
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermodynamically driven self-assembly of pyridinearene to hexameric capsules†
Anniina
Kiesilä
 a,
Ngong Kodiah
Beyeh
b,
Jani O.
Moilanen
a,
Rakesh
Puttreddy
a,
Sven
Götz
c,
Kari
Rissanen
a,
Perdita
Barran
d,
Arne
Lützen
c and
Elina
Kalenius
*a
a,
Ngong Kodiah
Beyeh
b,
Jani O.
Moilanen
a,
Rakesh
Puttreddy
a,
Sven
Götz
c,
Kari
Rissanen
a,
Perdita
Barran
d,
Arne
Lützen
c and
Elina
Kalenius
*a
aDepartment of Chemistry, Nanoscience Center, P. O. Box 35, FI-40014 University of Jyväskylä, Finland. E-mail: Elina.o.kalenius@jyu.fi
bOakland University, Department of Chemistry, 146 Library Drive, Rochester, Michigan 48309-4479, USA
cUniversity of Bonn, Kekulé-Institute of Organic Chemistry and Biochemistry, A, D-53121 Bonn, Germany
dMichael Barber Centre for Collaborative Mass Spectrometry, Manchester Institute of Biotechnology, School of Chemistry, The University of Manchester, Princess Street, Manchester, UK
First published on 2nd July 2019
Abstract
Pyridinearene macrocycles have previously shown unique host–guest properties in their capsular dimers including endo complexation of neutral molecules and exo complexation of anions. Here, we demonstrate for the first time the formation of hydrogen bonded hexamer of tetraisobutyl-octahydroxypyridinearene in all three states of matter – gas phase, solution and solid-state. Cationic tris(bipyridine)ruthenium(II) template was found to stabilize the hexamer in gas phase, whereas solvent molecules do this in condensed phases. In solution, the capsular hexamer was found to be the thermodynamically favoured self-assembly product and transition from dimer to hexamer occurred in course of time. The crystal structure of hexamer revealed 24 N–H⋯O direct intermolecular hydrogen bonds between the six pyridinearene macrocycles without any bridging solvent molecules. Hydrogen bond patterns correlate well with DFT computed structures. Thus, all structural chemistry methods (IM-MS, DOSY NMR, DFT, X-ray crystallography) support the same structure of the hexameric capsule that has a diameter of ca. 3 nm and volume of 1160 Å3.
Introduction
Capsular supramolecular structures are tempting species, as they enable encapsulation of guest molecules and their voids may act as confined spaces suitable for nanocatalysis.1 Resorcinarenes and pyrogallarenes (see Scheme 1), have been long known for their capability to spontaneously self-assemble into large hexameric capsules.2 Interestingly, the hydrogen bonding network in resorcinarene hexamers typically involve bridging solvent molecules, which might even be responsible for their unique catalytic activity.4 Both resorcinarenes and pyrogallarenes have demonstrated their ability to encapsulate various guest molecules in the voids of their capsular dimeric and hexameric assemblies.2,3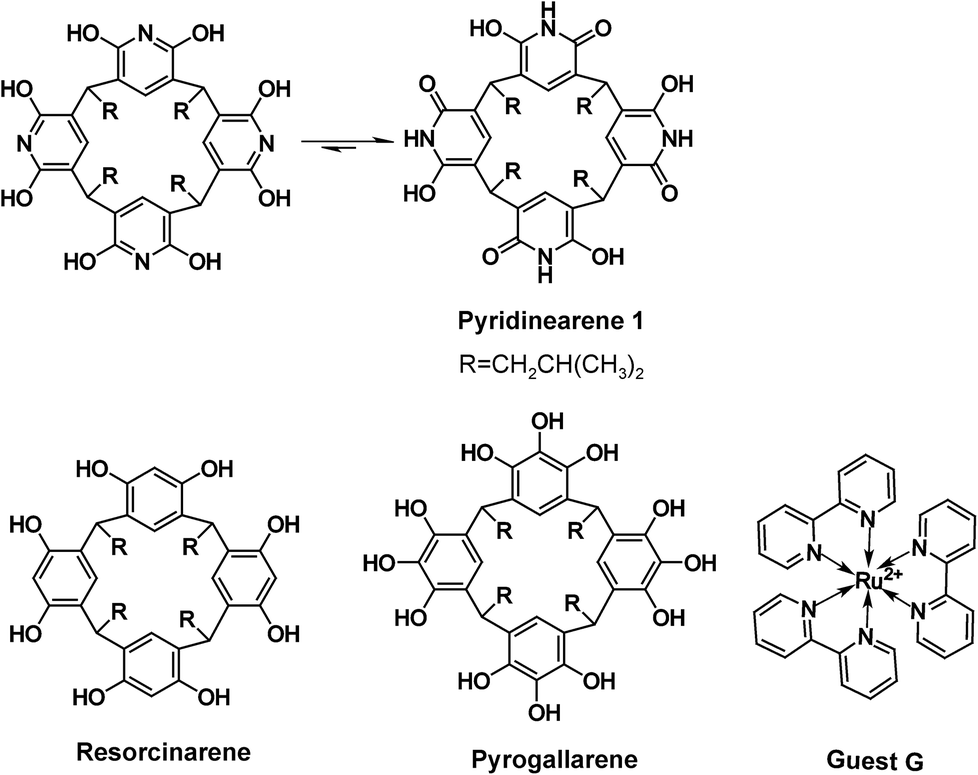 | ||
| Scheme 1 Structures of tetraisobutyl-pyridinearene 1, tris(bipyridine)ruthenium(II) guest (G), resorcinarene and pyrogallarene. For pyridinearene two tautomeric forms are shown, but equilibrium is greatly favoring hydroxy-oxo tautomer. | ||
Pyridinearene5 (1 in Scheme 1) is closely related to resorcinarene and pyrogallarene. Despite their obvious structural similarities, pyridinearenes display different binding properties due to amide–iminol tautomerism and electronic properties arising from the pyridine ring.6 From the two tautomeric forms, dihydroxy and hydroxy-oxo, only the latter has been observed in crystal structures.6 Recently, conformational analysis for the tautomers also revealed that the hydroxy-oxo tautomer (Scheme 1) is ∼150 kJ mol−1 lower in energy than the dihydroxy tautomer.7
The cavity of pyridinearene was earlier considered electron deficient and suitable to bind anionic guests.8,9 Recently, instead of encapsulation of anions, the pyridinearene dimers were shown to actually favour encapsulation of neutral guests of matching size, and hence maximizing dispersion interactions. In fact, pyridinearenes bind anions, but outside the cavity, in exo position between the lower rim alkyl chains via CH⋯anion interactions.6 Although the cavity of pyridinearenes is most suitable for neutral guests, also anion-driven encapsulation of cations inside the pyridinearene dimers was recently demonstrated.7
In one of the earliest reports concerning the self-assembly of pyridinearene, two species were observed in chloroform solution which were initially erroneously assigned as a mixture of monomers and dimeric assemblies.9 Cohen et al. later proved these as dimeric and hexameric species using diffusion ordered (DOSY) NMR spectroscopy experiments.10 In here, we report a detailed study of pyridinearene hexamer formation and structure in solution (NMR, DOSY NMR), in solid-state (X-ray crystallography) and in the gas phase (IM-MS, ESI-QTOF MS, DFT calculations).
Results and discussion
In the gas phase, by ESI-QTOF mass spectrometry, 1 typically ionizes well through deprotonation or anion complex formation (with PF6−, BF4− or I−). Despite many attempts, no traces of hexamer was detected in the negative mode. Similar hexameric capsules based on resorcinarene or pyrogallarene molecules have been earlier detected on positive mode by using a spherical and cationic transition metal complexes11 as the stabilizing templates.3 We, therefore, chose tris(bipyridine)ruthenium(II) (see Scheme 1, guest G) as a suitable complementary template. The cavity size for pyridinearene assemblies are roughly comparable to the size of corresponding assemblies of resorcinarenes and pyrogallarenes, but the affinity of pyridinearene towards cationic guests is lower. Nevertheless, when a sample of 1 (60 μM) with G (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6) was sprayed from an acetone/dichloromethane (1:1 v/v) mixture, a host–guest hexameric assembly [16 + G]2+ was detected in the mass spectrum (Fig. 1). In addition, a pentameric host–guest [15 + G]2+ and a dimeric [12 + H]+ complexes were also observed, but the hexameric assembly was clearly the dominating ion in the spectrum (Fig. 1). Ion [15 + G]2+ likely originates from the hexamer through fragmentation. Assemblies larger than the hexamer, namely heptamers or octamers were not observed. The abundance of hexameric ion and lack of other aggregates in the spectrum suggest that hexamer could be a hydrogen bonded capsule rather than an unspecific aggregated system or a polymer.‡
:6) was sprayed from an acetone/dichloromethane (1:1 v/v) mixture, a host–guest hexameric assembly [16 + G]2+ was detected in the mass spectrum (Fig. 1). In addition, a pentameric host–guest [15 + G]2+ and a dimeric [12 + H]+ complexes were also observed, but the hexameric assembly was clearly the dominating ion in the spectrum (Fig. 1). Ion [15 + G]2+ likely originates from the hexamer through fragmentation. Assemblies larger than the hexamer, namely heptamers or octamers were not observed. The abundance of hexameric ion and lack of other aggregates in the spectrum suggest that hexamer could be a hydrogen bonded capsule rather than an unspecific aggregated system or a polymer.‡
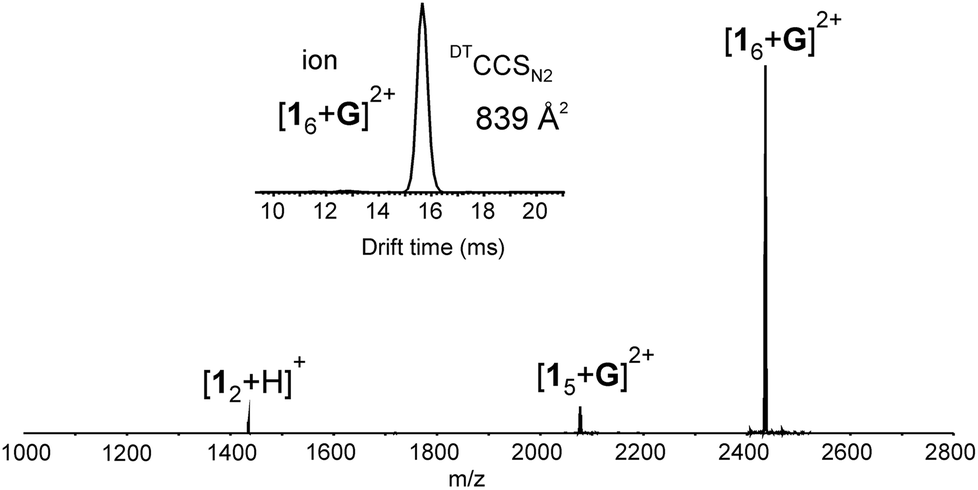 | ||
| Fig. 1 (+)ESI-MS spectrum of 1 (60 μM) and tris(bipyridine)ruthenium(II) (G) with 6:1 ratio measured from acetone/DCM mixture. Inset showing IMS arrival time distribution (ATD) for [16 + G]2+ ion. | ||
To get actual proof whether the studied system is really a capsular assembly, ion mobility mass spectrometry (IM-MS) was utilized to acquire ion mobility arrival time distribution (ATD) and to determine the structure related collision cross section (CCS) value for the hexamer. IM-MS has already demonstrated its power in structural analysis of different supramolecular systems.12 In the ATD only a single well-resolved peak was observed. This implies that the corresponding [16 + G]2+ ion has a relatively rigid well-defined structure. When a drift tube ion mobility (DT-IMMS) instrument employing helium as a drift gas was used, an experimental DTCCSHe value of 746.4 Å2 was obtained.§ If a spherical structure is assumed, this corresponds roughly to a diameter of 3.1 nm for the assembly. The theoretical DTMCCSHe was calculated on basis of the DFT calculated structure by using diffuse trajectory method and yielded a DTMCCSHe of 748.2 Å2,13¶ which is in good agreement with the experimental value. When more polarizable nitrogen was used as a drift gas, the DTCCSN2 value was determined as 838.7 Å2.|| This roughly corresponds to a sphere with a diameter of 3.3 nm. The theoretical DTMCCSN2 of 928.3 Å2 for the [16 + G]2+ ion indicate slightly bigger difference between experimental and theoretical values when N2 is used as a drift gas. In general, these CCS values are in good agreement with dimensions reported earlier for hexameric capsules of pyrogallarenes and resorcinarenes,14,15 and thus, verify that the gas phase structure is indeed a capsular hexameric assembly.
The self-assembly of 1 was further studied in solution by 1H and DOSY NMR (in CDCl3 at 298 K). In a 20 mM solution of 1, two different species were observed in 1H NMR spectra shortly after preparing the sample (Fig. 2a): one causing signals at 4.28 and 7.10 ppm (d/d′), and another (h/h′) with signals at 4.36 and 7.22 ppm. The ratio of these two (d:h) was 2:1. The diffusion coefficient determined for assembly d was 0.411 × 10−5 cm2 s−1, and 0.287 × 10−5 cm2 s−1 for assembly h. These values are in accordance with earlier results from Cohen,10 and confirm that the signal sets d/d′ and h/h′ correspond to the dimer and the hexamer of 1. These values are in the expected range when compared with the diffusion coefficients of the corresponding assemblies of resorcinarene and pyrogallarene, which were used as internal standards. Assuming spherical shapes of these assemblies this translates into diameters of 2.0 nm for the dimer and 2.8 nm for the hexamer using the Stokes–Einstein equation (see ESI†). These values are also well in line with the observed gas phase structures and determined CCS values. The DOSY NMR experiment in Fig. 2a was measured within 2 hours after sample preparation and shows the dimeric assembly as more abundant (2:1). The same NMR sample was stored and re-measured after seven days showing that the intensity of the signals for the dimer diminished dramatically, while at the same time signals for the hexamer increased and were now dominating (ratio d:h 1:10). This can also be seen in DOSY NMR, where only the signal of hexamer is observed after 7 days from sample preparation (Fig. 2b). This shows that pyridinearene forms indeed both dimeric and hexameric capsules in solution but the dimeric capsule is obviously only the kinetically favoured product while the hexameric capsule is the thermodynamic product, which abundance increases over time.
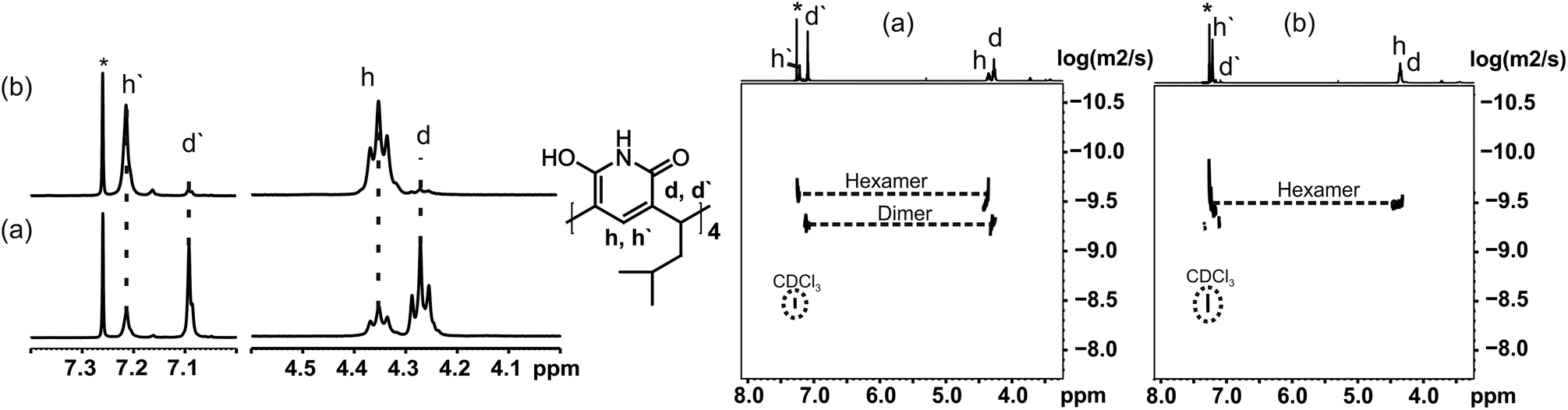 | ||
| Fig. 2 Left: 1H NMR of 1 (20 mM in CDCl3) (a) measured within 2 hours after sample preparation and (b) same sample after 7 days from sample preparation. Right: DOSY NMR of the same sample (a) measured within 2 hours form sample preparation and (b) after 7 days from sample preparation. Signals d and d′ correspond to the dimeric assembly and signals h and h′ to the hexameric assembly. | ||
Single crystals suitable for X-ray diffraction analysis were obtained by slow vapour diffusion of diisopropyl ether into the solution of 1 in chloroform. Block shaped crystals were formed and the hexamer structure (16) in the solid-state could be confirmed by X-ray crystallography (Fig. 3a). The asymmetric unit contains three crystallographically independent molecules of 1, and the resultant self-assembled hexameric structure contains 24 O–H⋯O and 24 N–H⋯O hydrogen bonds. As a result of hydroxy-oxo tautomer form, the 6-hydroxyl position in 6-hydroxy-2(1H)-pyridone units manifest four O–H⋯O circular intramolecular hydrogen bonds to give 1 bowl-like C4v conformation. The intermolecular N–H⋯O hydrogen bonding between the pyridone units in 16 provides a compact internal cavity with volume of 1160 Å3. This is smaller than corresponding hexameric capsules based on resorcinarenes and pyrogallarenes (volumes >1200 Å3, see ESI Fig. S4†).14,15 The DOSY derived hydrodynamic radii (2.80 nm) agrees well with the calculated value obtained from the X-ray crystal structure (2.82 nm). Of special note, the hydroxy-oxo tautomerism renders 1 a rigid cavity and definite directionality for hydrogen bonding. Robust C4v cavities are seamed together with direct N–H⋯O intermolecular hydrogen bonding to produce solvent-free hexamer 16. Solvent-free hydrogen bonding is also observed in the solid-state pyrogallarene hexamer structures.14 In the resorcinarene hexamers, due to flexible C4v cavities and steric reasons solvent molecules typically assist in capsule formation via intermolecular hydrogen bonding interactions, found between two resorcinarene units.15 Although chloroform is essential to obtain a stable 16 single-crystals its passive role in hydrogen bonding could be observed in the 3-D crystal lattice. The features of pyridinearene lead to unique and complementary alignment of the individual monomers in order to maximize the number of intermolecular hydrogen bonds. In the case of the resorcinarenes, this requires incorporation of solvent molecules as part of the intermolecular hydrogen bond network. Consequently, this causes the differences in the void size for three different hexamers.
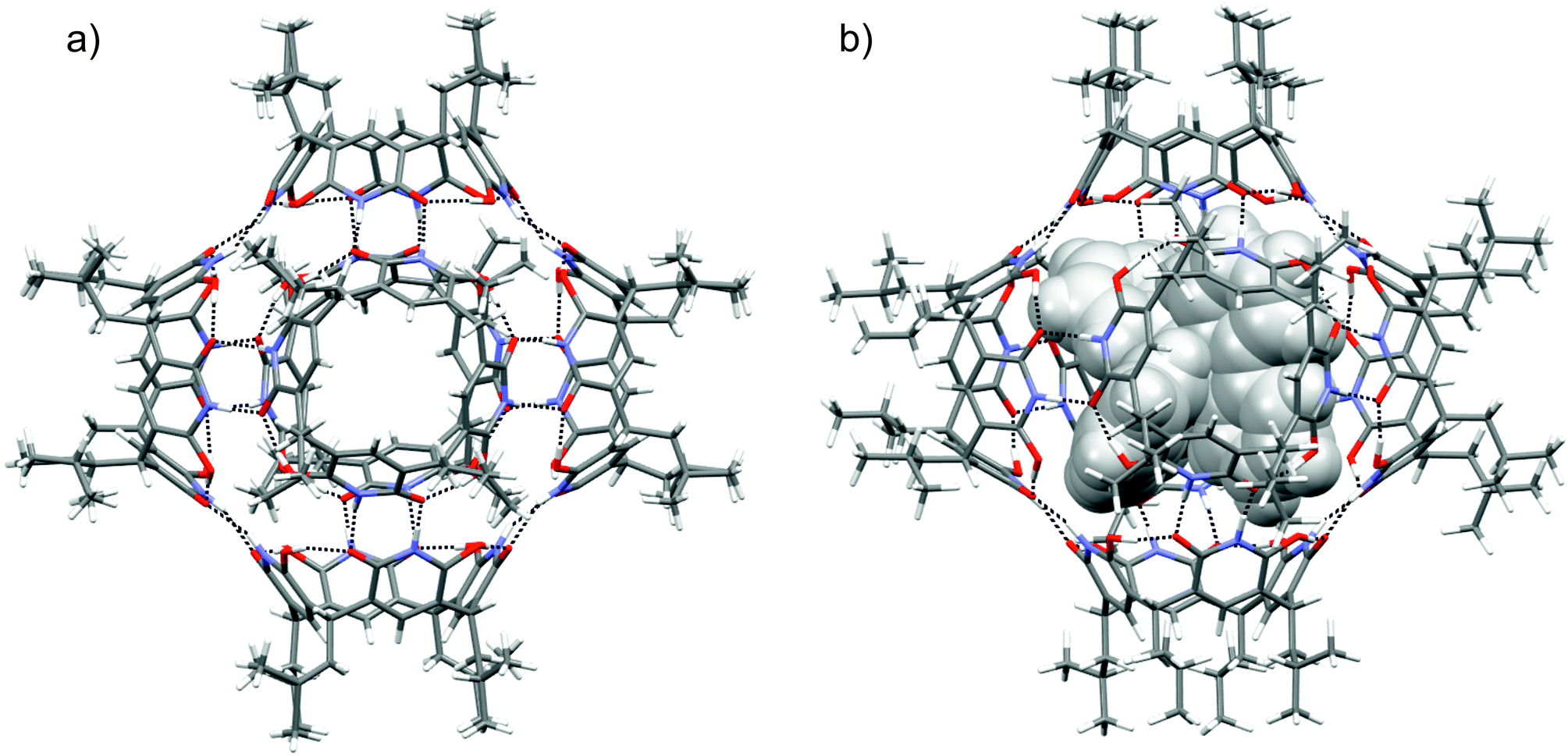 | ||
| Fig. 3 X-Ray crystal structure of 16 and (b) optimized structure of [16 + G]2+ at the RI-PBE-D3/def2-SVP level of theory in capped stick model. Tris(bipyridine)-ruthenium(II) (G) is shown in grey CPK model in (b). Black dashed lines represent hydrogen bonds. Color code: H = white, C = grey, N = blue, O = red. | ||
Unfortunately, all our attempts to obtain single crystals from [16 + G]2+ were unsuccessful. Therefore, we decided to carry out the computational analysis for studied systems at the RI-PBE-D3/def2-SVP//RI-PBE-D3/def2-TZVP level of theory in order to investigate the gas phase structure of 16 and [16 + G]2+ and to evaluate their interaction energies in detail.16–24
The optimized structures and hydrogen bonding patterns of 16 and [16 + G]2+ are illustrated in Fig. 3b (see also ESI†). Within 16 and [16 + G]2+, each monomer interacts with four neighbouring monomers through intermolecular N–H⋯O bonds whose distances vary from 1.703 to 1.716 Å and from 1.657 to 1.699 Å for 16 and [16 + G]2+, respectively (Table 1). Interestingly, as compared to the dimeric capsule, 12, the N–H⋯O bonds are slightly shorter in the hexamer. The total number of N–H⋯O bonds in one hexamer unit is 24. Furthermore, the seam of hydrogen bonds, consisting of four intramolecular O–H⋯O bonds, of each monomer 1, is retained during hexamer formation. However, small changes are observed in the O–H⋯O bonds of 1, 12, 16 and [16 + G]2+ if compared with each other: the H-bonds are slightly longer in the hexamers and the dimers than in the monomer. The elongation of the O–H⋯O bonds in the hexamers and dimers most likely arises from the small changes in electron density distribution around/on the oxygen atoms that function as a hydrogen bond acceptors in both, N–H⋯O and O–H⋯O, bonds in the hexamers and the dimers. Some electron density is therefore transferred from O–H⋯O bonds to N–H⋯O bonds upon the formation of dimers and hexamers. In the monomer, however, these acceptors oxygen atoms participates only in the formation of O–H⋯O bonds.
Due to the multiple intermolecular N–H⋯O bonds, 24 in total, the interaction energies of 16 and [16 + G]2+ are very high −717 kJ mol−1 and −1205 kJ mol−1, respectively (Table 1). These energy numbers suggest that the [16 + G]2+ is ∼500 kJ mol−1 more stable than empty hexamer 16 and underline the significance of template effect.3 If the interaction energies of 16 and [16 + G]2+ are compared to the interaction energies of 3 individual dimers 12 (3 × −197 kJ mol−1 = −590 kJ mol−1), it is clear that the formation of hexamer is more favourable than the formation of 3 individual dimers from six monomers. Although this very simplified gas phase model does not take into account all forces acting in the solution state, it not only supports the result obtained from MS studies, but is also in agreement with the NMR data: the hexamer 16 is thermodynamically more stable than the dimer 12 in gas phase and solution state.
Conclusions
To conclude, the pyridinearene hexamers were structurally characterized in all three states of matter, and hence, for the first time in gas-phase and solid-state. X-ray structure shows that the hexameric assembly is formed via directly linked, intermolecular hydrogen bonds without bridging solvent molecules, which are usually required in the solid-state structures of closely related resorcinarenes. This makes hexamer more compact and decreases the size of empty windows between monomer units, which is a significant feature when applications, especially related to catalysis in confined space, are considered. In chloroform solution the hexamer was found to be the thermodynamically favoured product compared to the corresponding kinetically favoured dimer. All analytical techniques used result in a structure with a diameter of ∼3 nm, and support the structural similarity of the pyridinearene hexamer in gas phase, solution and in solid state. As such, it is slightly more compact than the related hexamers of resorcinarenes and pyrogallarenes due to the different intermolecular hydrogen bonding network that implies to unique mutual alignment of the monomers in these assemblies. Interestingly, the use of a cationic, structurally complementary template increased the gas phase stability of the hexamer substantially, which could be rationalized by sophisticated DFT calculations.Having established the structural properties of these large hydrogen-bonded capsules now paves the avenue to explore its host–guest binding properties arising from its different and unique interactions and geometric features compared to the other similar assemblies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Academy of Finland for the funding (JOM: 285855, 315829, 320015, RP: 298817, EK: 284562, 278743 and 312514) as well as Prof. H. M. Tuononen, University of Jyväskylä, CSC-IT Center for Science in Finland, and the Finnish Grid and Cloud Infrastructure (persistent identifierurn:nbn:fi:research-infras-2016072533) for providing computational resources. NKB gratefully acknowledge the financial support from Oakland University, MI, USA.Notes and references
- D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349 CrossRef CAS PubMed; M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650 CrossRef PubMed; L. Catti, Q. Zhang and K. Tiefenbacher, Chem. – Eur. J., 2016, 22, 9060 CrossRef PubMed.
- M. Yamanaka, A. Shivanyuk and J. Rebek, J. Am. Chem. Soc., 2004, 126, 2939 CrossRef CAS PubMed; L. Avram and Y. Cohen, J. Am. Chem. Soc., 2002, 124, 15148 CrossRef PubMed; L. MacGillivray and J. Atwood, Nature, 1997, 389, 469 CrossRef.
- N. K. Beyeh, M. Kogej, A. Åhman, K. Rissanen and C. A. Schalley, Angew. Chem., Int. Ed., 2006, 45, 5214 CrossRef CAS PubMed.
- Q. Zhang, L. Catti and K. Tiefenbacher, Acc. Chem. Res., 2018, 51, 2107 CrossRef CAS PubMed.
- T. Gerkensmeier, J. Mattay and C. Näther, Chem. – Eur. J., 2001, 7, 465 CrossRef CAS.
- A. Kiesilä, L. Kivijärvi, N. K. Beyeh, J. O. Moilanen, M. Groessl, T. Rothe, S. Götz, F. Topić, K. Rissanen, A. Lützen and E. Kalenius, Angew. Chem., Int. Ed., 2017, 56, 10942 CrossRef PubMed.
- A. Kiesilä, J. O. Moilanen, A. Kruve, C. A. Schalley, P. Barran and E. Kalenius, Unpublished results.
- A. B. Rozhenko, W. W. Schoeller, M. C. Letzel, B. Decker and J. Mattay, New J. Chem., 2013, 37, 356 RSC.
- M. C. Letzel, B. Decker, A. B. Rozhenko, W. W. Schoeller and J. Mattay, J. Am. Chem. Soc., 2004, 126, 9669 CrossRef CAS PubMed.
- T. Evan-Salem and Y. Cohen, Chem. – Eur. J., 2007, 13, 7659 CrossRef CAS PubMed.
- G. Bianchini, A. Scarso, G. La Sorella and G. Strukul, Chem. Commun., 2012, 48, 12082–12084 RSC.
- E. Kalenius, M. Groessl and K. Rissanen, Nat. Chem. Rev., 2019, 3, 4 CrossRef CAS.
- C. Larriba and C. J. Hogan, Jr., J. Comput. Phys., 2013, 251, 344 CrossRef CAS; C. Larriba and C. J. Hogan Jr., J. Phys. Chem. A, 2013, 117, 3887 CrossRef PubMed.
- T. Gerkensmeier, W. Iwanek, C. Agena, R. Fröhlich, S. Kotila, C. Näther and J. Mattay, Eur. J. Org. Chem., 1999, 2257 CrossRef CAS.
- R. M. Payne and C. L. Oliver, CrystEngComm, 2018, 20, 1919 RSC.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 242, 652 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119 Search PubMed.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244 CrossRef PubMed.
- J. J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- V. Gabelica, et al. , Mass Spectrom. Rev., 2019 DOI:10.1002/mas.21585.
- V. Gabelica and E. Marklund, Curr. Opin. Chem. Biol., 2018, 42, 51 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Mass spectrometry, NMR, X-Ray crystallography and DFT calculations. CCDC 1899745. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ob01383k |
| ‡ Please note, that the template G is not necessary to observe the hexameric assembly in (+)ESI-MS. Without templating guest hexamer can be observed as [16 + Na]+ even in low (20 μM) concentration, but with relatively lower abundancy (see Fig. S1†). |
| § DTCCSHe states for CCS values obtained using drift tube instrument with helium gas, nomenclature recommended in V. Gabelica et al.25 |
| ¶ DTMCCSHe states for CCS values obtained using diffuse trajectory method with helium gas. |
| || According to Mason–Schamp equation, different gases result in different drift times, and thus to different CCS values. For more detailed explanation see V. Gabelica and E. Marklund.26 |
| This journal is © The Royal Society of Chemistry 2019 |