
Reactivity of borohydride incorporated in coordination polymers toward carbon dioxide†
Kentaro
Kadota
 a,
Easan
Sivaniah
ab and
Satoshi
Horike
*bcde
a,
Easan
Sivaniah
ab and
Satoshi
Horike
*bcde
aDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan
bInstitute for Integrated Cell-Material Sciences, Institute for Advanced Study, Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: horike@icems.kyoto-u.ac.jp
cAIST-Kyoto University Chemical Energy Materials Open Innovation Laboratory (ChEM-OIL), National Institute of Advanced Industrial Science and Technology (AIST), Yoshida-Honmachi, Sakyo-ku, Kyoto 606-8501, Japan
dDepartment of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan
eDepartment of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology, Rayong 21210, Thailand
First published on 6th April 2020
Abstract
Borohydride (BH4−)-containing coordination polymers converted CO2 into HCO2− or [BH3(OCHO)]−, whose reaction routes were affected by the electronegativity of metal ions and the coordination mode of BH4−. The reactions were investigated using thermal gravimetric analysis under CO2 gas flow, infrared spectroscopy, and NMR experiments.
Conversion of carbon dioxide (CO2) into valuable chemicals is a key to realize a sustainable society.1,2 In particular, it is essential to establish chemical reactions that transform CO2 into various types of chemical moieties under mild conditions.3 However, the inherent inertness of CO2 has hampered the utilization of CO2 in transformation reactions. To overcome the inertness, various catalytic and stoichiometric reactions have been widely studied in both solution and the solid-state, including metals, metal oxides,4 metal complexes,5,6 and metal-free organic molecules.7
Borohydride (BH4−), a hydride-based complex anion, has been commonly utilized as a reducing agent. In the solution phase, metal borohydrides (MBHs) stoichiometrically react with CO2 at ambient temperatures and pressures.8–11 BH4− in solution is able to convert CO2 into chemical species such as formate (HCO2−) and formylhydroborate ([BH4−x(OCHO)x]−, x = 1, 2 and 3) depending on the reaction conditions, e.g. counter cations, temperatures, solvents, and pressures.8,9,12 The solid-state reactivity of BH4− toward CO2 is also interesting from the viewpoint of heterogeneous catalysts and CO2 scrubbers. Nevertheless, limited studies have been performed on the solid-state reactivity of MBHs toward CO2.13,14 This is because slow diffusion of CO2 in dense MBHs results in low reactivity under mild conditions.14 Although porous structures are advantageous for the diffusion of CO2, MBHs with the porous structure are limited except for a few examples, e.g. γ-Mg(BH4)2.14,15
Coordination polymers (CPs) and metal–organic frameworks (MOFs) are crystalline solids constructed from metal ions and bridging organic linkers.16–18 Their open structures have offered an attractive platform for various gas–solid reactions, such as CO2 sorption19–21 and post-synthetic modification.22,23 In addition, rich structural and chemical tunability of CPs demonstrated the controlled reactivity of reactive species, e.g. radicals,24,25 imines,26 and photoactive metal complexes.27 CPs are a promising platform for solid–gas reactions between BH4− and CO2. BH4−-containing CPs are constructed from metal ions (e.g. Mg2+, Ca2+, Mn2+, Zn2+, and Th4+) and N-based neutral linkers show various types of the chemical environment of BH4−.28–30 Here, we investigate the reactivity of BH4−-containing CPs to convert CO2 into HCO2− or [BH3(OCHO)]− under mild conditions depending on their structures.
[M(BH4)2(pyz)2] (M-pyz, M = Mg2+, Ca2+, pyz = pyrazine)28,29 were selected to investigate the influence of metal ions on the reactivity of BH4− toward CO2. The metal ion center shows an octahedral geometry and the two BH4− ions coordinate in the axial positions (Fig. 1A). The extended structure of M-pyz comprises a 2D square grid constructed by [M4pyz4] units (Fig. 1B). The electronic properties and reactivity of BH4− are influenced by the electronegativity of counter metal ions.31,32 Attempts at the synthesis of isostructural M-pyz were made using a Mn2+-based MBH precursor. [Mn(BH4)2·3THF]·NaBH4 was prepared following the literature methods.33 The general synthetic method involves mechanochemical milling of the MBH precursor and pyz under Ar. Mg-pyz was previously synthesized in the solution phase, whereas the solvent-free conditions afford a highly crystalline product as well (Fig. S1, ESI†). The powder X-ray diffraction (PXRD) pattern of Mn-pyz shows a good agreement with that of Mg-pyz (Fig. S1, ESI†).
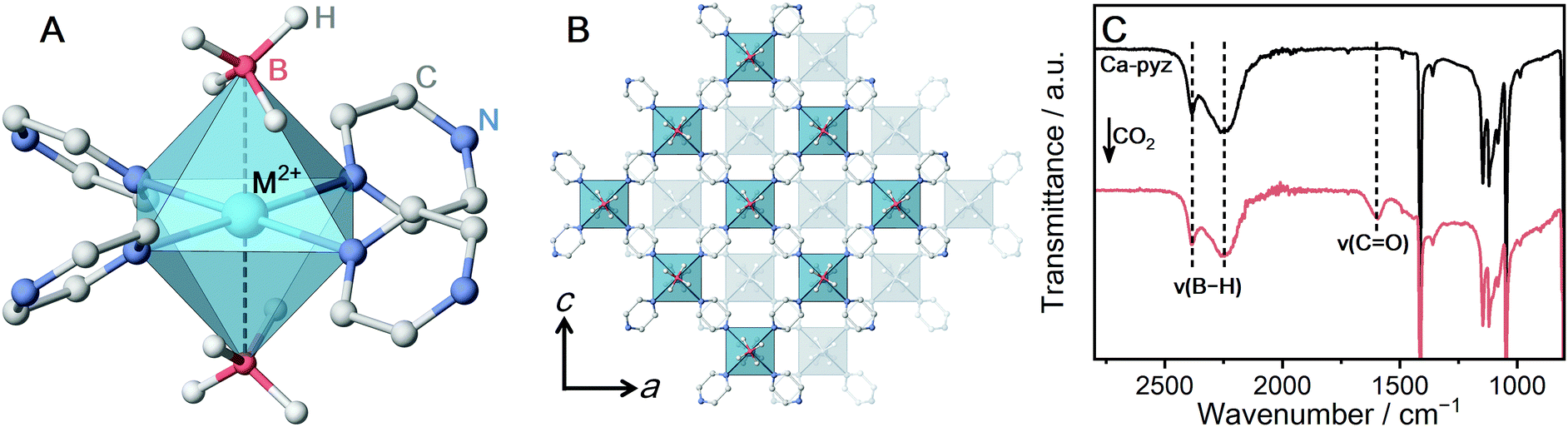 | ||
| Fig. 1 (A) Local coordination geometry of M-pyz (M = Mg2+, Ca2+, Mn2+). (B) ABAB stacking structure of the extended 2D layers of M-pyz (M = Mg2+, Ca2+, Mn2+). (C) IR spectra of Ca-pyz before and after CO2 adsorption at 25 °C. | ||
The solid-state synthesis of M-pyz proceeds without solvents at 25 °C within 30 min. The fast reaction kinetics in the solid-state is ascribed to the low melting point of pyz (52 °C). The lower melting point of reactants leads to higher molecular mobility, enhancing the reactivity in the solid-state.34 Mechanical milling is useful to synthesize CPs from MBHs because most of the MBHs are poorly soluble in common organic solvents. The thermal properties were characterized by thermal gravimetric analysis (TGA) under N2 (Fig. S2, ESI†). Each compound exhibits a weight loss at relatively low temperatures; 50, 70, and 70 °C for Mg-, Mn-, and Ca-pyz due to the low boiling point of pyz (115 °C). Isothermal TGA measurements at 40 °C under N2 indicate that Ca-pyz shows higher thermal stability than Mn-pyz (weight loss after 6 hours; 0.2 vs. 3.2 wt%, Fig. S3, ESI†). In the case of MBHs, electropositive metal ions construct MBHs with higher thermal stability.35 Meanwhile, in the case of BH4−-containing CPs, the strength of the coordination bonds is also essential. The Hard and Soft Acids and Bases (HSAB) theory reveals that electropositive metal ions (hard acids) form weaker coordination bonds with nitrogen-based linkers (soft bases) such as pyz. Therefore, the trend of thermal stability for M-pyz does not simply follow the electronegativity of metal ions (thermal stability: Mg < Mn < Ca, Pauling electronegativity: Ca < Mg < Mn).
To characterize the chemical environment of BH4− in the CP, solid-state 11B magic angle spinning (MAS) nuclear magnetic resonance (NMR) was carried out on non-paramagnetic Ca-pyz. The 11B NMR spectrum of Ca-pyz displays a peak at −36 ppm corresponding to the signal of BH4− (Fig. S4, ESI†). The total charge on BH4− is correlated with the chemical shift of 11B NMR: electron-rich BH4− shows a peak in a lower frequency.31 The low-frequency shift of the 11B peak indicates that BH4− in Ca-pyz is more electron-rich than Ca(BH4)2. In the framework of Ca-pyz, the Lewis acidity of Ca2+ was reduced by electron donation from the coordinating pyz molecules, which leads to the formation of electron-rich BH4−.29
CO2 adsorption measurement was carried out to evaluate the reactivity of Ca-pyz in gas–solid equilibrium. The CO2 isotherm at 25 °C displays irreversible adsorption (7 mL g−1 at 100 kPa), which is characteristic of chemisorption behavior (Fig. S5, ESI†).36 The IR spectrum of Ca-pyz after CO2 adsorption displays a new peak at 1600 cm−1, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (Fig. 1C). The solid-state 1H–13C cross-polarization (CP) MAS NMR spectrum of Ca-pyz after CO2 adsorption shows peaks at 170 and 145 ppm. The peaks correspond to the signals of HCO2− and pyz, respectively (Fig. S6, ESI†). The results indicate that BH4− in Ca-pyz reduces CO2 to HCO2− with the release of diborane (B2H6) as a by-product.10
O stretching (Fig. 1C). The solid-state 1H–13C cross-polarization (CP) MAS NMR spectrum of Ca-pyz after CO2 adsorption shows peaks at 170 and 145 ppm. The peaks correspond to the signals of HCO2− and pyz, respectively (Fig. S6, ESI†). The results indicate that BH4− in Ca-pyz reduces CO2 to HCO2− with the release of diborane (B2H6) as a by-product.10
The kinetic reactivity of M-pyz toward CO2 was evaluated using isothermal TGA under CO2 flow. Fig. 2 displays the TGA profiles of each powder sample (10 mg) under CO2 flow (0.1 MPa, 30 mL min−1) at 40 °C. Mg-pyz and Ca-pyz exhibit higher weight increases than Mn-pyz (25.5, 21.9, and 3.2 wt% after 400 min, respectively). Ca-pyz was amorphous after the CO2 reaction, as confirmed by PXRD (Fig. S7, ESI†). To identify the chemical species after the CO2 reaction, solution NMR was carried out on Ca-pyz dissolved in DMSO-d6. The solution 13C NMR spectrum of Ca-pyz after the CO2 reaction displays peaks at 167, 146, 53, 50, 47 and 44 ppm (Fig. S8, ESI†). The peaks at 167 and 146 ppm correspond to the 13C signals of HCO2− and pyz, respectively. The peak at 47 ppm is assigned to piperazine formed by the reduction of pyz by B2H6, whereas the rest of the peaks are not able to be assigned.29,37 The higher reactivity of Ca-pyz toward CO2 is attributed to the preferable electronic interaction between Ca2+ (hard acid) and HCO2− (hard base) rather than BH4− (soft base).
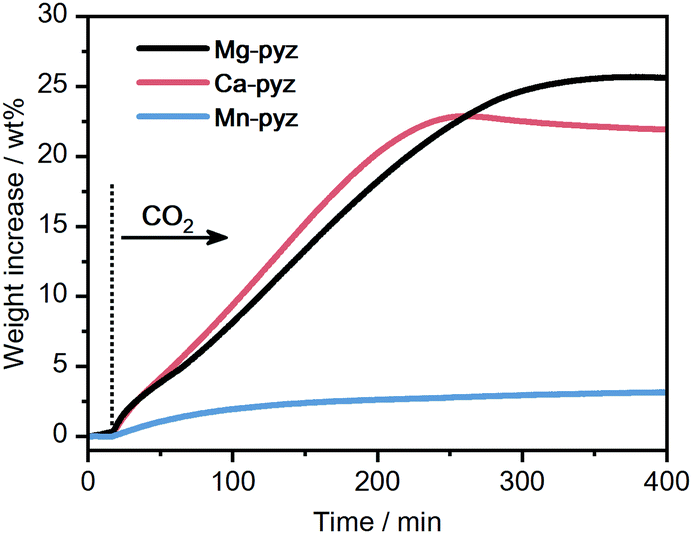 | ||
| Fig. 2 Isothermal TGA profiles of M-pyz (M = Mg2+, Ca2+, Mn2+) under CO2 flow (0.1 MPa, 30 mL min−1) at 40 °C. | ||
The formation of [BH4−x(OCHO)x]− from BH4− and CO2 was investigated at a BH4−-containing CP. Given that [BH4−x(OCHO)x]− is bulky than HCO2−, [Mn(BH4)2(dpe)1.5] (Mn-dpe, dpe = dipyridylethane) having voids was selected.28 The two BH4− ions coordinate to the Mn2+ center in a bidentate manner, which was confirmed by single-crystal X-ray diffraction (SC-XRD) in Fig. 3A. The extended structure of Mn-dpe comprises a 1D ladder constructed from [Mn4dpe4] units (Fig. 3B). The coordination mode of BH4− was confirmed by IR spectroscopy as well. The IR spectrum of Mn-dpe displays two stretching peaks in the B–H stretching region at 2378 and 2127 cm−1, respectively (Fig. 4B). The peak at 2378 cm−1 corresponds to the B–H bond coordinating to the Mn2+ center, whereas the peak at 2127 cm−1 corresponds to the non-coordinating B–H.38 In contrast to the broadened B–H stretching peak of Ca-pyz (Fig. 1C), Mn-dpe displays distinct two peaks of B–H stretching, which is originated from a stronger binding interaction between Mn2+ (soft acid) and BH4− (soft base).
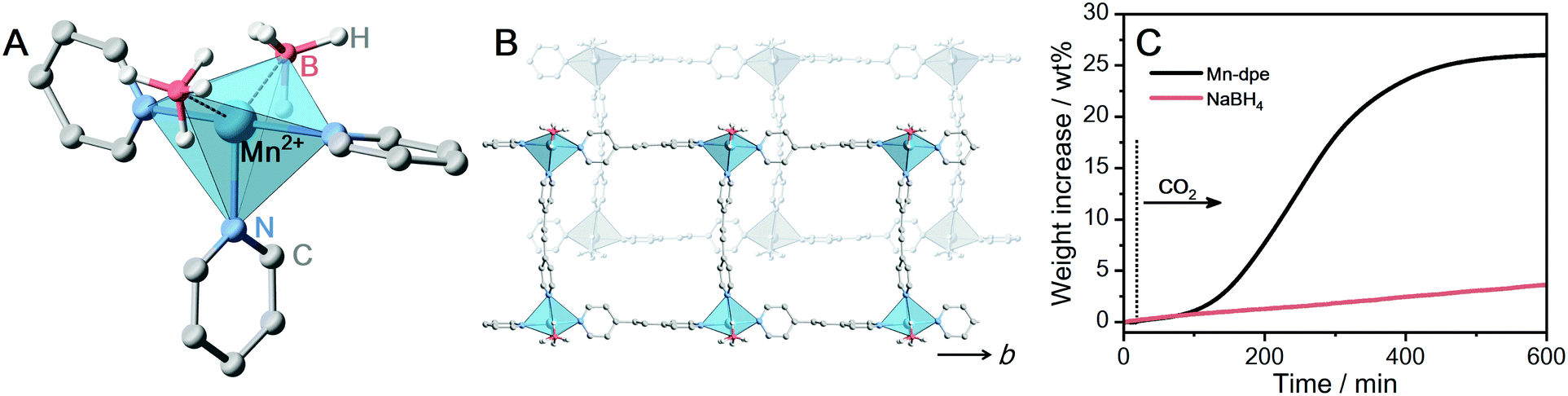 | ||
| Fig. 3 (A) Local coordination geometry of Mn-dpe. (B) Packing structure of the extended 1D ladders of Mn-dpe. (C) Isothermal TGA profiles of Mn-dpe and NaBH4 under CO2 flow (0.1 MPa, 30 mL min−1) at 40 °C. | ||
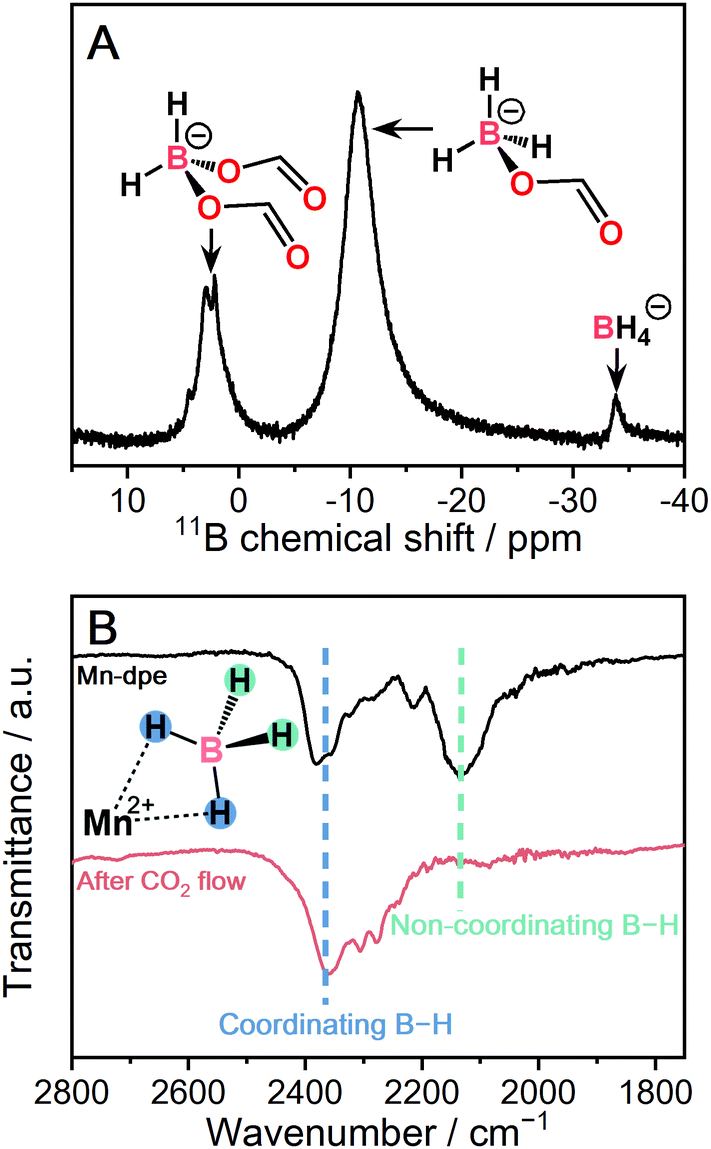 | ||
| Fig. 4 (A) Solution 11B{1H} NMR of digested Mn-dpe after CO2 reaction. (B) IR spectra of Mn-dpe before and after the CO2 reaction. | ||
The kinetic curve of the CO2 reaction with Mn-dpe was collected using the same procedure as M-pyz (Fig. 3B). Mn-dpe demonstrates a weight increase of 26.2 wt% after 600 min at 40 °C, which corresponds to a value of the 1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of reacted CO2 per BH4−. After the CO2 reaction, Mn-dpe shows small diffraction peaks different from the original peaks (Fig. S9, ESi†). Solution 11B NMR measurement was carried out to determine the chemical species after the CO2 reaction. The 11B{1H} NMR spectrum of digested Mn-dpe after the CO2 reaction displays the peaks at −33, −11, and 2.2 ppm in Fig. 4A. The broad peaks were observed due to the paramagnetic effect of Mn2+. The 11B peaks correspond to BH4−, [BH3(OCHO)]− and [BH2(OCHO)2]−, respectively.9,39 Successive CO2 insertions into the B–H bond of BH4− produce [BH4−x(OCHO)x]−, and the number of reacted CO2 molecules is affected by the reaction conditions such as pressure and temperature in the solution phase.8,9 The reaction of NaBH4 in acetonitrile with 0.1 MPa of CO2 for 10 minutes produces [BH(OCHO)3]− as a major product, and [BH3(OCHO)]− is not observed.9 This is because all the hydrogen atoms of BH4− dissociated in acetonitrile are available for the reaction with CO2. On the other hand, in the case of Mn-dpe, two of the hydrogen atoms of BH4− are pinned with the Mn2+ center by a coordination bond as confirmed by SC-XRD and IR spectroscopy. After the CO2 reaction, a non-coordinating B–H stretching peak was not observed, and this is because of the reaction with CO2 to form [BH3(OCHO)]− and [BH2(OCHO)2]− in Fig. 4B. The coordinating B–H stretching peak is preserved after the CO2 reaction, indicating the coordinating bonds between Mn2+ and [BH3(OCHO)]− or [BH2(OCHO)2]−. A sluggish kinetics of dense NaBH4 in the solid-state toward CO2 indicates that the open structure of Mn-dpe is essential for the diffusion of CO2 (Fig. 3C). Based on the results, the reaction between Mn-dpe and CO2 to produce [BH3(OCHO)]− and [BH2(OCHO)2]− is proposed (Fig. S11, ESI†). The results indicate that the anisotropic coordination geometry of BH4− in Mn-dpe affects the reaction route with CO2.
:1 molar ratio of reacted CO2 per BH4−. After the CO2 reaction, Mn-dpe shows small diffraction peaks different from the original peaks (Fig. S9, ESi†). Solution 11B NMR measurement was carried out to determine the chemical species after the CO2 reaction. The 11B{1H} NMR spectrum of digested Mn-dpe after the CO2 reaction displays the peaks at −33, −11, and 2.2 ppm in Fig. 4A. The broad peaks were observed due to the paramagnetic effect of Mn2+. The 11B peaks correspond to BH4−, [BH3(OCHO)]− and [BH2(OCHO)2]−, respectively.9,39 Successive CO2 insertions into the B–H bond of BH4− produce [BH4−x(OCHO)x]−, and the number of reacted CO2 molecules is affected by the reaction conditions such as pressure and temperature in the solution phase.8,9 The reaction of NaBH4 in acetonitrile with 0.1 MPa of CO2 for 10 minutes produces [BH(OCHO)3]− as a major product, and [BH3(OCHO)]− is not observed.9 This is because all the hydrogen atoms of BH4− dissociated in acetonitrile are available for the reaction with CO2. On the other hand, in the case of Mn-dpe, two of the hydrogen atoms of BH4− are pinned with the Mn2+ center by a coordination bond as confirmed by SC-XRD and IR spectroscopy. After the CO2 reaction, a non-coordinating B–H stretching peak was not observed, and this is because of the reaction with CO2 to form [BH3(OCHO)]− and [BH2(OCHO)2]− in Fig. 4B. The coordinating B–H stretching peak is preserved after the CO2 reaction, indicating the coordinating bonds between Mn2+ and [BH3(OCHO)]− or [BH2(OCHO)2]−. A sluggish kinetics of dense NaBH4 in the solid-state toward CO2 indicates that the open structure of Mn-dpe is essential for the diffusion of CO2 (Fig. 3C). Based on the results, the reaction between Mn-dpe and CO2 to produce [BH3(OCHO)]− and [BH2(OCHO)2]− is proposed (Fig. S11, ESI†). The results indicate that the anisotropic coordination geometry of BH4− in Mn-dpe affects the reaction route with CO2.
In conclusion, we demonstrated the reactivity of BH4− toward CO2 which is correlated with the crystal structures of BH4−-containing coordination polymers. The reactivity of [M(BH4)2(pyrazine)2] (M = Mg2+, Mn2+, Ca2+) and [Mn(BH4)2(dipyridylethane)1.5] toward CO2 at 40 °C was investigated by using isothermal TGA under CO2 flow, IR and NMR. BH4− in [Ca(BH4)2(pyrazine)2] converted CO2 into HCO2−. The BH4− pinned by coordination bonds with Mn2+ in [Mn(BH4)2(dipyridylethane)1.5] regulated the successive CO2 insertion reaction and produced [BH3(OCHO)]− as a major species. The structural diversity of coordination polymers provides a new approach to regulate the reaction routes between BH4− and CO2 in the solid-state.
The work was supported by the Japan Society of the Promotion of Science (JSPS) for a Grant-in-Aid for Scientific Research (B) (JP18H02032), the Challenging Research (Exploratory) (JP19K22200) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and Strategic International Collaborative Research Program (SICORP), the Adaptable and Seamless Technology Transfer Program through Target-driven R&D (A-STEP) from the Japan Science and Technology, Japan, Inamori Research Grants, and Tokuyama Science Foundation.
Conflicts of interest
The authors declare no conflict of interest.References
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- J. Artz, T. E. Muller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- J. L. White, M. F. Baruch, J. E. P. Iii, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- B. J. Cook, G. N. Di Francesco, K. A. Abboud and L. J. Murray, J. Am. Chem. Soc., 2018, 140, 5696–5700 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- G. La Monica, G. A. Ardizzoia, F. Cariati, S. Cenini and M. Pizzotti, Inorg. Chem., 1985, 24, 3920–3923 CrossRef CAS.
- I. Knopf and C. C. Cummins, Organometallics, 2015, 34, 1601–1603 CrossRef CAS.
- S. Murugesan, B. Stöger, M. Weil, L. F. Veiros and K. Kirchner, Organometallics, 2015, 34, 1364–1372 CrossRef CAS PubMed.
- J. G. Burr, W. G. Brown and H. E. Heller, J. Am. Chem. Soc., 1950, 72, 2560–2562 CrossRef CAS.
- K. Kadota, N. T. Duong, Y. Nishiyama, E. Sivaniah and S. Horike, Chem. Commun., 2019, 55, 9283–9286 RSC.
- J. Zhang and J. W. Lee, Carbon, 2013, 53, 216–221 CrossRef CAS.
- J. G. Vitillo, E. Groppo, E. G. Bardaji, M. Baricco and S. Bordiga, Phys. Chem. Chem. Phys., 2014, 16, 22482–22486 RSC.
- Y. Filinchuk, B. Richter, T. R. Jensen, V. Dmitriev, D. Chernyshov and H. Hagemann, Angew. Chem., Int. Ed., 2011, 50, 11162–11166 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O’Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS PubMed.
- E. González-Zamora and I. A. Ibarra, Mater. Chem. Front., 2017, 1, 1471–1484 RSC.
- M. Servalli, M. Ranocchiari and J. A. Van Bokhoven, Chem. Commun., 2012, 48, 1904–1906 RSC.
- V. Guillerm, H. Xu, J. Albalad, I. Imaz and D. Maspoch, J. Am. Chem. Soc., 2018, 140, 15022–15030 CrossRef CAS PubMed.
- H. Sato, R. Matsuda, K. Sugimoto, M. Takata and S. Kitagawa, Nat. Mater., 2010, 9, 661–666 CrossRef CAS PubMed.
- T. B. Faust and D. M. D'Alessandro, RSC Adv., 2014, 4, 17498–17512 RSC.
- T. Haneda, M. Kawano, T. Kawamichi and M. Fujita, J. Am. Chem. Soc., 2008, 130, 1578–1579 CrossRef CAS PubMed.
- S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2008, 130, 806–807 CrossRef CAS PubMed.
- K. Kadota, N. T. Duong, Y. Nishiyama, E. Sivaniah, S. Kitagawa and S. Horike, Chem. Sci., 2019, 10, 6193–6198 RSC.
- M. J. Ingleson, J. P. Barrio, J. Bacsa, A. Steiner, G. R. Darling, J. T. A. Jones, Y. Z. Khimyak and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2009, 48, 2012–2016 CrossRef CAS PubMed.
- J. McKinven, G. S. Nichol and P. L. Arnold, Dalton Trans., 2014, 43, 17416–17421 RSC.
- Z. Łodziana, P. Błoński, Y. Yan, D. Rentsch and A. Remhof, J. Phys. Chem. C, 2014, 118, 6594–6603 CrossRef.
- Y. Nakamori, H. Li, K. Miwa, S.-I. Towata and S.-I. Orimo, Mater. Trans., 2006, 47, 1898–1901 CrossRef CAS.
- V. D. Makhaev, A. P. Borisov, T. P. Gnilomedova, É. B. Lobkovskii and A. N. Chekhlov, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1987, 36, 1582–1586 CrossRef.
- A. Pichon and S. L. James, CrystEngComm, 2008, 10, 1839–1847 RSC.
- Y. Nakamori, K. Miwa, A. Ninomiya, H. Li, N. Ohba, S. Towata, A. Züttel and S. Orimo, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 045126 CrossRef.
- J. G. Bell, S. A. Morris, F. Aidoudi, L. J. McCormick, R. E. Morris and K. M. Thomas, J. Mater. Chem. A, 2017, 5, 23577–23591 RSC.
- B. Chatterjee and C. Gunanathan, J. Chem. Sci., 2019, 131, 118 CrossRef CAS.
- T. J. Marks and J. R. Kolb, Chem. Rev., 1977, 77, 263–293 CrossRef CAS.
- C. V. Picasso, D. A. Safin, I. Dovgaliuk, F. Devred, D. Debecker, H.-W. Li, J. Proost and Y. Filinchuk, Int. J. Hydrogen Energy, 2016, 41, 14377–14386 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc01753a |
| This journal is © The Royal Society of Chemistry 2020 |