
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Are the rates of dexter transfer in TADF hyperfluorescence systems optically accessible?†
Nils
Haase
*ab,
Andrew
Danos
 *c,
Christof
Pflumm
b,
Patrycja
Stachelek
c,
Wolfgang
Brütting
a and
Andrew P.
Monkman
c
*c,
Christof
Pflumm
b,
Patrycja
Stachelek
c,
Wolfgang
Brütting
a and
Andrew P.
Monkman
c
aInstitute of Physics, Experimental Physics IV, University of Augsburg, Universitätsstr. 1, 86135 Augsburg, Germany
bMerck KGaA, Display Solutions, Frankfurter Straße 250, 64293 Darmstadt, Germany. E-mail: nils.haase@merckgroup.com
cDepartment of Physics, Durham University, South Road, DH1 3LE, UK. E-mail: andrew.danos@durham.ac.uk
First published on 8th April 2021
Abstract
Seemingly not, but for unexpected reasons. Combining the triplet harvesting properties of TADF materials with the fast emission rates and colour purity of fluorescent emitters is attractive for developing high performance OLEDs. In this “hyperfluorescence” approach, triplet excitons are converted to singlets on the TADF material and transferred to the fluorescent material by long range Förster energy transfer. The primary loss mechanism is assumed to be Dexter energy transfer from the TADF triplet to the non-emissive triplet of the fluorescent emitter. Here we use optical spectroscopy to investigate energy transfer in representative emissive layers. Despite observing kinetics that at first appear consistent with Dexter quenching of the TADF triplet state, transient absorption, photoluminescence quantum yields, and comparison to phosphor-sensitised “hyperphosphorescent” systems reveal that this is not the case. While Dexter quenching by the fluorescent emitter is likely still a key loss mechanism in devices, we demonstrate that – despite initial appearances – it is inoperative under optical excitation. These results reveal a deep limitation of optical spectroscopy in characterizing hyperfluorescent systems.
New conceptsOptical spectroscopy is one of the key tools used to screen and assess new emissive materials for OLED applications. Pulsed optical methods allow selective excitation of the emitter in films representative of device emissive layers, frequently yielding insights into many of the photophysical processes relevant to device operation. As leading device architectures grow in complexity – for example in doubly-doped hyperfluorescent systems – such insights become increasingly valuable to identify and manage complex loss mechanisms. Contrary to this established trend though, here we show that optical methods are entirely unable to characterise the rates of Dexter transfer in hyperfluorescent films. The appearance of delayed fluorescence superficially resembling Dexter quenching is instead explained in terms of increased FRET rates in both the prompt and delayed fluorescence regimes. This unexpected limitation of optical methods is unprecedented, and establishes the need for new measurement techniques in addressing the limitations of this otherwise promising class of OLEDs. |
Introduction
In recent years, great progress has been made to develop organic light emitting diodes (OLEDs) beyond the 25% internal quantum efficiency limit of fluorescent emitter (FE) based devices.1 While red and green phosphorescent iridium-based OLEDs with internal efficiencies of near 100% have been successfully commercialised, suitable blue phosphorescent emitter (PE) materials remain elusive due to their intrinsic photo-electrochemical instability.2,3 Instead, triplet fusion enhanced blue OLED materials are still used in commercial applications. Unfortunately, the limited internal efficiency attainable by triplet fusion OLEDs (capped at ∼40%, or ∼60% in the absence of singlet fission) leads to high power consumption in these devices.4Uoyama et al. introduced thermally activated delayed fluorescence (TADF) as an alternative method to convert triplet into singlet excitons via reverse intersystem crossing (rISC), thereby utilising 100% of excitons while avoiding some of the limitations of PEs.5 However these TADF emitters also have drawbacks such as broad emission spectra and long excited state lifetimes, which make current blue TADF emitters not sufficiently mature for use in commercial display technologies.6,7
To overcome the disadvantages of both fluorescent and TADF emitters, Nakanotani et al. combined the two in a double-doped hyperfluorescent system.8 In this approach, high efficiency devices can be achieved with spectrally narrow emission and extended operational lifetimes.9–16 In these devices excitons are ideally formed on the TADF-sensitizer (TADF-S), which can then transmit singlet excitons via efficient Förster resonance energy transfer (FRET) to the FE. Triplet excitons formed on the TADF-S undergo rISC before also being transferred as singlets to the FE. Singlet excitons can also be formed by direct charge recombination on the FE, all of which undergo rapid fluorescence. In this way hyperfluorescent systems can enjoy both the high efficiencies associated with triplet harvesting of the TADF-S, as well as the narrow emission spectrum and device operational lifetime of the FE. Several recent reports have demonstrated the potential of this approach to achieve very high efficiency in deep blue OLEDs,17–21 generating accompanying theoretical interest as well.22–24
In practice, direct charge recombination on the FE will predominantly generate non-emissive triplet excitons. Dexter energy transfer (DET) from the high energy triplet states of the TADF-S to the lower energy FE triplet can further reduce emission efficiency. Due to some combination of these and other loss mechanisms, the external quantum efficiency (EQE) of hyperfluorescent devices is often smaller than those containing only the TADF-S.20,25–30 While rare examples of hyperfluorescence systems outperforming the corresponding TADF system have been reported,21,31 it is currently unclear how to identify and characterize the loss mechanisms in these OLEDs – so that improved performance can be purposefully engineered rather than discovered. Such characterisation of loss mechanisms is particularly challenging for in situ operating OLEDs, where excitation of different materials in the emissive layer occurs non-selectively. The finite charge transport rates and electrical circuit capacitance in operating OLEDs also makes it difficult to interpret time-resolved studies of their emission.
Even without full information about the loss mechanisms in hyperfluorescence, progress has been made to maximize performance using sensible approaches. To avoid aggregation quenching and prevent some DET, both the TADF-S and the FE are frequently (though not always32,33) doped into a host matrix (M) at suitable concentrations. While these are often 10–20% for the TADF-S, very low FE concentrations are typically used in devices to ensure large TADF-S to FE distances; at which DET is slow but FRET can still proceed rapidly. Indeed, recent reports have shown that designing TADF-S and FE with covalently bonded spacing groups can improve device performance.34,35 Exploiting deposition protocols that spatially segregate the TADF-S and FE also led to increased performance compared to simple coevaporation.27
Despite the growing interest in hyperfluorescent OLEDs, no detailed photophysical investigation of their energy transfer has been made up to now. Here we examine optically excited films in an attempt to quantify FRET and DET rates at different FE concentrations. Optical measurements of films were pursued instead of electrical excitation of devices, as these allow for the TADF-S to be excited selectively and all-at-once. Such experimental control enables precise measurement of emission decay lifetimes and avoids signal contamination from direct excitation of the FE (impossible to quantify in devices). Contrary to our expectations and the initial appearance of the data, we find that DET is inoperative under optical excitation, and that optical spectroscopy gives only limited insight into the quenching processes that are active under electrical excitation. The optical results are also highly susceptible to misinterpretation, in stark contrast to the established role of optical methods in characterizing similar optoelectronic materials and systems.
Hyperfluorescence system and mechanism
Energy diagram
Fig. 1 summarises the energy levels and rate constants that govern the hyperfluorescent system kinetics. For clarity, the non-radiative decay paths are not shown, but exist for all states shown. We also do not consider excitation of the host material, as in devices any host excitons are rapidly transferred to either the TADF-S or FE dopant by FRET and/or DET. In photoluminescence experiments the matrix material (with absorption in the deep UV) is typically not excited.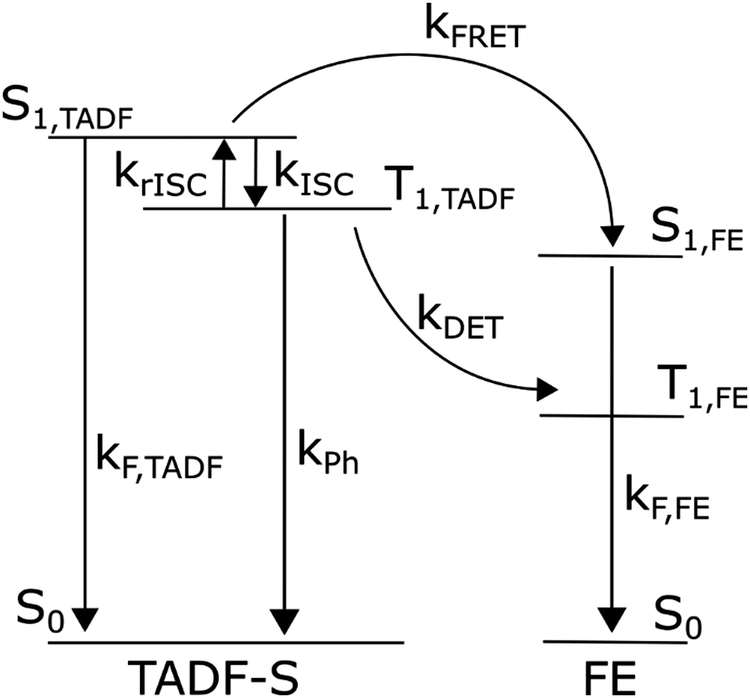 | ||
| Fig. 1 Energy diagram of a hyperfluorescent system including rates. Non-radiative decay paths are not included, nor energy levels of the host matrix. | ||
Upon photoexcitation some direct excitation of the FE is often unavoidable, although the ratio of TADF-S to FE excitation can be determined from material UVVIS spectra and film composition. Furthermore, as optical measurements can be precisely time-resolved in a manner that electroluminescence cannot (due to capacitive effects and residual charge recombination in devices), it is also straightforward to exclude the effects of direct FE excitation. Any delayed emission that occurs long after the FE natural decay lifetime can only arise due to energy transfer from the TADF-S, and is therefore valuable in characterizing energy transfer and loss pathways. In contrast, for electroluminescence measurements FE emission from both FRET and sustained/persistent direct excitation cannot be readily distinguished.
Returning to Fig. 1, an exciton generated directly on the FE dopant immediately decays radiatively, yielding rapid prompt emission from the FE (ignoring very small FE intersystem crossing). In contrast, an exciton generated on the TADF-S can either decay radiatively (prompt emission), or populate the triplet state of the TADF-S (T1,TADF) via intersystem crossing (ISC), or transfer its energy to the FE singlet via FRET. When the excited TADF-S triplet state is formed by ISC, it can later flip back via rISC, or decay non-radiatively to the ground state, or undergo DET to populate the dark triplet state of the FE.
Materials
Chemical structures of the investigated matrix material DPEPO (M), TADF-S material DDMA-TXO2 and FE material TBPe are shown in Fig. 2a, together with the extinction spectrum of the FE and the normalized photoluminescence (PL) spectra of the FE and M:TADF-S. This M:TADF-S system has been studied extensively by us36–42 and by others,43 including limited hyperfluorescence applications.9 The extinction and PL spectra of the FE are measured in low concentration toluene solution. In contrast, the PL spectrum of the TADF-S was measured in a coevaporated doped film of M:TADF-S(20%), since the spectra of TADF materials are strongly depended on the polarity of the surrounding environment.38 The TADF-S emission is characterized by a broad Gaussian spectrum, while the FE spectrum has vibronic emission bands. The triplet energy of the TADF-S is significantly higher than that of the FE (∼3 eV40 compared to ∼1.5 eV44,45), indicating that DET is energetically possible as implied by Fig. 1.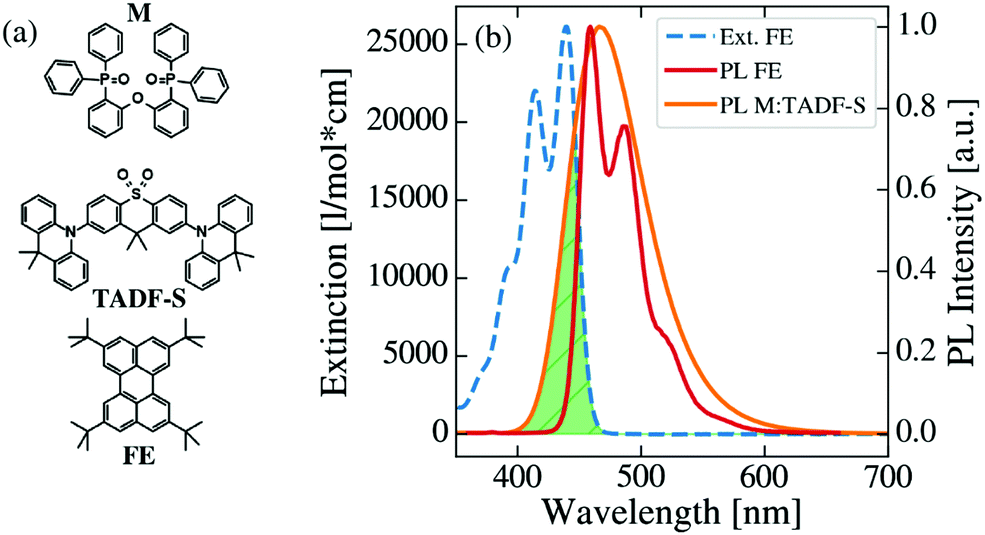 | ||
| Fig. 2 (a) Chemical structures of the host matrix DPEPO (M), the TADF sensitiser DDMA-TXO2 (TADF-S) and the fluorescent emitter TBPe (FE), (b) molar extinction spectrum of FE dissolved in toluene (blue), including the normalized photoluminescence spectra of TADF-S (orange) and FE (red). The overlap between TADF-S emission and FE extinction is highlighted in green resulting in a Förster radius RFRET of 3.0 nm. | ||
Energy transfer
The rate of FRET from DDMA-TXO2 to TBPe can be estimated from their spectra (Fig. 2) using Förster resonant energy transfer theory: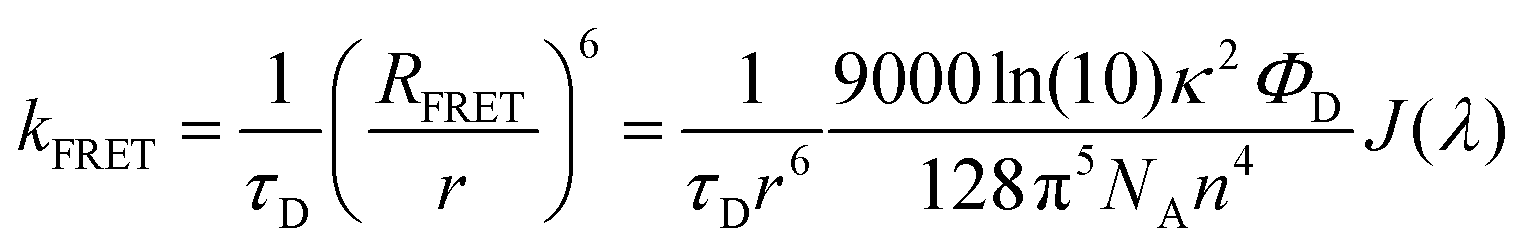 | (1) |
This RFRET value is inferior compared to analogous “hyperphosphorescent” systems consisting of a phosphorescent iridium or platinum complex sensitizer and a redshifted FE acceptor, with typical values in the range of 4–5.5 nm.46–49 However, for deep-blue FE emission, all known high efficiency FE materials will have relatively small FRET overlap due to the scarcity of suitable deep-blue PE or TADF sensitisers. For example the unicolor phosphor-sensitized fluorescent system introduced by Heimel et al. also has a small RFRET of 2.4 nm.50 In contrast, due to the broad Gaussian spectrum of the TADF-S, a reasonably high FRET rate can be ensured even though the emission maximum of the TADF-S is redshifted compared to the FE.
Results
Steady-state emission
Steady state photoluminescence spectra of evaporated films of M(100-20-x%):TADF-S(20%):FE(x%) were measured after 355 nm laser excitation. This ratio was chosen based on OLED performance optimization for these materials in previous studies.40 This wavelength was chosen as it excites the direct charge transfer absorbance band of the TADF-S, avoiding loss pathways associated with electron transfer that are not relevant to devices.51 Furthermore, this wavelength leads to minimal absorbance by the FE (Fig. S1a, ESI†), entirely avoids absorbance by the host matrix, and is widely available for time-resolved experiments in the form of higher harmonics of nanosecond–picosecond pulsed Nd:YAG lasers.Fig. S2a (ESI†) displays the normalized PL spectra for an FE concentration of 1%, together with the resolved TADF-S and FE components. For this concentration, 88% of the total emission originates from the FE. Total emission spectra and their TADF-S and FE components for films with other FE loadings are shown in Fig. S3 (ESI†). With further increase of the FE concentration a clear trend of decreasing TADF-S and increasing FE relative contributions to the total emission can be seen, plotted in Fig. S2b (ESI†). At an FE concentration of 2% nearly 98% of the total emission comes from the FE. This indicates an efficient energy transfer between the TADF-S and the FE, but cannot distinguish the increasing emission contribution due to direct FE excitation – discussed immediately below.
Direct FE absorption
To evaluate the extent of direct FE excitation, absorption spectra of 50 nm thick neat layers of pure M, pure TADF-S, and pure FE were measured (Fig. S1a, ESI†). A grey dashed line indicates the excitation wavelength (355 nm) used throughout subsequent time-resolved photoluminescence (TRPL) and photoluminescence quantum efficiency (PLQE) experiments. Due to the low absorption of DPEPO at 355 nm, direct excitation of the matrix material can be neglected. Instead, only TADF-S and FE show measurable absorptions of 4.6% and 5.3% through the 50 nm films. From these absorption values in pure films and the doping ratios of the mixed hyperfluorescent samples, the ratios of the initial excitation between TADF-S and FE can be calculated (Fig. S1b, ESI†). As expected, increasing the FE concentration results in a linear increase of the direct FE excitation fraction. By adding 5% of FE into the M:TADF-S(20%) sample, up to 22% of the initially generated excitons populate the singlet state of FE – although this results in almost 100% of emission emerging from the FE (Fig. S2b, ESI†), indicating efficient energy transfer.While direct FE excitation can therefore contribute significantly to the total steady-state emission spectra, we note that in time-resolved experiments direct excitation of the FE leads to rapid emission (∼6 ns lifetime for TBPe) with negligible potential impact on time-resolved spectra at delay times greater than ∼10 ns. For this reason, time-resolved studies were anticipated to be indispensable in characterizing the energy transfer processes in hyperfluorescence systems. Unlike steady-state methods, these can distinguish FE emission arising from direct (optical) or indirect (FRET sensitised) excitation due to the differences in their intrinsic timescales.
Time-resolved spectroscopy
To analyze energy transfer in the hyperfluorescent system, measurements were made of the time dependent spectra and emission decays of evaporated films of both M:TADF-S(20%):FE(x%) and M:TADF-S(x%):FE(1%) following ∼100 ps pulsed 355 nm laser excitation. A full concentration series was investigated for varying FE concentrations at a fixed TADF-S concentration (20%), as well as for different TADF-S concentrations at fixed FE concentration (1%).The first of these series is shown in the Fig. 3a. In these films the concentration of the TADF-S is kept constant at 20%, while the concentration of the FE is varied from 0% to 5%. Typical TADF kinetics consisting of a prompt (PF) and a delayed fluorescence (DF) component can be seen for all concentrations except for 5%, where the DF intensity dropped below instrument baseline after only ∼100 ns. The vibronic structure of the individual time-resolved spectra (Fig. S4, ESI†), indicates that the emission originates almost entirely from the FE at all times, in line with the steady state measurements (Fig. S2 and S3, ESI†).
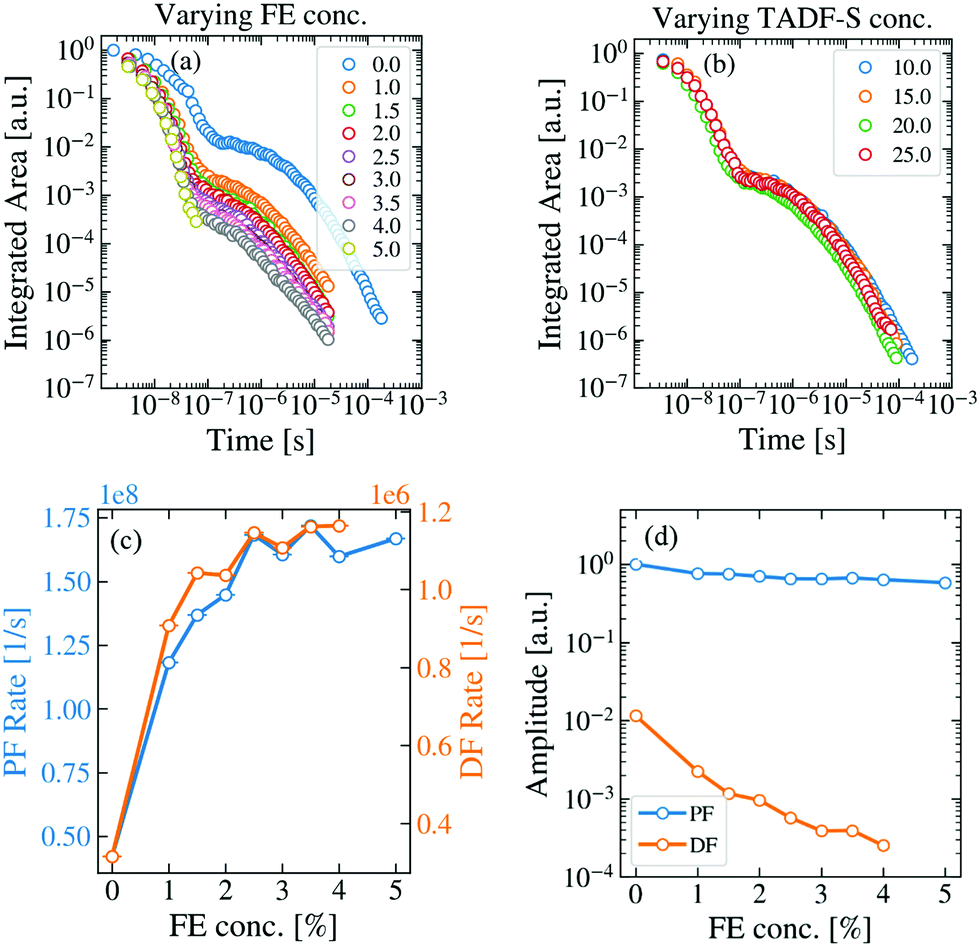 | ||
| Fig. 3 TRPL measurements of hyperfluorescent samples containing DPEPO (M), DDMA-TXO2 (TADF-S) and TBPe (FE). (a) The change in the TRPL kinetics of the FE-dominated emission (individual spectra in Fig. S4, ESI†) with increasing FE concentration at fixed TADF-S concentration of 20%. (b) Changing the TADF-S concentration has no significant effect on the TRPL kinetics (fixed 1% FE loading). The fitted exponential decay rates (c) and amplitudes (d) of the prompt and delayed emission components for the data in panel (a). | ||
Fig. 3b shows the effect of increasing TADF-S concentration from 10% to 25% at a constant FE concentration of 1%. Even though the TADF-S concentration is nearly tripled, the emission decays of the investigated samples are almost identical. This demonstrates that the FE is never saturated by energy transfer from the TADF-S, even at low loadings. This is as expected, as the fluorescence rate of the FE is orders of magnitude faster (∼1.7 × 108 s−1) than the rISC rate of the TADF-S (∼1.2 × 106 s−1), and so throughout the DF singlet excitons cannot be formed and transferred fast enough to accumulate on the FE.
The presence of DF emission in Fig. 3a immediately confirms that triplet excitons are formed in these films at FE concentrations below 5%, despite the use of optical excitation. Therefore, FRET does not fully outcompete ISC on the TADF-S, at least for the ∼1% FE loadings commonly reported for hyperfluorescence devices. It is therefore reasonable to expect that these triplet excitons formed by ISC would provide some insight towards energy transfer and loss mechanisms in devices (in which triplet excitons dominate).
By increasing the concentration of the FE, the PF and DF lifetimes both shorten compared to those observed for the TADF-S alone (rates increase, Fig. 3c). In addition, the DF amplitude decreases rapidly with increasing FE concentration (Fig. 3d), suggestive of a decreased initial triplet population and an active quenching process (such as DET) for triplet excitons in the DF region. At 5% loading of FE, the PF lifetime is similar to that of TBPe (∼6 ns) and no strong DF contribution is observed. This is consistent with FRET fully outcompeting ISC on the TADF-S at this concentration, with no triplet excitons available to contribute to emission at longer delay times.
The increase in PF decay rate can be simply explained. Even assuming selective excitation of the TADF-S, rapid FRET to the FE ensures fast exciton transfer from the slowly-decaying TADF-S singlet to the rapidly-decaying FE singlet. This effect accelerates the overall PF emission, limited by the intrinsic emission lifetime of the FE itself. For TBPe, this lifetime was measured separately to be ∼6 ns by time correlated single photon counting, corresponding to a maximum PF rate of ∼1.7 × 108 s−1. This value is in good agreement with the plateau value of the PF decay rate in the hyperfluorescence films. In practice, increased FE loading also leads to increasing direct FE excitation (Fig. S1b, ESI†), which results in a larger relative FE contribution to the measured PF signal, further decreasing the overall PF lifetime towards that of the FE.
In the DF time region, rapid FRET also acts upon singlet states on TADF-S molecules after they undergo rISC from triplet states. The transferred singlet excitons emit rapidly from the FE, but with a long DF lifetime uncharacteristic of the FE alone – rISC is the rate limiting step leading to emission. The DF spectra are still vibronically structured though, confirming that they emanate from the FE (Fig. S4, ESI†). However, the rapid FRET also competes with any preceding ISC on the TADF-S during the earlier PF regime, resulting in a smaller triplet population forming as the FE concentration is increased. This leads to a significant reduction in the DF intensity (and smaller fitted exponential DF amplitudes, Fig. 3d). The fitted amplitude of the PF is always close to 1 as the emission decays are all normalized to allow comparison.
The increased decay rate of the DF emission can be attributed to two possible causes. On the one hand, rapid FRET can interrupt repeated ISC/rISC cycling between the singlet and triplet states of the TADF-S, resulting in emission sooner than would otherwise be possible for the TADF-S alone.39,52 On the other hand, DET can quench triplet excitons from the TADF-S triplet to the FE at greater rates with increasing FE concentration. While FRET is desirable in the context of OLEDs, DET to the non-emissive FE triplet is a suspected loss mechanism. It is therefore imperative to distinguish which of these two processes (or what proportion of each) contributes to the observed DF lifetime shortening. Indeed, in a recent report53 similar changes in the DF lifetime have been wholly attributed to DET without sufficient justification. In order to understand the energy transfer processes active in hyperfluorescence films and OLEDs, it is necessary to develop methods that distinguish these two potential effects.
Photoluminescence quantum efficiency
To evaluate whether the DF lifetime reductions are a result of FRET – which would conserve overall emission efficiency – or dissipative DET to the FE triplet, the photoluminescence quantum efficiencies (PLQEs) of representative films were measured. 200 nm thick films of M:FE(x%) or M:TADF-S(20%):FE(x%) with varying FE concentration were thermally evaporated onto quartz or sapphire substrates. The excitation wavelength was set to 355 nm, matching the time-resolved measurements to ensure similar ratios of TADF-S and FE excitation. The results are shown in Fig. 4a. Consistent with previous reports, the PLQE of the TADF-only sample M:TADF-S(20%) is as high as 85 ± 5%. With increasing FE concentration no significant changes in the efficiency are observed for either the M:FE(x%) or the M:TADF-S(20%):FE(x%) samples. Instead, a constantly high PLQE between 85 ± 5% and 92 ± 5% is measured for all investigated films.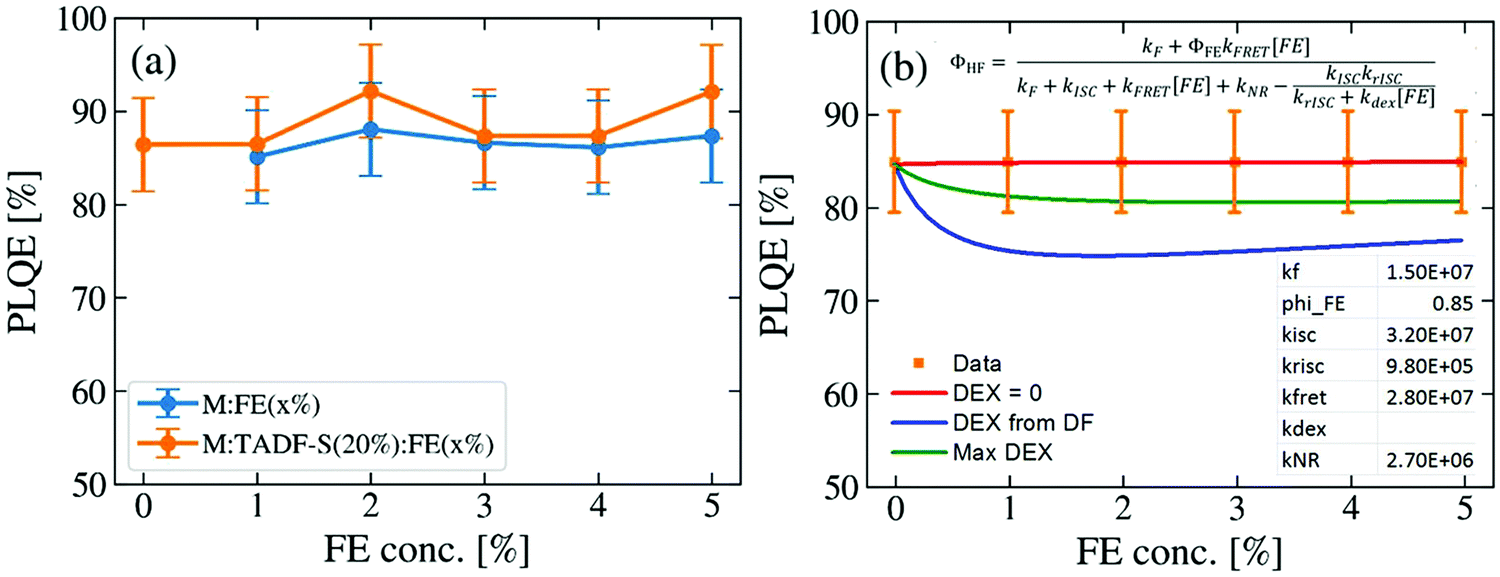 | ||
| Fig. 4 Photoluminescence quantum efficiencies of hyperfluorescence films at different FE concentrations: (a) experimental data, and (b) fitting to model equation (derivation, choice of fixed rates, and cases for kdex detailed in the ESI†). | ||
Numerical modelling of the PLQE results (shown in Fig. 4b, with derivation in the ESI†) indicate that if DET were significantly active, it would reveal itself as a dip in the overall PLQE at moderate FE concentrations. These concentrations correspond to those where a significant triplet population is formed by ISC on the TADF-S, and then quenched by DET to the FE triplet. In this scenario the overall film PLQE would rise again with further increases in FE concentration, and saturate towards the PLQE of the pure FE at very high concentrations. This saturation at high FE loadings occurs when FRET dominates ISC and no triplets are formed for DET to act upon, although the presence of DF in time-resolved measurements confirms that this saturation regime is not reached below 5% for our material system.
In order for our numerical modelling to qualitatively resemble the experimental results (i.e. no change in PLQE with FE concentration), the DET rate is fit to zero. This indicates that, despite confirming the generation of triplet excitons and observing DET-like behavior in the time-resolved emission decays, DET is entirely inactive in these optical experiments. Even deliberately using larger DET rates that either remain consistent with the PLQE error bars (‘Max DEX curve’) or taken from the ratio of PF (FRET) and DF (potentially DET) rate changes with varying FE concentration in Fig. 3c (‘DEX from DF’), in both cases yield DET rates too small to significantly compete with rISC. Discussion of these different cases and their derivations are included in the ESI.†
These PLQE results are particularly surprising given that hyperfluorescent OLEDs are known to suffer from strong quenching with increasing FE doping concentration. For example, in DPEPO:DMAC-DPS:TBPe devices, the presence of even 1% of TBPe results in a large loss in the optimised EQE from 19.5% for DPEPO:DMAC-DPS(10%) to 13.1% for DPEPO:DMAC-DPS(50%):TBPe(1%).27 This loss in performance is not reflected in experiments using optical excitation, again indicating that the device-relevant loss mechanisms are not active under optical excitation. In comparison, for analogous PE sensitized “hyperphosphorescent” systems, a clear correlation between losses in EQE and PLQE is verified. In these systems, both EQE and PLQE are found to decrease with increasing FE concentration, which can be explained by exciton trapping on the FE due to DET from the sensitizer.50,54
Transient absorption
In order to search exhaustively for signs of DET, ns-transient absorption spectra and kinetics were collected for M:TADF-S(20%) films with and without 2% co-doping of FE (355 nm pump). The experimental setup was as previously reported, and in agreement with our previously reported results the M:TADF-S(20%) film exhibited a long lived excited-state absorption onset at ∼600 nm, shown in Fig. S5 (ESI†).39 This excited state absorption decayed with similar kinetics to the triplet population generated by fitting of time resolved PL experiments, and so is attributed to the TADF-S triplet state.39 A high amplitude but short-lived negative feature was also observed at around 450 nm (Fig. S5a inset, ESI†), which is attributed to strong pump-induced PF emission from the TADF-S.As shown in Fig. S5b (ESI†), on addition of 2% FE the broad and long-lived TADF-S absorption vanishes, and is replaced by a much narrower and more rapidly decaying induced absorption centered at around 730 nm. This feature decays with similar kinetics as the film PL emission itself measured on the same system (Fig. S6 and S7, ESI†), which has the characteristic vibronic structure of the FE. In addition, the wavelength of the photoinduced absorption peak (730 nm) matches the energy of the S1 → S2 transition in TBPe, estimated from the two lowest-energy bands in its ground state absorption spectrum.55 Based on this peak position and the absence of any long-lived induced absorption signal that could be attributed to a triplet state, we instead attribute the observed signal to the S1 → S2 FE transition.
These transient absorption results are in strong contrast to the expected results if DET were active in these films. In that scenario, we would expect to observe lifetime quenching of the long lived TADF-S triplet signal over an extended duration, and the emergence of a new long-lived triplet absorption signal corresponding to TBPe at around 490 nm.45 Neither of these are observed, and the results are instead consistent with rapid FRET interrupting ISC and preventing the formation of a detectable triplet population in the hyperfluorescent films (with only singlet excited states detectable).
Discussion
EQE vs. PLQE for hyperfluorescence and hyperphosphorescence
In photophysical experiments only singlet states are initially excited, while electrically driven devices generate triplet excitons directly. Strong spin orbit coupling in PE molecules leads to ISC on timescales faster than 100 fs, outcompeting all other relevant processes by orders of magnitude.56,57 As a result, triplet states are almost instantly populated in PE materials independent of the mode of excitation. The rate of triplet formation also greatly outcompetes FRET to any FE dopant. Consequently, no differences in EQE and PLQE experiments are expected in hyperphosphorescent devices, assuming DET as the main loss channel.In contrast, TADF molecules have far slower ISC rates in the range of only 106–108 s−1.39,40,58,59 Thus the number of triplet states generated by optical excitation in hyperfluorescent materials depends strongly on the balance between kISC, kF,TADF and the energy transfer rate(s) to the FE. With increasing FE concentration kFRET increases, while kISC and kF,TADF are intrinsic to the M/TADF-S combination and expected to remain constant. This increasing FRET rate during the PF competes with ISC and reduces the number of triplet excitons generated in this time regime, resulting in a decreasing DF component (that still retains FE emission spectrum) as described above. Under electrical excitation, 75% triplet excitons are generated directly on the TADF-S without the need for ISC, leading to very different behavior in film PLQE and device EQE as the FE loading changes.
Explaining changes in DF lifetime
While the above explains the change in DF amplitude with FE concentration, the changes in DF lifetime were initially strongly suggestive of DET quenching. This lifetime change cannot be attributed to simple FRET, as we would then expect to see similar changes in decay rates in the DF and PF as the FE concentration changes. Instead these changes in rate are two orders larger in the PF than in the DF, observed both here and by others.9,53 The absence of any appreciable PLQE quenching at intermediate FE concentrations (and the lack of FE transient absorption signal) simultaneously indicate that DET is not active in optical measurements.With simple FRET and DET processes excluded, we are instead able to explain the DF lifetime trends seen in Fig. 3 by invoking a distribution of TADF-S–FE distances in the evaporated film. A diagram of this scenario is presented in Fig. 5. The central FE molecule is shown as a blue dot, and positions of TADF-S molecules for which the pictured FE is the nearest neighbor shown as orange dots. Such a distribution is unsurprising in evaporated films, with subsequent mechanistic insight likely applicable to hyperfluorescent systems generally.
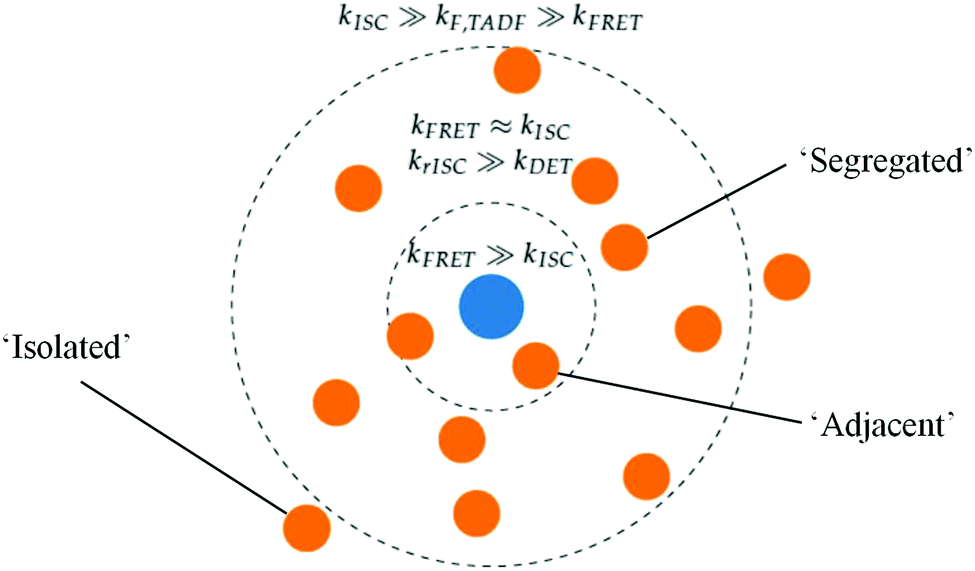 | ||
| Fig. 5 A distribution of distances between TADF-S (orange dots) and their nearest neighbour FE (blue dot) leads to different emission lifetimes and mechanisms in optically excited hyperfluorescence systems. Examples of adjacent, segregated, and isolated TADF-S FE pairs are indicated, determined by balances of emission and energy transfer rates at different molecular distances. | ||
If the distances from a TADF-S (orange) to its nearest-neighbor FE (blue) are normally distributed, this allows us to identify a subset of ‘segregated’ TADF-S molecules with only a distant neighboring FE. There will also be an ‘isolated’ population with no nearby FE. Additionally, TADF-S molecules with a nearby ‘adjacent’ FE partner will undergo rapid FRET and emit in the PF time region, generating no triplet states and contributing nothing to the DF emission.
As an illustrative example, we identify that all TADF-S molecules fall in the ‘adjacent’ category in the 5% FE film, with no triplets formed and no DF emission detectable. At lower FE concentrations, ‘segregated’ and ‘isolated’ TADF-S molecules will experience a slower or zero FRET rate by virtue of being more distant from their nearest FE. Their slower FRET rate allows these subsets to undergo ISC and contribute to DF emission. Whether this DF emission has the spectral shape of the TADF-S (isolated molecules with no FRET) or the FE (segregated molecules with FRET) depends on the balance of FRET and TADF emission rates for each individual molecule, and therefore on the distance to its nearest FE neighbor. From the dominance of FE emission in the measured DF spectra (Fig. S4, ESI†) we can conclude that isolated TADF-S molecules are rare even at 1% FE. Nonetheless the persistent TADF-S emission contribution in steady state spectra (Fig. S2 and S3, ESI†) means the isolated subset cannot be disregarded fully. In none of the three scenarios is DET from the TADF-S to FE triplets found to be relevant, as outlined below.
A distribution of TADF-S to FE distances implies a distribution of FRET rate in hyperfluorescence films. We subsequently identify a distance-controlled self-selection effect in the time-resolved emission spectra, with only the segregated and isolated TADF-S molecules contributing to the DF. Even amongst the group of segregated TADF-S molecules though, those that happen to have a slightly closer FE neighbor will undergo FRET faster and generate emission sooner. Therefore, the effective FRET rates in the films are also time-dependent, decreasing as all of the more closely spaced of the ‘segregated’ TADF-S molecules undergo FRET at earlier times. Similar time-dependence of FRET rates should be expected in the PF as well, with any single reported value being a representative average of the ‘adjacent’ population.
This framework then explains why the changes in DF rate (due to FRET) are smaller than the changes in PF rate (also due to FRET). Any emission in the DF can only arise from TADF-S molecules that are sufficiently distanced from their nearest-neighbour FE, so that ISC can compete with FRET and triplet excitons can form. Therefore, the rates of FRET in the DF (and their changes with FE concentration) will necessarily be smaller. There is no such self-selection in the PF, with much smaller intermolecular distances and much larger FRET rates contributing to the emission. We note that the differences between energy transfer rates in PF and DF are also (initially) highly suggestive of different mechanisms, specifically FRET in the PF and DET in the DF. It is only with PLQE and transient absorption results that we are able to rule out this seemingly-simpler interpretation.
Modelling intermolecular distances and energy transfer rates
To further support this interpretation, we return to the exponential decay rates plotted in Fig. 3c. For ease of analysis, we do not consider distributions or time-dependent dispersion in FRET rates across the PF and DF – only a single representative rate and representative nearest-neighbor distance for each region. We also note that emission in the PF and DF will arise from the adjacent and segregated TADF-S molecules respectively.In the PF, we can estimate the FE concentration at which the nearest-neighbor FE distance is equal to the calculated FRET radius (RFRET, previously calculated at 3 nm from spectra). This radius is defined kinetically as the distance at which FRET takes on 50% probability, and so the FRET rate must be equal in size to the sum of all other rates. Since in the PF region we attribute changes in the decay rate to FRET only, this condition corresponds to the FE concentration that doubles the initial PF rate. Interpolation of the data in Fig. 3c places this at approximately 0.4% loading of FE. Calculating the average TADF-S nearest-neighbour FE distance using this loading and assuming a uniform cubic lattice of FE molecules60,61 (and equal material densities for the M, TADF-S, and FE) gives a similar FRET radius (2.7 nm, from FE loading and kinetics) to that calculated from spectra. For comparison, estimated intermolecular distances at an FE loading of 0.3% return the spectrum-calculated value of RFRET (3 nm).
Finally, we can use the preceding analysis and the ratio of lifetime changes in PF and DF to determine the average distances between the segregated TADF-S molecules and their nearest-neighbour FE. The PF decay rate changes by a factor of ∼145 more than the rate of DF decay for the same FE loading, compared to their values for the plain TADF-S film (Fig. 3c). By eqn (1) the FRET rate varies with r−6, implying that the segregated TADF-S molecules are therefore on average 2.3 times further from an FE molecule than adjacent TADF-S molecules that give the PF (2.36 = 148). The adjacent PF molecules are taken as representative of the bulk average of intermolecular distances in the film, which can be estimated at different FE loadings by considering a uniform cubic lattice. In this way, we estimate the TADF-S–FE distances for both the bulk film (corresponding to ‘adjacent’ TADF-S molecules, responsible for the PF FRET rates) as well as distances for the segregated TADF-S molecules that give rise to the observed DF (Table 1).
| FE loading (%) | Estimated bulk distancea (nm) | Segregated TADF-S distanceb (nm) |
|---|---|---|
| a Estimated from FE loadings and assuming uniform cubic lattice. b Determined from ratio of changes in PF and DF decay rates, and estimated bulk distance. c Loading corresponding to a cubic lattice distance that matches spectrally calculated FRET distance. d FRET distance determined from spectra. e FE Loading corresponding to kinetically determined FRET distance. | ||
| 0.3c | 3.00d | 6.90 |
| 0.4e | 2.73 | 6.27 |
| 1 | 2.01 | 4.62 |
| 1.5 | 1.75 | 4.04 |
| 2 | 1.6 | 3.67 |
| 2.5 | 1.48 | 3.41 |
| 3 | 1.39 | 3.20 |
| 3.5 | 1.32 | 3.05 |
| 4 | 1.27 | 2.91 |
| 5 | 1.18 | 2.70 |
The distance values in Table 1 show that although the average bulk TADF-S–FE distance shrinks significantly with increasing FE loading, the segregated TADF-S molecules never falls below the ∼1 nm distance required for efficient DET. This is consistent with our other experimental results which also suggest that DET is not active in these films despite the appearance of DF emission. At 5% FE loading the calculated distance for the ‘segregated’ TADF-S molecules decreases to below RFRET, meaning that their ISC becomes interrupted by FRET in the PF region. Therefore, at this loading, there is no longer a distinction between adjacent and segregated TADF-S, and no DF emission should be expected as none of the TADF-S molecules are sufficiently far from their nearest neighbour FE to allow ISC to generate triplet excitons. This feature of the calculated distances is consistent with time-resolved emission decays, where at 5% the DF signal is no longer observed.
Thus, with optical measurements we do not and cannot gain information about DET from the TADF-S triplet states to the FE. Any molecule close enough to an FE to potentially undergo DET in a device will have their ISC interrupted by FRET in the PF, eliminating their contribution to the DF (when DET might be detectable). The TADF-S molecules that do contribute to the DF are a different subset, necessarily far enough from any FE that (slower) FRET does not outcompete ISC, also ensuring that they cannot undergo DET. Attempts to accelerate DET and make it easier to detect by increasing FE loading only decrease the subset of TADF molecules that form triplets. The subsequent decrease in DF lifetime is due to accelerated FRET, although at rates lower than observed in the PF due to self-selection effects.
In light of the failure of optical methods described here, it is clear that new methods of electrical excitation will be necessary to identify and fully understand the energy transfer mechanisms relevant to hyperfluorescent OLEDs. At present it is unclear how to address the problems of direct FE excitation from charge recombination, or how to overcome the slow electrical response time that obscures the fast decay mechanisms in pulsed OLEDs. Nonetheless, recently reported electron and hole-only device studies of hyperfluorescent devices strongly suggest that charge trapping by the FE plays a key role in determining device efficiency,29 even at 1% FE loading. Simultaneously, performance gains from efforts to physically distance the TADF-S and FE provide circumstantial evidence for DET as a loss mechanism.27,34,35 Both of these loss mechanisms are likely active to different extents in different hyperfluorescence systems.
Conclusion
Using a combination of time-resolved emission spectroscopy, transient absorption, and PLQE measurements we find that the rates of DET from a TADF sensitizer to the non-emissive triplet state of a fluorescence emitter – assumed to be the main loss mechanism in hyperfluorescent OLED devices – is inaccessible from photophysical experiments. The differences in DF lifetime for different FE concentrations seen here and elsewhere are instead attributed to a distribution of distances between the TADF-S and its nearest-neighbor FE in the film, and consequently a distribution of FRET rates. These results reveal a fundamental limitation in using optical excitation methods to characterize the loss mechanisms in hyperfluorescent systems.Author contributions
NH and AD performed the optical experiments, assisted by PS. NH and AD interpreted the results together with CP, WB, and APM. NH and AD wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the HyperOLED project from the European Unions's Horizon 2020 research and innovation program under grant agreement number 732013.References
- M. A. Baldo, D. O'brien, Y. You, A. Shoustikov, S. Sibley, M. Thompson and S. R. Forrest, Highly Efficient Phosphorescent Emission from Organic Electroluminescent Devices, Nature, 1998, 395, 151 CrossRef CAS.
- V. Jankus, P. Data, D. Graves, C. McGuinness, J. Santos, M. R. Bryce, F. B. Dias and A. P. Monkman, Highly Efficient TADF OLEDs: How the Emitter–Host Interaction Controls Both the Excited State Species and Electrical Properties of the Devices to Achieve near 100% Triplet Harvesting and High Efficiency, Adv. Funct. Mater., 2014, 24, 6178–6186 CrossRef CAS.
- V. Sivasubramaniam, F. Brodkorb, S. Hanning, H. P. Loebl, V. van Elsbergen, H. Boerner, U. Scherf and M. Kreyenschmidt, Fluorine Cleavage of the Light Blue Heteroleptic Triplet Emitter Firpic, J. Fluorine Chem., 2009, 130, 640–649 CrossRef CAS.
- D. Kondakov, T. Pawlik, T. Hatwar and J. Spindler, Triplet Annihilation Exceeding Spin Statistical Limit in Highly Efficient Fluorescent Organic Light-Emitting Diodes, J. Appl. Phys., 2009, 106, 124510 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence, Nature, 2012, 492, 234 CrossRef CAS PubMed.
- Y. J. Cho, S. K. Jeon, S.-S. Lee, E. Yu and J. Y. Lee, Donor Interlocked Molecular Design for Fluorescence-Like Narrow Emission in Deep Blue Thermally Activated Delayed Fluorescent Emitters, Chem. Mater., 2016, 28, 5400–5405 CrossRef CAS.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Ultrapure Blue Thermally Activated Delayed Fluorescence Molecules: Efficient Homo–Lumo Separation by the Multiple Resonance Effect, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- H. Nakanotani, T. Higuchi, T. Furukawa, K. Masui, K. Morimoto, M. Numata, H. Tanaka, Y. Sagara, T. Yasuda and C. Adachi, High-Efficiency Organic Light-Emitting Diodes with Fluorescent Emitters, Nat. Commun., 2014, 5, 4016 CrossRef CAS PubMed.
- D. H. Ahn, J. H. Jeong, J. Song, J. Y. Lee and J. H. Kwon, Highly Efficient Deep Blue Fluorescent Organic Light-Emitting Diodes Boosted by Thermally Activated Delayed Fluorescence Sensitization, ACS Appl. Mater. Interfaces, 2018, 10, 10246–10253 CrossRef CAS PubMed.
- D. Li, Y. Hu and L.-S. Liao, Triplet Exciton Harvesting by Multi-Process Energy Transfer in Fluorescent Organic Light-Emitting Diodes, J. Mater. Chem. C, 2019, 7, 977–985 RSC.
- S. Wang, Y. Zhang, W. Chen, J. Wei, Y. Liu and Y. Wang, Achieving High Power Efficiency and Low Roll-Off OLEDs Based on Energy Transfer from Thermally Activated Delayed Excitons to Fluorescent Dopants, Chem. Commun., 2015, 51, 11972–11975 RSC.
- D. Zhang, L. Duan, C. Li, Y. Li, H. Li, D. Zhang and Y. Qiu, High-Efficiency Fluorescent Organic Light-Emitting Devices Using Sensitizing Hosts with a Small Singlet–Triplet Exchange Energy, Adv. Mater., 2014, 26, 5050–5055 CrossRef CAS PubMed.
- T. Furukawa, H. Nakanotani, M. Inoue and C. Adachi, Dual Enhancement of Electroluminescence Efficiency and Operational Stability by Rapid Upconversion of Triplet Excitons in OLEDs, Sci. Rep., 2015, 5, 8429 CrossRef CAS PubMed.
- X. K. Liu, Z. Chen, C. J. Zheng, M. Chen, W. Liu, X. H. Zhang and C. S. Lee, Nearly 100% Triplet Harvesting in Conventional Fluorescent Dopant-Based Organic Light-Emitting Devices through Energy Transfer from Exciplex, Adv. Mater., 2015, 27, 2025–2030 CrossRef CAS PubMed.
- B. Zhao, Y. Miao, Z. Wang, W. Chen, K. Wang, H. Wang, Y. Hao, B. Xu and W. Li, Highly Efficient Orange Fluorescent OLEDs Based on the Energy Transfer from Bilayer Interface Exciplex, Org. Electron., 2016, 37, 1–5 CrossRef.
- C. Zhao, C. Li, Y. Li, Y. Qiu and L. Duan, Understanding the Operational Lifetime Expansion Methods of Thermally Activated Delayed Fluorescence Sensitized OLEDs: A Combined Study of Charge Trapping and Exciton Dynamics, Mater. Chem. Front., 2019, 3, 1181–1191 RSC.
- C.-Y. Chan, M. Tanaka, Y.-T. Lee, Y.-W. Wong, H. Nakanotani, T. Hatakeyama and C. Adachi, Stable Pure-Blue Hyperfluorescence Organic Light-Emitting Diodes with High-Efficiency and Narrow Emission, Nat. Photonics, 2021, 15, 203–207 CrossRef CAS.
- S. O. Jeon, K. H. Lee, J. S. Kim, S.-G. Ihn, Y. S. Chung, J. W. Kim, H. Lee, S. Kim, H. Choi and J. Y. Lee, High-Efficiency, Long-Lifetime Deep-Blue Organic Light-Emitting Diodes, Nat. Photonics, 2021, 1–8 Search PubMed.
- K. Stavrou, A. Danos, T. Ham, T. Hatakeyama and A. Monkman, Hot Vibrational States in a High-Performance Multiple Resonance Emitter and the Effect of Excimer Quenching on Organic Light-Emitting Diodes, ACS Appl. Mater. Interfaces, 2021, 13(7), 8643–8655 CrossRef CAS PubMed.
- Y. Wada, H. Nakagawa, S. Matsumoto, Y. Wakisaka and H. Kaji, Organic Light Emitters Exhibiting Very Fast Reverse Intersystem Crossing, Nat. Photonics, 2020, 1–7 CAS.
- D. Zhang, X. Song, A. J. Gillett, B. H. Drummond, S. T. Jones, G. Li, H. He, M. Cai, D. Credgington and L. Duan, Efficient and Stable Deep-Blue Fluorescent Organic Light-Emitting Diodes Employing a Sensitizer with Fast Triplet Upconversion, Adv. Mater., 2020, 32, 1908355 CrossRef CAS PubMed.
- H. Abroshan, V. Coropceanu and J.-L. Brédas, Hyperfluorescence-Based Emission in Purely Organic Materials: Suppression of Energy-Loss Mechanisms Via Alignment of Triplet Excited States, ACS Mater. Lett., 2020, 2, 1412–1418 CrossRef CAS.
- Y. Giret, J. Eng, T. Pope and T. Penfold, A Quantum Dynamics Study of the Hyperfluorescence Mechanism, J. Mater. Chem. C, 2021, 9, 1362–1369 RSC.
- M. Jakoby, B. S. Richards, U. Lemmer and I. A. Howard, Investigations of Singlet and Triplet Diffusion in Thermally Activated Delayed-Fluorescence Emitters: Implications for Hyperfluorescence, Phys. Rev. B, 2019, 100, 045303 CrossRef CAS.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Efficient Blue Organic Light-Emitting Diodes Employing Thermally Activated Delayed Fluorescence, Nat. Photonics, 2014, 8, 326 CrossRef CAS.
- I. HoáLee and J. YeobáLee, High Efficiency Blue Fluorescent Organic Light-Emitting Diodes Using a Conventional Blue Fluorescent Emitter, J. Mater. Chem. C, 2015, 3, 8834–8838 RSC.
- S. H. Han and J. Y. Lee, Spatial Separation of Sensitizer and Fluorescent Emitter for High Quantum Efficiency in Hyperfluorescent Organic Light-Emitting Diodes, J. Mater. Chem. C, 2018, 6, 1504–1508 RSC.
- J. H. Kim, K. H. Lee and J. Y. Lee, Design of Thermally Activated Delayed Fluorescent Sensitizers for High Efficiency over 20% and Long Lifetime in Yellow Fluorescent Organic Light-Emitting Diodes, J. Mater. Chem. C, 2020, 8, 5265–5272 RSC.
- Y. Chen, Q. Sun, Y. Dai, D. Yang, X. Qiao and D. Ma, High Efficiency Blue and Color-Stable Hybrid Warm White Organic Light-Emitting Diodes Based on a Thermally Activated Delayed Fluorescent Material as Assistant Host, J. Mater. Chem. C, 2020, 8, 13777–13785 RSC.
- L.-S. Cui, A. J. Gillett, S.-F. Zhang, H. Ye, Y. Liu, X.-K. Chen, Z.-S. Lin, E. W. Evans, W. K. Myers and T. K. Ronson, Fast Spin-Flip Enables Efficient and Stable Organic Electroluminescence from Charge-Transfer States, Nat. Photonics, 2020, 1–7 Search PubMed.
- J. H. Kim, K. H. Lee and J. Y. Lee, Design of Thermally Activated Delayed Fluorescent Assistant Dopants to Suppress the Nonradiative Component in Red Fluorescent Organic Light-Emitting Diodes, Chem. – Eur. J., 2019, 25, 9060–9070 CrossRef CAS PubMed.
- H. Abroshan, Y. Zhang, X. Zhang, C. Fuentes-Hernandez, S. Barlow, V. Coropceanu, S. R. Marder, B. Kippelen and J. L. Brédas, Thermally Activated Delayed Fluorescence Sensitization for Highly Efficient Blue Fluorescent Emitters, Adv. Funct. Mater., 2020, 2005898 CrossRef CAS.
- H. H. Cho, A. S. Romanov, M. Bochmann, N. C. Greenham and D. Credgington, Matrix-Free Hyperfluorescent Organic Light-Emitting Diodes Based on Carbene–Metal–Amides, Adv. Opt. Mater., 2021, 9, 2001965 CrossRef CAS.
- D. Liu, K. Sun, G. Zhao, J. Wei, J. Duan, M. Xia, W. Jiang and Y. Sun, Spatial Separation of a TADF Sensitizer and Fluorescent Emitter with a Core-Dendron System to Block the Energy Loss in Deep Blue Organic Light-Emitting Diodes, J. Mater. Chem. C, 2019, 7, 11005–11013 RSC.
- J. S. Jang, S. H. Han, H. W. Choi, K. S. Yook and J. Y. Lee, Molecular Design of Sensitizer to Suppress Efficiency Loss Mechanism in Hyper-Fluorescent Organic Light-Emitting Diodes, Org. Electron., 2018, 59, 236–242 CrossRef CAS.
- E. Aksoy, A. Danos, C. Varlikli and A. P. Monkman, Navigating CIE Space for Efficient TADF Downconversion WOLEDs, Dyes Pigm., 2020, 183, 108707 CrossRef CAS.
- T. Cardeynaels, S. Paredis, A. Danos, D. Vanderzande, A. P. Monkman, B. Champagne and W. Maes, Benzo [1, 2-B: 4, 5-B'] Dithiophene as a Weak Donor Component for Push-Pull Materials Displaying Thermally Activated Delayed Fluorescence or Room Temperature Phosphorescence, Dyes Pigm., 2021, 186, 109022 CrossRef CAS.
- P. L. dos Santos, J. S. Ward, M. R. Bryce and A. P. Monkman, Using Guest–Host Interactions to Optimize the Efficiency of TADF OLEDs, J. Phys. Chem. Lett., 2016, 7, 3341–3346 CrossRef CAS PubMed.
- N. Haase, A. Danos, C. Pflumm, A. Morherr, P. Stachelek, A. Mekic, W. Brütting and A. P. Monkman, Kinetic Modeling of Transient Photoluminescence from Thermally Activated Delayed Fluorescence, J. Phys. Chem. C, 2018, 122, 29173–29179 CrossRef CAS.
- P. Stachelek, J. S. Ward, P. L. dos Santos, A. Danos, M. Colella, N. Haase, S. J. Raynes, A. S. Batsanov, M. R. Bryce and A. P. Monkman, Molecular Design Strategies for Color Tuning of Blue TADF Emitters, ACS Appl. Mater. Interfaces, 2019, 11, 27125–27133 CrossRef CAS PubMed.
- J. S. Ward, A. Danos, P. Stachelek, M. A. Fox, A. S. Batsanov, A. P. Monkman and M. R. Bryce, Exploiting Trifluoromethyl Substituents for Tuning Orbital Character of Singlet and Triplet States to Increase the Rate of Thermally Activated Delayed Fluorescence, Mater. Chem. Front., 2020, 4, 3602–3615 RSC.
- I. Wright, A. Danos, S. Montanaro, A. S. Batsanov, A. P. Monkman and M. R. Bryce, Conformational Dependence of Triplet Energies in Rotationally-Hindered N- and S-Heterocyclic Dimers: New Design and Measurement Rules for High Triplet Energy OLED Host Materials, Chem. – Eur. J., 2021 DOI:10.1002/chem.202100036.
- I. Lee and J. Y. Lee, Molecular Design of Deep Blue Fluorescent Emitters with 20% External Quantum Efficiency and Narrow Emission Spectrum, Org. Electron., 2016, 29, 160–164 CrossRef CAS.
- Y. Y. Cheng, B. Fückel, T. Khoury, R. G. C. R. Clady, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, Entropically Driven Photochemical Upconversion, J. Phys. Chem. A, 2011, 115, 1047–1053 CrossRef CAS PubMed.
- C. Ye, V. Gray, J. Mårtensson and K. Börjesson, Annihilation Vs. Excimer Formation by the Triplet Pair in Triplet–Triplet Annihilation Photon Upconversion, J. Am. Chem. Soc., 2019, 141, 9578–9584 CrossRef CAS PubMed.
- B. W. D’Andrade, M. A. Baldo, C. Adachi, J. Brooks, M. E. Thompson and S. R. Forrest, High-Efficiency Yellow Double-Doped Organic Light-Emitting Devices Based on Phosphor-Sensitized Fluorescence, Appl. Phys. Lett., 2001, 79, 1045–1047 CrossRef.
- T. Virgili, D. G. Lidzey and D. D. Bradley, Efficient Energy Transfer from Blue to Red in Tetraphenylporphyrin-Doped Poly (9, 9-Dioctylfluorene) Light-Emitting Diodes, Adv. Mater., 2000, 12, 58–62 CrossRef CAS.
- G. Cerullo, S. Stagira, M. Zavelani-Rossi, S. De Silvestri, T. Virgili, D. Lidzey and D. Bradley, Ultrafast Förster Transfer Dynamics in Tetraphenylporphyrin Doped Poly (9, 9-Dioctylfluorene), Chem. Phys. Lett., 2001, 335, 27–33 CrossRef CAS.
- C. Nguyen, G. Ingram and Z.-H. Lu, Quantifying Interdopant Exciton Processes in Organic Light Emitting Diodes, J. Phys. Chem. C, 2017, 121, 3304–3309 CrossRef CAS.
- P. Heimel, A. Mondal, F. May, W. Kowalsky, C. Lennartz, D. Andrienko and R. Lovrincic, Unicolored Phosphor-Sensitized Fluorescence for Efficient and Stable Blue OLEDs, Nat. Commun., 2018, 9, 4990 CrossRef PubMed.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos and M. R. Bryce, The Role of Local Triplet Excited States and D-A Relative Orientation in Thermally Activated Delayed Fluorescence: Photophysics and Devices, Adv. Sci., 2016, 3, 1600080 CrossRef PubMed.
- T. Penfold, F. Dias and A. Monkman, The Theory of Thermally Activated Delayed Fluorescence for Organic Light Emitting Diodes, Chem. Commun., 2018, 54, 3926–3935 RSC.
- C. Zhang, Y. Lu, Z. Liu, Y. Zhang, X. Wang, D. Zhang and L. Duan, A π–D and π–A Exciplex-Forming Host for High-Efficiency and Long-Lifetime Single-Emissive-Layer Fluorescent White Organic Light-Emitting Diodes, Adv. Mater., 2020, 2004040 CrossRef CAS PubMed.
- L. Paterson, A. Mondal, P. Heimel, R. Lovrincic, F. May, C. Lennartz and D. Andrienko, Perspectives of Unicolored Phosphor-Sensitized Fluorescence, Adv. Electron. Mater., 2019, 1900646 CrossRef CAS.
- J. H. Yun, C. W. Lee, K. S. Yook and J. Y. Lee, Alkyl Free Design of Anthracene Based Host Material for Solution Processed Blue Fluorescent Organic Light-Emitting Diodes, Synth. Met., 2016, 217, 216–219 CrossRef CAS.
- G. J. Hedley, A. Ruseckas and I. D. Samuel, Ultrafast Luminescence in Ir(ppy)3, Chem. Phys. Lett., 2008, 450, 292–296 CrossRef CAS.
- T. Hofbeck and H. Yersin, The Triplet State of fac-Ir(ppy)3, Inorg. Chem., 2010, 49, 9290–9299 CrossRef CAS PubMed.
- R. Huang, N. A. Kukhta, J. S. Ward, A. Danos, A. S. Batsanov, M. R. Bryce and F. B. Dias, Balancing Charge-Transfer Strength and Triplet States for Deep-Blue Thermally Activated Delayed Fluorescence with an Unconventional Electron Rich Dibenzothiophene Acceptor, J. Mater. Chem. C, 2019, 7, 13224–13234 RSC.
- N. A. Kukhta, H. F. Higginbotham, T. Matulaitis, A. Danos, A. N. Bismillah, N. Haase, M. K. Etherington, D. S. Yufit, P. R. McGonigal and J. V. Gražulevičius, Revealing Resonance Effects and Intramolecular Dipole Interactions in the Positional Isomers of Benzonitrile-Core Thermally Activated Delayed Fluorescence Materials, J. Mater. Chem. C, 2019, 7, 9184–9194 RSC.
- M. Colella, A. Danos and A. P. Monkman, Less Is More: Dilution Enhances Optical and Electrical Performance of a TADF Exciplex, J. Phys. Chem. Lett., 2019, 10, 793–798 CrossRef CAS PubMed.
- N. J. Davis, R. W. MacQueen, D. A. Roberts, A. Danos, S. Dehn, S. Perrier and T. W. Schmidt, Energy Transfer in Pendant Perylene Diimide Copolymers, J. Mater. Chem. C, 2016, 4, 8270–8275 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0mh01666g |
| This journal is © The Royal Society of Chemistry 2021 |