
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid photoinduced charge injection into covalent polyoxometalate–bodipy conjugates†
Fiona A.
Black
a,
Aurélie
Jacquart
b,
Georgios
Toupalas
b,
Sandra
Alves
b,
Anna
Proust
b,
Ian P.
Clark
c,
Elizabeth A.
Gibson
 *a and
Guillaume
Izzet
*b
*a and
Guillaume
Izzet
*b
aChemistry: School of Natural and Environmental Science, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: Elizabeth.gibson@ncl.ac.uk
bSorbonne Université, CNRS, Institut Parisien de Chimie Moléculaire, IPCM, 4 Place Jussieu, F-75005 Paris, France. E-mail: guillaume.izzet@sorbonne-universite.fr
cCentral Laser Facility, Research Complex at Harwell, Science and Technology Facilities Council, Rutherford Appleton Laboratory, Harwell Oxford, Didcot, Oxfordshire OX11 0QX, UK
First published on 1st June 2018
Abstract
Controlled design of photoactive hybrids would provide highly active materials for solar energy conversion and photo(electro) catalysis. We describe the kinetics of photoinduced electron transfer in a series of photoactive hybrids based on Keggin-type polyoxometalates (POMs) covalently grafted to bodipy photosensitizers. We show how the electronic properties and corresponding dynamics of these hybrids can be readily tuned by varying the POM metal ion, the anchoring functionalization and the spacer length. Ultrafast visible and IR transient absorption spectroscopy, supported by spectroelectrochemical measurements, reveals that photoinduced electron transfer from the bodipy chromophore to the organosilyl POM derivative occurs as rapidly as τ = 54 ps to generate a long-lived (τ = 4.8 ns) charge-separated (CS) state, making this system appropriate for applications in photoelectrochemical devices.
Introduction
The direct generation of chemical fuels from sunlight is a major scientific challenge for the development of a sustainable economy and production of hydrogen through water splitting. In green plants, photosynthesis operates in four steps: (1) light collection, (2) charge separation, (3) charge accumulation and (4) conversion to chemical fuel. Charge accumulation is a key step as most of the redox reactions for fuel generation such as H2 production from water are multi-electronic processes.1 While various noble-metal-based complexes (e.g. Pd, Pt…) have been widely employed as reduction catalysts in artificial photosynthetic systems, polyoxometalates have recently emerged in this field owing to their electron reservoir abilities and the activity of their reduced forms in the hydrogen evolution reaction.2–4 POMs form a remarkable class of well-defined molecular nano-scale oxoclusters of the early transition metals with an unmatched diversity of structures and properties.5 Their implementation in artificial photosynthetic devices requires their association with a visible-range antenna since POMs only absorb in the UV region of the solar spectrum. Owing to their anionic nature, POMs have mainly been associated with photosensitizer (PS) through electrostatic interactions.6–11 By comparison, few covalent organic–inorganic POM-based hybrids have been developed.12–17 While the covalent functionalization of POMs is synthetically more demanding, this approach enables fine control between the different subunits of the system, which is required for tuning the kinetics of photoinduced electron transfer between the excited chromophore and the POM.3We previously reported the synthesis and photophysical properties of different POM–PS conjugates.18–23 Among them heteroleptic carbocyclometalated iridium(III)-POM dyads offered the most promising photophysical performance.23 In these compounds, photoinduced charge-separated excited states of various lifetimes (ranging from nanoseconds to hundreds of nanoseconds, the longest values reported for covalently bonded photosensitized POMs) were observed by transient absorption spectroscopy. Furthermore, in the presence of a sacrificial electron donor (triethylamine) and a proton source (acetic acid), the system is capable of photo-accumulating two electrons on the POM and produces hydrogen.20 However, the POM-Ir(III) system suffered from the presence of a noble metal (Ir) and the low stability of the iridium complex in the presence of strong acid, which is necessary to improve the electron reservoir properties of the POM.
Furthermore, in order to incorporate the POM–PS hybrids into a photocathode, the functionalization of the PS by an appropriate anchoring group (carboxylic acid, phosphonate…) is a required step, which has been very scarcely developed with carbocyclometalated iridium(III) complexes.24 In this context, bodipy fluorophores have been identified as adequate candidates because of their tunable photophysical properties, high chemical stability, easy and versatile chemical modification such as the addition of a grafting function.25 Here, we present the first examples of Keggin-type POMs that are covalently bound to one or two bodipy units via organic tethers of different lengths (Scheme 1). The series of three hybrids display distinct redox behaviour both on the POM and on the bodipy units. The photophysical properties of these hybrids are reported and discussed on the basis of the required kinetics that should be achieved for their implementation in a photoelectrocatalytic device.
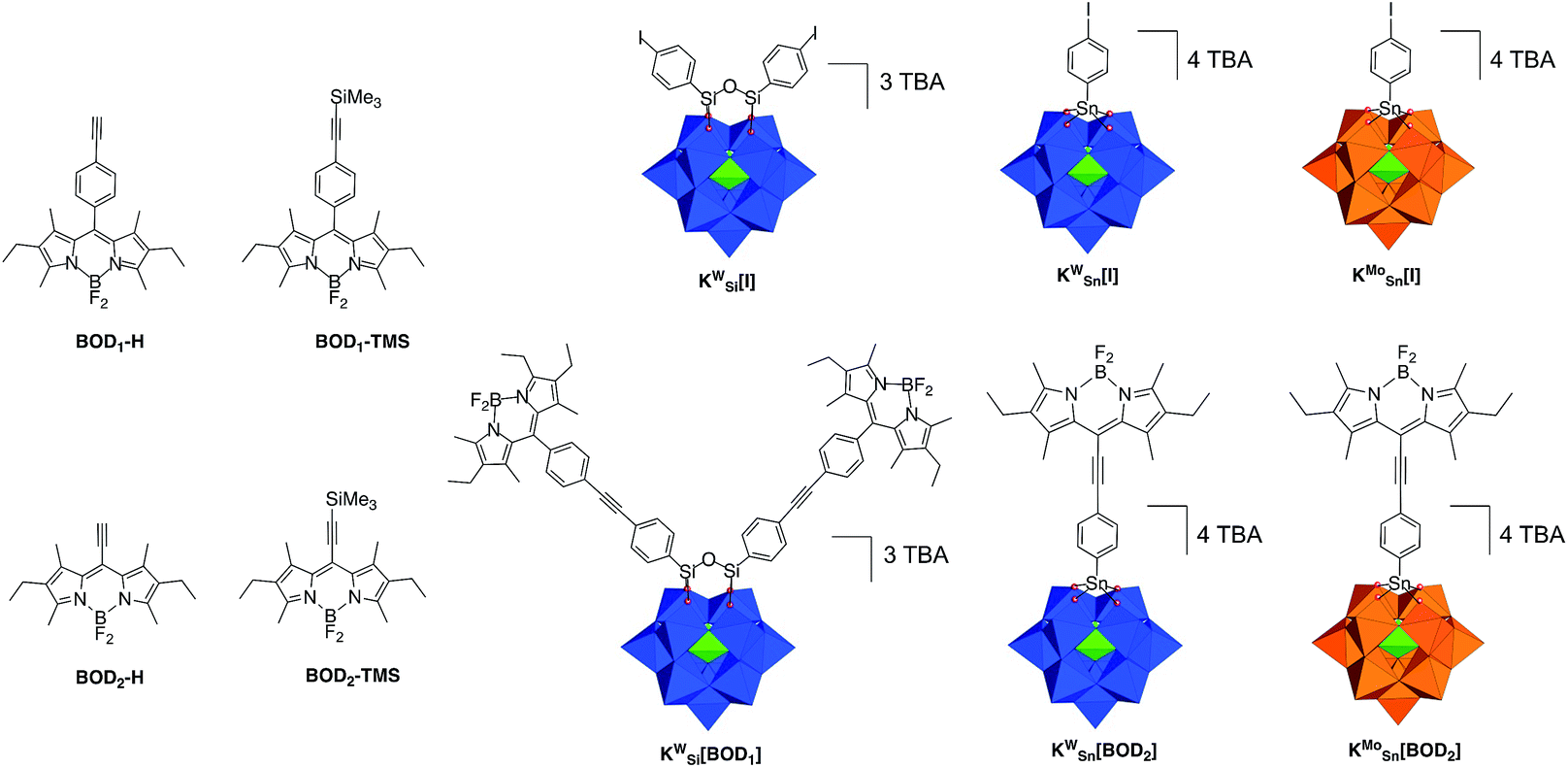 | ||
| Scheme 1 Molecular drawings of the bodipy reference compounds (BOD1-H, BOD1-TMS, BOD2-H, BOD2-TMS), the iodoaryl-terminated POM-based platforms (KSiW[I], KSnW[I], KSnMo[I]) and the POM–bodipy conjugates described in this study (KSiW[BOD1], KSnW[BOD2], KSnMo[BOD2]). Color code: WO6 octahedra, blue; MoO6 octahedra, blue; PO4 tetrahedra, green. | ||
Results and discussion
Synthesis of the different POM–bodipy conjugates
We previously developed three Keggin-based polyoxometalate hybrid platforms bearing one or two remote iodo aryl functions. These hybrids vary according to the nature of the metal ion (polyoxotungstate vs. polyoxomolybdate) and the nature of the primary functionalization (silyl vs. tin).21,26,27 The organosilyl POM derivative (noted KSiW[I]‡) is more easily reducible than its organotin counterparts due to a lower charge. Furthermore, the reduction processes are more accessible on the polyoxomolybdate organotin derivative (noted KSnMo[I]) compared to its isocharged tungstate analogue (noted KSnW[I]), as classically observed for polyoxometalates.27,28 The electron acceptor abilities of these hybrids thus follow the trend: KSiW[I] > KSnMo[I] > KSnW[I].3In a first attempt we tried to couple the organosilyl polyoxotungstate platform KSiW[I] (displaying the best electron accepting properties) with both alkyne terminated bodipy BOD1-H or BOD2-H in order to investigate the effect of the spacer length on the photophysical properties using the same Keggin-based platform. However, while KSiW[BOD1] turned out to be stable, the hybrid resulting from the coupling between KSiW[I] and BOD2-H unexpectedly slightly decomposed upon precipitation. Consequently, we were not able to obtain KSiW[BOD2] with the required purity. The organotin functionalization of POMs leads to the formation of robust hybrids since the lacuna of monovacant POMs is generally well-suited for the incorporation of one Sn4+ atom.3 We thus investigated the functionalization of the organotin polyoxotungstate and polyoxomolybdate platforms KSnW[I] and KSnMo[I] with BOD2-H. In both cases stable hybrids of appropriate purity were obtained. The coupling reactions between the POM hybrid platforms bearing the iodoaryl moieties (i.e.KSiW[I], KSnMo[I] and KSnW[I]) were performed following conditions that we previously established. Typically, Sonogashira C–C cross coupling occurs at 70 °C in ca. 30 min under microwave activation, in DMF containing triethylamine (20 equiv.), using [Pd(PPh)3Cl2] (8–10%) as catalyst source and Cu I (8–10%) as co-catalyst. An excess of the alkyne terminated-bodipy is required for the total conversion of the starting POM. For the synthesis of KSnW[BOD2] and KSnMo[BOD2] a second loading of BOD2-H into the reaction mixture was performed after 15 min, due to the inevitable decomposition of BOD2-H under these conditions.
Electrochemistry
The redox properties of the POM-bodipy dyads, their related POM hybrids and bodipy references were investigated by cyclic voltammetry and, in cases of irreversible redox processes, by differential pulse voltammetry in deoxygenated dichloromethane (DCM) with tetrabutylammonium hexafluorophosphate (TBAPF6) as the supporting electrolyte in a standard three-electrode cell, composed of a glassy carbon working electrode, a platinum counter electrode, and a saturated calomel reference electrode (SCE) (Fig. 1 and Table 1).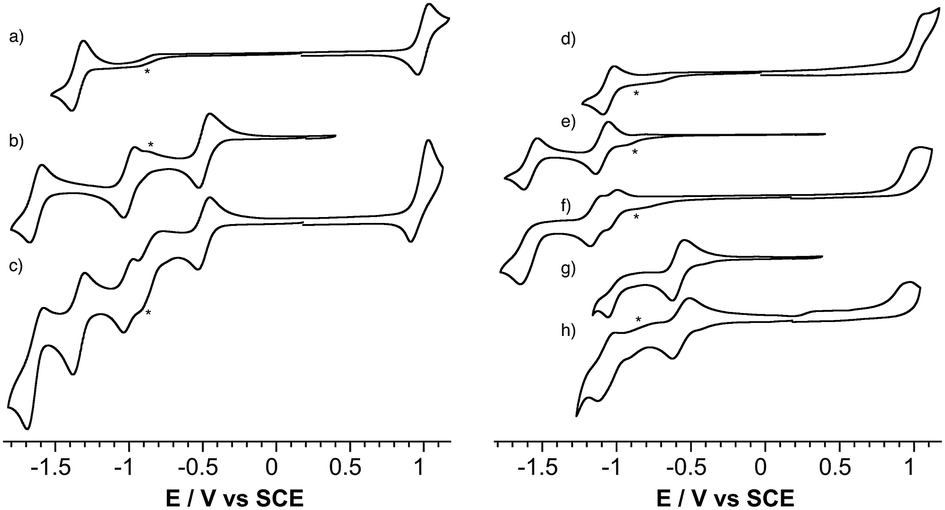 | ||
| Fig. 1 Cyclic voltammograms of ca. 0.5 mM solutions of reported compounds in DCM containing 0.1 M of TBAPF6: (a) BOD1-TMS, (b) KSiW[I], (c) KSiW[BOD1], (d) BOD2-TMS (e) KSnW[I], (f) KSnW[BOD2], (g) KSnMo[I], (h) KSnMo[BOD2]. Scan rate, 20 mV s−1; working electrode, glassy carbon; reference electrode, SCE. The minor peaks noted with an asterisk are observed in the blank and correspond to residual traces of oxygen and/or solvent impurities. | ||
| Compound | [BOD]+/[BOD] | [BOD]/[BOD]− | POM/POM + 1 e | POM + 1 e/POM + 2 e |
|---|---|---|---|---|
| a The standard potentials of irreversible processes were determined by differential pulse voltammetry experiments (Fig. S7–S14). | ||||
| BOD1-TMS | 1.00 (80) | −1.35 (80) | ||
| KSiW[I] | −0.49 (70) | −1.00 (70) | ||
| KSiW[BOD1] | 0.98 | −1.34 (80) | −0.49 (80) | −1.00 (70) |
| BOD2-TMS | 1.02 | −1.05 (80) | ||
| KSnW[I] | −1.10 (80) | −1.58 (90) | ||
| KSnW[BOD2] | 1.02 | −1.00 | −1.11 | −1.60 |
| KSnMo[I] | −0.58 (80) | −1.03 | ||
| KSnMo[BOD2] | 0.98 | −1.00 | −0.57 (110) | −1.04 |
First, it is noted that the redox potentials of the starting POM-based platforms are considerably shifted in DCM compared to acetonitrile,3 which outlines a very important effect of the organic solvent in the reduction processes of the polyanions. As previously mentioned when one compares the reduction potentials of the polyoxotungstates KSiW[I]versusKSnW[I], it can be seen that the latter is more difficult to reduce by more than half a volt due to a one-charge difference between both oxoclusters. BOD1-TMS and BOD2-TMS both feature reversible monoelectronic oxidation and reduction processes. While the oxidation potential for each are rather similar, reduction of BOD2-TMS occurs at a significantly more positive potential than reduction of BOD1-TMS (ΔEred = 300 mV). Indeed, in BOD1-TMS the phenyl unit is twisted for steric reasons, which decouples it from the π system of the bodipy unit,29 while in BOD2-TMS, the π system of the bodipy unit extends over the alkynyl moieties, favouring its reduction. The redox properties of KSiW[BOD1] are very close to those of the reference compounds KSiW[I] and BOD1-TMS, as is often reported with our POM hybrid platforms.3,19,23,26,30 The shape of the oxidation waves of KSiW[BOD1] is characteristic of adsorption of the oxidized species at the working electrode. The redox properties of KSnW[BOD2] and KSnMo[BOD2] are also similar to those of the reference compounds KSnW[I], KSnMo[I] and BOD2-TMS. As for BOD2-TMS, the oxidation of KSnW[BOD2] and KSnMo[BOD2] are irreversible. In the reduction part, KSnW[BOD2] displays two quasi-reversible processes followed by an irreversible process. The first one is attributed of the reduction of the bodipy unit while the two others correspond to monoelectronic reductions of the POM framework. In the reduction part, KSnMo[BOD2] displays two apparent quasi-reversible processes. The first one at −0.57 V vs. SCE is attributed to the reduction of the polyoxomolybdate framework, while the second at −1.04 V vs. SCE is attributed to quasi simultaneous reduction of the bodipy and the one-electron reduced POM (note that in differential pulse voltammetry experiment these two reduction processes are slightly separated with a maximum at −1.04 V and a shoulder at ca. −1.00 V vs. SCE attributed to the one-electron reduced POM and bodipy reduction respectively, Fig. S14†). The reduction potential comparison of KSnMo[BOD2]versusKSnW[BOD2] thus suggests that the polyoxomolybdate core of KSnMo[BOD2] is more readily reduced than the polyoxotungstate core of KSnW[BOD2] by the BOD2 excited state.
Electronic absorption and photophysical properties
The electronic absorption spectra for bodipy reference compounds and POM–bodipy hybrids dissolved in DCM are shown in Fig. 2. The absorption spectra of the hybrids are dominated by the bodipy absorption in the visible range, since the POM itself only absorbs in the UV part. The absorption profiles show a typical sharp bodipy-centred S0–S1 absorption with a maximum at 527 nm for BOD1-TMS and KSiW[BOD1]. For BOD2-TMS, KSnW[BOD2] and KSnMo[BOD2], the absorption profile is red-shifted due to an increase of the π system resulting from the presence of the alkynyl group at the meso-position. Note that the maximum bodipy absorption of KSnW[BOD2] and KSnMo[BOD2] is slightly red-shifted (5 nm) compared to BOD2-TMS due to the participation of the aryl tin unit into the π system of the bodipy unit. A summary of the photophysical data is presented in Table 2.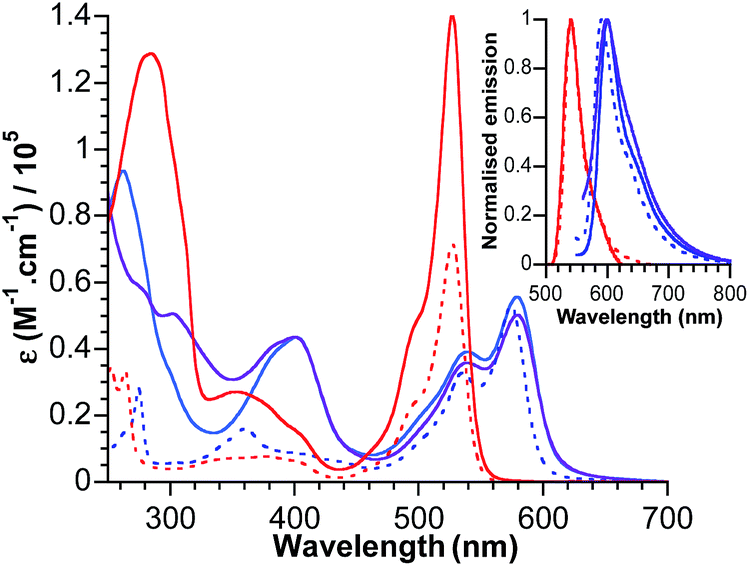 | ||
| Fig. 2 Absorbance and normalised emission (inset) of BOD1-TMS (dotted red), BOD2-TMS (dotted blue), KSiW[BOD1] (plain red), KSnW[BOD2] (plain blue) and KSnMo[BOD2] (plain purple) in DCM. | ||
| Absorption λmax (nm) | Emission λmax (nm) | E 0–0 (eV) | Φ FL | |
|---|---|---|---|---|
| BOD1-TMS | 527 | 540 | 2.33 | 0.67 |
| BOD2-TMS | 574 | 590 | 2.14 | 0.52 |
| KSiW[BOD1] | 527 | 541 | 2.33 | 0.07 |
| KSnW[BOD2] | 579 | 600 | 2.10 | 0.22 |
| KSnMo[BOD2] | 579 | 597 | 2.11 | 0.06 |
The fluorescence quantum yield for KSiW[BOD1] and KSnMo[BOD2] decrease by ca. 90% of that of BOD1-TMS and BOD2-TMS, respectively. This quenching of the bodipy fluorescence is consistent with electron transfer to the polyoxometalate. For KSnW[BOD2], the fluorescence quantum yield decreases by ca. 60% relative to BOD2-TMS, suggesting charge-transfer (if operating) from bodipy to POM is less efficient (note that the presence of the aryl tin unit that participates into the π system of the bodipy unit slightly modifies its electronic properties and may account for the decrease in quantum yield of KSnW[BOD2]).
To probe the fate of the bodipy excited state in these hybrids, transient optical and infrared absorption spectroscopy was performed on DCM solutions of the hybrids. The results are summarised in Table 3. To extract the intermediate, global analysis of the spectra was performed using the program OPTMUS, in which the transients at all detection wavelengths are analysed simultaneously with a single set of exponential functions.31 The transient absorption spectra of KSiW[BOD1] at various time delays after excitation at 540 nm are shown in Fig. 3. At short delay times, a bleach centered at 520 nm forms, consistent with the depletion of the bodipy ground state. The first transient species resembles the excited bodipy (spectra for BOD1-TMS and BOD2-TMS are provided in the ESI for comparison, Fig. S16 and 17†) and decays with τ1 = 54 ps, as a second transient species with a sharp absorption at 400 nm grows in over the same timescale and decays with τ2 = 4.8 ns. The absorption profile of this second transient is consistent with the oxidised bodipy generated by spectroelectrochemical methods (Fig. S15†). Furthermore, this transient also features a broad absorption starting at 600 nm and extending past 700 nm, which is characteristic of a reduced polyoxometalate, and is hence assigned to the charge-separated state, [BOD+-POM(+1 e−)].
| −ΔGCS (eV) | −ΔGrec (eV) | τ CS (ps) | τ rec (ns) | |
|---|---|---|---|---|
| KSiW[BOD1] | 0.86 | 1.47 | 54 | 4.8 |
| KSnW[BOD2] | −0.03 | 2.13 | — | — |
| KSnMo[BOD2] | 0.56 | 1.55 | 180 | 0.52 |
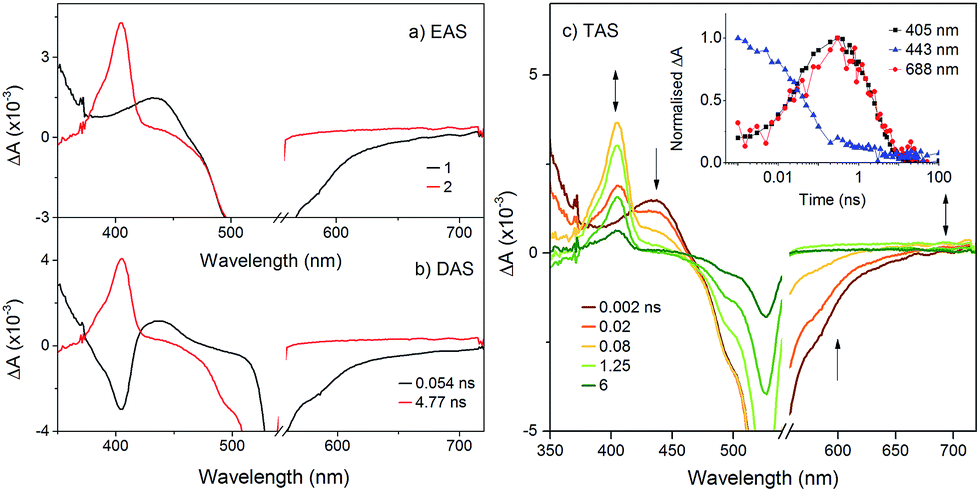 | ||
| Fig. 3 Transient absorption spectra of KSiW[BOD1] in DCM following excitation at 540 nm: (a) evolution associated difference spectra and; (b) decay associated difference spectra derived from global analysis; (c) transient absorption difference spectra at selected time delays after excitation, with single-point decays at 405 nm, 443 nm and 688 nm (inset). | ||
The transient spectra for KSnMo[BOD2] in DCM were more complex since a three component model was needed to accurately fit the relaxation dynamics of this hybrid. The first component (τ1 = 180 ps) in the evolution-associated difference spectra (EAS) has the same general shape as the BOD2-TMS excited state (Fig. S17†), and contains a negative signal from the stimulated emission. The second component (τ2 = 520 ps) is red-shifted (by 6 nm) relative to the first one and does not contain a contribution from the stimulated emission. We have assigned this to the charge-separated state, [BOD+-POM(+1 e−)] since the electron transfer from the BOD2 to the polyoxomolybdate core of KSnMo[BOD2] is thermodynamically favourable and the absorption profile of this second transient is consistent with the oxidised bodipy (Fig. S15†) (Fig. 4).
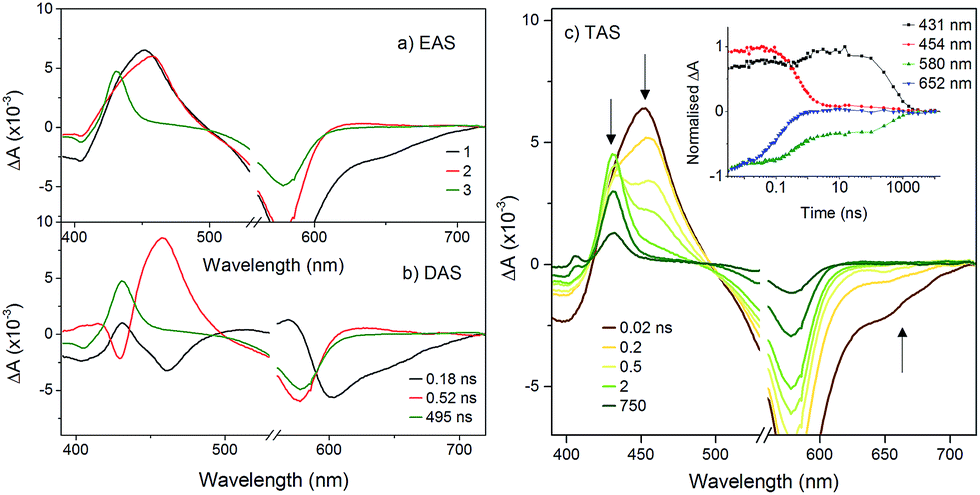 | ||
| Fig. 4 Transient absorption spectra of KSnMo[BOD2] in DCM following excitation at 540 nm: (a) evolution associated difference spectra and; (b) decay associated difference spectra derived from global analysis; (c) transient absorption difference spectra at selected time delays after excitation, with single-point decays at 431 nm, 454 nm, 580 nm and 652 nm (inset). | ||
This second species decays to form a long-lived component (τ3 = 495 ns), attributed to a bodipy triplet excited state. We have previously observed this behaviour for a triphenylamine–bodipy conjugate, arising from photoinduced charge-separation followed by recombination to the triplet state.32
The transient absorption spectra of KSnW[BOD2] in DCM were much simpler (Fig. 5) since electron transfer from the BOD2 to the polyoxotungstate core is thermodynamically not favourable. These transient spectra are initially similar in shape to those of the early transient spectra of KSnMo[BOD2], in agreement with a bodipy centred excited state25 and decays with τ1 = 3.4 ns. A second minor transient signal at time delays exceeding 10 ns, which resembles that of the long-lived species observed with KSnMo[BOD2], is observed (τ2 = 410 ns). This suggests a small concentration of the bodipy centered triplet state is formed, possibly caused by a heavy atom effect from the appended POM.
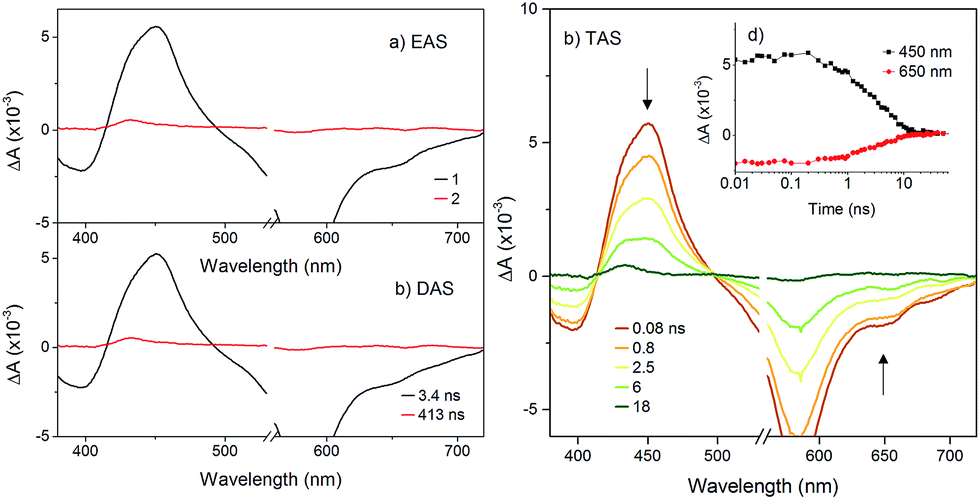 | ||
| Fig. 5 Transient absorption spectra of KSnW[BOD2] in dichloromethane following excitation at 540 nm: (a) evolution associated difference spectra and; (b) decay associated difference spectra derived from global analysis; (c) transient absorption difference spectra at selected time delays after excitation, with single-point decays at 450 nm and 650 nm (inset). | ||
To compare our results with those we obtained previously for all-organic bodipy systems,32,33 we also performed time-resolved infrared spectroscopy (Fig. S22†). The dynamics agree with those recorded in the visible region. In the spectra for KSnMo[BOD2] a sharp, long lived band at 1530 cm−1 is present. The shape and the lifetime of this band are in agreement with our previous work and supports our assignment of the long-lived species as the bodipy triplet excited state.33 The fact that the charge-separated state recombines via a bodipy-centered triplet state in KSnMo[BOD2] but directly to the ground state in KSiW[BOD1] is probably due to the slightly higher energy of the CS state (Fig. 6) and the lower energy of the bodipy triplet state in KSnMo[BOD2] compared to KSiW[BOD1]. Phosphorescence from bodipy is rarely observed, but Harriman et al. recorded λphos = 750 nm (1.65 eV) in a molecular dyad system incorporating a structurally similar bodipy to BOD1 appended to zinc terpyridine.34 Assuming that the triplet energy is similar, this places it 150–200 meV higher than the charge-separated state in KSiW[BOD1].
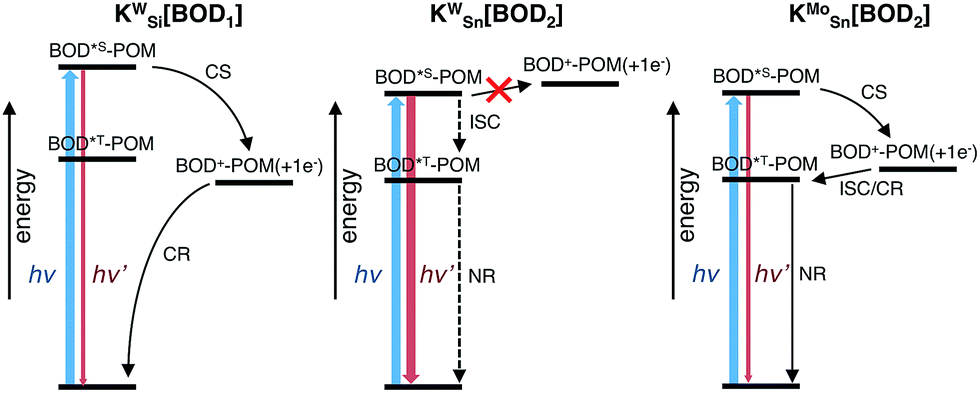 | ||
| Fig. 6 Energy level diagram of POM–bodipy hybrids in dichloromethane. CS: charge separation; CR: charge recombination; ICS: intersystem crossing; NR: non-radiative decay. The dotted arrows indicate poorly effective deactivation pathways. KSiW[BOD1]: E(BOD*S-POM) = 2.33 eV; E(BOD+-POM(+1 e−)) = 1.47 eV; E(BOD*T-POM) ∼1.65 eV. KSnW[BOD2]: E(BOD*S-POM) = 2.10 eV; E(BOD+-POM(+1 e−)) = 2.13 eV; E(BOD*T-POM) ∼1.45 eV. KSnMo[BOD2]: E(BOD*S-POM) = 2.11 eV; E(BOD+-POM(+1 e−)) = 1.55 eV; E(BOD*T-POM) ∼1.45 eV. | ||
As the POM and the bodipy are considerably electronically decoupled, it is possible to separate processes centered on each component of the POM–bodipy hybrids and determine the driving forces for charge-separation and recombination, as presented in Table 3.
For KSiW[BOD1] and KSnMo[BOD2] there is a large driving force for charge-separation (−ΔGCS, 0.86 and 0.56 eV respectively), which occurs with a significantly faster rate for KSiW[BOD1] than KSnMo[BOD2]. This is consistent with charge separation in the Marcus normal region (as described for the POM–iridium hybrids).23 In KSiW[BOD1], the organic spacer between the POM and the bodipy introduces an additional phenyl ring compared to KSnMo[BOD2] increasing thus the POM-bodipy distance of ca. 4 Å. Furthermore, in KSiW[BOD1], the organic spacer is electronically decoupled from the π system of the bodipy unit and thus acts as an insulator.
By contrast, in KSnMo[BOD2] the π system of the bodipy unit extends over the aryl tin ring i.e. at the vicinity of the polyoxomolybdate core. According to Marcus theory, the rate constant for an electron transfer process of a supramolecular system in the non-adiabatic limit (i.e. when its different elements are poorly electronically coupled) can be expressed by the following equation:35,36
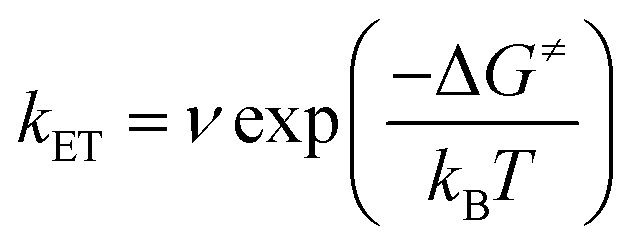
Conclusions
New POM–bodipy conjugates were synthesized through post-functionalization of organosilyl and organotin POM derivatives. An advantage of these systems over e.g. dye-sensitized TiO2, is that the electronic properties of both the donor and acceptor can be specifically tuned. In these photoactive hybrids the redox properties of the POM, the bodipy and the spacer length were modified in order to evaluate the effect of these parameters on the kinetics of photoinduced electron transfer. The transient absorption spectroscopy unequivocally indicates the occurrence of photoinduced electron transfer from the bodipy to the POM for hybrids displaying the best electron accepting properties, with kinetics up to ca. 2 × 1010 s−1, constituting the first example of charge-separated state on noble metal-free covalent POM–PS conjugates. While POMs are drawing a growing attention in the field of artificial photosynthesis and molecular electronics, fundamental insights on their kinetics of intramolecular electron injection into the POM unit within POM–PS conjugates are scare.18,23,39,40 For instance the effects of the solvent and their associated counter-ions on their reorganization energy remain unexplored, the only experimental studies being yet limited to outer-sphere chemical and electrochemical reduction of POMs.41,42 The present system should allow filling this shortfall owing to their modular design. The long lifetime of the charge separated state is also an exciting prospect for integrating these systems into photocathodes, since electron-hopping between POM units or charge-transfer to a substrate or catalyst would be competitive with recombination.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank STFC for access to the CLF ULTRA facility at the Rutherford Appleton Laboratories, the EPSRC and Newcastle University for a studentship for Fiona A. Black (EPJ5002881) and COST Action CM1202, Perspect H2O. Murielle Chavarot-Kerlidou is acknowledged for fruitful discussions.Notes and references
- Y. Pellegrin and F. Odobel, Coord. Chem. Rev., 2011, 255, 2578–2593 CrossRef.
- J. J. Walsh, A. M. Bond, R. J. Forster and T. E. Keyes, Coord. Chem. Rev., 2016, 306, 217–234 CrossRef.
- G. Izzet, F. Volatron and A. Proust, Chem. Rec., 2017, 250–266 CrossRef PubMed.
- C. Streb, Dalton Trans., 2012, 41, 1651–1659 RSC.
- Special issue devoted to polyoxometalates: L. Cronin and A. Müller, Chem. Soc. Rev., 2012, 41(22), 7325–7648 RSC.
- J. L. Xie, B. F. Abrahams and A. G. Wedd, Chem. Commun., 2008, 576–578 RSC.
- N. Fay, V. M. Hultgren, A. G. Wedd, T. E. Keyes, R. J. Forster, D. Leane and A. M. Bond, Dalton Trans., 2006, 4218–4227 RSC.
- T. E. Keyes, E. Gicquel, L. Guerin, R. J. Forster, V. Hultgren, A. M. Bond and A. G. Wedd, Inorg. Chem., 2003, 42, 7897–7905 CrossRef PubMed.
- X. L. Wang, Z. B. Han, E. B. Wang, H. Zhang and C. W. Hu, Electroanalysis, 2003, 15, 1460–1464 CrossRef.
- J. Fielden, J. M. Sumliner, N. N. Han, Y. V. Geletii, X. Xiang, D. G. Musaev, T. Q. Lian and C. L. Hill, Chem. Sci., 2015, 6, 5531–5543 RSC.
- M. Orlandi, R. Argazzi, A. Sartorel, M. Carraro, G. Scorrano, M. Bonchio and F. Scandola, Chem. Commun., 2010, 46, 3152–3154 RSC.
- F. Odobel, M. Severac, Y. Pellegrin, E. Blart, C. Fosse, C. Cannizzo, C. R. Mayer, K. J. Eliott and A. Harriman, Chem.–Eur. J., 2009, 15, 3130–3138 CrossRef PubMed.
- M. P. Santoni, A. K. Pal, G. S. Hanan, A. Proust and B. Hasenknopf, Inorg. Chem., 2011, 50, 6737–6745 CrossRef PubMed.
- S. Schonweiz, S. A. Rommel, J. Kubel, M. Micheel, B. Dietzek, S. Rau and C. Streb, Chem.–Eur. J., 2016, 22, 12002–12005 CrossRef PubMed.
- C. Allain, D. Schaming, N. Karakostas, M. Erard, J. P. Gisselbrecht, S. Sorgues, I. Lampre, L. Ruhlmann and B. Hasenknopf, Dalton Trans., 2013, 42, 2745–2754 RSC.
- S. Schonweiz, M. Heiland, M. Anjass, T. Jacob, S. Rau and C. Streb, Chem.–Eur. J., 2017, 23, 15370–15376 CrossRef PubMed.
- Y. Luo, M. Wachtler, K. Barthelmes, A. Winter, U. S. Schubert and B. Dietzek, Chem. Commun., 2018, 54, 2970–2973 RSC.
- C. Bosch-Navarro, B. Matt, G. Izzet, C. Romero-Nieto, K. Dirian, A. Raya, S. I. Molina, A. Proust, D. M. Guldi, C. Marti-Gastaldo and E. Coronado, Chem. Sci., 2014, 5, 4346–4354 RSC.
- B. Matt, C. Coudret, C. Viala, D. Jouvenot, F. Loiseau, G. Izzet and A. Proust, Inorg. Chem., 2011, 50, 7761–7768 CrossRef PubMed.
- B. Matt, J. Fize, J. Moussa, H. Amouri, A. Pereira, V. Artero, G. Izzet and A. Proust, Energy Environ. Sci., 2013, 6, 1504–1508 Search PubMed.
- B. Matt, J. Moussa, L. M. Chamoreau, C. Afonso, A. Proust, H. Amouri and G. Izzet, Organometallics, 2012, 31, 35–38 CrossRef.
- B. Matt, S. Renaudineau, L. M. Chamoreau, C. Afonso, G. Izzet and A. Proust, J. Org. Chem., 2011, 76, 3107–3112 CrossRef PubMed.
- B. Matt, X. Xiang, A. L. Kaledin, N. N. Han, J. Moussa, H. Amouri, S. Alves, C. L. Hill, T. Q. Lian, D. G. Musaev, G. Izzet and A. Proust, Chem. Sci., 2013, 4, 1737–1745 RSC.
- A. Sinopoli, C. J. Wood, E. A. Gibson and P. I. P. Elliott, Dyes Pigm., 2017, 140, 269–277 CrossRef.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef PubMed.
- V. Duffort, R. Thouvenot, C. Afonso, G. Izzet and A. Proust, Chem. Commun., 2009, 6062–6064 RSC.
- C. Rinfray, S. Renaudineau, G. Izzet and A. Proust, Chem. Commun., 2014, 50, 8575–8577 RSC.
- M. T. Pope, in Polyoxometalate Molecular Science, ed. J. J. Borras-Almenar, E. Coronado, A. Müller and M. Pope, Kluwer Academic Publishers, Dordrecht, 2003 Search PubMed.
- M. Galletta, S. Campagna, M. Quesada, G. Ulrich and R. Ziessel, Chem. Commun., 2005, 4222–4224 RSC.
- A. Parrot, G. Izzet, L. M. Chamoreau, A. Proust, O. Oms, A. Dolbecq, K. Hakouk, H. El Bekkachi, P. Deniard, R. Dessapt and P. Mialane, Inorg. Chem., 2013, 52, 11156–11163 CrossRef PubMed.
- C. Slavov, H. Hartmann and J. Wachtveitl, Anal. Chem., 2015, 87, 2328–2336 CrossRef PubMed.
- J. F. Lefebvre, X. Z. Sun, J. A. Calladine, M. W. George and E. A. Gibson, Chem. Commun., 2014, 50, 5258–5260 RSC.
- F. A. Black, C. A. Clark, G. H. Summers, I. P. Clark, M. Towrie, T. Penfold, M. W. George and E. A. Gibson, Phys. Chem. Chem. Phys., 2017, 19, 7877–7885 RSC.
- A. Harriman, J. P. Rostron, M. Cesario, G. Ulrich and R. Ziessel, J. Phys. Chem. A, 2006, 110, 7994–8002 CrossRef PubMed.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265–322 CrossRef.
- P. Ceroni and V. Balzani, in The exploration of supramolecular systems and nanostructures by photochemical techniques, ed. P. Ceroni, Springer, New York, 2011, ch. 2 Search PubMed.
- F. Odobel and Y. Pellegrin, J. Phys. Chem. Lett., 2013, 4, 2551–2564 CrossRef.
- D. Dini, Y. Halpin, J. G. Vos and E. A. Gibson, Coord. Chem. Rev., 2015, 304, 179–201 CrossRef.
- C. C. Zhao, Z. Q. Huang, W. Rodriguez-Cordoba, C. S. Kambara, K. P. O'Halloran, K. I. Hardcastle, D. G. Musaev, T. Q. Lian and C. L. Hill, J. Am. Chem. Soc., 2011, 133, 20134–20137 CrossRef PubMed.
- P. L. Veya and J. K. Kochi, J. Organomet. Chem., 1995, 488, C4–C8 CrossRef.
- I. A. Weinstock, Chem. Rev., 1998, 98, 113–170 CrossRef PubMed.
- S. X. Guo, A. W. A. Mariotti, C. Schlipf, A. M. Bond and A. G. Wedd, J. Electroanal. Chem., 2006, 591, 7–18 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: General methods, reaction procedures, characterization of the hybrids, differential pulse voltammograms, spectroelectrochemical measurements and transient absorption spectra. See DOI: 10.1039/c8sc00862k |
| ‡ Acronyms used for the POM hybrids: K or D refers to the Keggin or the Dawson-type anion, Mo or W as superscript indicates the metal of the POM backbone, Si, or Sn, as subscript relates to the primary functionalization and the term in brackets corresponds to the remote organic moieties. |
| This journal is © The Royal Society of Chemistry 2018 |