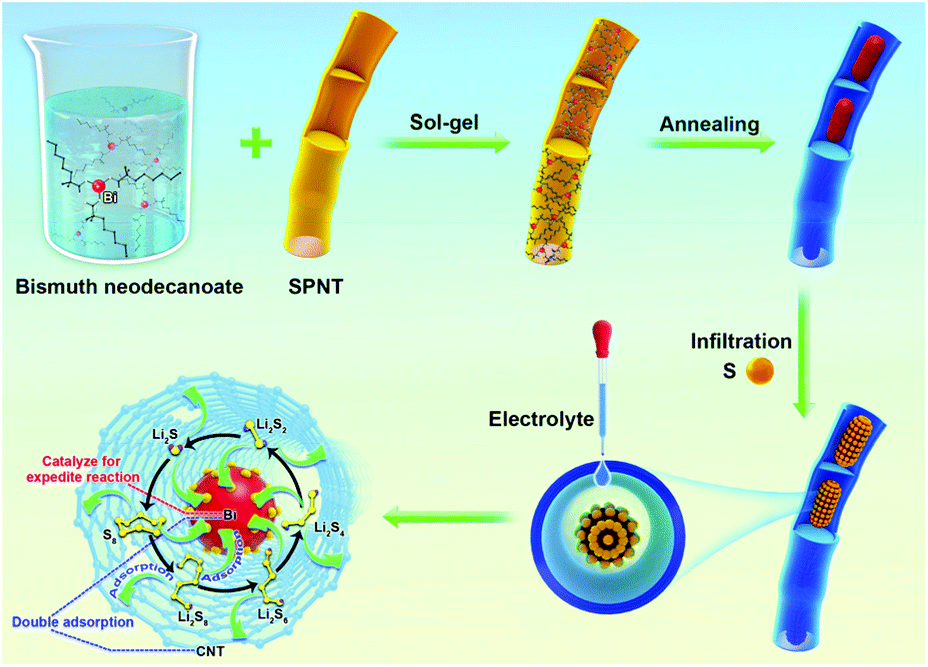 | ||
| Scheme 1 Fabrication process of the CNTs@Bi/S. | ||
Based on using BNT2, BNT3 and BNT4 hosts, the obtained CNTs@Bi/S composites are marked as BNTS2, BNTS3 and BNTS4, respectively.
The composition and crystal structures of BNT1, BNT2, BNT3 and BNT4 were examined by XRD. As shown in Fig. 1a, all the characteristic diffraction peaks of the four samples can be indexed to the rhombohedral Bi (JCPDS no. 85-1329).41 There is little bismuth trioxide in the product calcinated at 550 °C, owing to the incomplete conversion of Bi3+ into Bi at low temperature. The formation mechanism of Bi and chemical reaction route during the calcination are shown in Fig. S1.† It is worth mentioning that the intensity of peaks becomes stronger with the increase of the calcination temperature, except for BNT4, which is attributed to liquid–liquid phase mutation starting to form other phases at 740 °C, while the density and volume expansion coefficient of liquid bismuth decrease with rising temperature after 800 °C.42 The diffraction peak of BNT3 is strongest in all samples, indicating the good crystallinity. The carbon contents of the as-prepared samples were determined via thermogravimetric analyses (TGA). As shown in Fig. S2,† there is weight loss deriving from carbon combustion and weight gain originating in the formation of Bi2O3 occurring between 330 °C and 550 °C, in which the theoretical augmentation of O content is 11%. Therefore, the total carbon contents of BNT1, BNT2, BNT3 and BNT4 can be calculated as 59%, 62%, 68% and 72%, respectively. Carbon content is increased with temperature due to more carbon being consumed as a reducing agent at lower temperatures.
 | ||
| Fig. 1 XRD patterns of BNT1, BNT2, BNT3 and BNT4 (a), SEM and TEM images of BNT1 (b, c, d), BNT2 (f, g, h), BNT3 (i, j, k) and BNT4 (m, n, o), HRTEM image of BNT3 (e), selected area diffraction of BNT3 (l). | ||
The morphologies of BNT1, BNT2, BNT3 and BNT4 were clearly characterized by SEM. As shown in Fig. 1b, f, i and m, all the prepared samples present 1D structures, while their surfaces are smooth and have no agglomeration. Then, the 1D structures are further confirmed by TEM images. As shown in Fig. 1c, g, j and n, it is clearly seen that the samples take on a nanotube structure, and Bi nanoparticles/nanorods are completely encapsulated in the cavities of CNTs, displaying a cowpea-like structure. The structure features are further shown in Fig. 1d, h and o, and it can be seen that the bismuth nanoparticles with spherical-shape (diameter of 30–40 nm) become smaller as the temperature rises. Notably, Bi in BNT3 presents clearly a rod-shaped structure (diameter about 20 nm, length about 50–80 nm) in Fig. 1k, which is attributed to liquid–liquid phase mutation and Peierls distortion at about 740 °C, resulting in massive volume shrinkage and discontinuous change in the density of molten bismuth.42–44 It is worth mentioning that the short Bi nanorods in BNT3 with a large contact area, can facilitate the reaction kinetics, and provide more attachment points for sulfur compared to the spherical shape nanoparticles. Additionally, such an encapsulated structure can furnish adequate void space to adapt the volume variation originating from the cycling process, maintain the integrity of the electrode, avoiding the side reaction between the electrolyte and Bi.
The high-resolution TEM (HRTEM) image of BNT3 in Fig. 1e shows the large lattice fringes spacing of 0.328 nm, matching well with the (012) plane of the rhombohedral Bi. The large crystal spacing is very suitable for the insertion/extraction of Li+, which ensures that the electrode structure is not damaged during the long cycling lifespan. There are no lattice fringes of carbon, indicating the amorphous feature of carbon. The selected area electron diffraction (SAED) pattern from Fig. 1l shows that the crystal structure of Bi nanorods is polycrystalline, and the presented diffraction rings can be well-indexed to the rhombohedral phase of Bi. Furthermore, the energy dispersive spectroscopy (EDS) mapping images of BNT3 show the distribution of Bi and C in the CNTs@Bi composites in the Fig. S3.† It can be clear found that the Bi element are located in the cavities of CNTs, further indicating that the bismuth nanorods are encapsulated in the cavities of CNTs. For understanding the melting point of all the samples, the DSC curves of all the samples were tested and shown in Fig. S4a and c.† The melting points of Bi in BNT1, BNT2, BNT3 and BNT4 are about 271 °C, which are consistent with the reported literature.36 It is worth mentioning that there are the diffraction peaks of Bi and Bi2O3 in the XRD of BNT1, so there are two melting points of 271.6 and 726 °C for Bi and Bi2O3 (shown in Fig. S4b, d and e†), respectively.
The XRD patterns of BNTS2, BNTS3 and BNTS4 are shown in Fig. S5.† All strong characteristic peaks can be indexed to sulfur (JCPDS no. 99-0100), verifying the successful combination of sulfur and the host. The SEM images in Fig. 2a, b and c show the morphologies of BNTS2, BNTS3 and BNTS4, respectively. The surfaces of all samples remain smooth and no appreciable agglomeration of insulating sulfur are found, which can enlarge the contacting area between the electrolyte and the active materials. The TEM images in Fig. 2d, e and f further show the encapsulated structure, which can provide enough void space to accommodate serious volume change during cycling. Notably, such a structure can tightly grasp LIPs under the impedence of CNT walls and chemisorption of Bi. In addition, comparing to the TEM images (Fig. 1h and o) of BNT2 and BNT4, the nanospheres in the cavities of CNTs are changed to the tadpole-like shape in the BNTS2 and BNTS4 after loading sulfur. It is worth noting that there are still nanorods in the cavity of CNTs for BNTS3 (Fig. 2h), which are longer (30–60 nm) compared to the TEM image of BNT3 (Fig. 1k). This can be ascribed to the strong adsorption of bismuth to molten sulfur at around 155 °C, and loading of a large amount of sulfur.
 | ||
| Fig. 2 SEM images of BNTS2, BNTS3 and BNTS4 (a, b, c), TEM images of BNTS2, BNTS3 and BNTS4 (d, e, f), the inserts (g, h, i) are HRTEM images of BNTS2, BNTS3 and BNTS4, respectively. Line-scans of BNTS3 (j), and TGA curves of BNTS2, BNTS3 and BNTS4 (k). | ||
To further reveal the binding form of S and Bi, the HRTEM images of BNTS2, BNTS3 and BNTS4 are shown in Fig. S6.† From Fig. S6a and b,† the lattice fringes can be clearly seen in the head but are not visible on the tail, suggesting the uneven distribution of sulfur on the surface of Bi nanoparticles for BNTS2. Moreover, most of sulfur is located on the tail. By contrast, the lattice fringes of BNTS3 and BNTS4 are blurry shown in Fig. S6c and d,† indicating that sulfur is uniformly coated on the surface of Bi nanorods and nanospheres. What's more, the BNTS3 composite still preserves the nanorod structure in the cavities, in which the large contacting area may be beneficial to promote the catalysis and adsorption of Bi for LIPs. From the line scanning image of BNTS3 in Fig. 2j, it can be seen that the appearance sequence of the element signal is C, S and Bi from the surface to cavities of CNTs, and the peak of S located in the cavity of CNT is strongest, which proves that lots of sulfur infiltrated through the walls of CNTs and covered on the surface of Bi nanorods. The mapping scanning images of BNTS3 in Fig. S7† present the distribution of Bi, C and S elements, and it can be seen that S is concentrated in the part where Bi is located, which further proves that sulfur is evenly distributed on the surface of Bi nanorods. In addition, a small amount of sulfur can be found in the walls of CNTs. The sulfur content of three samples was analyzed by thermogravimetric analyses under a N2 atmosphere. As shown in Fig. 2k, the sulfur contents of BNTS2, BNTS3 and BNTS4 were calculated as 74.9%, 77.3% and 84.0%, respectively. It is worth noting that the terminal temperature for the weightlessness of BNTS3 is higher than those of BNTS2 and BNTS4, suggesting the strongest action between Bi and sulfur in BNT3.
Before discussing performance of Li–S batteries, the 1D cowpea-like CNTs@Bi composites (BNT1, BNT2, BNT3 and BNT4) are first applied in LIBs. As anodes for LIBs, the cycle performance of samples was tested in the voltage window of 0.01 and 3 V as shown in Fig. 3a. The capacities of BNT1, BNT2, BNT3 and BNT4 are 226.8, 251.6, 394.7 and 353.7 mA h g−1 after 100 cycles at 0.1 A g−1, respectively. In which, the capacity of BNT3 is highest, which is attributed to the good crystallinity that can optimize the advantage of the layer structure allowing the insertion/extraction of Li+ and avoid the structural collapse of the electrode. What is more, the Bi nanorods can make more active materials contact with electrolytes to promote the lithiation/delithiation reaction. In addition, the sharp capacity fading can be clearly seen in the initial cycling, which is due to the formation of the stable solid electrolyte interphase (SEI). The rate performances of composites were further investigated with various current densities from 0.05 to 3 A g−1 as shown in Fig. 3b. Among the four samples, the BNT3 electrode displays the highest capacity of 522.6, 333.5, 283.6, 254.5, and 207 mA h g−1 at 0.05, 0.2, 0.5, 1, and 3 A g−1, respectively. When the current density is returned to 0.05 A g−1, the specific capacity can return back to the initial capacity. The marked structural stability of BNT3 further proves that the encapsulated structure can offer adequate void space to alleviate the volume variation at a high current density.
 | ||
| Fig. 3 Cycle performances of BNT1, BNT2 BNT3 and BNT4 over 0.01–3.0 at 0.1 A g−1 (a) and their rate performances (b), cycle performances of BNT3 and BNT4 over 0.01–3.0 V at 2 A g−1 (c), cyclic voltammograms at a scan rate of 0.1 mV s−1 (d). | ||
The cycling stability of BNT3 and BNT4 anodes were discussed at 2 A g−1. As shown in Fig. 3c, a decent capacity with 290 mA h g−1 of BNT3 can be obtained after 500 cycles higher than BNT4 (262 mA h g−1), and the excellent performance of BNT3 further proves that the encapsulated structure containing nanorods can tolerate the mechanical strain caused by volume change during cycling. Even when the current density is increased to 5 A g−1, the capacity of BNT3 can still maintain 170 mA h g−1 after 1000 cycles shown in Fig. S8,† and the coulombic efficiency is 100%. The excellent electrochemical performance profits from the design of the encapsulated structure, (i) the large specific surface area and abundant pores can not only provide more active sites, but also shorten the ion transport pathway to further accelerate the ion transmission rate, and (ii) such an encapsulated structure can supply sufficient spacing to release the volume variation and avoid the collapse of the electrode structure during cycling, resulting in a long lifespan.
Fig. 3d shows the first three CV curves of BNT3 at a scan rate of 0.1 mV s−1 within the potential range of 0.01–3 V. The irreversible broad peaks at 0.4 and 1.4 V in the first cathodic cycle can be ascribed to the decomposition of the electrolyte and the formation of the SEI layer on the electrode surface.45,46 Two peaks are located at 0.77 and 0.70 V, which can be due to the formation of LiBi and Li3Bi, respectively.47,48 The obvious oxidation peak at 0.86 V is owed to the reversible process of the Li–Bi alloy reaction.49 The weak redox peak at 1.78/2.3 V is attributed to the conversion of small amounts of Bi3+ into metallic Bi.50 It is worth mentioning that the CV curves of subsequent scans are completely overlapped after the first scan, indicating the high reversibility of lithiation/delithiation. Fig. S9† shows the first galvanostatic charge–discharge curve of BNT3 at 0.1 A g−1. The plateaus appeared at 0.75–0.77 and 0.85 V are consistent with the redox peak in the CV, and the initial discharge capacity is 792 mA h g−1, which is almost twice the theoretical capacity. In addition, the initial coulombic efficiency is 84.8%, and the large irreversible capacity loss is ascribed to the decomposition of the electrolyte and the formation of the SEI layer.51
Based on the excellent electrochemical performance of the 1D cowpea-like CNTs@Bi composites in LIBs, we further made the BNT2, BNT3 and BNT4 composites as the S hosts in Li–S batteries. The cycling performances of BNTS2, BNTS3 and BNTS4 cathodes with the voltage window of 1.7 to 2.8 V are shown in Fig. 4a. A high initial capacity of 1169 mA h g−1 can be obtained at 0.1 C (1 C = 1.675 A g−1) for the BNTS3 cathode, and a capacity of 708 mA h g−1 can be retained after 200 cycles at the current rate of 1 C with a coulombic efficiency of 99.4%. By contrast, the capacities of BNTS2 and BNTS4 are only 422.2 and 482 mA h g−1. The desirable performance of BNTS3 is attributed to the uniform distribution of sulfur on the surface of Bi nanorods, which can enhance the catalytic efficiency of Bi for LIPs and accelerate the reaction dynamics. It is worth mentioning that there is a slope when the cycle starts at 1 C, which is attributed to the activation process of the active material that the electrolyte penetrates into the cavity from the surface of CNTs and active sulfur.
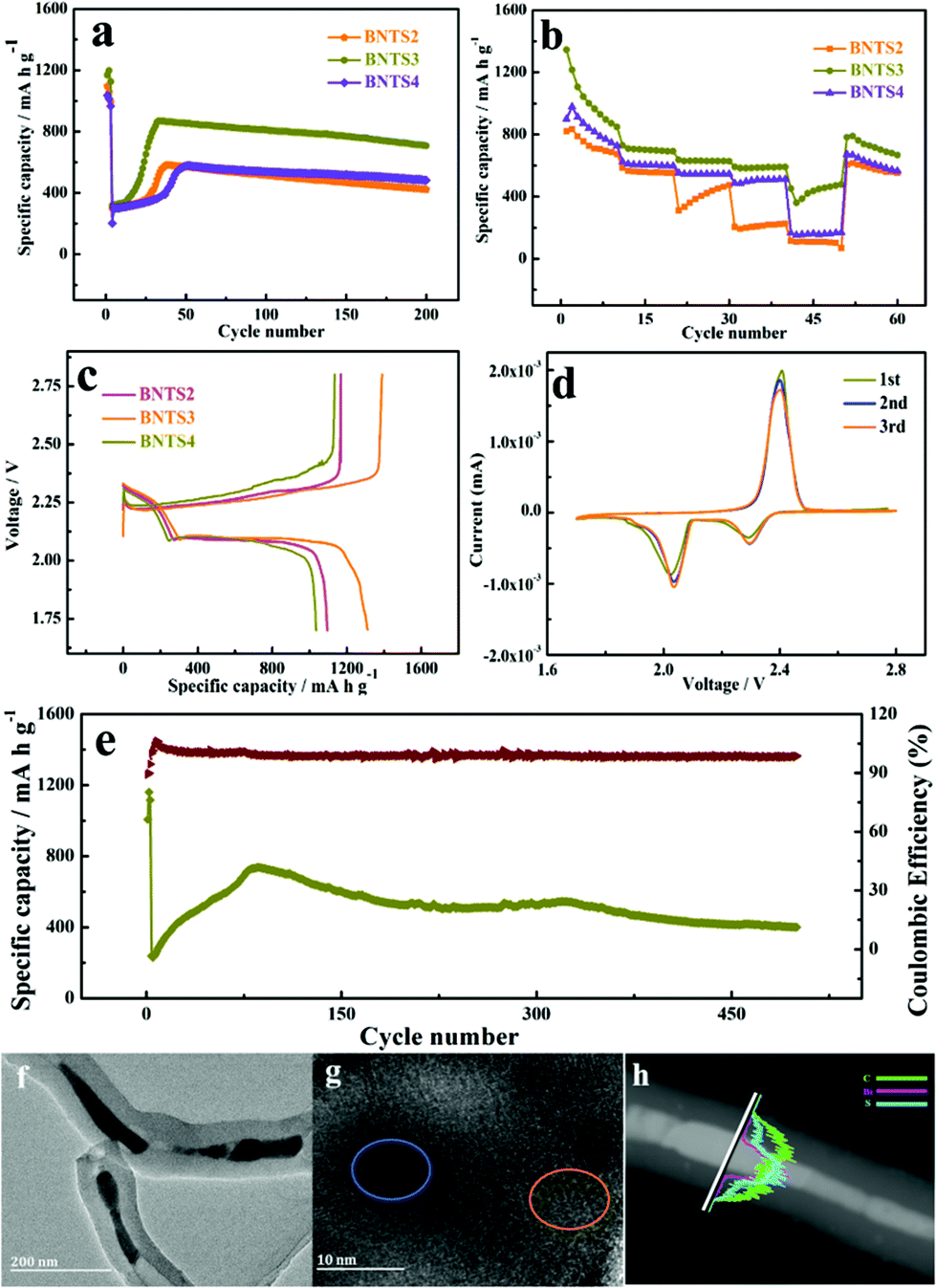 | ||
| Fig. 4 Cycle performances of BNTS2, BNTS3 and BNTS4 over 1.7–2.8 V at 1 C (a), rate performances of BNTS2, BNTS3 and BNTS4 (b), the first cycle charge–discharge profiles of BNTS2, BNTS3 and BNTS4 at 0.05 C (c) and cyclic voltammograms at a scan rate of 0.1 mV s−1 (d), and cycle performance of BNTS3 at 2 C (e), TEM (f), HRTEM (g) and line-scans (h) of BNTS3 after 500 cycles at 2 C. | ||
The rate capabilities of the BNTS2, BNTS3 and BNTS4 cathodes were measured at various current densities from 0.05 to 3 C. As shown in Fig. 4b, the BNTS3 cathode can achieve capacities of 1345, 730.7, 636.6, 628.1, and 590.5 mA h g−1 at 0.05, 0.2, 0.5, 1, and 3 C, respectively. What is more, even after cycling at high current density, the discharge capacity can be backed when the current rate is returned to 0.05 C again, which confirms the superior rate performance of the BNST3 cathode resulting from the high structural stability during cycling. In contrast, the BNTS2 and BNTS4 cathodes exhibit only the capacities of 101.9 and 166.2 mA h g−1 at 3 C. The superior rate performance of the BNTS3 cathode is benefited by the uniform distribution of sulfur, which can promote ion transmission rate and ensure high sulfur utilization.
The 1st cycle charge–discharge voltage profiles of BNTS2, BNTS3 and BNTS4 cathodes at 0.05 C are presented in Fig. 4c. The BNTS3 cathode shows obvious discharge voltage plateau at about 2.3 and 2.0 V, which corresponds to the transformation of S8/LIPs and LIPs/Li2S, respectively. The initial discharge capacity of BNTS3 (1312 mA h g−1) is higher than those of BNTS2 and BNTS4, and the initial coulomb efficiency of BNTS3 can be obtained as high as 94.4%. It is worth noting that there is a low valley at the second voltage platform in the place marked in black, which is ascribed to the rapid conversion of soluble Li2S4 to insoluble Li2S2.52,53 The first three CV curves of the BNTS3 cathodes were measured at a scan rate of 0.1 mV s−1 in the potential range of 1.7 to 2.8 V. As shown in Fig. 4d, there are two distinctly cathodic peaks at 2.31 and 2.02 V, which are attributed to the reduction of sulfur (S8) to soluble intermediate LIPs (Li2Sn, 4 ≤ n ≤ 8), and further insoluble Li2S, respectively.54,55 For the anodic scan, the anodic peak at 2.43 V is relative to the reverse oxidation of short-chain LIPs to S8.56 Notably, the first three CV curves are highly overlapped, indicating a little capacity fading and the high reversibility.
Additionally, a long-life cycling measurement at 2 C is shown in Fig. 4e. The capacity of 400.4 mA h g−1 can be attained after 500 cycles, while the Coulomb efficiency can be up to 98.76%, verifying the superior stability of the BNTS3 cathode. To reveal the architectural feature of BNTS3 after long cycling, the electrode structure after 500 cycles at 2 C was characterised by XRD, Raman, TEM and HRTEM. As shown in Fig. S10a,† the peaks located at 16.3°, 18.1°, 22.7°, 25.7° are ascribed to the characteristic diffraction peaks of S; and the characteristic diffraction peaks of Li2S are located at 26.6°, 31.2°, 44.8° and 65.5°, suggesting that a large amount of S and Li2S covers the surface of the Bi nanorods. In the Raman pattern (Fig. S10b†), the value of ID/IG is higher than in previous work,57 implying the lower graphitization degree and the conductivity due to the coating of S and LIPs. From the TEM image (Fig. 4e and f), it can be seen that the integrated structure still remains after 500 cycles, indicating the excellent structural stability of BNTS3. Moreover, from the HRTEM image (Fig. 4g), it can be found that the lattice fringes can be seen in some places (marked in yellow) due to the consumption of sulfur, and some places (marked in blue) are still blurred because of the coverage of sulfur/LIPs. Additionally, the order of the appearance of the element signal (Fig. 4h) is the same as in Fig. 2j. The above phenomena demonstrate that the diffusion and dissolution of LIPs are powerfully restricted by the collaboration of the physical barrier and the chemical sorbent. The satisfactory electrochemical performance is attributed to the art-designed structure of BNTS3, which can not only enhance the conductivity, but also provide more active sites and accelerate the ion transmission rate owing to the large contact area. Meanwhile, there is sufficient void space to release the severe volume variation to ensure the integrity of electrode structure during cycling, resulting in a long favourable lifespan. Importantly, such an encapsulated structure allows the electrolyte to penetrate into the cavities of CNTs and form LIPs with sulfur, meanwhile the diffusion of LIPs can be effectively restrained under the hindrance of walls of CNTs and the strong chemical adsorption of Bi, so the shuttle effect is effectively suppressed. Furthermore, the high catalytic activity of Bi boosts the conversion between soluble LIPs and Li2S, resulting in the high utilization of active materials.
In order to prove the immobilization between the host materials and LIPs, the visualized adsorption behavior is shown in Fig. 5a and b. After 200 cycles at 1 C, the BNTS3 electrode was dipped in the electrolyte, and the colorless solution soon turned to light yellow due to the dissolution of the few LIPs (Fig. 5a). After 6 hours, the yellow solution returned to colorless owing to the adsorption of the BNT3 host. As shown in Fig. 5b, when adding BNT3 (15 mg) into the 0.1 M Li2S6 solution, the solution changed from dark brown to colorless after 5 h. The results indicate that the CNTs@Bi host exhibits a strong absorption for LIPs even in high concentration Li2S6 solution compared with the reported literature.20,28,30,52,58–60 Both experiments indicate that the composites with the encapsulated structure have strong adsorption capability for LIPs under double adsorption: one is the physical retainment of abundant pores, and the other is chemisorption as shown in Scheme 1.
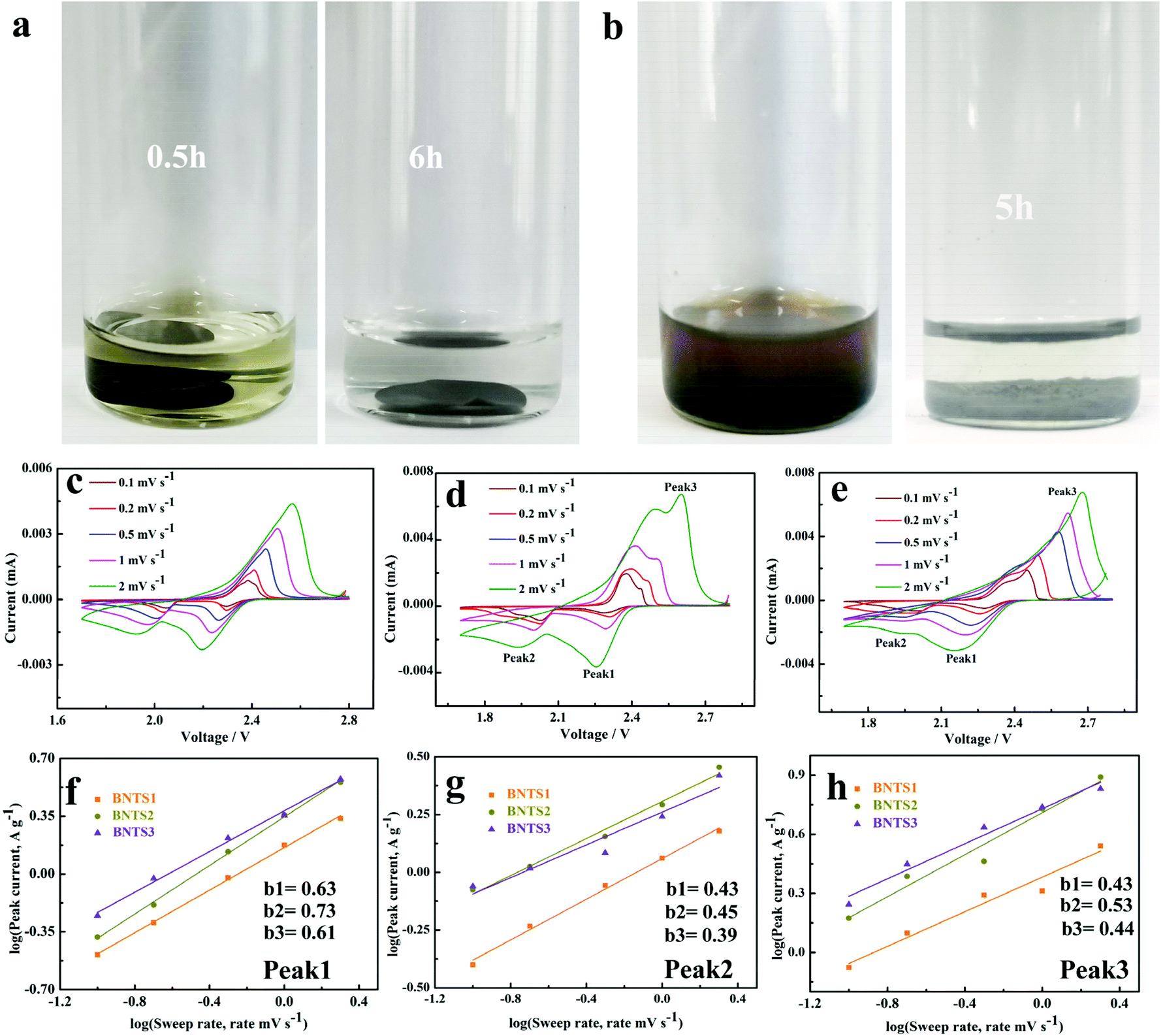 | ||
Fig. 5 Adsorption ability tests of BNTS3 (a and b), CV curves at different scan rates of BNTS2 (c), BNTS3 (d) and BNTS4 (e), plots of the corresponding log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) i vs. logv at peak 1 (f), peak 2 (g), and peak 3 (h). i vs. logv at peak 1 (f), peak 2 (g), and peak 3 (h). | ||
The CV curves of BNTS2, BNTS3 and BNTS4 electrodes at various scan rates from 0.1 to 5 mV s−1 are shown in Fig. 5c, d and e. All the cathodes exhibit one oxidation peak (peak 3) and two reduction peaks (peak 1 and peak 2). To further investigate the electrochemical kinetics of the cathodes, the relationship between the peak current (i) and the scan rate (v) can be analyzed as i = avb, where a is a constant and b varies from 0.5 to 1.0. Typically, when b = 0.5 and 1, it represents diffusion-controlled and capacity-controlled behaviors, respectively.61 As linearly fitted in Fig. 5f, g and h, all b values between 0.43–0.73 are close to 0.5, indicating the diffusion-controlled behavior. The b values of peak 1 (Fig. 5f) correspond to the conversion of S8 to the long-chain LIPs and those of peak 3 (Fig. 5h) relate to the reaction of LIPs to S8. Additionally, both b values (peak 1: 0.73, peak 3: 0.53) of the BNTS3 cathode are higher than those of BNTS2 (peak 1: 0.63, peak 3: 0.43) and BNTS4 (peak 1: 0.61, peak 3: 0.44), suggesting the rapid conversation between S8 and the long-chain LIPs in the BNTS3 cathode due to the large contacting area of Bi nanorods. The b values of all the cathodes for peak 2 (Fig. 5g), corresponding to the conversion of Li2S4 to solid Li2S, are similar, and the reaction kinetics are much slower than peak 1.62
Conclusions
In summary, we designed a cowpea-like CNT@Bi composite by sol–gel and calcination method, in which the Bi nanoparticles/nanorods were completely encapsulated in the cavities of CNTs. As an anode of LIBs, the BNT3 composite can exhibit a high capacity (394.7 mA h g−1 after 100 cycles at 0.1 A g−1), superior cycling stability (170 mA h g−1 after 1000 cycles at 5 A g−1), and excellent rate performance. The outstanding properties are ascribed that there is enough space to tolerate the volume change ensuring the electrode integrity. In addition, as the sulfur host, a high sulfur loading can be attained owing to the adsorbing ability of Bi for sulfur, and the sulfur is evenly distributed on the Bi surface without aggregation, ensuring high conductivity of the electrode. Furthermore, LIPs are firmly grasped under the collaboration of the retained porous carbon structure and the chemical adsorption of Bi, resulting in a weak shuttle effect. Meanwhile, the catalytic activity of Bi can promote the transformation between LIPs and Li2S, increasing the utilization of sulfur. Therefore, the CNTs@Bi/S cathode shows excellent cycling stability, high capacity and good rate performance. Our work in this article comes up with a new strategy for constructing advanced electrodes for LIBs and Li–S batteries.Author contributions
X. Y. Zeng, Y. K. Tang contributed equally to this work. X. Y. Zeng, Y. K. Tang, and L. Liu designed the research, analyzed data and wrote the paper. X. Y. Zeng and Q. T. Ma carried out the electrochemical measurements. Y. Gao, M. Qian and D. Z. Jia carried out the materials characterization.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51902277, 51672235), the Science and Technology Talents Training Project of Urumqi (2019), the Xinjiang Tianshan Youth Doctoral Project (2019Q061), the Natural Science Foundation of Xinjiang Uygur Autonomous Region of China (2019D01C044), the National Ten Thousand Talents Program (2017), and the Graduate Science Innovation Project of Xinjiang Uygur Autonomous Region (XJ2020G037).References
- K. Kim, H. Ma, S. Park and N. S. Choi, ACS Energy Lett., 2020, 5, 1537–1553 CrossRef CAS.
- Q. L. Wei, Y. Q. Fu, G. X. Zhang, D. C. Yang, G. M. Meng and S. H. Sun, Nano Energy, 2019, 55, 234–259 CrossRef CAS.
- X. F. Yang, J. Luo and X. L. Sun, Chem. Soc. Rev., 2020, 49, 2140–2195 RSC.
- A. Fotouhi, D. J. Auger, K. Propp, S. Longo and M. Wild, Renewable Sustainable Energy Rev., 2016, 56, 1008–1021 CrossRef CAS.
- M. Liu, N. P. Deng, J. G. Ju, L. L. Fan, L. Q. Wang, Z. J. Li, H. J. Zhao, G. Yang, W. M. Kang, J. Yan and B. W. Cheng, Adv. Funct. Mater., 2019, 29 Search PubMed.
- M. Jana, R. Xu, X. B. Cheng, J. S. Yeon, J. M. Park, J. Q. Huang, Q. Zhang and H. S. Park, Energy Environ. Sci., 2020, 13, 1049–1075 RSC.
- H. Yin, Q. W. Li, M. L. Cao, W. Zhang, H. Zhao, C. Li, K. Huo and M. Zhu, Nano Res., 2017, 10, 2156–2167 CrossRef CAS.
- Z. W. Seh, Y. M. Sun, Q. F. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- S. J. Ruan, Z. C. Huang, W. D. Cai, C. Ma, X. J. Liu, J. T. Wang, W. M. Qiao and L. C. Ling, Chem. Eng. J., 2020, 385, 123840–123851 CrossRef.
- X. Pu, G. Yang and C. Yu, Adv. Mater., 2014, 26, 7456–7461 CrossRef CAS PubMed.
- R. P. Fang, K. Chen, L. C. Yin, Z. H. Sun, F. Li and H. M. Cheng, Adv. Mater., 2019, 31, 1800863–1800884 CrossRef PubMed.
- J. B. Li, C. Y. Chen, Y. W. Chen, Z. H. Li, W. F. Xie, X. Zhang, M. F. Shao and M. Wei, Adv. Energy Mater., 2019, 9, 1901935–1901944 CrossRef CAS.
- J. C. Jiang, Q. N. Fan, Z. Zheng, M. R. Kaiser, Q. F. Gu, S. L. Chou, K. Konstantinov and J. Z. Wang, ACS Appl. Energy Mater., 2020, 3, 6447–6459 CrossRef CAS.
- D. Chen, X. Y. Yue, X. L. Li, J. Bao, Q. Q. Qiu, X. J. Wu, X. Zhang and Y. N. Zhou, ACS Appl. Mater. Interfaces, 2020, 12, 2354–2361 CrossRef CAS PubMed.
- T. Li, X. Bai, U. Gulzar, Y. J. Bai, C. Capiglia, W. Deng, X. F. Zhou, Z. Q. Liu, Z. F. Feng and R. Proietti Zaccaria, Adv. Funct. Mater., 2019, 29, 1901730–1901786 CrossRef.
- L. L. Zhang, D. B. Liu, Z. Muhammad, F. Wan, W. Xie, Y. Q. Wang, L. Song, Z. Q. Niu and J. Chen, Adv. Mater., 2019, 31, 1903955–1903963 CrossRef CAS PubMed.
- L. Z. Fang, Z. G. Feng, L. Cheng, R. E. Winans and T. Li, Small Methods, 2020, 4, 2000315–2000331 CrossRef CAS.
- Z. Z. Du, X. J. Chen, W. Hu, C. H. Chuang, S. Xie, A. Hu, W. S. Yan, X. H. Kong, X. J. Wu, H. G. Ji and L. J. Wan, J. Am. Chem. Soc., 2019, 141, 3977–3985 CrossRef CAS PubMed.
- Q. F. Zhang, Z. S. Qiao, X. R. Cao, B. H. Qu, J. Yuan, T. E. Fan, H. F. Zheng, J. Q. Cui, S. Q. Wu, Q. S. Xie and D. L. Peng, Nanoscale Horiz., 2020, 5, 720–729 RSC.
- Y. F. Huang, S. X. Chen, Z. L. Wu, J. Wang, Q. Deng, Z. L. Zeng and S. G. Deng, Electrochim. Acta, 2020, 343, 136148–136157 CrossRef CAS.
- L. Sun, M. Y. Li, Y. Jiang, W. B. Kong, K. L. Jiang, J. P. Wang and S. S. Fan, Nano Lett., 2014, 14, 4044–4049 CrossRef CAS PubMed.
- M. Liu, D. Zhou, H. R. Jiang, Y. X. Ren, F. Y. Kang and T. S. Zhao, Nano Energy, 2016, 28, 97–105 CrossRef CAS.
- H. B. Din, J. Zhou, A. M. Rao and B. G. Lu, Natl. Sci. Rev., 2020, nwaa276 Search PubMed.
- H. B. Ding, J. Wang, L. Fan, Z. M. Liu, X. X. Jia, X. Z. Yu and B. G. Lu, Chem. Eng. J., 2020, 395, 125147–125152 CrossRef CAS.
- L. Zhou, D. L. Danilov, R. A. Eichel and P. H. L. Notten, Adv. Energy Mater., 2020, 2001304–1001352 Search PubMed.
- H. D. Liu, Z. L. Chen, L. Zhou, X. Li, K. Pei, J. Zhang, Y. Song, F. Fang, R. Che and D. Sun, J. Mater. Chem. A, 2019, 7, 7074–7081 RSC.
- S. P. Li, X. Chen, F. Hu, R. Zeng, Y. H. Huang, L. X. Yuan and J. Xie, Electrochim. Acta, 2019, 304, 11–19 CrossRef CAS.
- P. Zeng, C. Liu, X. F. Zhao, C. Yuan, Y. G. Chen, H. P. Lin and L. Zhang, ACS Nano, 2020, 14, 11558–115469 CrossRef CAS PubMed.
- L. Fan, S. H. Chen, J. Y. Zhu, R. F. Ma, S. P. Li, R. Podila, A. M. Rao, G. Z. Yang, C. X. Wang, Q. Liu, Z. Xu, L. Yuan, Y. H. Huang and B. G. Lu, Adv. Sci., 2018, 5, 1700934–1700942 CrossRef PubMed.
- B. Long, Z. P. Qiao, J. N. Zhang, S. Q. Zhang, M. S. Balogun, J. Lu, S. Song and Y. X. Tong, J. Mater. Chem. A, 2019, 7, 11370–11378 RSC.
- H. B. Ding, Q. F. Zhang, Z. M. Liu, J. Wang, R. F. Ma, L. Fan, T. Wang, J. G. Zhao, J. M. Ge, X. L. Lu, X. Z. Yu and B. G. Lu, Electrochim. Acta, 2018, 284, 314–320 CrossRef CAS.
- Y. C. Hao, Y. Guo, L. W. Chen, M. Shu, X. Y. Wang, T. A. Bu, W. Y. Gao, N. Zhang, X. Su and X. J. N. C. Feng, Nat. Catal., 2019, 2, 448–456 CrossRef CAS.
- X. N. Li, J. W. Liang, Y. Lu, Z. G. Hou, W. Q. Zhang, Y. C. Zhu and Y. T. Qian, J. Power Sources, 2016, 329, 379–386 CrossRef CAS.
- H. F. Xu, S. B. Yang and B. Li, J. Mater. Chem. A, 2020, 8, 149–157 RSC.
- Y. Zhang, Q. Wang, B. Wang, Y. Mei and P. C. Lian, Ionics, 2017, 23, 1407–1415 CrossRef CAS.
- X. H. Liu, S. L. Zhang, S. Y. Guo, B. Cai, S. A. Yang, F. K. Shan, M. Pumera and H. B. Zeng, Chem. Soc. Rev., 2020, 49, 263–285 RSC.
- M. Pumera and Z. Sofer, Adv. Mater., 2017, 29, 1605299–1605306 CrossRef PubMed.
- W. W. Hong, P. Ge, Y. L. Jiang, L. Yang, Y. Tian, G. Q. Zou, X. Y. Cao, H. S. Hou and X. B. Ji, ACS Appl. Mater. Interfaces, 2019, 11, 10829–10840 CrossRef CAS PubMed.
- H. Yang, R. Xu, Y. Yao, S. F. Ye, X. F. Zhou and Y. Yu, Adv. Funct. Mater., 2019, 29, 1809195–1809204 CrossRef.
- T. C. Jiang, F. X. Bu, X. X. Feng, I. Shakir, G. L. Hao and Y. X. Xu, ACS Nano, 2017, 11, 5140–5147 CrossRef CAS PubMed.
- L. B. Wang, C. C. Wang, F. J. Li, F. J. Cheng and J. Chen, Chem. Commun., 2017, 54, 38–41 RSC.
- Y. Greenberg, E. Yahel, E. N. Caspi, C. Benmore, B. Beuneu, M. P. Dariel and G. Makov, Europhys. Lett., 2009, 86, 36004–36010 CrossRef.
- Y. Shu, D. Yu, W. T. Hu, Y. B. Wang, G. Y. Shen, Y. S. Kono, B. Xu, J. L. He, Z. Y. Liu and Y. J. Tian, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3375–3380 CrossRef CAS PubMed.
- M. Emuna, S. Matityahu, E. Yahel, G. Makov and Y. Greenberg, J. Chem. Phys., 2018, 148, 034505–034511 CrossRef CAS PubMed.
- J. Y. Hwang, S. T. Myung and Y. K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- C. P. Han, Y. B. He, M. Liu, B. H. Li, Q. H. Yang, C. P. Wong and F. Y. Kang, J. Mater. Chem. A, 2017, 5, 6368–6381 RSC.
- H. C. Yuan, Y. Q. Jin, X. N. Chen, J. L. Lan, Y. H. Yu and X. P. Yang, ACS Sustainable Chem. Eng., 2019, 7, 6033–6042 CrossRef CAS.
- Y. T. Zhong, B. Li, S. M. Li, S. Y. Xu, Z. H. Pan, Q. M. Huang, L. D. Xing, C. S. Wang and W. S. Li, Nanomicro Lett., 2018, 10, 56–69 Search PubMed.
- W. Wang, L. Liu, P. F. Wang, T. T. Zuo, Y. X. Yin, N. Wu, J. M. Zhou, Y. Wei and Y. G. Guo, Chem. Commun., 2018, 54, 1714–1717 RSC.
- R. Dai, Y. H. Wang, P. Da, H. M. Wu, M. Xu and G. F. Zheng, Nanoscale, 2014, 6, 13236–13241 RSC.
- P. X. Xiong, P. X. Bai, A. Li, B. F. Li, M. R. Cheng, Y. P. Chen, S. P. Huang, Q. Jiang, X. H. Bu and Y. H. Xu, Adv. Mater., 2019, 31, 1904771–1904778 CrossRef CAS PubMed.
- Z. P. Ye, Y. Jiang, T. Feng, Z. H. Wang, L. Li, F. Wu and R. J. Chen, Nano Energy, 2020, 70, 104540–104549 CrossRef.
- W. W. Sun, Y. J. Li, S. K. Liu, Q. P. Guo, Y. H. Zhu, X. B. Hong, C. M. Zheng and K. Xie, Nano Res., 2020, 13, 2143–2148 CrossRef CAS.
- J. R. He, Y. F. Chen and A. Manthiram, Energy Environ. Sci., 2018, 11, 2560–2568 RSC.
- B. Yu, Y. F. Chen, Z. G. Wang, D. J. Chen, X. Q. Wang, W. L. Zhang, J. R. He and W. D. He, J. Power Sources, 2020, 447, 227364–227374 CrossRef CAS.
- J. Yu, J. W. Xiao, A. R. Li, Z. Yang, L. Zeng, Q. F. Zhang, Y. J. Zhu and L. Guo, Angew. Chem. Int. Ed. Engl., 2020, 59, 13071–13078 CrossRef CAS PubMed.
- Y. K. Tang, L. Liu, X. C. Wang, H. J. Zhou and D. Z. Jia, RSC Adv., 2014, 4, 44852–44857 RSC.
- X. Chen, T. Z. Hou, K. A. Persson and Q. Zhang, Mater. Today, 2019, 22, 142–158 CrossRef CAS.
- H. F. Xu, Q. B. Jiang, B. K. Zhang, C. Chen and Z. Lin, Adv. Mater., 2020, 32, 1906357–1906365 CrossRef CAS PubMed.
- T. Liu, X. F. Li, H. J. Nie, C. Xu and H. M. Zhang, J. Power Sources, 2015, 286, 73–81 CrossRef CAS.
- J. W. Gallaway, G. G. Yadav, D. E. Turney, M. Nyce, J. Huang, Y.-c. Karen Chen-Wiegart, G. Williams, J. Thieme, J. S. Okasinski, X. Wei and S. Banerjee, J. Electrochem. Soc., 2018, 165, A2935–A2947 CrossRef CAS.
- J. Y. Lee, G. D. Park, J. H. Choi and Y. C. Kang, Nanoscale, 2020, 12, 2142–2153 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, materials characterization, electrochemical characterization, and some characterization of prepared samples. See DOI: 10.1039/d1nr01456k |
| This journal is © The Royal Society of Chemistry 2021 |