
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterisation of estrogen receptor alpha (ERα) expression in breast cancer cells and effect of drug treatment using targeted nanoparticles and SERS†
Anastasia
Kapara
 ab,
Valerie G.
Brunton
b,
Duncan
Graham
a and
Karen
Faulds
*a
ab,
Valerie G.
Brunton
b,
Duncan
Graham
a and
Karen
Faulds
*a
aPure and Applied Chemistry, Technology and Innovation Centre, University of Strathclyde, 99 George Street, Glasgow, Scotland G1 1RD, UK. E-mail: karen.faulds@strath.ac.uk
bEdinburgh Cancer Research UK Centre, University of Edinburgh, Crewe Road South, Edinburgh, Scotland EH4 2XU, UK
First published on 3rd September 2020
Abstract
The detection and identification of estrogen receptor alpha (ERα), one of the main biomarkers in breast cancer, is crucial for the clinical diagnosis and therapy of the disease. Here, we use a non-destructive approach for detecting and localising ERα expression at the single cell level using surface enhanced Raman spectroscopy (SERS) combined with functionalised gold nanoparticles (AuNPs). Antibody functionalised nanotags (ERα-AuNPs) showed excellent biocompatibility and enabled the spatial and temporal understanding of ERα location in breast cancer cell lines with different ERα expression status. Additionally, we developed an approach based on the percentage area of SERS response to qualitatively measure expression level in ERα positive (ERα+) breast cancer cells. Specifically, the calculation of relative SERS response demonstrated that MCF-7 cells (ERα+) exhibited higher nanotag accumulation resulting in a 4.2-times increase in SERS signal area in comparison to SKBR-3 cells (ERα−). These results confirmed the strong targeting effect of ERα-AuNPs towards the ERα receptor. The functionalised ERα-AuNP nanotags were also used to investigate the activity of fulvestrant, the first-in-class approved selective estrogen receptor degrader (SERD). SERS mapping confirmed that ERα degradation occurred after fulvestrant treatment since a weaker SERS signal, and hence accumulation of nanotags, was observed in MCF-7 cells treated with fulvestrant. Most importantly, a correlation coefficient of 0.9 between the SERS response and the ERα expression level, obtained by western blot, was calculated. These results confirmed the strong relationship between the two approaches and open up the possibilities of using SERS as a tool for the estimation of ERα expression levels, without the requirement of destructive and time-consuming techniques. Therefore, the potential of using SERS as a rapid and sensitive method to understand the activity of SERDs in breast cancer is demonstrated.
Introduction
Breast cancer is a major disease and the leading cause of oncologic mortality and morbidity among women worldwide.1,2 In 2018 there were over 2 million new cases of breast cancer representing about 25 per cent of all cancers in women. Incidence rates vary widely across the world, from 27 per 100000 in Middle Africa and Eastern Asia to 92 per 100000 in Northern America.1 Current statistics also suggest than one in eight women will develop breast cancer at some point in their lifetime and more than 71![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 new cases are expected to be diagnosed by 2035 in the UK.3 Approximately 75% of primary breast tumours are diagnosed as being positive for a hormone receptor, which can either be progesterone or estrogen receptor alpha (ERα).4 ERα is a ligand-activated transcription factor, which regulates the expression of sequences containing specific hormone response elements responsible for body functions in the reproductive system,5 immune system,6 bones7 and brain.8 In the UK, the phenotype of around three-quarters of all breast cancers is characterised by the presence of ERα.3,9,10 Therefore, ERα is a key receptor biomarker whose status plays a pivotal role in the classification of breast cancer subtypes, since its overexpression is related to increased proliferation and metastasis in breast cancer11 which makes it an important marker for prediction of the likelihood of a patient developing metastatic disease.12 Therefore, the accurate assessment of ERα status is essential for diagnosis and treatment decision making for breast cancer patients.
000 new cases are expected to be diagnosed by 2035 in the UK.3 Approximately 75% of primary breast tumours are diagnosed as being positive for a hormone receptor, which can either be progesterone or estrogen receptor alpha (ERα).4 ERα is a ligand-activated transcription factor, which regulates the expression of sequences containing specific hormone response elements responsible for body functions in the reproductive system,5 immune system,6 bones7 and brain.8 In the UK, the phenotype of around three-quarters of all breast cancers is characterised by the presence of ERα.3,9,10 Therefore, ERα is a key receptor biomarker whose status plays a pivotal role in the classification of breast cancer subtypes, since its overexpression is related to increased proliferation and metastasis in breast cancer11 which makes it an important marker for prediction of the likelihood of a patient developing metastatic disease.12 Therefore, the accurate assessment of ERα status is essential for diagnosis and treatment decision making for breast cancer patients.
Currently, the most commonly used methods for assessing and characterising ERα in human breast cancer cell lines are immunofluorescence13,14 western blotting15 and reverse transcription polymerase chain reaction (RT-PCR).16 Although these methods can be useful for measuring the ERα expression, there are still technical limitations as these approaches are destructive, require processed/fixed samples17,18 and are not suitable for use in vivo. Immunofluorescence uses fluorescent dyes which usually have high background signals19 and limited multiplexing capabilities. Additionally, western blot and RT-PCR involve time-consuming experimental steps which require cell lysis and cellular subfractionation.20,21
Surface-enhanced Raman spectroscopy (SERS) has attracted considerable interest as a non-invasive optical technique with unique advantages, such as narrow spectral bands that increase the multiplexing capabilities, high sensitivity, selectivity and specificity.22–24 Brolo et al. focused on quantitatively detecting single molecules based on SERS signal intensity.25 Additionally, Schultz et al. showed that SERS can be used to distinguish specific to non-specific nanoparticle receptor binding.26 Therefore, it is clear that SERS is a powerful method for the detection of single molecules and enables the spatial and temporal understanding of where specific molecules are located at a single cell level. SERS nanotags have been successfully synthesised for various bioanalytical measurements of cancer cells27,28 including breast cancer.29–31 The design of these SERS nanotags usually involves attaching Raman reporters to the surface of gold (Au) or silver (Ag) nanoparticles (NPs) and functionalising with a specific targeting biomolecule, such as monoclonal antibodies, drugs or DNA sequences.32 Poly-ethylene glycol (PEG) can also be attached to the nanoparticle surface to avoid dissociation of the functionalised biomolecules, decrease the toxic effects,33 reduce non-specific cellular internalisation,34 facilitate longer circulation time in biological fluids35 and reduce aggregation of nanotags in biological solutions.36 These nanotags can be introduced to unprocessed samples and they can be used for a range of applications including in vitro, ex vivo and in vivo measurements using SERS. These advantages make SERS a powerful bioanalytical tool for early tumour identification and characterisation.
Here, anti-ERα antibody conjugated AuNPs were developed for characterising and distinguishing breast cancer cells with different ERα statuses, using image evaluation of the SERS response. In parallel, we were able to investigate the activity of fulvestrant, a commercially available ERα degrader in breast cancer. To our knowledge, this is the first study where the assessment of the SERS response per cell has been utilised to characterise the ERα cancer phenotype at a single cell level and inform about fulvestrant drug activity. This is a first step towards using SERS for both diagnosis and investigation of the efficacy of selective estrogen receptor degrader (SERD) drugs in breast cancer. This has a potential future use in personalised clinical approaches where patient derived samples could be screened for assessing the medication activity to avoid drug resistance outcomes.
Results and discussion
Nanoparticle synthesis and characterisation of ERα-AuNPs SERS nanotags
Bare AuNPs were synthesised by standard citrate reduction of gold.37 The functionalisation, characterisation and stability of ERα-AuNPs SERS nanotags have been previously reported by our group.38 Briefly, anti-ERα antibody was attached to the AuNP gold surface via carbodiimide crosslinking chemistry. The coupling chemistry was achieved after the attachment of 1,2-bis(4-pyridyl) ethylene (BPE) Raman reporter to the AuNPs surface. The nanotags did not demonstrate any aggregation and maintained their strong and characteristic SERS signal.38Characterisation of breast cancer cells and effects of SERS nanotags on cell viability
To validate the ability of SERS nanotags to identify and distinguish between breast cancer cells with different ERα status, two breast cancer cell lines, ERα-positive MCF-7 cells and ERα-negative SKBR-3 cells were chosen. To confirm the ERα expression level in each cell line before SERS experiments were carried out, western blots were performed. Western blot data showed that the ERα protein had detectable expression levels only in the MCF-7 cells and not in the SKBR-3 cells (ESI, Fig. S1†). Immunofluorescent labelling of ERα also showed that ERα was highly expressed in MCF-7 cells, in contrast to SKBR-3 cells where there was no detectable ERα expression (ESI, Fig. S2†). Before introducing the ERα-AuNP nanotags into the cells, their cytotoxicity was assessed in MCF-7 cells and SKBR-3 cells. 60 pM of nanotags, with and without antibody bioconjugation, were incubated with the cells for 2 h before the measurement of cell viability. The viability in MCF-7 cells treated with ERα-AuNP nanotags was over 97%. There was a small reduction in viability when MCF-7 cells were treated with AuNPs functionalised with the BPE Raman reporter alone (96% cell viability). Finally, the lowest viability was observed when the cells were treated with bare AuNPs (85% cell viability) (ESI, Fig. S3†). Similarly, SKBR-3 cells treated with ERα-AuNPs exhibited 99% viability in contrast to PEGylated BPE-AuNPs (94% cell viability) and BPE-AuNPs (81% cell viability) (ESI, Fig. S4†). These results indicated that the ERα-AuNP nanotags showed good biocompatibility and did not cause any detectable cell toxicity. Therefore, ERα-AuNPs can be used as an ERα targeting nanotags in live cells without affecting cell viability.Identification and characterisation of ERα positive breast cancer cells using SERS
To characterise the breast cancer cell phenotype based on the ERα expression, MCF-7 cells and SKBR-3 cells were incubated with the ERα-AuNP nanotags under the same conditions (60 pM, 2 h). SERS mapping of the cells after incubation with the nanotags showed that the ERα-AuNP nanotags accumulated more in MCF-7 cells compared to SKBR-3 cells (Fig. 1A). Additionally, the average SERS signal from ten cells, in three independent biological replicates, showed that MCF-7 cells had a higher SERS intensity from the nanotags compared to SKBR-3 cells (Fig. 1B). Specifically, the average SERS intensity of the BPE peak at 1610 cm−1 was statistically significantly higher in MCF-7 cells compared to SKBR-3 cells (4.5-times higher) (Fig. 1C). Therefore, the greater SERS signal in MCF-7 cells confirmed that ERα-AuNPs specifically interacted with the ERα receptor for their cellular uptake. In contrast, the low uptake of ERα-AuNP nanotags in SKBR-3 cells was probably due to non-specific interactions. This non-specific interaction was expected to be caused by the EPR effect i.e. by non-receptor-based uptake mechanisms. These data agree with our previous work where we found that ERα-AuNPs enter MCF-7 cells using an ERα receptor-mediated endocytosis process.38 To further investigate the location of the nanotags within the cells, 3D SERS mapping experiments were performed throughout the whole cell depth (30 μm in total) (ESI, Fig. S5†). The location of the nanotags was found by creating a false colour image using the Windows-based Raman Environment (WiRE™ – Renishaw plc) 4.4 software package on a Renishaw InVia microspectrometer. Direct classical least square analysis (DCLS) was used to match the cell spectra with the reference spectrum of BPE Raman reporter, which corresponded to ERα-AuNP nanotags. The minimum and maximum look up table (LUT) thresholds were set to exclude any poorly correlating or noisy spectra (min = 0.4). The 3D Raman mapping waterfall plot of average SERS spectra at different z-axis points treated with ERα-AuNPs showed that that the SERS signal in MCF-7 comes from within the cells rather than the surface (ESI, Fig. S6†). These data agree with previous 3D SERS mapping performed from our group in MCF-7 cells treated with the nanotags.38 The series of SERS spectra detected from z-steps in the cell volume confirmed that the nanotags were concentrated in certain intracellular locations, suggesting the presence of ERα within MCF-7 cells.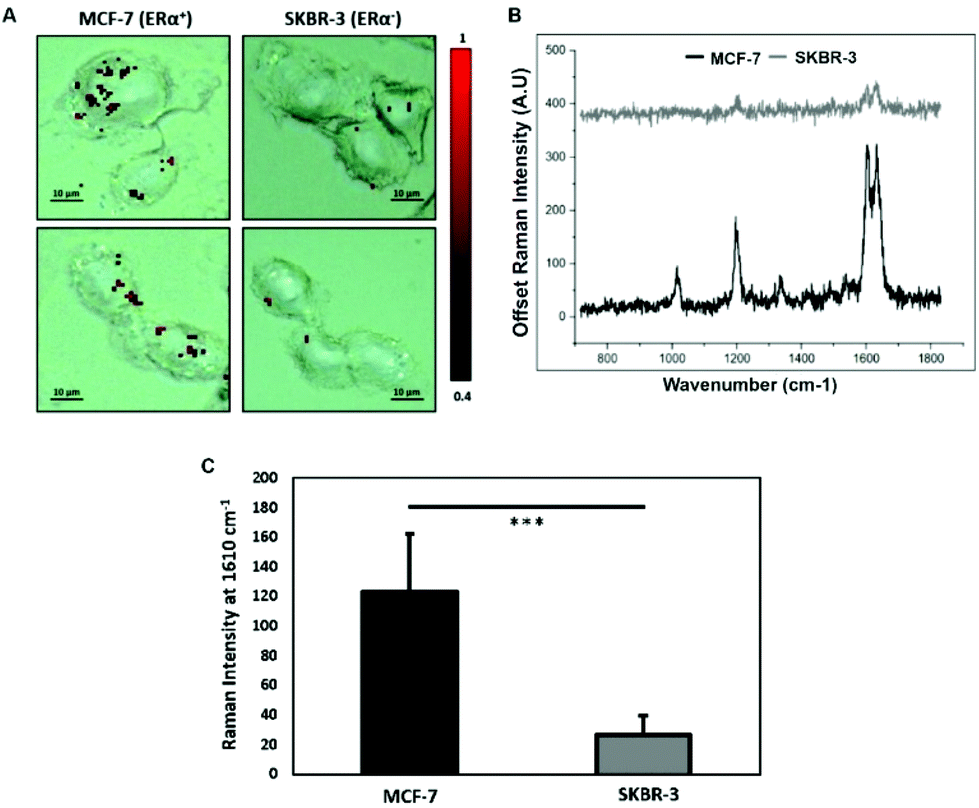 | ||
| Fig. 1 ERα-AuNP nanotags accumulation in MCF-7 cells compared to SKBR-3 cells. (A) SERS map of MCF-7 cells and SKBR-3 cells incubated with 60 pM ERα-AuNP nanotags for 2 h at 37 °C. Images were generated with a 50× magnification NIR APO Nikon water immersion objective with 1.0 NA, laser power of 1.2 mW (10% power) at the sample, from a HeNe 633 nm excitation source with a 0.1 s acquisition time per point, and a 1200 L mm−1 grating in high confocality mode. The false colour images representing the location of ERα-AuNPs were generated using the Windows-based Raman Environment (WiRE™ – Renishaw plc) 4.4 software package on a Renishaw InVia microspectrometer and direct classical least square analysis (DCLS) based on a BPE Raman reporter reference spectrum. The minimum and maximum look up table (LUT) thresholds were set to exclude any poorly correlating or noisy spectra (min = 0.4). Results are representative of 3 independent experiments. Scale bars = 10 μm. (B) Average SERS spectra of MCF-7 cells (black) and SKBR-3 cells (grey) incubated with ERα-AuNPs (60 pM for 2 h). Average SERS intensity was calculated from ten cells in three separate experiments. (C) Average SERS intensity at 1610 cm−1 (most intense peak of the BPE Raman reporter). The average of ten samples from three independent biological replicates is shown. Error bars presented as mean ± S.D. * Significant difference (p < 0.05) in a Student's t test. | ||
SERS was found to be more effective to estimate ERα expression and localisation in comparison to the widely used immunofluorescence. Specifically, here we performed both techniques for the characterisation of ERα positive breast cancer cells (ESI, Fig. S2†). Immunofluorescence was found to be a highly laborious technique that involved the fixation, permeabilisation and blocking of the cells before incubation with the primary and secondary antibodies. SERS avoided all these steps by being a quick, non-destructive technique that required only a primary antibody for the characterisation of MCF-7 cells. Overall, immunofluorescence staining took 24 h whereas SERS took only 2 hours for the slide development. This clearly demonstrates that SERS and ERα-AuNPs nanotags can be utilised to investigate the ERα status of breast cancer cells, as well as to distinguish ERα positive breast cancer cells from ERα negative breast cancer cells with high sensitivity and specificity.
Calculation of relative SERS response value in breast cancer cells
A method for the estimation of the relative uptake of SERS nanotags into cells has been previously described by our group.38 Briefly, we have developed a direct, rapid and non-destructive method to calculate the percentage of SERS responsive pixels per cell and provide an indication of the relative value for the uptake of nanotags per condition. Although, this is not a direct quantification of the total number of nanotags in the cells, it is an effective way to estimate the SERS signal per cell area, quantify the number of pixels that correspond to the SERS signal, identify the localisation of the nanotags, and allowing relative comparison between different samples. Here, the evaluation of SERS response in cells showed that there was a statistically significant higher percentage area of SERS response in MCF-7 cells in comparison to SKBR-3 cells (4.2-times higher) (Fig. 2). The different phenotype of MCF-7 cells and SKBR-3 cells was also confirmed using the average SERS intensity of the peak at 1610 cm−1 (Fig. 1C). However, since the average signal can be influenced by the formation of aggregates in the cell area, the evaluation of SERS response using pixel counting is considered more informative since it takes into account only the location of the nanoparticles’ signal and it is not influenced by SERS intensity. Additionally, it was observed that the percentage of SERS response from targeted ERα-AuNP nanotags was 3.8-times higher compared to untargeted PEG5000-AuNPs (no antibody attached) in MCF-7 cells (1.68% and 0.44% respectively). In contrast, there was no statistically significant difference between the ERα-AuNPs and PEG5000-AuNPs (0.4% and 0.6% respectively) in the SKBR-3 cells (Fig. 2) suggesting a non-specific interaction. These results confirmed the strong targeting effect of the ERα-AuNP nanotags towards the ERα positive MCF-7 cells in comparison to the negative SKBR-3 cells, whilst the nanotags without the antibody on their surface had no targeting effect in both cell lines. This is an important finding since it demonstrates that the targeted nanotags have much greater uptake compared to non-specific uptake by the enhanced permeability and retention (EPR) effect.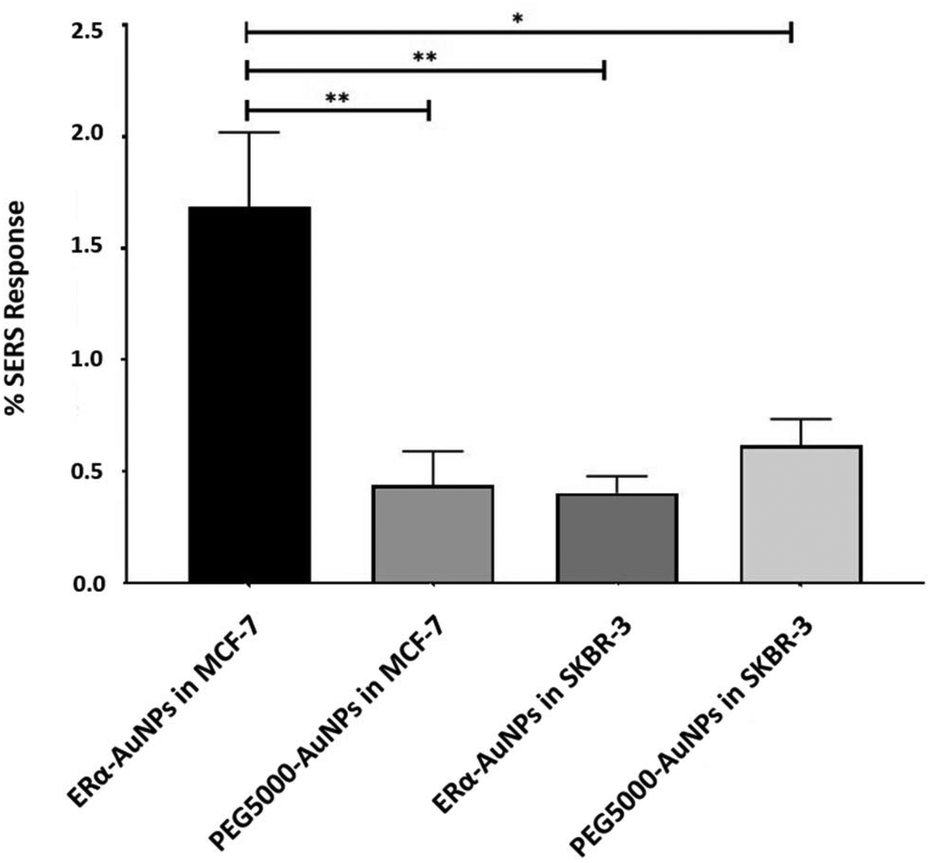 | ||
| Fig. 2 Calculation of relative SERS response showed statistically significant higher uptake in MCF-7 cells than SKBR-3 cells treated with ERα-AuNPs (60 nM, 2 h) and PEG5000-AuNPs (60 nM, 2 h). The calculations suggest that the binding and internalisation of the ERα-AuNPs is more efficient at targeting MCF-7 cells. Percentage of ERα-AuNPs area in MCF-7 cells estimated using the Fiji image processing package by calculating the red pixel number, corresponding to the ERα-AuNPs, and the cell area that was mapped. This was an estimation of the percentage of SERS responsive pixels per cell and not a quantification of the total number of nanoparticles in the cells. The average of ten samples from three independent biological replicates is shown. Error bars presented as mean ± S.D. *Significant difference (p < 0.05) in a one-way analysis of variance (ANOVA) with post-hoc Tukey's test. | ||
Assessment of fulvestrant activity using SERS in breast cancer
Approximately 75% of primary breast tumours in women test positive for a hormone receptor.3 The hormone receptor can be either progesterone (PR) and/or estrogen receptor alpha (ERα).3 In the UK, the phenotype of around three-quarters of all breast cancers is characterised by the presence of ERα.4,5 Currently, both ERα and PR positive patients are treated with the same hormonal therapy.6 Therefore, the assessment of the drug activity of an anti-ERα treatment will also be beneficial for patients with PR positive breast cancer. Fulvestrant is the first commercially available drug that is a selective estrogen receptor degrader (SERD).39,40 SERD is a class of drugs that bind to the ERα receptor and causes its degradation and, thus, its down regulation.41 However, like all hormonal therapy drugs, fulvestrant is known to have side effects, such as decreased white blood cells, abnormal liver function and increased risk of infections.40,42 The ability to understand the efficacy of fulvestrant is important for improving the way that the drug is used clinically. Therefore, we used our SERS approach to determine the activity of fulvestrant in MCF-7 cells. Specifically, MCF-7 cells were treated with increasing concentrations of fulvestrant (0.01 μM to 1 μM) for 24 h before lysing and western blot analysis. The results showed that increasing the concentration of fulvestrant decreased the expression of the ERα protein in MCF-7 cells compared to DMSO (vehicle control) treated cells (Fig. 3A and B). Since 500 nM and 1 μM fulvestrant resulted in the lowest ERα expression, these concentrations were used for the SERS experiments. Specifically, for SERS mapping, MCF-7 cells were treated with DMSO, 500 nM and 1 μM fulvestrant for 24 h before the addition of ERα-AuNPs nanotags (60 pM) for 2 h.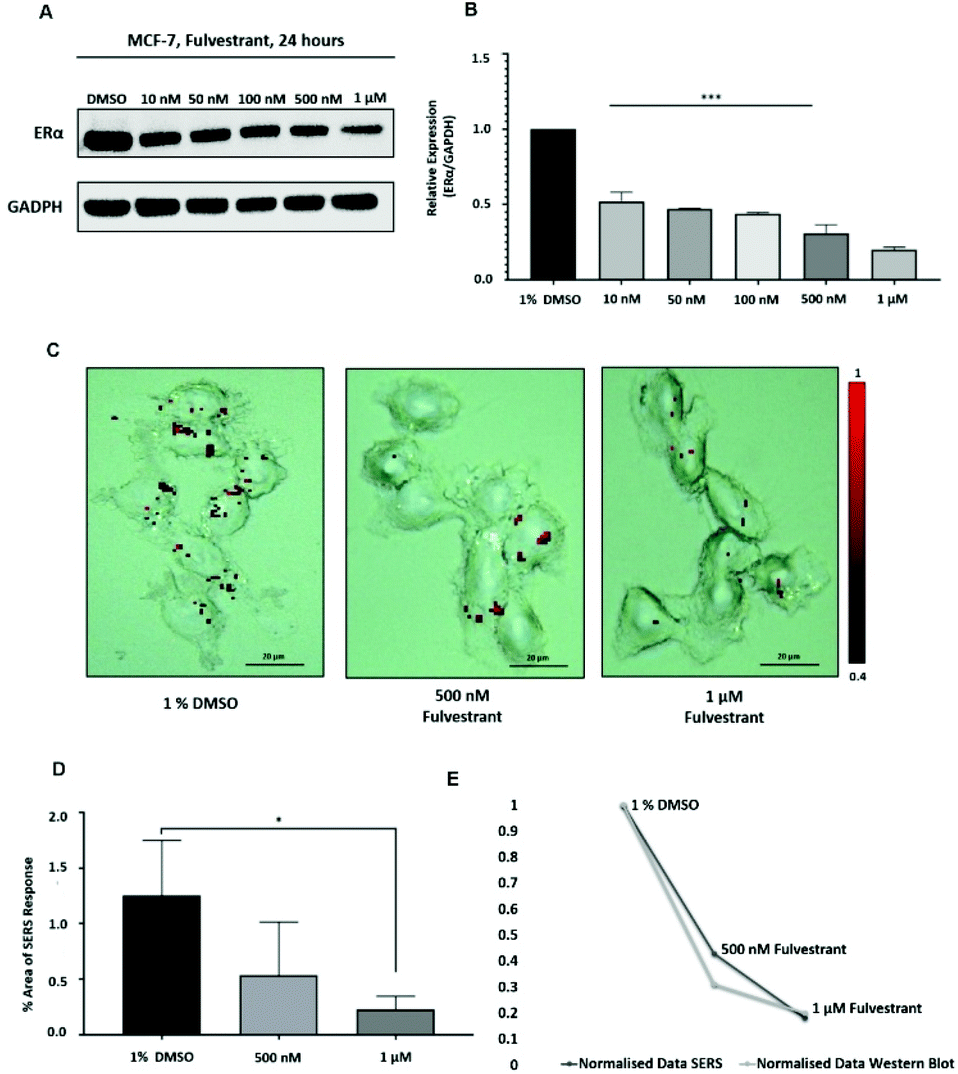 | ||
| Fig. 3 ERα-AuNP nanotags can be used to assess fulvestrant activity in MCF-7 cells. (A) ERα expression in MCF-7 cells treated with different concentrations of fulvestrant (0.01 μM to 1 μM) for 24 h. Cell lysates were prepared from breast cancer cell lines and western blot analysis carried out using a secondary antibody to ERα, β-actin was used as a loading control. (B) Quantitative analysis of the relative expression of ERα and GAPDH (housekeeping protein). The results are expressed as means ± SD of triplicate determinations. P < 0.001 versus 1% DMSO (vesicle control). Statistical analysis was conducted using one-way ANOVA. (C) SERS map of MCF-7 cells treated with fulvestrant (500 nM and 1 μM) for 24 h and ERα-AuNP nanotags (60 pM, 2 h). The images were generated with a 20× magnification NIR APO Nikon objective, laser power of 1.2 mW (10% power), from a HeNe 633 nm excitation source with a 0.1 s acquisition time per point, and a 1200 L mm−1 grating in high confocality mode. The false colour images representing the location of ERα-AuNPs were generated using the Windows-based Raman Environment (WiRE™ – Renishaw plc) 4.4 software package on a Renishaw InVia microspectrometer and direct classical least square analysis (DCLS) based on the BPE Raman reporter reference spectrum. The minimum and maximum look up table (LUT) thresholds were set to exclude any poorly correlating or noisy spectra (min = 0.4) Scale bars = 20 μm. (D) Area of SERS response in MCF-7 cells treated with fulvestrant (500 nM and 1 μM) for 24 h and then with ERα-AuNP nanotags (60 pM) for 2 h. The average of ten samples from three independent biological replicates is shown. Error bars presented as mean ± S.D. (E) Correlation of normalised area of SERS response and normalised relative expression of ERα in untreated MCF-7 cells and MCF-7 cells treated with fulvestrant (500 nM and 1 μM) for 24 h. | ||
The SERS results clearly correlated with the western blot experiment since a decreased accumulation of nanotags was observed after treatment with 500 nM fulvestrant compared to DMSO (vehicle control). Additionally, a further reduction in the nanotag accumulation was observed with 1 μM fulvestrant in comparison to 500 nM fulvestrant treatment (Fig. 3C). The calculation of the area of SERS response in MCF-7 cells showed that there was a statistically significant decrease (5.4-times reduction) between the MCF-7 cells treated with 1% DMSO (vehicle control) and the ones treated with 1 μM fulvestrant (Fig. 3D). Moreover, 500 nM fulvestrant treatment led to a 2.3 times decrease in the area of SERS response compared to 1% DMSO. These results clearly demonstrate that the amount of targeted nanotag uptake by cells, and the resultant SERS response, correlates with ERα expression levels and is a potentially powerful method for understanding the activity of SERD drugs by providing real time measurements of activity at the single cell level. Most importantly, it was shown that the SERS response and the ERα quantification data from the western blot had a very strong relationship (Fig. 3E). Specifically, the correlation coefficient between the normalised data from SERS and western blot was found to be 0.9 which indicates positive linear correlation between the two variables.43 Therefore, this correlation opens up the potential of using SERS as a tool to predict ERα protein expression levels without the need for destructive and time-consuming western blot analysis.
Conclusions
A novel method based on the assessment of SERS response was successfully demonstrated for characterising ERα-positive breast cancer cells. This provided a method to follow the effects of a drug treatment in breast cancer cells using the ERα-AuNP nanotags. SERS is a non-destructive method that has the advantages of being rapid with high sensitivity and specificity for targeting ERα overexpression at a single breast cancer cell level. However, most importantly, SERS was used for both characterising the breast cancer cell phenotype as well as understanding fulvestrant activity, a SERD treatment for ERα positive breast cancer. The calculation of SERS response confirmed higher nanotag accumulation in ERα-positive cells (MCF-7) compared to ERα-negative cells (SKBR-3). The obvious differences in SERS intensity detected from the two different breast cancer cell lines confirmed the specificity and strong targeting effect of ERα-AuNP nanotags against ERα. These results were more effective in comparison to other experimental approaches that use fluorescence for ERα analysis since they are usually related to high background signals, time consuming experiments, and use of a primary and a secondary antibody for the imaging analysis. The accurate evaluation of the percentage area of SERS response provided an understanding of ERα expression levels before and after fulvestrant treatment. The SERS response calculations verified the reduction in ERα expression levels after treatment with the ERα degrader, fulvestrant. Above all, a direct correlation between the SERS response and western blot experiments, which quantified ERα expression, was observed. This work, therefore, highlights that AuNP based SERS imaging could be of great importance for diagnostic accuracy, involving molecular characterisation and intracellular imaging, and understanding of drug activity into different cell types. Future prospective studies may also involve using SERS as a complementary approach for monitoring patient derived breast cancer cells for evaluating the drug efficacy and propose a personalised drug treatment to avoid drug resistance outcomes.Experimental
Materials
Anti-estrogen receptor alpha antibody (ab16660) was purchased from Abcam (330 Cambridge Science Park, Cambridge, CB4 0FL, UK). Anti-mouse IgG HRP-linked antibody (7076S) and anti-rabbit IgG HRP-linked antibody (7074S) were purchased from Cell Signalling Technology (Hamilton House, Mabledon Place, London, WC1H 9BB, UK). Sodium tetrachloroaurate dihydrate, (N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride) (EDC), N-hydroxysulfosuccinimide sodium salt (NHS), poly (ethylene glycol) 2-mercaptoethyl ether acetic acid (HS-PEG5000-COOH), 1,2-bis(4-pyridyl) ethylene (BPE), 4-(2-hydroxyethyl)-1-piperazineethancesulfonic acid (HEPES), and 2-(N-morpholino) ethanesulfonic acid (MES) were obtained from Sigma-Aldrich Ltd (The Old Brickyard, New Road, Gillingham, Dorset, SP8 4XT, UK). LIVE/DEAD Viability/Cytotoxicity Assay Kit was purchased from ThermoFisher Scientific (3 Fountain Dr, Inchinnan, Renfrew PA4 9RF, UK). Milli-Q deionized water was used after purification by Milli-Q purification system. All glassware was cleaned in aqua regia (3 HCl:1 HNO3).
Nanoparticle synthesis and functionalisation
Citrate reduced gold (Au) nanoparticles were synthesised according to the Turkevich, Stevenson and Hillier method.12 Briefly, sodium tetrachloroaurate dihydrate solution (10 mL, 15 mM) in 490 mL deionised water was boiled under continuous stirring. Sodium citrate tribasic dihydrate solution (7.5 mL, 26 mM) was then added. The mixture was boiled with stirring for 1 h. Differential light scanning analysis (DLS) of AuNPs showed that the average size of AuNPs was 52.5 ± 0.79 nm with a narrow size distribution. Scanning electron microscopy (SEM) of AuNPs agreed with the DLS measurements and confirmed that AuNPs had a spherical shape with a diameter distribution ranging from 40 to 50 nm. For the carbodiimide crosslinking conjugation, 74 μL of EDC solution (1 mg mL−1 in 10 mM MES, pH 6.0) was mixed with 40 μL of HS-PEG5000-COOH (12.5 μM in dH20) followed by the addition of 217 μL of NHS (1 mg mL−1 in 10 mM MES, pH 6.0) and 20 μL of anti-ERα antibody (2.5 mg mL−1 in dH20). The final solution was incubated in 40 μL of 10 mM HEPES buffer pH 7.0 on a shaker plate for 18 h at room temperature. 10 μL of 1,2-bis(4-pyridyl) ethylene (BPE) (0.1 μM) was added to bare AuNPs (0.03 nM, 990 μL) and the solution was incubated on the shaker plate for 30 min followed by centrifugation at 6000 rpm for 20 min. The solution of EDC-NHS-PEG5000-mAb was added dropwise to the pelleted BPE-AuNPs. The nanotags were incubated on a shaker plate for 3 h. Excess free protein was removed by centrifugation at 8000 rcf for 10 min and was used for protein concentration estimation analysis.Nanoparticle characterisation
Extinction spectra were measured using an Agilent Cary 60 UV-Visible (UV-vis) spectrophotometer with Win UV scan V.2.00 software. The instrument was allowed to equilibrate to RT before using poly(methyl methacrylate) (PMMA) disposable plastic micro cuvettes with 500 μL sample volumes to scan wavelengths from 300–800 nm. Where required, samples were diluted to give extinction values of less than one to adhere to the Beer–Lambert law, to allow calculation of the concentration of AuNPs. Dynamic light scattering (DLS) and ζ-potential were measured using a Malvern Zetasizer Nano ZS with 800 μL of the sample in a PMMA disposable micro cuvette with Zetasizer μV and APS v.6.20 software. Polystyrene latex beads (40 nm) were used as a standard to validate the calibration of the system before running samples. Measurements were taken in triplicate. A scanning electron microscope (SEM) FEI Quanta 250 FEG-ESEM was used to image at an accelerating voltage of 30 kV and typically a spot size of 4 was selected, and an Everhart–Thornley detector collected secondary electrons. For the solution measurements of the nanotags, SERS analysis was carried out on a Snowy Range CBEx 2.0 handheld Raman spectrometer (Snowy Range Instruments, Laramie WY USA) equipped with a 638 nm laser with a maximum laser power of 30 mW. Samples were deposited in glass vials for interrogation. The sample volumes were 600 μL and spectra were collected using 100% laser power at the sample with a 0.05 s accumulation time. The software used to acquire spectra was peak 1.1.112. Resulting spectra were baseline corrected in Matlab 2014b.44Cell culture conditions
MCF-7 cells (ATCC® HTB-22™) and SKBR-3 cells (ATCC® HTB-30™) were obtained from American Type Culture Collection (ATCC) (Queens Road, Teddington, Middlesex, TW11 0LY, UK). Cells were maintained between passage number 5 and 30. The human breast cancer cells were cultured in Rosewell Park Memorial Institute medium (RPMI 1640) supplemented with 1% penicillin/streptomycin (10000 units per mL), 1% fungizone, and 10% heat-inactivated fetal bovine serum (FBS). Cells were incubated under humidified 37 °C and 5% CO2 in a humidified incubator. Cells at a confluence of ca. 90% growing in a T175 flask were trypsinised and re-suspended in medium to give a concentration of ca. 1 × 106 cells per mL.
ERα-AuNPs and fulvestrant treatment
MCF-7 cells (1 × 106 cells per mL) were seeded onto sterile 22 mm square glass coverslips with culture medium. ERα-AuNP nanotags (60 pM, 2 h) were incubated with the cells at 37 °C, 5% CO2 in a humidified incubator. The coverslips were washed in PBS three times and fixed in 4% paraformaldehyde for 15 min. For assessing fulvestrant activity using SERS, MCF-7 cells (1 × 106 cells per mL) were seeded onto sterile 22 mm square glass coverslips and treated with 1% DMSO (control) or with fulvestrant (500 nM and 1 μM) for 24 h. ERα-AuNPs (60 pM) were then added to the cells for 2 h. The coverslips were washed in PBS three times and fixed in 4% paraformaldehyde for 15 min. The fixed cells were washed in PBS and dH20 and left to air dry before mounting on a standard glass microscope slide for SERS imaging.Western blot experiments after fulvestrant treatment
The cells (1 × 106 per mL) were plated in 10 cm diameter dishes with 10 mL of RPMI and left for 24 h before fulvestrant was added with increasing concentrations (0.01 μM to 1 μM) for 24 h. The next day the media was aspirated from the culture. The cells were washed in ice cold PBS twice and were lysed with 200 μL of ice cold RIPA buffer (#10017003, Thermo Fisher), containing a tablet of protease and phosphatase inhibitor (#A32959, Pierce). The cells were immediately scraped off the plate and their extracts were transferred to a microcentrifuge tube. The samples were kept on ice for 15 min and were centrifuged at 14000 rpm for 15 min at 4 °C. The cell lysate supernatant was then used for protein quantification using the BCA assay. The cell lysate samples (20 μL, 1 mg mL−1) were diluted with 5× SDS loading buffer, heated to 95 °C for 5 min and microcentrifuged for 5 min at RT. 20 μL of the denatured cell lysate was loaded into a 12% gel (Mini Protean TGX stain free Pre-cast gels, #456-8085, Bio-Rad) and run at 140 V for 40 min. A prestained molecular weight marker (5 μL) was also loaded into the gel to determine the molecular weights of the proteins of interest. The gel was electrotransferred to a 0.2 μm nitrocellulose membrane (#170-4159, Bio-Rad) with the BioRad TransBlot Turbo Transfer System using the Midi gel 10 min transfer setting. The membrane was blocked with 5% BSA blocking buffer for 1 h at room temperature. After blocking, the membrane was incubated at 4 °C overnight whilst rocking with the appropriate primary antibody diluted in 5 mL 5% w/v BSA, 1× TBS, 0.1% Tween 20. The next day the membrane was washed three times for 5 min each with 15 mL of TBST buffer (1.5% Tween 20 in 1× TBS). The membrane was incubated with the appropriate secondary antibody in 10 ml of 5% BSA blocking buffer with gentle agitation for 1 h at room temperature and was washed three times for 5 min each with 15 mL of TBST buffer. For the detection of the proteins, the membrane was incubated with 1:1 of Pierce™ ECL western blotting substrate (#32106, Thermo fisher) for 1 min. Finally, a Bio-Rad ChemiDoc MP Imaging System-Universal Hood III with Image Lab V.4.1 software was used to image and quantify the protein levels on the membrane.
Fluorescence microscopy
MCF-7 cells were seeded onto a 6-well plate (1 × 106 cells per mL) and incubated at 37 °C and 5% CO2 for 24 h. After 24 h the cells were washed in PBS three times and fixed in 4% paraformaldehyde for 15 min at room temperature. The fixed cells were permeabilised with 0.1% Triton X-100 in PBS for 10 min. The cells were then washed in PBS three times for 5 min. Then, the cells were blocked with 1% BSA, 22.52 mg mL−1 glycine in 0.1% PBS-Tween for 1 h. The cells were then incubated with the diluted antibody (ab16660) (1/200 dilution in 1% BSA in PBS-Tween) overnight at 4 °C. The cells were then washed in PBS three times for 5 min. Finally, the cells were incubated with the secondary antibody (Alexa Fluor® 647) (1/200 dilution in 1% BSA) for 1 h at room temperature in the dark. The cells were then washed in PBS three times for 5 min and 1 μg mL−1 of DAPI (DNA stain) was added for 10 min. The images were generated using a Leica Microsystems TCS SP8 with continuous wave visible lasers and Leica DMi8 inverted microscope and DFC 7000T and TL LED cameras. The software was Leica Application Suite X V.3.1.5.16308 to carry out live/dead studies a 63× magnification HC PL APO water objective with a 1.2 NA was used. Intensity and area of fluorescence were measured using Image J (National Institute of Health (NIH)) software with Fiji plug-in to measure the area of fluorescent stain.45Solution measurements of ERα-AuNPs
The SERS nanotags were assessed by SERS analysis which was carried out on a Snowy Range CBEx 2.0 handheld Raman spectrometer (Snowy Range Instruments, Laramie WY USA) equipped with a 638 nm laser with a maximum laser power of 30 mW. Samples were deposited in glass vials for interrogation. The sample volume was 600 μL and spectra were collected using 100% laser power at the sample with a 0.05 s accumulation time. The software used to acquire spectra was peak 1.1.112. Resulting spectra were baseline corrected in Matlab 2014b typically using polynomial smoothing of 1 × 107 and asymmetry of 1 × 10−3.44Raman cell mapping
The intracellular uptake of the nanotags was examined using Raman cell mapping. A Renishaw InVia Raman confocal microscope was used to create initial depth profiles, to establish the focal plane of the fixed cells in correlation with the white light images. The MCF-7 cells and SKBR-3 cells that were imaged had a typical cell size of around 20–25 microns. Cells with similar height characteristics were selected for the 3D SERS mapping experiments to allow more direct volume comparisons. Therefore, cells that were mapped were all approximately 15 μm in height. Before each map, the cells were analysed under the microscope, and the z = 0 was set accordingly. 3D SERS maps were collected in edge Streamline HR high confocality mode at 1 μm resolution in the X and Y directions and 3 μm between Z-stacks. A 50× magnification NIR APO Nikon water immersion objective with a 1.0 NA was used on the samples at a laser power of 1.2 mW (10% power) at the sample, from a HeNe 633 nm excitation source with a 0.1 s acquisition time per point, and a 1200 L mm−1 grating in high confocality mode. Windows-based Raman Environment (WiRE™ – Renishaw plc) 4.4 software package was used to pre-process the data for cosmic ray removal and baseline subtraction. The image was generated using direct classical least square analysis (DCLS) based on BPE or PPY reference spectrum. Therefore, the false colour was generated only when there was a good spectral fit between the reference and the collected spectra. All SERS experiments contained n = 10 biological replicates and experiments were in triplicate.Calculation of relative SERS response value in breast cancer cells
The evaluation of SERS response in breast cancer cells has been previously described by our group.38 Briefly, the spectra from the SERS cell mapping analysis were baselined and cosmic rays were removed using the Windows-based Raman Environment (WiRE™ – Renishaw plc) 4.4 software package. The images were generated using direct classical least square analysis (DCLS) based on the Raman reporter reference spectrum. DCLS fitted the unknown data (collected during cell mapping) to a linear combination of the specified component spectrum (Raman reporter reference spectrum). If there was a good spectral fit between the Raman reporter reference and the collected spectra a gradient red false colour was assigned. Associated with each false colour image was a look up table (LUT). The minimum and maximum values of the LUT indicates the degree of spectral fit. Afterwards, the gradient red false colour was converted to monochromatic red colour, without affecting the intracellular SERS signal, using the Windows-based Raman Environment (WiRE™ – Renishaw plc) 4.4 software package.Fiji image processing package45 was used for the quantification of the pixel numbers of ERα-AuNPs and the cell area after SERS mapping. Specifically, the cellular area was selected by masking everything outside of it. The image was then colour split to the monochromatic red channel, where only the red pixels were present. Then, the red pixels were extracted above 200-threshold to count only the pixels that correspond to the nanotags and not from any cellular component. Finally, the percentage of the red pixel area (corresponding to SERS response) versus the full cell area was calculated. The percentage of the cellular SERS signal was determined by calculating the area of cells with SERS response divided by the surface area.
Statistical analysis
Statistical analysis was carried out on GraphPad Prism 8.1.2 (GraphPad Software, Inc., San Diego, CA). The Student's t-test was used for comparison of two variables and one-way analysis of variance (ANOVA) with post-hoc Tukey's test for comparison of three or more groups. Differences between groups were significant at a P value of <0.05.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (EPSRC) and Medical Research Council (MRC) through the CDT in Optical Medical Imaging (OPTIMA), grant number EP/L016559/1. Research data associated with this paper will become available through the following link: https://doi.org/10.15129/7d12a867-232a-41f3-817a-82bfcb1d6349.References
- J. Ferlay, I. Soerjomataram, R. Dikshit and E. All, Int. J. Cancer, 2015, 136, 359–386 CrossRef.
- L. A. Torre, R. L. Siegel, E. M. Ward and A. Jemal, Cancer Epidemiol. Biomarkers Prev., 2016, 25, 16–27 CrossRef.
- Cancer Research UK, Breast Cancer Incidence (Invasive Statistics), Cancer Research UK, 2016 Search PubMed.
- S. B. Giordano and W. Gradishar, Curr. Opin. Obstet. Gynecol., 2017, 29, 12–17 CrossRef.
- H. R. Lee, T. H. Kim and K. C. Choi, Lab. Anim. Res., 2012, 28, 71–76 CrossRef.
- S. Kovats, Cell. Immunol., 2015, 294, 63–69 CrossRef CAS.
- A. B. Khalid and S. A. Krum, Bone, 2016, 87, 130–135 CrossRef CAS.
- L. A. Bean, L. Ianov and T. C. Foster, Neuroscientist, 2014, 20, 534–545 CrossRef.
- J. Richman and M. Dowsett, Nat. Rev. Clin. Oncol., 2019, 16, 296–311 CrossRef CAS.
- E. V. Jensen, G. Cheng, C. Palmieri, S. Saji, S. Makela, S. Van Noorden, T. Wahlstrom, M. Warner, R. C. Coombes and J. A. Gustafsson, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 15197–15202 CrossRef CAS.
- M. C. Louie and M. B. Sevigny, Am. J. Cancer Res., 2017, 7, 1617–1636 CAS.
- N. Cabioglu, Y. Gong, R. Islam, K. R. Broglio, N. Sneige, A. Sahin, A. M. Gonzalez-Angulo, P. Morandi, C. Bucana and G. N. Hortobagyi, Ann. Oncol., 2007, 18, 1021–1029 CrossRef CAS.
- P. Meng, M. Vaapil, A. Tagmount, A. Loguinov, C. Vulpe and P. Yaswen, Breast Cancer Res. Treat., 2019, 176, 131–140 CrossRef CAS.
- J. K. Lee, J. C. Garbe, L. Vrba, M. Miyano, B. W. Futscher, M. R. Stampder and M. A. LaBarge, Front. Cell Dev. Biol., 2015, 3, 13 Search PubMed.
- C. H. J. Ford, A. L. B. Maie, A. L. A. Bushra and F. Issam, Exp. Ther. Med., 2011, 2, 537–544 CrossRef.
- M. Al-Bader, S. Al-Saji, C. H. J. Ford, I. Francis and B. Al-Ayadhy, Anticancer Res., 2010, 30, 4147–4156 CAS.
- A. Thompson, K. Brennan, A. Cox, J. Gee, D. Harcourt, A. Harris, M. Harvie, I. Holen, A. Howell and R. Nicholson, Breast Cancer Res., 2008, 10, R6 CrossRef.
- L. Pusztai, C. Mazouni, K. Anderson, Y. Wu and W. F. Symmans, Oncologist, 2006, 11, 868–877 CrossRef CAS.
- S. Schlücker, ChemPhysChem, 2009, 10, 1344–1354 CrossRef.
- J. J. Bass, D. J. Wilkinson, D. Rankin, B. E. Phillips, N. J. Szewczyk, K. Smith and P. J. Atherton, Scand. J. Med. Sci. Sports, 2017, 27, 4–25 CrossRef CAS.
- C. J. Smith and A. M. Osborn, FEMS Microbiol. Ecol., 2009, 67, 6–20 CrossRef CAS.
- J. A. Dougan and K. Faulds, Analyst, 2012, 137, 545–554 RSC.
- S. Laing, K. Gracie and K. Faulds, Chem. Soc. Rev., 2016, 45, 1901–1918 RSC.
- S. McAughtrie, K. Faulds and D. Graham, J. Photochem. Photobiol., C, 2014, 21, 40–53 CrossRef CAS.
- C. D. L. de Albuquerque, R. G. Sobral-Filho, R. J. Poppi and A. G. Brolo, Anal. Chem., 2018, 90, 1248–1254 CrossRef CAS.
- S. Sloan-Dennisona and Z. D. Schultz, ORCID logo *a.
- C. Sun, M. Gao and X. Zhang, Anal. Bioanal. Chem., 2017, 409, 4915–4926 CrossRef CAS.
- L. Li, M. Liao, Y. Chen, B. Shan and M. Li, J. Mater. Chem., 2019, 7, 815–822 RSC.
- R. Davis, J. Campbell, S. Burkitt, Z. Qiu, S. Kang, M. Mehraein, D. Miyasato, H. Salinas, J. Liu and C. Zavaleta, Nanomaterials, 2018, 8, 953 CrossRef.
- M. K. Yu, J. Park and S. Jon, Theranostics, 2012, 2, 3–44 CrossRef CAS.
- S. Lee, H. Chon, J. Lee, J. Ko, B. H. Chung, D. W. Lim and J. Choo, Biosens. Bioelectron., 2014, 51, 238–243 CrossRef CAS.
- R. Thiruppathi, S. Mishra, M. Ganapathy, P. Padmanabhan and B. Gulyás, Adv. Sci., 2017, 4, 1600279 CrossRef.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS.
- I. Canton and G. Battaglia, Chem. Soc. Rev., 2012, 41, 2718–2739 RSC.
- J. Suh, K. L. Choy, S. K. Lai, J. S. Suk, B. C. Tang, S. Prabhu and J. Hanes, Int. J. Nanomed., 2007, 2, 735–741 CrossRef CAS.
- T. Gillich, C. Acikgöz, L. Isa, A. D. Schlüter, N. D. Spencer and M. Textor, ACS Nano, 2013, 7, 316–329 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hiller, Faraday Discuss., 1951, 11, 55–75 RSC.
- A. Kapara, V. Brunton, D. Graham and K. Faulds, Chem. Sci., 2020, 11, 5819–5829 RSC.
- J. D. Croxtall and K. McKeage, Drugs, 2011, 71, 363–380 CrossRef CAS.
- M. R. Nathan and P. Schmid, Oncology Ther., 2017, 5, 17–29 CrossRef.
- H. K. Patel and T. Bihani, Pharmacol. Ther., 2018, 186, 1–24 CrossRef CAS.
- C. C. O'Sullivan and K. J. Ruddy, Curr. Breast Cancer Rep., 2016, 8, 183–192 CrossRef.
- S. Williams, N. Z. Med. J., 1996, 109, 39 Search PubMed.
- M. U. Guide, MATLAB and Statistics Toolbox Release 2014b, The MathWorks Inc., Natick, 2014 Search PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfelf, B. Schmid, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 671–675 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0an01532f |
| This journal is © The Royal Society of Chemistry 2020 |