
Vacuum ultraviolet photochemistry of the conformers of the ethyl peroxy radical†
Zuoying
Wen‡
a,
Xiaoxiao
Lin‡
 a,
Xiaofeng
Tang
*a,
Bo
Long
b,
Chengcheng
Wang
a,
Cuihong
Zhang
a,
Christa
Fittschen
c,
Jiuzhong
Yang
d,
Xuejun
Gu
a and
Weijun
Zhang
*a
a,
Xiaofeng
Tang
*a,
Bo
Long
b,
Chengcheng
Wang
a,
Cuihong
Zhang
a,
Christa
Fittschen
c,
Jiuzhong
Yang
d,
Xuejun
Gu
a and
Weijun
Zhang
*a
aLaboratory of Atmospheric Physico-Chemistry, Anhui Institute of Optics and Fine Mechanics, HFIPS, Chinese Academy of Sciences, Hefei, 230031 Anhui, China. E-mail: tangxf@aiofm.ac.cn; wjzhang@aiofm.ac.cn
bSchool of Materials Science and Engineering, Guizhou Minzu University, Guiyang, 550025 Guizhou, China
cUniversity Lille, CNRS, UMR 8522, PC2A – Physicochimie des Processus de Combustion et de l’Atmosphère, F-59000 Lille, France
dNational Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, 230029 Anhui, China
First published on 7th September 2021
Abstract
We study the conformers of the ethyl peroxy radical (C2H5O2), the simplest peroxy radical having more than one conformer, by combining synchrotron radiation vacuum ultraviolet (VUV) photoionization mass spectrometry with theoretical calculations. The ethyl peroxy radical is formed in a microwave discharge flow tube through the reaction of the ethyl radical (C2H5) with oxygen molecules, where C2H5 is generated via the hydrogen-abstraction reaction of ethane with fluorine atoms. Two kinds of C2H5+, originating from photoionization of C2H5 and from dissociative photoionization of C2H5O2, whose cation is not stable, have been identified and separated in photoionization mass spectra. The photoionization spectrum corresponding to C2H5O2 is obtained and assigned with Franck–Condon calculations. The present findings show that the gauche conformer (G-C2H5O2) of C2H5O2 has favorable Franck–Condon factors in the ionization transitions, whereas the contribution of the trans conformer (T-C2H5O2) to the photoionization spectrum is minor or negligible due to its large geometric changes in the photoionization process. Moreover, the reason for the instability of C2H5O2+ and its detailed dissociation mechanisms have been unraveled with the aid of the calculated potential energy curves.
1. Introduction
Peroxy radicals (RO2) are very important reaction intermediates that play crucial roles in the low temperature oxidation of organic compounds in combustion and atmospheric chemistry.1–3 In the atmosphere, peroxy radicals are formed via the reaction of alkyl radicals (R) with oxygen molecules. Then the formed peroxy radicals can undergo bimolecular reactions with various species, such as NOx (NO and NO2), HO2 and other peroxy radicals, self-reaction and unimolecular reaction. These key processes make an important contribution for the production of secondary pollutants such as ozone and secondary organic aerosols (SOA).2,3 Thus the direct measurement of peroxy radicals is a fundamental issue in atmospheric chemistry.The spectroscopy and structure of peroxy radicals are of considerable importance for understanding their reactivity, sources and sinks in the atmosphere. In the past decades, benefiting from the constant development of analytical methods such as absorption spectroscopy,4,5 cavity ring-down spectroscopy (CRDS),6–9 photoionization mass spectrometry (PIMS)10,11 and photoelectron photoion coincidence spectroscopy (PEPICO),12,13 a great deal of advance has been made to unravel their spectroscopy and structures. Among them, PIMS and PEPICO in particular combined with tunable synchrotron radiation as the photoionization light source have attracted attention and are considered as universal and sensitive methods to probe and analyze such elusive species.14–16 For example, Meloni et al. studied the vacuum ultraviolet (VUV) photoionization of alkylperoxy radicals at the Advanced Light Source by using PIMS and found most of their cations unstable except for the methyl peroxy radical (CH3O2).10 The ionization energy (IE) of CH3O2 and the appearance energy (AE) of the CH3+ fragment ion in dissociative photoionization of CH3O2 were measured using PEPICO by Voronova et al. at the Swiss Light Source and Tang et al. at SOLEIL, France.12,13
With the increase in mass, the structure of peroxy radicals becomes complex, having several isomers or conformers. For instance, the ethyl peroxy radical has two conformers, the gauche conformer (G-C2H5O2) with its CCOO dihedral angle of ±60° and the trans conformer (T-C2H5O2) with the CCOO dihedral angle of ±180°, and the propyl peroxy radical has two isomers, 1-C3H7O2 and 2-C3H7O2, each of which also has several conformers.6 The knowledge on the isomers or conformers of peroxy radicals is very limited, and identifying them is still a challenge. Recently, using the method of PEPICO at SOLEIL, two isomers of C3H7O2 were successfully identified and assigned in our publication.17
Here we focus on the ethyl peroxy radical, C2H5O2, and its two conformers, G-C2H5O2 and T-C2H5O2. The ethyl peroxy radical is the simplest peroxy radical having more than one conformer, and has attracted attention.18–21 Previous theoretical calculations showed that G-C2H5O2 is the global minimum with an energy of ∼80 cm−1 lower than T-C2H5O2.20,21 Upon photoionization, the nascent C2H5O2+ cation is not stable and dissociates to C2H5+ and O2 fragments.10,11 To explain the instability of C2H5O2+, Meloni et al. suggested the cation has a hyper-conjugation structure.10 The AE of the C2H5+ and O2 fragments was measured at 10.0 ± 0.1 eV in the photoionization spectrum by these authors.
In this work, VUV photoionization of C2H5O2 has been investigated in detail by using synchrotron-based PIMS complemented by high-level theoretical calculations on the IEs, structures and spectra. After distinguishing two different sources of C2H5+, originating from the photoionization of the ethyl radical (C2H5) and from the dissociative photoionization of C2H5O2, the photoionization spectrum corresponding to C2H5O2 is acquired and has been assigned with Franck–Condon calculations. The present findings will demonstrate that the G-C2H5O2 conformer has favorable Franck–Condon factors in the ionization transition, whereas the contribution of T-C2H5O2 to the photoionization spectrum is minor or negligible due to its large geometric changes in the photoionization process.
2. Methods
The experiments were carried out on the combustion and flame beamline at National Synchrotron Radiation Laboratory (NSRL) in Hefei, China.22 A fast flow tube was employed as the reactor to generate free radicals and initiate their chemical reactions.23 A home-made photoionization orthogonal acceleration reflectron time-of-flight (TOF) mass spectrometer was installed to probe and analyze reactants, reaction intermediates and products inside the fast flow tube.24 The detailed configurations of the synchrotron beamline, the fast flow tube and the TOF mass spectrometer have already been introduced before and only a brief description is presented here.22–24Briefly, on the beamline, synchrotron photons emitted from an undulator were dispersed by a monochromator equipped with two gratings, 200 and 400 l mm−1, covering the photon energy range of 5–21 eV. A gas filter presently filled with Ar gas was adopted to suppress high-order harmonic radiations from the undulator, and the absolute photon energy was calibrated within an accuracy of ±10 meV using the resonant absorption lines of Ar in the gas filter.25 The synchrotron photon flux was measured using a photodiode (SXUV-100, IRD Inc.) and used to normalize ion signals in the photon energy scans.
The fast flow tube mainly including a main tube and a coaxial movable injector was installed inside the source chamber of the TOF mass spectrometer.23 Fluorine atoms were generated from 1% diluted F2 gas in helium using a 2.45 GHz microwave discharge generator (GMS-200W, Sairem, France) and introduced into the fast flow tube to initiate chemical reactions. Other reactant gases such as ethane and oxygen, together with helium bath gas, were injected via the arms of the main tube or the injector. The total pressure inside the flow tube was monitored using a capacity gauge and fixed at 2 Torr by using a closed-loop feedback throttle valve.
After passing through one skimmer (1 mm diameter), the gas mixture in the fast flow tube was sampled and entered into the ionization chamber of the TOF mass spectrometer, crossing a synchrotron photon beam at a right angle and then being ionized. A cage-shaped photoionization source connected with an Einzel lens was developed to extract and focus ions, and then to enhance the detection sensitivity of the setup.24 The TOF mass spectrometer with an orthogonal acceleration and reflectron structure was employed to analyze the mass of ions. The total ion flight length was ∼1 m and the mass resolving power of the TOF mass spectrometer was measured to be M/ΔM ∼ 2000 (FWHM, the full width at half maximum).24
To provide further insights into the experimental data, theoretical calculations have been performed to study the adiabatic ionization energies (AIEs), the potential energy curves and the Franck–Condon factors, using the Gaussian 16 package and MOLPRO 2015 software.26,27 The geometric structures of the two conformers of C2H5O2 and their cations were optimized by using the M06-2X/aug-cc-pVTZ theoretical methods. The AIEs of the two conformers were calculated using the explicitly correlated coupled cluster single-double and perturbative triple excitations approach, (R)CCSD(T)-F12/aug-cc-pVTZ.27 The potential energy curves of the low-lying electronic states of neutral C2H5O2 and its cation along the C–O coordinates have been calculated at the M06-2X/aug-cc-pVTZ level. In order to assign the photoionization spectrum of C2H5O2, the Franck–Condon factors in the ionization transitions have been calculated at the M06-2X/aug-cc-pVTZ level too, using the time-independent adiabatic Hessian Franck–Condon model in the Gaussian program.26
3. Results and discussion
The experiments were performed in sequence and can be divided into two parts. At first, the experiment was carried out without adding oxygen gas into the fast flow tube, and the ethyl radical (C2H5) was produced via the hydrogen-abstraction reaction of ethane with fluorine atoms.| F + C2H6 → C2H5 + HF | (1) |
Secondly, with the addition of oxygen gas, the nascent ethyl radical reacted with oxygen to generate the products, C2H5O2, ethylene (C2H4) and the hydroperoxy radical (HO2), via the following two reactions.19,28
| C2H5 + O2 → C2H5O2 | (2a) |
| → C2H4 + HO2 | (2b) |
The photoionization mass spectrum in the absence of oxygen in the fast flow tube was acquired at the photon energy of hν = 9.6 eV and is presented as a blue line in Fig. 1. An intense peak is observed at m/z = 29 and has been assigned as C2H5 produced from the above mentioned hydrogen-abstraction reaction (1). Another mass peak at m/z = 30 is observed in the mass spectrum too, and can be ascribed to the 13C2H5 isotopic ethyl radical with an intensity of about 4% of the m/z = 29 peak. The IE of ethane locates at 11.52 eV,29 above the present photon energy of 9.6 eV, indicating that ethane does not contribute to the m/z = 30 peak.
 | ||
| Fig. 1 Photoionization TOF mass spectra acquired without (in blue, hν = 9.6 eV) and with (in red, hν = 9.6 eV; in black, hν = 11.0 eV) the addition of oxygen into the fast flow tube. | ||
The photoionization spectrum of C2H5 has been measured by scanning the synchrotron photon energy with a step size of 0.05 eV, and is presented as black squares in Fig. 2(a). The overall shape of the photoionization spectrum is similar to the literature results of Ruscic et al.,30 and the difference might be due to the different internal energies of the produced ethyl radicals and the different photo-flux calibrations in the two experiments. Presently the cationic signal appears at hν = 8.33 ± 0.05 eV in the photoionization spectrum, in accordance with the IE of C2H5.29,31
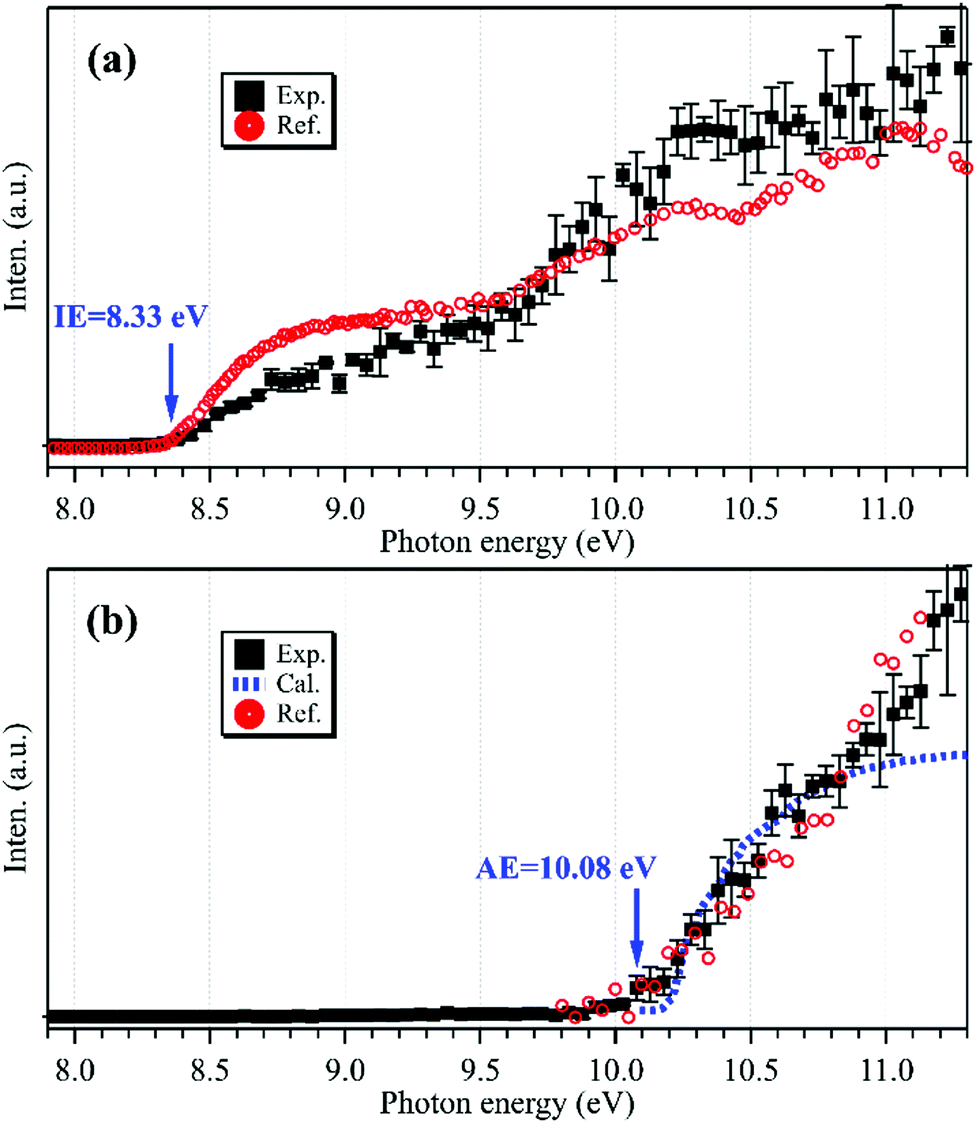 | ||
| Fig. 2 Photoionization spectra of the m/z = 29 cations acquired without (a) and with (b) the addition of oxygen gas into the flow tube, together with the reference data of C2H5 (a, in red) reprinted from Ruscic et al.30 (with the permission of AIP Publishing), the reference data of C2H5O2 (b, in red) adapted from Meloni et al.10 (Copyright 2021 American Chemical Society), and the calculated results of C2H5O2 (b, in blue). | ||
After adding oxygen gas into the flow tube, the intensity of the m/z = 29 mass peak declines, nearly approaching the baseline of the mass spectrum, as shown using a red line in Fig. 1. In addition, almost no other peaks can be observed in the mass spectrum. Previous results have shown that the reaction of C2H5 with oxygen is fast with a rate constant of 2 × 10−12 cm3 molecule−1 s−1 at 298 K,32 and can generate the products of C2H5O2, C2H4 and HO2. The C2H5O2+ cation is not stable and the AE of the C2H5+ and O2 fragments was measured at 10.0 ± 0.1 eV,10 above the present photon energy of 9.6 eV. For the other products, C2H4 and HO2, their IEs locate at 10.5138 and 11.359 eV,29 respectively, and thus they are not observed in the mass spectrum.
The synchrotron photon energy was increased to hν = 11.0 eV, and the photoionization mass spectrum was acquired and is presented as a black line in Fig. 1. Similar to the previous results,10,11 still no signal could be observed at m/z = 61 (C2H5O2+) in the mass spectrum. But, presently the m/z = 29 mass peak appears again and this reappearance should have a different source as that of the above C2H5 obtained in the absence of O2. In addition, the product of C2H4 (m/z = 28) from the reaction of C2H5 with oxygen has been observed in the mass spectrum. Some other peaks are observed in the mass spectrum too and can be ascribed to the secondary reactions occurring in the flow tube.33 For example, the m/z = 30 mass peak is ascribed to the secondary reaction product of HCHO whose IE is measured at 10.83 ± 0.05 eV in the photoionization spectrum (Fig. S1, ESI†), and the m/z = 62 mass peak can be assigned as C2H5OOH, the product of the reaction of C2H5O2 with HO2.
To explain the reappearance of the m/z = 29 peak in the mass spectrum, its photoionization spectrum was acquired (with the addition of oxygen) and is presented as black squares in Fig. 2(b), together with the literature data of Meloni et al.10 Unlike the case in Fig. 2(a), presently the m/z = 29 cation does not appear until at hν = 10.08 ± 0.05 eV in the photoionization spectrum, agreeing well with the literature, AE(C2H5+) = 10.0 ± 0.1 eV in dissociative photoionization of C2H5O2 measured by Meloni et al.,10 and thus should be ascribed to the contribution of C2H5O2.
The neutral C2H5O2 at the X2A′′ ground electronic state has an open-shell electronic configuration of (11a′)2(12a′)2(3a′′)2(13a′)2(4a′′)1.34 Theoretical calculations show that removing an electron from the 13a′ orbital or from the 4a′′ orbital through photoionization can lead to the X3A′′ ground electronic state or the a1A′ first excited electronic state of C2H5O2+, respectively. The geometric structures of the two conformers of C2H5O2, G-C2H5O2 and T-C2H5O2, in the X2A′′ ground state are optimized at the M06-2X/aug-cc-pVTZ level of theory and presented in Fig. 3, together with the optimized structures of their individual cations in the X3A′′ and a1A′ electronic states after checking their individual wavefunction stablity.26 It is shown that G-C2H5O2 takes a C1 point-group symmetry and T-C2H5O2 has a Cs symmetry. For consistency, in the following text we do not change and will still use the symbols X2A′′, X3A′′ and a1A′ (instead of X2A, X3A and a1A) to describe the electronic states of G-C2H5O2 and its cation. We can see that upon photoionization the structure of the two conformers, i.e. their C–O bond length or the CCOO dihedral angle, changes somewhat. This will be discussed in detail in the following paragraphs.
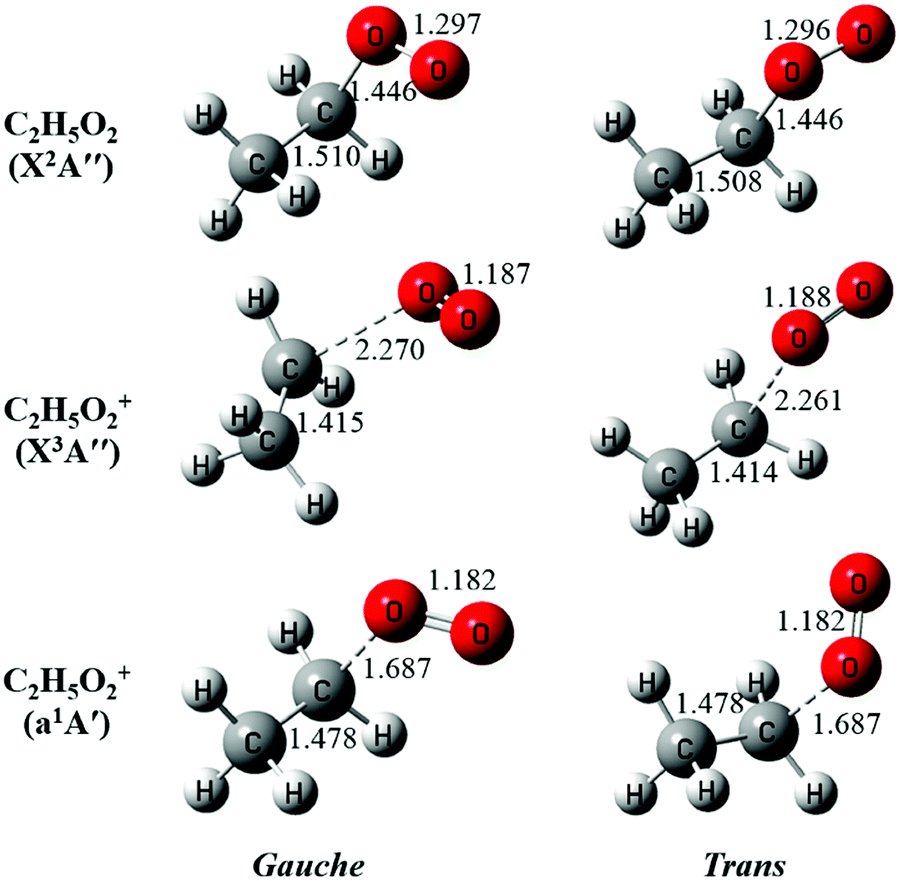 | ||
| Fig. 3 The optimized structures of the gauche and the trans conformers of C2H5O2 and their cations. The bond lengths are in Å. | ||
The potential energy curves of the low-lying electronic states of the neutral ethyl peroxy radical (the minimal conformer G-C2H5O2) and its cation have been calculated at the M06-2X/aug-cc-pVTZ level of theory, and are presented in Fig. 4.26 Concretely, their structures have been re-optimized at each C–O coordinate and then their energies have been computed. It is shown that the neutral X2A′′ ground state is bound with a C–O equilibrium bond length of 1.446 Å. The cationic X3A′′ ground state of C2H5O2+ is calculated to be a quasi-bound state with a very shallow well and a substantially elongated C–O bond at 2.270 Å, which is out of the Franck–Condon region reached by ionization transitions, as shown using a green rectangle in Fig. 4. The singlet a1A′ first excited state of C2H5O2+ is a bound state with a C–O bond length at 1.687 Å, close to that of the neutral X2A′′ ground state, and should be majorly populated in the photoionization process.
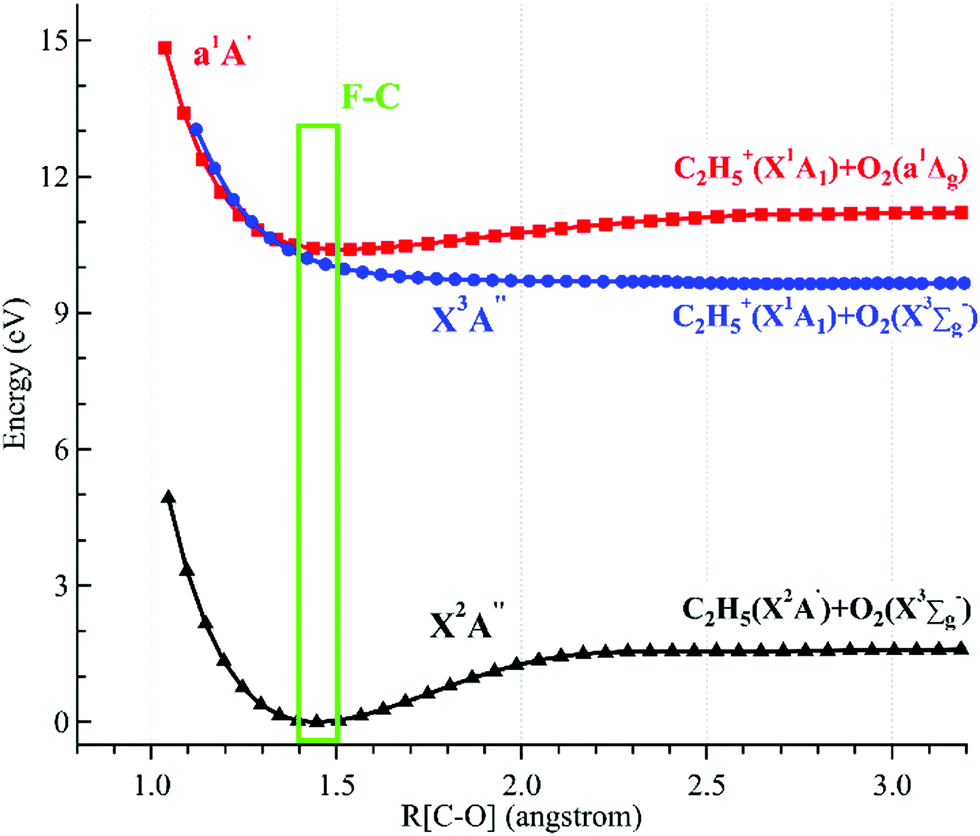 | ||
| Fig. 4 Potential energy curves of the ethyl peroxy radical (G-C2H5O2) and its cation along the C–O coordinates. The Franck–Condon (F–C) region is displayed using a green rectangle. | ||
Theoretical calculations in particular using energy and charge distribution analyses show that the cationic X3A′′ ground state adiabatically correlates to the C2H5+(X1A1) + O2(X3Σg−) dissociation limit, and the a1A′ first excited state of C2H5O2+ correlates to the C2H5+(X1A1) + O2(a1Δg) dissociation limit, not to the C2H5(X2A′) + O2+(X2Πg) dissociation limit.10 In addition, as shown in Fig. 4, the potential energy curve of the a1A′ excited state crosses that of the X3A′′ ground state at ∼1.3 Å and their mutual interaction, e.g. via the spin–orbit coupling, can cause the singlet a1A′ electronic state to pre-dissociate into C2H5+ and O2 fragments along the potential energy curve of the triplet X3A′′ ground state. This pre-dissociation should be a major reason why the C2H5O2+ cation is not observed and why only the C2H5+ fragment ion is observed in the mass spectra.10,11 In the schematic potential energy diagram of Meloni et al.,10 the a3A′′ repulsive state, which also adiabatically correlates to the C2H5+(X1A1) + O2(X3Σg−) dissociation limit, was proposed to cross with the a1A′ excited state at a larger C–O coordinate and this pre-dissociation might also cause the C2H5O2+ cation to be unstable. Presently the vertical ionization energy (VIE) of the second triplet electronic state, A3A′′, of G-C2H5O2+ is calculated to be at 12.24 eV at the TD-M062X/aug-cc-pVTZ level of theory,26 as listed in Table 1.
| State | Species | IE/AE (eV) | Method | Reference | |
|---|---|---|---|---|---|
| a The adiabatic ionization energy. b The vertical ionization energy. c The appearance energy. | |||||
| Calc. | X3A′′ | C2H5O2+ | 9.58a | QCISD(T)/∞ | 10 |
| G-C2H5O2+ | 9.60a/10.40b | CCSD(T)-F12/aug-cc-pVTZ | This work | ||
| T-C2H5O2+ | 9.59a/10.39b | CCSD(T)-F12/aug-cc-pVTZ | This work | ||
| a1A′ | C2H5O2+ | 10.01a | QCISD(T)/∞ | 10 | |
| G-C2H5O2+ | 10.13a/10.42b | CCSD(T)-F12/aug-cc-pVTZ | This work | ||
| T-C2H5O2+ | 10.13a/10.47b | CCSD(T)-F12/aug-cc-pVTZ | This work | ||
| b1A′′ | G-C2H5O2+ | 11.80b | TD-M062X/aug-cc-pVTZ | This work | |
| T-C2H5O2+ | 11.89b | TD-M062X/aug-cc-pVTZ | This work | ||
| A3A′′ | G-C2H5O2+ | 12.24b | TD-M062X/aug-cc-pVTZ | This work | |
| T-C2H5O2+ | 12.18b | TD-M062X/aug-cc-pVTZ | This work | ||
| Exp. | C2H5+ | 10.0 ± 0.1c | PIMS | 10 | |
| C2H5+ | 10.08 ± 0.05c | PIMS | This work | ||
The AIEs and the VIEs of the two conformers of C2H5O2, corresponding to their individual cationic X3A′′ and a1A′ electronic states, have been calculated at the (R)CCSD(T)-F12/aug-cc-pVTZ level of theory and are listed in Table 1 too, together with the literature data.10 The calculated AIEs of the two conformers are very close, as the difference between their two cationic states is within 0.01 eV. In addition, the experimental AE of the C2H5+ and O2 fragments is close to the calculated AIE of the a1A′ electronic state of C2H5O2+, in good accordance with the above results of the potential energy curves.
In order to assign the photoionization spectrum in Fig. 2(b), the Franck–Condon factors for the photoionization of C2H5O2 have also been calculated at the M062X/aug-cc-pVTZ level.26 Theoretical calculations show that the G-C2H5O2 conformer has favorable Franck–Condon factors in the photoionization transition, from the neutral X2A′′ ground electronic state to the cationic a1A′ first excited state, whereas the Franck–Condon factors of the T-C2H5O2 conformer are calculated to be too low to be reliable. The Franck–Condon factors for the transitions to the cationic X3A′′ ground state can be neglected, and its large VIE–AIE difference (0.8 eV), as listed in Table 1, is also an indicator for the negligible ionization cross section. The simulated photoelectron spectrum (PES) of G-C2H5O2 is generated from the Franck–Condon factors by convolving the stick spectrum with a Gaussian function (HWHM = 200 cm−1, half width at half-maximum), shown in Fig. S2 (ESI†). The simulated photoionization spectrum is then obtained by the integration of the PES and presented as a blue dotted line in Fig. 2(b). The simulated photoionization spectrum has been energetically shifted (∼90 meV) to match the experimental data. We can see that the simulated photoionization spectrum satisfactorily agrees with the experimental data, although some differences can be observed at higher energy. One possible reason for the discrepancy is that, besides the a1A′ electronic state, the excitations of other higher excited states of G-C2H5O2+ such as the b1A′′ and A3A′′ states listed in Table 1 might also have contributed to the photoionization spectrum at higher energy. Another possible reason is that the experimental photoionization spectrum is directly obtained from the signal of the C2H5+ fragment ion and thus its dissociation mechanisms might also have influences on the ion intensity, all of which need to be theoretically investigated in detail in the future.
The above favorable Franck–Condon transition can be understood from the optimized geometric change in the photoionization processes too. Previous theoretical predictions showed that the G-C2H5O2 conformer is the minimum with an energy of ∼80 cm−1 lower than T-C2H5O2.18–21 So under the present experimental conditions (∼298 K), both the G-C2H5O2 and T-C2H5O2 conformers should have already been prepared and populated in the near effusive gas beam sampling from the fast flow tube at 2 Torr, and then both of them should have contributed to the photoionization spectrum. But, as shown in Fig. 3, for the T-C2H5O2 conformer, its C–O bond length of 1.446 Å at the neutral X2A′′ ground state should be increased upon ionization to 2.261 Å at the cationic X3A′′ ground state, or its CCOO dihedral angle of ±180° at the X2A′′ ground state should be changed to ±60° at the cationic a1A′ excited state. Our theoretical calculations on the torsional potential energy curves (not presented here) also show that the vertical transition of the T-C2H5O2 conformer from the neutral X2A′′ ground state is to the top of the torsional barrier of the cationic a1A′ excited state, whereas the vertical transition of the G-C2H5O2 conformer is close to the minima of the a1A′ state. The calculated VIE difference (0.05 eV) of the a1A′ excited state of the two conformers, as listed in Table 1, can somewhat reflect the height of the torsional barrier,34 which also needs to be studied in detail in the future. These large geometric changes will result in poor Franck–Condon factors and thus will not favor their ionization transitions.
4. Conclusions
In conclusion, we have studied the VUV photoionization of C2H5O2 by using a home-made photoionization TOF mass spectrometer at Hefei synchrotron complemented with theoretical calculations. The ethyl peroxy radical was produced in a microwave discharge fast flow tube via the reaction of C2H5 with oxygen molecules, whereby C2H5 was generated via the hydrogen-abstraction of ethane with fluorine atoms. The present experiments and theoretical calculations show that the C2H5O2+ cation is not stable and dissociates to C2H5+ and O2 fragments in the photoionization process. Thus, two kinds of C2H5+, one from the photoionization of C2H5 and one from the dissociative photoionization of C2H5O2, have been clearly identified and confirmed in the photoionization mass spectra. The calculated potential energy curves show that the C2H5O2+ cation is firstly prepared at the a1A′ first excited electronic state and then pre-dissociates to the C2H5+ and O2 fragments via the spin–orbital coupling of the a1A′ state with the X3A′′ ground state, which is a quasi-bound state with a very shallow well and adiabatically correlates to the C2H5+ and O2 dissociation limit.The photoionization spectrum corresponding to C2H5O2 is obtained and assigned with the aid of the Franck–Condon calculations. Of the two possible conformers for C2H5O2, our present findings show that the G-C2H5O2 conformer has favorable Franck–Condon factors, from the neutral X2A′′ ground state to the cationic a1A′ first excited electronic state, whereas the contribution of the T-C2H5O2 conformer to the photoionization spectrum is minor or negligible due to its large geometric changes in the photoionization process. The present work provides a prototype using synchrotron-based photoionization mass spectrometry to probe the spectroscopy and structure of peroxy radicals plus their isomers or conformers involved in complex reactions. Some other complex peroxy radicals are being investigated and will be reported in forthcoming papers.
Author contributions
Conception and design: X. T.; acquisition of data: Z. W., X. T., C. W., C. Z., J. Y., and X. G.; theoretical calculation: X. L., B. L., and W. Z.; analysis and interpretation of data: Z. W., X. L., and X. T.; writing—original draft preparation: X. T.; writing—review and editing: all authors; all authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21773249, 91961123, 42120104007, U1832184), the International Partnership Program of Chinese Academy of Sciences (No. 116134KYSB20170048) and the Key Program of Research and Development of Hefei Science Center, CAS (No. 2020HSCKPRD001). C. F. is grateful to the Chinese Academy of Sciences President's International Fellowship Initiative (No. 2018VMA0055). The authors are grateful to Dr Jean-Christophe Loison at Univ. Bordeaux, for helpful discussions on the Franck–Condon factors. The authors also would like to thank the NSRL staff for running the synchrotron facility and providing beamtime.References
- C. K. Westbrook, Chemical kinetics of hydrocarbon ignition in practical combustion systems, Proc. Combust. Inst., 2000, 28, 1563–1577 CrossRef CAS.
- P. S. Monks, Gas-phase radical chemistry in the troposphere, Chem. Soc. Rev., 2005, 34, 376–395 RSC.
- J. J. Orlando and G. S. Tyndall, Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 2012, 41, 6294–6317 RSC.
- G. Chettur and A. Snelson, Alkylperoxy and alkyl radicals. 4. Matrix IR spectra and UV photolysis of ethylperoxy and ethyl radicals, J. Phys. Chem., 1987, 91, 3483–3488 CrossRef CAS.
- G. S. Tyndall, R. A. Cox, C. Granier, R. Lesclaux and G. K. Moortgat, et al., Atmospheric chemistry of small organic peroxy radicals, J. Geophys. Res.: Atmos., 2001, 106, 12157–12182 CrossRef CAS.
- E. N. Sharp, P. Rupper and T. A. Miller, The structure and spectra of organic peroxy radicals, Phys. Chem. Chem. Phys., 2008, 10, 3955–3981 RSC.
- D. B. Atkinson and J. W. Hudgens, Chemical kinetic studies using ultraviolet cavity ring-down spectroscopic detection: self-reaction of ethyl and ethylperoxy radicals and the reaction O2 + C2H5 → C2H5O2, J. Phys. Chem. A, 1997, 101, 3901–3909 CrossRef CAS.
- D. B. Atkinson and J. L. Spillman, Alkyl peroxy radical kinetics measured using near-infrared CW-cavity ring-down spectroscopy, J. Phys. Chem. A, 2002, 106, 8891–8902 CrossRef CAS.
- C. Zhang, M. Shamas, M. Assali, X. Tang and W. Zhang, et al., Absolute absorption cross section of the à < --X electronic transition of the ethyl peroxy radical and rate constant of its cross reaction with HO2, Photonics, 2021, 8, 296 CrossRef.
- G. Meloni, P. Zou, S. J. Klippenstein, M. Ahmed and S. R. Leone, et al., Energy-resolved photoionization of alkylperoxy radicals and the stability of their cations, J. Am. Chem. Soc., 2006, 128, 13559–13567 CrossRef CAS PubMed.
- H. Fu, Y. Hu and E. Bernstein, Generation and detection of alkyl peroxy radicals in a supersonic jet expansion, J. Chem. Phys., 2006, 125, 014310 CrossRef CAS PubMed.
- K. Voronova, K. M. Ervin, K. G. Torma, P. Hemberger and A. Bodi, et al., Radical thermometers, thermochemistry, and photoelectron spectra: a PEPICO study of the methyl peroxy radical, J. Phys. Chem. Lett., 2018, 9, 534–539 CrossRef CAS PubMed.
- X. Tang, X. Gu, X. Lin, W. Zhang and G. A. Garcia, et al., Vacuum ultraviolet photodynamics of the methyl peroxy radical studied by double imaging photoelectron photoion coincidences, J. Chem. Phys., 2020, 152, 104301 CrossRef CAS PubMed.
- F. Qi, Combustion chemistry probed by synchrotron VUV photoionization mass spectrometry, Proc. Combust. Inst., 2013, 34, 33–63 CrossRef CAS.
- A. Bodi, P. Hemberger, D. L. Osborn and B. Sztaray, Mass-resolved isomer-selective chemical analysis with imaging photoelectron photoion coincidence spectroscopy, J. Phys. Chem. Lett., 2013, 4, 2948–2952 CrossRef CAS.
- X. Tang, X. Lin, G. A. Garcia, J.-C. Loison and C. Fittschen, et al., Threshold photoelectron spectroscopy of the methoxy radical, J. Chem. Phys., 2020, 153, 031101 CrossRef CAS PubMed.
- X. Tang, X. Lin, G. A. Garcia, J.-C. Loison and Z. Gouid, et al., Identifying isomers of peroxy radicals in the gas phase: 1-C3H7O2 vs. 2-C3H7O2, Chem. Commun., 2020, 56, 15525–15528 RSC.
- A. M. Launder, J. M. Turney, J. Agarwal and H. F. Schaefer, Ethylperoxy radical: approaching spectroscopic accuracy via coupled-cluster theory, Phys. Chem. Chem. Phys., 2017, 19, 15715–15723 RSC.
- J. C. Rienstra-Kiracofe, W. D. Allen and H. F. Schaefer, The C2H5 + O2 reaction mechanism: high-level ab initio characterizations, J. Phys. Chem. A, 2000, 104, 9823–9840 CrossRef CAS.
- G. M. Just, P. Rupper, T. A. Miller and W. L. Meerts, High-resolution cavity ringdown spectroscopy of the jet-cooled ethyl peroxy radical C2H5O2, J. Chem. Phys., 2009, 131, 184303 CrossRef PubMed.
- P. Rupper, E. N. Sharp, G. Tarczay and T. A. Miller, Investigation of ethyl peroxy radical conformers via cavity ringdown spectroscopy of the Ã-
![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) electronic transition, J. Phys. Chem. A, 2007, 111, 832–840 CrossRef CAS PubMed.
electronic transition, J. Phys. Chem. A, 2007, 111, 832–840 CrossRef CAS PubMed. - Z. Zhou, X. Du, J. Yang, Y. Wang and C. Li, et al., The vacuum ultraviolet beamline/endstations at NSRL dedicated to combustion research, J. Synchrotron Radiat., 2016, 23, 1035–1045 CrossRef CAS PubMed.
- Z. Wen, X. Tang, C. Wang, C. Fittschen and T. Wang, et al., A vacuum ultraviolet photoionization time-of-flight mass spectrometer with high sensitivity for study of gas-phase radical reaction in a flow tube, Int. J. Chem. Kinet., 2019, 51, 178–188 CrossRef CAS.
- Z. Wen, X. Tang, C. Fittschen, C. Zhang and T. Wang, et al., Online analysis of gas-phase radical reactions using vacuum ultraviolet lamp photoionization and time-of-flight mass spectrometry, Rev. Sci. Instrum., 2020, 91, 043201 CrossRef CAS PubMed.
- NIST Atomic Spectra Database, https://www.nist.gov/pml/atomic-spectra-database, Accessed June 6, 2021.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria and M. A. Robb, et al., Gaussian 09, Revision A02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- H.-J. Werner and P. J. Knowles, MOLPRO version 2015, http://www.molpro.net Search PubMed.
- J. D. DeSain, S. J. Klippenstein, J. A. Miller and C. A. Taatjes, Measurements, theory, and modeling of OH formation in ethyl+ O2 and propyl+ O2 reactions, J. Phys. Chem. A, 2003, 107, 4415–4427 CrossRef CAS.
- NIST Chemistry WebBook, http://webbook.nist.gov/chemistry/, Accessed June 6, 2021.
- B. Ruscic, J. Berkowitz, L. Curtiss and J. Pople, The ethyl radical: Photoionization and theoretical studies, J. Chem. Phys., 1989, 91, 114–121 CrossRef CAS.
- B. Gans, G. A. Garcia, S. Boye-Peronne, J.-C. Loison and S. Douin, et al., Absolute photoionization cross section of the ethyl radical in the range 8-11.5 eV: Synchrotron and vacuum ultraviolet laser measurements, J. Phys. Chem. A, 2011, 115, 5387–5396 CrossRef CAS PubMed.
- R. X. Fernandes, K. Luther, G. Marowsky, M. P. Rissanen and R. Timonen, et al., Experimental and modeling study of the temperature and pressure dependence of the reaction C2H5+ O2 (+ M)→ C2H5O2 (+ M), J. Phys. Chem. A, 2015, 119, 7263–7269 CrossRef CAS PubMed.
- E. P. Clifford, J. T. Farrell, J. D. DeSain and C. A. Taatjes, Infrared frequency-modulation probing of product formation in alkyl + O2 reactions: I. The reaction of C2H5 with O2 between 295 and 698 K, J. Phys. Chem. A, 2000, 104, 11549–11560 CrossRef CAS.
- G. E. Quelch, M. M. Gallo and H. F. Schaefer III, Aspects of the reaction mechanism of ethane combustion. Conformations of the ethylperoxy radical, J. Am. Chem. Soc., 1992, 114, 8239–8247 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp02655k |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2021 |