
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Materials science based on synthetic polysaccharides
Martina
Delbianco
 *a and
Peter H.
Seeberger
*ab
*a and
Peter H.
Seeberger
*ab
aDepartment of Biomolecular Systems, Max Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: martina.delbianco@mpikg.mpg.de; peter.seeberger@mpikg.mpg.de
bDepartment of Chemistry and Biochemistry, Freie Universität Berlin, Arnimallee 22, 14195 Berlin, Germany
First published on 5th March 2020
Abstract
Supramolecular architectures, based on synthetic peptides or DNA, are the essence of modern bionanotechnology. Carbohydrates, the most abundant biopolymers in Nature tend to form hierarchical architectures. Limited access to pure and well-defined carbohydrates hampered the molecular level understanding of polysaccharides, preventing the production of tailor-made materials. Automated Glycan Assembly produces now well-defined natural and unnatural oligosaccharides for detailed structural characterization. Defined glycans can assemble into supramolecular materials with different morphologies, depending on their chemical structure. Here, we describe how synthetic oligo- and polysaccharides help to establish structure–property correlations to guide the development of novel polysaccharide materials.
1. Introduction
Natural polymers are a reservoir of renewable and biocompatible materials with minimal environmental impact. Nanotechnology based on rationally designed peptide and oligonucleotide sequences that assemble into supramolecular architectures has found applications in the medical and energy fields.1–3 Chemical modifications that induce particular conformations gave rise to new materials with defined morphologies, such as particles, fibers, and gels.4 Automated synthesis technologies, computational methods, and analytical techniques, were key to these advances.5,6Carbohydrates comprise 80% of biomass and have a strong tendency to aggregate into hierarchical architectures with different physical properties that renders them ideal bio-materials.7 The versatility of carbohydrate materials has resulted in nanomaterials for tissue engineering and cell growth8 as well as in bulk industrial production in the paper and textile industries.9 However, polysaccharide materials have some major drawbacks. Complex polysaccharides are less well understood at the molecular level than other biomaterials and tailor-made carbohydrate materials are still beyond reach (Fig. 1). Isolation from natural sources often requires extensive purification and harsh treatments resulting in poly-disperse samples, making the analysis, reproducible production, and quality control of such materials very difficult (Fig. 2). Chemical modification, commonly used to tune physical and mechanical performance, is only poorly regioselective and increases the sample polydispersity even further. Few detailed structure–function correlations have been established.
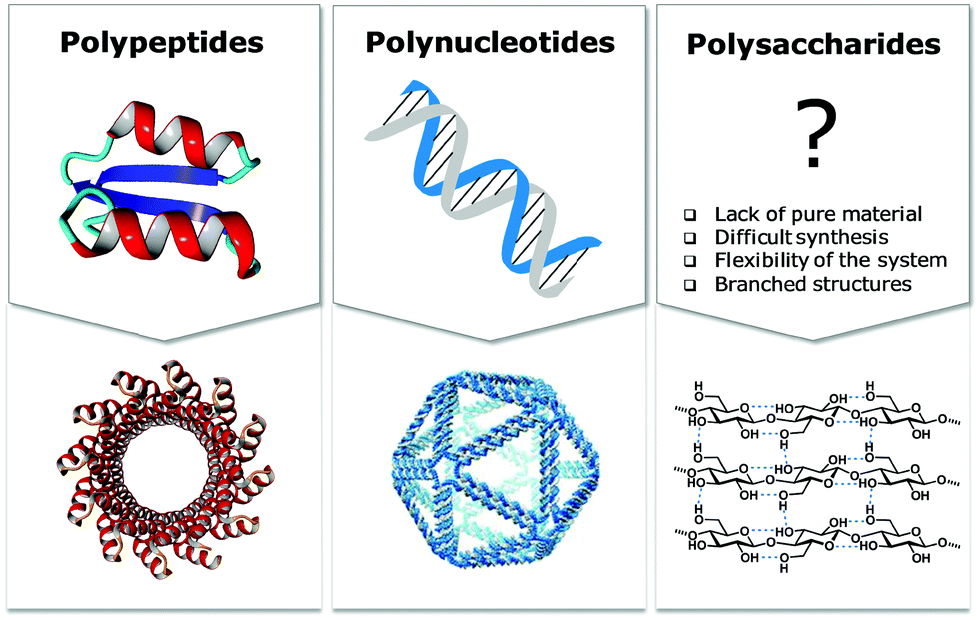 | ||
| Fig. 1 Schematic representation of three major classes of biomolecules, their conformation, and their application in materials science. The DNA origami structure was reproduced from ref. 5 with permission from Science. | ||
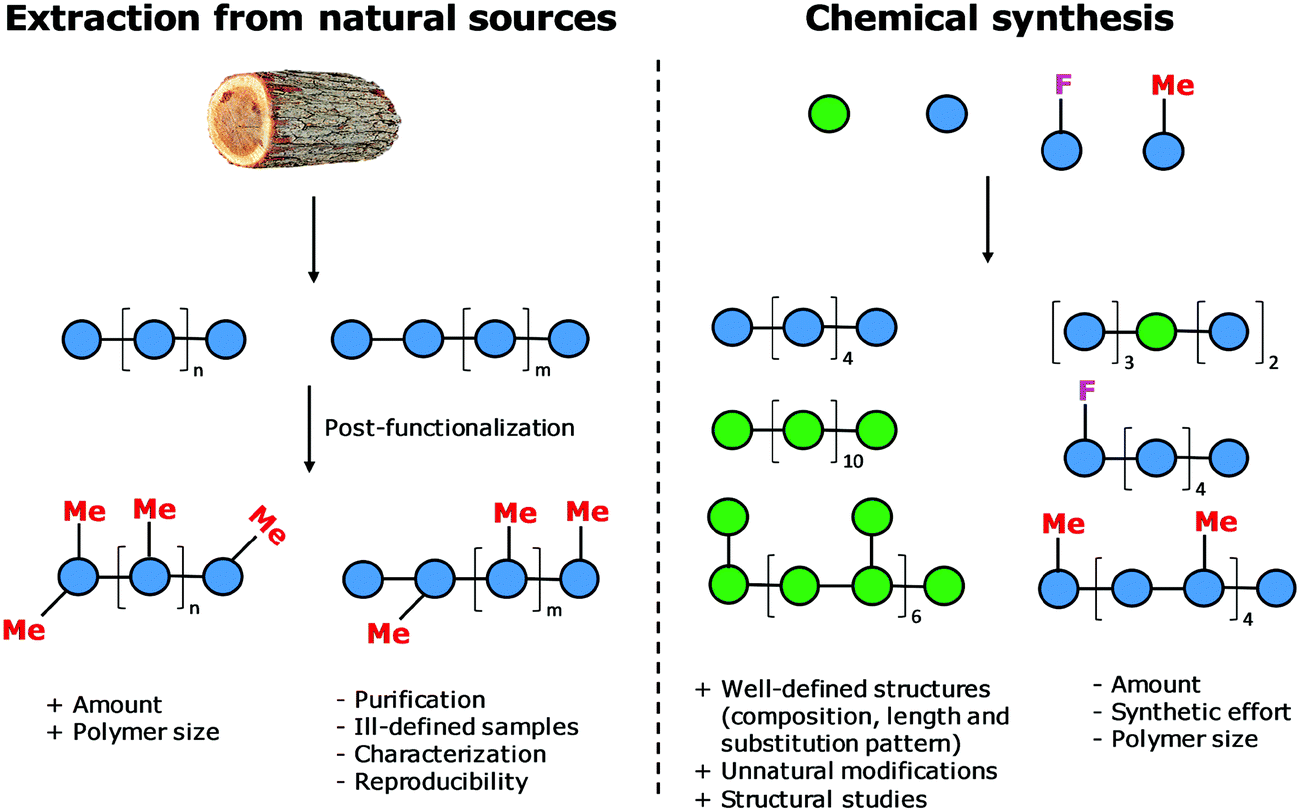 | ||
| Fig. 2 Comparison of extraction and chemical synthesis used to produce polysaccharide materials. | ||
A better understanding of polysaccharides at the molecular level is key to overcome the drawbacks of traditional extraction and processing methods. New means to better control polysaccharide modifications, in terms of degree and substitution patterns, are essential for biological experiments, where batch-to-batch reproducibility and quality control are essential. The structural diversity resulting from the different monosaccharide units and different connectivities poses a severe bottleneck. Branching and structural flexibility render polysaccharides versatile molecules, but makes them difficult to study. Chemical synthesis can help to overcome issues with polysaccharide purity, but until recently has required immense efforts by specialists. Synthetic carbohydrates have been used mainly as tools to explore biological and medical applications, where small quantities are sufficient.
Here, we discuss the synthesis, structural characterization, and supramolecular assembly of carbohydrate materials.
2. Synthesis of polysaccharides
The standard approach for the creation of polysaccharide materials follows a “post-functionalization” approach, whereby chemical reactions are performed using native polysaccharides (Fig. 2). Poor control over the degree and pattern of substitution yields mixtures of ill-defined compounds10 such that polymerization or enzymatic reactions are attractive alternatives.11–13 Still, the production of modified cellulosic materials by biosynthetic engineering in bacteria is limited due to poor product solubility and narrow substrate scope. Total synthesis offers a unique opportunity to access well-defined structures, has a broader reaction scope, but is too laborious to provide analogues for systematic studies.Automated Glycan Assembly (AGA) enables rapid access to a multitude of biologically relevant glycans, reducing effort and time.14–17 AGA relies on the iterative coupling of monosaccharide building blocks (BBs) on a solid support. Each BB is equipped with a leaving group and a temporary protecting group that is easily removed after glycosylation to release a free hydroxyl group that serves as the new glycosyl acceptor in the next coupling cycle. Long purification processes are replaced by simple washing steps to decrease the synthesis time (each elongation step requires 1.5 h).18 Upon completion of the assembly, the protected glycan target is released from the solid support, before subsequent final deprotection affords the desired product. The step-wise elongation results in well-defined polysaccharides. The automated assembly of a 50-mer18,19 and most recently a 100-mer polymannoside (achieved in four and eight days respectively) set the stage to explore the properties of well-defined carbohydrate materials. A collection of oligo- and polysaccharides, including natural and non-natural structures, provided the ideal starting point to study fundamental properties of carbohydrates (Fig. 3).20 Polysaccharides of different lengths (hexamer and dodecamers) were prepared from different monosaccharide units (i.e. mannose, glucose, and glucosamine). Interestingly, for particular oligomers longer than six units, the formation of insoluble compounds during post-AGA manipulations poses a severe bottleneck, resulting in low isolated yields. Differences in aggregation tendency and solubility are associated with the conformations adopted by the different oligosaccharides in solution. Molecular dynamics (MD) simulations and NMR experiments revealed that some polymers form helices (e.g. β-1,6 oligoglucosides), while others adopt rod-like structures (e.g. β-1,4 oligoglucosides). Notably, the lack of a rigid three-dimensional structure observed for polymannosides is associated with the highest isolated yields. Modifications in specific positions of the oligosaccharide chains permitted tuning of three-dimensional structures and properties (i.e. solubility), simplifying the synthetic process. In particular, the introduction of a single mannose unit in the oligoglucoside chain distorted the helical conformation, resulting in much more soluble analogues.
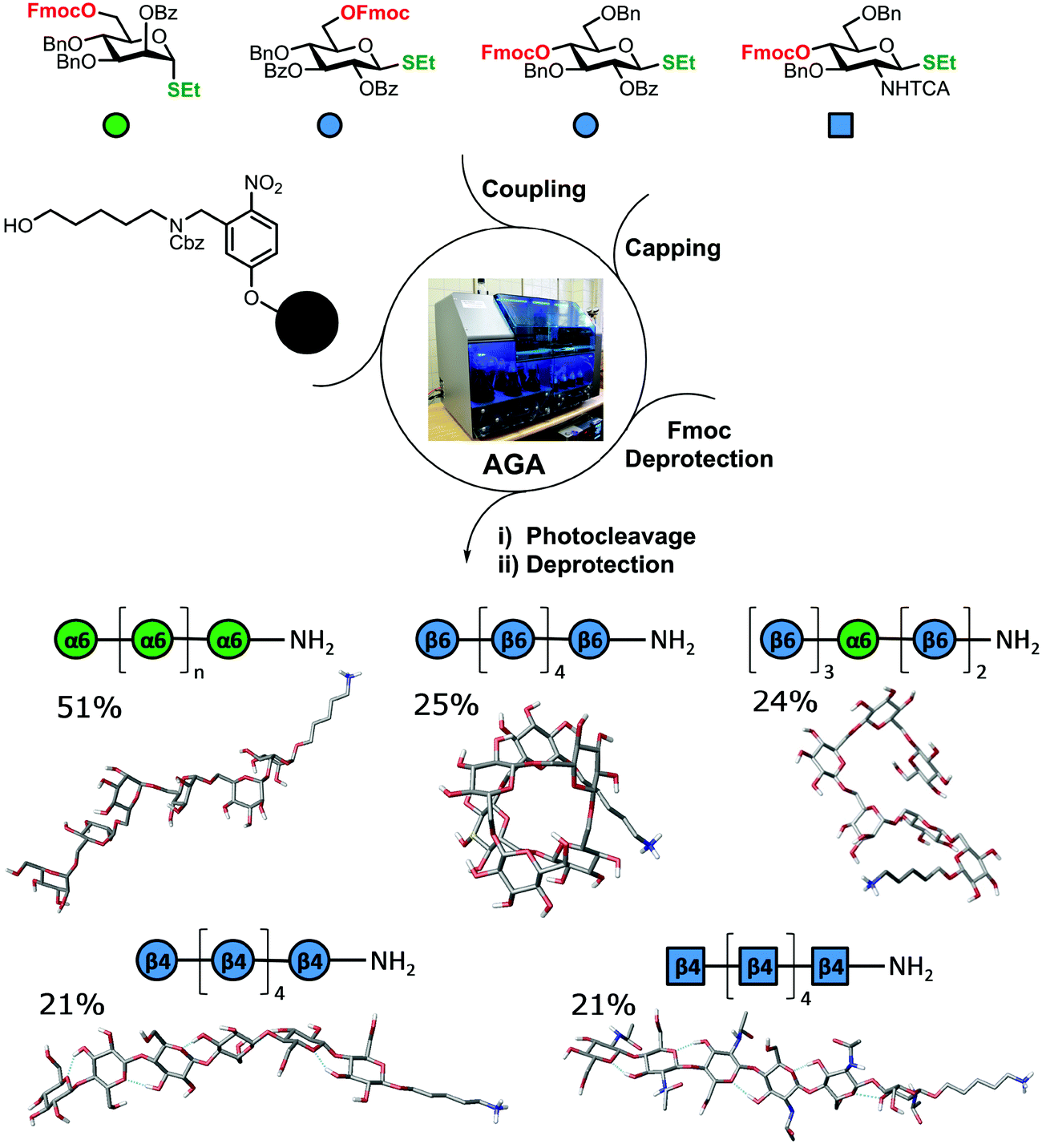 | ||
| Fig. 3 AGA of a collection of well-defined oligosaccharides revealing that some polymers form helices (β-1,6 glucosides), some adopt rod-like structures (β-1,4 oligomers), while others lack a defined conformational structure (α-1,6 mannosides). Isolated yields after AGA, deprotection, and purifications are shown. | ||
Similarly, tailor-made methylated, deoxygenated, and deoxyfluorinated cellulose analogues, in addition to well-defined carboxymethyl cellulose, were synthesized by AGA to tune the otherwise rigid three-dimensional structure of cellulose. The compounds were prepared with full control over the length, pattern and degree of substitution (Fig. 4).21 The unnatural modifications were designed to selectively disrupt hydrogen-bond networks and/or alter the electronic properties, aiming to improve the poor solubility of cellulose and establish structure-properties correlations. Such systematic hydrogen bond manipulations tuned the geometry and flexibility of the oligomer with significant improvements in the isolated yields. All unnatural hexasaccharide analogues were obtained in yields of 24–72% compared to the 18% yield for the natural analogue A6. Analogues with the same degree of substitution, but different substitution patterns, showed remarkably different geometries, stressing the importance of a synthetic approach to establish definitive structure–property correlations.
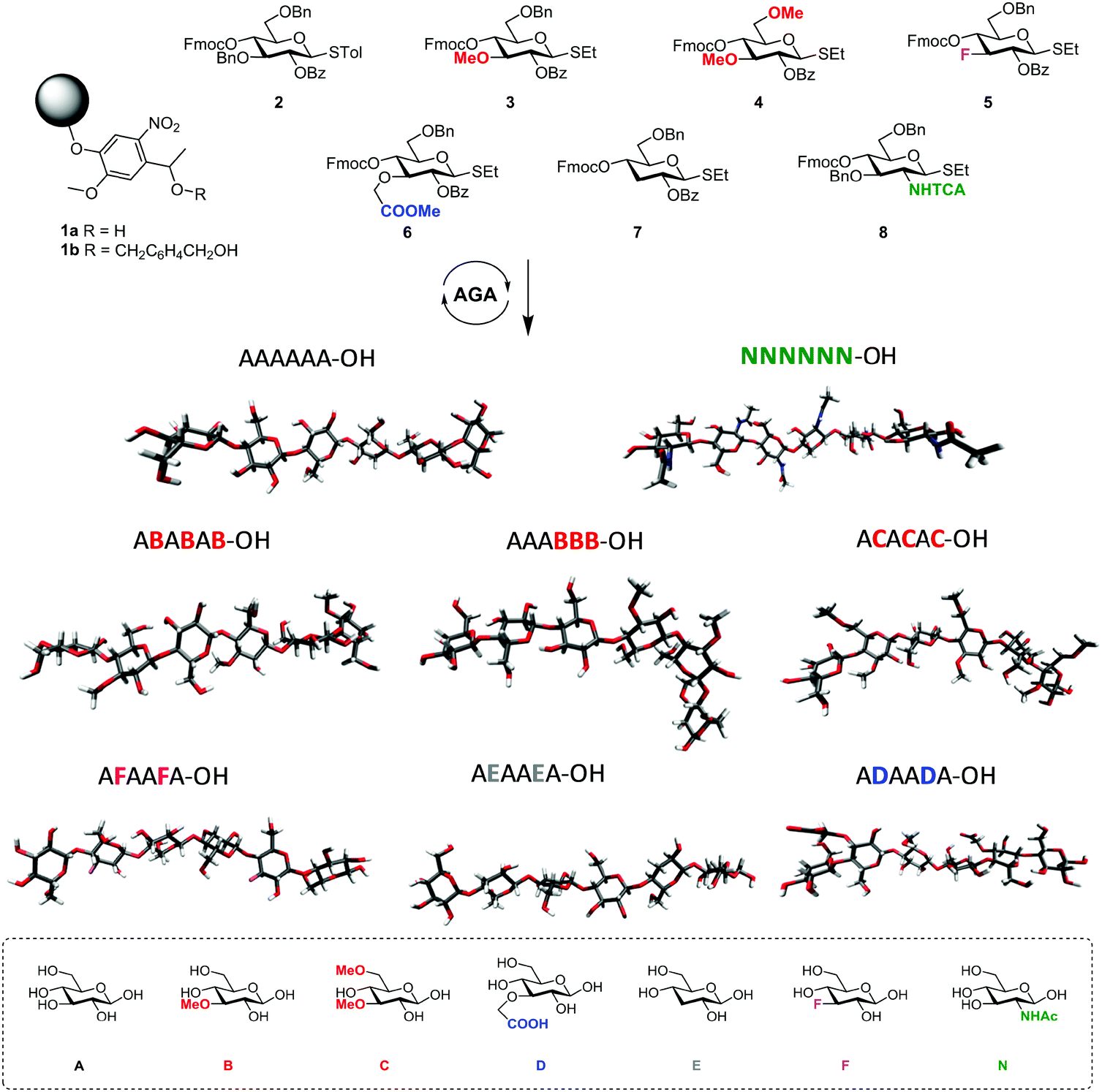 | ||
| Fig. 4 AGA of a collection of tailor-made cellulose derivatives. Modifications were designed to disrupt particular H-bonds and alter the rigidity of natural cellulose oligomers.21 | ||
3. Structural characterization
Large collections of well-defined polysaccharides are the basis for their characterization at the molecular level. Still, the intrinsic complexity of polysaccharides and the lack of suitable analytical techniques, renders the structural characterization of glycans extremely challenging.22 Sufficiently large single polysaccharide crystals are hard to obtain limiting the use of X-ray crystallography. Moreover, the conformation of most polysaccharides in solution remains elusive, as the intrinsic flexibility of polysaccharides limits the use of conventional techniques, that produce averaged information. MD simulations can help the prediction of oligosaccharide conformation in water,23 but a lack of validation standards has slowed down the development of reliable simulation packages. In recent years, new force fields, optimized for carbohydrates, have become more accurate and user friendly.24 Still, MD analyses are not used routinely in the synthetic laboratory to guide carbohydrate syntheses as most force fields do not support non-natural modifications.The most common strategy to study glycan conformation remains NMR spectroscopy. Scalar J-couplings and residual dipolar couplings (RDCs) could be extremely informative25 and chemical shift degeneracy of polysaccharides can be overcome elegantly with the introduction of isotopic labels (i.e.13C and 19F)20 or paramagnetic lanthanide complexes.26 To this end, synthetic analogues that bear labels or tags are particularly valuable to simplify the NMR analysis.27 The specific substitution of some hydroxyl groups with the isosteric fluorine shed light on the role of the replaced hydroxyl group in the overall conformation stabilization21 and interaction with protein receptors.28 The targeted OH/F substitution is a tool to map the hydrogen bonds required to stabilize a particular conformation. On the other hand, deoxyfluorination can also tune the oligosaccharide interactions with water molecules, suggesting that OH/F substitutions can have a profound effect on the glycan conformation. The combination of MD simulations with NMR experiments is often the best solution for structural analysis at the molecular level. Synthetic oligosaccharide collections followed by MD and NMR analyses confirmed that a particular conformation (i.e. a full helical turn) was essential for good interaction with the antibody, with important implications in vaccine design.29
4. Polysaccharide materials
Synthetic chemistry can manipulate the conformation of oligosaccharides at the molecular level. From a material science standpoint, it is important to understand the effect of a defined chemical substitution on the overall material properties. To this end, electron microscopy (EM), atomic force microscopy, X-ray powder diffraction, and solid state NMR analysis are valuable tools for the biophysical characterization of carbohydrate materials and their mechanical properties.30,31 To date, such techniques are mainly applied to natural polysaccharide materials, where large quantities are readily available. A molecular level understanding of how synthetic carbohydrate materials are formed is required to make tunable materials. For example, a systematic hydrogen bond manipulation helped to create unnatural cellulose materials with increased water-solubility and reduced crystallinity as single site substitutions disrupted the natural cellulose conformation.21 The industrial production of cellulose derivatives, such as cellulose esters and ethers, resulted in dramatic changes in the material properties.32 Limited control over the degree of modification prevents detailed structure–property correlations to be established and generates unreproducible results. Synthetic well-defined polysaccharides allowed for definitive correlations between the increased flexibility of the single chain and the reduced aggregation, precipitation, and regular packing upon drying. Importantly, not only the nature but also the pattern of substitution plays a crucial role, with a higher cellulose-like character observed for the regularly substituted analogues as compared to randomly functionalized structures (Fig. 5). Such precision substitution could only be obtained following a synthetic approach.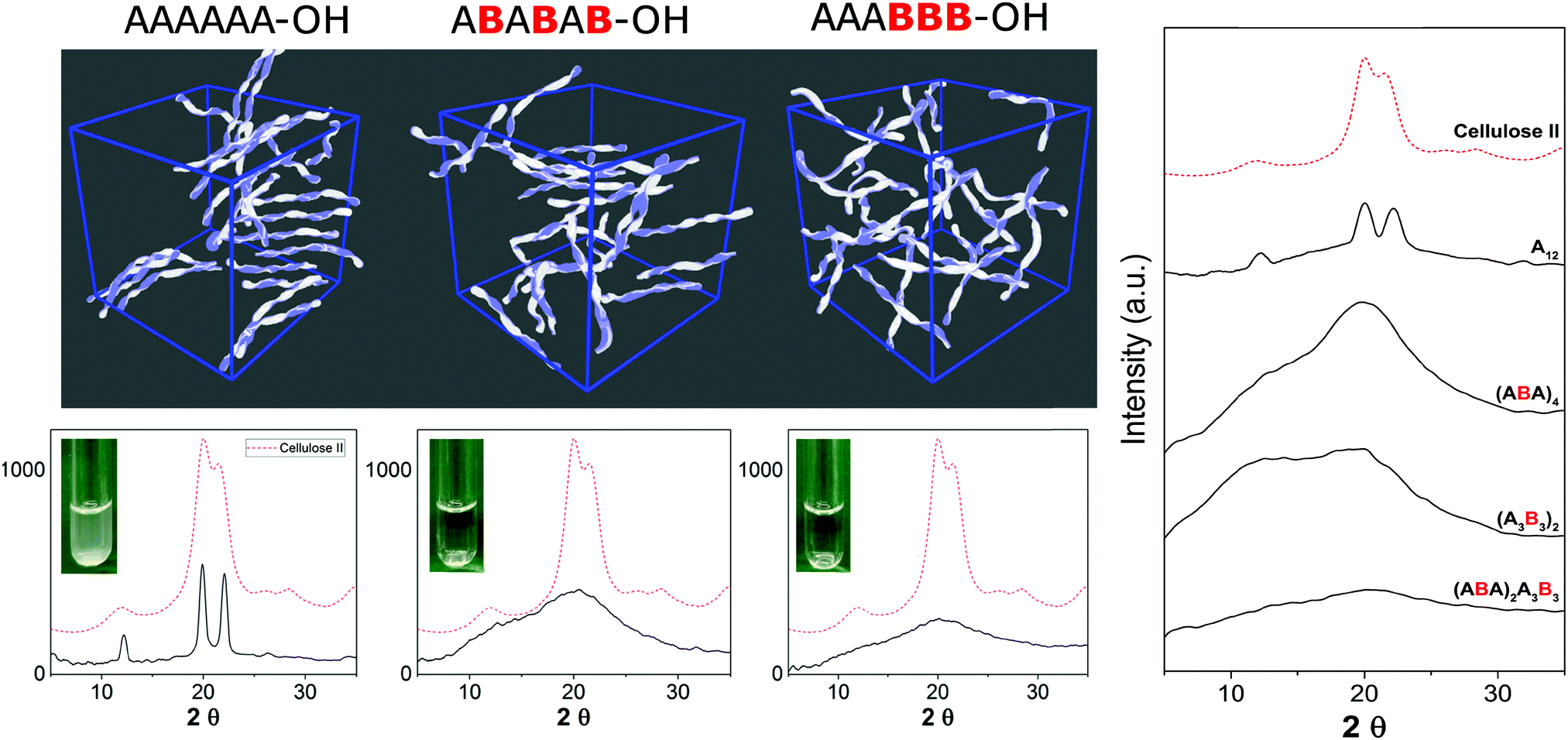 | ||
| Fig. 5 Methylated cellulose oligomers exhibit different solubility and aggregation tendencies depending on the methylation pattern, as suggested by MD simulations and confirmed by XRD analysis. Reproduced from ref. 21 under the terms of the Creative Commons Attribution License. Copyright 2019 Y. Yu et al. Published by Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Next, fundamental insights can guide the creation of novel synthetic oligosaccharides materials. Much like peptides and oligonucleotides, oligosaccharide self-assembly can generate synthetic materials for nanotechnology applications. Recently, self-assembly of synthetic oligosaccharides was first observed into nanostructures with distinct morphologies, depending on chain length and modifications (Fig. 6).33 The combination of hydrogen bonding and π–π interactions involving benzyl ether modifications drove the supramolecular assembly. Different assembly conditions produced remarkably diverse morphologies. Supramolecular aggregation induced by slow dialysis resulted in spherical particles for all of the studied compounds (Fig. 6, 9–14a). A much faster aggregation process, based on solvent-switch, showed much more diversity. Fibers, gels, or spherical particles are obtained depending on the length and modification pattern (Fig. 6, 9–14b). These differences likely are a consequence of the different conformations adopted by the oligosaccharides in solution. Unexpected excitation-dependent intrinsic optical properties are also observed, as a consequence of supramolecular aggregation.
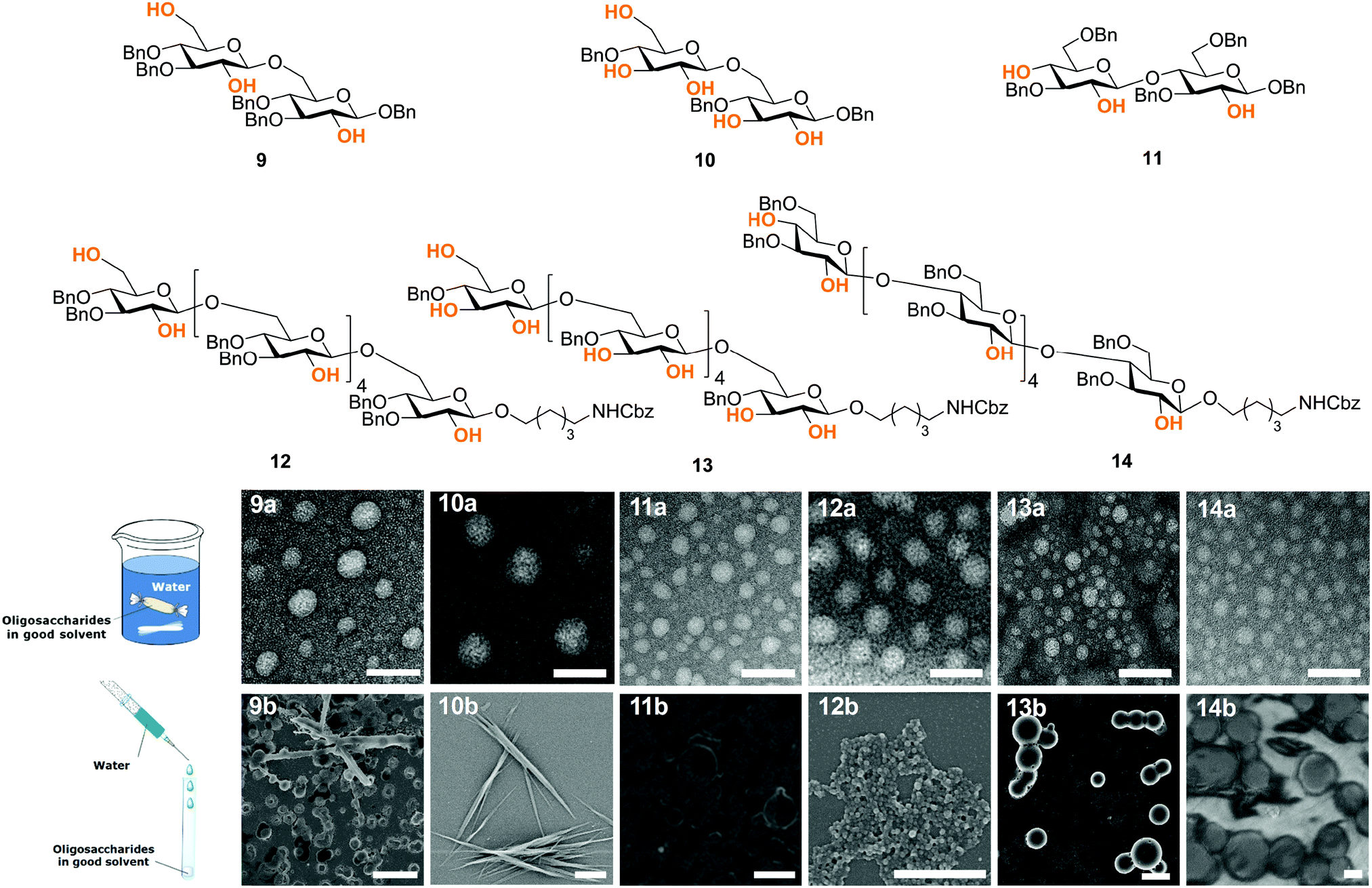 | ||
| Fig. 6 Synthetic oligosaccharides self-assemble with different morphologies depending on the length, substitution and assembly method. TEM images (scale bars: 100 nm) of sample prepared with dialysis (9–14a), SEM images (scale bars: 2 μm) of samples prepared by solvent-switch method (9–14b). | ||
5. Conclusions and perspective
Reliable automated techniques grant unprecedented access to well-defined oligo- and polysaccharides as important tools for fundamental studies of polysaccharides structure and properties. The field of synthetic carbohydrate materials is still in its infancy, yet synthetic oligomers have helped to gain fundamental insights into the forces that guide polysaccharide aggregation in nature. Fine-tuning of single molecule conformations will enable the creation of designer supramolecular materials with adjustable mechanical and physical properties, with applications in drug delivery and tissue engineering. Systems mimicking the extracellular environment, a key area for cellular interaction and communication, can be imagined. The systematic approach outlined here will have to be brought to bear on more naturally occurring polysaccharides and their derivatives. The introduction of charges is crucial to exploit polysaccharide materials to their full potential. Synthesis scale up will have to be addressed for the production of these synthetic materials. Supramolecular materials, based on the self-assembly of simple peptides, have found applications in nanotechnology, due to their intrinsic dynamic nature.34,35 Synthetic oligosaccharide materials are expected to combine the benefits of dynamic supramolecular materials and biocompatible polysaccharides, with important implications in nanotechnology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Max-Planck Society, the Minerva Fast Track Program, and the MPG-FhG Cooperation Project Glyco3Dysplay, for generous financial support and Theodore Tyrikos-Ergas for the help with graphics. Open Access funding provided by the Max Planck Society.References
- F. Lapenta, J. Aupič, Ž. Strmšek and R. Jerala, Chem. Soc. Rev., 2018, 47, 3530–3542 RSC.
- Q. Luo, C. Hou, Y. Bai, R. Wang and J. Liu, Chem. Rev., 2016, 116, 13571–13632 CrossRef CAS PubMed.
- F. Hong, F. Zhang, Y. Liu and H. Yan, Chem. Rev., 2017, 117, 12584–12640 CrossRef CAS PubMed.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- R. Veneziano, S. Ratanalert, K. Zhang, F. Zhang, H. Yan, W. Chiu and M. Bathe, Science, 2016, 352, 1534 CrossRef CAS PubMed.
- P.-S. Huang, S. E. Boyken and D. Baker, Nature, 2016, 537, 320 CrossRef CAS PubMed.
- M. Delbianco, P. Bharate, S. Varela-Aramburu and P. H. Seeberger, Chem. Rev., 2016, 116, 1693–1752 CrossRef CAS PubMed.
- S. Gim, Y. Zhu, P. H. Seeberger and M. Delbianco, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2019, 0, e1558 Search PubMed.
- M. Kostag, M. Gericke, T. Heinze and O. A. El Seoud, Cellulose, 2019, 26, 139–184 CrossRef CAS.
- S. C. Fox, B. Li, D. Xu and K. J. Edgar, Biomacromolecules, 2011, 12, 1956–1972 CrossRef CAS PubMed.
- J.-i. Kadokawa, Chem. Rev., 2011, 111, 4308–4345 CrossRef CAS PubMed.
- R. Xiao and M. W. Grinstaff, Prog. Polym. Sci., 2017, 74, 78–116 CrossRef CAS.
- W. Thongsomboon, D. O. Serra, A. Possling, C. Hadjineophytou, R. Hengge and L. Cegelski, Science, 2018, 359, 334–338 CrossRef CAS PubMed.
- M. Guberman and P. H. Seeberger, J. Am. Chem. Soc., 2019, 141, 5581–5592 CrossRef CAS PubMed.
- H. S. Hahm, F. Broecker, F. Kawasaki, M. Mietzsch, R. Heilbronn, M. Fukuda and P. H. Seeberger, Chem, 2017, 2, 114–124 CAS.
- B. Schumann, H. S. Hahm, S. G. Parameswarappa, K. Reppe, A. Wahlbrink, S. Govindan, P. Kaplonek, L. A. Pirofski, M. Witzenrath, C. Anish, C. L. Pereira and P. H. Seeberger, Sci. Transl. Med., 2017, 9, eaaf5347 CrossRef PubMed.
- M. Panza, S. G. Pistorio, K. J. Stine and A. V. Demchenko, Chem. Rev., 2018, 118, 8105–8150 CrossRef CAS PubMed.
- Y. Yu, A. Kononov, M. Delbianco and P. H. Seeberger, Chem. – Eur. J., 2018, 24, 6075–6078 CrossRef CAS PubMed.
- K. Naresh, F. Schumacher, H. S. Hahm and P. H. Seeberger, Chem. Commun., 2017, 53, 9085–9088 RSC.
- M. Delbianco, A. Kononov, A. Poveda, Y. Yu, T. Diercks, J. Jiménez-Barbero and P. H. Seeberger, J. Am. Chem. Soc., 2018, 140, 5421–5426 CrossRef CAS PubMed.
- Y. Yu, T. Tyrikos-Ergas, Y. Zhu, G. Fittolani, V. Bordoni, A. Singhal, R. J. Fair, A. Grafmüller, P. H. Seeberger and M. Delbianco, Angew. Chem., Int. Ed., 2019, 58, 1433–7851 Search PubMed.
- M. R. Wormald, A. J. Petrescu, Y.-L. Pao, A. Glithero, T. Elliott and R. A. Dwek, Chem. Rev., 2002, 102, 371–386 CrossRef CAS PubMed.
- R. J. Woods, Chem. Rev., 2018, 118, 8005–8024 CrossRef CAS.
- K. N. Kirschner, A. B. Yongye, S. M. Tschampel, J. González-Outeiriño, C. R. Daniels, B. L. Foley and R. J. Woods, J. Comput. Chem., 2008, 29, 622–655 CrossRef CAS PubMed.
- P. Valverde, J. I. Quintana, J. I. Santos, A. Ardá and J. Jiménez-Barbero, ACS Omega, 2019, 4, 13618–13630 CrossRef CAS PubMed.
- A. Canales, I. Boos, L. Perkams, L. Karst, T. Luber, T. Karagiannis, G. Domínguez, F. J. Cañada, J. Pérez-Castells, D. Häussinger, C. Unverzagt and J. Jiménez-Barbero, Angew. Chem., Int. Ed., 2017, 56, 14987–14991 CrossRef CAS.
- T. Tyrikos-Ergas, G. Fittolani, P. H. Seeberger and M. Delbianco, Biomacromolecules, 2020, 21, 18–29 CrossRef CAS PubMed.
- T. Diercks, A. S. Infantino, L. Unione, J. Jiménez-Barbero, S. Oscarson and H.-J. Gabius, Chem. – Eur. J., 2018, 24, 15761–15765 CrossRef CAS.
- Q. Zhang, A. Gimeno, D. Santana, Z. Wang, Y. Valdés-Balbin, L. M. Rodríguez-Noda, T. Hansen, L. Kong, M. Shen, H. S. Overkleeft, V. Vérez-Bencomo, G. A. van der Marel, J. Jiménez-Barbero, F. Chiodo and J. D. C. Codée, ACS Cent. Sci., 2019, 5, 1407–1416 CrossRef CAS PubMed.
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- M. Diener, J. Adamcik, A. Sánchez-Ferrer, F. Jaedig, L. Schefer and R. Mezzenga, Biomacromolecules, 2019, 20, 1731–1739 CrossRef CAS PubMed.
- H. C. Arca, L. I. Mosquera-Giraldo, V. Bi, D. Xu, L. S. Taylor and K. J. Edgar, Biomacromolecules, 2018, 19, 2351–2376 CrossRef CAS PubMed.
- Y. Yu, S. Gim, D. Kim, Z. A. Arnon, E. Gazit, P. H. Seeberger and M. Delbianco, J. Am. Chem. Soc., 2019, 141, 4833–4838 CrossRef CAS PubMed.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2015, 15, 13 CrossRef PubMed.
- R. Freeman, M. Han, Z. Álvarez, J. A. Lewis, J. R. Wester, N. Stephanopoulos, M. T. McClendon, C. Lynsky, J. M. Godbe, H. Sangji, E. Luijten and S. I. Stupp, Science, 2018, 362, 808–813 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |