
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric trehalose analogues to probe disaccharide processing pathways in mycobacteria†
Hadyn L.
Parker
 a,
Ruben M. F.
Tomás
b,
Christopher M.
Furze
a,
Collette S.
Guy
ab and
Elizabeth
Fullam
*a
a,
Ruben M. F.
Tomás
b,
Christopher M.
Furze
a,
Collette S.
Guy
ab and
Elizabeth
Fullam
*a
aSchool of Life Sciences, University of Warwick, Coventry, CV4 7AL, UK. E-mail: e.fullam@warwick.ac.uk
bDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK
First published on 30th April 2020
Abstract
The uptake and metabolism of the disaccharide trehalose by Mycobacterium tuberculosis is essential for the virulence of this pathogen. Here we describe the chemoenzymatic synthesis of new azido-functionalised asymmetric trehalose probes that resist degradation by mycobacterial enzymes and are used to probe trehalose processing pathways in mycobacteria.
Introduction
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), is a major global health challenge. TB is now the leading cause of death from a single infectious agent world-wide with 1.5 million deaths reported in 2018 alone, more than HIV and malaria combined.1 The incidences of drug-resistant strains of TB are escalating which increases the complexity of effective treatment regimens and the implementation of control measures. Therefore, there is an urgent need to develop new interventions to combat this global pathogen.One of the distinguishing features of Mtb is a highly complex cell envelope that provides a very effective barrier against many antibiotics and further complicates TB drug-regimens, whilst lipid components of the cell envelope play important roles in its pathogenicity.2–4 The Mtb cell wall core consists of mycolic acids covalently linked to arabinogalactan that is attached to peptidoglycan, known as the mAGP complex.4 Additional non-covalently linked lipids and lipoglycans intercalate within the lipid environment to form an outer ‘myco-membrane’ and include the trehalose containing glycolipids, trehalose mono- and dimycolates (TMM and TDM respectively).3,5 TMM and TDM are essential components of the mycobacterial cell envelope for both the viability and virulence of the Mtb pathogen.3 It has recently been reported that ‘free’ trehalose released from these glycolipids through the catalytic action of the antigen 85 (Ag85) complex, is reused through uptake via the Mtb LpqY-SugABC transporter and this recycling process is critical for virulence.6 Given that Mtb resides within a nutrient poor environment this recycling strategy provides a route for Mtb to scavenge an essential carbon source that can subsequently be utilised as a key metabolite in a number of biosynthetic processes.6–8 For these reasons, targeting trehalose uptake and metabolism in Mtb has recently gained attention as an attractive route for the development of new anti-tubercular therapeutic and diagnostic agents.
To develop effective trehalose targeting strategies in mycobacteria chemical tools are required to increase our understanding of Mtb trehalose metabolism. Recent studies have incorporated bioorthogonal handles and fluorescent moieties into the trehalose scaffold with retention of the α,α1,1-glycosidic bond and these probes have been used for live imaging of mycobacteria and for the rapid detection of Mtb in sputum.9–13 The diverse panel of unnatural trehalose-based probes has been an important step towards providing insights into the molecular pathways of trehalose metabolism in Mtb. However, enzymes that are present in both Mtb and the human host can metabolise trehalose which has the potential to lead to a loss in the labelling efficacy and potential off-target effects of the modified trehalose probes.14–17 In Mtb there are two main pathways that metabolise trehalose. The Mtb trehalase enzyme catalyses the hydrolysis of the α,α1,1-glycosidic bond of trehalose to form two molecules of glucose (Fig. 1).15 Alternatively, the Mtb trehalose synthase (TreS) enzyme can isomerise trehalose through rearrangement of the α,α1,1-linkage to form the α1,4-linked glucose disaccharide maltose (Fig. 1).18 Therefore, there is a need for an alternative approach and the identification of enzymatically-stable chemical probes that can still be utilised by the mycobacterial trehalose processing machinery. We therefore reasoned that asymmetric trehalose analogues, in which one glucose moiety is replaced with either mannose (mannotrehalose (2)) or galactose (galactotrehalose (3)) and the corresponding 6-azido modified versions (5–6), are an appealing route to generate trehalose probes with improved metabolic profiles that are resistant to degradation in mycobacteria (Scheme 1). Here, we report the chemoenzymatic synthesis of asymmetric trehalose derivatives and their evaluation as mycobacterial probes.
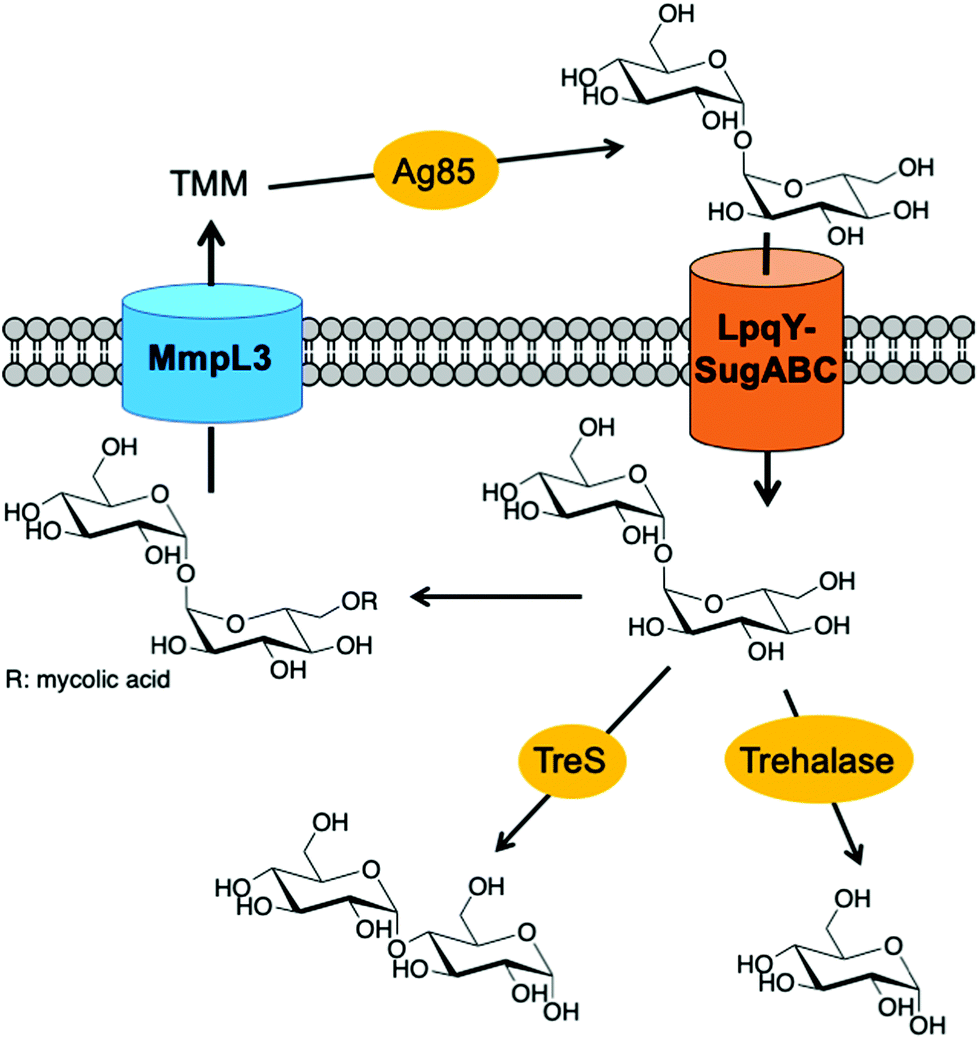 | ||
| Fig. 1 Pathways involved in trehalose uptake and metabolism in mycobacteria. TMM: trehalose monomycolate. | ||
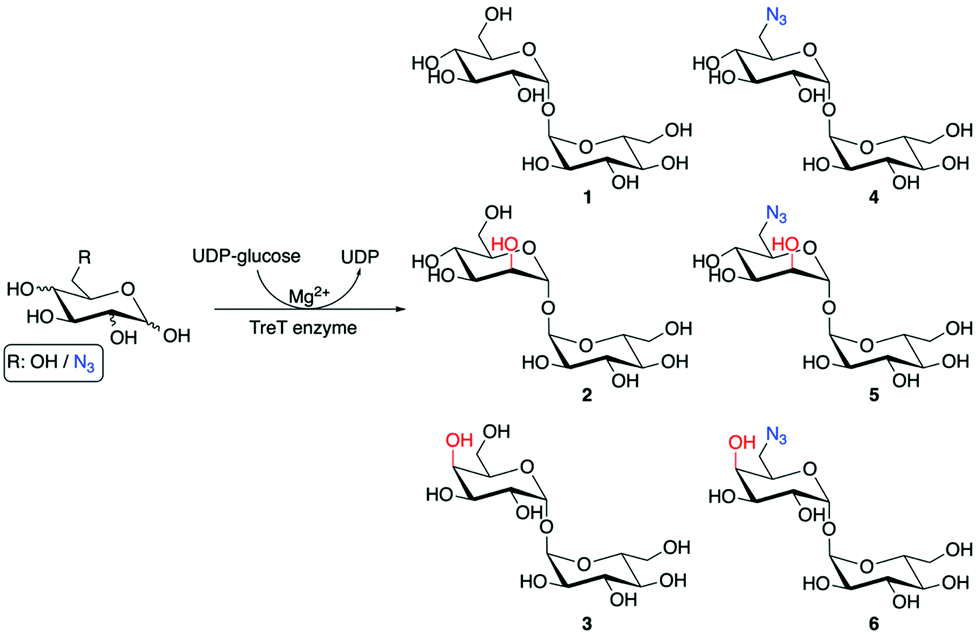 | ||
| Scheme 1 Chemoenzymatic synthesis of trehalose analogues. | ||
Results and discussion
To obtain mannotrehalose (2), galactotrehalose (3) and the corresponding 6-azido-modified derivatives 5–6 a modified chemoenzymatic method previously reported by Swarts et al. was employed utilising the trehalose synthase (TreT) enzyme from Thermoproteus tenax (Scheme 1).19 Enzymatic reactions were performed at 70 °C for 2 h in 50 mM HEPES, 300 mM NaCl, pH 8.0 and the equivalents of the glucose analogue![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :UDP-glucose varied. The exact ratios and full chemoenzymatic synthesis details are specified in the ESI Methods.† Previous studies report that glucose analogues modified at the 4-position are poor substrates of TreT.19 Here we found that altering the ratio of galactose:UDP-glucose from 1:4 to 1.5:1 improved the conversion of galactose to the corresponding galactotrehalose analogue and increased the yield of 3 from 12% to 28%. Employing this approach resulted in the generation of trehalose analogues 2–6 in a single-step from commercially available glucose analogues (Scheme 1). The reaction products were readily purified by size-exclusion chromatography and HPLC to obtain compounds 2–6, and the purified products analysed by 1H and 13C NMR and mass spectrometry to confirm the formation of the correct structure and retention of the α,α1,1-glycosidic bond (ESI Fig. S4–S13†). These results expand on the known substrate profile tolerated by TreT to include 6-azido-mannose and 6-azido-galactose enabling access to unnatural azido-modified mannotrehalose (5) and galactotrehalose (6) disaccharides respectively via a chemoenzymatic route, thereby expanding the trehalose probe toolkit.
:UDP-glucose varied. The exact ratios and full chemoenzymatic synthesis details are specified in the ESI Methods.† Previous studies report that glucose analogues modified at the 4-position are poor substrates of TreT.19 Here we found that altering the ratio of galactose:UDP-glucose from 1:4 to 1.5:1 improved the conversion of galactose to the corresponding galactotrehalose analogue and increased the yield of 3 from 12% to 28%. Employing this approach resulted in the generation of trehalose analogues 2–6 in a single-step from commercially available glucose analogues (Scheme 1). The reaction products were readily purified by size-exclusion chromatography and HPLC to obtain compounds 2–6, and the purified products analysed by 1H and 13C NMR and mass spectrometry to confirm the formation of the correct structure and retention of the α,α1,1-glycosidic bond (ESI Fig. S4–S13†). These results expand on the known substrate profile tolerated by TreT to include 6-azido-mannose and 6-azido-galactose enabling access to unnatural azido-modified mannotrehalose (5) and galactotrehalose (6) disaccharides respectively via a chemoenzymatic route, thereby expanding the trehalose probe toolkit.
Having chemoenzymatically synthesised mannotrehalose (2), galactotrehalose (3) and the azido-derivatives (5–6) we evaluated whether the trehalose analogues were resistant to degradation by the mycobacterial trehalase and TreS enzymes. We first incubated 30 mM of either trehalose (1), trehalose analogues 2–3 or azido-trehalose analogues (4–6) with the recombinant trehalase enzyme from Mycobacterium smegmatis, or with buffer alone containing no enzyme, over 19 h and the reaction products analysed by thin-layer chromatography (TLC) (Fig. 2). As expected,15 within 1 h we observed the rapid catalytic degradation of trehalose (1) to glucose by the M. smegmatis trehalase enzyme (ESI Fig. S1A†). Further characterisation revealed that azido-trehalose (4) is also a substrate of the mycobacterial trehalase enzyme although the rate of hydrolysis of azido-trehalose (4) is lower than that observed for the endogenous trehalose substrate (ESI Fig. S1B†). These results provide the first evidence that 6-azido-trehalose probes have the potential to be degraded by mycobacterial enzymes leading to the generation of glucose and azido-glucose monosaccharides that could be used as a substrate to regenerate 6-azido-trehalose or diverted into alternative, non-trehalose specific processing pathways. Further experiments are required to determine the importance of 6-azido-trehalose degradation and the impact on metabolic labelling in mycobacteria. In contrast, only very low levels of hydrolysis of the epimeric trehalose analogues mannotrehalose (2), galactotrehalose (3) and no hydrolysis of the corresponding azido-derivatives 5–6 were observed under the same test conditions at the extended 19 h time point (Fig. 2). Taken together these findings indicate that the manno- and galacto-trehalose derivatives (2–3 and 5–6) are poorly tolerated by the mycobacterial trehalase enzyme highlighting that stereochemical inversion of the 2- and 4-hydroxy groups results in highly hydrolytically stable trehalose analogues.
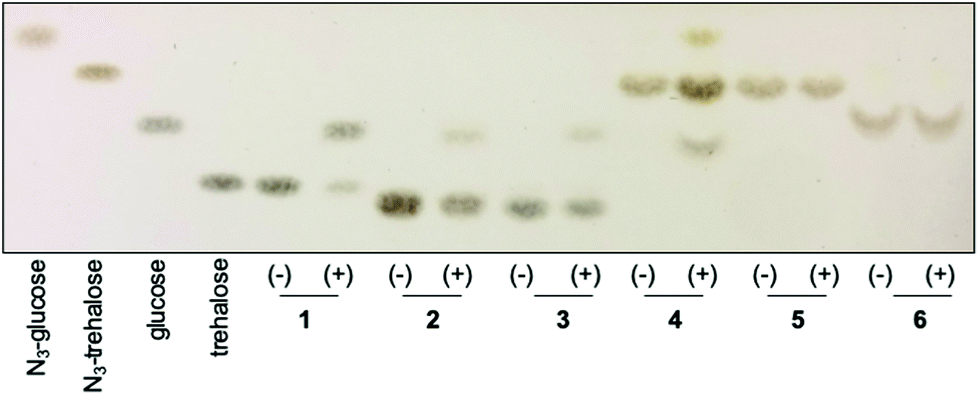 | ||
| Fig. 2 Activity of M. smegmatis trehalase with trehalose 1 and trehalose derivatives (2–6). Compounds were incubated in the absence (−) or presence (+) of the M. smegmatis trehalase enzyme for 19 h. Mannotrehalose (2), galactotrehalose (3), azido-trehalose (4), azido-mannotrehalose (5) and azido-galactotrehalose (6). | ||
Next, we investigated whether the mycobacterial TreS enzyme is able to isomerise the asymmetric trehalose analogues 2–6. 20 mM of trehalose (1) or trehalose analogues 2–6 were incubated with the TreS enzyme from M. smegmatis and the reaction monitored by TLC. In this instance, it was clearly observed that trehalose was isomerised to maltose by TreS after 30 min (ESI Fig. S2†), whereas we were unable to detect any product from the TreS reaction under the same conditions with either mannotrehalose 2, galactotrehalose 3 or the azido-derivatives 4–6 over a 4.5 h period (Fig. 3). The reversible isomerisation of trehalose by the mycobacterial TreS enzyme proceeds via the formation of a glucosyl-TreS enzyme intermediate followed by release and reorientation of glucose to enable either the 1- or 4-hydroxy group to reattack to generate either trehalose or maltose respectively.17 The absence of any observed isomerisation of 2–3 and the azido-derivatives 4–6 coupled with the lack of glucose build up indicates that the mycobacterial TreS enzyme is sensitive to the stereochemistry of both halves of the molecule and that modification at the 6-position with an azido-group is not tolerated. These results demonstrate the high stability of all trehalose analogues (2–6) to isomerisation by the mycobacterial TreS.
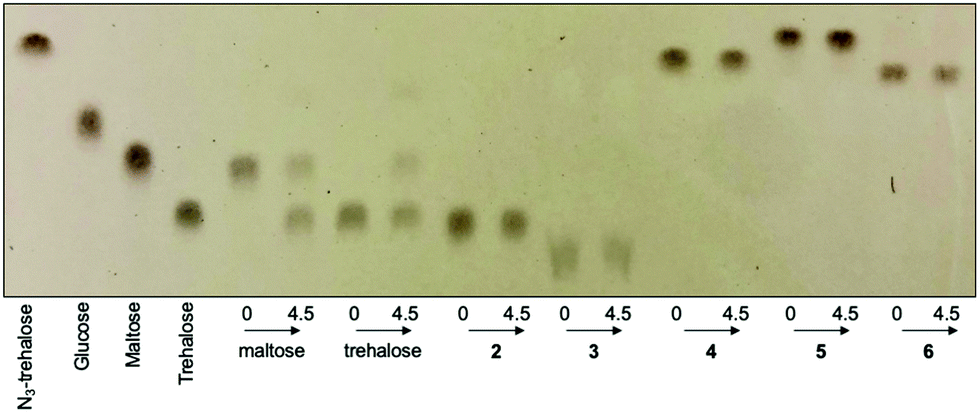 | ||
| Fig. 3 Activity of M. smegmatis TreS with maltose, trehalose (1) and trehalose derivatives (2–6). Compounds were incubated with M. smegmatis TreS enzyme for 0 h or 4.5 h. Mannotrehalose (2), galactotrehalose (3), azido-trehalose (4), azido-mannotrehalose (5) and azido-galactotrehalose (6). | ||
Having demonstrated that mannotrehalose (2), galactotrehalose (3) and the corresponding azido-analogues (5–6) are hydrolytically stable to the mycobacterial trehalase and resistant to isomerisation by the mycobacterial TreS enzyme we evaluated whether the asymmetric trehalose analogues 2 and 3 support the growth of mycobacteria. M. smegmatis and Mycobacterium bovis BCG were grown to mid-exponential phase, starved overnight in PBS containing 0.05% tyloxapol and then grown in defined minimal-media supplemented with either 2.9 mM trehalose (1) or trehalose analogues 2 and 3 as sole carbon sources (Fig. 4). In agreement with previous studies,20 both M. smegmatis and M. bovis BCG were able to grow in the presence of trehalose alone. Notably, in direct contrast, both mannotrehalose (2) and galactotrehalose (3) were unable to support the growth of M. smegmatis or M. bovis BCG (Fig. 4). Our metabolic labelling experiments (see later) indicate that azido-analogues of 2 and 3 modified at the 6-position (5–6) can be taken up by mycobacteria. These findings therefore support the notion that within a nutrient limited environment the degradation of trehalose is required to support mycobacterial growth. Interestingly, recent studies have shown that Clostridioides difficile can efficiently use mannotrehalose (2) as a sole carbon source,21 highlighting the divergence of trehalose derivative processing pathways that are available in different microorganisms. Moreover, 6-azido-trehalose (4) has previously been found to have very weak antimycobacterial activities and high minimum inhibitory concentrations (MICs) of 100–>500 μg mL−1 against Mycobacterium aurum and Mycobacterium tuberculosis9,22,23 Therefore, we used the resazurin-reduction assay24 to evaluate the ability of mannotrehalose (2) and galactotrehalose (3) to inhibit the growth of Mtb in liquid media and found that compounds 2–3 did not inhibit the growth of Mtb at concentrations as high as 5 mg mL−1.
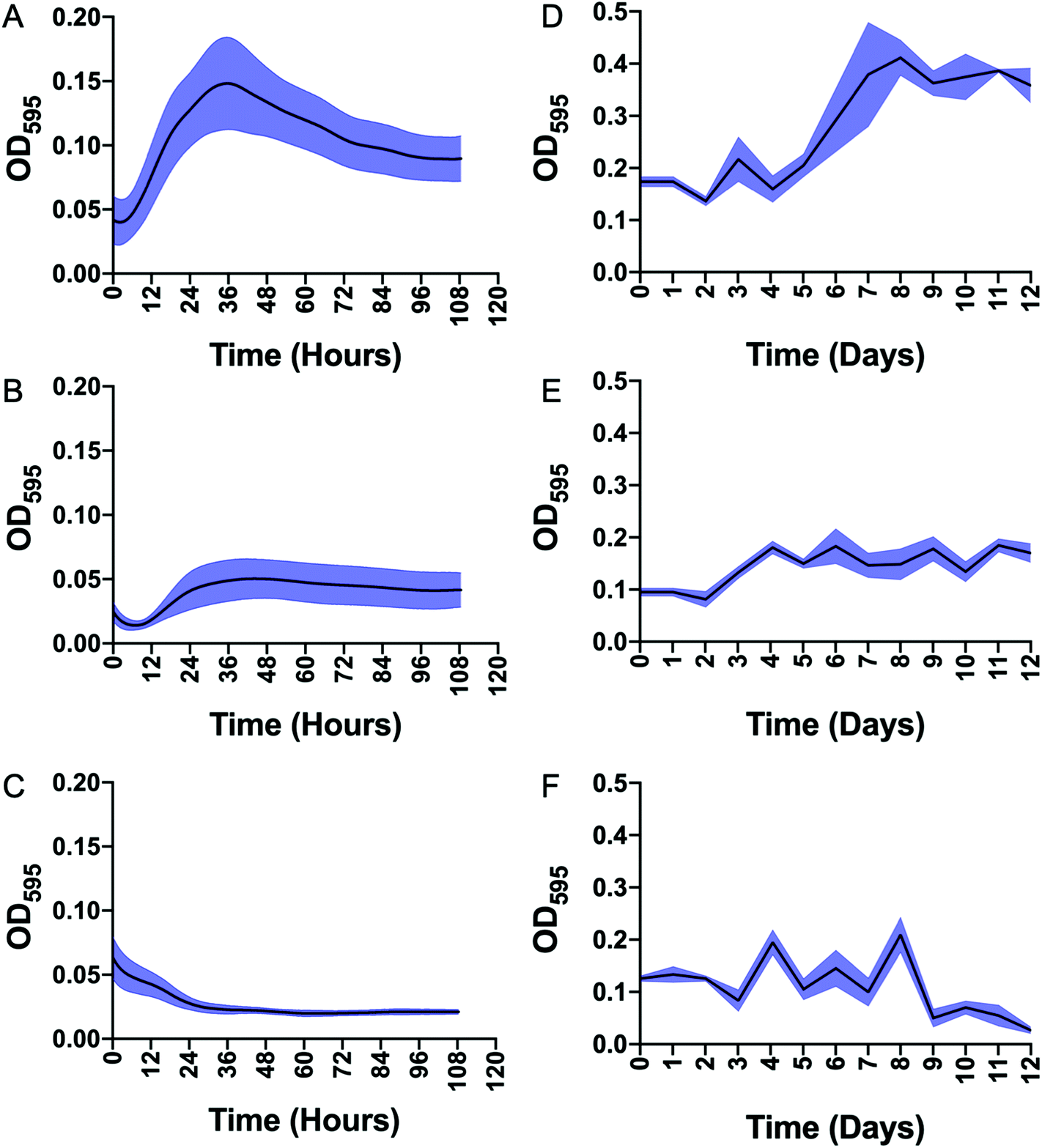 | ||
| Fig. 4 Growth of Mycobacterium smegmatis and Mycobacterium bovis BCG starved in minimal media and supplemented with either trehalose (1), mannotrehalose (2) or galactotrehalose (3). A–C: M. smegmatis plus trehalose (A), mannotrehalose (B), galactotrehalose (C). D–F: M. bovis BCG plus trehalose (D), mannotrehalose (E), galactotrehalose (F). OD595 is optical density at 595 nm. The error bars represent plus/minus SEM of three biological replicates are shown in blue. | ||
Next, we evaluated whether the trehalose analogues 2 and 3 could be taken up by mycobacteria. M. smegmatis was incubated with 100 μM of the corresponding azido-derivatives of either mannotrehalose (5), galactotrehalose (6) or trehalose (4) for 4 h. After this time the mycobacteria were pelleted, washed with PBS containing 0.05% Tween 80 (PBST) to remove any free sugar and successful labelling was visualised using the azide-reactive fluorophore, dibenzocyclooctyne-PEG4-tetramethylrhodamine (DBCO-PEG4-TAMRA), through strain-promoted copper-free azide–alkyne cycloaddition. In agreement with previous studies, fluorescence microscopy revealed fluorescent labelling of the mycobacterial cells that had been treated with azido-trehalose (4).11 Interestingly, we also observed fluorescent labelling of M. smegmatis cells that had been treated with azido-mannotrehalose (5) and azido-galactotrehalose (6) compared to control mycobacteria cells indicating that these asymmetric trehalose analogues also label mycobacteria. The fluorescent signal from the higher-resolution Airyscan detector was mostly localised at the cell-surface which is consistent with the presence of trehalose glycolipids within the myco-membrane (Fig. 5A). In addition, these results were further corroborated by flow cytometry analysis. Previous studies have found that azido-trehalose analogues modified at either the 2- or 6-hydroxyl positions label mycobacteria at low concentrations (5–25 μM), whereas higher concentrations (25–500 μM) were required when the azido-group is at either the 3- or 4-hydroxly position.11 Here, we found that azido-galactotrehalose (6) was readily incorporated into M. smegmatis with an efficiency similar to that of azido-trehalose (4), indicating that an inverted hydroxyl group at the 4-position is also well tolerated. In contrast, a smaller increase in the number of cells labelled with azido-mannotrehalose (5) was observed (Fig. 5). These results suggest that either mycobacteria are unable to rapidly import the azido-mannotrehalose analogue (5) with an inverted 2-hydroxyl group or, instead, that 5 cannot be utilised as efficiently by the mycobacterial biosynthetic processing machinery as other azido-containing trehalose derivatives. Azido-trehalose derivatives can be incorporated into the trehalose mycolates through two pathways: either the disaccharide is first imported into the cell via the LpqY-SugABC trehalose recycling transporter or incorporated into the lipids using extracellular biosynthetic machinery via the Ag85 complex.6,22 Therefore, to establish the mechanism of incorporation of 5 and 6, we repeated these metabolic labelling experiments in mutant strains of M. smegmatis that lack a functional trehalose transporter.6 Here, we observed that fluorescent labelling of M. smegmatis was almost completely abolished in the presence of the trehalose analogues 4–6 in strains that lack either the substrate binding domain LpqY or the nucleotide binding domain SugC of the LpqY-SugABC transporter. Fluorescent labelling was restored in the complemented strain (Fig. 5B) indicating that in M. smegmatis azido-mannotrehalose (5) and azido-galactotrehalose (6) are also imported and incorporated into the trehalose mycolates via the LpqY-SugABC trehalose recycling pathway.
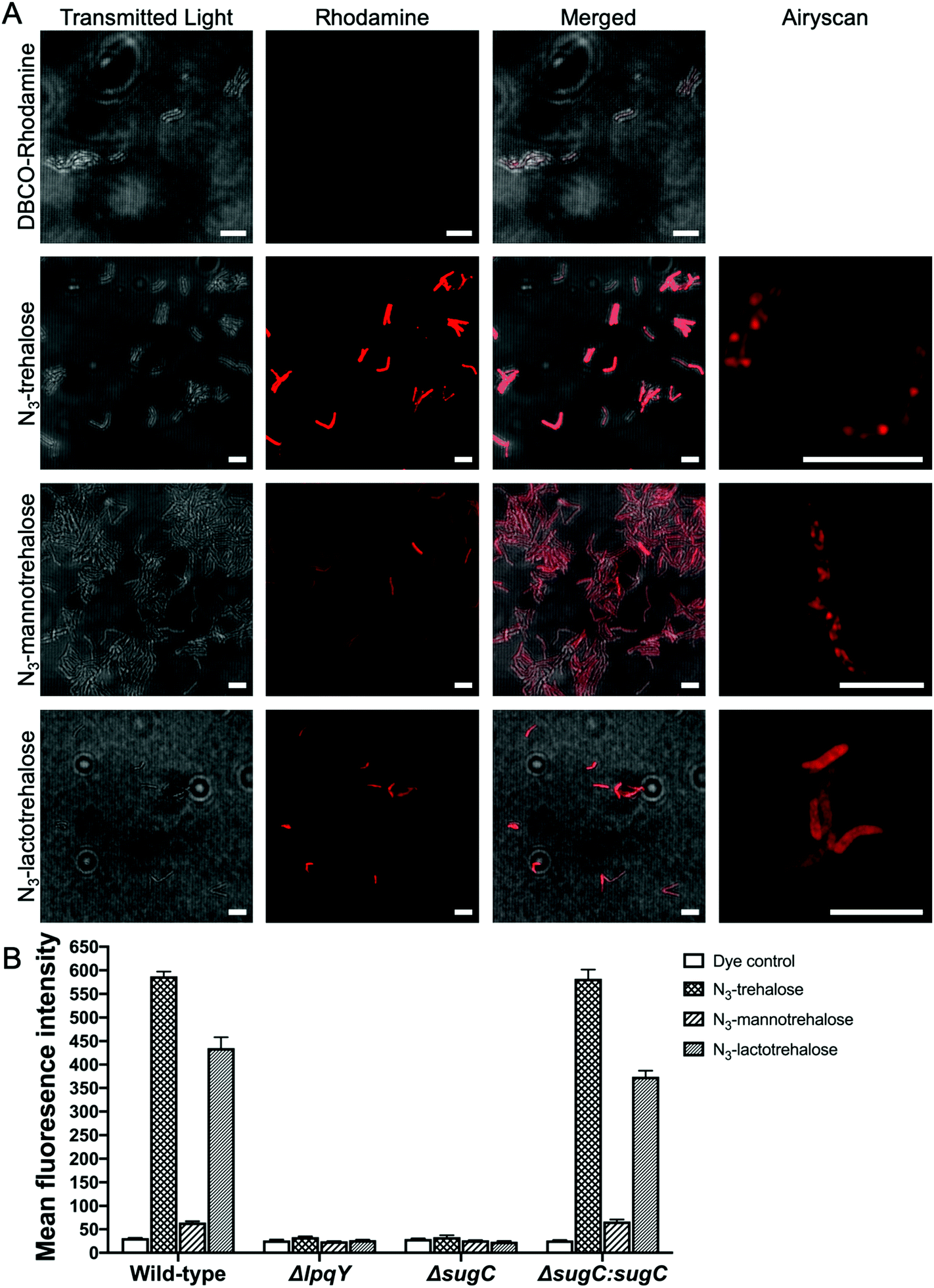 | ||
| Fig. 5 Labelling of M. smegmatis with azido-trehalose (4), azido-mannotrehalose (5), azido-galactotrehalose (6) and reacted with DBCO-PEG4-TAMRA. (A) Fluorescence microscopy. Scale bars 5 μm. (B) Flow cytometry analysis. | ||
Finally, to investigate the mycobacterial pathways impacted in response to the addition of trehalose analogues whole cell proteomics was employed. M. bovis BCG was grown in the presence of 1.5 mM of either trehalose (1), mannotrehalose (2) or galactotrehalose (3) for 48 h and the proteins were isolated using a modified Triton X-114 extraction protocol.25 A list of all of the proteins identified, annotations and fold changes are in ESI List 1.† Interestingly, we did not observe any changes in proteins that are involved in trehalose uptake (LpqY) or metabolism (TreS, trehalase) indicating, importantly, that low concentrations of trehalose and modified derivatives do not alter the trehalose biosynthetic machinery of mycobacteria (ESI List 1, Fig. S3†).
Conclusions
In summary, we have taken advantage of the ability to rapidly access new modified trehalose probes through a single-step chemoenzymatic route. In combination with bioorthogonal azide–alkyne click chemistry, we have shown that asymmetric trehalose derivatives are metabolised by mycobacteria and incorporated into the mycobacterial cell envelope via the trehalose recycling pathway. In direct contrast to trehalose (1), mannotrehalose (2), galactotrehalose (3) and the corresponding 6-azido analogues (5–6) that retain the α,α1,1-glycosidic linkage are resistant to degradation/isomerisation by mycobacterial enzymes. We anticipate that these metabolically stable probes will be useful tools for the investigation of trehalose processing in mycobacteria and related microorganisms.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded through a Sir Henry Dale Fellowship to EF jointly funded by the Wellcome Trust and Royal Society (104193/Z/14/Z and 104193/Z/14/B), a research grant from the Royal Society (RG120405), a research grant from the Leverhulme Trust (RP-2019-087) the Midlands Doctoral Training Partnership for a studentship to HLP (BB/M01116X/1) and the Molecular Analytical Science Doctoral Training Programme for a studentship to RMFT (EP/L015307). We thank Dr Ben Swarts (Central Michigan University, USA) for providing the TreT construct and Professor Rainer Kalscheuer (HHU Dusseldorf, Germany) for kindly providing the mutant strains. We thank Professor Matthew Gibson for critical reading of this manuscript. We acknowledge the contribution of the WPH Proteomics research technology platform in the School of Life Sciences, University of Warwick and Dr Andrew Bottrill for technical assistance. We thank Ian Hands-Portman for technical assistance with the microscopy studies.References
- WHO Global Tuberculosis Report, https://www.who.int/tb/publications/global_report/en/.
- K. A. Abrahams and G. S. Besra, Parasitology, 2018, 145, 116–133 CrossRef PubMed.
- M. Jackson, Cold Spring Harb. Perspect Med., 2014, 4, a021105 CrossRef PubMed.
- P. J. Brennan and H. Nikaido, Annu. Rev. Biochem., 1995, 64, 29–63 CrossRef CAS PubMed.
- P. J. Brennan, Tuberculosis, 2003, 83, 91–97 CrossRef CAS PubMed.
- R. Kalscheuer, B. Weinrick, U. Veeraraghavan, G. S. Besra and W. R. Jacobs Jr., Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21761–21766 CrossRef CAS PubMed.
- A. Nobre, S. Alarico, A. Maranha, V. Mendes and N. Empadinhas, Microbiology, 2014, 160, 1547–1570 CrossRef CAS PubMed.
- D. G. Russell, H. C. Mwandumba and E. E. Rhoades, J. Cell Biol., 2002, 158, 421–426 CrossRef CAS PubMed.
- K. M. Backus, H. I. Boshoff, C. S. Barry, O. Boutureira, M. K. Patel, F. D'Hooge, S. S. Lee, L. E. Via, K. Tahlan, C. E. Barry 3rd and B. G. Davis, Nat. Chem. Biol., 2011, 7, 228–235 CrossRef CAS PubMed.
- M. Kamariza, P. Shieh, C. S. Ealand, J. S. Peters, B. Chu, F. P. Rodriguez-Rivera, M. R. Babu Sait, W. V. Treuren, N. Martinson, R. Kalscheuer, B. D. Kana and C. R. Bertozzi, Sci. Transl. Med., 2018, 10, eaam6310 CrossRef PubMed.
- B. M. Swarts, C. M. Holsclaw, J. C. Jewett, M. Alber, D. M. Fox, M. S. Siegrist, J. A. Leary, R. Kalscheuer and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 16123–16126 CrossRef CAS PubMed.
- H. N. Foley, J. A. Stewart, H. W. Kavunja, S. R. Rundell and B. M. Swarts, Angew. Chem., Int. Ed., 2016, 55, 2053–2057 CrossRef CAS PubMed.
- H. L. Hodges, R. A. Brown, J. A. Crooks, D. B. Weibel and L. L. Kiessling, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5271–5276 CrossRef CAS PubMed.
- K. A. De Smet, A. Weston, I. N. Brown, D. B. Young and B. D. Robertson, Microbiology, 2000, 146(Pt 1), 199–208 CrossRef CAS PubMed.
- J. D. Carroll, I. Pastuszak, V. K. Edavana, Y. T. Pan and A. D. Elbein, FEBS J., 2007, 274, 1701–1714 CrossRef CAS PubMed.
- R. Ishihara, S. Taketani, M. Sasai-Takedatsu, M. Kino, R. Tokunaga and Y. Kobayashi, Gene, 1997, 202, 69–74 CrossRef CAS PubMed.
- R. Zhang, Y. T. Pan, S. He, M. Lam, G. D. Brayer, A. D. Elbein and S. G. Withers, J. Biol. Chem., 2011, 286, 35601–35609 CrossRef CAS PubMed.
- Y. T. Pan, V. Koroth Edavana, W. J. Jourdian, R. Edmondson, J. D. Carroll, I. Pastuszak and A. D. Elbein, Eur. J. Biochem., 2004, 271, 4259–4269 CrossRef CAS PubMed.
- B. L. Urbanek, D. C. Wing, K. S. Haislop, C. J. Hamel, R. Kalscheuer, P. J. Woodruff and B. M. Swarts, ChemBioChem, 2014, 15, 2066–2070 CrossRef CAS PubMed.
- J. Marrero, C. Trujillo, K. Y. Rhee and S. Ehrt, PLoS Pathog., 2013, 9, e1003116 CrossRef CAS PubMed.
- N. D. Danielson, J. Collins, A. I. Stothard, Q. Q. Dong, K. Kalera, P. J. Woodruff, B. J. DeBosch, R. A. Britton and B. M. Swarts, Chem. Commun., 2019, 55, 5009–5012 RSC.
- J. T. Belisle, V. D. Vissa, T. Sievert, K. Takayama, P. J. Brennan and G. S. Besra, Science, 1997, 276, 1420–1422 CrossRef CAS PubMed.
- J. M. Wolber, B. L. Urbanek, L. M. Meints, B. F. Piligian, I. C. Lopez-Casillas, K. M. Zochowski, P. J. Woodruff and B. M. Swarts, Carbohydr. Res., 2017, 450, 60–66 CrossRef CAS PubMed.
- J. C. Palomino, A. Martin, M. Camacho, H. Guerra, J. Swings and F. Portaels, Antimicrob. Agents Chemother., 2002, 46, 2720–2722 CrossRef CAS PubMed.
- M. Li, C. Muller, K. Frohlich, O. Gorka, L. Zhang, O. Gross, O. Schilling, O. Einsle and C. Jessen-Trefzer, Cell Chem. Biol., 2019, 26, 852–862 CrossRef CAS PubMed , e856.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob00253d |
| This journal is © The Royal Society of Chemistry 2020 |