
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic strategies towards mechanically interlocked oligomers and polymers
Nadia
Hoyas Pérez
and
James E. M.
Lewis
 *
*
Department of Chemistry, Imperial College London, Molecular Sciences Research Hub, 80 Wood Lane, London W12 0BZ, UK. E-mail: james.lewis@imperial.ac.uk; Web: http://lewisgroup.org.uk
First published on 25th August 2020
Abstract
Mechanically interlocked molecules have fascinated chemists for decades. Initially a tantalising synthetic challenge, interlocked molecules have continued to capture the imagination for their aesthetics and, increasingly, for their potential as molecular machines and use in materials applications. Whilst preliminary statistical attempts to prepare these molecules were exceedingly inefficient, a raft of template-directed strategies have now been realised, providing a vast toolbox from which chemists can access interlocked structures in excellent yields. For many envisaged applications it is desirable to move away from small, discrete interlocked molecules and turn to oligomers and polymers instead, either due to the need for multiple mechanical bonds within the desired material, or to exploit an extended scaffold for the organisation and arrangement of individual mechanically interlocked units. In this tutorial-style review we outline the synthetic strategies that have been employed for the synthesis of mechanically interlocked oligomers and polymers, including oligo-/polymerisation of (pseudo)interlocked precursors, metal–organic self-assembly, the use of orthogonal template motifs, iterative approaches and grafting onto polymer backbones.
1 Introduction
Mechanically interlocked molecules (MIMs), such as rotaxanes (Fig. 1a) and catenanes (Fig. 1b), have fascinated chemists ever since they were first purportedly proposed by Willstätter over one hundred years ago.1 Interest in MIMs has continued to grow to the extent that the 2016 Nobel prize in chemistry, awarded for seminal contributions to the design and synthesis of molecular machines, was in part awarded to two pioneers in the field of interlocked molecules.2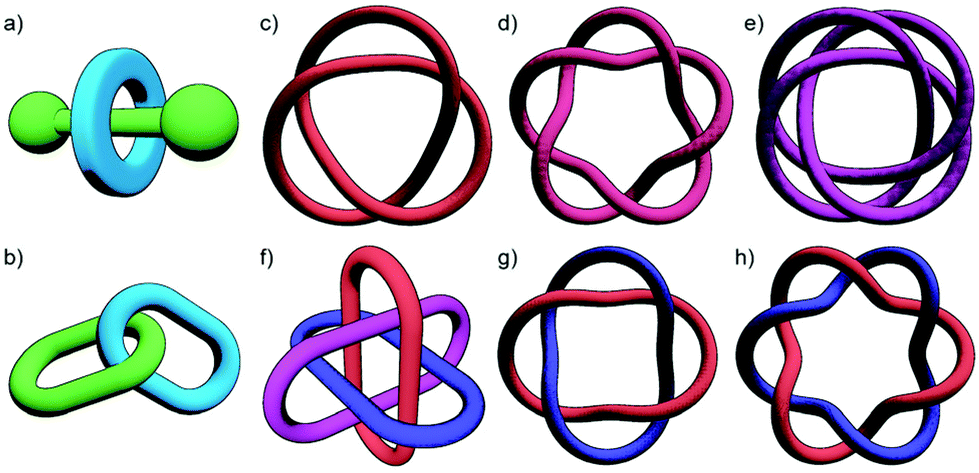 | ||
| Fig. 1 Examples of mechanically interlocked molecules: (a) [2]rotaxane; (b) [2]catenane; (c) trefoil knot; (d) pentafoil knot; (e) 819 knot; (f) Borromean rings; (g) Solomon link (double-braided [2]catenane); (h) Star of David [2]catenane (621 link). | ||
Initial efforts to prepare these species were either extremely low yielding – using Wasserman's statistical synthesis approach,3 Harrison and Harrison published the preparation of a [2]rotaxane in 6% yield after 70 iterations of the reaction!4 – or a synthetic tour de force, as exemplified through Schill and Lüttringhaus’ ingenious multi-step covalent approach to the synthesis of a [2]catenane.5
The introduction of template methodologies – Sauvage's passive metal template (PMT),6 Stoddart's use of π–π interactions,7 and Vögtle and Hunter's hydrogen-bond approach8 – finally allowed access to interlocked species in good yield and using relatively simple starting materials. The field was further revolutionised by Leigh's introduction9 of the active metal template (AMT) approach,10 in which metal ions act to both pre-organise precursors and catalyse covalent bond formation to mechanically trap components.
With a range of synthetic strategies available, efforts to synthesise more and more elaborate interlocked molecules remain ongoing. Some of the MIMs realised to date (Fig. 1c–h) include the trefoil knot,11 pentafoil knot,12 an 819 knot,13 Borromean rings,14 Solomon link,15 and a Star of David catenane (621 link).16 Alongside these impressive synthetic efforts, applications of these molecules have begun to be investigated. Some notable examples include the use of MIMs as information ratchets,17 drug delivery vectors,18 a sequence-specific peptide synthesiser,19 mechanically planar chiral ligands for asymmetric catalysis,20 and as components of monolayers to control the wettability of a surface.21
One of the most enticing properties of MIMs is the ability of their sub-components to move relative to each other (Fig. 2). Both translational (shuttling) and rotational (pirouetting) motion have been exploited in the functioning of molecular switches and machines based on mechanically interlocked components.22 It is this very same characteristic that makes small molecule MIMs so exciting a prospect that has encouraged chemists to explore incorporating them into polymeric materials. The archetypal examples of this is the slide-ring gel, first reported by Okumura and Ito. Prepared through cross-linking of macrocycles between polyrotaxane strands, the resultant gel was capable of absorbing 400 times its dry weight in water and possessed good tensile strength. This was due to the ability of the macrocycles to move along the polymeric axles and delocalise stress, allowing tension to be distributed not only across a single polymer chain, but also those held adjacent by the mechanical bond.23
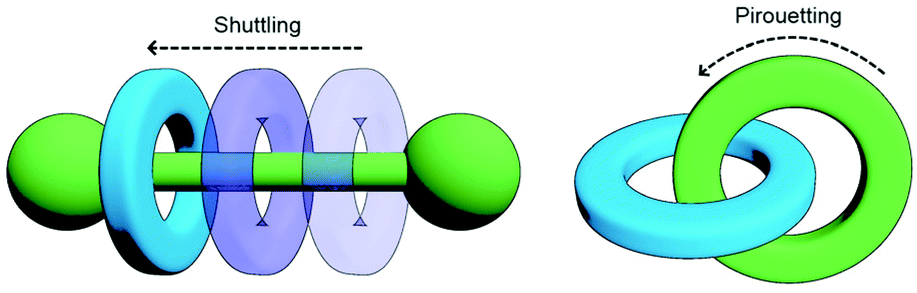 | ||
| Fig. 2 Changes in co-conformation of MIMs as a result of shuttling or pirouetting of sub-components relative to each other. | ||
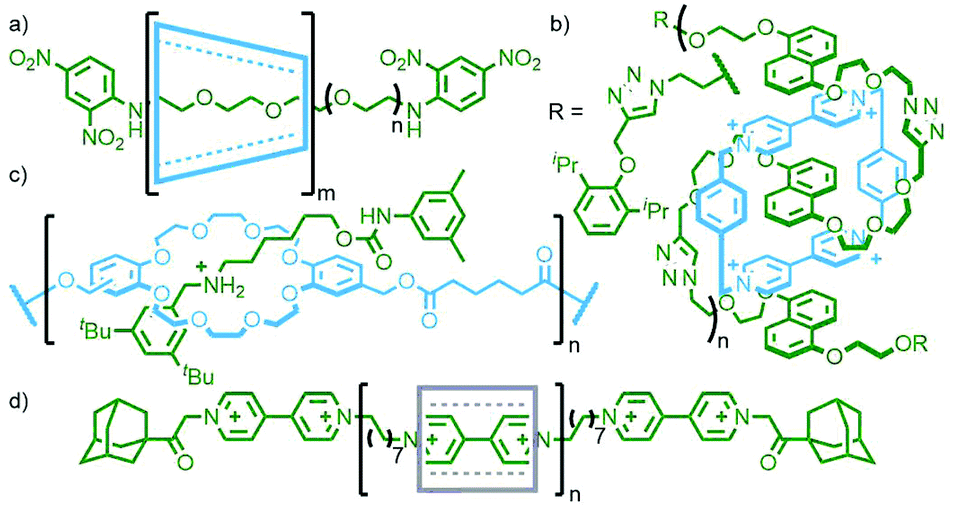
Polymeric MIMs can be classified dependent on both the interlocked unit in the structure and the manner in which the mechanical bond is incorporated into the polymer (Fig. 3). These include different types of main chain polyrotaxanes (Fig. 3a–d), side chain polyrotaxanes (Fig. 3e and f), daisy chain [an]- and [cn]polyrotaxanes (Fig. 3g and h, respectively), linear, radial and network polycatenanes (Fig. 3i, j and k, respectively), main chain polycatenanes (Fig. 3l) and side chain polycatenanes (Fig. 3m and n).
 | ||
| Fig. 3 Cartoon diagrams of various polymeric rotaxanes and catenanes. Main chain polyrotaxanes which may consist of (a) a polymeric axle with multiple threaded macrocycles, (b) a [2]rotaxane polymerised through the macrocycle, (c) a [3]rotaxane polymerised through both macrocycles, or (d) a [2]rotaxane polymerised through both the macrocycle and axle. Side chain polyrotaxanes in which either (e) the axle or (f) macrocycle components are pendant from the polymer chain. (g) [an]- and (h) poly[c2]-daisy chain rotaxanes. Poly[n]catenanes can be (i) linear, (j) radial, or (k) branched/network. (l) Main chain polycatenanes consist of polymer strands joined through a topological link. Side chain polycatenanes can have one of the macrocycle components covalently (m) appended off the polymer chain, or (n) incorporated into it. | ||
Excellent reviews on polyrotaxanes and polycatenanes have been previously published with an emphasis often placed on specific types of polyMIMs,24 select components,25 or an analysis of function.26 Herein, we instead seek to provide a broad and general overview of the synthetic approaches that have been applied to preparing oligomeric and polymeric mechanically interlocked systems (polypseudo-MIMs will not included). This will not be comprehensive; instead, relevant examples from the literature will be used to highlight different synthetic strategies. For clarity and utility we have separated methodologies for preparing oligomeric and polymeric MIMs, with oligomers broadly defined for convenience as molecular systems for which the precise number of interlocked components is known (and is greater than two). MIMs of this nature are often termed higher order to distinguish them from simpler, more common systems, such as [2]catenanes and -rotaxanes. Systems for which there is a polydispersity of products will be considered polymeric.
2 Synthesis of mechanically interlocked oligomers
In some instances it may be preferable to have a mechanically interlocked oligomer either with a particular number of components, or for which the precise number of components is known. In this section we outline three main approaches to such oligomeric MIMs. The first is a one-pot approach with pre-organised precursors designed to react to give a specific product. We term this the orthogonal template strategy as it uses multiple templating interactions simultaneously – in order to be effective these must be specific and not interfere with each other in a deleterious fashion, i.e. they must be orthogonal. This approach is operationally quite simplistic, with a number of different components being able to be brought together in a single reaction. However, this also limits the range of products that can be obtained, as these need to be considered when designing the precursor materials. An alternative, and (at least potentially) more flexible, approach is the iterative reaction strategy, in which an additional unit is added with each reaction cycle. Although generally less expedient, this method does allow for different components to be incorporated at each iteration which might not otherwise be mutually compatible and hence amenable to orthogonal templation.The third approach described is oligomerisation of (pseudo)interlocked precursors. This can be further subdivided: if irreversible covalent chemistry is used the oligomeric product will be formed under kinetic control; if labile metal–organic interactions are used then conditions can be tuned for thermodynamic control. The former is likely to lead to a mixture of products that will require separation. However, whilst self-assembly is liable to give more homogenous metallo-oligomers in high yields, the products will tend to be less robust than their organic counterparts.
2.1 Orthogonal templates
To construct relatively complex interlocked molecules in a minimum number of steps, multiple copies of templates used for lower order MIMs can be incorporated into the building blocks. As an exemplar of this concept, Cronin, Stoddart and co-workers observed that an acyclic diamine could undergo imine condensation with 2,6-pyridinedicarboxaldehyde about a bis-ammonium axle to quantitatively form a [3]rotaxane (Fig. 4). By using oligo-ammonium axle components, the same clipping methodology was able to furnish main chain [12]-, [16]- and [20]rotaxanes in excellent yields (typically >90%). Thus, by simply extending the number of template moieties in the axle (combined with an exceedingly efficient cyclisation reaction) higher order rotaxanes could be obtained.27 | ||
| Fig. 4 Cronin and Stoddart's imine clipping approach to main chain oligo[n]rotaxanes. By incorporating multiple ammonium template motifs into an oligomeric axle, high yielding dynamic covalent chemistry was demonstrated to afford oligorotaxanes with up to 20 interlocked components. | ||
To incorporate multiple different components, however, requires the simultaneous use of multiple templates. This is a sophisticated approach that necessitates orthogonality between template motifs – the intercomponent interactions need to be specific so as not to cause interference.
An early example of this approach was the synthesis of a sequence-specific [3]rotaxane by Schalley and co-workers, employing what was referred to as ‘cascade stoppering’. An axle component with two ammonium binding sites was prepared with a phenylene spacer unit between them. Combining with two crown ether macrocycles of different sizes (dibenzo-24-crown-8 – DB24C8 – and benzo-21-crown-7 – B21C7) resulted in integrative self-sorting of the components to give a pseudo[3]rotaxane. The inability of the smaller B21C7 macrocycle to pass over the phenylene spacer ensured that, following stoppering (with a group small enough for any errant DB24C8 to pass over), the heterocircuit [3]rotaxane product consisted exclusively of one of the possible sequence isomers (Fig. 5a).28
 | ||
| Fig. 5 (a) Integrative self-sorting of a pseudo[3]rotaxane via cascade stoppering ensured exclusive formation of a single sequence-specific isomer of a heterocircuit [3]rotaxane. This approach has subsequently been extended to the one-pot synthesis of (b) a [4]rotaxane prepared from six components, and (c) a doubly-threaded [7]rotaxane from eleven components. | ||
This methodology has recently been expanded by Qu and co-workers to prepare a [4]rotaxane, in one pot from six components, incorporating a [c2]daisy chain motif in over 50% isolated yield (Fig. 5b),29 and Liu and co-workers to synthesise an astonishing doubly-threaded [7]rotaxane (Fig. 5c) from eleven components in 42% yield!30 Whilst the remainder of this discussion will continue to focus on templates that operate under thermodynamic control, it should be noted that it is possible to achieve self-sorting through kinetic control. This has been demonstrated by Neal and Goldup with the AMT synthesis of heterocircuit [3]rotaxanes. Through careful choice of macrocycle and stoppering groups, specific sequence isomers of heterocircuit [3]rotaxanes were formed to the exclusion of alternatives by exploiting the relative kinetic stabilities of the potential products.31
Although a potentially powerful approach, cascade stoppering relies upon carefully tuned steric interactions, which may not always be simple to include in a molecular design. An alternative would be to use two (or more) distinct intercomponent interactions, wherein selective formation of specific host–guest assemblies enables one-pot syntheses of multi-component MIMs with high structural fidelity.
Qu and co-workers were able to achieve this through an extension of Schalley's cascade stoppering with DB24C8 and B21C7 through inclusion of a third crown ether macrocycle, bis(paraphenylene)-34-crown-10 (BPP34C10), known to bind electron poor paraquat-based guests, chiefly through π–π interactions,32 with much greater affinity than DB24C8. In this manner a sequence-specific heterocircuit [6]rotaxane was obtained in almost 50% yield from the CuAAC reaction of a DB24C8/BPP34C10 pseudo[4]rotaxane with a B21C7 pseudo[2]rotaxane, based on the selective binding preferences of the three macrocycles for the three axle recognition motifs (Fig. 6).33
 | ||
| Fig. 6 Qu's eight-component synthesis of a heterocircuit [6]rotaxane with three different crown ether macrocycles (DB24C8, blue; BPP34C10, red; B21C7, orange). | ||
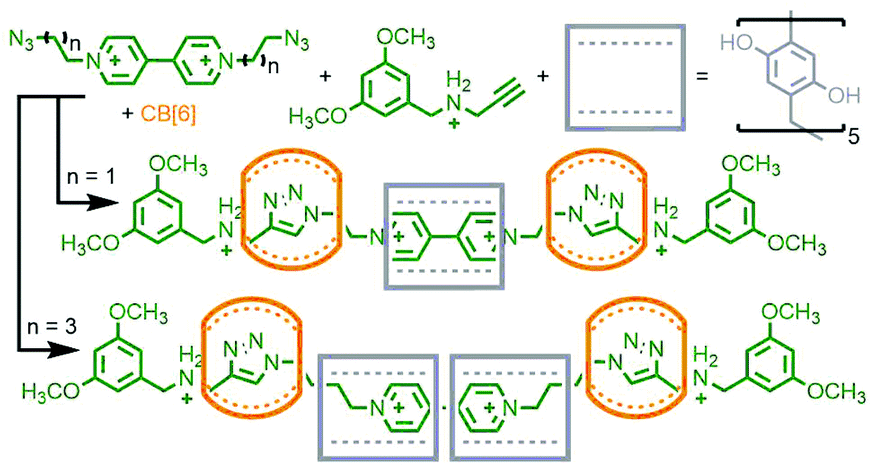
Stoddart and co-workers also investigated the potential of orthogonal interactions by using two macrocycles with different guest-binding properties: cucurbit[n]urils (CB[n]s) and cyclodextrins (CDs). CDs are a versatile class of macrocycles that are water soluble yet possess a hydrophobic cavity. This facet can be exploited to drive host–guest formation via the hydrophobic effect.34 In contrast CB[n]s bind strongly to cationic guests, such as those with ammonium units.35 An interesting property of CB[6], first noticed in the early 1980s by Mock and co-workers, is that upon formation of a heteroternary assembly with azide and alkyne guests, regiospecific formation of 1,4-disubstituted-1,2,3-triazoles by confinement-accelerated 1,3-dipolar cycloaddition of the reagents is effected.36 Through the use of bulky N-tert-butyl ammonium reagents, too large to pass through the cavity of CB[6], the same group were able to form a kinetically stable [2]rotaxane product using this CB-azide–alkyne cycloaddition (CB-AAC) reaction.37
Based on this property, a bitolyl building block was designed such that the core aromatic unit was bound by β- or γ-CD in aqueous media, whilst propargylammonium groups at the termini were encapsulated by CB[6]. Addition of a suitable azide blocking group allowed the CB-AAC reaction to proceed, resulting in the desired heterocircuit [4]rotaxanes in essentially quantitative yield (Fig. 7a). Intriguingly, the presence of the CD macrocycle appeared to accelerate the CB-AAC reaction over 100-fold, apparently arising from hydrogen bonding interactions between the two macrocycles.38
 | ||
| Fig. 7 Combining non-covalent templates with the CB[6]-catalysed azide–alkyne cycloaddition: (a) Stoddart's synthesis of a heterocircuit [4]rotaxane with β- and γ-CDs, and (b) Au-Yeung's [6]catenane synthesised using a Cu(I)-phenanthroline PMT. | ||
The CB-AAC reaction is also amenable to cyclisation reactions between appropriate di-functionalised building blocks. Under pseudo-high dilution conditions, Au-Yeung and co-workers were able to cyclise two bis(propargylammonium) units, held orthogonal to each other by phenthroline ligands coordinated to a Cu(I) ion, with a bis-azide. The resulting molecule, formed in an astounding 91% yield (as determined by LCMS), can be described as a heterocircuit [6]catenane, composed of two interlocked [3]catenanes (Fig. 7b). Omitting the Cu(I) template allowed isolation of the individual [3]catenane component. Alternatively, addition of a pre-formed phenanthroline macrocycle with Cu(I) to the reaction formed a pseudo[2]rotaxane complex, resulting in a radial [4]catenane constituted of three different macrocycles.39
Although extensive research has given us an acute knowledge of various template motifs, seemingly innocent structural modifications are still capable of profoundly affecting reaction outcomes in unpredictable ways, as demonstrated by the following example from Stoddart and co-workers. Building on the above work on CD-accelerated CB-AAC reactions, an investigation was undertaken to explore the potential for using Ogoshi's pillar[n]arenes40 in a similar fashion. An N,N′-bis(azidoalkyl)-4,4′-bipyridinium was reacted in the presence of pillar[5]arene, with which it is capable of forming a pseudorotaxane assembly, with a propargylammonium stopper and CB[6]. With an ethyl or propyl linker between the pyridinium and azide units heterocircuit [4]rotaxanes with two CB[6] macrocycles and one pillar[5]arene were obtained in ≥95% yield; when repeated with the butyl-linked bis-azide, the seemingly insignificant addition of a single methylene unit was found to effect exclusive formation of a [5]rotaxane with two pillar[5]arene macrocycles encircling the bipyridinium core of the axle (Fig. 8).41
 | ||
| Fig. 8 Stoddart's orthogonal CB-AAC-mediated formation of heterocircuit [n]rotaxanes. Changing from an ethyl/propyl to a butyl linker between the azide and pyridinium units served to effect quantitative switching of the reaction product from a [4]rotaxane to a [5]rotaxane. | ||
2.2 Iterative approaches
One of the key advantages of the previously described orthogonal template strategies is that a considerable amount of molecular complexity can be built up in a single step, allowing rapid access to intricate multi-component MIMs. However, the exact structure of the target molecule would need to be known in order to design the building blocks with precision, allowing little in the way of adaptability. In contrast an iterative approach, in which a set number of components would be added with each reaction, could potentially allow for the introduction of different components and enable structure diversification at a late stage in the synthetic process.An early example of this was reported by Tsivgoulis and Hadziioannou. A [2]rotaxane was prepared with functional groups on the axle and macrocycle – an alcohol and a carboxylic acid, respectively – rendered unreactive by orthogonal protecting groups. Selective removal of either the alcohol's tBuPh2Si protecting group with Bu4NF or the allyl group of the carboxylic acid with Pd(PPh3)4 yielded [2]rotaxanes with complementary reactive groups. An esterification reaction between the two selectively deprotected products gave the corresponding [3]rotaxane (Fig. 9); this MIM could again be selectively deprotected, and reaction of the two resultant products successfully yielded the desired [5]rotaxane.42 This orthogonal protecting group strategy is elegant in its simplicity, although versatility may be hindered by the need for deprotection and coupling conditions that do not interfere with each other or the structure of the mechanically interlocked components.
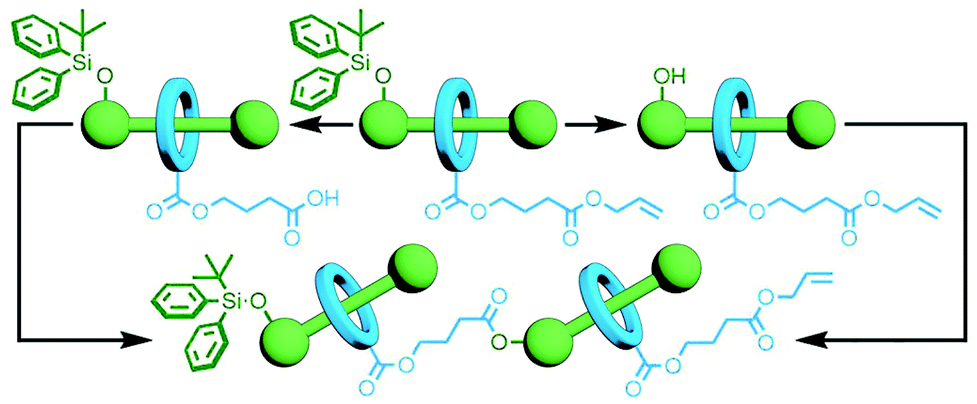 | ||
| Fig. 9 Orthogonal protecting group strategy towards the iterative synthesis of oligo[n]rotaxanes: the tBuPh2Si alcohol protecting group can be selectively removed with Bu4NF, and the allyl ester can be cleaved with Pd(PPh3)4. Selective ester formation between the macrocycle of one rotaxane and the axle of another can be ensured in this manner. | ||
An alternative method, utilised by Stoddart and co-workers, is to exploit the difference in reactivity profiles between reagents. The clipping of tetracationic cyclobis(paraquat-4,4′-biphenylene) around two tris-p-phenylene-51-crown-15 (TPP51C15) macrocycles in MeCN at ambient temperature and pressure gave the corresponding [3]catenane, albeit in a modest 3.5% yield. Under more forcing conditions (DMF, 11–14 kbar) it was possible to subsequently effect interlocking of the smaller cyclobis(paraquat-p-phenylene) (CBPQT4+), giving both the heterocircuit [4]catenane, in 22% yield, and trace amounts (<0.5%) of the [5]catenane (Fig. 10a).43
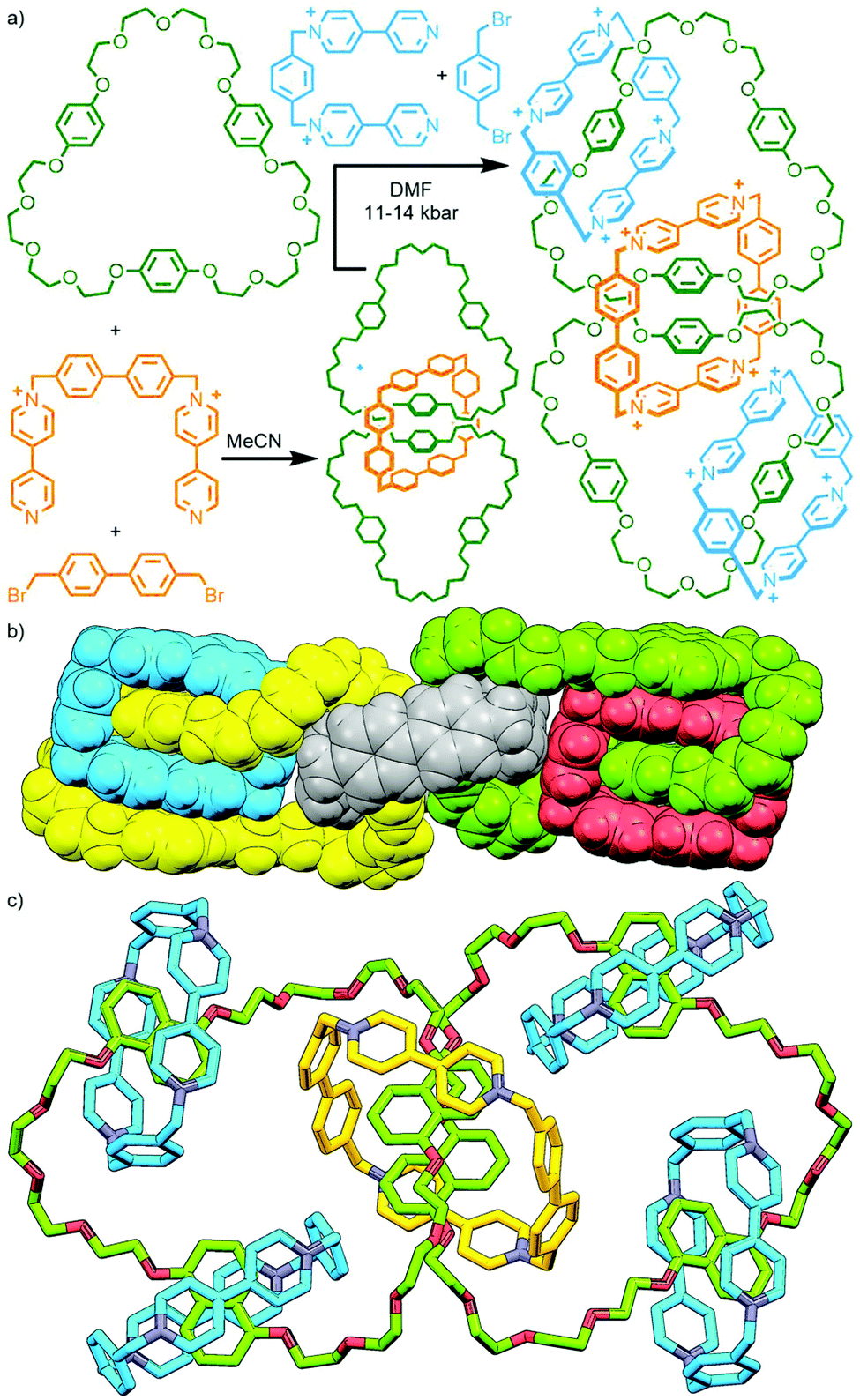 | ||
| Fig. 10 Stoddart's iterative synthesis of oligo[n]catenanes: (a) two-step synthesis of a [5]catenane via a [3]catenane intermediate, and SCXRD structures of (b) the [5]catenane olympiadane, and (c) a fully saturated [7]catenane. | ||
As 1,5-dihydroxynaphthylene derivatives were known to bind within the cavity of CBPQT4+ with higher affinity than hydroquinone derivatives, TPP51C15 was replaced with tris-naphtho-57-crown-15 (TNP57C15) to give an analogous [3]catenane intermediate. Subsequent clipping of CBPQT4+ was able to be effected under milder conditions (ambient temperature and pressure), giving the [4]- and [5]catenane products in appreciable yields (31% and 5%, respectively). The latter compound is famously known by the name ‘olympiadane’, after its resemblance to the five interlocked rings constituting the symbol of the Olympic Games (its SCXRD structure is shown, with appropriate colouring of the macrocycles, in Fig. 10b).
Repeating the second step of this synthetic route under high pressure conditions (12 kbar in DMF) enabled clipping of additional CBPQT4+ macrocycles around the remaining naphthalene binding sites. This led to a mixture of products that could be separated and isolated by column chromatography, giving an increased yield of olympiadane (30%) as well as a [6]catenane (28% yield) and the fully saturated (i.e. all recognition sites occupied) [7]catenane (26%; Fig. 10c).44 Thus through careful design of components and judicious choice of reaction conditions, [n]catenanes with n = 3–7 were able to be obtained in good yield, with control over the incorporation of different macrocyclic components.
One drawback of this approach was that the inter-component interactions used to drive formation of the mechanical bonds persisted in the resultant interlocked structure. This effectively removed them from use as template sites and once fully saturated, as in the [7]catenane above, subsequent reaction becomes impossible.
Leigh and co-workers devised an ingenious method to avoid this by incorporating a single, reusable binding site into a rotaxane axle (Fig. 11a). The rotaxane design was based on a previously established Pd-PMT approach.45 An asymmetric axle was prepared incorporating a pyridine station at one end, and a collecting chain at the other. Binding of the palladium complex of an acyclic pyridine dicarboxamide ligand (Fig. 11b), followed by cyclisation and demetallation, gave the [2]rotaxane product (62% yield). Importantly, the demetallation step removed the template interaction between macrocycle and axle, liberating the pyridine binding site. Subsequent iterations of this reaction sequence added, due to the lone binding site, a single macrocycle each time, allowing successive access to the [3]- and [4]rotaxanes.46 Due to the proximity of the pyridine station to the adjacent stoppering group, binding of the incipient macrocycle ensured that extant macrocycles were located on the collecting chain only. This was subsequently exploited for the sequence specific addition of different macrocycles, resulting in the ability to uniquely form and isolate specific diastereoisomers of a heterocircuit [3]rotaxane (Fig. 11c).47
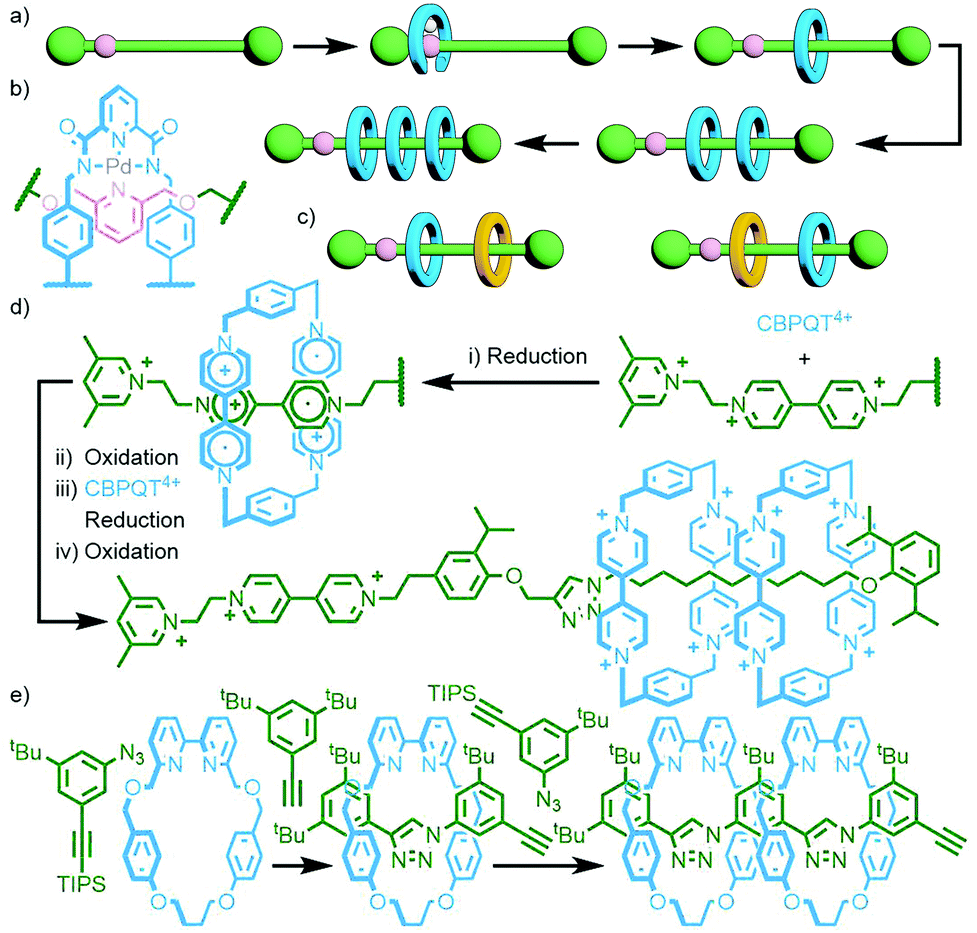 | ||
| Fig. 11 (a) Leigh's iterative approach to sequentially add macrocycles to a pre-formed axle, using a (b) palladium-based PMT motif. (c) Through careful design of the axle it was possible to add different macrocycles in a specific sequence allowing access to heterocircuit [3]rotaxane isomers. (d) Stoddart's redox-driven molecular pump. Reduction of CBPQT4+ and the bipyridinium station of the axle results in threading due to attractive interactions between the radical cations. Subsequent oxidation forces the CBPQT4+ onto the collecting chain. Repetition of the redox cycle traps a second macrocycle. (e) Goldup's AMT iterative strategy in which each reaction both adds a macrocycle and extends the axle, theoretically allowing indefinite growth of the oligo[n]rotaxane. | ||
A thematically similar approach, utilising an energy ratchet mechanism, was reported recently by Stoddart and co-workers. The pump operates through initial reduction of the CBPQT4+ macrocycle and bipyridinium station of the axle to CBPQT2(+˙) and the bipyridine radical cation, respectively. The stable radical trication host–guest complex48 formed between these two allows the macrocycle to pass over a pyridinium valve. Subsequent oxidation results in coulombic repulsion between the tetracationic macrocycle and both the pyridinium valve and bipyridinium station, forcing it over an isopropylphenyl speed bump onto the collecting chain where it is kinetically trapped, giving a [2]rotaxane. Another round of redox pumping traps a second macrocycle (Fig. 11d).49 Exploiting the precision of this mechanism, Astumian, Li, Stoddart and co-workers positioned two of the molecular pumps at either end of a PEG collecting chain. Thus, with each round of redox pumping, two CBPQT4+ macrocycles were added to the polymer chain. Due to the high efficiency of the molecular pumps, iterative redox cycles could be performed in a one-pot manner. In this way homocircuit [5]-, [7]-, [9]- and [11]rotaxanes were obtained without the need for intermediate purification steps.50
An inherent issue with these designs is the finite size of the axles, which limits the potential number of macrocycles that can be incorporated into the main chain oligo[n]rotaxane structures. A solution to this was presented by Goldup and co-workers, using an efficient AMT51 variant of this iterative approach, in which the addition of a macrocycle component occurred with concomitant lengthening of the axle, theoretically presenting the opportunity for indefinite, controlled growth. The key building block was a stoppering unit with both azide and TIPS-protected alkyne moieties. The CuAAC-AMT reaction of this unit with a terminal stopper, followed by deprotection of the TIPS-alkyne, gave a [2]rotaxane with an alkyne unit on one end, capable of undergoing another CuAAC-AMT reaction. Thus, re-submission of this product to the reaction conditions gave the [3]rotaxane product (Fig. 11e). The need to remove the TIPS protecting group between AMT iterations ensured that only a single macrocycle was added each time. It was shown to be possible to generate up to a [6]rotaxane, with no loss of efficiency for each iteration. This method was also suitable for the formation of heterocircuit systems. By using a different macrocycle in three back-to-back iterations, a [4]rotaxane was prepared with the three different macrocycles incorporated in a specific sequence.52
2.3 Oligomerisation
Shortly after their initial report on the use of a Cu(I) metal ion template to synthesise a [2]catenane, Sauvage and co-workers applied this PMT to the synthesis of a [3]catenane (in 58% yield) through cyclodimerisation of a pseudo[2]rotaxane precursor. It was noted that an additional product, formed in 22% yield, was likely to be the corresponding radial [4]catenane resulting from cyclotrimerisation of the precursor.53 Repeating this reaction with an alternative pseudo[2]rotaxane (featuring a smaller, 27-membered, macrocycle) allowed isolation of the [3]- (23%), [4]- (23%), and [5]catenane (10%) products (Fig. 12), as well as a mixture of higher order structures (identified by MS to include the [6]- and [7]catenanes).54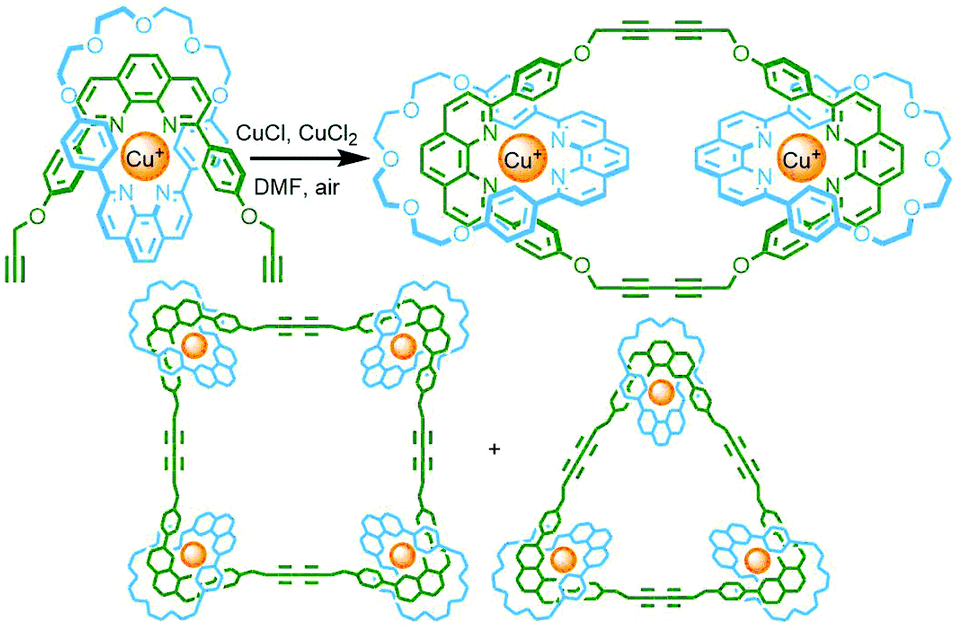 | ||
| Fig. 12 Sauvage's cyclooligomerisation via oxidative homocoupling of a pseudo[2]rotaxane. The [3]-, [4]-, and [5]catenane products could be separated and isolated, with a mixture of higher order catenanes also obtained. | ||
To limit this polydispersity of products, Anderson and co-workers employed a template-directed strategy for the cyclo-oligomerisation of an alkyne-terminated bis-porphyrin [2]rotaxane. A hexapyridyl template was used to pre-organise the rotaxane through coordination to the axial sites of the Zn(II)-porphyrin,55 with subsequent oxidative homocoupling giving the cyclotrimerised radial [4]catenane in 62% yield (with displacement of the template able to be effected through addition of excess DABCO as competing ligand). Interestingly, the “figure-of-eight” [7]catenane, incorporating two copies of the template molecule, was also isolated from the reaction mixture, albeit in only 6% yield (Fig. 13).56
 | ||
| Fig. 13 Anderson's template-directed synthesis of a radial[4]catenane. Addition of DABCO as a competing ligand resulted in displacement of the hexapyridyl template to give the “free” catenane. As a by-product of the reaction the double-template [7]catenane was also isolated. | ||
Alternatively, dynamic covalent chemistry may be used to enable ‘error correction’ and arrive at the thermodynamically favoured structure. In this respect, this approach has more in common with self-assembled metallo-MIMs (vide infra) than the covalent systems described above. The potential power of this method was demonstrated by Wang and co-workers who were able to isolate a [5]catenane in 58% yield, through simple precipitation, from the imine condensation reaction between a 90° diaminocarbazole unit and a diformyl bis-pyridinium axle threaded through CB[7] (Fig. 14).57
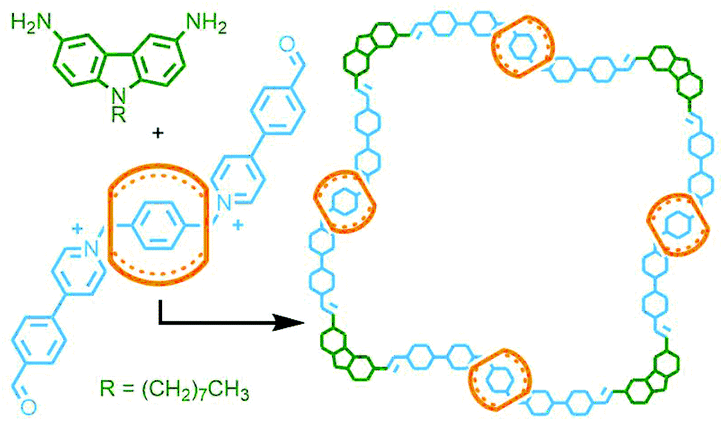 | ||
| Fig. 14 Wang's dynamic covalent approach to the synthesis of a radial [5]catenane via imine condensation of an aldehyde-functionalised CB[7] pseudorotaxane with a carbazole-based diamine. | ||
2.4 Self-assembly
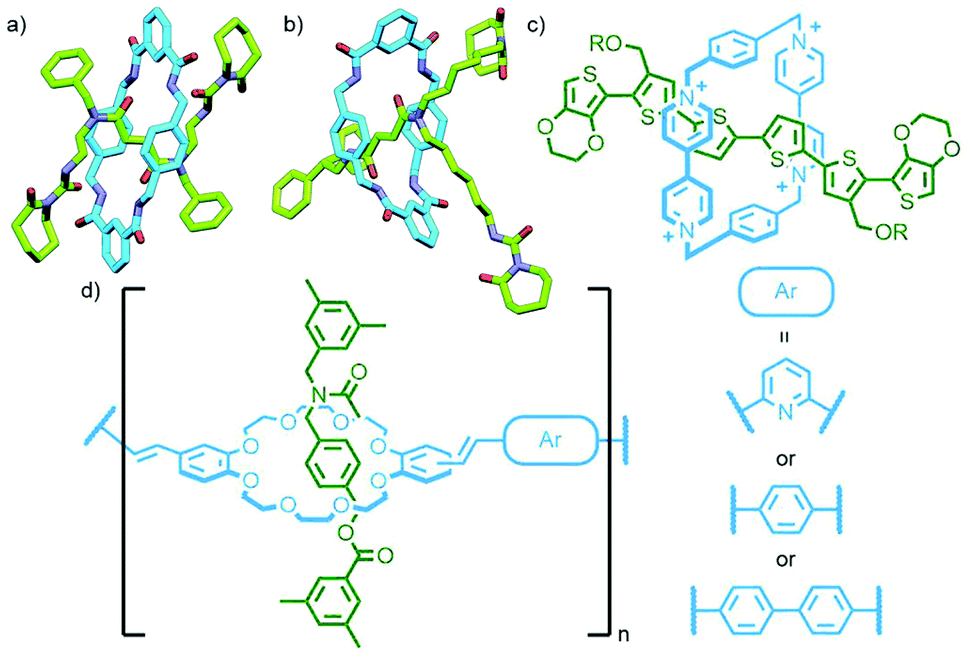
Metallo-supramolecular self-assembly has become a topic of intense research interest over the last few decades due to the ability to generate astounding and aesthetically beautiful structures in high yields by combining metal ions with relatively simple ligands.58 It is therefore perhaps unsurprising that (pseudo)MIMs have begun to be explored as ligands59 and assembled into discrete (and polymeric – vide infra) metal–organic structures. Metal–organic self-assembly is a particularly attractive option as the metal nodes can simultaneously link MIMs together and act as stoppers to prevent dethreading; furthermore, by operating under thermodynamic control greater product heterogeneity and yields are often possible.Self-assembly of a pseudorotaxane was first demonstrated by Kim and co-workers who observed the quantitative formation of a radial [4]catenane, termed a molecular necklace, from the combination of a dipyridyl bis-ammonium axle and CB macrocycle with cis-protected Pt(II) ions (Fig. 15a).60 Subsequently radial [5]catenanes were accessed through altering the substitution of the pyridyl ligand components61 and dimerisation of a pseudo[3]rotaxane ligand.62 This approach has been used by Stang and co-workers to generate radial [3]-, [4]- and [7]catenanes as platinum complexes of ligands derived from pseudorotaxanes of dipyridyl 1,2-bis(pyridinium)ethane/paraquat axles and crown ether macrocycles/cryptands.63
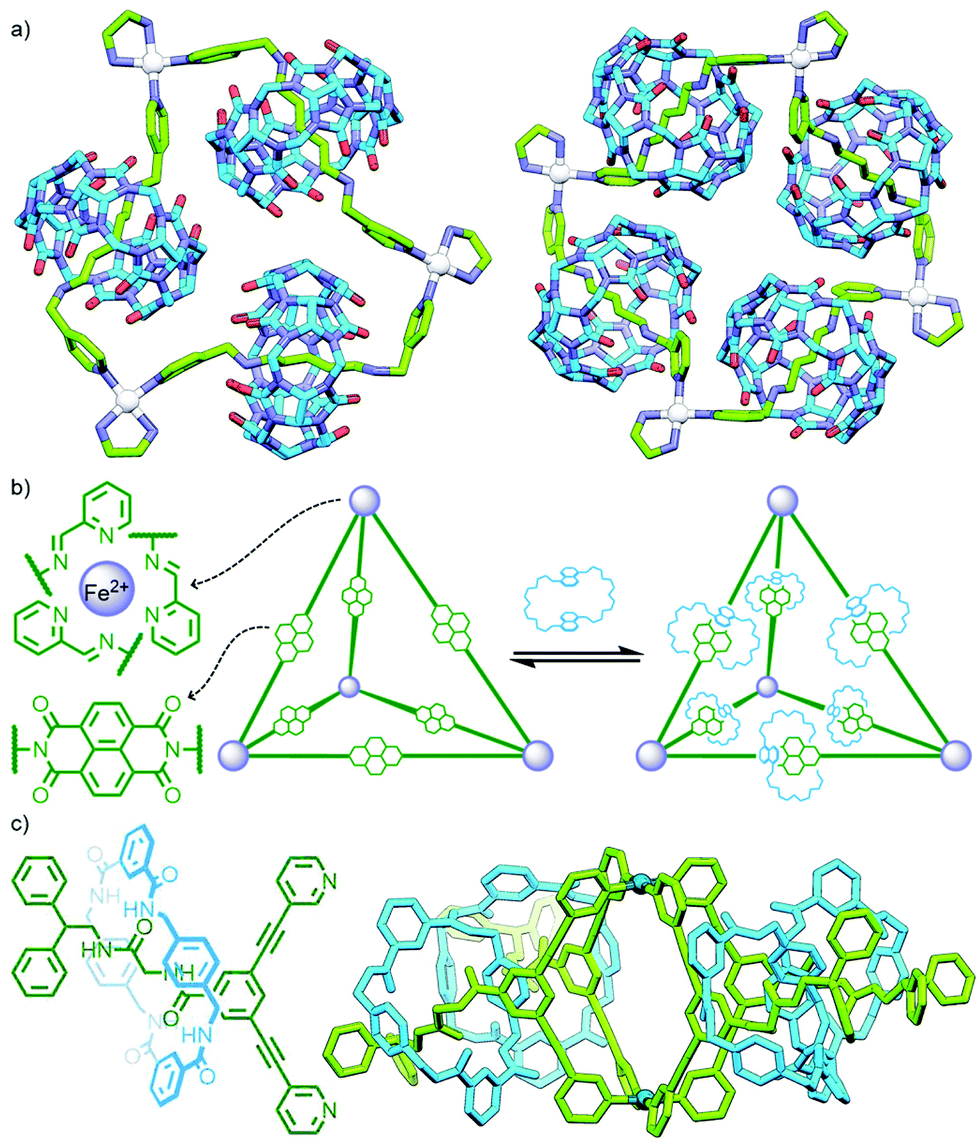 | ||
| Fig. 15 (a) SCXRD structures of radial metallo[4]- and [5]catenanes reported by Kim and co-workers. (b) Schalley, Sanders and Nitschke's formation of a 3D metallo[7]catenane from naphtho-crown ether macrocycles and an Fe4L6 tetrahedron with NDI-functionalised ligands. (c) Lewis’ [2]rotaxane ligand capable of self-assembling into a metallo-[5]rotaxane with a Pd2L4 cage core (PM6 model). | ||
An interesting variant on this approach is the formation of dynamic covalent libraries (DCLs) of 3D metallo[n]catenanes by Schalley, Sanders, Nitschke and co-workers. Addition of the macrocycle dinaphtho-38-crown-10 (DNP38C10) to a tetrahedral Fe4L6 cage – assembled from bis(pyridylimine) ligands with a naphthalenediimide (NDI) core – resulted in self-assembly of a dynamic mixture of [n]catenanes arising from the favourable π–π interactions between the NDI units and threaded macrocycles. Through addition of a large excess of macrocycle it was possible to effect quantitative formation of the [7]catenane structure (Fig. 15b).64 A similar Zn4L6 tetrahedron assembled from smaller NDI ligands was found to only form up to a [3]catenane as a result of steric encumberance between macrocycles.65
In contrast to these DCL systems, Lewis and co-workers recently synthesised a permanently interlocked [2]rotaxane incorporating a dipyridyl ligand motif into the structure as one of the stoppering units. Addition of Pd(II) ions resulted in self-assembly of the ligand into a Pd2L4 cage architecture, peripherally decorated with rotaxanes, forming a porous metallo-[5]rotaxane (Fig. 15c).66
With the previously described tetrahedral cage structures from Schalley, Sanders, Nitschke and co-workers, the axle ligand components used were relatively rigid. Thus, formation of the interlocked assembly had minimal effect on the structural framework. Loeb and co-workers have investigated the difference in coordination complexes formed between rotaxanes and their non-interlocked axle counterparts when the latter possesses a degree of flexibility. A non-interlocked dipyridyl axle was found to form a dinuclear complex (Fig. 16a) upon coordination to cis-protected [Pt(dppp)]2+ as it was able to adopt a bent conformation (Fig. 16c) (dppp = 1,3-bis(diphenylphosphino)propane). In contrast when encircled by a 24-crown-6-ether a linear conformation of the axle was enforced (Fig. 16d); as a result, self-assembly of the rotaxane ligand formed a trimeric structure – a radial metallo-[4]catenane (Fig. 16b).67
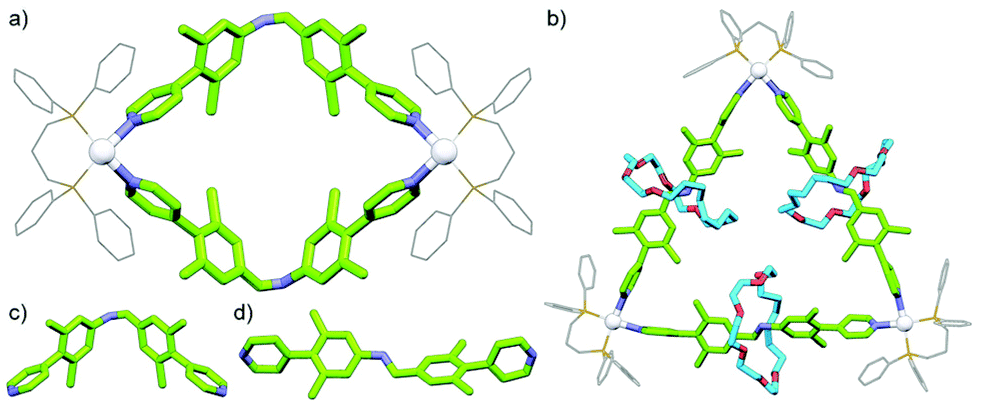 | ||
| Fig. 16 SCXRD structures of Loeb's (a) [Pt2(dppp)2(L)2]4+ metallacycle and (b) [Pt3(dppp)3(L)3]6+ radial metallo[4]catenane. The dipyridyl axle component was able to adopt (c) a bent conformation, but when encircled by the crown ether macrocycle (d) a linear conformation was enforced. | ||
Metal complexes have been used as stoppering units for rotaxanes since some of the earliest work in the field.68 They are versatile building blocks which, through careful design, can act as both inert blocking groups and reactive moieties for self-assembly. This dual functionality has been demonstrated elegantly through the work of Yang and co-workers. A [2]rotaxane was prepared with a pillar[5]arene macrocycle encircling an axle with a 1,3-bis(triisopropylsilylethynyl)phenyl stopper at one end, and a Pt(II)–acetylide complex at the other (Fig. 17). Trimerisation of this rotaxane through reaction of the Pt(II) with 1,3,5-triethynylbenzene resulted in formation of a [4]rotaxane. Removal of the TIPS protecting groups from this product and subsequent reaction with the rotaxane precursor yielded the second generation dendritic [10]rotaxane. Two further iterations gave the [46]rotaxane; the high fidelity of the coupling reaction was demonstrated through the low polydispersity of the dendritic assemblies as determined by gel permeation chromatography (GPC) analysis.69 Alternatively, through iterative reaction of an analogous, but non-interlocked, precursor, a dendritic framework could be constructed and subsequently terminated with rotaxane units, yielding [7]-, [13]- and [25]rotaxanes in yields of 60%, 49% and 41%, respectively.70 More recent work has shown that it is possible to utilise this strategy with stimuli-responsive building blocks, allowing the construction of dendrimers capable of exhibiting expansion/contraction behaviour.71
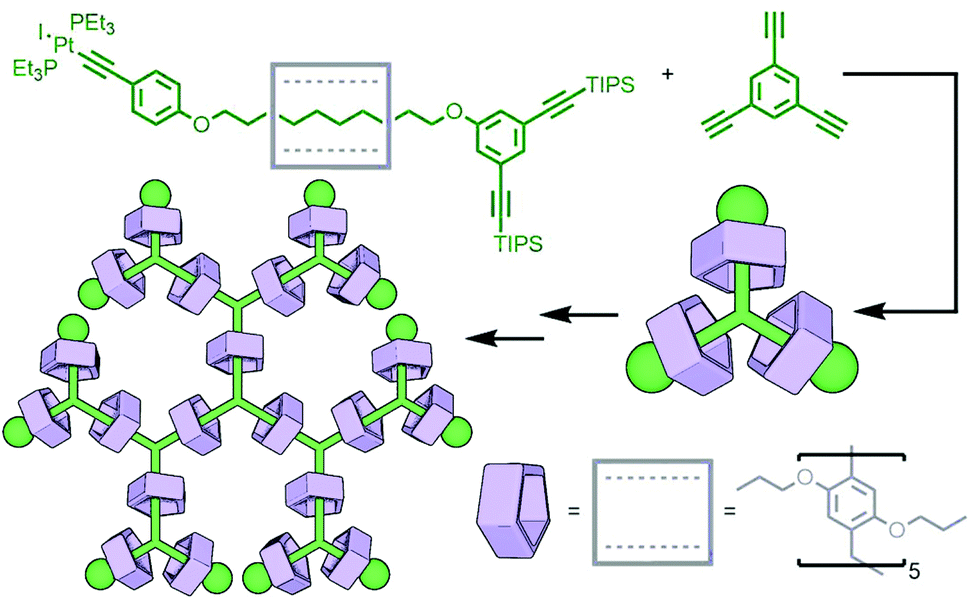 | ||
| Fig. 17 Yang's iterative approach to rotaxane metallo-dendrimers. Coupling of a Pt(II)-acetylide rotaxane to 1,3,5-triethynylbenzene gave the first generation dendrimer, a [4]rotaxane. Two subsequent iterations of deprotection and coupling gave the third generation dendritic [22]rotaxane. | ||
2.5 Conclusions
It is now thirty years since Stoddart suggested that non-covalent templates could be used to assemble multi-component interlocked molecules towards “the construction of a molecular abacus”.72 Since then, not only have a multitude of template motifs been elaborated, but also numerous compatible methods of assembling (pseudo)MIMs into larger arrayed structures have been established. As outlined above, these have their benefits and their disadvantages. Generally speaking, there is a trade-off between robustness and the yield/specificity of product formation. However, by judicious choice of methodology, the desired properties of the target species can be incorporated into the molecular design. In this manner, oligomeric MIMs can be engineered with precision control over the number and arrangement of the building blocks.3 Synthesis of mechanically interlocked polymers
Polymeric materials incorporating mechanical bonds, either in the polymer backbone itself or as pendant units, are highly attractive targets sought after for use in various applications.73 Ostensibly there are two ways to obtain these materials – either forming mechanical bonds about a pre-formed polymer, or polymerisation of an (pseudo)interlocked monomer unit. Conceptually we have divided synthetic methodologies for polymeric interlocked materials into five key stretegies: (i) polymerisation of MIMs, or their (ii) pseudo-interlocked counterparts (host–guest assemblies); (iii) functionalisation of a polymer chain with MIMs (grafting); (iv) trapping of a pseudopoly-MIM through either stoppering or clipping reactions, and finally (v) formation of coordination polymers upon self-assembly of metal ions with (pseudo)interlocked ligands.3.1 Polymerisation of MIMs
Following several decades of research there is now a plethora of effective template motifs for the synthesis of MIMs, allowing access to a wide range of structures. This has enabled the synthesis of MIMs incorporating reactive functionalities suitable for polymerisation reactions. Through using polymerisable monomers in which the mechanical bond is kinetically stable, it can be ensured that the ratio of monomer units to mechanical bonds incorporated into the resultant polymer is known precisely. In designing MIM monomers for polymerisation there are three main factors to consider to obtain the desired polymer structure: (i) the number of reactive groups to incorporate; (ii) which MIM sub-components to append these to; (iii) whether the functional groups are designed to react with themselves, each other, or an external linker.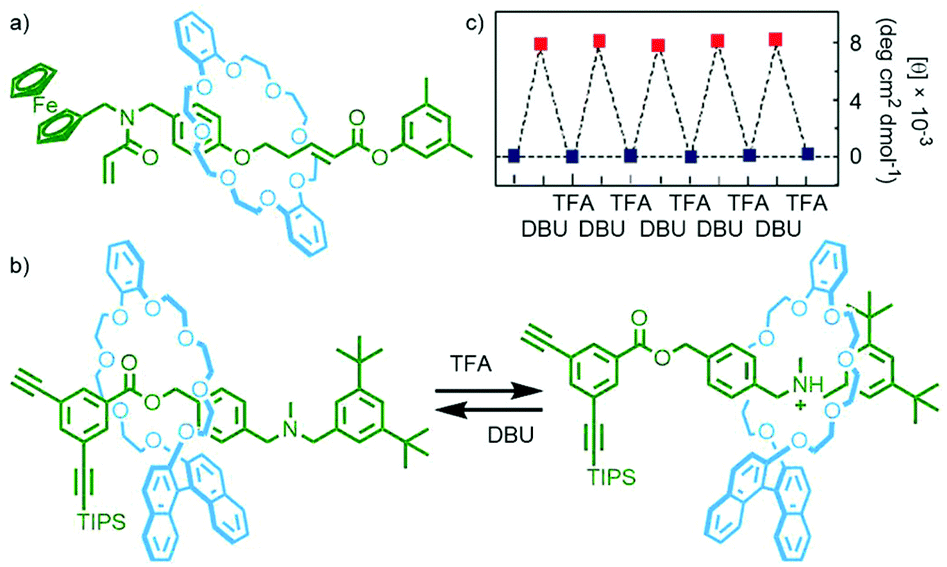 | ||
| Fig. 18 (a) Osakada's acrylamide [2]rotaxane monomer; (b) Takata's acid/base-switchable [2]rotaxane acetylene monomer with chiral BINOL-based crown ether macrocycle; (c) change in CD intensity (500 nm) of the switchable polyacetylene upon sequential addition of base and acid. Adapted from ref. 75 with permission from the Royal Society of Chemistry. | ||
Homopolymerisation of this rotaxane was found not to proceed, likely due to the steric bulk of the macrocycle. This idea was supported by the work of Takata and co-workers who were able to incorporate an acid/base-controlled switchable molecular shuttle, featuring a chiral BINOL-based macrocycle, into a helical polyacetylene. In the protonated state the macrocycle was located remotely from the acetylene stopper (and subsequently polymer chain), and adjacent when neutral (Fig. 18b). Polymerisation of the neutral rotaxane could not be achieved, whilst efficient formation of the desired polymer was observed when the amine station was protonated.
In apolar CHCl3 no Cotton effect could be observed for the polyacetylene chain by circular dichroism (CD) spectroscopy, indicative of a racemic helix, in the cationic polymer. Addition of DBU to neutralise the ammonium stations and induce shuttling of the macrocycle proximal to the polymer chain resulted in the appearance of a Cotton effect, indicative of single-handed helicity in the polymer. This switching between a racemic and single-handed helix could be effected through sequential addition of TFA and DBU, respectively (Fig. 18c).75
Alternatively, if more than one reactive group is introduced into a rotaxane structure, the resultant polymeric architecture will depend upon the relative locations of these groups. Leigh and co-workers demonstrated this through the preparation of [2]rotaxanes with tetralactam macrocycles in which the axles contained two blocked isocyanate moieties – either one on each (Fig. 19a) or both on a single stopper (Fig. 19b). Reaction of these with a diamine co-monomer gave a main chain poly[n]rotaxane (n ≈ 46) or side chain poly[n]rotaxane (n ≈ 96), respectively.76
 | ||
| Fig. 19 SCXRD structures of Leigh's [2]rotaxane monomers with blocked isocyanate groups on (a) both stoppers, and (b) one stopper; (c) Ikeda's sexithiophene rotaxane monomer, used to prepare electrochromic films through electrochemical polymerisation; (d) Takata's main chain poly[n]rotaxane prepared through Mizoroki–Heck coupling of a [2]rotaxane with a dialkene-functionalised macrocycle. | ||
Ikeda and co-workers were able to generate an electrochromic poly[n]rotaxane film through electrochemical polymerisation of a [2]rotaxane monomer with a sexithiophene axle component (Fig. 19c).77 In comparison to the non-interlocked axle polymer, the polyrotaxane films were found to be more homogenous, and possessed improved electrochromic properties as a result of the mechanical insulation afforded by the macrocyclic components.78 Although the polymerisation of a symmetrical [2]rotaxane with reactive groups on each stopper is perhaps the conceptually simplest approach to main chain poly[n]rotaxanes, it appears to be underutilised in the literature.
Conversely, if both reactive groups are present on the macrocycle of a [2]rotaxane precursor, the product obtained following polymerisation might be considered somewhat more difficult to categorise. If the reactive handles are at distal ends of the macrocycle, as in the case of a divinyl-functionalised rotaxane reported by Sato and Takata, then polymerisation will yield a main chain poly[n]rotaxane in which the threaded macrocycles are part of the polymer chain (Fig. 3b). In this example, Mizoroki–Heck coupling of the rotaxane with various aryl dihalides gave the corresponding polyrotaxanes (Fig. 19d) in good yields (83–98%), with co-polymerisation of two different rotaxane monomers also achieved.79 Alternatively if the functional groups were to be located proximal to each other, the distinction between a main chain (Fig. 3b) and side chain (Fig. 3f) polyrotaxane becomes more difficult to make.
If reactive groups are placed on both the axle and macrocycle components of a rotaxane monomer, the resultant polymer architecture will depend on whether these groups are complementary. If the groups are the same then there will theoretically be an equal chance of forming macrocycle–macrocycle, axle–axle and macrocycle–axle linkages, and will result in a main chain polyrotaxane of the type seen in Fig. 3d. Examples of this include work from Tamaoki and co-workers wherein a bis-isocyanate linker was reacted with a [2]rotaxane incorporating alcohol functional groups on both the axle and macrocycle components (Fig. 20a),80 and a CuAAC variant reported by Wang and co-workers in which a bis-alkyne rotaxane (Fig. 20b) was reacted with a bis-azido polycaprolactone linker.81
 | ||
| Fig. 20 [2]Rotaxane monomers with equivalent functional groups on the axle and macrocycle components. (a) Tamaoki's alcohol-functionalised rotaxane that was reacted with a bis-isocyanate linker, and (b) Wang's bis-alkyne rotaxane that was polymerisered by CuAAC chemistry with a bis-azide linker. | ||
Alternatively, if complementary functional groups are used that cannot react with themselves, only each other, then [an]daisy chain rotaxanes (Fig. 3g) will be obtained. Such a polymer was reported by Takata and co-workers by incorporating a terminal alkyne onto the macrocycle and an aryl iodide onto the axle of a [2]rotaxane (Fig. 21a). Submission of this precursor to Sonogashira reaction conditions ([Pd(PPh3)2Cl2], CuI, THF/piperidine) ensured exclusive formation of bonds between macrocycle and axle components.82
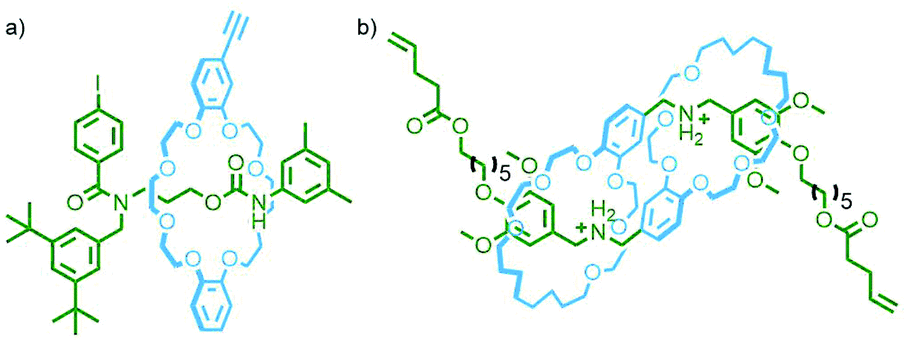 | ||
| Fig. 21 (a) [2]Rotaxane monomer with aryl iodide and alkyne functional groups on the axle and macrocycle components, respectively. Polymerisation under Sonogashira reaction conditions yielded an [an]daisy chain poly[n]rotaxane. (b) A [c2]daisy chain rotaxane with alkene units, tethered to the stoppering units, that could be polymerised via alkene metathesis. | ||
It is also possible to prepare polymeric [c2]daisy chain rotaxanes (Fig. 3h) through polymerisation of monomeric[c2]daisy chains with functional groups on each of the stoppering units. This approach has been shown to be compatible with both alkene metathesis83 (Fig. 21b) and CuAAC chemistry.84
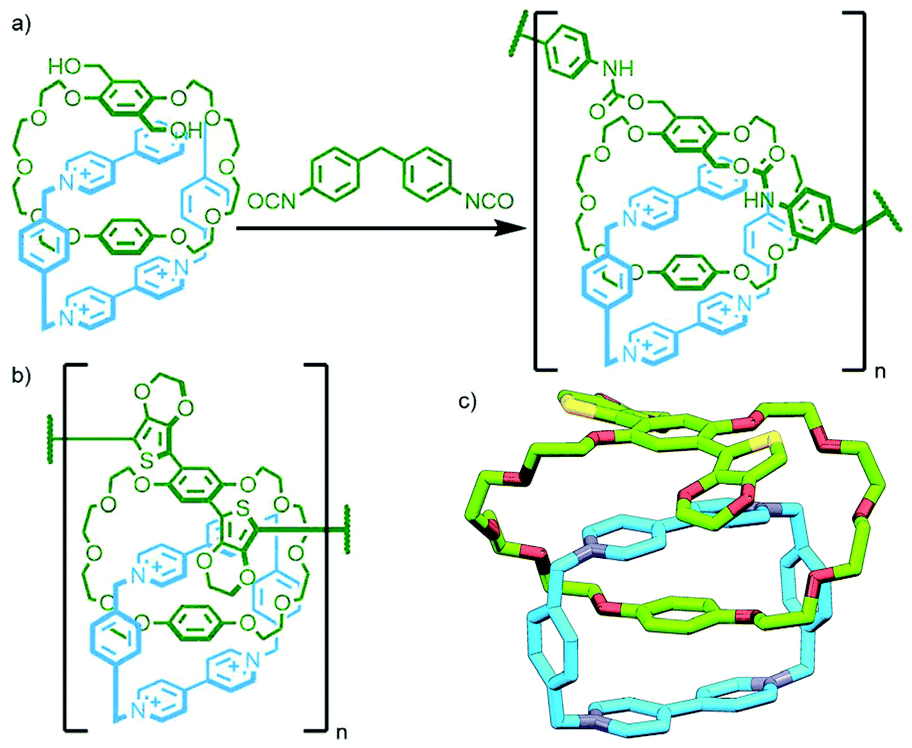 | ||
| Fig. 22 (a) Stoddart's polymerisation of a xylylenediol-functionalised [2]catenane upon reaction with 4,4′-methylenebis(phenyl isocyanate), and (b) Swager's electrochemically polymerised thiophene-based side chain poly[n]catenane; (c) SCXRD structure of the bis(thiophene) [2]catenane monomer. | ||
The first polymerisation of a [2]catenane with reactive groups on both macrocycles was reported by Geerts and co-workers in 1995. Based on the octaamide catenanes reported by Vögtle and Hunter,8 a [2]catenane with aryl bromide moieties on each macrocycle (Fig. 23a) was prepared and various palladium-catalysed cross-couplings (Sonogashira, Suzuki and Stille) attempted. In two instances oligomeric materials were obtained, albeit with low degrees of polymerisation (up to 5 by GPC, although larger oligomers were observed by MS) as a result of the poor reactivity of the bulky catenane substrate.87
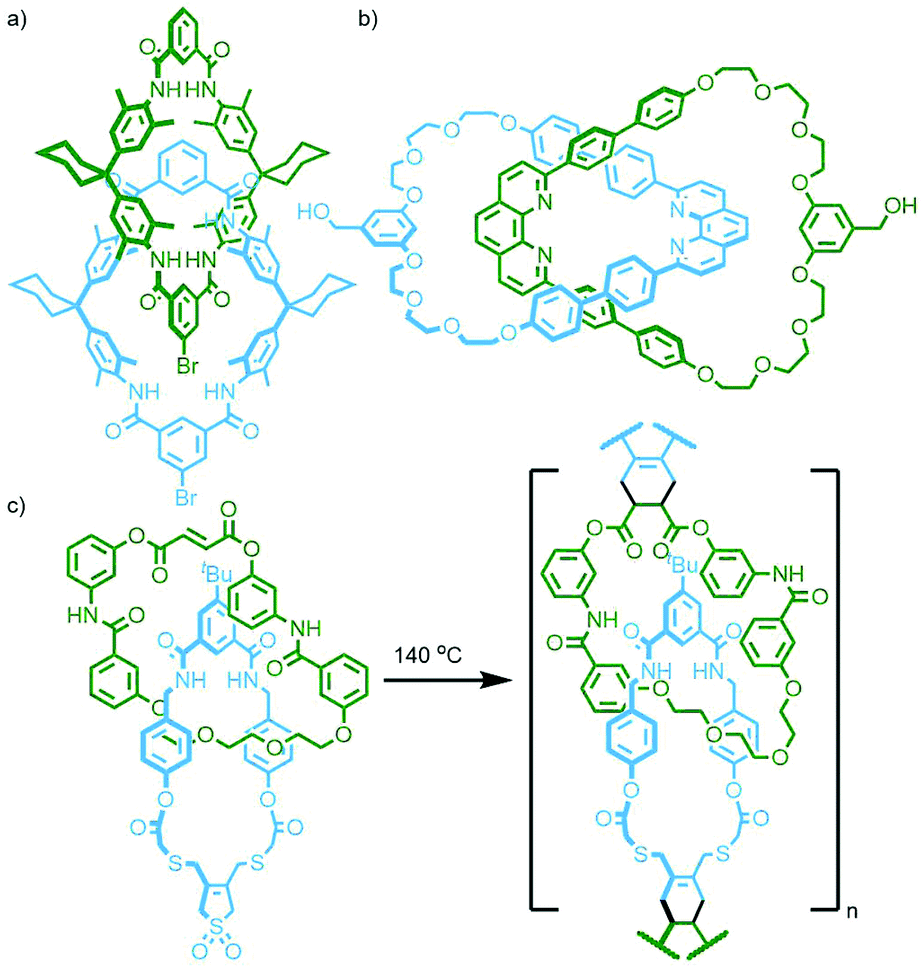 | ||
| Fig. 23 (a) Geerts’ bis(aryl bromide) functionalised octaamide [2]catenane, used to prepare main chain poly[n]catenanes through palladium-catalysed cross couplings; (b) Kern, Sauvage and Geerts’ bis-phenanthroline catenane with alcohol functionality used in polyesterification reactions; (c) Takata's heterocircuit [2]catenane with dienophile and masked-diene functionality for Diels–Alder polymerisation. | ||
Subsequent work by Kern, Sauvage, Geerts and co-workers with a larger, more flexible and more readily soluble [2]catenane precursor bearing alcohol groups on each macrocycle produced main chain poly[n]catenanes following ester condensation with a terephthalic acid derivative. Interestingly, if the Cu(I) complex of the catenane monomer was used in the polymerisation, mainly linear oligomers/polymers were obtained; in contrast the demetallated catenane (Fig. 23b) appeared to give mostly cyclic oligomers.88
Since this early work a variety of reactions have been used to polymerise difunctional [2]catenane monomers: Sonogashira coupling between a catenane with aryl iodide units and diyne linkers;89 CuAAC reaction between a bis-azide catenane and diyne linker,90 and incorporation of catenanes into polycarbonate91 and poly(ethylene terephthalate)92via solid-state polymerisation have been reported.
All of these previous examples involve the polymerisation of homocircuit catenanes through reaction with an appropriate linker. Takata and co-workers reported the polymerisation of a heterocircuit [2]catenane in which each macrocycle incorporated one component for a Diels–Alder reaction: a fumaric acid ester as the dienophile, and a sulfolene as a masked diene. Heating the catenane, either neat or in 1,2-dichlorobenzene as solvent, at 140 °C resulted in elimination of SO2 to form the diene in situ, and rapid formation of the Diels–Alder adduct gave the poly[n]catenane (Fig. 23c).93
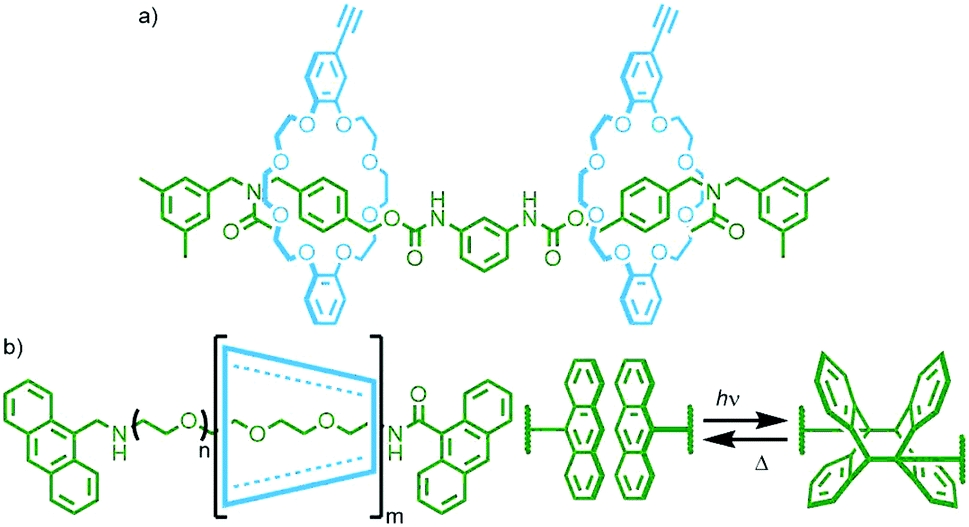 | ||
| Fig. 24 (a) Takata's [3]rotaxane polymerised through reaction with bis nitrile-N-oxide linkers; (b) Harada's CD/PEG poly[n]rotaxane with anthracene stoppering units that reversibly dimerise upon photoirradiation, resulting in larger main chain poly[n]rotaxanes and radial poly[n]catenanes which disassemble upon heating. | ||
A notable example by Okada and Harada entailed the photopolymerisation of α-CD/PEG main chain poly[n]rotaxanes utilising anthracene units as stoppering moieties (Fig. 24b). Irradiation of a DMSO solution (2 × 10−2 mM) of the poly[n]rotaxane with visible light (λ > 340 nm) resulted in photodimerisation of the anthracene units, giving larger polymers with molecular weights in excess of 107 g mmol−1. Performing the reaction under more dilute conditions (7 × 10−3 mM) gave additional products, proposed to be radial poly[n]catenanes arising from intramolecular cyclisation reactions. Due to the reversibility of the anthracene dimerisation reaction, simply heating a DMSO solution of the polymers at 120 °C resulted in over 90% reversion to the starting poly[n]rotaxanes.96 This motif was also used by Huang and co-workers for the reversible assembly/disassembly of a poly[n]rotaxane from a [3]rotaxane incorporating two axle components, each with one anthracene stopper.97
3.2 Polymerisation of pseudo-MIMs
The polymerisation of MIM monomers has the advantage that the exact nature of the interlocked component(s) being incorporated into the polymer is known and as such, assuming the integrity of the mechanical bond is maintained under the reaction conditions, the ratio of repeating units to mechanical bonds can be known with precision. However, the synthesis of mechanically interlocked monomers containing functional groups suitable for polymerisation may be arduous, limiting the amount of material that can be conveniently accessed.A commonly used route to discrete rotaxanes is to form a pseudorotaxane, or host–guest complex, between a macrocycle and a linear guest, before covalently appending stoppering groups to secure the mechanical bond; this is known as capping. This approach can be applied to main chain polyrotaxanes, wherein self-assembled pseudo-interlocked monomers are polymerised in the presence of a suitable bulky reagent, resulting in simultaneous in situ formation and stoppering of a pseudopolyrotaxane (Fig. 25). Through careful design of the components and choice of functional groups for the polymerisation reaction other forms of mechanically interlocked polymers can be accessed. A limitation of this approach is that the ratio of recognition sites to interlocked macrocycles is likely to be less than unity. However, the simplicity and relative ease of access to starting materials for this method makes it a highly attractive one that has been exploited extensively in the synthesis of polymeric MIMs.
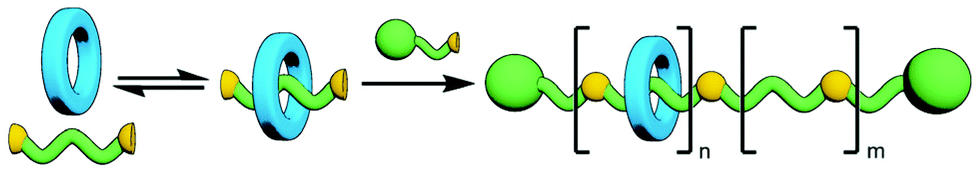 | ||
| Fig. 25 Polymerisation of a pseudo[2]rotaxane, derived from the self-assembly of a reactive axle unit with a macrocycle, in the presence of a stoppering reagent results in a main chain poly[n]rotaxane. Due to the equilibrium formation of the pseudorotaxane precursor, the number of trapped macrocycles, n, is less than the total number of recognition sites, n + m. | ||
In 1989 Gibson and co-workers reported the synthesis of a polyester from the condensation of sebacoyl chloride with 1,10-decanediol using macrocyclic 30-crown-10 as solvent, with addition of 3,3,3-triphenylpropionyl chloride as a stoppering group prior to the end of the reaction. By using the macrocycle as solvent it was hoped that serendipitous threading would take place in the vein of Harrison and Harrison's statistical synthesis of a [2]rotaxane,4 with kinetic trapping of the threaded crown ethers effected by the bulky stopper groups. Purification of the resultant polyrotaxane by precipitation ensured removal of any remaining non-interlocked macrocycle, and the repeating unit/macrocycle ratio was determined to be 6.0 by integration of signals in the NMR spectrum.98 Gradual de-threading of the polymer over time indicated that this was in fact a metastable pseudopolyrotaxane. Subsequent use of larger substituted trityl groups in a similar polyrotaxane, prepared by transesterification, was shown to completely inhibit de-threading.99
As in the development of methods for the synthesis of their discrete counterparts, it was rapidly recognised that statistical methods are extremely limiting for the preparation of polymeric MIMs. To increase the threading efficiency in forming pseudorotaxane monomers, template motifs were required.
In 1987 Stoddart and co-workers had reported the formation of a host–guest adduct between bis-(p-phenylene)-34-crown-10 (BPP34C10) and paraquat100 (and related crown-ether macrocycles101). These arose from favourable charge transfer interactions between the electron poor guest and electron rich aromatic units of the macrocycle, a motif that was able to be exploited for the template-directed synthesis of catenanes102 and rotaxanes.103 Using this template, Gibson and co-workers used the pseudo[2]rotaxane formed between BPP34C10 and N,N′-bis(2-hydroxyethyl)-4,4′-bipyridinium as a monomer in the synthesis of a polyurethane-based poly[n]rotaxane (Fig. 26). Due to the strong host–guest interactions, 80% of the bipyridinium moieties in the polymer were encircled by BPP34C10 macrocycles.104
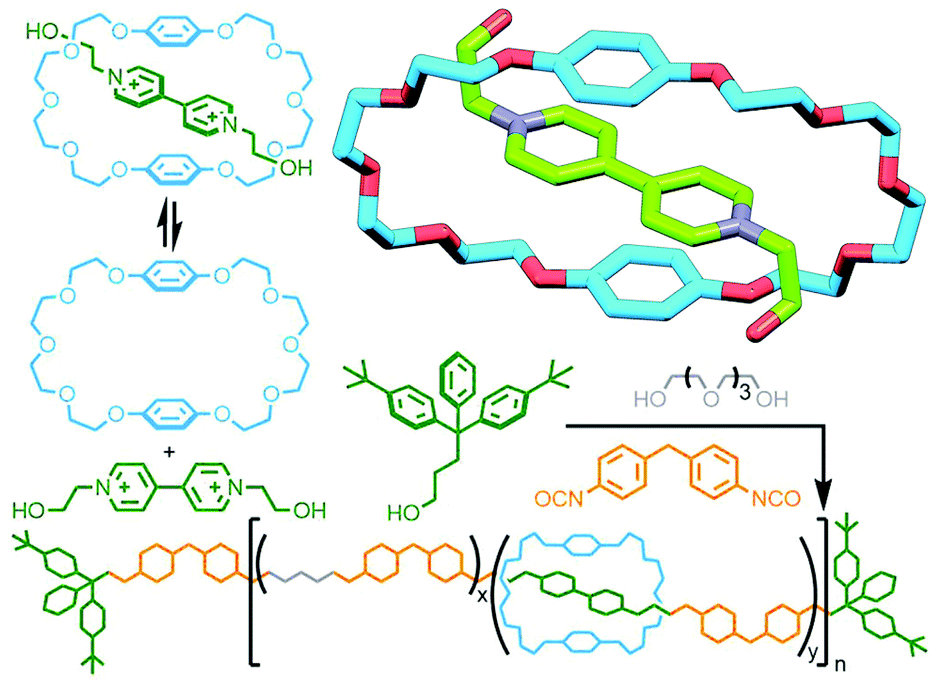 | ||
| Fig. 26 Gibson's polymerisation of a pseudo[2]rotaxane precursor assembled from BPP34C10 macrocycle and paraquat-based diol. | ||
Since this early example, a variety of pseudorotaxane motifs have been used as monomers for the synthesis of main chain poly[n]rotaxanes. In theory any combination of template interaction and polymerisation reaction can be used, provided one does not interfere to any great extent with the other. Below we have described select examples from the literature to display the utility of this approach, categorised based on the macrocyclic component employed.
Grubbs and co-workers used acyclic diene metathesis polymerisation of a DB24C8 host–guest adduct with dienyl ammonium salts in the presence of a bulky chain-transfer agent to produce main chain polyrotaxanes with threading efficiencies of up to 82% (Fig. 27a).108 Through use of this strategy with an alternative dipyridyl-24-crown-8 macrocycle, Li, Wu and co-workers were able to obtain a main chain poly[n]rotaxane with switchable properties. Addition of acid to the polymer resulted in protonation of the macrocycle pyridyl units, causing them to shuttle away from the ammonium stations as a result of charge repulsion, with deprotonation by NEt3 allowing them to shuttle back. The pyridine units also allowed for supramolecular cross-linking of polymer chains upon addition of [Pd(PhCN)2Cl2] through formation of metal–ligand bonds, with reversion to the individual polymer strands triggered through addition of a competing PPh3 ligand to displace the Pd(II) ions.109
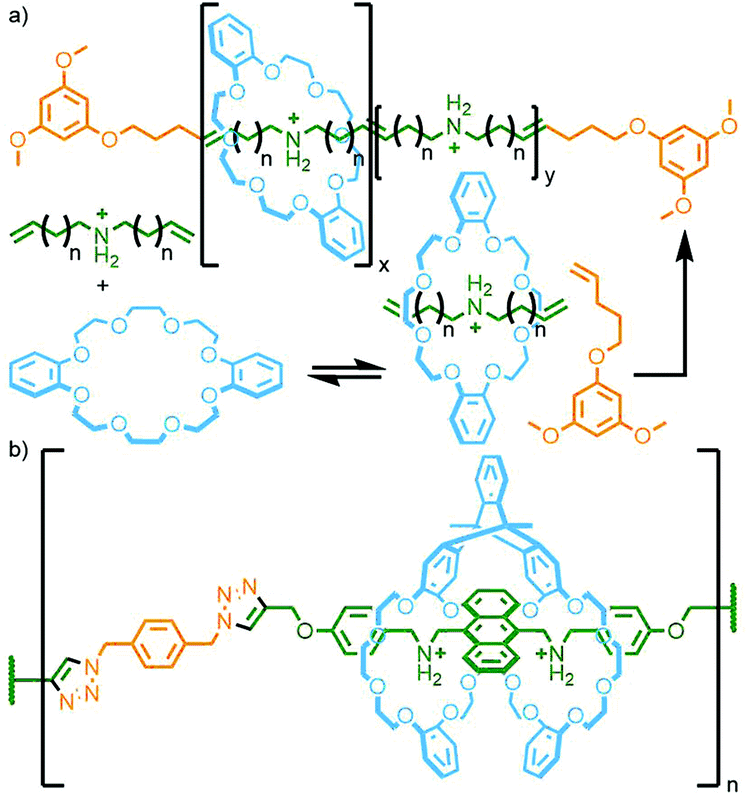 | ||
| Fig. 27 (a) Grubbs’ ADMET polymerisation of a diene pseudorotaxane; (b) Chen's main chain poly[n]rotaxane prepared from the CuAAC polymerisation of a pseudosuitane precursor. | ||
An interesting variant of the crown-ether/ammonium host–guest motif was reported by Chen and co-workers in which a ‘pseudosuitane’ assembled from a bis-macrocycle was polymerised by CuAAC reaction with a bis-azide, with an anthracene core acting as a stoppering unit to prevent dethreading (Fig. 27b).110
Through incorporating alkene tethers on both a dialkyl-ammonium half-axle component and a substituted DB24C8 macrocycle, Oishi and co-workers formed a cross-linked rotaxane polymer, by alkene metathesis, composed of all three possible hetero- and homo-dimers of the two starting materials (Fig. 28a).111 In this example the macrocycle and axle components were able to react with themselves as well as each other. If the functional groups on each component are only able to react with the other then an [an]daisy chain polyrotaxane will result. This was demonstrated by Huang and co-workers through polyesterification of a crown ether/1,2-bis(pyridinium)ethane pseudorotaxane (Fig. 28b).112 Alternatively, if the pseudorotaxane axle contains only a single polymerisable reactive group, with the other end possessing a suitable stoppering unit (this species sometimes referred to as a semi-rotaxane), then a side-chain polyrotaxane will be obtained. Takata and co-workers have used such a design with DB24C8 and an ammonium half-axle through radical alkene polymerisation, reporting up to 51% macrocycle incorporation of the theoretical maximum (Fig. 28c).113
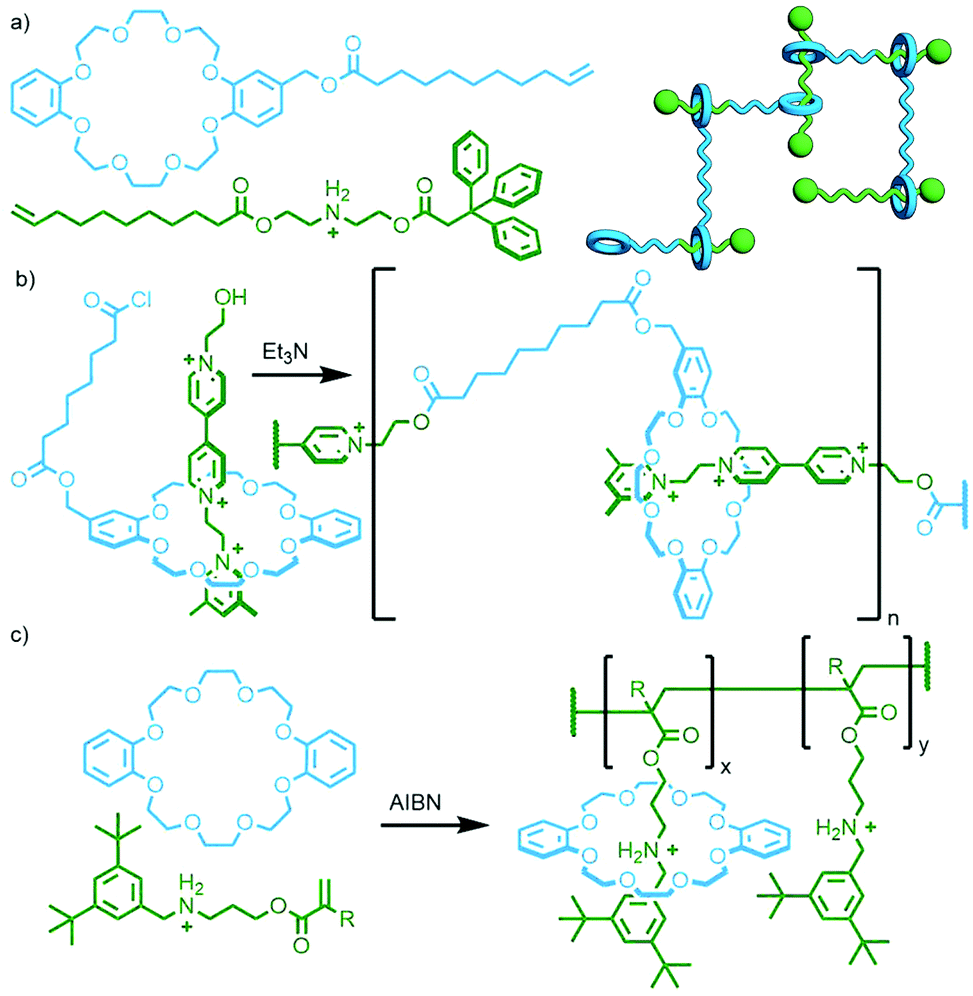 | ||
| Fig. 28 (a) Macrocycle and axle monomers used by Oishi, and a cartoon representation of the resultant polymer containing both homo- and hetero-dimers. (b) Huang's synthesis of an [an]daisy chain rotaxane through ester condensation. (c) Side-chain poly[n]rotaxane reported by Takata, prepared from the radical polymerisation of a pseudorotaxane with an acrylate moiety at one of the axle component. | ||
 | ||
| Fig. 29 (a) Yamaguchi, Osakada and Yamamoto's benzimidazole-based poly[n]rotaxane, and (b) Simionescu's imine poly[n]rotaxane. | ||
In the same year Simionescu and co-workers polymerised pseudorotaxanes of α- and β-CD with 1,6-diaminohexane via imine condensation with a slight excess of terephthaldehyde, catalysed by toluenesulfonic acid. Addition of p-aminophenyltriphenylmethane served to stopper the resultant pseudointerlocked polymers, giving main chain polyrotaxanes with CD![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :monomer ratios of 1:6 and 1:7 for α- and β-CD, respectively (Fig. 29b).116 Farcas and Grigoras subsequently showed that a diformylcarbazole derivative was sufficiently bulky to negate the need for an additional stoppering unit upon condensation with these pseudorotaxanes.117
:monomer ratios of 1:6 and 1:7 for α- and β-CD, respectively (Fig. 29b).116 Farcas and Grigoras subsequently showed that a diformylcarbazole derivative was sufficiently bulky to negate the need for an additional stoppering unit upon condensation with these pseudorotaxanes.117
Anderson and co-workers were able to form a pseudorotaxane through encapsulation of a 4,4′-diphenyldiboronic ester within β-CD. Suzuki coupling of this assembly with an iodonaphthalene gave the desired [2]rotaxane, although the stoppering unit appeared to impinge on the bound β-CD resulting in a low 4% isolated yield. However, when the reaction was repeated in the presence of a 4,4′-diiododiphenyl moiety, the resulting conjugated poly-p-phenylene main chain polyrotaxane was successfully obtained in 45% yield (Fig. 30a).118 This strategy was also shown to work with Ni-catalysed Yamamoto coupling between β-CD encapsulated dibromothiophene and dibromofluorene in the presence of 9-bromoanthracene (Fig. 30b), with macrocycle coverage for both polymers estimated to be 60% based on data from small angle neutron scattering measurements.119 Sonogashira coupling has been shown to be effective as well: Shinohara and co-workers polymerised pseudorotaxane complexes of α-CD with p-diiodobenzene and p-diethynylbenzene in the presence of 10% 9,10-diethynylanthracene to prevent dethreading.120
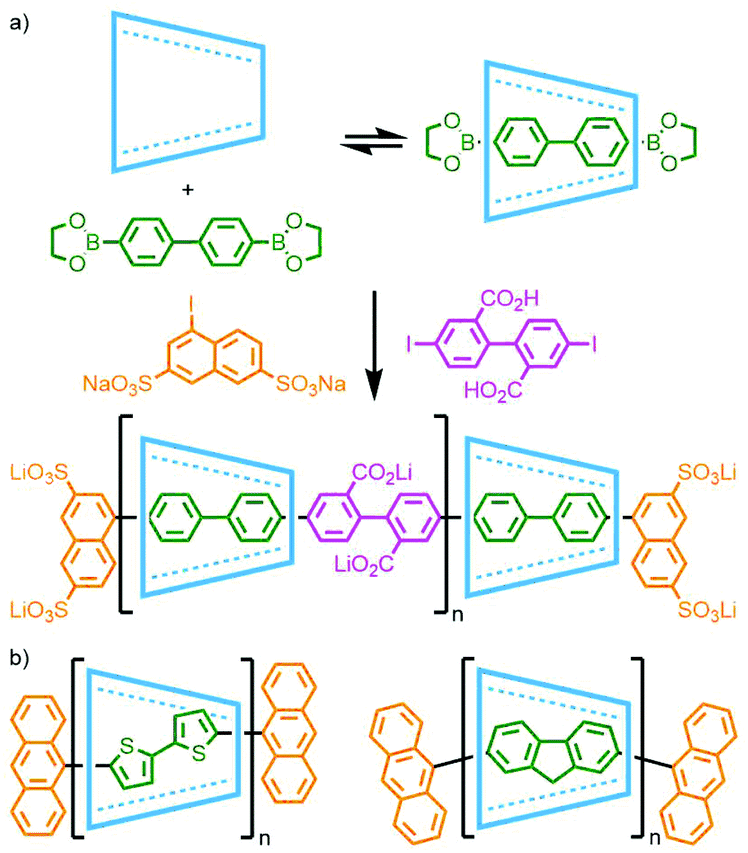 | ||
| Fig. 30 Polymerisation of CD-pseudorotaxanes through (a) Suzuki coupling, and (b) Yamamoto coupling. | ||
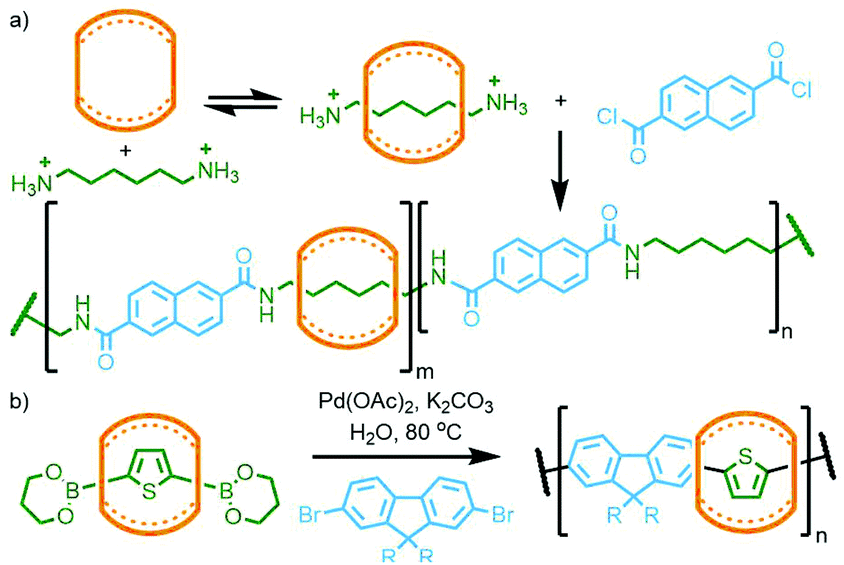 | ||
| Fig. 31 (a) Buschmann's amide condensation and (b) Tuncel's Suzuki coupling approaches to CB-polyrotaxanes. | ||
Using Mock's CB-AAC reaction, in 1999 Tuncel and Steinke showed that bis-azide/alkyne units with mesityl cores were able to react with the corresponding tert-butyl azide/alkyne stoppering reagent, catalysed by CB[6], resulting in [3]rotaxanes isolated in excellent yield. As the mesityl units were sufficiently large to act as stoppers, polymerisation between the bis-azide and bis-alkyne building blocks gave main chain poly[n]rotaxanes (Fig. 32).125
 | ||
| Fig. 32 Steinke's CB-AAC approach to the synthesis of main chain poly[n]rotaxanes. | ||
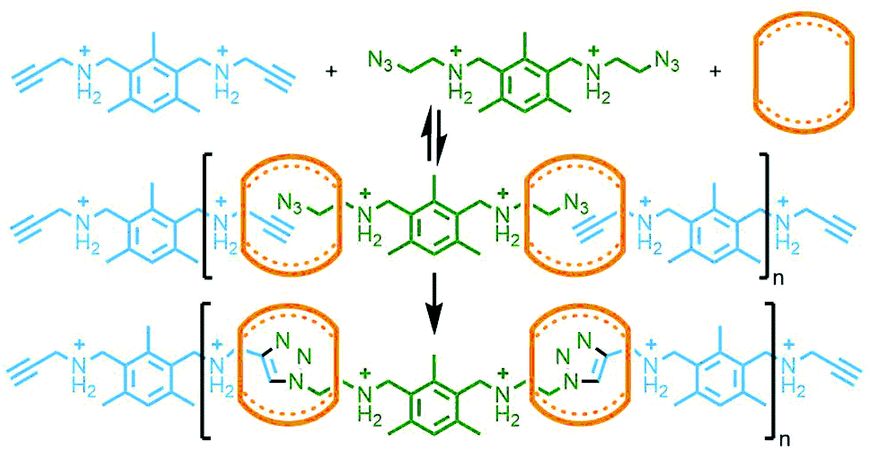
In 2011 Takata and co-workers prepared a bis-macrocycle with pyridinedicarboxamide coordinating units which, upon reaction with Pd(OAc)2, gave the neutral dipalladium(II) complex (Fig. 33). Addition of 4-vinylpyridine was found to result in displacement of coordinated solvent from the metal ions. Radical polymerisation of 4-vinylpyridine with a small amount (0.5 mol%) of this complex, with 4-tert-butylstyrene as co-monomer to prevent dethreading, afforded a polyrotaxane gel. The effect of the mechanical bonds in the polymer network was demonstrated through the preparation of two control gels, one without 4-tert-butylstyrene as co-monomer, and one with an acyclic analogue of the bis-macrocycle complex. In contrast to the polyrotaxane gel, dilution of these two control polymers in DMF led to dissociation of the components with a resultant gel–sol transition.128 In a subsequent study, several vinyl monomers were polymerised in the presence of the 4-vinylpyridine adduct of the palladium(II) bis-macrocycle complex (0.5 mol%) and a bulky tris(4-tert-butylphenyl)methyl monomer (10 mol%). For the polymer derived from butyl acrylate, decreasing the content of the bulky monomer to 2 mol% resulted in a gel with increased swellability, attributed to the increased mobility of the macrocycle components along the polyvinyl chains.129
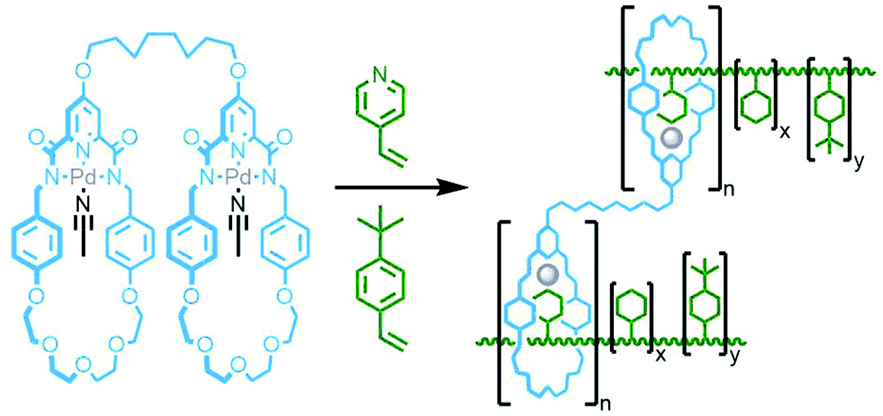 | ||
| Fig. 33 Takata's bis-macrocycle palladium(II) complex with in situ formation of a bis-pseudorotaxane complex with 4-vinylpyridine resulting in a cross-linked rotaxane polymer during radical polymerisation in the presence of a bulky 4-tert-butylstyrene co-monomer to inhibit dethreading. | ||
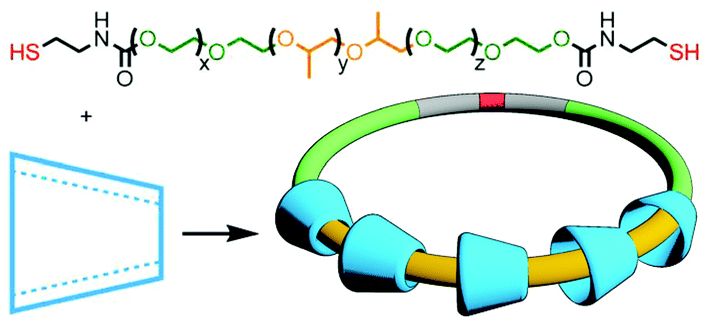
The extensive work of the Beer group over the last two decades has shown the viability of using anions as alternative templates to metal ions for the formation of (pseudo)MIMs.130 For example, a combination of isophthalamide and pyridiniumdicarboxamide ligands is able to form a heteroleptic aniono-assembly about halide ions (Fig. 34a).131 Due to their intramolecular nature, cyclisation reactions are inherently more prone to non-productive side reactions than intermolecular couplings. Nonetheless, both clipping132 and stoppering133 reactions of anion-templated pseudointerlocked precursors have been used to prepare MIMs.
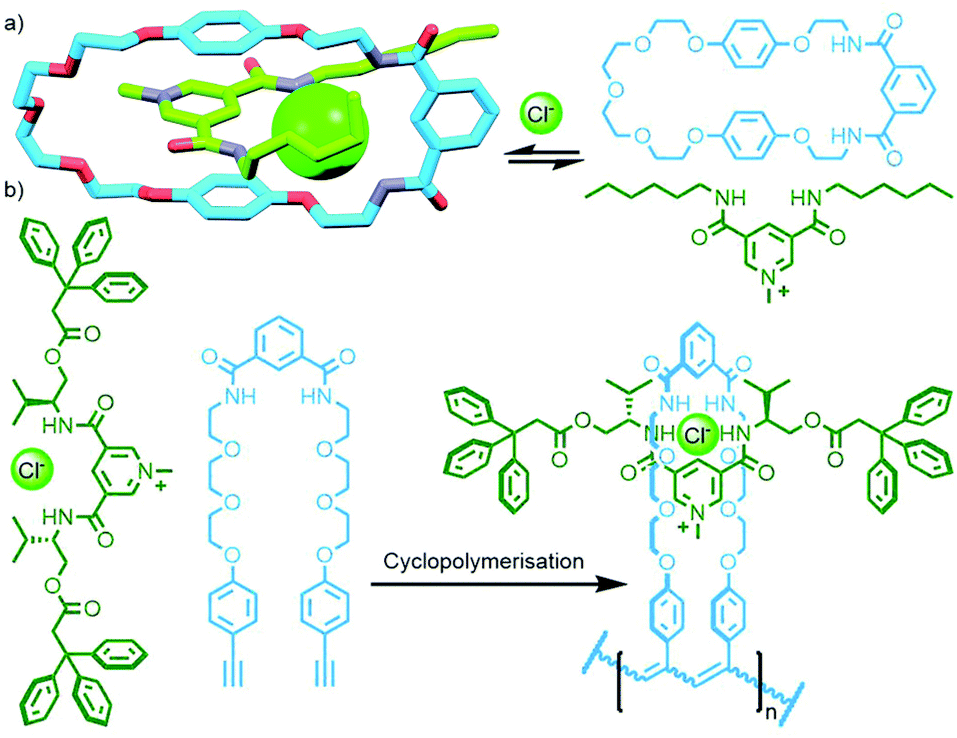 | ||
| Fig. 34 (a) Formation of a pseudorotaxane from the self-assembly of an isophthalamide macrocycle and pyridiniumdicarboxamide axle using an anion template. (b) Kakuchi's cyclopolymerisation to form a chloride-templated polyrotaxane. | ||
Using the design principles delineated through this work, Kakuchi and co-workers prepared a chloride-templated assembly consisting of an axle component with trityl stoppers, and an acyclic macrocycle precursor with terminal alkyne units. Cyclopolymerisation134 of this precursor resulted in simultaneous cyclisation of the acyclic unit and mechanical bond formation between the axle and incipient macrocycle, as well as polymerisation of the rotaxane structure (Fig. 34b). The cyclisation efficiency was determined to be very high (>99%) with up to 26% of the macrocycles successfully threaded by an axle in the isolated polymers (DOSY NMR was used to confirm the interlocked nature of the components).135
In summary, a wide range of pseudorotaxane assemblies have been shown to be viable monomer precursors for the synthesis of main chain poly[n]rotaxanes, provided the reaction conditions do not significantly disrupt the host–guest interactions. It is interesting to note that, despite their continued popularity for the synthesis of discrete MIMs, fuelled by their predictability and robustness, metal ion templates have rarely been used in preparing mechanically interlocked polymers, perhaps due to the potential difficulties for subsequent removal of the metal ions from the resultant polymers.
3.3 Post-synthetic polymer functionalisation (grafting)
In the previous examples, polymer formation, trapping of the mechanical bond, or both, occurred as the final synthetic step. Alternatively, pre-formed polymers may be post-synthetically functionalised with MIMs. Woisel and co-workers prepared [2]catenane and [2]rotaxane monomers with alkyne-functionalised CBPQT4+ macrocycles and were able to achieve an estimated 50% incorporation of these MIMs onto the backbone of an azide-functionalised polystyrene using CuAAC chemistry (Fig. 35).136 Subsequently Grzybowski, Stoddart and co-workers demonstrated the efficacy of this CuAAC approach for grafting an electrochemically active catenane onto a polymer side chain, importantly showing that it retained its ability to operate as a molecular switch.137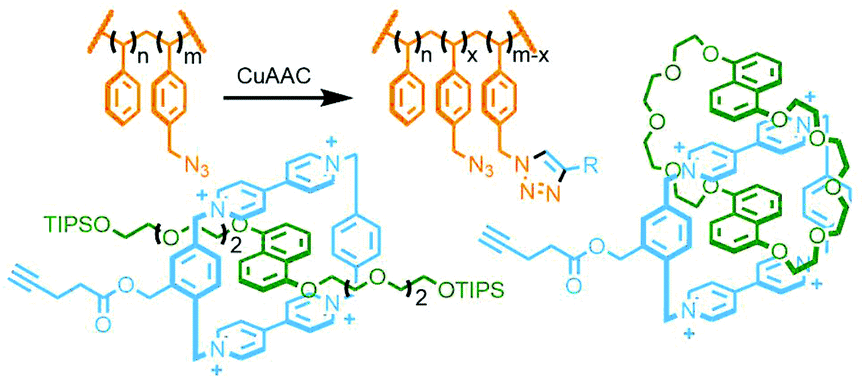 | ||
| Fig. 35 Woisel's CuAAC approach to grafting rotaxane and catenane units onto the side chains of an azide-functionalised polystyrene. | ||
3.4 Capping/clipping approaches
As with discrete MIMs, capping – the addition of stoppers to a pseudorotaxane to kinetically trap the macrocycle(s) on the axle – and clipping – the cyclisation of a macrocycle precursor about an axle – are commonly employed strategies for the synthesis of poly[n]rotaxanes (Fig. 36). Thus, unsurprisingly, many of the template motifs used for the synthesis of discrete MIMs can, and have, be used for preparing polymeric analogues.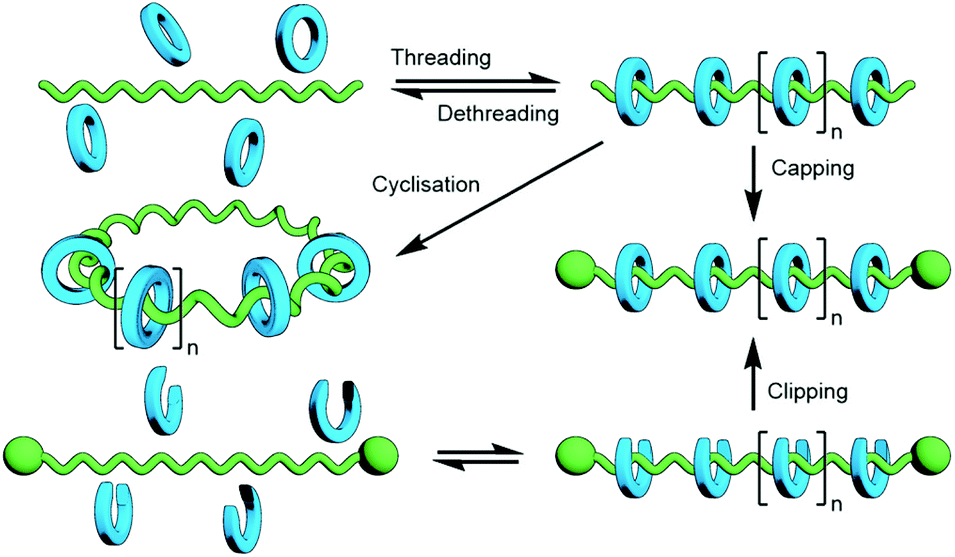 | ||
| Fig. 36 Capping and clipping are commonly employed methods to prepare main chain poly[n]rotaxanes. Although an inherently more challenging reaction, the intramolecular cyclisation of a pseudo[n]rotaxane will give rise to a radial [n]catenane. | ||
 | ||
| Fig. 37 Main chain poly[n]rotaxanes prepared via the capping approach: (a) Harada's seminal PEG/α-CD polyrotaxane; (b) Stoddart's donor–acceptor system; (c) an alternative design in which the macrocycle is incorporated in the polymer backbone by Takata; (d) Ogoshi's pillarene polyrotaxane with viologen-based axle. | ||
The threading of PEG was found to be specific for α-CD; neither β- nor γ-CD formed similar inclusion complexes. However, both of these larger macrocycles were able to encircle poly(propylene glycol) (PPG).140 Unsurprisingly, addition of appropriate stoppering groups to a β-CD/PPG polypseudorotaxane gave the permanently interlocked structure.141 Likewise, poly[n]rotaxanes generated from inclusion complexes of poly(butadiene) (PBD) and poly(dimethylsiloxane) (PDMS) have also been reported.142
Following on from Harada's seminal work, the combination of PEG and α-CD remains the most popular choice for preparing poly[n]rotaxanes via this capping approach. However, there have been numerous reports using alternative template motifs. Stoddart and co-workers used a CuAAC-derived polymer with 1,5-dioxynaphthalene units to thread CBPQT4+, with a final CuAAC used to stopper the system (Fig. 37b);143 Wenz and Keller observed threading of α-CD onto poly(iminooligomethylene) chains, introducing nicotinyl groups to effectively prevent dethreading;144 diamino-PEG could also be used as an axle component with CB[7], as shown by Resmerita and co-workers;145 Ogoshi and co-workers synthesised a polyrotaxane composed of an adamantane-stoppered viologen polymer threaded with pillar[5]arene macrocycles (Fig. 37d);146 poly(hexamethyleneammonium) was found to be a suitable axle for trapping DB24C8 with very high efficiency by Takata and co-workers.147
A much less common approach to main chain poly[n]rotaxanes is the capping of axle components threaded through a polymer constructed from multiple macrocyclic components. Takata and co-workers first demonstrated the feasibility of this strategy through self-assembly of a poly-DB24C8 chain with an ammonium half-axle that was subsequently stoppered (Fig. 37c).148
 | ||
| Fig. 38 Higashi, Arima and Li's formation of a radial poly[n]catenane through oxidative cyclisation of a dithiol block co-polymer threaded with CD macrocycles. | ||
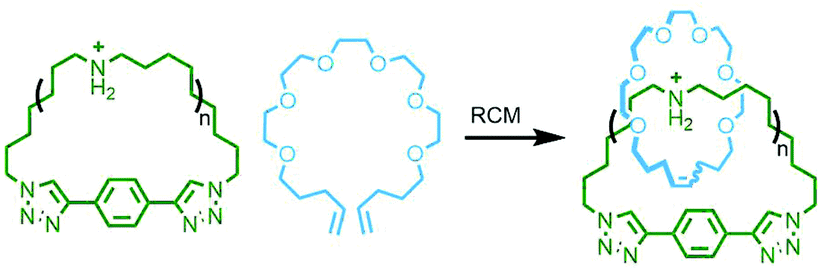 | ||
| Fig. 39 Grubbs’ RCM clipping approach to radial poly[n]catenanes. | ||
In 2017 Rowan and co-workers reported the synthesis and characterisation of main chain poly[n]catenanes obtained from ring-closing alkene metathesis of a Zn(II) coordination polymer, assembled from an alternating mixture of two components: a macrocycle and an acyclic diolefin, both incorporating two tridentate coordination sites (Fig. 40). Following demetallation and isolation of the resultant polymeric material from non-interlocked by-products and smaller oligomers, the mixture was fractionated by preparative GPC. Through a combination of GPC coupled to multiangle light scattering, MS and NMR analysis, including comparison with model compounds, the polymeric mixture was found to contain linear, cyclic and branched poly[n]catenanes. Of the latter, polymers with molecular weights of up to 200 kg mol−1 were identified, corresponding to poly[130]catenanes!151
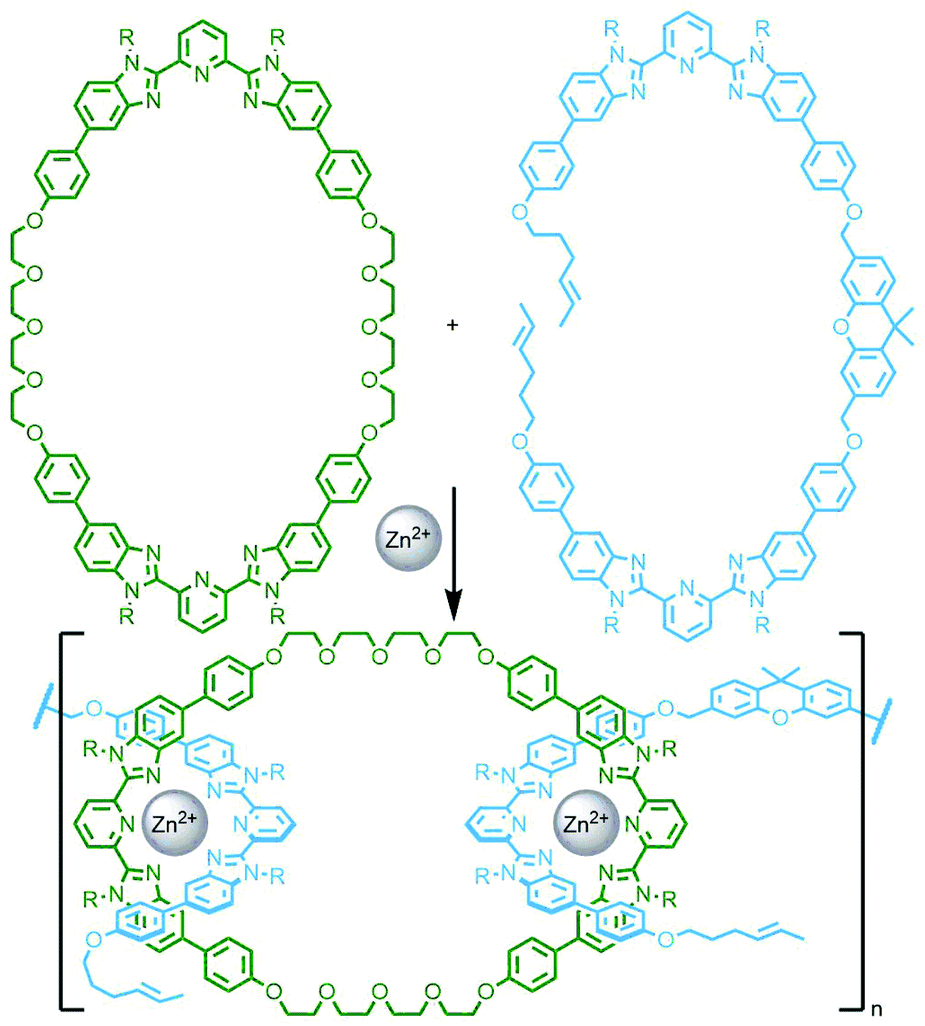 | ||
| Fig. 40 Rowan's synthesis of main chain poly[n]catenanes through ring closing alkene metathesis of a zinc(II) coordination polymer assembled from bis-tridentate macrocyclic (green) and acyclic (blue) building blocks. | ||
3.5 Coordination polymers
As highlighted above, metal ions/complexes can be extremely useful units for organising (pseudo)MIMs into oligomeric metallo[n]rotaxanes and -catenanes. Just as these can be obtained from judiciously chosen building blocks, careful design can also allow the synthesis of polymeric materials incorporating mechanically bonded components.152In the mid-1990s the early work in the field was dominated by the group of Kim who explored the effect of both metal source and ligand structure on the self-assembly of CB-based pseudorotaxanes (Fig. 41). A bis(4-pyridyl) axle was observed to form a one-dimensional polymer with Cu(NO3)2 (with Cu(OH2)3 nodes stoppering and connecting interlocked ligands)153 and a two-dimensional network with Ag(NO3).154 The first three-dimensional rotaxane coordination polymer, latterly termed metal–organic rotaxane frameworks (MORFs) by Loeb,155 was obtained from the self-assembly of a bis(benzoate) axle with Tb(NO3)3.156 Likewise, Loeb and co-workers showed that pseudo[2]rotaxanes derived from 1,2-bis(pyridinium)ethane axle ligands and DB24C8 macrocycles could be used to assemble one-, two- and three-dimensional coordination polymers.157
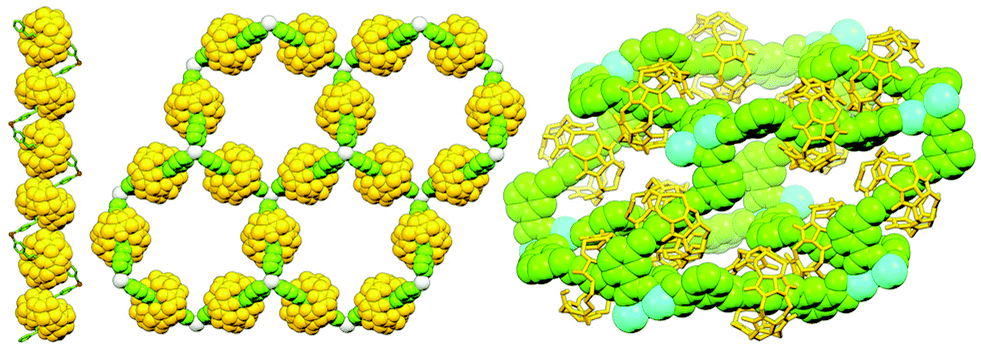 | ||
| Fig. 41 SCXRD structures of 1D, 2D and 3D coordination polymers derived from CB[6]-based pseudo[2]rotaxanes with Cu(II), Ag(I) and Tb(III) ions, respectively. | ||
In the majority of MORFs the coordinating moieties are located on the axle component, likely due to a combination of the commercial availability of many macrocycles and the relative synthetic ease of preparing functionalised acyclic components. Despite this, there are several examples in the literature of (pseudo)[2]rotaxane coordination polymers with ligands on the macrocycle,158 or on both components (Fig. 42a).159
 | ||
| Fig. 42 (a) A [2]rotaxane reported by Loeb incorporating ligand units in both the axle and macrocycle components. (b) Stoddart and Yaghi's [2]catenane ligand used in the synthesis of a 2D MOF. | ||
For the same reasons outlined above, catenanes as ligands in coordination polymers are relatively scarce. However, Ag(I)-based one-dimensional coordination polymers have been reported by the groups of Lewis – who utilised a [2]catenane ligand with monodentate pyridine donors in each macrocycle160 – and Stoddart, who employed a handcuff [3]catenane with bipyridine-functionalised terminal macrocycles.161 Furthermore, by using relatively rigid carboxylate ligands with pendant catenane units, Stoddart, Yaghi and co-workers have successfully prepared both two- and three-dimensional MOFs (Fig. 42b).162
Due to the efficiency of solid-state packing, incorporation of MIMs into crystalline coordination networks tends to inhibit relative motion of the components. This is something of a disappointment given that mechanical dynamism within ordered arrays is an oft touted reason for studying these materials in the first place.163 However, through careful ligand design, Schurko, Loeb and co-workers have managed to generate several MORFs in which movement of uncoordinated components within interstitial spaces can be realised. Using rigid axle components, rotaxane ligands within crystalline MOFs have been demonstrated to exhibit both pirouetting164 and shuttling165 motion of interlocked crown ether macrocycles in the solid state (Fig. 43).
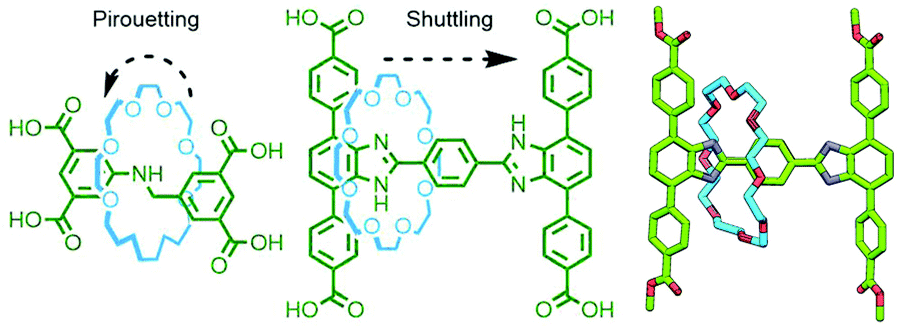 | ||
| Fig. 43 Schurko and Loeb's [2]rotaxane ligands that form MORFs in which the macrocycle components are able to undergo pirouetting (left) and shuttling (middle; SCXRD structure of methyl ester on right) motion in the solid state. | ||
4 Conclusions
The development of synthetic methods for accessing mechanical bonds has revolutionised the chemical space in which the scientific community can operate. Progress in this area has been exponential: from the early, painstaking, syntheses of MIMs in the 1960s – half a century after they were first proposed – it was two decades until Sauvage's seminal metal template-directed preparation of a [2]catenane was reported. Within another ten years the field had exploded, with various non-covalent templates described, multi-component MIMs and molecular switches realised, and the first examples of polyrotaxanes synthesised. Today a multitude of efficient synthetic strategies have been established, and increasingly attention is being paid to potential applications for these molecules in areas as diverse as drug166 and gene delivery,167 and as components in hydrogels168 and batteries.169In this review we have sought to outline the key synthetic strategies used to prepare higher order discrete MIMs, and their polymeric counterparts. Clearly there remain issues to be solved. For many systems controlling the polymer length, and even the nature of the mechanical connectivity (as in the elegant synthesis of polycatenanes reported by Rowan and co-workers151), can be extremely difficult. However, with the range of methods available it is fast becoming possible to design interlocked molecules with precision engineering of their properties. Given the previous rate of expansion of our knowledge and understanding of mechanically interlocked systems it is clear that sophisticated technologies and materials exploiting the mechanical bond are within our grasp.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by an Imperial College Research Fellowship (JEML) and a Royal Society Research Grant (RG170321). We thank Professor Matthew J. Fuchter for useful discussions and access to equipment and resources.References
- H. L. Frisch and E. Wasserman, J. Am. Chem. Soc., 1961, 83, 3789–3795 CrossRef CAS.
- (a) D. A. Leigh, Angew. Chem., Int. Ed., 2016, 55, 14506–14508 CrossRef CAS PubMed; (b) J. P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080–11093 CrossRef CAS PubMed; (c) J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed; (d) B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060–11078 CrossRef CAS PubMed.
- E. Wasserman, J. Am. Chem. Soc., 1960, 82, 4433–4434 CrossRef CAS.
- I. T. Harrison and S. Harrison, J. Am. Chem. Soc., 1967, 89, 5723–5724 CrossRef CAS.
- G. Schill and A. Lüttringhaus, Angew. Chem., Int. Ed. Engl., 1964, 3, 546–547 CrossRef.
- (a) C. O. Dietrich-Buchecker, J. P. Sauvage and J. P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095–5098 CrossRef CAS; (b) C. O. Dietrich-Buchecker, J. P. Sauvage and J. M. Kern, J. Am. Chem. Soc., 1984, 106, 3043–3045 CrossRef CAS. For selected reviews see: (c) J. C. Chambron, J. P. Collin, V. Heitz, D. Jouvenot, J. M. Kern, P. Mobian, D. Pomeranc and J. P. Sauvage, Eur. J. Org. Chem., 2004, 1627–1638 CrossRef CAS; (d) J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed; (e) J. E. M. Lewis, P. D. Beer, S. J. Loeb and S. M. Goldup, Chem. Soc. Rev., 2017, 46, 2577–2591 RSC; (f) A. Inthasot, S. T. Tung and S. H. Chiu, Acc. Chem. Res., 2018, 51, 1324–1337 CrossRef CAS PubMed.
- (a) P. R. Ashton, T. T. Goodnow, A. E. Kaifer, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vicent and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1989, 28, 1396–1399 CrossRef; (b) G. Barin, A. Coskun, M. M. G. Fouda and J. F. Stoddart, ChemPlusChem, 2012, 77, 159–185 CrossRef CAS.
- (a) F. Vögtle, S. Meier and R. Hoss, Angew. Chem., Int. Ed. Engl., 1992, 31, 1619–1622 CrossRef; (b) C. A. Hunter, J. Am. Chem. Soc., 1992, 114, 5303–5311 CrossRef CAS.
- (a) V. Aucagne, K. D. Hänni, D. A. Leigh, P. J. Lusby and D. B. Walker, J. Am. Chem. Soc., 2006, 128, 2186–2187 CrossRef CAS PubMed; (b) V. Aucagne, J. Berná, J. D. Crowley, S. M. Goldup, K. D. Hänni, D. A. Leigh, P. J. Lusby, V. E. Ronaldson, A. M. Z. Slawin, A. Viterisi and D. B. Walker, J. Am. Chem. Soc., 2007, 129, 11950–11963 CrossRef CAS PubMed.
- (a) J. D. Crowley, S. M. Goldup, A. L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC; (b) M. Denis and S. M. Goldup, Nat. Rev. Chem., 2017, 1, 61 CrossRef CAS.
- C. O. Dietrich-Buchecker and J. P. Sauvage, Angew. Chem., Int. Ed. Engl., 1989, 28, 189–192 CrossRef.
- V. Marcos, A. J. Stephens, J. Jaramillo-Garcia, A. L. Nussbaumer, S. L. Woltering, A. Valero, J. F. Lemonnier, I. J. Vitorica-Yrezabal and D. A. Leigh, Science, 2016, 352, 1555–1559 CrossRef CAS PubMed.
- J. J. Danon, A. Krüger, D. A. Leigh, J. Lemonnier, A. J. Stephens, I. J. Vitorica-Yrezabal and S. L. Woltering, Science, 2017, 355, 159–162 CrossRef CAS PubMed.
- K. S. Chichak, S. J. Cantrill, A. R. Pease, S. H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS PubMed.
- J. F. Nierengarten, C. O. Dietrich-Buchecker and J. P. Sauvage, J. Am. Chem. Soc., 1994, 116, 375–376 CrossRef CAS PubMed.
- D. A. Leigh, R. G. Pritchard and A. J. Stephens, Nat. Chem., 2014, 6, 978–982 CrossRef CAS PubMed.
- V. Serreli, C.-F. Lee, E. R. Kay and D. A. Leigh, Nature, 2007, 445, 523–527 CrossRef CAS PubMed.
- R. Barat, T. Legigan, I. Tranoy-Opalinski, B. Renoux, E. Péraudeau, J. Clarhaut, P. Poinot, A. E. Fernandes, V. Aucagne, D. A. Leigh and S. Papot, Chem. Sci., 2015, 6, 2608–2613 RSC.
- B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef CAS PubMed.
- A. W. Heard and S. M. Goldup, Chem, 2020, 6, 994–1006 CAS.
- J. Berná, D. A. Leigh, M. Lubomska, S. M. Mendoza, E. M. Pérez, P. Rudolf, G. Teobaldi and F. Zerbetto, Nat. Mater., 2005, 4, 704–710 CrossRef PubMed.
- (a) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed; (b) C. Pezzato, C. Cheng, J. F. Stoddart and R. D. Astumian, Chem. Soc. Rev., 2017, 46, 5491–5507 RSC; (c) L. Zhang, V. Marcos and D. A. Leigh, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9397–9404 CrossRef CAS PubMed; (d) A. W. Heard and S. M. Goldup, ACS Cent. Sci., 2020, 6, 117–128 CrossRef CAS PubMed.
- (a) Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS; (b) K. Ito, Polym. J., 2007, 39, 489–499 CrossRef CAS.
- (a) Z. Niu and H. W. Gibson, Chem. Rev., 2009, 109, 6024–6046 CrossRef CAS PubMed; (b) A. Harada, A. Hashidzume, H. Yamaguchi and Y. Takashima, Chem. Rev., 2009, 109, 5974–6023 CrossRef CAS PubMed; (c) R. Jürgen and M. Marcel, Chem. Soc. Rev., 2013, 42, 44–62 RSC; (d) H. Y. Au-Yeung, C. C. Yee, A. W. Hung Ng and K. Hu, Inorg. Chem., 2018, 57, 3475–3485 CrossRef CAS PubMed.
- (a) J. Araki and K. Ito, Soft Matter, 2007, 3, 1456–1473 RSC; (b) A. Hashidzume, H. Yamaguchi and A. Harada, Eur. J. Org. Chem., 2019, 3344–3357 CrossRef CAS.
- (a) M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS PubMed; (b) S. Mena-Hernando and E. M. Pérez, Chem. Soc. Rev., 2019, 48, 5016–5032 RSC; (c) T. Takata, ACS Cent. Sci., 2020, 6, 129–143 CrossRef CAS PubMed.
- M. E. Belowich, C. Valente, R. A. Smaldone, D. C. Friedman, J. Thiel, L. Cronin and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 5243–5261 CrossRef CAS PubMed.
- W. Jiang, H. D. F. Winkler and C. A. Schalley, J. Am. Chem. Soc., 2008, 130, 13852–13853 CrossRef CAS PubMed.
- X. Fu, Q. Zhang, S. J. Rao, D. H. Qu and H. Tian, Chem. Sci., 2016, 7, 1696–1701 RSC.
- Z. J. Zhang, H. Y. Zhang, H. Wang and Y. Liu, Angew. Chem., Int. Ed., 2011, 50, 10834–10838 CrossRef CAS PubMed.
- E. A. Neal and S. M. Goldup, Angew. Chem., Int. Ed., 2016, 55, 12488–12493 CrossRef CAS PubMed.
- B. L. Allwood, N. Spencer, H. Shahriari-Zavareh, J. F. Stoddart and D. J. Williams, J. Chem. Soc., Chem. Commun., 1987, 1064–1066 RSC.
- S. J. Rao, Q. Zhang, J. Mei, X. H. Ye, C. Gao, Q. C. Wang, D. H. Qu and H. Tian, Chem. Sci., 2017, 8, 6777–6783 RSC.
- A. Harada, Y. Takashima and M. Nakahata, Acc. Chem. Res., 2014, 47, 2128–2140 CrossRef CAS PubMed.
- Z. Han, Q. Zhou and Y. Li, J. Inclusion Phenom. Macrocyclic Chem., 2018, 92, 81–101 CrossRef CAS.
- W. L. Mock, T. A. Irra, J. P. Wepsiec and T. L. Manimaran, J. Org. Chem., 1983, 48, 3619–3620 CrossRef CAS.
- W. L. Mock, T. A. Irra, J. P. Wepsiec and M. Adhya, J. Org. Chem., 1989, 54, 5302–5308 CrossRef CAS.
- C. Ke, R. A. Smaldone, T. Kikuchi, H. Li, A. P. Davis and J. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 381–387 CrossRef CAS PubMed.
- K. Wang, C. C. Yee and H. Y. Au-Yeung, Chem. Sci., 2016, 7, 2787–2792 RSC.
- (a) T. Ogoshi, S. Kanai, S. Fujinami, T. A. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed; (b) T. Ogoshi, T. A. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed.
- C. Ke, N. L. Strutt, H. Li, X. Hou, K. J. Hartlieb, P. R. McGonigal, Z. Ma, J. Iehl, C. L. Stern, C. Cheng, Z. Zhu, N. A. Vermeulen, T. J. Meade, Y. Y. Botros and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 17019–17030 CrossRef CAS PubMed.
- (a) M. P. L. Werts, M. Van Den Boogaard, G. Hadziioannou and G. M. Tsivgoulis, Chem. Commun., 1999, 623–624 RSC; (b) M. P. L. Werts, M. Van Den Boogaard, G. M. Tsivgoulis and G. Hadziioannou, Macromolecules, 2003, 36, 7004–7013 CrossRef CAS.
- D. B. Amabilino, P. R. Ashton, A. S. Reder, N. Spencer and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1994, 33, 433–437 CrossRef.
- (a) D. B. Amabilino, P. R. Ashton, S. E. Boyd, J. Y. Lee, S. Menzer, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1997, 36, 2070–2072 CrossRef CAS; (b) D. B. Amabilino, P. R. Ashton, V. Balzani, S. E. Boyd, A. Credi, J. Y. Lee, S. Menzer, J. F. Stoddart, M. Venturi and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 4295–4307 CrossRef CAS.
- A. M. Fuller, D. A. Leigh, P. J. Lusby, I. D. H. Oswald, S. Parsons and D. B. Walker, Angew. Chem., Int. Ed., 2004, 43, 3914–3918 CrossRef CAS PubMed.
- A. M. L. Fuller, D. A. Leigh and P. J. Lusby, Angew. Chem., Int. Ed., 2007, 46, 5015–5019 CrossRef CAS PubMed.
- A. M. L. Fuller, D. A. Leigh and P. J. Lusby, J. Am. Chem. Soc., 2010, 132, 4954–4959 CrossRef CAS PubMed.
- A. Trabolsi, N. Khashab, A. C. Fahrenbach, D. C. Friedman, M. T. Colvin, K. K. Cotí, D. Benítez, E. Tkatchouk, J. C. Olsen, M. E. Belowich, R. Carmielli, H. A. Khatib, W. A. Goddard, M. R. Wasielewski and J. F. Stoddart, Nat. Chem., 2010, 2, 42–49 CrossRef CAS PubMed.
- (a) C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS PubMed; (b) C. Pezzato, M. T. Nguyen, C. Cheng, D. J. Kim, M. T. Otley and J. F. Stoddart, Tetrahedron, 2017, 73, 4849–4857 CrossRef CAS.
- Y. Qiu, B. Song, C. Pezzato, D. Shen, W. Liu, L. Zhang, Y. Feng, Q. H. Guo, K. Cai, W. Li, H. Chen, M. T. Nguyen, Y. Shi, C. Cheng, R. D. Astumian, X. Li and J. F. Stoddart, Science, 2020, 368, 1247–1253 CrossRef CAS PubMed.
- H. Lahlali, K. Jobe, M. Watkinson and S. M. Goldup, Angew. Chem., Int. Ed., 2011, 50, 4151–4155 CrossRef CAS PubMed.
- J. E. M. Lewis, J. Winn, L. Cera and S. M. Goldup, J. Am. Chem. Soc., 2016, 138, 16329–16336 CrossRef CAS PubMed.
- C. O. Dietrich-Buchecker, A. Khemiss and J. P. Sauvage, J. Chem. Soc., Chem. Commun., 1986, 1376–1378 RSC.
- F. Bitsch, C. O. Dietrich-Buchecker, A. K. Khémiss, J. P. Sauvage and A. Van Dorsselaer, J. Am. Chem. Soc., 1991, 113, 4023–4025 CrossRef CAS.
- This template motif had previously been shown effective for cyclotrimerisation of the non-interlocked axle component, giving a hexaporphyrin nanoring: M. Hoffmann, J. Kärnbratt, M. H. Chang, L. M. Herz, B. Albinsson and H. L. Anderson, Angew. Chem., Int. Ed., 2008, 47, 4993–4996 CrossRef CAS PubMed.
- M. J. Langton, J. D. Matichak, A. L. Thompson and H. L. Anderson, Chem. Sci., 2011, 2, 1897–1901 RSC.
- Y. Shen, Q. Zhou, L. Zou and Q. Wang, ChemistrySelect, 2017, 2, 11977–11980 CrossRef CAS.
- (a) M. Fujita, Chem. Soc. Rev., 1998, 27, 417 RSC; (b) S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853–907 CrossRef CAS PubMed; (c) B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022–2043 CrossRef CAS; (d) S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825–841 CrossRef CAS; (e) M. D. Ward, Chem. Commun., 2009, 4487–4499 RSC; (f) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed; (g) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC; (h) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed; (i) R. A. S. Vasdev, D. Preston and J. D. Crowley, Dalton Trans., 2017, 46, 2402–2414 RSC; (j) J. E. M. Lewis and J. D. Crowley, ChemPlusChem, 2020, 85, 815–827 CrossRef CAS PubMed; (k) W.-X. Gao, H.-J. Feng, B.-B. Guo, Y. Lu and G.-X. Jin, Chem. Rev., 2020, 120, 6288–6325 CrossRef PubMed.
- J. E. M. Lewis, M. Galli and S. M. Goldup, Chem. Commun., 2017, 53, 298–312 RSC.
- D. Whang, K. M. Park, J. Heo, P. Ashton and K. Kim, J. Am. Chem. Soc., 1998, 120, 4899–4900 CrossRef CAS.
- K. M. Park, S. Y. Kim, J. Heo, D. Whang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2002, 124, 2140–2147 CrossRef CAS PubMed.
- S.-G. Roh, K.-M. Park, G.-J. Park, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 1999, 38, 637–641 CrossRef PubMed.
- (a) S. Li, J. Huang, F. Zhou, T. R. Cook, X. Yan, Y. Ye, B. Zhu, B. Zheng and P. J. Stang, J. Am. Chem. Soc., 2014, 136, 5908–5911 CrossRef CAS PubMed; (b) Y. Ye, S. P. Wang, B. Zhu, T. R. Cook, J. Wu, S. Li and P. J. Stang, Org. Lett., 2015, 17, 2804–2807 CrossRef CAS PubMed.
- S. P. Black, A. R. Stefankiewicz, M. M. J. Smulders, D. Sattler, C. A. Schalley, J. R. Nitschke and J. K. M. Sanders, Angew. Chem., Int. Ed., 2013, 52, 5749–5752 CrossRef CAS PubMed.
- S. P. Black, D. M. Wood, F. B. Schwarz, T. K. Ronson, J. J. Holstein, A. R. Stefankiewicz, C. A. Schalley, J. K. M. Sanders and J. R. Nitschke, Chem. Sci., 2016, 7, 2614–2620 RSC.
- K. K. G. Wong, N. Hoyas Pérez, A. J. P. White and J. E. M. Lewis, Chem. Commun., 2020 10.1039/d0cc04780e.
- E. Viljoen, K. Zhu and S. J. Loeb, Chem. – Eur. J., 2016, 22, 7479–7484 CrossRef CAS PubMed.
- H. Ogino, J. Am. Chem. Soc., 1981, 103, 1303–1304 CrossRef CAS.
- W. Wang, L. J. Chen, X. Q. Wang, B. Sun, X. Li, Y. Zhang, J. Shi, Y. Yu, L. Zhang, M. Liu and H. B. Yang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5597–5601 CrossRef CAS PubMed.
- Y. X. Wang, Q. F. Zhou, L. J. Chen, L. Xu, C. H. Wang, X. Li and H. B. Yang, Chem. Commun., 2018, 54, 2224–2227 RSC.
- (a) X. Q. Wang, W. J. Li, W. Wang, J. Wen, Y. Zhang, H. Tan and H. B. Yang, J. Am. Chem. Soc., 2019, 141, 13923–13930 CrossRef CAS PubMed; (b) W.-J. Li, W. Wang, X.-Q. Wang, M. Li, Y. Ke, R. Yao, J. Wen, G.-Q. Yin, B. Jiang, X. Li, P. Yin and H.-B. Yang, J. Am. Chem. Soc., 2020, 142, 8473–8482 CrossRef CAS PubMed.
- M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vicent and D. J. Williams, J. Chem. Soc., Chem. Commun., 1991, 630–634 RSC.
- (a) T. Higashi, K. Motoyama and H. Arima, J. Drug Delivery Sci. Technol., 2013, 23, 523–529 CrossRef CAS; (b) A. Tamura and N. Yui, Polym. J., 2017, 49, 527–534 CrossRef CAS; (c) Y. Arisaka and N. Yui, J. Mater. Chem. B, 2019, 7, 2123–2129 RSC.
- Y. Suzaki, S. Murata and K. Osakada, Chem. Lett., 2009, 38, 356–357 CrossRef CAS.
- F. Ishiwari, K. Nakazono, Y. Koyama and T. Takata, Chem. Commun., 2011, 47, 11739–11741 RSC.
- T. J. Kidd, T. J. A. Loontjens, D. A. Leigh and J. K. Y. Wong, Angew. Chem., Int. Ed., 2003, 42, 3379–3383 CrossRef CAS PubMed.
- T. Ikeda, M. Higuchi and D. G. Kurth, J. Am. Chem. Soc., 2009, 131, 9158–9159 CrossRef CAS PubMed.
- T. Ikeda and M. Higuchi, Langmuir, 2011, 27, 4184–4189 CrossRef CAS PubMed.
- T. Sato and T. Takata, Macromolecules, 2008, 41, 2739–2742 CrossRef CAS.
- A. Belaissaoui, S. Shimada, A. Ohishi and N. Tamaoki, Tetrahedron Lett., 2003, 44, 2307–2310 CrossRef CAS.
- P. Wang, Z. Gao, M. Yuan, J. Zhu and F. Wang, Polym. Chem., 2016, 7, 3664–3668 RSC.
- H. Sasabe, N. Inomoto, N. Kihara, Y. Suzuki, A. Ogawa and T. Takata, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4154–4160 CrossRef CAS.
- E. N. Guidry, J. Li, J. F. Stoddart and R. H. Grubbs, J. Am. Chem. Soc., 2007, 129, 8944–8945 CrossRef CAS PubMed.
- (a) P. G. Clark, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 13631–13633 CrossRef CAS PubMed; (b) L. Fang, M. Hmadeh, J. Wu, M. A. Olson, J. M. Spruell, A. Trabolsi, Y. W. Yang, M. Elhablrl, A. M. Albrecht-Gary and J. F. Stoddart, J. Am. Chem. Soc., 2009, 131, 7126–7134 CrossRef CAS PubMed.
- C. Hamers, F. M. Raymo and J. F. Stoddart, Eur. J. Org. Chem., 1998, 2109–2117 CrossRef CAS.
- D. L. Simone and T. M. Swager, J. Am. Chem. Soc., 2000, 122, 9300–9301 CrossRef CAS.
- Y. Geerts, D. Muscat and K. Müllen, Macromol. Chem. Phys., 1995, 196, 3425–3435 CrossRef CAS.
- (a) J. L. Weidmann, J. M. Kern, J. P. Sauvage, Y. Geerts, D. Muscat and K. Müllen, Chem. Commun., 1996, 1243–1244 RSC; (b) J. Weidmann, J. Kern, J. Sauvage, D. Muscat, S. Mullins, W. Köhler, C. Rosenauer, H. J. Räder, K. Martin and Y. Geerts, Chem. – Eur. J., 1999, 5, 1841–1851 CrossRef CAS.
- T. Hagiwara, Y. Murano, Y. Watanabe, T. Hoshi and T. Sawaguchi, Tetrahedron Lett., 2012, 53, 2805–2808 CrossRef CAS.
- T. Hagiwara, M. Yamazaki, T. Suzuki, T. Sawaguchi and S. Yano, Polymer, 2011, 52, 5426–5430 CrossRef CAS.
- (a) C. A. Fustin, C. Bailly, G. J. Clarkson, P. De Groote, T. H. Galow, D. A. Leigh, D. Robertson, A. M. Z. Slawin and J. K. Y. Wong, J. Am. Chem. Soc., 2003, 125, 2200–2207 CrossRef CAS PubMed; (b) C. A. Fustin, C. Bailly, G. J. Clarkson, T. H. Galow and D. A. Leigh, Macromolecules, 2004, 37, 66–70 CrossRef CAS.
- C. A. Fustin, G. J. Clarkson, D. A. Leigh, F. Van Hoof, A. M. Jonas and C. Bailly, Macromolecules, 2004, 37, 7884–7892 CrossRef CAS.
- N. Watanabe, Y. Ikari, N. Kihara and T. Takata, Macromolecules, 2004, 37, 6663–6666 CrossRef CAS.
- T. Sato and T. Takata, Polym. J., 2009, 41, 470–476 CrossRef CAS.
- Y. G. Lee, Y. Koyama, M. Yonekawa and T. Takata, Macromolecules, 2010, 43, 4070–4080 CrossRef CAS.
- M. Okada and A. Harada, Macromolecules, 2003, 36, 9701–9703 CrossRef CAS.
- P. Wei, X. Yan and F. Huang, Chem. Commun., 2014, 50, 14105–14108 RSC.
- P. R. Lecavalier, P. T. Engen, Y. X. Shen, S. Joardar, T. Ward and H. W. Gibson, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1989, 30, 189–190 CAS.
- C. Wu, M. C. Bheda, C. Lim, Y. X. Shen, J. Sze and H. W. Gibson, Polym. Commun., 1991, 32, 204–207 CAS.
- B. L. Allwood, N. Spencer, H. Shahriari-Zavareh, J. F. Stoddart and D. J. Williams, J. Chem. Soc., Chem. Commun., 1987, 1064–1066 RSC.
- (a) P. R. Ashtona, E. J. T. Chrystal, J. P. Mathias, K. P. Parry, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, Tetrahedron Lett., 1987, 28, 6367–6370 CrossRef; (b) J.-Y. Ortholand, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1989, 28, 1394–1395 CrossRef.
- P. R. Ashton, T. T. Goodnow, A. E. Kaifer, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vicent and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1989, 28, 1396–1399 CrossRef.
- (a) P. L. Anelli, N. Spencer and J. F. Stoddart, J. Am. Chem. Soc., 1991, 113, 5131–5133 CrossRef CAS PubMed; (b) P. R. Ashton, M. Grognuz, A. M. Z. Slawin, J. F. Stoddart and D. J. Williams, Tetrahedron Lett., 1991, 32, 6235–6238 CrossRef CAS.
- Y. X. Shen, C. Lim and H. W. Gibson, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1991, 32, 166–167 CAS.
- P. R. Ashton, P. J. Campbell, P. T. Glink, D. Philp, N. Spencer, J. F. Stoddart, E. J. T. Chrystal, S. Menzer, D. J. Williams and P. A. Tasker, Angew. Chem., Int. Ed. Engl., 1995, 34, 1865–1869 CrossRef CAS.
- S. J. Loeb, J. Tiburcio, S. J. Vella and J. A. Wisner, Org. Biomol. Chem., 2006, 4, 667–680 RSC.
- D. Castillo, P. Astudillo, J. Mares, F. J. González, A. Vela and J. Tiburcio, Org. Biomol. Chem., 2007, 5, 2252–2256 RSC.
- N. Momčilović, P. G. Clark, A. J. Boydston and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 19087–19089 CrossRef PubMed.
- S. Li, G. H. Weng, W. Lin, Z. B. Sun, M. Zhou, B. Zhu, Y. Ye and J. Wu, Polym. Chem., 2014, 5, 3994–4001 RSC.
- F. Zeng, Z. Meng, Y. Han and C. F. Chen, Chem. Commun., 2014, 50, 7611–7613 RSC.
- K. Yamabuki, Y. Isobe, K. Onimura and T. Oishi, Chem. Lett., 2007, 36, 1196–1197 CrossRef CAS.
- M. Zhang, S. Li, S. Dong, J. Chen, B. Zheng and F. Huang, Macromolecules, 2011, 44, 9629–9634 CrossRef CAS.
- T. Takata, H. Kawasaki, N. Kihara and Y. Furusho, Macromolecules, 2001, 34, 5449–5456 CrossRef CAS.
- I. Yamaguchi, K. Osakada and T. Yamamoto, J. Am. Chem. Soc., 1996, 118, 1811–1812 CrossRef CAS.
- O. Noll and H. Ritter, Macromol. Chem. Phys., 1998, 199, 791–794 CrossRef CAS.
- C. I. Simionescu, M. Grigoras, A. Farcas and A. Stoleru, Macromol. Chem. Phys., 1998, 199, 1301–1306 CrossRef CAS.
- A. Farcas and M. Grigoras, High Perform. Polym., 2001, 13, 149–158 CrossRef CAS.
- P. N. Taylor, M. J. O'Connell, L. A. McNeill, M. J. Hall, R. T. Aplin and H. L. Anderson, Angew. Chem., Int. Ed., 2000, 39, 3456–3460 CrossRef CAS PubMed.
- M. Van Den Boogaard, G. Bonnet, P. Van't Hof, Y. Wang, C. Brochon, P. Van Hutten, A. Lapp and G. Hadziioannou, Chem. Mater., 2004, 16, 4383–4385 CrossRef CAS.
- K. I. Shinohara, T. Suzuki, T. Kitami and S. Yamaguchi, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 801–809 CrossRef CAS.
- W. L. Mock and N. Y. Shih, J. Org. Chem., 1983, 48, 3618–3619 CrossRef CAS.
- C. Meschke, H.-J. Buschmann and E. Schollmeyer, Macromol. Rapid Commun., 1998, 19, 59–63 CrossRef CAS.
- C. Meschke, H. J. Buschmann and E. Schollmeyer, Polymer, 1999, 40, 945–949 CrossRef CAS.
- (a) A. Farcas, P. H. Aubert, J. Mohanty, A. I. Lazar, S. Cantin and W. M. Nau, Eur. Polym. J., 2015, 62, 124–129 CrossRef CAS; (b) M. Idris, M. Bazzar, B. Guzelturk, H. V. Demir and D. Tuncel, RSC Adv., 2016, 6, 98109–98116 RSC.
- D. Tuncel and J. H. G. Steinke, Chem. Commun., 1999, 1509–1510 RSC.
- (a) P. L. Vidal, M. Billon, B. Divisia-Blohorn, G. Bidan, P. L. Vidal, B. Divisia-Blohorn, J. M. Kern and J. P. Sauvage, Chem. Commun., 1998, 629–630 RSC; (b) P. L. Vidal, B. Divisia-Blohorn, G. Bidan, J. M. Kern, J. P. Sauvage and J. L. Hazemann, Inorg. Chem., 1999, 38, 4203–4210 CrossRef CAS; (c) J. P. Sauvage, J. M. Kern, G. Bidan, B. Divisia-Blohorn and P. L. Vidal, New J. Chem., 2002, 26, 1287–1290 RSC.
- (a) S. S. Zhu, P. J. Carroll and T. M. Swager, J. Am. Chem. Soc., 1996, 118, 8713–8714 CrossRef CAS; (b) S. S. Zhu and T. M. Swager, J. Am. Chem. Soc., 1997, 119, 12568–12577 CrossRef CAS.
- M. Ogawa, A. Kawasaki, Y. Koyama and T. Takata, Polym. J., 2011, 43, 909–915 CrossRef CAS.
- M. Ogawa, H. Sogawa, Y. Koyama and T. Takata, Polym. J., 2015, 47, 580–584 CrossRef CAS.
- (a) P. D. Beer, M. R. Sambrook and D. Curiel, Chem. Commun., 2006, 2105–2117 RSC; (b) M. S. Vickers and P. D. Beer, Chem. Soc. Rev., 2007, 36, 211–225 RSC; (c) G. T. Spence and P. D. Beer, Acc. Chem. Res., 2013, 46, 571–586 CrossRef CAS PubMed.
- J. A. Wisner, P. D. Beer and M. G. B. Drew, Angew. Chem., Int. Ed., 2001, 40, 3606–3609 CrossRef CAS PubMed.
- (a) J. A. Wisner, P. D. Beer, M. G. B. Drew and M. R. Sambrook, J. Am. Chem. Soc., 2002, 124, 12469–12476 CrossRef CAS PubMed; (b) M. R. Sambrook, P. D. Beer, J. A. Wisner, R. L. Paul and A. R. Cowley, J. Am. Chem. Soc., 2004, 126, 15364–15365 CrossRef CAS PubMed.
- C. J. Serpell, R. Chall, A. L. Thompson and P. D. Beer, Dalton Trans., 2011, 40, 12052 RSC.
- D. Pasini and D. Takeuchi, Chem. Rev., 2018, 118, 8983–9057 CrossRef CAS PubMed.
- T. Isono, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3184–3192 CrossRef CAS.
- M. Bria, J. Bigot, G. Cooke, J. Lyskawa, G. Rabani, V. M. Rotello and P. Woisel, Tetrahedron, 2009, 65, 400–407 CrossRef CAS.
- M. A. Olson, A. B. Braunschweig, L. Fang, T. Ikeda, R. Klajn, A. Trabolsi, P. J. Wesson, D. Benítez, C. A. Mirkin, B. A. Grzybowski and J. F. Stoddart, Angew. Chem., Int. Ed., 2009, 48, 1792–1797 CrossRef CAS PubMed.
- A. Harada and M. Kamachi, Macromolecules, 1990, 23, 2821–2823 CrossRef CAS.
- A. Harada, J. Li and M. Kamachi, Nature, 1992, 356, 325–327 CrossRef CAS.
- A. Harada and M. Kamachi, J. Chem. Soc., Chem. Commun., 1990, 1322–1323 RSC.
- H. Fujita, T. Ooya, M. Kurisawa, H. Mori, M. Terano and N. Yui, Macromol. Rapid Commun., 1996, 17, 509–515 CrossRef CAS.
- K. Kato, H. Komatsu and K. Ito, Macromolecules, 2010, 43, 8799–8804 CrossRef CAS.
- (a) W. Zhang, W. R. Dichtel, A. Z. Stieg, D. Benítez, J. K. Gimzewski, J. R. Heath and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6514–6519 CrossRef CAS PubMed; (b) Z. Zhu, C. J. Bruns, H. Li, J. Lei, C. Ke, Z. Liu, S. Shafaie, H. M. Colquhoun and J. F. Stoddart, Chem. Sci., 2013, 4, 1470–1483 RSC.
- G. Wenz and B. Keller, Angew. Chem., Int. Ed. Engl., 1992, 31, 197–199 CrossRef.
- A. M. Resmerita, K. I. Assaf, A. I. Lazar, W. M. Nau and A. Farcas, Eur. Polym. J., 2017, 93, 323–333 CrossRef CAS.
- T. Ogoshi, Y. Nishida, T. A. Yamagishi and Y. Nakamoto, Macromolecules, 2010, 43, 7068–7072 CrossRef CAS.
- K. Nakazono, T. Ishino, T. Takashima, D. Saeki, D. Natsui, N. Kihara and T. Takata, Chem. Commun., 2014, 50, 15341–15344 RSC.
- Y. Kohsaka, G. I. Konishi and T. Takata, Polym. J., 2007, 39, 861–873 CrossRef CAS.
- T. Higashi, K. Morita, X. Song, J. Zhu, A. Tamura, N. Yui, K. Motoyama, H. Arima and J. Li, Commun. Chem., 2019, 2, 78 CrossRef.
- P. G. Clark, E. N. Guidry, W. Y. Chan, W. E. Steinmetz and R. H. Grubbs, J. Am. Chem. Soc., 2010, 132, 3405–3412 CrossRef CAS PubMed.
- Q. Wu, P. M. Rauscher, X. Lang, R. J. Wojtecki, J. J. De Pablo, M. J. A. Hore and S. J. Rowan, Science, 2017, 358, 1434–1439 CrossRef CAS PubMed.
- (a) K. Kim, Chem. Soc. Rev., 2002, 31, 96–107 RSC; (b) S. J. Loeb, Chem. Soc. Rev., 2007, 36, 226–235 RSC; (c) V. N. Vukotic and S. J. Loeb, Chem. Soc. Rev., 2012, 41, 5896–5906 RSC.
- D. Whang, Y. M. Jeon, J. Heo and K. Kim, J. Am. Chem. Soc., 1996, 118, 11333–11334 CrossRef CAS.
- D. Whang and K. Kim, J. Am. Chem. Soc., 1997, 119, 451–452 CrossRef CAS.
- S. J. Loeb, Chem. Commun., 2005, 1511–1518 RSC.
- E. Lee, J. Heo and K. Kim, Angew. Chem., Int. Ed., 2000, 39, 2699–2701 CrossRef CAS PubMed.
- (a) G. J. E. Davidson and S. J. Loeb, Angew. Chem., Int. Ed., 2003, 42, 74–77 CrossRef CAS PubMed; (b) D. J. Hoffart and S. J. Loeb, Angew. Chem., Int. Ed., 2005, 44, 901–904 CrossRef CAS PubMed.
- D. J. Mercer, V. N. Vukotic and S. J. Loeb, Chem. Commun., 2011, 47, 896–898 RSC.
- N. C. Frank, D. J. Mercer and S. J. Loeb, Chem. – Eur. J., 2013, 19, 14076–14080 CrossRef CAS PubMed; A. Noor, S. C. Moratti and J. D. Crowley, Chem. Sci., 2014, 5, 4283–4290 RSC; G. Gholami, K. Zhu, J. S. Ward, P. E. Kruger and S. J. Loeb, Eur. J. Inorg. Chem., 2016, 4524–4529 CrossRef; G. Gholami, G. Baggi, K. Zhu and S. J. Loeb, Dalton Trans., 2017, 46, 2462–2470 RSC.
- J. E. M. Lewis, Org. Biomol. Chem., 2019, 17, 2442–2447 RSC.
- C. Hamers, O. Kocian, F. M. Raymo and J. F. Stoddart, Adv. Mater., 1998, 10, 1366–1369 CrossRef CAS.
- (a) Q. Li, W. Zhang, O. Š. Miljanić, C. B. Knobler, J. F. Stoddart and O. M. Yaghi, Chem. Commun., 2010, 46, 380–382 RSC; (b) Q. Li, C. H. Sue, S. Basu, A. K. Shveyd, W. Zhang, G. Barin, L. Fang, A. A. Sarjeant, J. F. Stoddart and O. M. Yaghi, Angew. Chem., Int. Ed., 2010, 49, 6751–6755 CrossRef CAS PubMed.
- H. Deng, M. A. Olson, J. F. Stoddart and O. M. Yaghi, Nat. Chem., 2010, 2, 439–443 CrossRef CAS PubMed.
- (a) V. N. Vukotic, K. J. Harris, K. Zhu, R. W. Schurko and S. J. Loeb, Nat. Chem., 2012, 4, 456–460 CrossRef CAS PubMed; (b) K. Zhu, V. N. Vukotic, C. A. Okeefe, R. W. Schurko and S. J. Loeb, J. Am. Chem. Soc., 2014, 136, 7403–7409 CrossRef CAS PubMed; (c) V. N. Vukotic, C. A. O'Keefe, K. Zhu, K. J. Harris, C. To, R. W. Schurko and S. J. Loeb, J. Am. Chem. Soc., 2015, 137, 9643–9651 CrossRef CAS PubMed.
- K. Zhu, C. A. O'Keefe, V. N. Vukotic, R. W. Schurko and S. J. Loeb, Nat. Chem., 2015, 7, 514–519 CrossRef CAS PubMed.
- C. Moon, Y. M. Kwon, W. K. Lee, Y. J. Park and V. C. Yang, J. Controlled Release, 2007, 124, 43–50 CrossRef CAS PubMed.
- T. Ooya, H. S. Choi, A. Yamashita, N. Yui, Y. Sugaya, A. Kano, A. Maruyama, H. Akita, R. Ito, K. Kogure and H. Harashima, J. Am. Chem. Soc., 2006, 128, 3852–3853 CrossRef CAS PubMed.
- A. Bin Imran, K. Esaki, H. Gotoh, T. Seki, K. Ito, Y. Sakai and Y. Takeoka, Nat. Commun., 2014, 5, 5124 CrossRef CAS PubMed.
- S. Choi, T. W. Kwon, A. Coskun and J. W. Choi, Science, 2017, 357, 279–283 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |