
Doxorubicin and PD-L1 siRNA co-delivery with stem cell membrane-coated polydopamine nanoparticles for the targeted chemoimmunotherapy of PCa bone metastases†
Xupeng
Mu‡
 a,
Meng
Zhang‡
a,
Anhui
Wei
b,
Fei
Yin
c,
Yan
Wang
d,
Kebang
Hu
*e and
Jinlan
Jiang
*a
a,
Meng
Zhang‡
a,
Anhui
Wei
b,
Fei
Yin
c,
Yan
Wang
d,
Kebang
Hu
*e and
Jinlan
Jiang
*a
aScientific Research Center, China-Japan Union Hospital, Jilin University, Changchun, China. E-mail: jiangjinlan@jlu.edu.cn
bCollege of Pharmacy, Jilin University, Changchun, China
cDepartment of Orthopedics, China-Japan Union Hospital, Jilin University, Changchun, China
dDepartment of Hepatobiliary surgery, China-Japan Union Hospital, Jilin University, Changchun, China
eDepartment of Urology, The First Hospital of Jilin University, Changchun, China. E-mail: hukb@jlu.edu.cn
First published on 22nd April 2021
Abstract
Programmed cell death ligand 1 (PD-L1) blockade has achieved great success in cancer immunotherapy. PD-L1 siRNA can restore the immune anti-tumor activity of T cells by downregulating the level of PD-L1 on tumor cells, but the efficiency of PD-1/PD-L1 monotherapy is relatively low. Doxorubicin (DOX) can induce tumor cell apoptosis, and then increase the release of tumor antigen. But the expression of PD-L1 in tumor tissues treated with DOX will be enhanced adaptively. Therefore, DOX combination with PD-L1 siRNA can produce a good synergistic anti-tumor effect. In this study, stem cell membrane (SCM) camouflaged polydopamine nanoparticles carrying DOX and PD-L1 siRNA (PDA–DOX/siPD-L1@SCM) were constructed for targeting prostate cancer (PCa) bone metastases. PDA–DOX/siPD-L1@SCM NPs could effectively enhance blood retention and improve accumulation at tumor sites. In vitro and in vivo studies demonstrated that PDA–DOX/siPD-L1@SCM NPs showed excellent performance in synergistic chemoimmunotherapy for PCa bone metastases. Hence, this study provided an effective strategy for developing biomimetic multifunctional nanoparticles for PCa bone metastasis treatment.
Introduction
Immunotherapy has become the most promising research direction for cancer treatment in recent years. Tumor cells can promote the formation of the immunosuppressive tumor microenvironment and inhibit the function of immune cells by upregulating the expression of programmed death 1 (PD-1) and its ligand PD-L1, thus leading to tumor immune escape.1,2 Overexpression of PD-L1 is an important mechanism for immune evasion in many malignant tumors and is usually associated with a poor prognosis.3 PD-1/PD-L1 inhibitors block the negative regulatory signal by blocking the binding of PD-1 and PD-L1, and reactivating the immune response of T cells to the tumor, so as to achieve the anti-tumor effect. In recent years, compared to chemotherapy drugs, anti-PD-L1 drugs approved by FDA have better therapeutic effects and less side effects. However, anti-PD-L1 drugs still have immune toxicity and side effects and are very easy to produce drug resistance due to the mutation of the target protein.4 Therefore, in order to reduce the toxicity and drug resistance, and to enhance the anti-tumor effect, it is necessary to target PD-L1 at the gene level, such as RNA or DNA. At present, there are a few studies on siRNA targeting PD-L1. For example, by the combination of PD-L1 siRNA with tumor targeting polymers, micelles can specifically downregulate the expression of PD-L1 at the mRNA level in tumor cells, activate the killing effect of T lymphocytes, and inhibit the proliferation of tumor cells.5–7 In addition to the efficacy of antibody drugs, siRNA drugs can also avoid failure due to the mutation of PD-L1 protein. However, due to the low efficiency of immunotherapy for solid tumors such as prostate cancer, the combination of gene immunotherapy and drug therapy has become a hot spot in solid tumor research.Prostate cancer is one of the major tumors that seriously endanger men's health. Most patients have metastasis at the initial diagnosis. One of the most common metastatic sites of prostate cancer is bone, which affects the hematopoietic function of bone marrow and destroys the bone structure. Bone metastasis becomes one of the main causes of death in patients with advanced prostate cancer.8 After distant metastasis of prostate cancer, the effect of surgical treatment is very limited. Therefore, chemotherapy and other therapies have gradually become the most important palliative therapy for patients with metastasis of prostate cancer.
As one of the commonly used chemotherapeutic drugs for prostate cancer, doxorubicin (DOX) can induce tumor cell apoptosis and increase the release of tumor antigen, thus enhancing the immunogenicity of tumor cells and ultimately upregulating the function of CD8+ T cells. So DOX can transform an immune desert or immune rejection tumor (cold tumor) into an inflammatory tumor (hot tumor) and increase the sensitivity of the tumor to immune checkpoint treatment.9 However, the expression of PD-L1 in tumor tissues treated with DOX will be increased adaptively.10,11 Therefore, the combination of DOX and immunosuppressant PD-L1 siRNA can enhance the body's own immunity. Although DOX can prolong the overall survival time of patients with prostate cancer, it will cause related toxic side effects. During the course of administration, DOX can be easily cleared by the liver or kidneys, which limits the effect of tumor inhibition.12 Meanwhile, due to its polyanionic and instability character, it is difficult for siRNA to enter solid tumor cells by a passive diffusion mechanism.13 Therefore, an efficient drug delivery system should be used to improve the collaborative therapy efficiency of prostate cancer. Nanomaterials provide a new method to solve the synergistic anti-tumor problem of conventional chemotherapy drugs and gene drugs, due to their various properties and advantages. Dopamine has good hydrophilicity, biocompatibility, stability and biodegradability and can spontaneously polymerize to polydopamine (PDA) nanoparticles (NPs) in aerobic alkaline solutions.14 In addition, PDA has a large number of catechol and amino functional groups, which can bind many functional molecules to the surface, making it a potential drug carrier material.15,16
Due to the risk of immune rejection caused by NPs, researchers are trying to modify the surface of NPs. In recent years, nanomaterials coated with cell membranes have been used for drug and gene delivery, and great progress has been made in cancer treatment.17,18 Of all the cells, mesenchymal stem cells (MSCs) have demonstrated tropism toward tumor microenvironments. However, the specific mechanism of the tumor tropism of MSCs is complicated, mainly involving chemokine-receptor interaction.19 Tumor cells and tumor-associated stromal cells could produce a lot of chemokines, including platelet-derived growth factor (PDGF), stromal cell-derived factor-1 (SDF-1), and vascular endothelial growth factor (VEGF), which could attract MSCs to migrate.20 It has been found that NPs camouflaged with mesenchymal stem cell membranes (SCMs) could migrate to the tumor sites and effectively inhibit the growth of the tumors.17,18 The targeting and immune camouflage properties of mesenchymal stem cell membrane (SCM) surface proteins greatly improve the target and biocompatibility of NPs, which provide a new targeted carrier for drugs and genes in vivo.
Herein, we developed mesenchymal stem cell membrane-coated PDA NPs as an efficient tumor-targeting delivery platform, which encapsulated DOX and siPD-L1 (PDA-DOX/siPD-L1@SCM NPs) for the chemoimmunotherapy of PCa bone metastases. DOX and PD-L1 siRNA can be efficiently loaded onto the surface of PDA NPs via π–π stacking interactions. PDA–DOX/siPD-L1@SCM NPs not only reduce the toxicity of DOX but also improve the efficiency of PD-L1 siRNA in vivo, resulting in a better synergistic therapeutic effect for PCa bone metastases.
Experimental section
Materials and reagents
Dulbecco's modified Eagle medium (DMEM) with high glucose, fetal bovine serum (FBS), penicillin–streptomycin, and DMEM-F12 were purchased from ThermoFisher (Waltham, MA, USA). Cell counting kit-8 (CCK-8), DAPI, TUNEL apoptosis assay kit, and BCA protein assay kit were obtained from Beyotime (China). Dopamine hydrochloride and doxorubicin hydrochloride (DOX) were purchased from Millipore-Sigma (Darmstadt, Germany). Other reagents used in this work were obtained from Solarbio (China) and Aladdin-Reagent (China). SiRNA targeting PD-L1 (S1: 5′-CUGGGAGCCAUCUUAUUAUTT-3′; S2: 5′-GCCGAAGUCAUCUGGACAATT-3′; S3: 5′-CCAGCACACUGAGAAUCAATT-3′) and siRNA-NC labeled with FAM (siRNAFAM) were ordered from GenePharma (China). The primers were ordered from Sangon (China). Anti-PD-L1, anti-ki67, and anti-β-actin were purchased from Proteintech (IL, USA). IRDye®680 goat anti-mouse IgG was obtained from Li-COR Biosciences Inc (NE, USA).PC-3 prostate cancer cells and human umbilical cord–derived mesenchymal stem cells (MSCs) were purchased from American Type Culture Collection (Manassas, VA, USA). All cells were cultured according to the instructions.
Synthesis and characterization of PDA NPs
PDA NPs were synthesized according to the previous report.21 Briefly, 40 mL of ethanol and 90 mL of water were mixed at room temperature, followed by the addition of 2 mL of an aqueous solution of ammonia (28–30%) and stirred for 30 min. Dopamine hydrochloride (0.5 g) in water (10 mL) was added to the above mixture in a dropwise manner. The reaction was allowed to proceed for 6 h until the color of the solution changed to dark brown. Finally, the products were obtained by centrifugation and washed with deionized water.Drug loading
PDA (1 mg) was suspended in 10× Tris-buffer (0.5 mL) and mixed with DOX solution (2 mL, 1 mg mL−1). After stirring at room temperature for 24 h, the obtained PDA–DOX NPs were purified by centrifugation and washed with deionized water. The DOX in the supernatant was collected, and the concentration of unloaded DOX was analyzed by using the calibration curve of DOX at a wavelength of 480 nm in the UV-vis spectrum. The loading efficiency of DOX was calculated as follows: LE (%) = (weight of loaded DOX/weight of the NPs) × 100%. The Fourier transform-infrared (FTIR) spectra of PDA and PDA–DOX were obtained using a Bruker Vertex 70 FTIR spectrometer (Bruker, Karlsruhe, Germany).Loading siRNA onto PDA–DOX NPs
50 nM siRNA (siPD-L1 or siRNAFAM) was mixed with varying PDA concentrations (0, 0.02, 0.04, 0.08, 0.16, and 0.32 mg mL−1). After magnetically stirring for 30 min at 4 °C, the prepared PDA–DOX/siRNA NPs were centrifuged at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min and then the fluorescence spectra of the supernatant were measured using a Hitachi F-4600 fluorescence spectrophotometer (Hitachi, Japan).
000 rpm for 10 min and then the fluorescence spectra of the supernatant were measured using a Hitachi F-4600 fluorescence spectrophotometer (Hitachi, Japan).
Preparation of the MSC membrane
The mesenchymal stem cell membrane-derived vesicles were prepared according to the method previously reported.14 When MSCs almost covered the entire culture flasks, the cells were harvested and resuspended in a hypotonic lysis buffer (1 mM NaHCO3, 1 mM PMSF, and 0.2 mM EDTA) at 4 °C overnight, followed by sonicating on ice for 5 min. After centrifuging at 2000g for 10 min at 4 °C, the resulting supernatant was collected and centrifuged again at 15000g for 30 min. The obtained cell membrane precipitate was re-suspended in PBS.
PDA–DOX/siPD-L1@SCM NP synthesis and characterization
To prepare the MSC membrane–derived vesicles, the obtained stem cell membranes were extruded using a mini extruder (Avanti Polar Lipids, USA) through 400 nm and 200 nm porous polycarbonate membranes (10 times), respectively. Then, the prepared PDA–DOX/siPD-L1 NP cores were co-extruded with vesicles through a 200 nm polycarbonate membrane for 10 times. The resultant PDA–DOX/siPD-L1@SCM NPs were stored in PBS at 4 °C before use.To quantify the obtained nanoparticles and the loss of DOX during the physical extrusion process, 2 mL of PDA–DOX (1 mg mL−1) was physically extruded through 400 nm and 200 nm porous polycarbonate membranes 10 times. The same amount of PDA–DOX NPs without extrusion was used as the control group. Then the obtained PDA–DOX@SCM NPs and PDA–DOX NPs were immersed in PBS at pH 5.0 for 24 h. The concentration of DOX released was determined at 480 nm by UV-Vis and the amounts of DOX were calculated. Then we could quantify the obtained nanoparticles according to the loss of DOX.
The structure of the resulting NPs was observed using a transmission electron microscope (TEM, Jeol, Akishima Tokyo, Japan). Particle size and zeta potential were measured by dynamic light scattering (DLS) using a Malvern Zetasizer Nano-ZS instrument (Malvern Instruments, Malvern, UK).
Characterization of cell membrane protein
The samples of stem cell membrane derived vesicles and PDA–DOX/siRNA@SCM NPs were lysed with RIPA lysis buffer (Beyotime, Shanghai, China), and then the protein concentration was quantified using a BCA protein kit (Beyotime, Shanghai, China). After being mixed with a loading buffer and boiled for 5 min, the samples (100 μg per well) were loaded into each well of SDS-PAGE gel. After electrophoresis, the gel was stained using Coomassie Brilliant Blue (Beyotime, Shanghai, China) and destained in acetic acid overnight before imaging.In vitro DOX release
Briefly, PDA–DOX NPs, and PDA–DOX@SCM NPs were suspended in PBS, sealed in a dialysis bag (MWCO = 8000 Da), and then immersed in a PBS solution at pH 5.0, or 7.4 at 37 °C with moderate shaking for different periods of time. At designated time intervals, DOX in the release medium was collected and analyzed by UV-Vis spectroscopy to measure the concentration of released DOX. For each measurement, the release medium was replaced with an equal volume of fresh PBS to keep the volume constant.Cell viability assay
The in vitro cytotoxicity of PDA–DOX/siRNA@SCM NPs was evaluated using the CCK-8 assay. The 3 × 104 PC-3 cells were first seeded in a 96-well plate and incubated overnight. Then, the media were replaced with fresh media containing different concentrations of nanocomposites and were incubated for another 24 h. Finally, CCK-8 (10 μL) was added to each well for 3 h and then the absorbance of the sample at 450 nm was read using a microplate reader (ELx-800, BioTek Instruments, USA).Cellular uptake and internalization of PDA–DOX/siRNA@SCM NPs
The cellular uptake of PDA–DOX/siRNA@SCM NPs was investigated using both fluorescence microscopy and flow cytometry. For cellular uptake experiments, PC-3 cells were treated with free DOX + siRNAFAM, PDA–DOX/siRNA NPs, or PDA–DOX/siRNA@SCM NPs in a complete DMEM medium for 4 h, respectively. Thereafter, the cells were washed with PBS, and fixed with 4% paraformaldehyde and DAPI to stain the nuclei. The stained cells were observed under a confocal laser scanning microscope (IX5-RFACA, Olympus, Japan) at excitation wavelengths of 488, 525, and 633 nm.For flow cytometry, the PC-3 cells were treated with free-siRNA, PDA–siRNA NPs, PDA–siRNA@SCM NPs or DOX, PDA–DOX NPs, and PDA–DOX@SCM NPs as described above in 6-well plates. After 4 h of incubation, the cells were harvested, resuspended in PBS and quantitatively analyzed using an FACS Calibur flow cytometer (BD Biosciences, USA). The untreated cells were used as the control for background calibration.
Gene silencing efficiency of PD-L1 siRNA via PDA/siPD-L1@SCM NPs
The PC-3 cells were seeded in 6-well plates (2 × 105 cells per well) and then incubated with naked siPD-L1 or PDA–siPD-L1@SCM NPs (siPD-L1 concentration of 100 nM) for 24 h. Lipofectamine 2000 (Lipo2k) carrying 100 nM of siPD-L1 was used as the positive control. Total RNA was extracted from the transfected cells using an RNeasy mini kit (Qiagen, CA) according to the manufacturer's protocol. The PD-L1 mRNA was detected by quantitative real-time PCR (qRT-PCR) with an ABI StepOne Plus system (Applied Biosystems, USA) assisted with an SYBR Green Master Mix kit (Thermo Fisher, USA).The PC-3 cells were treated as described above for qRT-PCR analysis and were harvested at 72 h after incubation with various formulations. For western blot analysis, proteins were extracted using the RIPA lysis buffer (Beyotime, China), and the protein concentrations of different samples were measured using a BCA protein assay kit (Beyotime, China). Subsequently, equal amounts of protein (100 μg) were separated on 10% SDS-PAGE and transferred onto nitrocellulose membranes (Millipore, MA, USA). After blocking with 5% nonfat dry milk for 1 h, the membranes were incubated overnight at 4 °C with the primary anti-PD-L1 or anti-β-actin antibodies (1:1000). After washing three times with a TBST buffer, the membranes were further incubated with IRDye®680 goat anti-mouse secondary antibodies (1:10000) for 1 h. The images were analyzed using an Odyssey infrared imaging system (LI-COR Biosciences, NE, USA).
Animal model
BALB/c nude mice (male, 4–6 weeks old) were purchased from the Vital River Company (Beijing, China). All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Jilin University and approved by the Animal Ethics Committee of Jilin University (Changchun, China). The bone metastasis model of prostate cancer in mice was established by injecting PC-3 cells into the bone marrow cavity of the tibia. Specifically, the mice were anesthetized with pentobarbital sodium and exposed to the right tibia under sterile conditions. Then, 30 μL of PBS containing 5 × 105 PC-3 cells was injected into the tibial bone marrow cavity. Subsequently, the injection site was sealed with bone wax and the wound was sutured. About 21 days later, spontaneous elevations of the right hind limb were observed, which indicated that the mouse model of prostate cancer bone metastasis was successfully established.Pharmacokinetics and biodistribution in vivo
To study the pharmacokinetics of NPs in the blood and their biodistribution in major organs and tumor tissues, 100 μL of DOX, PDA–DOX NPs or PDA–DOX@SCM NPs (DOX dose: 5 mg kg−1) were injected into the BALB/c nude mice with PC-3 bone metastasis via the tail vein. At a predetermined time point, blood was collected from the mouse eyeballs and the DOX fluorescence intensity of the blood was analyzed using a microplate reader (Bio-Rad, CA, USA). The total weight of blood was estimated to be 6% of the total mice body weight.22 The tissues and main organs (heart, liver, spleen, lung, kidneys and tumor) were collected, weighed, and homogenized in a lysis buffer (Sigma-Aldrich, MO, USA). Afterward, the lysate of each tissue sample was centrifuged at 1200 rpm for 5 min and the DOX in the supernatant was measured at 480 nm using a microplate reader (Bio-Rad, CA, USA).Antitumor efficacy and histological examinations in vivo
When the tumor volume reached about 100 mm3, the tumor-bearing mice were randomly divided into 7 groups (n = 5 per group) and treated with PBS, free siPD-L1, PDA–siPD-L1, DOX, PDA–DOX, PDA–DOX/siPD-L1 NPs and PDA–DOX/siPD-L1@SCM NPs (DOX dose: 5 mg kg−1; siPD-L1 dose: 0.31 mg kg−1). Tumor volume and body weight were measured every other day. Tumor volume (V) was calculated using the formula: V = L × W2/2 (W and L are the means of the shortest and the longest diameters, respectively). Mice were euthanized and then the major organs and tumor forming legs were collected 4 days after the last treatment. The soft tissue of the tumor forming legs was carefully removed, placed in 4% paraformaldehyde and scanned by Micro CT (Bruker, Skycan1172, Kontich, Belgium) or by X-ray (In vivo Imaging Instruments, PerkinElmer, Waltham, USA). The major organs and tumors were sectioned for histological examination by hematoxylin–eosin (H&E) staining or immunohistochemical staining. Tumor damage was also detected by TUNEL staining to further explore the effect of the co-delivered NPs.Statistical analysis
All experimental data were expressed as means ± standard deviations. The statistical analyses were assessed using Student's t-test or one-way analysis of variance.Results and discussion
Construction and characterization of PDA–DOX/siRNA@SCM NPs
The synthetic process of PDA–DOX/siRNA@SCM NPs is schematically demonstrated in Fig. 1, and was mainly divided into the following three steps: synthesizing PDA NPs, loading DOX and siRNA onto PDA NPs, and then coating PDA–DOX/siRNA NPs with obtained MSC-vesicles. PDA NPs were synthesized through spontaneous in situ polymerization under weakly alkaline conditions.18 As shown in the TEM image (Fig. 2A), the as-prepared PDA NPs had an average diameter of approximately 95 nm.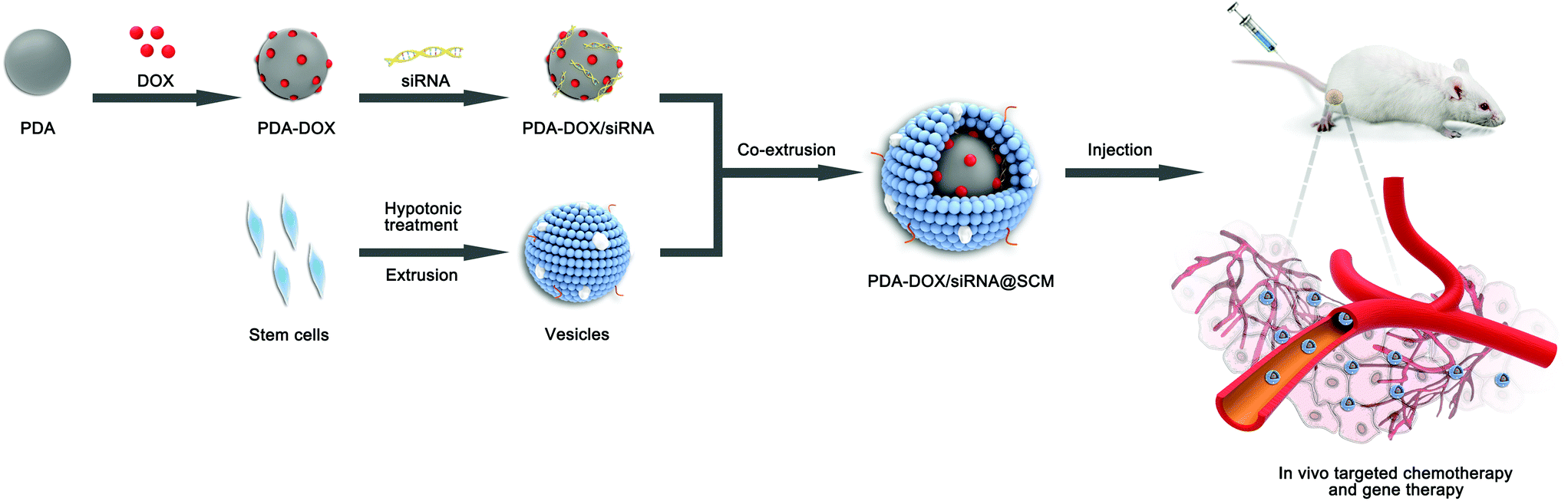 | ||
| Fig. 1 Schematic of the preparation of PDA–DOX/siRNA@SCM NPs and the tumor-targeted combination therapy for PCa bone metastases in vivo. | ||
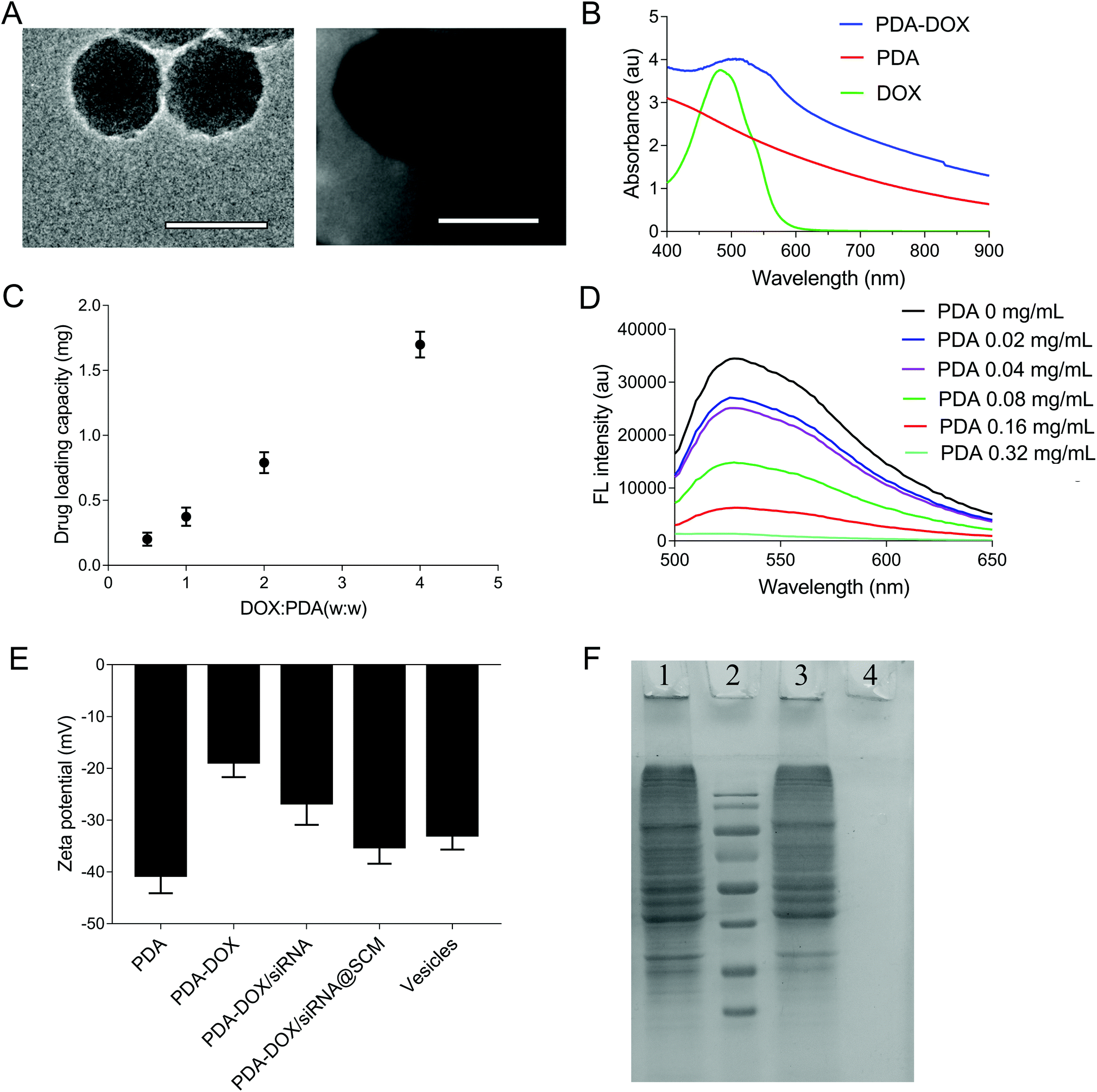 | ||
| Fig. 2 Characterization of PDA–DOX/siRNA@SCM NPs. (A) The TEM images of synthesized PDA (left) and PDA–DOX/siRNA@SCM (right). Scale bar = 100 nm. (B) The UV-vis absorbance spectra of DOX, PDA NPs, and PDA–DOX NPs. (C) The DOX loading capacities of PDA NPs under various proportions (DOX:PDA). (D) Fluorescence quenching of 50 nM siRNAFAM in the presence of PDA NPs with a series of concentrations (0, 0.02, 0.04, 0.08, 0.16, 0.32 mg mL−1). (E) Surface zeta potential of PDA, PDA–DOX, PDA–DOX/siRNA, PDA–DOX/siRNA@SCM and MSC-membrane (SCM) vesicles. (F) SDS-PAGE protein analysis staining with Coomassie Brilliant Blue. Lane 1: MSC-membrane (SCM) vesicles; lane 2: protein ladder; lane 3: PDA–DOX/siRNA@SCM NPs; lane 4: PDA NPs. | ||
Given that PDA contains numerous surface functional groups, DOX and siRNA could bind to the surface of PDA NPs via π–π stacking interactions. PDA NPs were mixed with DOX at different ratios and incubated under slightly alkaline conditions overnight, and DOX loading can be conveniently observed by monitoring the decrease of UV-vis intensity. As shown in Fig. 2B, the UV-vis absorption spectrum showed that the DOX characteristic peak shifted from approximately 480 nm to about 500 nm, confirming the successful DOX loading. Meanwhile, in the FTIR spectra, the adsorption peaks at 1042 cm−1 and 1511 cm−1 were attributed to the bending vibration of the C–H bond and the weak bending vibration of the C–N bond of PDA. After loading DOX, the additional peaks of DOX at 995 cm−1, 1121 cm−1, 1415 cm−1, 1570 cm−1 were seen on the curve of PDA/DOX, and were ascribed to the bending vibration of the C–C bonds in the ketone group, the C–H vibration of the methyl ether bond, the C–H vibrational and respiratory vibrational peaks of the aromatic rings, respectively (Fig. S1†). The absorption peaks that belonged to DOX were observed in the FTIR spectrum of PDA–DOX NPs, which also verified the synthesis of PDA–DOX NPs. With the increase in feeding DOX:PDA weight ratios, the DOX loading also increased. The maximal loading capacity reached about 300% (DOX:PDA, w/w) (Fig. 2C). Subsequently, the siRNA adsorption capacity of the prepared PDA NPs was measured by the fluorescence intensity of FAM-labeled siRNA while mixing FAM-siRNA with different concentrations of PDA NPs. The fluorescence was almost entirely quenched when the concentration of PDA NPs was 0.32 mg mL−1 (Fig. 2D), indicating that most of the siRNA was adsorbed onto the PDA NPs at 0.32 mg mL−1 and PDA displayed a high fluorescence quenching efficiency. The zeta potential was measured to investigate the surface modification loading of PDA NPs. When PDA was loaded with DOX, an increase of zeta potential (about 21.9 mV) was observed. After siRNA was absorbed onto the NPs, the zeta potential decreased to about −26.6 mV, which was attributed to the negative charge of siRNA (Fig. 2E).
The SCM-derived vesicles were prepared by physically extruding the membrane through 400 nm and 200 nm porous polycarbonate membranes according to the reported procedure.14 Subsequently, fresh membrane vesicles were mixed with the as-synthesized PDA–DOX/siRNA NPs and co-extruded through the 400 nm and 200 nm porous membrane for 10 passes. The resulting PDA–DOX/siRNA@SCM NPs were obtained by centrifugation and further characterized. Meanwhile, the UV-vis results showed that about 5% DOX was lost from PDA–DOX@SCM NPs during the extrusion process (Fig. S2†). The extrusion process of stem cell membranes had little effect on DOX loading. The TEM image clearly showed that the PDA–DOX/siRNA@SCM NPs have a core–shell structure, with the PDA core and an outer membrane shell. The diameter of the PDA–DOX/siRNA@SCM NPs was about 125 nm after DOX loading and SCM coating (Fig. 2A). After coating with the SCM, the zeta potential of PDA NPs changed to about −35.1 mV, which was close to that of natural stem cell vesicles (−32.8 mV; Fig. 2E) and also confirmed the successful SCM coating. To verify whether the prepared PDA–DOX/siRNA@SCM NPs have stem cell membrane proteins, SDS-PAGE was used to examine the protein contents of the PDA–DOX/siRNA@SCM NPs (Fig. 2F) and natural stem cell membranes. The results showed that the cell membrane proteins of PDA–DOX/siRNA@SCM NPs were very close to the MSC-membrane vesicles after extrusion treatment, indicating that the membrane proteins were mostly retained during the preparation process. Overall, these measurements indicated the successful translocation of natural stem cell membranes onto PDA NPs and the successful construction of the multifunctional PDA–DOX/siRNA@SCM NPs.
Cellular internalization of PDA–DOX/siRNA@SCM NPs
Due to having tumor-specific targeting components on the cell surface, the prepared PDA–DOX/siRNA@SCM NPs should effectively target the tumor sites. To assess the targeting capability of the prepared PDA–DOX/siRNA@SCM NPs, equivalent amounts of free DOX + siRNA, PDA–DOX/siRNA NPs and PDA–DOX/siRNA@SCM NPs were incubated with PC-3 cells, respectively. As shown in Fig. 3A, compared to free DOX + siRNA and the PDA–DOX/siRNA treated group, the cells cultured with PDA–DOX/siRNA@SCM NPs showed a higher and faster accumulation rate, and then the fluorescence intensity enhanced obviously within 4 h. Meanwhile, flow cytometry analysis was employed to quantitatively detect the cellular uptake of two NPs. In order to eliminate the interference, PC-3 cells were transfected with PDA–DOX@SCM or PDA–siRNA@SCM NPs, respectively. For PDA–DOX@SCM NPs, the cellular uptake efficiency of DOX was up to 96.2% which was around 1.5 times higher than that of PDA–DOX NPs (75.5%). Moreover, for PDA–siRNA@SCM NPs, the cellular uptake efficiency of siRNAFAM was up to 91.5% which was also higher than that of PDA–siRNA NPs (69.3%) (Fig. 3B). These results were consistent with the CLSM data. According to the above analysis, SCM coating could enhance the tumor targeting capability and cellular uptake efficiency of PDA–DOX/siRNA@SCM NPs.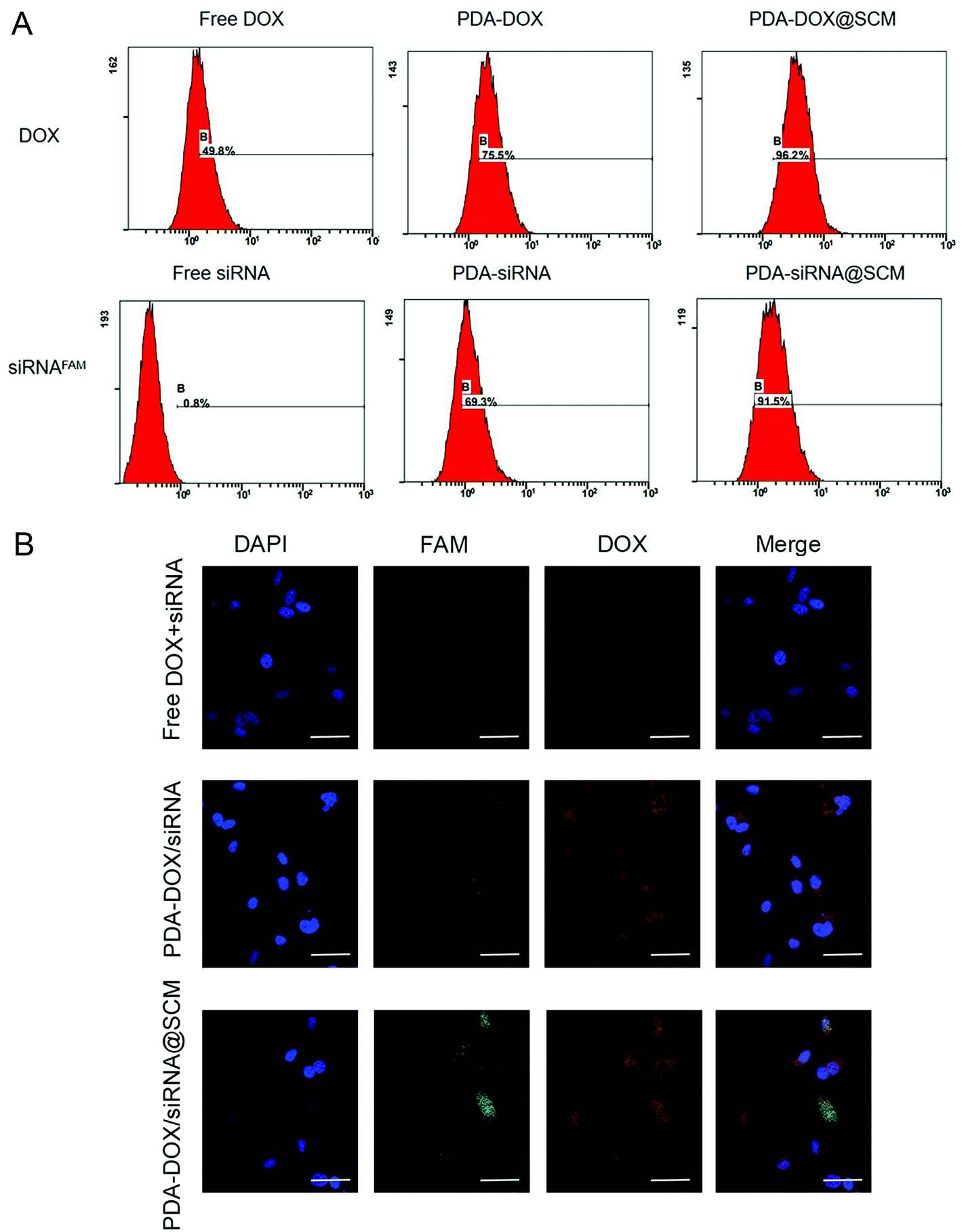 | ||
| Fig. 3 Enhanced in vitro cancer-cell accumulation of PDA–DOX/siRNA@SCM. (A) The cell uptake efficiency of different formulations in PC-3 cells measured by flow cytometric analysis. (B) The CLSM images of the PC-3 cells treated with free DOX + siRNAFAM, PDA–DOX/siRNA NPs and PDA–DOX/siRNA@SCM NPs for 4 h. Scale bar = 50 μm. | ||
In vitro siRNA transfection
We examined the expression of the PD-L1 gene in PC-3 cells following the delivery of siPD-L1 through PDA–siPD-L1@SCM NPs. The expression level of PD-L1 mRNA in PC-3 cells was analyzed by qRT-PCR 24 h after transfection. As shown in Fig. 4A, the PDA–siPD-L1@SCM NP treatment significantly decreased the expression of PD-L1 mRNA in PC-3 cells compared with free siPD-L1, which was similar to that of Lipo2k/siPD-L1 positive control. PDA–siPD-L1@SCM NPs lowered the PD-L1 mRNA expression approximately up to 50% from free siPD-L1 at an siPD-L1 concentration of 100 nM. Meanwhile, western blot analysis showed that the treatment with PDA–siPD-L1@SCM NPs led to markedly decreased PD-L1 protein expression in PC-3 cells, and the knockdown efficiency was very close to the mRNA expression data (Fig. 4B).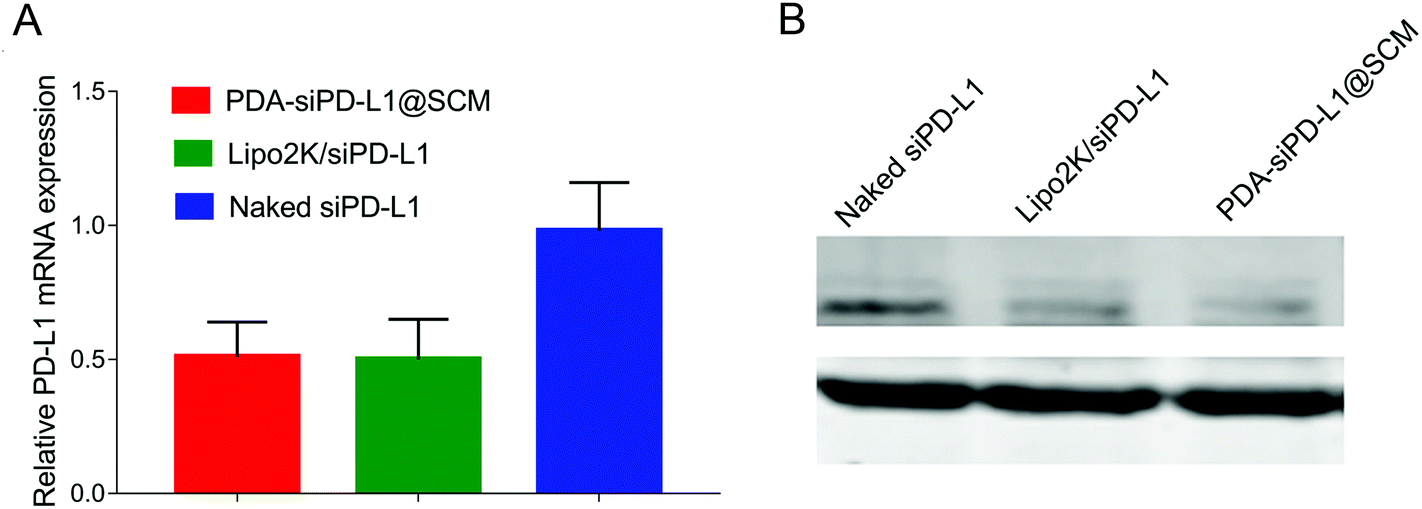 | ||
| Fig. 4 In vitro gene silencing effects. The relative PD-L1 mRNA (A) and PD-L1 protein (B) expression in PC-3 cells after transfection with naked siPD-L1, Lipo2k/siPD-L1, or PDA–siPD-L1@SCM NPs, which were detected by qRT-PCR and western blot analysis. | ||
Drug release in vitro and combined antitumor therapeutic efficacy
The drug release abilities of our engineered PDA–DOX@SCM NPs were tested. As shown in Fig. 5A, the cumulative release ratio of DOX was slow at pH 5.0 and 7.4 within 24 h, owing to the strong π–π interactions and electrostatic attractions. The PDA–DOX@SCM NPs released approximately 24% of DOX at pH 5.0 and the release of DOX was more than twice that at pH 7.4. The decrease in pH accelerated drug release may be attributed to the protonation of amino groups on the PDA surface or in DOX molecules weakening the π–π stacking interaction under acidic conditions. Therefore, the PDA–DOX@SCM NPs displayed a cumulative drug release ability, which could be beneficial for long-term drug treatment and could reduce the frequency of drug administration.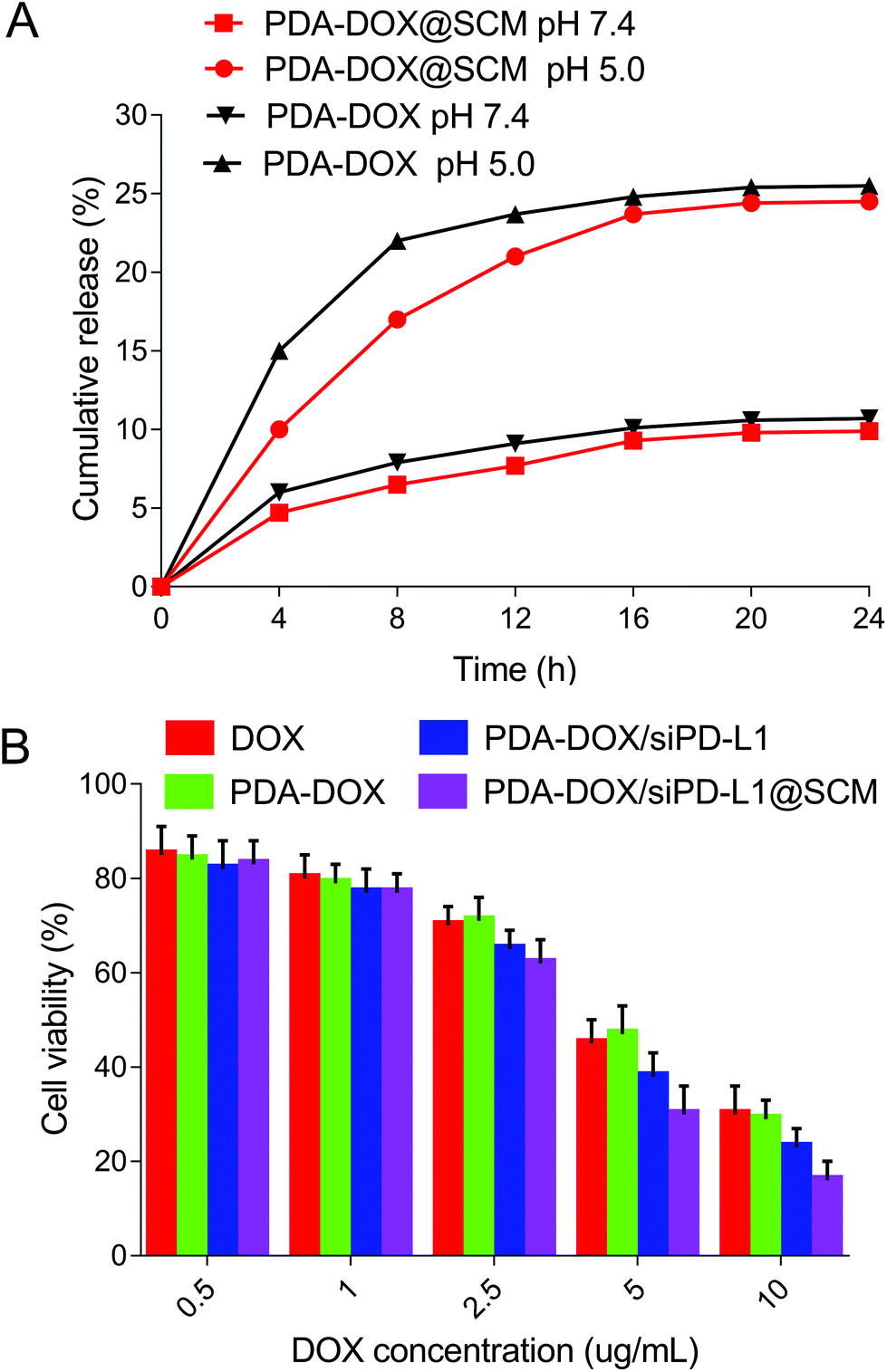 | ||
| Fig. 5 In vitro anti-tumor evaluation. (A) The release profile of DOX from PDA–DOX and PDA–DOX@SCM NPs at different pH within 24 h. (B) The cytotoxicity of NPs at various DOX concentrations against PC-3 cells after 24 h incubation. | ||
About the biosafety of the PDA@SCM nanocomposite, our research group has carried out relevant tests about PDA@SCM NPs.17 The results indicated that the camouflage properties attributed to the SCM improved the biocompatibility of PDA NPs against normal tissue cells and showed good compatibility with blood.17
To assess the cancer cell targeted drug delivery efficacy of PDA–DOX/siRNA@SCM NPs, the nanosystem was evaluated for in vitro cytotoxicity by using the CCK8 assay. As can be seen in Fig. 5B, all formulations displayed a typical dose-dependent cytotoxicity to the PC-3 cells for 24 h. Free-DOX displayed considerable anti-tumor activity, and this cytotoxicity was enhanced by delivering DOX and siPD-L1 using PDA–DOX/siPD-L1@SCM NPs to enhance cellular accumulation. Compared to free-DOX and PDA–DOX, the PDA–DOX/siPD-L1 NPs and PDA–DOX/siPD-L1@SCM NPs exhibited greater cellular damage, which was associated with the synergistic antitumor effect due to chemotherapy and gene therapy.
In vivo antitumor efficacy
On the basis of the above results in vitro, the antitumor efficacy of PDA–DOX/siPD-L1@SCM nanocomposites in the PCa bone metastasis tumor-bearing mice was further studied. We firstly examined the biodistribution of DOX in vivo. Tumor-bearing nude mice were injected with each formulation through the tail vein and the major organs and tumors were harvested 24 h after injection. As shown in Fig. 6A, the tumor tissues from PDA–DOX@SCM-treated mice showed a higher DOX concentration than the DOX and PDA–DOX-treated groups, which was attributed to the tumor targeting ability of the SCM on the surface of NPs. Furthermore, compared to the DOX-treated group, the PDA–DOX@SCM-treated group exhibited less accumulation of DOX in the liver and heart, suggesting that the PDA–DOX@SCM NPs could reduce the phagocytosis of nanoparticles by the body organs and heart toxicity. The pharmacokinetics were also further investigated by detecting the DOX concentrations in plasma at predetermined time points. As shown in Fig. S3,† free DOX was rapidly removed after systemic circulation. In contrast, the PDA–DOX@SCM NPs showed prolonged blood circulation. This might be due to the stable structure of the SCM-coated NPs under physiological conditions. These results indicated that the PDA–DOX@SCM NPs could reduce the clearance of DOX from the bloodstream, and then achieve a higher drug accumulation in the tumor tissues, which further verified the tumor-targeting properties of SCM-coated PDA NPs.17,18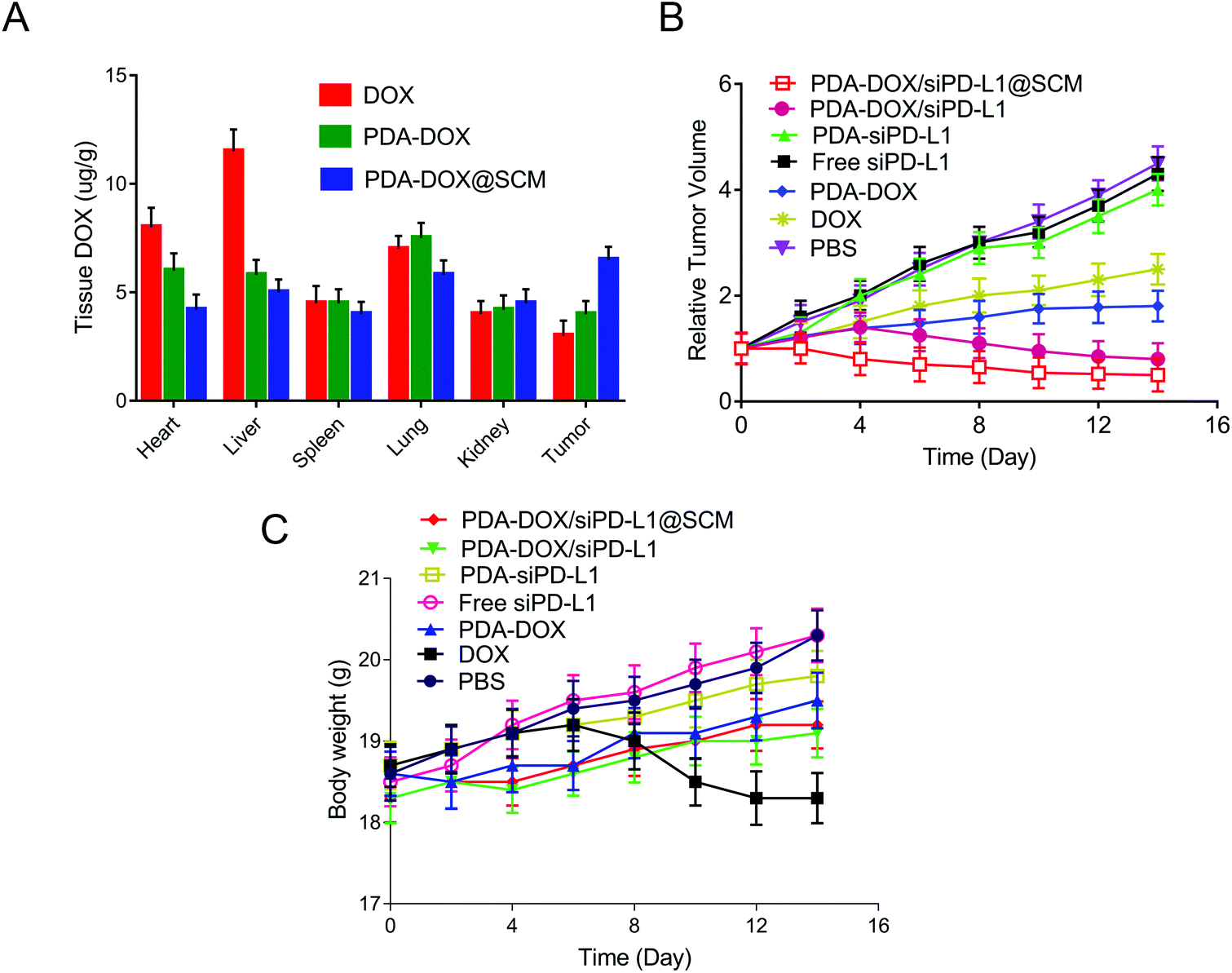 | ||
| Fig. 6 The in vivo anti-tumor performance of PDA–DOX/siPD-L1@SCM NPs. (A) The biodistribution of various NPs in mice at 24 h after the injection. (B) The tumor growth curves of different treatment groups during 14 days. (C) The relative body weight changes of mice with different treatments during 14 days. | ||
Subsequently, the anti-tumor efficacy was dynamically monitored by measuring the tumor volume. As shown in Fig. 6B, compared with the groups treated with PBS, DOX and PDA–DOX could only moderately inhibit the tumor growth. But this anti-tumor efficacy was significantly enhanced with the simultaneous delivery of DOX and siPD-L1 because of the combined effects of tumor chemotherapy and gene therapy. Furthermore, PDA–DOX/siPD-L1@SCM displayed the highest tumor growth inhibition, demonstrating the superiority of tumor targeted chemoimmunotherapy. Moreover, except for free DOX, no noticeable body weight loss was observed in the NP treated mice, suggesting a satisfactory biocompatibility of the synthetic NPs (Fig. 6C). Free DOX induced a slight decrease in the body weight over time, due to the side effects and its induction of acute toxicity. However, the nano-delivery systems could effectively alleviate the toxicity of DOX. These results indicated that PDA–DOX/siPD-L1@SCM possessed an efficient synergistic chemoimmunotherapy effect for PCa bone metastasis treatment.
The size of prostate tumor tissue in the hindlimb of nude mice was evaluated by X-ray in different treatment groups. As shown in Fig. 7A, the X-ray results showed that there was a huge high-density prostate cancer tissue shadow around the bone tissue of the upper tibia in the PBS group. In contrast, there was a certain degree of prostate cancer tissue shadow in free siPD-L1, PDA–siPD-L1, DOX, PDA–DOX and PDA–DOX/siPD-L1-treated mice. The PDA–DOX/siPD-L1@SCM-treated mice showed almost no prostate cancer tissue shadow, indicating that the PDA–DOX/siPD-L1@SCM treatment could effectively inhibit the growth of PCa bone metastases in nude mice. Furthermore, we used a micro CT to scan the bone tissue of the hind limb tibia of nude mice in different treatment groups to analyze the damage of prostate cancer to bone tissue. It can be seen that the tibia of the mice treated with PBS was severely damaged, showing obvious osteolytic lesions and cortical damage. However, in the DOX group and PDA–DOX group, the tibia of nude mice showed moderate damage and cortical defects. In the PDA–DOX/siPD-L1 group, the tibial bone was less damaged and the cortical bone became thinner. In the PDA–DOX/siPD-L1@SCM group, the bone cortex of the tibia remained almost intact. These results indicated that PDA–DOX/siPD-L1@SCM could effectively inhibit PCa bone metastases and weaken the invasion of metastatic PCa cancer to bone.
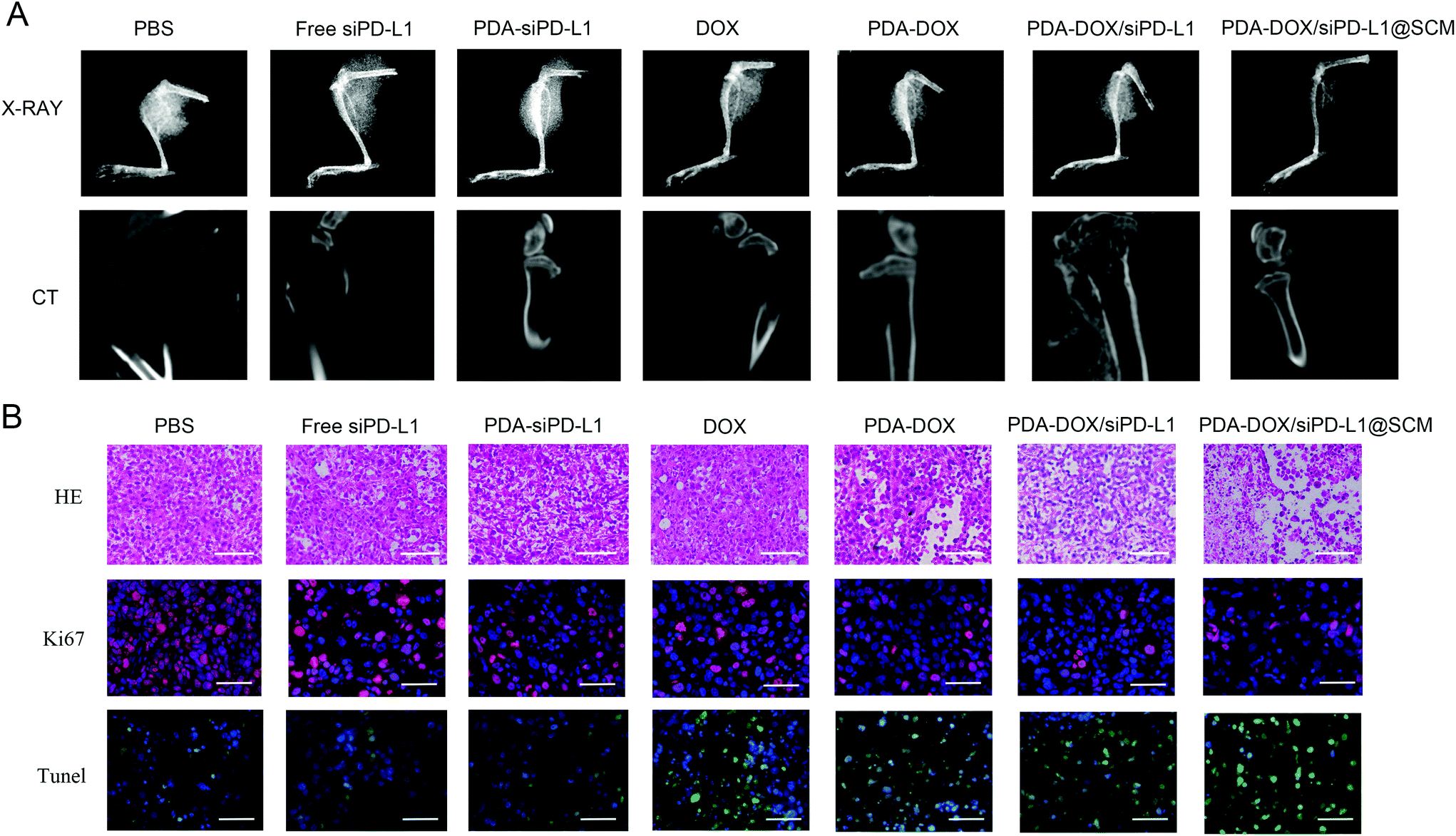 | ||
| Fig. 7 The histological analysis of the tumor tissues after treatment. (A) The visualization of tumor growth inhibition and degree of bone injury using CT and X-ray imaging after treatment. (B) The H&E, TUNEL staining, and immunofluorescence of Ki67 protein in tumor tissues harvested from different groups. Scale bar = 50 μm. | ||
The H&E and TUNEL stained tumor sections revealed that the PDA–DOX/siPD-L1@SCM treatment induced significant cell apoptosis and necrosis of the tumor tissues when compared to the control group, indicating the excellent therapeutic efficacy of PDA–DOX/siPD-L1@SCM NPs (Fig. 7B). Furthermore, immunohistochemical analyses showed diminished expression of cell proliferation Ki67 protein in the tumors treated with PDA–DOX/siPD-L1@SCM NPs (Fig. 7B). The results indicated that the targeted PDA–DOX/siPD-L1@SCM NPs could induce the apoptosis of tumor cells and then significantly inhibit the growth of PCa bone metastases. In addition, no obvious pathological changes were found in the section of the major organs in the PDA–DOX/siPD-L1@SCM group, indicating that there was no noticeable side effect on the treated mice during the treatment period (Fig. S4†).
Conclusions
In summary, we have successfully developed a multifunctional platform (PDA–DOX/siRNA@SCM NPs) for combined tumor chemotherapy and gene therapy. This robust nanocarrier showed a high capability to simultaneously transport siRNA and DOX into PC-3 cells. The western blot experiment revealed that the PD-L1 protein on the surface of PC-3 cells was effectively suppressed by codelivered PD-L1 siRNA. The yielded PDA–DOX/siRNA@SCM NPs share the biological functions of the stem cell membrane, which facilitated the remarkable stability and tumor-targeting capability both in vitro and in vivo. The in vivo biodistribution demonstrated that the PDA–DOX/siRNA@SCM NPs could specifically target and accumulate in the tumor sites. In addition, the tumor growth was effectively inhibited due to the combined tumor chemotherapy and gene therapy. Overall, the SCM-functionalized NPs represent a new class of nanocarriers for effective targeted drug delivery.Author contributions
X. Mu and M. Zhang conducted the experiments and analyzed the data. A. Wei participated in analyzing the results. F. Yin and Y. Wang provided useful suggestions to this work. K. Hu and J. Jiang designed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Science and Technology Development Plan Projects of Jilin Province (Grant No. 20200201558JC), the Special Project of Health Research Talents in Jilin Province (Grant No. 2019SC2062), and the Health Technology Innovation Project of Jilin Province (Grant No. 2019J028).References
- D. S. Chen and I. Mellman, Nature, 2017, 541, 321–330 CrossRef CAS PubMed.
- M. Poggio, T. Hu, C. Pai, B. Chu, C. D. Belair, A. Chang, E. Montabana, U. E. Lang, Q. Fu, L. Fong and R. Blelloch, Cell, 2019, 177, 414–427 CrossRef CAS PubMed.
- S. R. Gordon, R. L. Maute, B. W. Dulken, G. Hutter, B. M. George, M. N. McCracken, R. Gupta, J. M. Tsai, R. Sinha, D. Corey, A. M. Ring, A. J. Connolly and I. L. Weissman, Nature, 2017, 545, 495–499 CrossRef CAS PubMed.
- W. Hobo, T. J. Hutten, N. P. Schaap and H. Dolstra, Br. J. Haematol., 2018, 181, 38–53 CrossRef PubMed.
- G. Kwak, D. Kim, G. H. Nam, S. Y. Wang, I. S. Kim, S. H. Kim, I. C. Kwon and Y. Yeo, ACS Nano, 2017, 11, 10135–10146 CrossRef CAS.
- S. Y. Kwak, S. Lee, H. D. Han, S. Chang, K. Kim and H. J. Ahn, Mol. Pharmaceutics, 2019, 16, 4940–4953 CrossRef CAS PubMed.
- D. Wang, T. Wang, J. Liu, H. Yu, S. Jiao, B. Feng, F. Zhou, Y. Fu, Q. Yin, P. Zhang, Z. Zhang, Z. Zhou and Y. Li, Nano Lett., 2016, 16, 5503–5513 CrossRef CAS PubMed.
- J. L. Sottnik, J. Dai, H. Zhang, B. Campbell and E. T. Keller, Cancer Res., 2015, 75, 2151–2158 CrossRef CAS PubMed.
- M. F. Sanmamed and L. Chen, Cell, 2018, 175, 313–326 CrossRef CAS PubMed.
- D. Samanta, Y. Park, X. Ni, H. Li, C. A. Zahnow, E. Gabrielson, F. Pan and G. L. Semenza, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E1239–E1248 CrossRef CAS PubMed.
- N. Qiu, Y. Liu, Q. Liu, Y. Chen, L. Shen, M. Hu, X. Zhou, Y. Shen, J. Gao and L. Huang, Biomaterials, 2020, 269, 120604 CrossRef PubMed.
- C. Teng, B. Zhang, Z. Yuan, Z. Kuang, Z. Chai, L. Ren, C. Qin, L. Yang, X. Han and L. Yin, Nanoscale, 2020, 12, 23756–23767 RSC.
- Y. Wang, J. Luo, I. Truebenbach, S. Reinhard, P. M. Klein, M. Höhn, S. Kern, S. Morys, D. M. Loy, E. Wagner and W. Zhang, ACS Biomater. Sci. Eng., 2020, 6, 1074–1089 CrossRef CAS PubMed.
- B. Bao, L. Tong, Y. Xu, J. Zhang, X. Zhai, P. Su, L. Weng and L. Wang, Nanoscale, 2019, 11, 14727–14733 RSC.
- J. Song, H. Liu, M. Lei, H. Tan, Z. Chen, A. Antoshin, G. F. Payne, X. Qu and C. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 8915–8928 CrossRef CAS PubMed.
- Y. Yao, D. Zhao, N. Li, F. Shen, J. O. Machuki, D. Yang, J. Li, D. Tang, Y. Yu, J. Tian, H. Dong and F. Gao, Anal. Chem., 2019, 91, 7850–7857 CrossRef CAS PubMed.
- M. Zhang, F. Zhan, T. Liu, P. Shao, L. Duan, J. Yan, X. Mu and J. Jiang, Int. J. Nanomed., 2020, 15, 10183–10197 CrossRef CAS.
- X. Hao, B. Xu, H. Chen, X. Wang, J. Zhang, R. Guo, X. Shi and X. Cao, Nanoscale, 2019, 11, 4904–4910 RSC.
- Y. Shi, L. Du, L. Lin and Y. Wang, Nat. Rev. Drug Discovery, 2017, 16, 35–52 CrossRef CAS PubMed.
- N. Yang, Y. Ding, Y. Zhang, B. Wang, X. Zhao, K. Cheng, Y. Huang, M. Taleb, J. Zhao, W. Dong, L. Zhang and G. Nie, ACS Appl. Mater. Interfaces, 2018, 10, 22963–22973 CrossRef CAS PubMed.
- N. Celebi, M. Y. Aydin, F. Soysal, N. Yıldız and K. Salimi, ACS Appl. Nano Mater., 2020, 3(11), 11543–11554 CrossRef CAS.
- L. Rao, L. Bu, J. Xu, B. Cai, G. Yu, X. Yu, Z. He, Q. Huang, A. Li, S. Guo, W. Zhang, W. Liu, Z. Sun, H. Wang, T. Wang and X. Zhao, Small, 2015, 46, 6225–6236 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr08024a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |