
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of orthogonally protected and functionalized bacillosamines†
Jeanine
van Mechelen
,
Jim
Voorneveld
,
Hermen S.
Overkleeft
,
Dmitri V.
Filippov
 ,
Gijsbert A.
van der Marel
* and
Jeroen D. C.
Codée
*
,
Gijsbert A.
van der Marel
* and
Jeroen D. C.
Codée
*
Bioorganic synthesis group, Leiden University, Einsteinweg 55, 2333 CC Leiden, Leiden, The Netherlands. E-mail: jcodee@chem.leidenuniv.nl
First published on 25th March 2020
Abstract
2,4-Diamino-2,4,6-trideoxyglucose (bacillosamine) is a monosaccharide found in many pathogenic bacteria, variation in the functionalities appended to the amino groups occurs depending on the species the sugar is derived from. We here report the first synthesis of bacillosamine synthons that allow for the incorporation of two different functionalities at the C-2-N-acetyl and C-4-amines. We have developed chemistry to assemble a set of conjugation ready Neisseria meningitidis C-2-N-acetyl bacillosamine saccharides, carrying either an acetyl or (R)- or (S)-glyceroyl at the C-4 amine. The glyceroyl bacillosamines have been further extended at the C-3-OH with an α-D-galactopyranose to provide structures that occur as post-translational modifications of N. meningitidis PilE proteins, which make up the bacterial pili.
Introduction
Bacillosamine (1, Bac, Fig. 1) is a characteristic bacterial monosaccharide that is present in various bacterial capsular polysaccharides and glycopeptides.1 It is a 2,4-diamino-2,4,6-trideoxyhexose (DATDH), that is most commonly functionalized with N-acetyl groups at C-2 and C-4 (2,4-di-NAcBac, 2). This residue is found at the reducing end of oligosaccharide chains of both O- and N-linked glycoproteins of Campylobacter jejuni2 and Neisseria gonorrhoeae.3 Recently, a differently modified bacillosamine has been discovered in Neisseria meningitidis,4 where it is decorated, besides a single acetamide, with an N-glyceroyl group, thought to reside at the C4-amine (3, Fig. 1).5 This Bac-residue represents one of various post-translational modifications (PTMs) that can occur on N. meningitides pilE proteins, the constituent protein monomers of the type IV pili. It can be further decorated with different mono- or disaccharides (see Fig. 1). The role of these PTMs is currently not known but it is clear that the type IV pili play an important role in the pathogenicity of the bacterium. Also, the exact structure of the Bac-residue present on the N. meningitides pili remains elusive as the stereochemistry of the glyceroyl group has not been established.5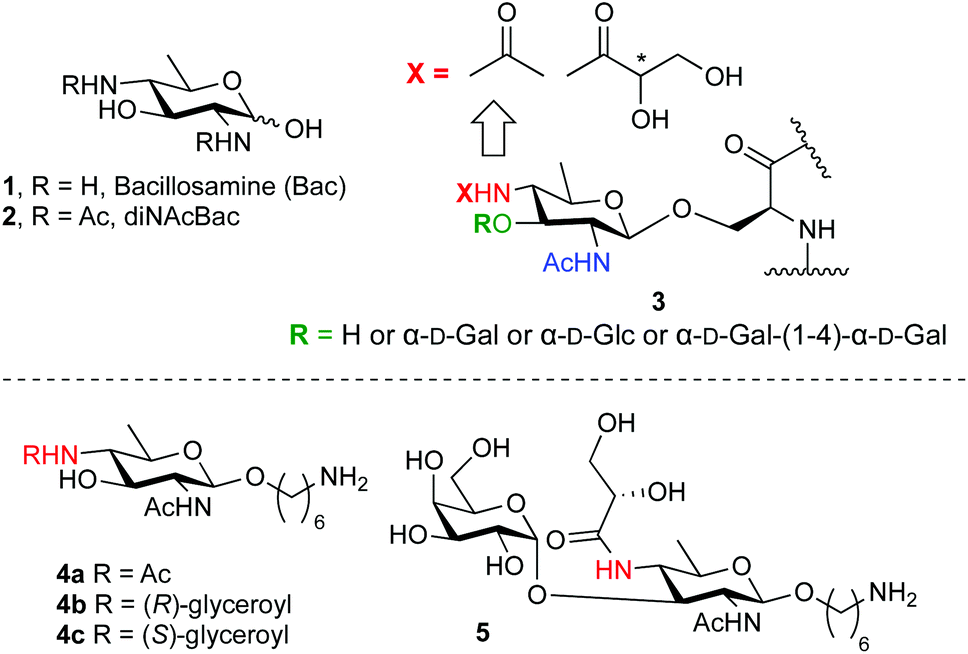 | ||
| Fig. 1 Bacillosamine (1), di-N-acetylbacillosamine (2), bacillosamine PTMs found in N. meningitidis pili (3) and the synthetic targets of the current study (4 and 5). | ||
To aid in the structure elucidation of the functionalized Bac residues in the N. meningitidis pili, its detection and the role of these differentially decorated bacillosamines in virulence, we reasoned that the availability of synthetic Bac-residues would provide powerful tools. We therefore set out to develop an effective route of synthesis for an orthogonally protected bacillosamine synthon, of which all functional groups can be site selectively addressed. We here report the first synthesis of such an orthogonally protected bacillosamine building block and we show the synthesis of spacer-equipped bacillosamines bearing different acyl functionalities (4b–4c, Fig. 1) and the extension of these towards a relevant conjugation-ready disaccharide (5, Fig. 1).
Results and discussion
Different syntheses of bacillosamine6 and bacillosamine containing oligosaccharides7 and glycopeptides8 have been reported, but none of these allow for the discrimination of the C-2 and C-4 amino groups and therefore the development of a new synthetic route was required. We thus set out to target key synthon 7, bearing two orthogonally protected amino groups at C-2 and C-4, as well as a 2-napthylmethyl ether that masks the C-3-OH and an anomeric selenophenyl group for glycosylation purposes (see Scheme 1). Retrosynthetically, this building block can be derived from a suitably protected 6-deoxy galactosamine derivative 8, by a substitution reaction of the C-4-O-mesylate. Selenophenyl galactosamine 8 can be readily accessed from D-fucal through an azidoselenation event9 and subsequent transformation of the azide into the protected C-2-aminosugar.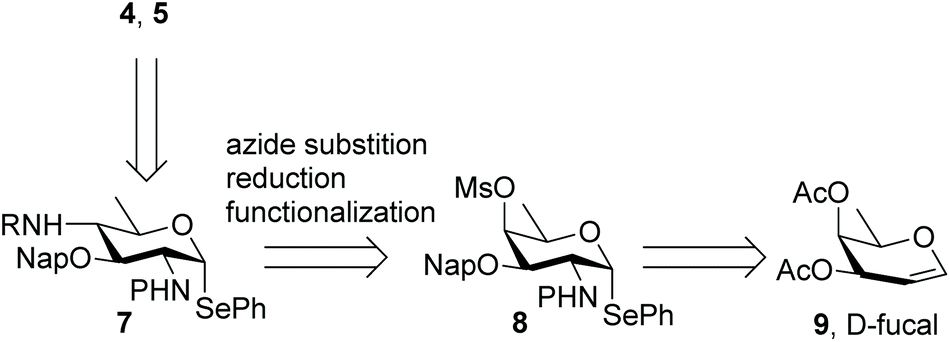 | ||
| Scheme 1 Retrosynthetic analysis for the assembly of target compounds 4 and 5. | ||
Accordingly, 3,4-di-O-acetyl-D-fucal 910 was subjected to azidoselenation conditions to provide azidofucose 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 (see Scheme 2). Saponification of the two acetates in 10 was then followed by regioselective installation of the C-3-naphthylmethyl ether and subsequent mesylation of the C-4-OH. Because we aimed to use azide as nucleophile for the introduction of the C-4-amine functionality, we had to transform the C-2-azide in 11 to achieve orthogonality. Although amines are commonly avoided during manipulations on carbohydrate building blocks, we reasoned that inorganic azide anion should be sufficiently nucleophilic to outcompete the C-2 amine in mesylate displacement. We therefore decided to reduce the azido group in 11 to the corresponding amine and invert the C-4-O-mesylate in the resulting fucosamine, without masking the C-2-amine. Thus, fucosazide 11 was exposed to 1,3-propanedithiol to deliver fucosamine 12, which was treated with NaN3 and TBABr in DMF at elevated temperature to provide the desired bacillosamine 13, bearing a fully orthogonal functional group pattern, in 81%. Next, the C-2-amine was masked with a trichloroacetyl (TCA) group to provide fully protected bacillosamine 14. Reduction of the azide with 1,3-propanedithiol then set the stage for the differentiation of the C-4-amino group into either an acetamide (towards 4a) or the two glyceroyl substituted bacillosamines 4b and 4c. During the reduction of the azide in 14, the TCA moiety was partially reduced as well to the corresponding C-2-dichloroacetamide (DCA), yielding compound 15. As the DCA group can be removed under similar conditions as the TCA, the synthetic strategy did not need any adjustment and we continued with 4-N-acylation. Trichloroacetylation provided bacillosamine synthon 16a while reaction of the C-4-amine with either R- or S-isopropylidene-glyceric acid under the agency of HCTU, provided bacillosamines 16b and 16c. Next, the three building blocks were equipped with an azidohexanol spacer, in glycosylation reactions promoted by N-iodosuccinimide (NIS) and trimethylsilyl trifluoromethanesulfonate (TMSOTf) to provide 17a, 17b and 17c. The products were obtained as single diastereoisomers and no oxazoline side products were detected, indicating that the dichloroacetyl groups provided effective anchimeric assistance.12 With the fully protected spacer-equipped bacillosamines in hand, deprotection of the intermediates was undertaken. We first tried to unmask the TCA and DCA groups in 17a using Cs2CO3 in DMF,13 but this did not lead to the desired C-2,4-diamine. Instead bicyclic urea 18 was formed.14 We therefore switched to a hydrogenation reaction, aiming to unmask the spacer azide, C-2-N-DCA, C-3-O-Nap and C-4-N-TCA groups in one step. After the first hydrogenation event using Pearlman's catalyst, LC-MS analysis of the crude reaction mixture revealed the presence of three chlorides and therefore the mixture was resubjected to the hydrogenation conditions until all chlorides had been removed. This delivered spacer-equipped di-N-acetyl bacillosamine 4a in 63% yield. After the acidic hydrolysis of the glyceroyl isopropylidene ketal in 17b and 17c, deprotection under the same conditions delivered the C-2-N-acetyl-C-4-N-glyceroyl bacillosamines 4b and 4c in 79% and 81% yield, respectively.
11 (see Scheme 2). Saponification of the two acetates in 10 was then followed by regioselective installation of the C-3-naphthylmethyl ether and subsequent mesylation of the C-4-OH. Because we aimed to use azide as nucleophile for the introduction of the C-4-amine functionality, we had to transform the C-2-azide in 11 to achieve orthogonality. Although amines are commonly avoided during manipulations on carbohydrate building blocks, we reasoned that inorganic azide anion should be sufficiently nucleophilic to outcompete the C-2 amine in mesylate displacement. We therefore decided to reduce the azido group in 11 to the corresponding amine and invert the C-4-O-mesylate in the resulting fucosamine, without masking the C-2-amine. Thus, fucosazide 11 was exposed to 1,3-propanedithiol to deliver fucosamine 12, which was treated with NaN3 and TBABr in DMF at elevated temperature to provide the desired bacillosamine 13, bearing a fully orthogonal functional group pattern, in 81%. Next, the C-2-amine was masked with a trichloroacetyl (TCA) group to provide fully protected bacillosamine 14. Reduction of the azide with 1,3-propanedithiol then set the stage for the differentiation of the C-4-amino group into either an acetamide (towards 4a) or the two glyceroyl substituted bacillosamines 4b and 4c. During the reduction of the azide in 14, the TCA moiety was partially reduced as well to the corresponding C-2-dichloroacetamide (DCA), yielding compound 15. As the DCA group can be removed under similar conditions as the TCA, the synthetic strategy did not need any adjustment and we continued with 4-N-acylation. Trichloroacetylation provided bacillosamine synthon 16a while reaction of the C-4-amine with either R- or S-isopropylidene-glyceric acid under the agency of HCTU, provided bacillosamines 16b and 16c. Next, the three building blocks were equipped with an azidohexanol spacer, in glycosylation reactions promoted by N-iodosuccinimide (NIS) and trimethylsilyl trifluoromethanesulfonate (TMSOTf) to provide 17a, 17b and 17c. The products were obtained as single diastereoisomers and no oxazoline side products were detected, indicating that the dichloroacetyl groups provided effective anchimeric assistance.12 With the fully protected spacer-equipped bacillosamines in hand, deprotection of the intermediates was undertaken. We first tried to unmask the TCA and DCA groups in 17a using Cs2CO3 in DMF,13 but this did not lead to the desired C-2,4-diamine. Instead bicyclic urea 18 was formed.14 We therefore switched to a hydrogenation reaction, aiming to unmask the spacer azide, C-2-N-DCA, C-3-O-Nap and C-4-N-TCA groups in one step. After the first hydrogenation event using Pearlman's catalyst, LC-MS analysis of the crude reaction mixture revealed the presence of three chlorides and therefore the mixture was resubjected to the hydrogenation conditions until all chlorides had been removed. This delivered spacer-equipped di-N-acetyl bacillosamine 4a in 63% yield. After the acidic hydrolysis of the glyceroyl isopropylidene ketal in 17b and 17c, deprotection under the same conditions delivered the C-2-N-acetyl-C-4-N-glyceroyl bacillosamines 4b and 4c in 79% and 81% yield, respectively.
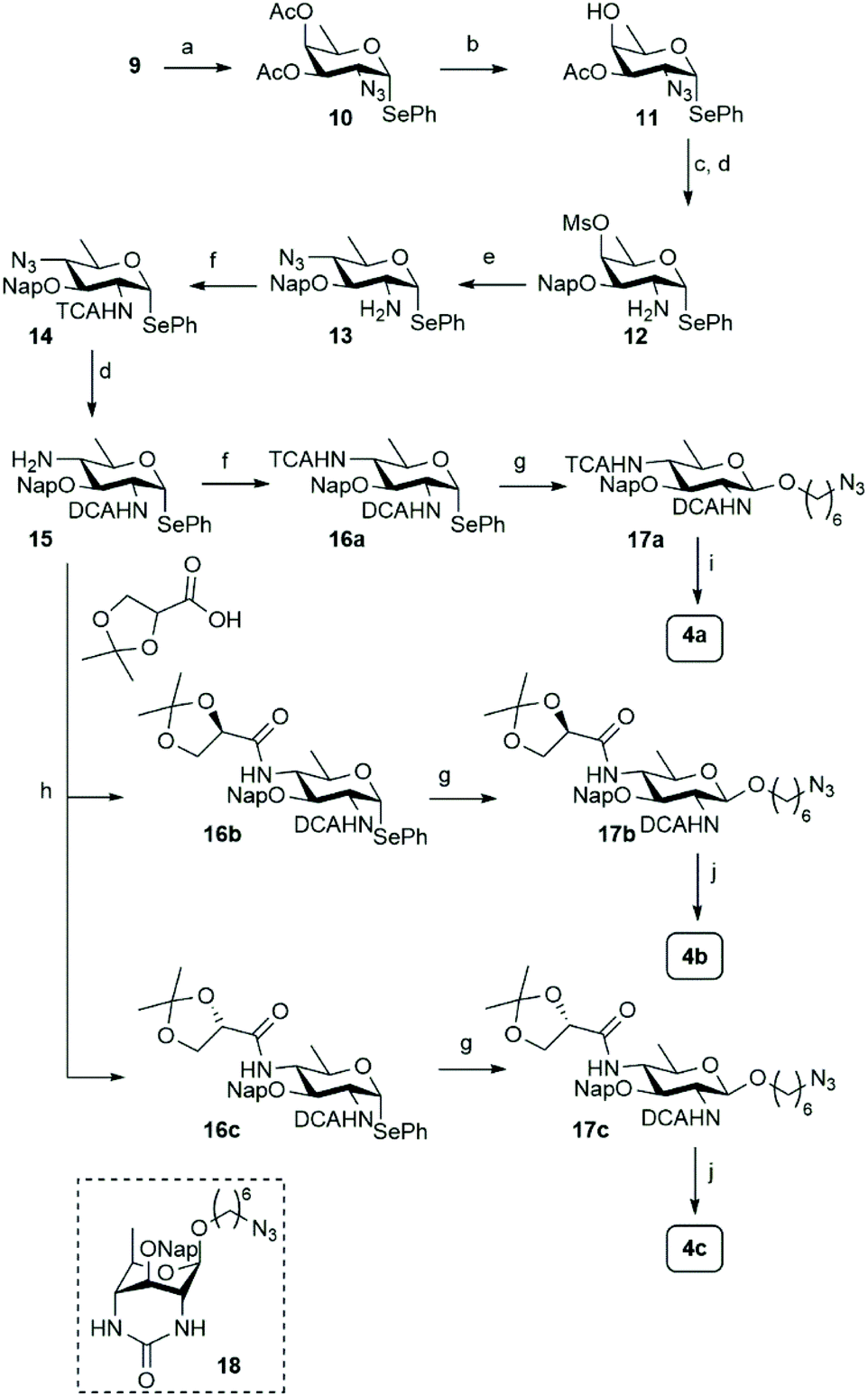 | ||
| Scheme 2 Synthesis of differentially functionalized bacillosamines. Reagents and conditons: (a) (PhSe)2, PhI(OAc)2, TMS-N3, DCM, −20 °C to rt., 93%. (b) (i) NaOMe, MeOH. (ii) Bu2SnO, toluene, 110 °C. (iii) TBABr, NapBr, 82%. (c) MsCl, pyr. 0 °C to rt., 93%. (d) 1,3-Propanedithiol, TEA, DMF, 92% for 12, 82% for 15. (e) NaN3, TBABr, DMF, 80 °C, 81%. (f) TCA-Cl, DCM:pyr., 0 °C, 89% for 14, 88% for 16a. (g) 6-Azidohexanol, NIS, TMSOTf, DCM, 0 °C, 70% for 17a, 74% for 17b, 74% for 17c. (h) 2,2-Dimethyl-1,3-dioxolane-4-carboxylic acid (R for 16b, S for 16c), HCTU, DIPEA, DMF, 0 °C to rt., 83% for 16b, 80% for 16c. (i) Pd(OH)2/C 20 wt%, H2. H2O/THF/tBuOH, 63%. (j) (i) H2O/THF/AcOH, 80 °C. (ii) Pd(OH)2/C 20 wt%, H2, H2O/THF/tBuOH, 79% for 4b, 81% for 4c. | ||
With a successful route of synthesis towards differentially functionalized bacillosamines established, we set out to probe the generation of a N. meningitidis disaccharides, featuring an additional α-D-galactopyranose attached to the C-3-OH of the bacillosamine. To install the required 1,2-cis-galactosyl linkage we used a 4,6-di-tert-butyl silylidene protected galactose donor 19 as 4,6-silylidene functionalized galactopyranosides are amongst the most reliable donors for the installation of cis-galactosyl linkages.15 Because the presence of amides in glycosyl acceptors can lead to problematic glycosylation reactions16 we decided to attach the α-galactoside prior to the introduction of the N-glyceroyl. Thus, we prepared C-4-azide acceptor 20 by DDQ-mediated unmasking of the C-3-hydroxyl function in 14. In a chemoselective glycosylation reaction, imidate donor 1917 and bacillosamine selenoglycoside acceptor 20 were then reacted to provide, in a completely stereoselective manner, the desired disaccharide 21 in 91% yield. Before installation of the azidohexanol spacer, the C-4-N-glyceroyl groups had to be incorporated and we therefore reduced the C-4-azide in 21, which again led to the conversion of the C-2-N-TCA amide into the corresponding DCA-group. Amine 22 was obtained in 48% yield and condensed with (S)-isopropylidene-glyceric acid using HCTU as the condensation agent to furnish the glyceroyl bacillosamine disaccharide 23 in 30%. It was observed that both the reduction and the amide bond forming reactions proceeded significantly slower in the disaccharide than the corresponding reactions on the monosaccharides described above. This may be attributed to increased steric hindrance of the appended galactopyranose with the neighboring C4-nitrogen. Next the azidohexanol spacer was introduced using NIS/TMSOTf to activate the seleno disaccharide to afford the fully protected disaccharide 24 in 57% yield. At this stage, all protecting groups in disaccharide 24 were removed by the following procedure. First, the silylidene group was removed by treatment of HF·Pyr in THF yielding the diol in 64% yield. Next, the isopropylidene group in the glyceroyl moiety was hydrolysed with AcOH/H2O/THF. Finally, the hydrogenation procedure, as described above, was undertaken to remove the benzyl ethers and transform the dichloroacetamide into the corresponding acetamide and the spacer azide into a primary amine. The hydrogenation reaction was monitored by LC-MS analysis, and repeated when required. In this manner disaccharide 5 was obtained in 41% yield (Scheme 3).
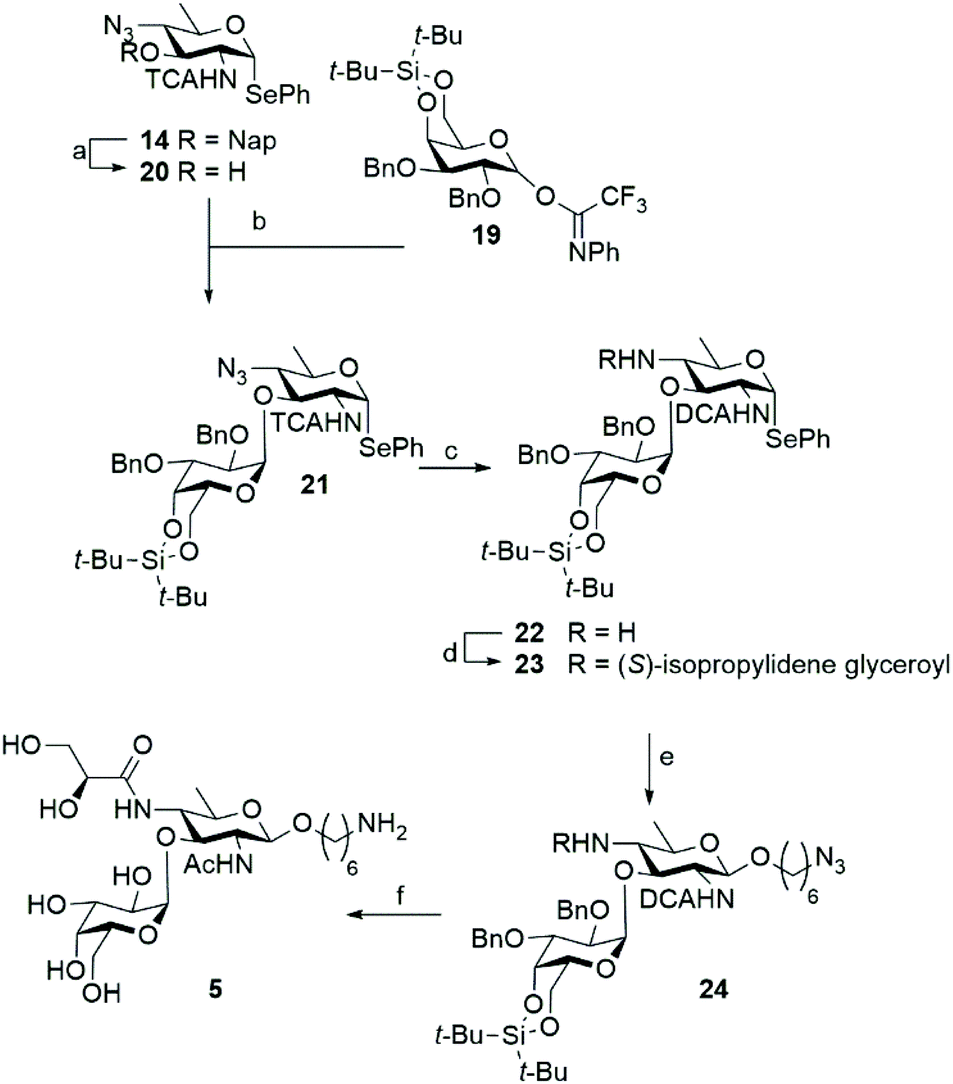 | ||
| Scheme 3 Synthesis of galactosylated C4-N-glyceroyl bacillosamines. Reagents and conditions: (a) DDQ, DCM/H2O, 80%. (b) Compound 20, TMSOTf, DCM, 0 °C, 91%. (c) 1,3-Propanedithiol, TEA, DMF, 48%. (d) (S)-2,2-Dimethyl-1,3-dioxolane-4-carboxylic acid for, HCTU, DIPEA, DMF, 0 °C to rt., 30%. (e) 6-Azidohexanol, NIS, TMSOTf, DCM, 0 °C, 57%, (f) i) HF·pyr, THF, 64% (ii) H2O/THF/AcOH, 80 °C. (iii) Pd(OH)2/C 20 wt%, H2, H2O/THF/tBuOH, 41%. | ||
Conclusions
In conclusion, we have here disclosed the first synthesis towards an orthogonally protected bacillosamine synthon, that allows for the differentiation of the two amino functionalities, and its use in the generation of a N. meningitidis PilE disaccharide bearing a C-3-α-D-galactose appendage and a C-4-N-glyceroyl. The chemistry developed here can be used to generate differentially functionalised bacillosamines and pave the way to the assembly of larger bacillosamine containing oligosaccharides as well as glycopeptides. The set of conjugation ready bacillosamines can be used to generate antibodies directed at these N. meningitidis PilE post-translational modifications for both therapeutic and diagnostic purposes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by The Netherlands Organization for Scientific Research (NWO).Notes and references
- N. Sharon, Glycobiology, 2007, 17, 1150–1155 CrossRef CAS; E. Vinogradov, L. Nossova, A. Swierzko and M. Cedzyński, Carbohydr. Res., 2004, 339, 2045–2047 CrossRef PubMed.
- N. M. Young, J. R. Brisson, J. Kelly, D. C. Watson, L. Tessier, P. H. Lanthier, H. C. Jarrell, N. Cadotte, F. St Micheal, E. Aberg and C. M. Szymanski, J. Biol. Chem., 2002, 277, 42530–42539 CrossRef CAS.
- F. E. Aas, A. Vik, J. Vedde, M. Koomey and W. Egge-Jacobsen, Mol. Microbiol., 2007, 65, 607–624 CrossRef CAS; M. D. Hartley, M. J. Morrison, F. E. Aas, B. Borud, M. Koomey and B. Imperiali, Biochemistry, 2011, 50, 4936–4948 CrossRef.
- E. Stimson, M. Virji, S. Barker, M. Panico, I. Blench, J. Saunders, G. Payne, E. R. Moxon, A. Dell and H. R. Morris, Biochem. J., 1996, 33, 29–33 CrossRef PubMed.
- (a) J. Chamot-Rooke, G. Mikaty, C. Malosse, M. Soyer, A. Dumont, J. Gault, A. F. Imhaus, P. Martin, M. Trellet, G. Clary, P. Chafey, L. Camoin, M. Nilges, X. Nassif and G. Duménil, Science, 2011, 331, 778–782 CrossRef CAS; (b) J. Gault, M. Ferber, S. Machata, A. F. Imhaus, C. Malosse, A. Charles-Orszag, C. Millien, G. Bouvier, B. Bardiaux, G. Péhau-Arnaudet, K. Klinge, I. Podglajen, M. C. Ploy and G. Duménil, PLoS Pathog., 2015, 1–24 CAS.
- E. Bedini, D. Esposito and M. Parrilli, Synlett, 2006, 825–830 CrossRef CAS; E. Weerapana, K. J. Glover, M. M. Chen and B. Imperiali, J. Am. Chem. Soc., 2005, 127, 13766–13767 CrossRef.
- M. N. Amin, A. Ishiwata and Y. Ito, Carbohydr. Res., 2006, 341, 1922–1929 CrossRef CAS.
- M. Emmadi and S. S. Kulkarni, Nat. Prod. Rep., 2014, 31, 870–879 RSC.
- R. J. Roblez-Diaz, J. Org. Chem., 1993, 58, 6122–6125 CrossRef.
- L. Somsák, I. Németh and J. Carbohydrate, Chem. – Eur. J., 1993, 12, 679–684 Search PubMed.
- (a) Y. V. Mironov, A. A. Sherman and N. E. Nifantiev, Tetrahedron Lett., 2004, 45, 9107–9110 CrossRef CAS; (b) B. Hagen, S. Ali, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, J. Org. Chem., 2017, 82, 848–868 CrossRef CAS.
- S. Kusumoto, K. Yamamoto, M. Imoto, M. Inage, M. Tsuijmoto, S. Kotani and T. Shiba, Bull. Chem. Soc. Jpn., 1986, 59, 1411–1417 CrossRef.
- D. Urabe, K. Sugino, T. Nishikawa and M. Isobe, Tetrahedron Lett., 2004, 45, 9405–9407 CrossRef.
- This product was not purified and therefore no yield has been determined. The spectroscopic data sufficed to establish the structure.
- (a) A. Imamura, H. Ando, S. Korogi, G. Tanabe, O. Muraoka, H. Ishida and M. Kiso, Tetrahedron Lett., 2003, 44, 6725–6728 CrossRef; (b) A. Imamura, H. Ando, H. Ishida and M. Kiso, Org. Lett., 2005, 7, 4415–4418 CrossRef PubMed; (c) A. Imamura, A. Kimura, H. Ando, H. Ishida and M. Kiso, Chem. – Eur. J., 2006, 12, 8862–8870 CrossRef PubMed; (d) B. Hagen, J. H. M. van Dijk, Q. Zhang, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Org. Lett., 2017, 19, 2514–2517 CrossRef PubMed.
- D. Crich and V. Dudkin, J. Am. Chem. Soc., 2001, 123, 6819–6825 CrossRef CAS PubMed.
- B. Liu, J. van Mechelen, R. J. B. H. N. van den Berg, A. M. C. H. van den Nieuwendijk, J. M. F. G. Aerts, G. A. van der Marel, J. D. C. Codée and H. S. Overkleeft, Eur. J. Org. Chem., 2019, 118–129 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and experimental data. See DOI: 10.1039/d0ob00256a |
| This journal is © The Royal Society of Chemistry 2020 |