
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of platinum, palladium and rhodium complexes of α-aminophosphine ligands
Erika
Bálint
 *,
Ádám
Tajti
,
Anna
Tripolszky
and
György
Keglevich
*,
Ádám
Tajti
,
Anna
Tripolszky
and
György
Keglevich
Department of Organic Chemistry and Technology, Budapest University of Technology and Economics, 1521 Budapest, Hungary. E-mail: ebalint@mail.bme.hu; Fax: +36 1 46336648; Tel: +36 1 4631111/3653
First published on 26th February 2018
Abstract
α-Aminophosphine-type ligands are of interest as building blocks of transition metal complexes. This review focuses on the utilization of α-aminophosphines as monodentate and bidentate ligands in platinum, palladium and rhodium complexes. Besides the linear derivatives, the applications of cyclic α-aminophosphines as ligands are also summarized. Various aspects, such as synthesis, structure and applications, as well as the catalytic activity of these complexes are discussed.
 Erika Bálint | Erika Bálint was born in 1986 in Budapest. She graduated from the Budapest University of Technology and Economics in 2009 as a chemical engineer. She obtained her PhD in 2013 under the supervision of Prof. György Keglevich, and became a research associate at the same University. Her main research interests embrace organophosphorus and microwave chemistry including the synthesis of transition metal complexes. She is the co-author of approximately 55 papers and book chapters. |
 Ádám Tajti | Ádám Tajti was born in 1989 and graduated from the Budapest University of Technology and Economics in 2015 as a chemical engineer. He has been a member of the Organophosphorus Research Group since 2011. Since 2015, he has been a PhD student at BUTE. His research topics include microwave-assisted synthesis of organophosphorus derivatives and continuous flow MW reactors. |
 Anna Tripolszky | Anna Tripolszky was born in 1992. She graduated at the Budapest University of Technology and Economics in 2017 as a pharmaceutical engineer. She has been a member of the Organophosphorus Research Group since 2013. Since 2017, she has been a PhD student at the same university. Her interest lies in organophosphorus chemistry with a focus on the synthesis of aminophosphine oxide derivatives and their utilization in transition metal complexes. |
1. Introduction
Phosphorus(III) ligands, such as phosphines, phosphinines, phosphites and phosphinites, are a highly important class of ligands.1,2 α-Aminophosphines form a significant group within the large family of phosphine ligands and play an important role in the synthesis of P(III)-transition metal complexes, which are widely applied catalysts in homogeneous catalytic reactions.3–5Among the transition metal complexes, derivatives of the platinum group (such as platinum, palladium, rhodium and ruthenium) present special properties, as compared to other metals. From a catalytic point of view, complexes of platinum, palladium and rhodium are the most important. Industrially relevant examples of these species include Wilkinson's catalyst (Rh(PPh3)3Cl)6 or tetrakis(triphenylphosphine)-palladium(0).7 Complexes of ruthenium show unique coordination and medicinal properties, which can be considered as a separate research topic.8–11 Besides the biologically active Ru derivatives, Pt complexes can also be used as anticancer agents.12–14
Phosphine ligands containing an amine group offer new functionalization possibilities of the transition metal complexes. Although a large amount of data has accumulated on α-aminophosphines (P–C–N), the related field has not been summarized. Reviews on similar compounds, such as phosphinoamines (P–N),15 β-aminophosphines (P–C2–N)16 and miscellaneous aminophosphines (P–Cn–N), have been published previously.17
As the most common synthetic routes, α-aminophosphines may be prepared by the three-component condensation of an amine, an oxo-compound and a secondary phosphine,18–23 by the reaction between amines and hydroxymethyl phosphines,24–28 and by the deoxygenation of α-aminophosphine oxides.29,30 These derivatives can be functionalized further on the nitrogen atom, and they may be good starting materials for polymer-immobilized P-ligands.31–33
Synthetic methods for α-aminophosphines incorporating platinum, palladium and rhodium complexes have been developing since the 1980s. The purpose of this review is to summarize the most important results of this special field of organometallic chemistry. The utilization of the linear and cyclic α-aminophosphines as mono- and bidentate ligands in the synthesis of transition metal complexes comprising platinum-, palladium or rhodium is described. The general structures of the latter compounds are shown in Scheme 1. In addition, the application of several complexes as catalysts is also presented.
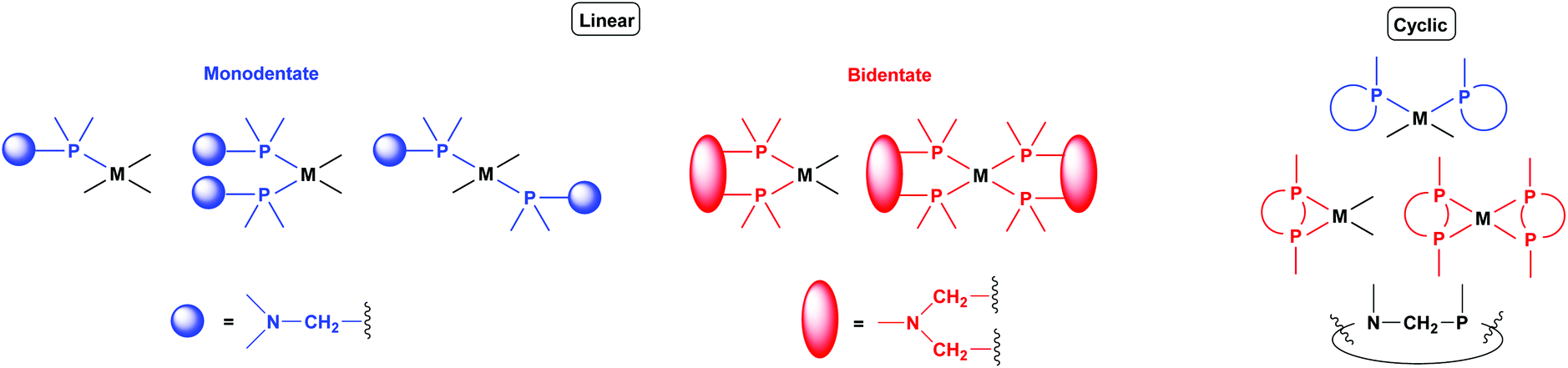 | ||
| Scheme 1 General representation of transition metal complexes containing α-aminophosphine ligand. | ||
2. Utilization of α-aminophosphines as monodentate ligands
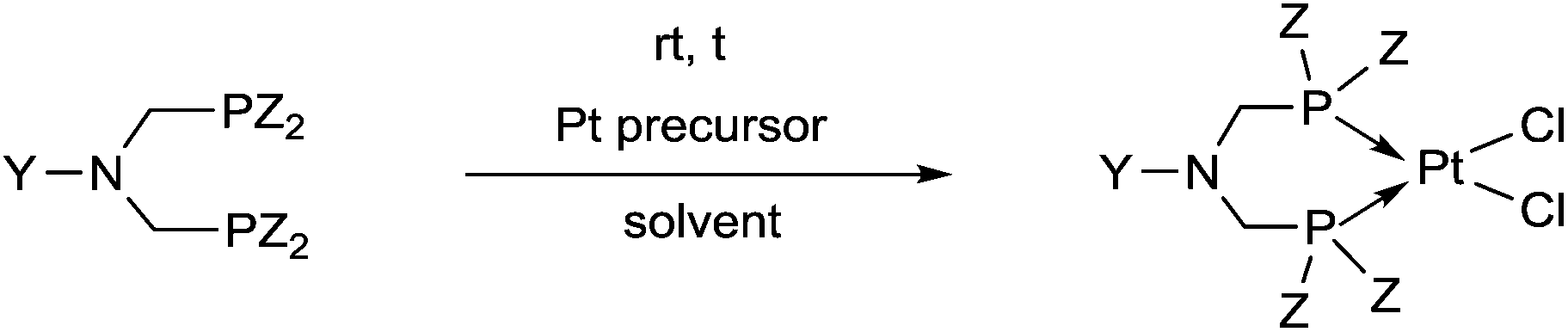
2.1. Synthesis of platinum and palladium complexes
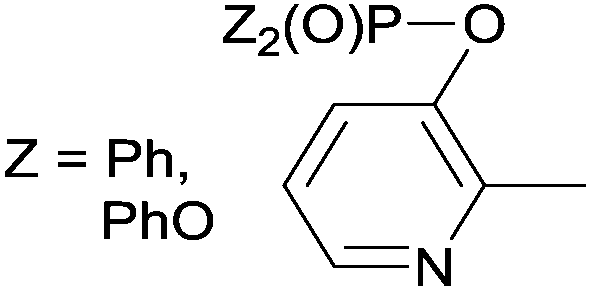
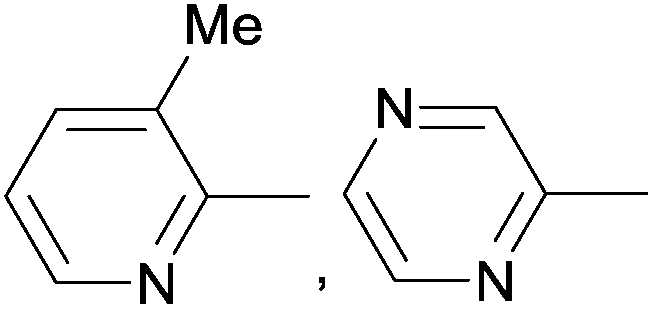
Due to their similar valence structure and reactivity, the complexes of platinum and palladium are discussed together.According to the literature, diphenylphosphinomethylamines are the most widely used monodentate α-aminophosphine ligands. A common procedure to prepare cis-oriented PtII and PdII complexes involves the reaction of the latter species with PtII- or PdII(cod)Cl2 (cod = cycloocta-1,5-diene) at room temperature in DCM (dichloromethane) as the solvent (Table 1). This ligand family was utilized for the first time by Davis in 1993 (Table 1/entry 1). The corresponding N-tBu PtII complex was synthesized in a yield of 69%. The cis conformation of the product was proved by 31P NMR spectroscopy based on the stereospecific Pt–P coupling (3710 Hz). Amino alcohol-functionalized α-aminophosphines were also proved to be efficient ligands in the synthesis of PtII complexes (Table 1/entry 2). The corresponding bis(phosphine)PtCl2 compounds were obtained in yields of 58–74% after 15 min. The synthesis of both PtII and PdII complexes of N-4-pyridyl aminophosphines was also reported (Table 1/entry 3). It should be noted that in the case of PtII(cod)Cl2, the reaction was carried out in DCM at the boiling point. The products obtained could be easily converted to water-soluble complexes by quaternization of the N atom of the pyridine rings by HCl. α-Aminophosphines containing a halogenated pyridine moiety were coordinated to PtII in a reaction time of 15 min (Table 1/entry 4). X-Ray investigation of the 5-Cl-2-pyridyl derivative revealed dimers in the crystal structure, which were held together by two H-bonds between each N–H⋯Cl pair. Derivatives containing phosphate or phosphinate moieties on the hetaryl ring were also tried out as ligands in the complexation (Table 1/entry 5). In the X-ray structure of the products, there were two N–H⋯Cl–Pt intramolecular H-bonds, as well as P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moieties oriented “away” to the central metal atom (Fig. 1). Other N-hetaryl (3-Me-pyridyl and 2-pyrazinyl) aminophosphines were also proved to be useful ligands in complexations (Table 1/entry 6). The PdII complexes synthesized were tested in the Heck reaction of styrene and aryl bromides (Scheme 2). The complexes (A and B) showed different catalytic activities, which was explained by the investigation of the reaction mechanism by DFT calculations.
O moieties oriented “away” to the central metal atom (Fig. 1). Other N-hetaryl (3-Me-pyridyl and 2-pyrazinyl) aminophosphines were also proved to be useful ligands in complexations (Table 1/entry 6). The PdII complexes synthesized were tested in the Heck reaction of styrene and aryl bromides (Scheme 2). The complexes (A and B) showed different catalytic activities, which was explained by the investigation of the reaction mechanism by DFT calculations.
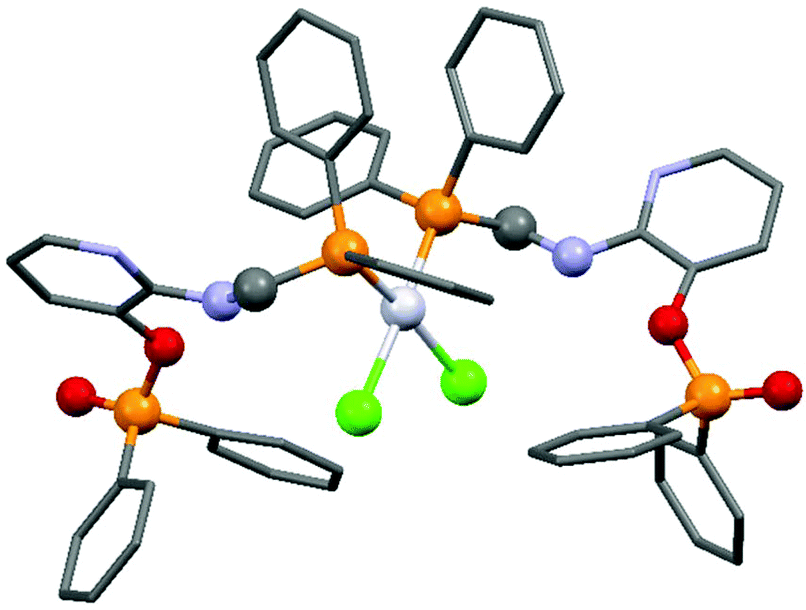 | ||
| Fig. 1 X-Ray structure of cis-dichloro-bis(diphenyl(3-(diphenylphosphinato)-2-pyridylaminomethyl)phosphine)-platinum(II) [CCDC 150040].38 | ||
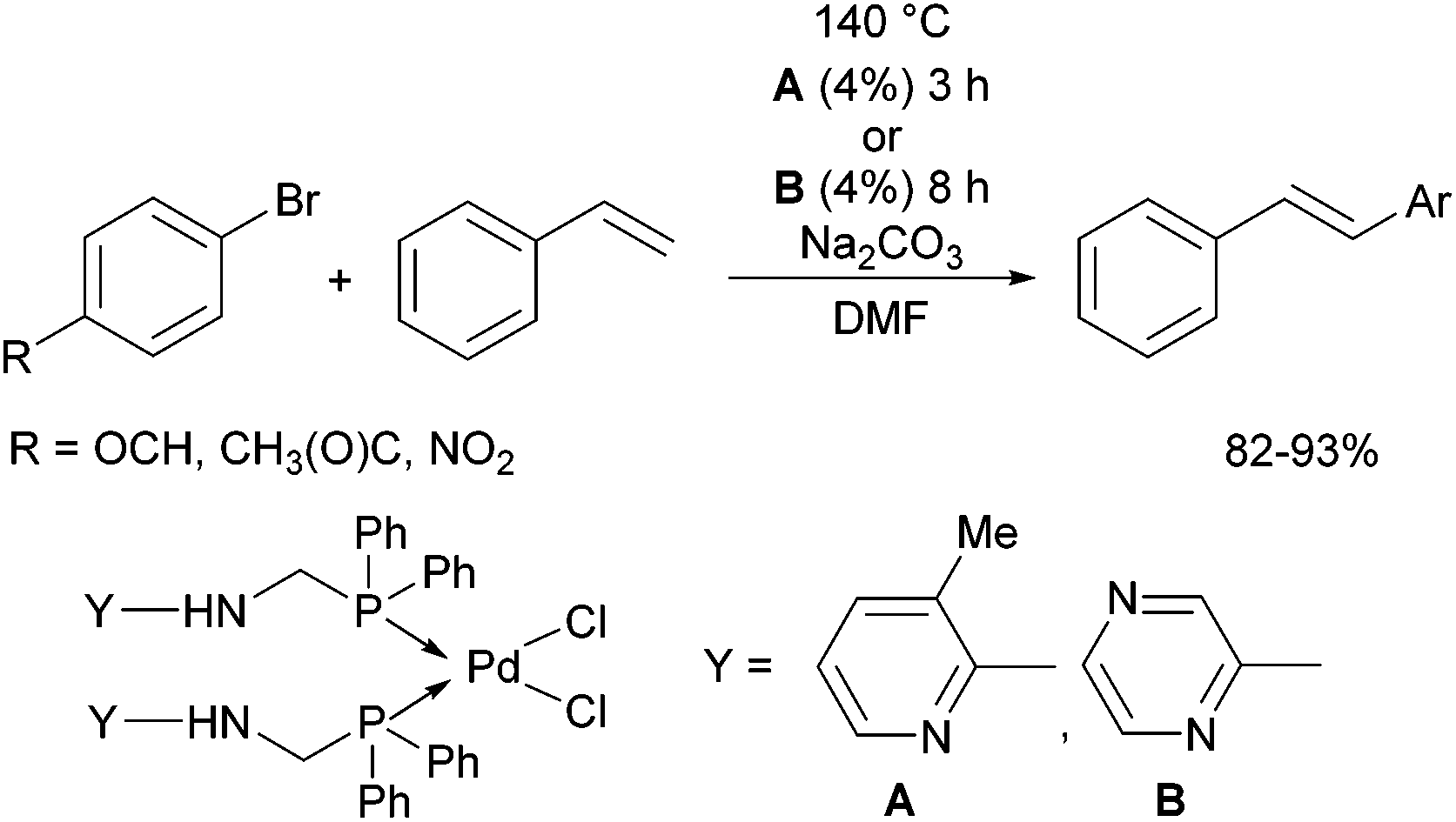 | ||
| Scheme 2 Pd complexes of N-hetaryl α-aminophophines as catalysts in Heck reactions. | ||
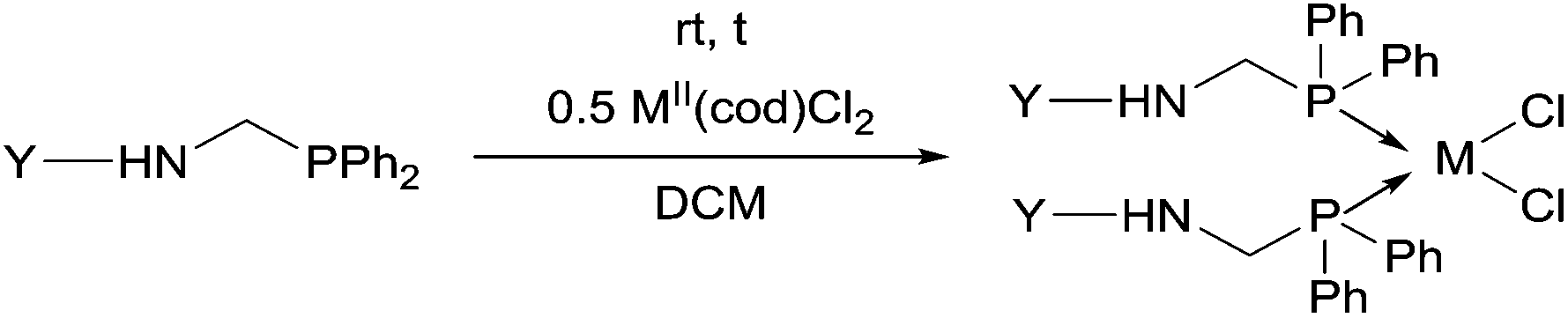
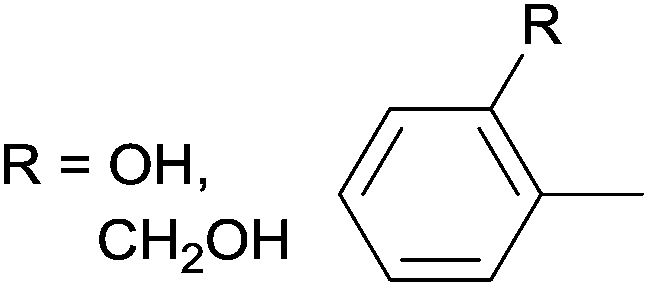
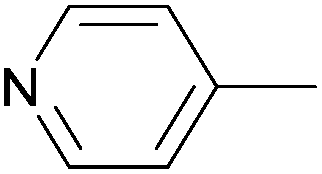
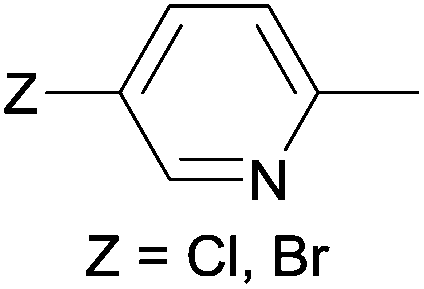
α-Aminophosphines reacted easily with PtII(cod)Cl2 or PdII(cod)Cl2 at room temperature to furnish the PtII or PdII complexes in yields of ca. 60–90%. The reaction conditions required did not depend on the different (alkyl, aryl or hetaryl) substituents of the N atom.
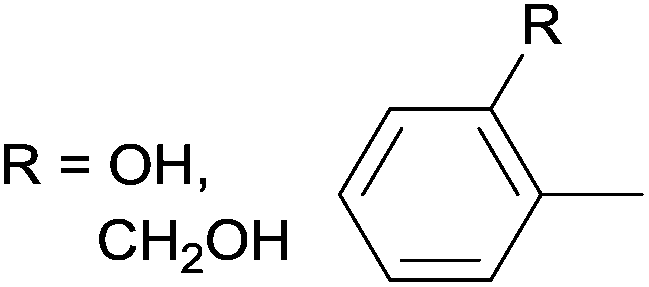
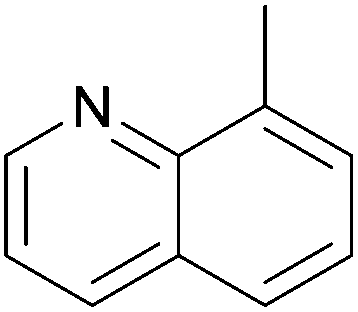
In a few cases, both of the cis (major) and the trans (minor) complexes of bis(α-aminophosphine)PdCl2 derivatives could be observed by NMR spectroscopy in solution, while the solid products showed only the cis conformation (Table 2). Starting from amino alcohol-functionalized α-aminophosphines, complexation afforded the products in yields of 68–93% after 15 min (Table 2/entry 1). IR spectroscopy revealed cis conformation in the solid phase, while in solution, the ratio of cis and trans complexes was 82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :18 and 69:31 as determined by 31P NMR. In the case of an N-quinolinyl derivative, the formation of the two isomers was also corroborated, but the composition was not reported (Table 2/entry 2). The complex was synthesized in a yield of 73%. In the reaction of halogenated diphenylphosphinomethylanilines with PtII(cod)Cl2, the corresponding complexes were obtained in yields of 77–85% (Table 2/entry 3). The cis conformation in the solid phase was also confirmed by X-ray diffraction measurements besides IR spectroscopy. The cis:trans ratio (71:29) was only mentioned in the case of the 5-Cl-aniline derivative.
:18 and 69:31 as determined by 31P NMR. In the case of an N-quinolinyl derivative, the formation of the two isomers was also corroborated, but the composition was not reported (Table 2/entry 2). The complex was synthesized in a yield of 73%. In the reaction of halogenated diphenylphosphinomethylanilines with PtII(cod)Cl2, the corresponding complexes were obtained in yields of 77–85% (Table 2/entry 3). The cis conformation in the solid phase was also confirmed by X-ray diffraction measurements besides IR spectroscopy. The cis:trans ratio (71:29) was only mentioned in the case of the 5-Cl-aniline derivative.

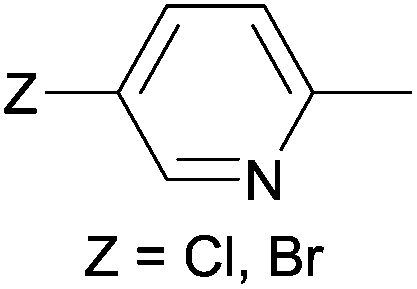
In the examples where the cis:trans ratio in the solution was given, the cis product was present as the major component. Furthermore, the cis:trans composition was similar starting from both aryl and hetaryl derivatives.
In the reaction of diphenylphosphinomethyl-4-methylaniline with PtII(cod)Cl2, a mixture of two complexes was obtained based on 31P NMR. According to the chemical shifts and the Pt–P couplings, a bis(phosphinomethyl)amine derivative was also formed as a by-product besides the expected PtII complex (Scheme 3).41
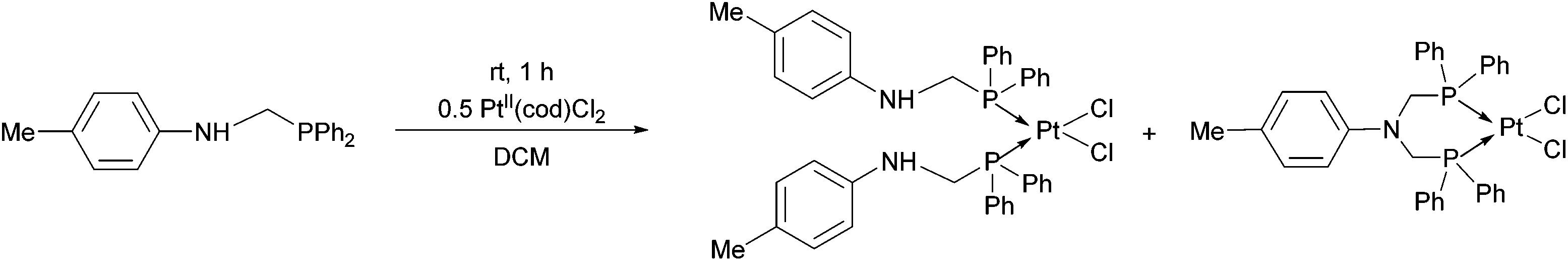 | ||
| Scheme 3 The complexation of diphenylphosphinomethyl-4-methylaniline with PtII(cod)Cl2. | ||
When an N-quinoline-α-aminophosphine was reacted with PtII(cod)Cl2 at room temperature, the corresponding cis complex was obtained in a yield of 60% (Scheme 4).40 Removing one of the chlorine atoms of the PtII complex with AgBF4, the N atom of the hetaryl ring was coordinated to the PtII.
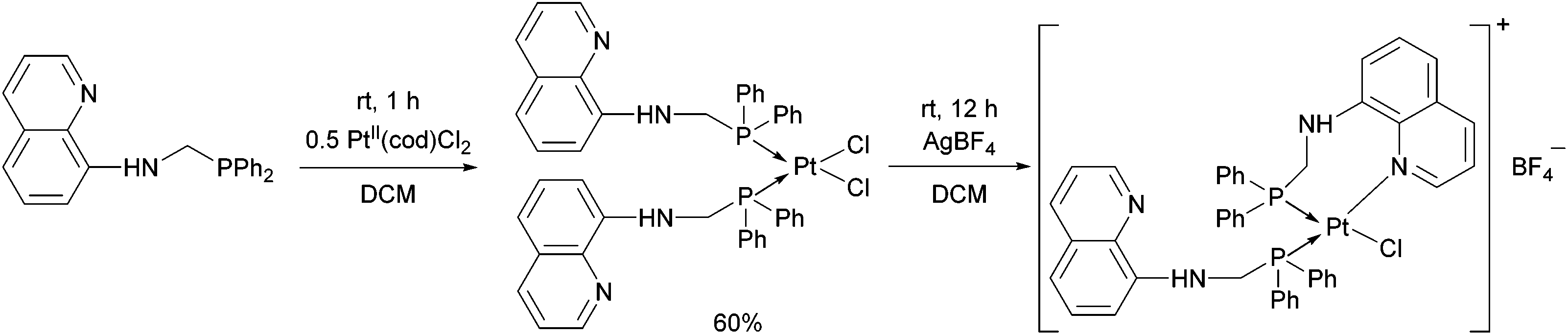 | ||
| Scheme 4 The reaction of an N-quinoline-α-aminophosphine with PtII(cod)Cl2. | ||
An α-aminophosphine containing a 2-pyridyl-piperazine moiety was also tested as a ligand for PtII (Scheme 5).42 The product was obtained in a quantitative yield after a reaction time of 1 h. By reacting the product with AgClO4, similarly to the previous example, the nitrogen atom of the hetero ring was coordinated to the PtII centre.
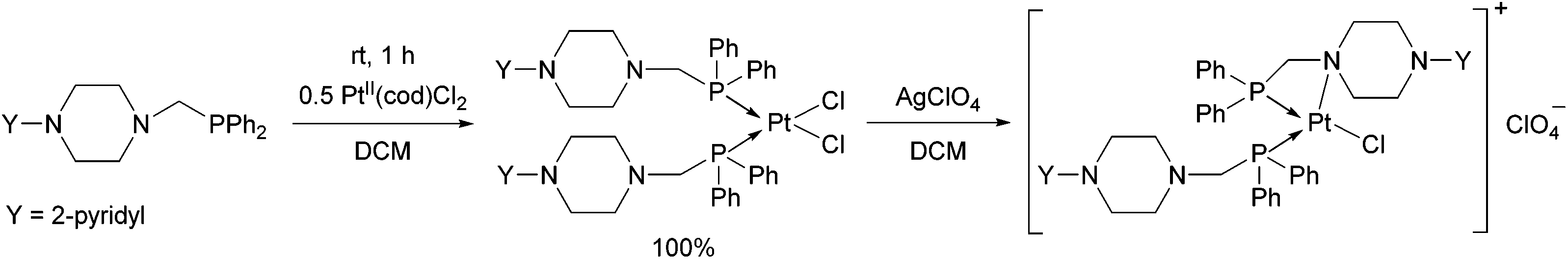 | ||
| Scheme 5 Platinum(II) complexes of α-aminophosphine containing a 2-pyridyl-piperazine moiety. | ||
If an N atom is present at a suitable position of the PtII complexes, the parallel coordination of the N and cleavage of a Pt–Cl bond can be accomplished by adding silver salts.
The reaction of diphenylphosphinomethyldimethylamine with PtII(nbd)Me2 (nbd = γ4-2,5-norbornadiene) was performed at room temperature for 1 h in benzene as the solvent (Scheme 6).43 The cis complex was obtained in a yield of 79%.
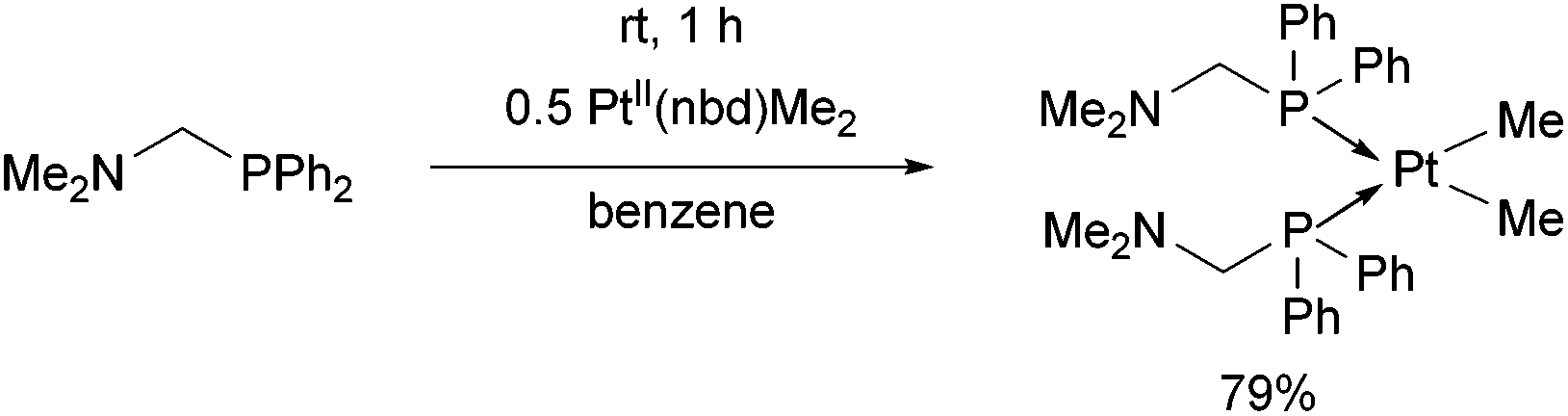 | ||
| Scheme 6 Synthesis of bis[(N,N-dimethylamino)-methyldiphenylphosphino]dimethyl-platinum(II). | ||
By applying different platinum(II) precursors, the conformation of PtII complexes of 2-(N-diphenylphosphinomethyl-N-benzyl)-aminopyridine could be fine-tuned. Complexation with PtII(cod)Cl2 afforded the corresponding cis product, whereas by applying PtII(cod)(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)2, a complex with a trans conformation could be synthesized (Scheme 7).44 The trans product could also be prepared by reaction of the cis derivative with sodium phenylacetilide; however, in this case the yield was only 46%. The related structures were proved by X-ray diffraction measurements (Fig. 2).
CPh)2, a complex with a trans conformation could be synthesized (Scheme 7).44 The trans product could also be prepared by reaction of the cis derivative with sodium phenylacetilide; however, in this case the yield was only 46%. The related structures were proved by X-ray diffraction measurements (Fig. 2).
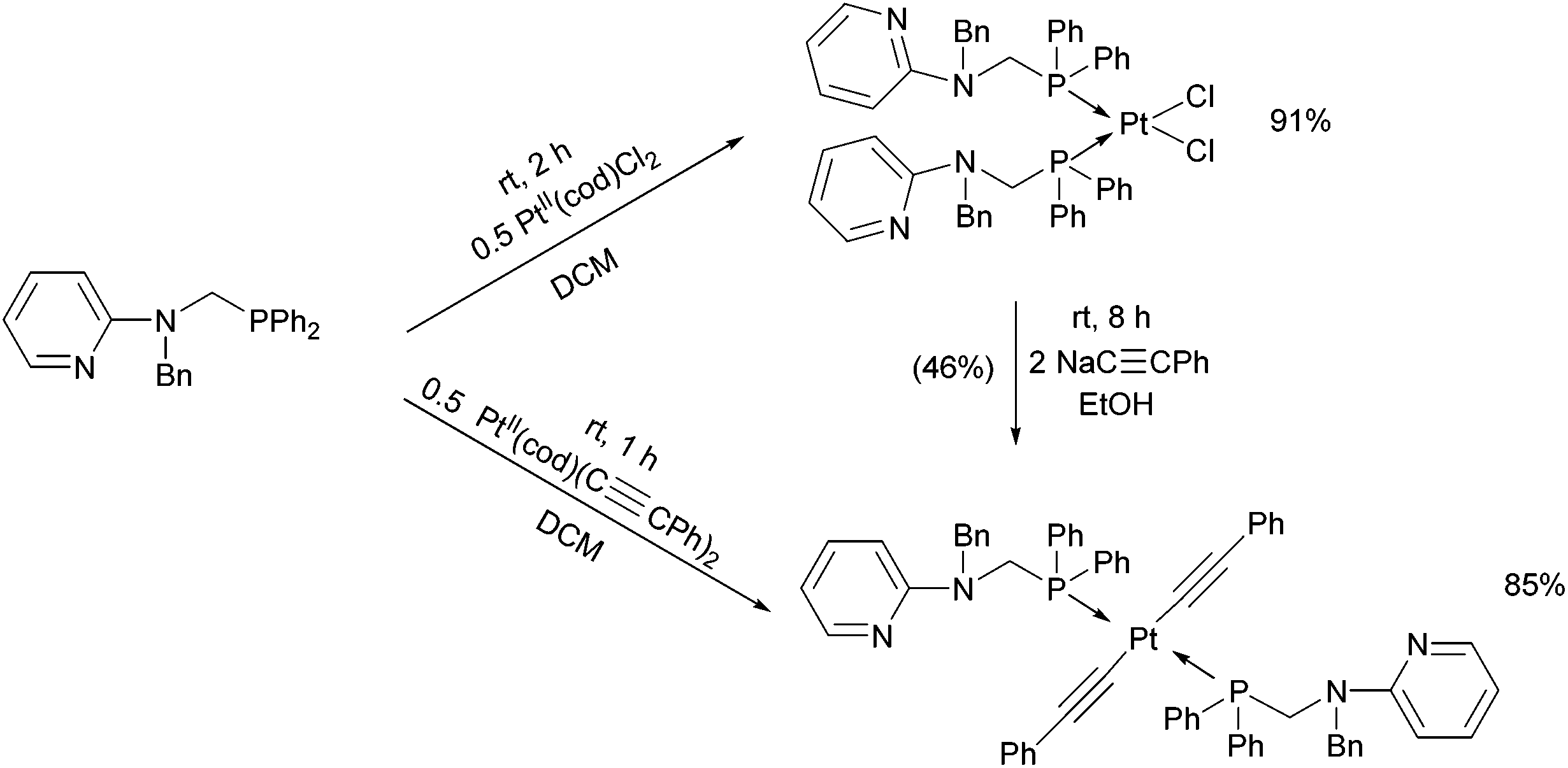 | ||
| Scheme 7 Fine-tuning the conformation of the P-ligands in the PtII complexes by varying the PtII precursors. | ||
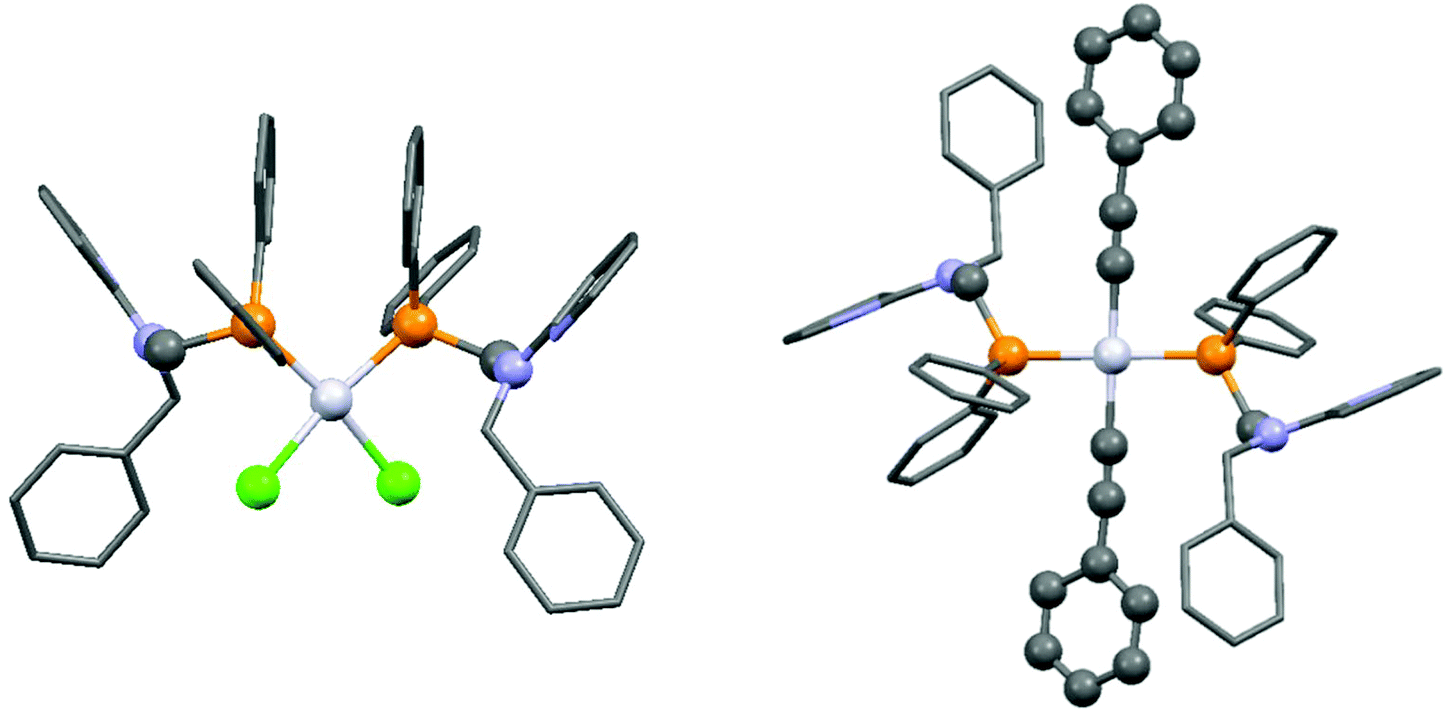 | ||
| Fig. 2 X-Ray structures of PtII complexes incorporating 2-(N-diphenylphosphinomethyl-N-benzyl)aminopyridine [CCDC 197274, 197275].44 | ||
In contrast to the previous cases, the trans PdII complex was obtained by the reaction of 2-(N-dicyclohexylphosphinomethyl-N-methyl)aminopyridine with PdII(cod)Cl2 after 24 h (Scheme 8).45 The different reactivity may be explained by the presence of two cyclohexyl groups on the phosphorus.
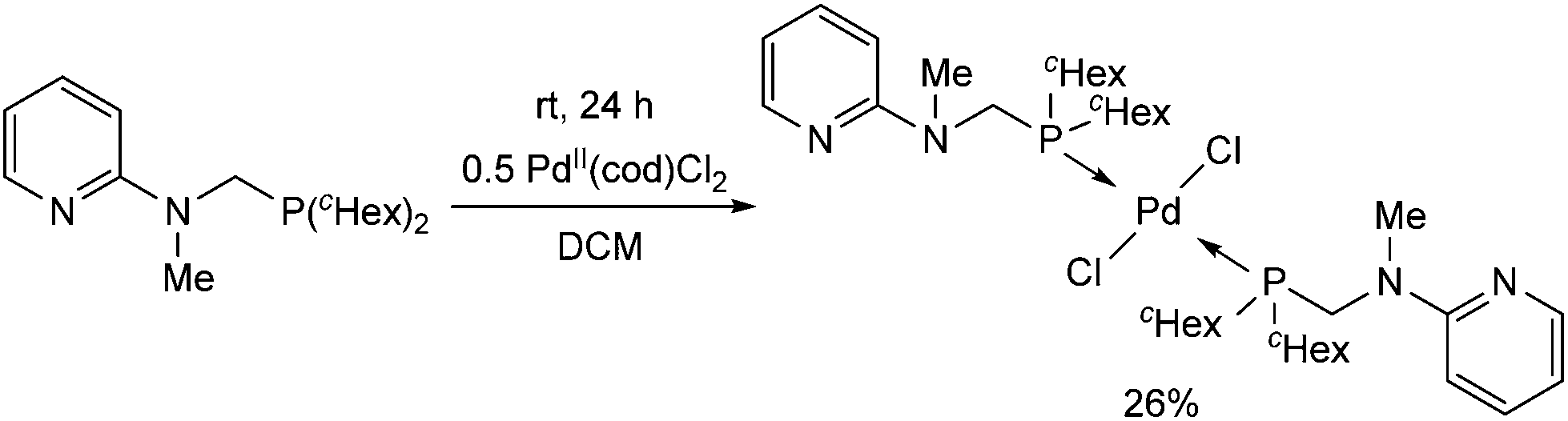 | ||
| Scheme 8 The reaction of 2-(N-dicyclohexylphosphinomethyl-N-methyl)aminopyridine with PtII(cod)Cl2. | ||
Another PdII complex containing a pyridyl moiety was synthesized from an N-4-pyridyl α-aminophosphine at room temperature after 45 min by applying [PdII(2-MeC3H4)Cl]2 as the precursor (Scheme 9).36
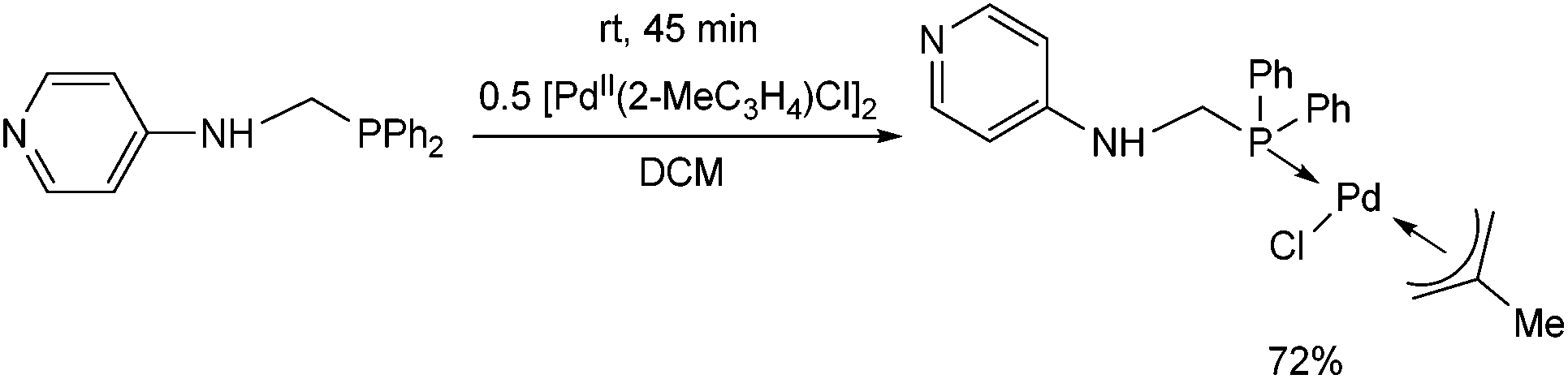 | ||
| Scheme 9 Complexation of an N-pyridyl α-aminophosphine with [PdII(2-MeC3H4)Cl]2. | ||
Reactions of diphenylphosphinomethylamines with [MII(triphos)OTf](OTf) (M = Pt, Pd) led to the corresponding tetracoordinated PdII complexes in yields of 52–56% at room temperature (Scheme 10).46 The complexes were tested as catalysts in electrochemical proton reduction, and showed modest activities.
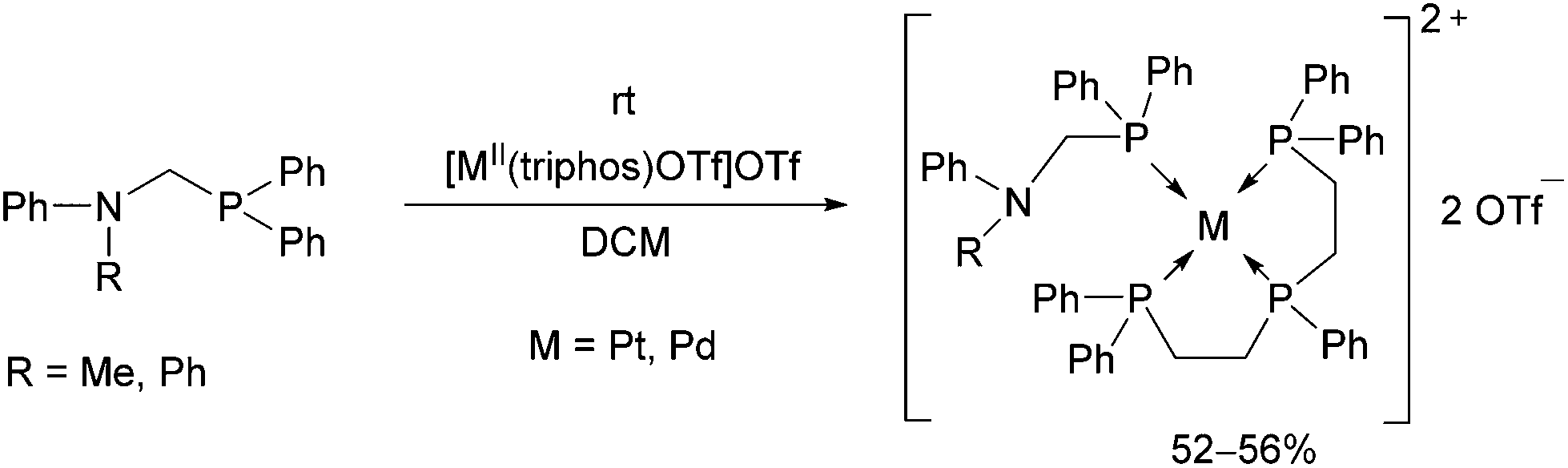 | ||
| Scheme 10 Synthesis of [Pt(triphos)P(Ph2)CN(Ph)(R)](OTf)2 complexes. | ||
Tris(aminomethyl)phosphines were also efficient P-ligands in the synthesis of PtII complexes (Scheme 11).47 The reactions were carried out by applying K2PtCl4 as the precursor in water. Due to the sterically demanding ligands, the trans isomers were the only products. The structure of the complexes was evaluated by X-ray measurements and DFT calculations. According to in vitro investigations, the complex containing morpholine moieties was able to induce apoptosis.
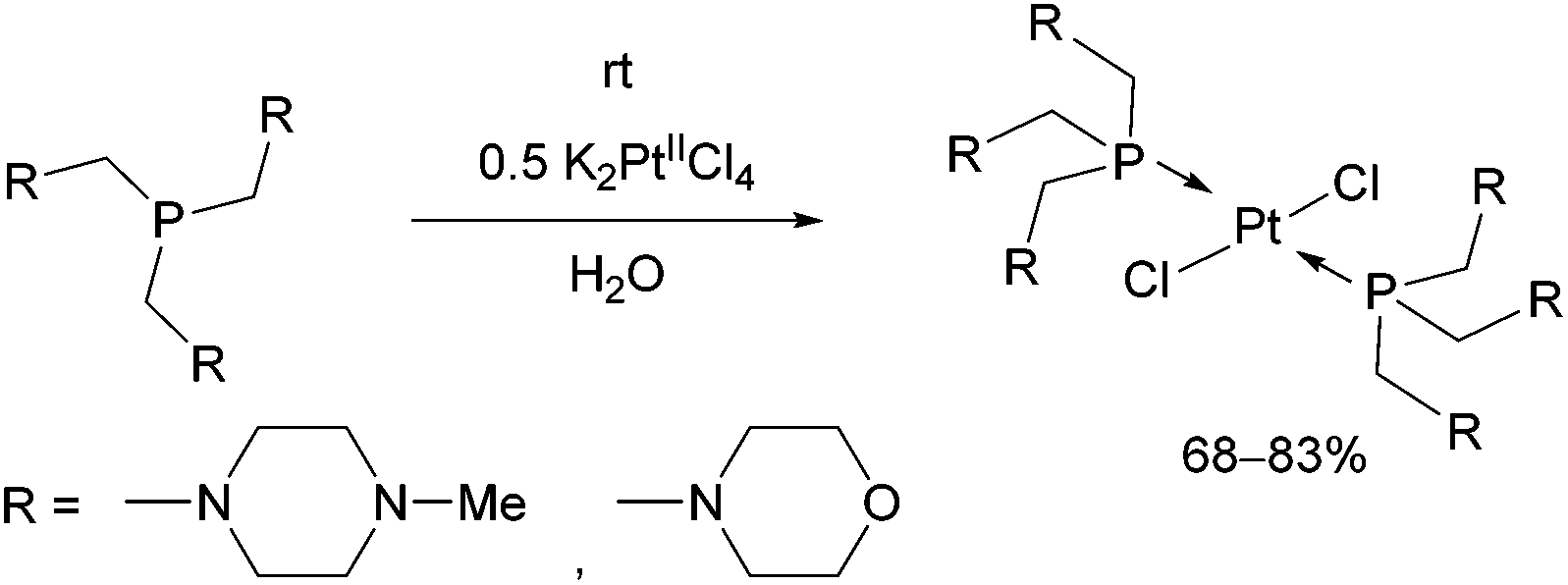 | ||
| Scheme 11 Reaction of tris(aminomethyl)phosphines with K2PtIICl4. | ||
2.2. Synthesis of rhodium complexes
In the case of monodentate α-aminophosphine ligands, [RhIIICpCl2]2 was the most widely used precursor in the synthesis of rhodium(III) complexes. In most instances, the RhIII complexes were prepared at room temperature using DCM as the solvent. Complexation of α-aminophosphines containing a hydroxy group led to full conversion after 15 minutes, furnishing the desired products in yields of 63–90% (Scheme 12).35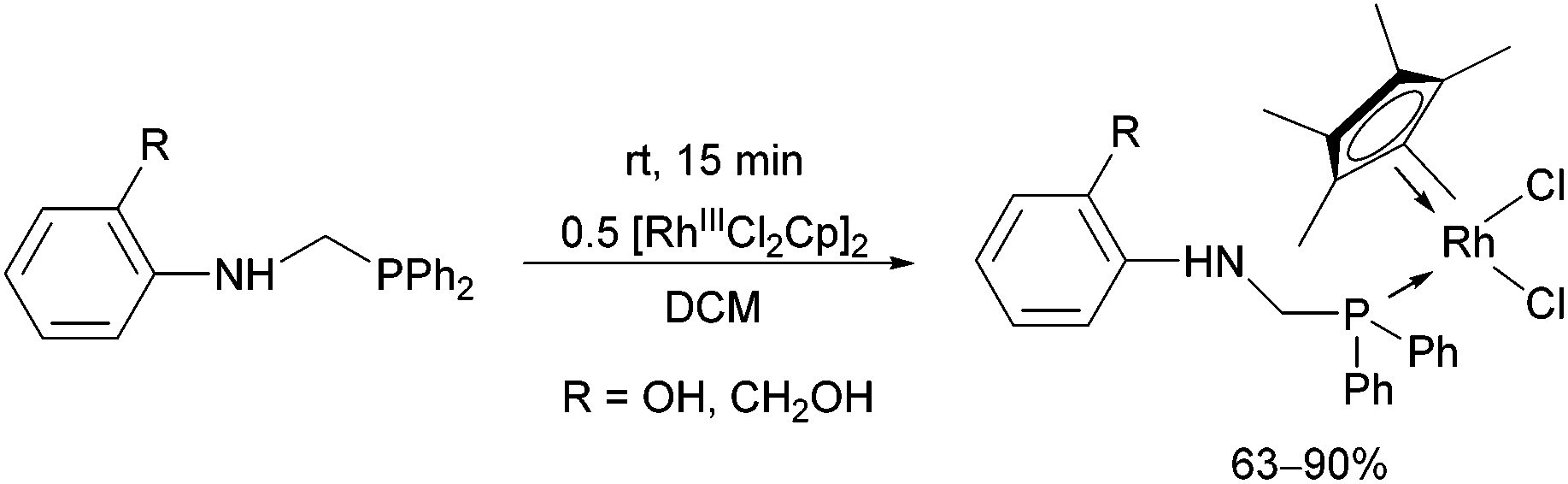 | ||
| Scheme 12 The complexation of α-aminophosphines containing a hydroxy function. | ||
Complexation of N-diphenylphosphinomethyl(2-diphenylphosphino)aniline was investigated in THF (tetrahydrofuran) as the solvent (Scheme 13).48 This special ligand was also able to act as a bidentate P-ligand via the coordination of the phosphine function to the RhIII by reaction with AgClO4.
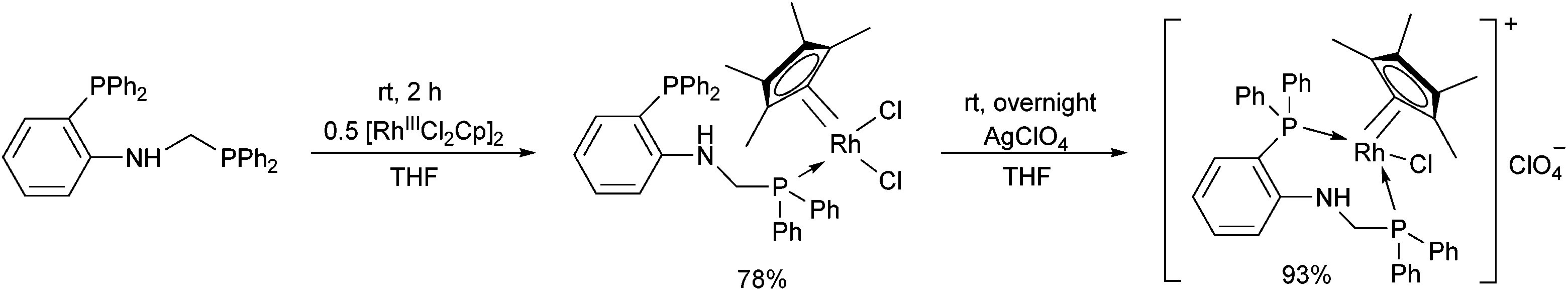 | ||
| Scheme 13 The use of N-diphenylphosphinomethyl(2-diphenylphosphino)aniline as a monodentate or a bidentate ligand. | ||
The reaction of α-aminophosphines containing a 5-chloro- or 5-bromopyridyl moiety with [RhIIICl2Cp]2 was also studied (Scheme 14).34,37 The N atom of the pyridine ring could also be coordinated to the metal centrum by a reaction of the resulting RhIII complex with AgBF4. The incorporation of a suitably disposed halogeno group offers the possibility for further functionalization of the products.
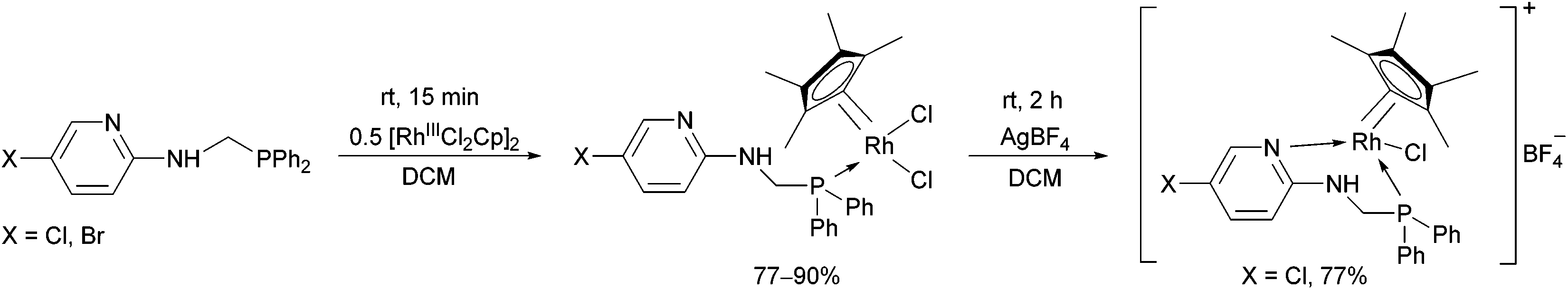 | ||
| Scheme 14 The reaction of α-aminophosphines containing a 5-halogeno-pyridyl moiety with [RhIIICl2Cp]2. | ||
Due to their versatility, N-pyridyl-functionalized aminophosphines represent an important class among P-ligands.49 Derivatives bearing a >P(O)O-function on the hetaryl ring (a pyridyl phosphate or phosphinate) were also used as P-ligands to obtain RhIII complexes in good yields (85–90%) after a reaction time of 30 min (Scheme 15).38
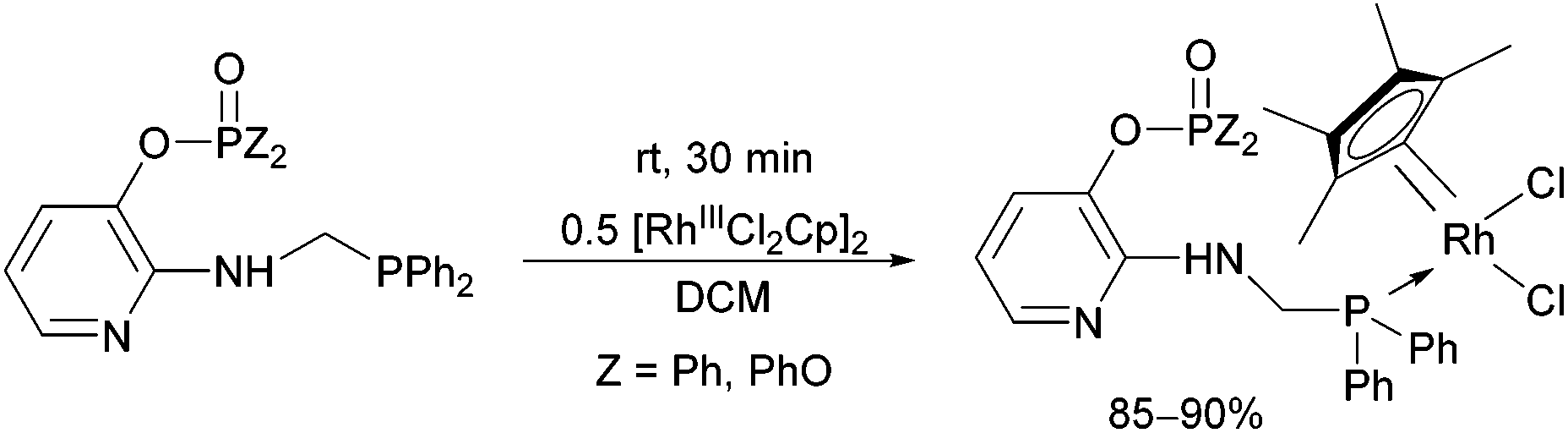 | ||
| Scheme 15 The utilization of N-pyridyl-substituted aminophosphines as P-ligands. | ||
8-(Diphenylphosphino)methylaminoquinoline (8-dppmaq) was also tried out in the complexation with [RhIIICpCl2]2 as the metal precursor (Scheme 16).40 An X-ray study of the product revealed an intramolecular H bond between the N atom of the quinoline and the H atom of the NH function (Fig. 3). When the resulting RhIII complex was reacted with two equivalents of AgBF4, the two N atoms of the aminoquinoline could also be coordinated to the metal centrum. The in situ formed RhI catalyst from the same ligand and Rh(acac)CO2 as a RhI precursor was proved to be efficient in the hydroformylation of hex-1-ene.
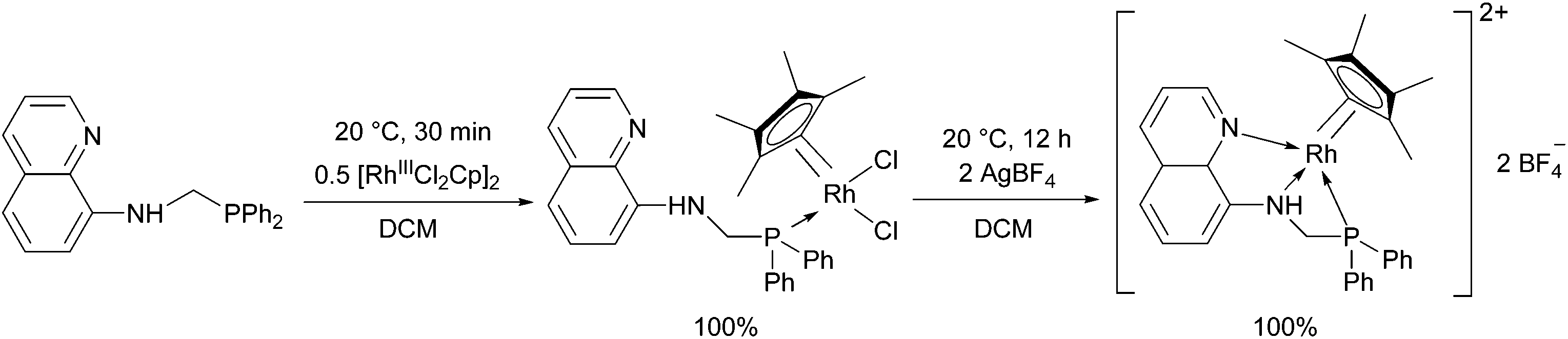 | ||
| Scheme 16 The complexation of 8-(diphenylphosphino)methylaminoquinoline. | ||
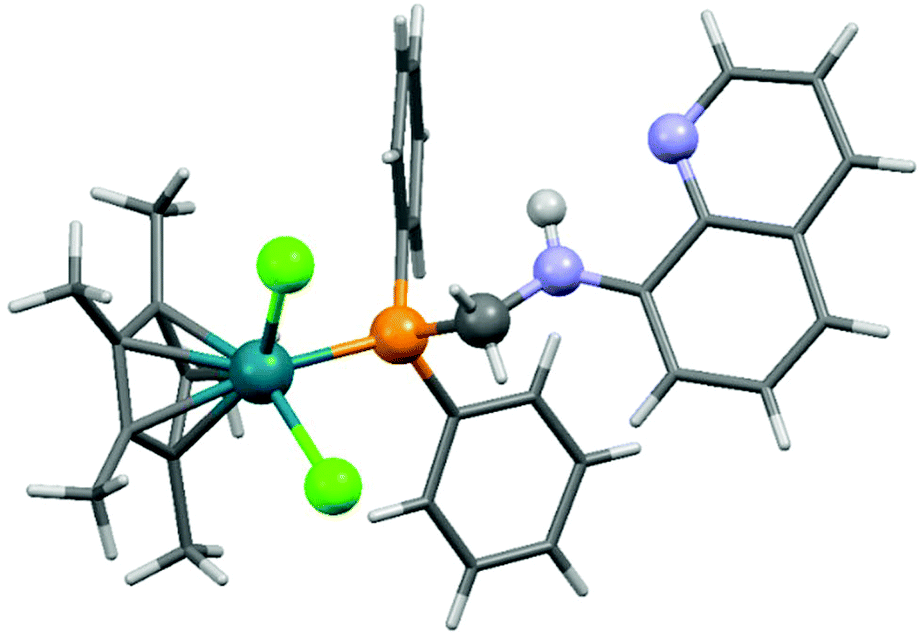 | ||
| Fig. 3 X-Ray structure of [CpRhCl2(8-dppmaq)] [CCDC 177446].40 | ||
[RhIIICl2Cp]2 also served as a RhIII precursor in the complexation of 9-(diphenylphosphinomethyl)adenine (Scheme 17).50 After a reaction time of 1 h, the complex was prepared in a yield of 73%. It was found that the corresponding pincer-type complex, where the adenine ring is also a ligand, could not be obtained, because the complex was not electron-rich enough for the oxidative addition.
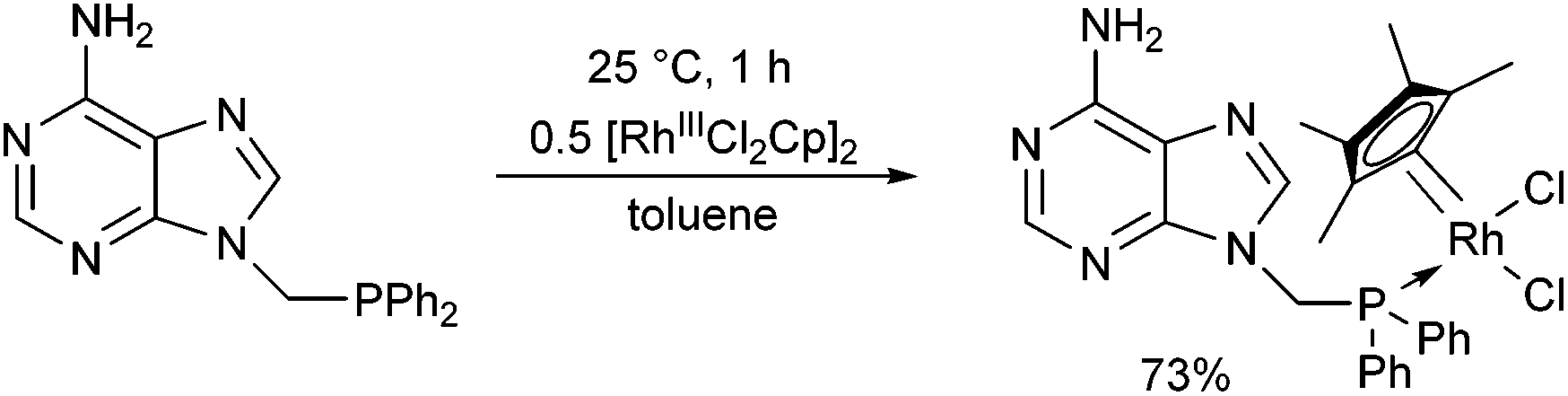 | ||
| Scheme 17 The use of 9-(diphenylphosphinomethyl)adenine as a monodentate ligand. | ||
Dicyclohexylphosphinomethylaniline was also subjected to complexation (Scheme 18).51 After a reaction time of 0.5 h, the corresponding RhI complex was obtained in a yield of 86%. An X-ray study of the product revealed a square planar geometry around the metal center (Fig. 4).
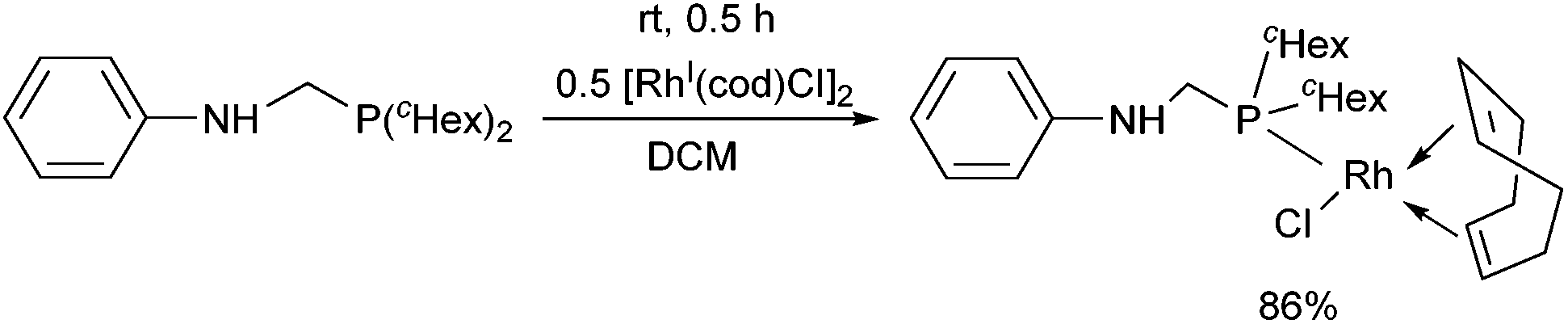 | ||
| Scheme 18 Complexation of the dicyclohexylphosphinomethylaniline with [RhI(cod)Cl2]2. | ||
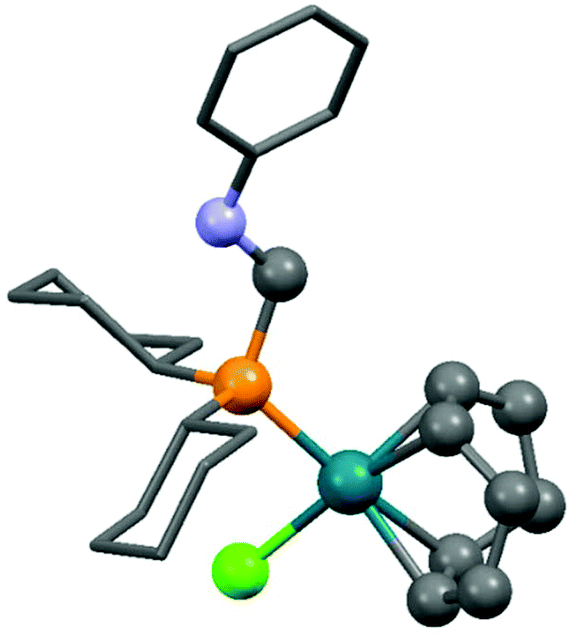 | ||
| Fig. 4 X-Ray structure of (cHex2PCH2NHPh)Rh(cod)Cl [CCDC 748467].51 | ||
N-(Diphenylphosphinomethyl)-4-aminopyridine was also reacted with [RhI(cod)Cl2]2 as the rhodium(I) precursor (Scheme 19).36 The RhI complex was prepared at room temperature using DCM as the solvent. Attempts to synthesize bi-metallodendrimers from the corresponding RhI complex were not successful.
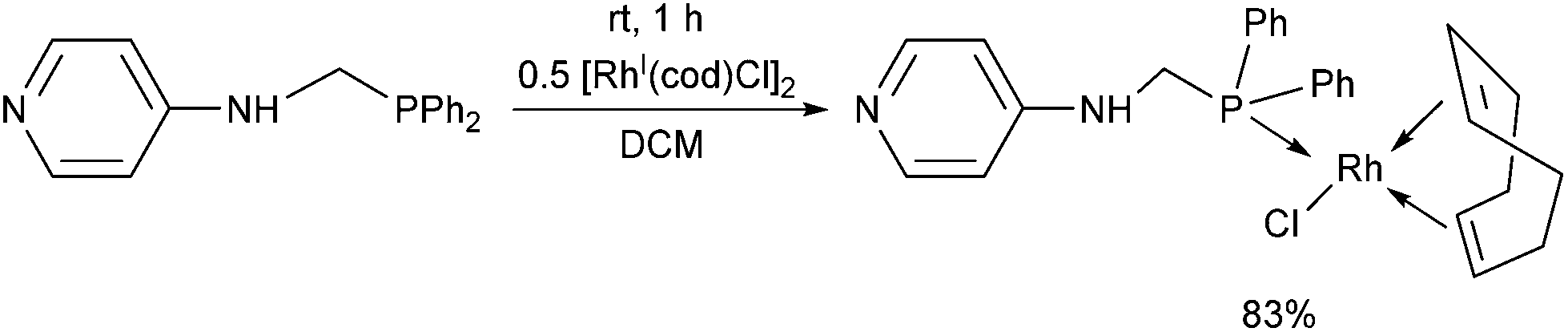 | ||
| Scheme 19 The reaction of phosphinomethylamine ligands with [RhI(cod)Cl2]2. | ||
Complexes containing two α-aminophosphine ligands could be obtained in the reactions of two equivalents of diphenylphosphinomethylamines with one equivalent of the RhI precursor (Table 3). Starting from diphenylphosphinomethyl-diethylamine and [RhI(CO)2Cl]2 or [RhI(CO)(CH2CH2)Cl]2, the complexations were carried out at room temperature for 2 h using DCM as the solvent (Table 3/entry 1). The products were characterized by NMR spectroscopy, but the yields were not reported. According to a recent study, the reaction of diphenylphosphinomethyldiphenylamine with [RhI(CO)2Cl]2 was complete after 0.5 h using toluene as the solvent (Table 3/entry 2). An X-ray study of the corresponding RhI complex confirmed the trans geometry (Fig. 5).
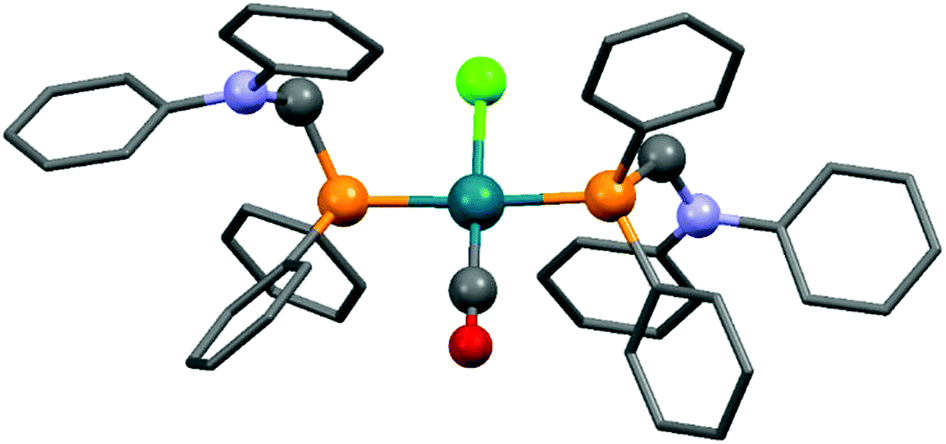 | ||
| Fig. 5 X-Ray structure of Rh(Ph2PCH2NPh2)2(CO)Cl [CCDC 1265045].53 | ||

In a special case, tris[(arylamino)methyl]phosphines were used as monodentate P-ligands in the synthesis of RhI complexes by applying [RhI(CO)2Cl]2 as the metal precursor (Scheme 20).54 The reactions were performed in deuterated dichloromethane at room temperature to allow an NMR characterization study immediately after the reaction. As suggested by the IR spectra of the complexes, despite the rather long distance from the P-center, the effect of the different aryl substituents was significant on the C–O stretching frequencies.
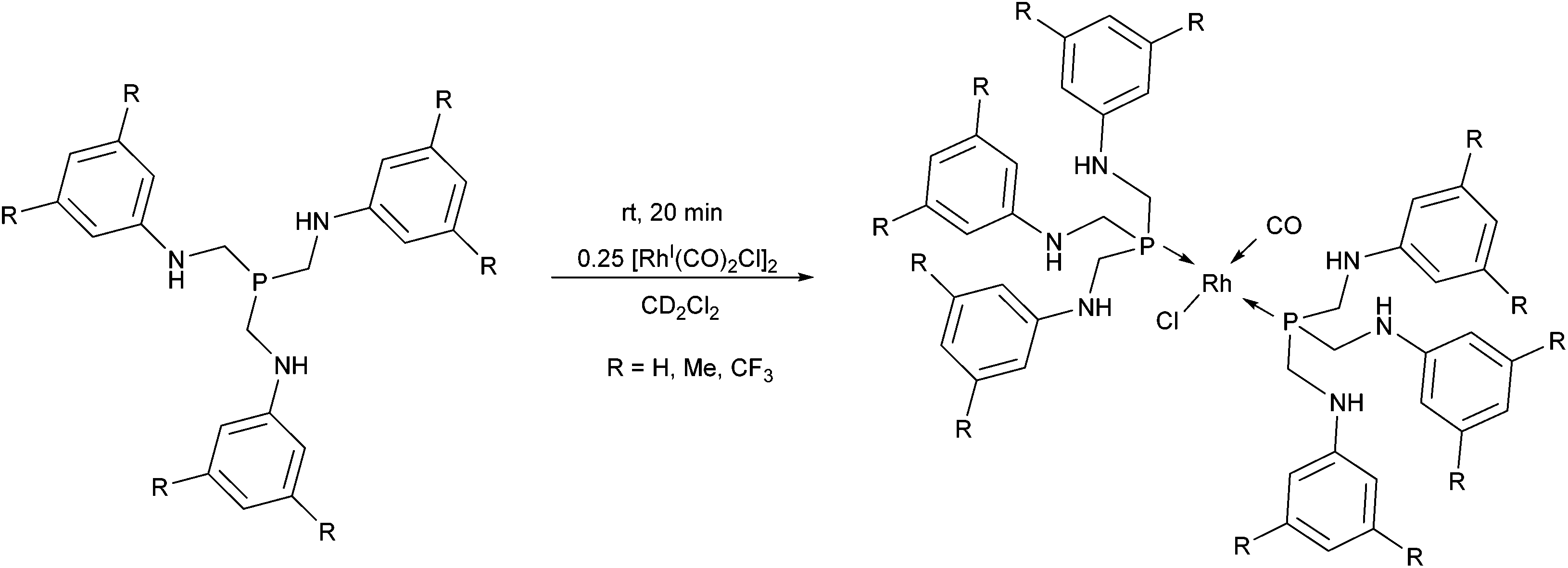 | ||
| Scheme 20 Rhodium complexes incorporating two tris[(arylamino)methyl]phosphine derivatives as the ligands. | ||
2.3. General methods for the preparation of complexes containing monodentate α-aminophosphines as ligands
Based on the various synthetic methods reported, we wished to provide a brief summary of the preparation of Pt, Pd and Rh complexes incorporating α-aminophosphines as monodentate P-ligands (I–III) (Table 4). According to the literature data, most of the reactions can be carried out at room temperature using DCM as the solvent. Pt and Pd complexes with one monodentate P-ligand (I) may be prepared using [MII(triphos)OTf][OTf] (M = Pt, Pd) as the metal precursor to afford the complexes in yields of 52–56%. Similar Rh complexes may be synthesized by reaction of α-aminophosphines with 0.5 equivalents of [RhIIICl2Cp]2 in good to quantitative yields. Pt and Pd complexes bearing two α-aminophosphine ligands in cis conformation (II) may be obtained easily using 0.5 equivalents of MII(cod)Cl2 (M = Pt, Pd) to furnish the products in yields of 58–92%. The trans oriented Pt, Pd and Rh complexes (III) may be prepared by reaction of bulky α-aminophosphines with 0.5 equivalents of PtII(cod)(CCPh)2, 0.5 equivalents of PdII(cod)Cl2, or 0.25 equivalents of [RhI(CO)2Cl]2, respectively.
| Type of complexes | M | Precursor | t | Average yield [%] |
|---|---|---|---|---|
|
|
||||
| n.a.: Not available. | ||||
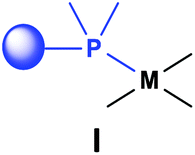
|
Pt | [PtII(triphos)OTf][OTf] | n.a. | 52–56 |
| Pd | [PdII(triphos)OTf][OTf] | n.a. | 55–56 | |
| Rh | 0.5 [RhIIICl2Cp]2 | 15 min–2 h | 63–100 | |
|
Pt | 0.5 PtII(cod)Cl2 | 15–30 min | 58–89 |
| Pd | 0.5 PdII(cod)Cl2 | 15–30 min | 63–92 | |
|
Pt | 0.5 PtII(cod)(CDCPh)2 |
1 h | 85 |
| Pd | 0.5 PdII(cod)Cl2 | 24 h | 26 | |
| Rh | 0.25 [RhI(CO)2Cl]2 | 0.5–2 h | 67 | |
3. Utilization of α-aminophosphines as bidentate ligands
Among α-aminophosphines, bidentate derivatives are the most widely applied as ligands in the synthesis of platinum(II), palladium(II) or rhodium(II) complexes.3.1. Synthesis of platinum and palladium complexes
Based on the literature data, one of the most important types of bidentate α-aminophosphine ligands is the family of bis(phosphinomethyl)amines. The synthesis of cyclic platinum complexes containing simple alkyl or aryl bis(phosphinomethyl)amine ligands at room temperature is summarized in Table 5. A series of cis-oriented [bis(diphenylphosphinomethyl)amine]dichloroplatinum(II) complexes was prepared by our group using dichlorodibenzonitrile platinum(II) (Table 5/entry 1). The complexation was extended by applying bis(aminophosphine) ligands bearing benzyl or 4-methylphenyl groups on the phosphorus atoms (Table 5/entry 2). The dependence of the energetics of the complexations on the substituents and the stereostructure of the complexes was evaluated by B3LYP/6-31G(d,p) calculations. The six-membered metallocycle with two benzyl groups on each P atom adopts a half-chair conformation, while the P-aryl species take up a chair-like conformation. This was also confirmed by X-ray investigations (Fig. 6). PtII(cod)Cl2 was also applied as a precursor for the synthesis of cis chelate Pt complexes (Table 5/entries 3 and 4). In these cases, the reactions were carried out in DCM or in THF, and the corresponding PtII complexes were obtained in yields of 70–85%.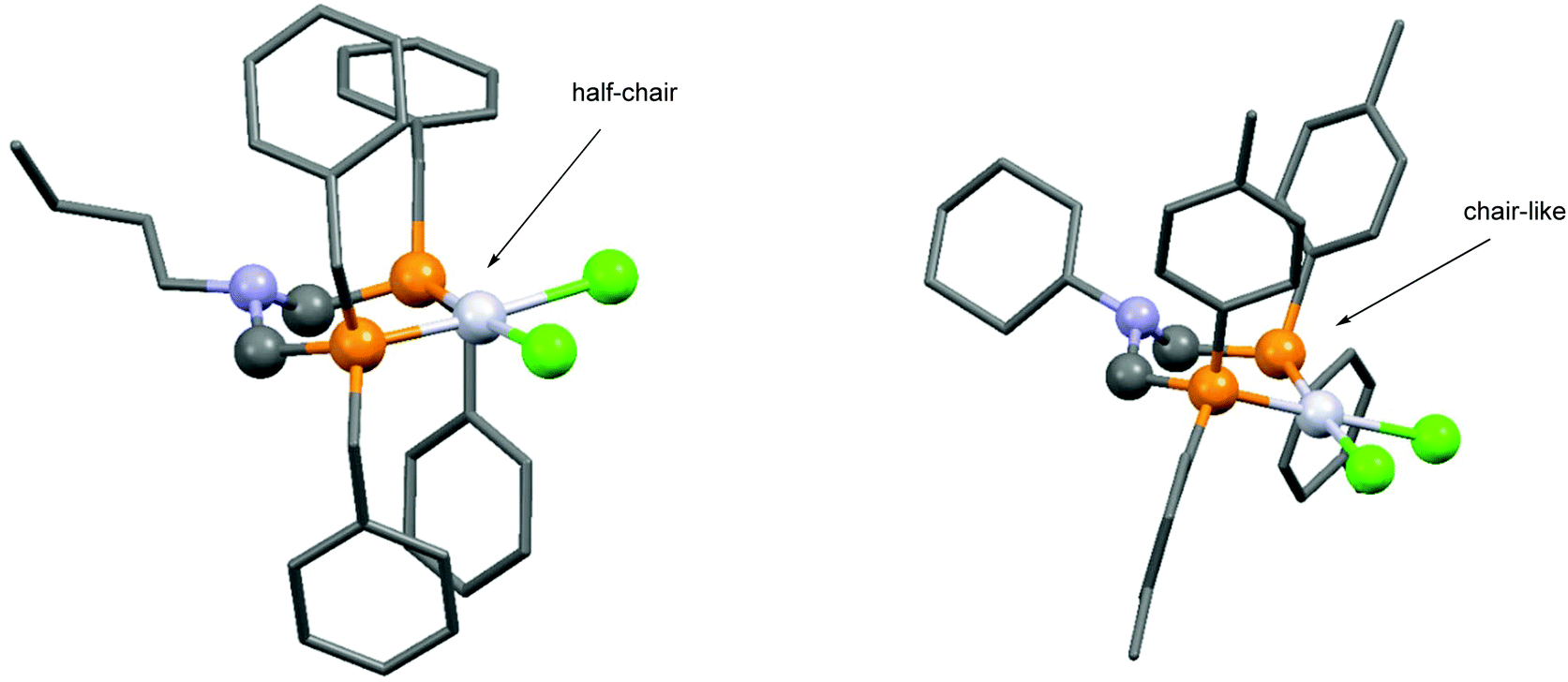 | ||
| Fig. 6 X-Ray structures of N,N-[bis(dibenzylphosphinomethyl)butylamine]-dichloroplatinum(II) [CCDC 1414765] and N,N-[bis(ditolylphosphinomethyl)cyclohexylamine]-dichloroplatinum(II) [CCDC 1416216].30 | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Y | Z | Pt precursor | Solvent | t [h] | Yield [%] | Ref. |
| 1 | n Pr, nBu, cHex, Bn, Ph, 4-MeC6H4, 4-MeOC6H4 | Ph | PtII(PhCN)2Cl2 | Benzene | 24 | 38–60 | 29 and 55 |
| 2 | n Bu, cHex, Bn | Bn, 4-MeC6H4 | PtII(PhCN)2Cl2 | Benzene | 12 | 52–75 | 30 |
| 3 | Ph | c Hex, Ph | PtII(cod)Cl2 | THF, DCM | 0.5–24 | 70–85 | 56 |
| 4 | t Bu, 4-MeC6H4 | Ph | PtII(cod)Cl2 | DCM | 1 | 74–79 | 41 |
On the above basis, the complexation of bis(phosphinomethyl)amines was efficient, independently of the substituents on the N and P atoms, with both types of PtII precursors affording cyclic cis-oriented complexes.
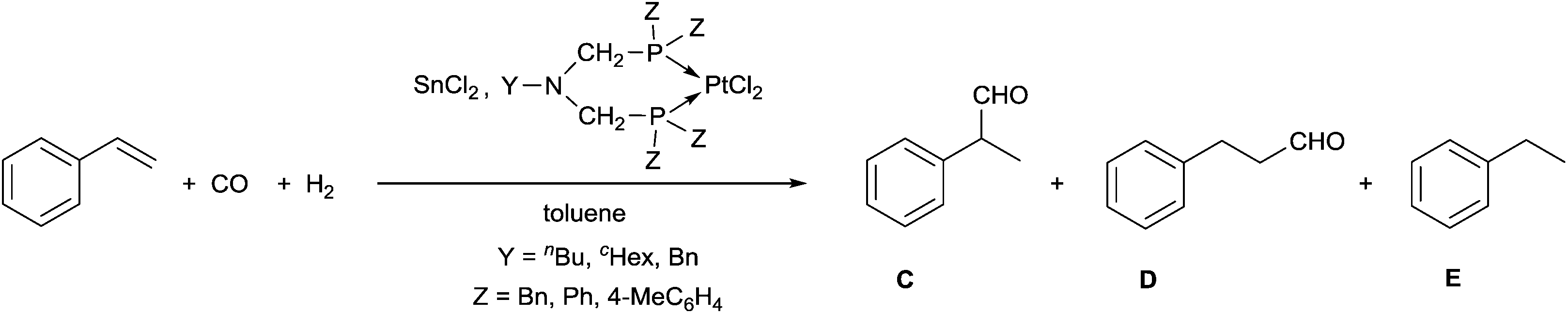
A few related PtII complexes were tested as catalysts in the hydroformylation of styrene, where tin(II) chloride was used as a cocatalyst, and toluene served as a solvent (Table 6). Comparing the effect of the substituents on the P atoms, it can be seen that the P-aryl complexes were more active than the P-benzyl derivatives (Table 6/entries 1, 2 and 6–8 vs. entries 3–5). Regarding the N-substituent, a benzyl group on the nitrogen atom increases the activity, as compared to the butyl and cyclohexyl groups (Table 6/entry 2 vs. entry 1, and entry 8 vs. entries 6, 7). As regards the chemoselectivity, the complexes had a similar effect; however, from the point of view of regioselectivity, the best precatalysts were the P-phenyl complexes giving the branched aldehyde (C) in regioselectivities of 74 and 77% (Table 6, entries 1 and 2).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Y | Z | Temperature [°C] | t [h] | Conversion [%] | R c [%]a | R br [%]b | Ref. |
| a Chemoselectivity towards aldehydes (C, D). [(C + D)/(C + D + E) × 100]. b Regioselectivity towards branched aldehyde (C). [C/(C + D) × 100]. c Reaction conditions: Pt/SnCl2/styrene = 1/1/100; p(CO) = p(H2) = 40 bar. d Reaction conditions: Pt/SnCl2/styrene = 1/2/200; p(CO) = p(H2) = 40 bar. | ||||||||
| 1c | c Hex | Ph | 60 | 20 | ∼100 | 78 | 74 | 29 |
| 2c | Bn | Ph | 60 | 5 | 87 | 76 | 77 | |
| 3d | n Bu | Bn | 100 | 3 | 32 | 72 | 65 | 30 |
| 4d | c Hex | Bn | 100 | 3 | 50 | 74 | 61 | |
| 5d | Bn | Bn | 100 | 3 | 52 | 70 | 65 | |
| 6d | n Bu | 4-MeC6H4 | 100 | 8 | 98 | 75 | 63 | 30 |
| 7d | c Hex | 4-MeC6H4 | 100 | 6 | 98 | 74 | 63 | |
| 8d | Bn | 4-MeC6H4 | 100 | 3 | 98 | 79 | 56 | |
The complexation of a chiral (S)-α-phenylethylamine functionalized α-aminophosphine with PtII(PhCN)2Cl2 has been described by our group (Scheme 21).57 It was observed that besides the chiral bidentate Pt complex expected, a bicyclic derivative was also formed in a small amount as a by-product. Based on the 31P NMR spectrum, the ratio of the two complexes was 85:15. The X-ray investigation of the bicyclic complex revealed a highly solvated complex salt structure, as well as a pseudo-centrosymmetric disposition of most atoms of a chiral molecular complex in a chiral crystal lattice (Fig. 7).
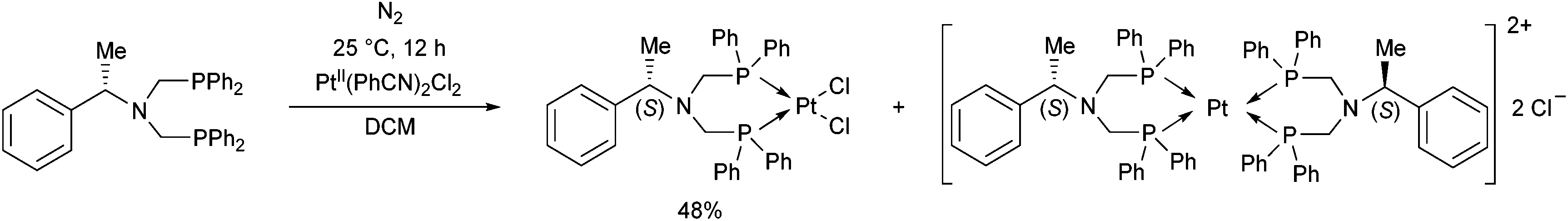 | ||
| Scheme 21 Synthesis of a chiral platinum(II) complex. | ||
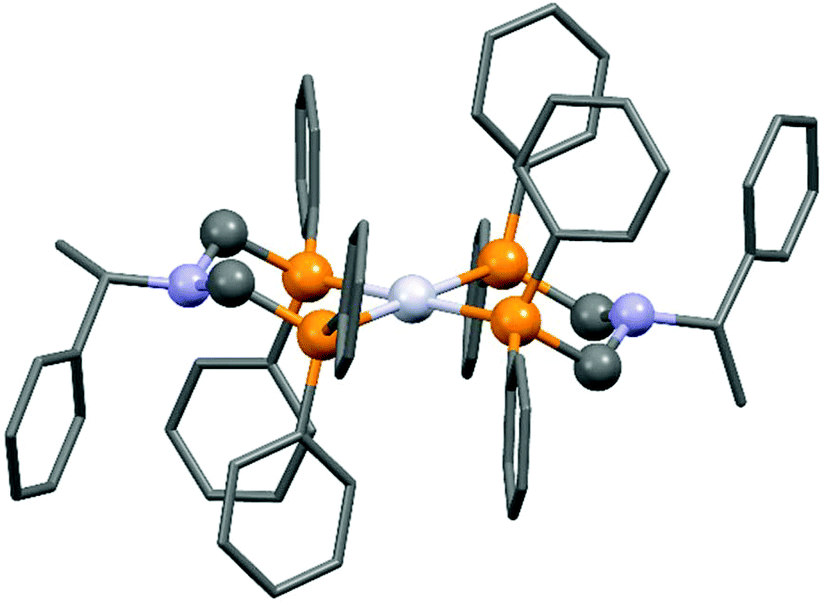 | ||
| Fig. 7 X-Ray structure of the chiral bicyclic platinum(II) complex [CCDC 1547003].57 | ||
Bis(phosphinomethyl)amines were also proved to be efficient ligands in the synthesis of palladium(II) complexes (Table 7). The complexation of bis(diphenylphosphinomethyl)aniline with PdII(PhCN)2Cl2 in DCM afforded the corresponding complex in a yield of 79% after 5 min (Table 7/entry 1). According to the X-ray structure of the complex, a flattened boat conformation was observed. Similar PdII complexes containing tert-butyl or cyclohexyl groups on the two P atoms were also prepared. In these cases, higher temperature (65–80 °C) and longer reaction time (18–20 h) were necessary (Table 7/entry 2). Starting from N-aliphatic or sulfonated phosphines and PdII(cod)Cl2, the corresponding complexes were obtained at room temperature after 1–2 h in yields of 81–88% (Table 7/entry 3). By treatment of the same precursor with phosphines containing a substituted Ph-ring in boiling DCM, several new bidentate PdII chelate complexes were obtained, which were effective catalysts in the Heck reaction of olefins and aryl halides (Table 7/entry 4). Non-dendritic bisphosphines were also used as Pd ligands (Table 7/entry 5). The complexes synthesized were tested as catalysts in the Heck reaction of 4-iodotoluene and methyl acrylate. In the case of bis(diphenylphosphinomethyl) amino-2-pyridine as the ligand, PdIICl2 was applied as the precursor (Table 7/entry 6). The complexation performed at room temperature resulted in the formation of the PdII complex in a high yield (94%).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Y | Z | Pd precursor | Solvent | Temperature, t | Yield [%] | Ref. |
| 1 | Ph | Ph | PdII(PhCN)2Cl2 | DCM | rt, 5 min | 79 | 53 |
| 2 | Ph | t Bu, cHex | PdII(PhCN)2Cl2 | THF, toluene | 65–80 °C, 18–20 h | 85–99 | 56 |
| 3 | Me, tBu, 3-NaSO3-C6H4 | Ph | PdII(cod)Cl2 | DCM | rt, 1–2 h | 81–88 | 58 |
| 4 |
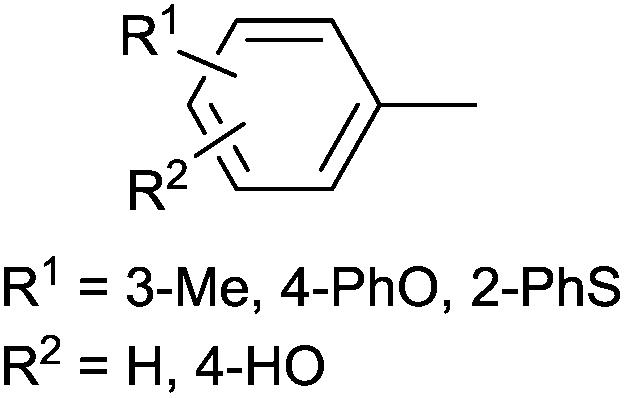
|
Ph | PdII(cod)Cl2 | DCM | 40 °C, 3–4 h | 65–80 | 59 |
| 5 |
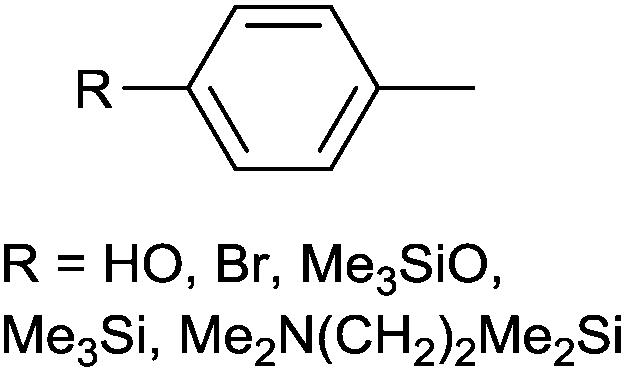
|
Ph | PdII(cod)Cl2 | DCM | rt, 3 h | 75–87 | 60 |
| 6 |
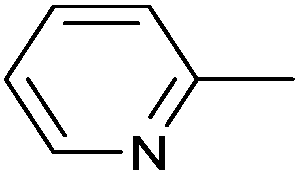
|
Ph | PdIICl2 | DCM:MeCN (1:1) |
rt, 5 min | 94 | 61 and 62 |
Starting from different types of PdII precursors, the palladium(II) complexes of bis(phosphinomethyl)amines could be prepared efficiently. Depending on the substituents of the P and N atoms of the ligand, the complexations required different reaction conditions. The catalytic activity of several PdII complexes was tested in the Heck reactions of aryl halides and alkyl acrylates (Table 8). In the reactions, potassium phosphate or triethylamine was used as the base in N-methylpyrrolidone (NMP) or in acetonitrile. It could be observed that the performance of the catalysts depended on the N-substituents. The N-aliphatic or the N-(sodium benzenesulfonyl) complexes were less active than the PdII complexes bearing an aryl group on the nitrogen atom (Table 8/entry 1 vs. entry 2). Complexes of non-dendritic bisphosphines showed a modest activity and stability in the Heck reaction of 4-iodotoluene and methyl acrylate (Table 8/entry 3).
The PdII complex of bis(diphenylphosphinomethyl)amino-2-pyridine (bdppmapy) was applied as a catalyst in the decarboxylative C–C coupling of 4-picolinic acid with aromatic bromides, and showed a good catalytic performance (Scheme 22).61 The reactions were carried out at 130 °C in N,N-dimethylacetamide (DMA) as the solvent. The catalytic activity of the bdppmapy-Pd complex was compared to that of other PdII complexes containing alkyl and aryl phosphines in cross-coupling of 4-picolinic acid and 2,4-dimethoxy bromobenzene. From the catalytic results, the bdppmapy-Pd complex was the most effective, as the yield of the product was 78%. In the case of other phosphine-PdII complexes, the yields were in the range of 15–48%.
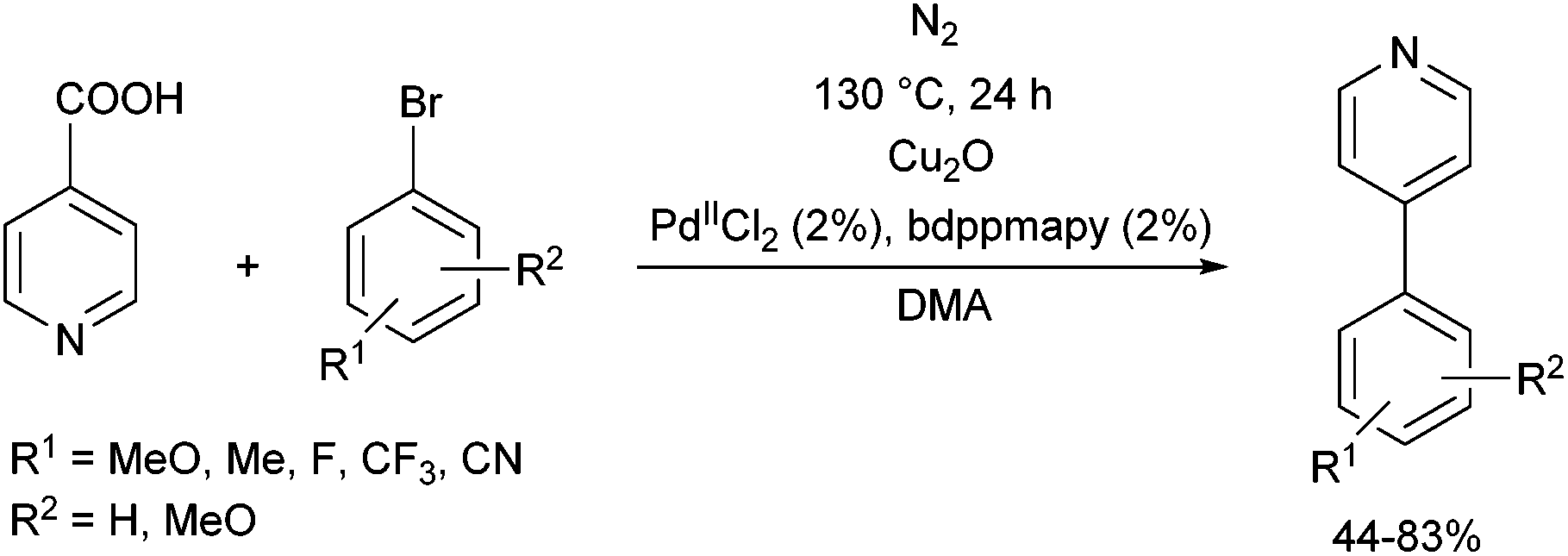 | ||
| Scheme 22 Pd-Catalyzed cross-coupling of 4-picolinic acid and aryl bromides. | ||
The effect of different phosphine ligands on the Suzuki–Miyaura cross-coupling of 4-bromoacetophenone and 4-methoxyphenylboronic acid was also investigated, where the α-aminophosphine-based PdII complexes were also found to be more efficient than the alkyl and aryl phosphine complexes (yields of 99% vs. 79–85%, respectively).62 The coupling reaction was extended to other aryl halides and arylboronic acids (Scheme 23). A wide range of biaryl compounds were obtained in yields of 65–99% under mild conditions.
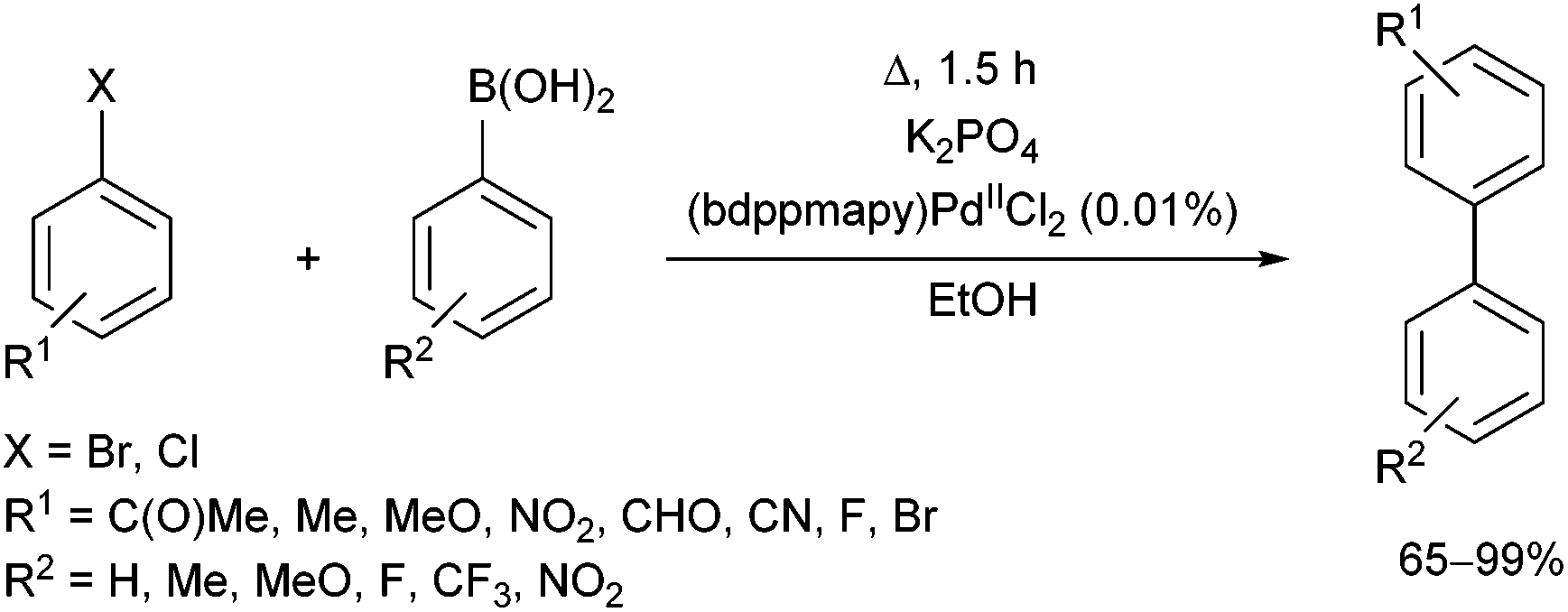 | ||
| Scheme 23 Suzuki–Miyaura coupling of aryl halides with arylboronic acids. | ||
The complexation of α-aminophosphines bearing a benzoic acid moiety using PdII(cod)MeCl as the metal precursor was performed at room temperature for 15 min (Scheme 24).63 The metathesis of one of the complexes (R1 = H, R2 = MeO) with sodium bromide and iodide was also elaborated, giving (methyl)bromopalladium(II) and (methyl)iodopalladium(II) derivatives.
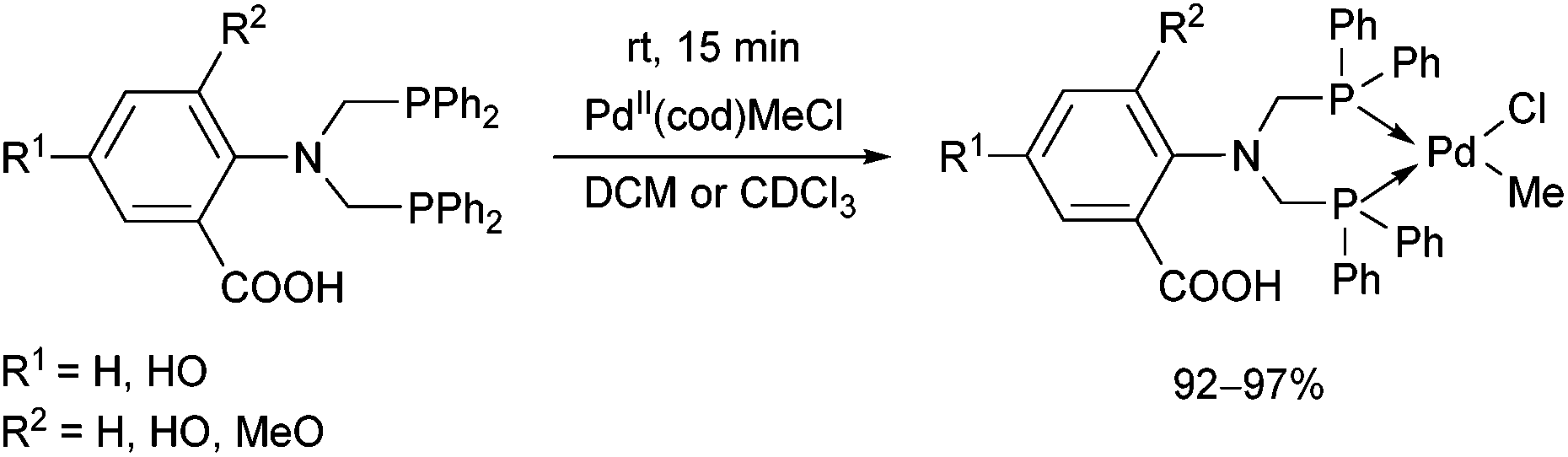 | ||
| Scheme 24 Preparation of mononuclear PdII complexes from α-aminophosphines bearing a benzoic acid moiety. | ||
N-Phenylselenoalkyl-bis(aminophosphines), a special family of ligands, were also utilized as bidentate P-ligands in the synthesis of PtII and PdII complexes (Scheme 25).64 According to X-ray studies, the products were of cis configuration, and the metal centre was in a nearly square planar geometry.
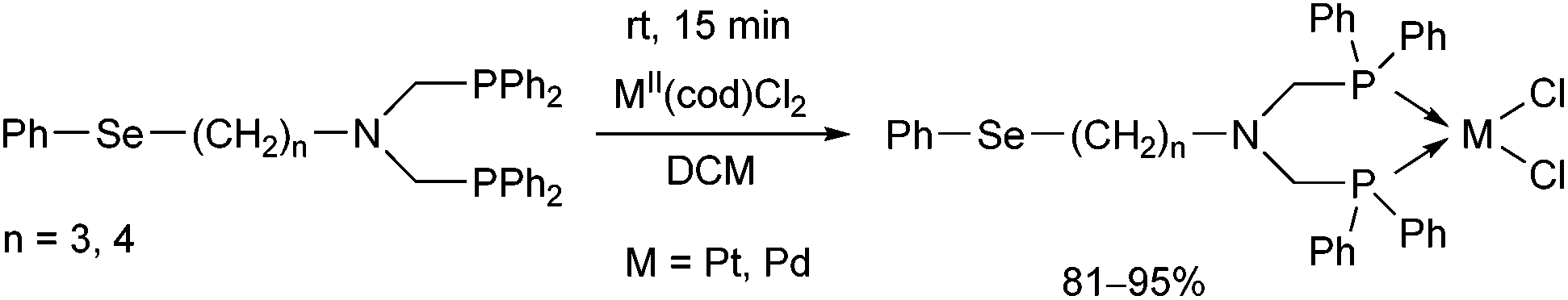 | ||
| Scheme 25 Utilization of N-phenylselenoalkyl-bis(aminophosphines) as ligands in the synthesis of cis chelate transition metal complexes. | ||
The coordination of bis(diphenylphosphinomethyl)amino derivatives of adenine to transition metals was also investigated (Scheme 26).65 A series of bidentate chelate complexes was synthesized in good yields using various PtII and PdII(cod) precursors. It was observed that all complexes retained the free adenine moiety for complementary hydrogen bonding.
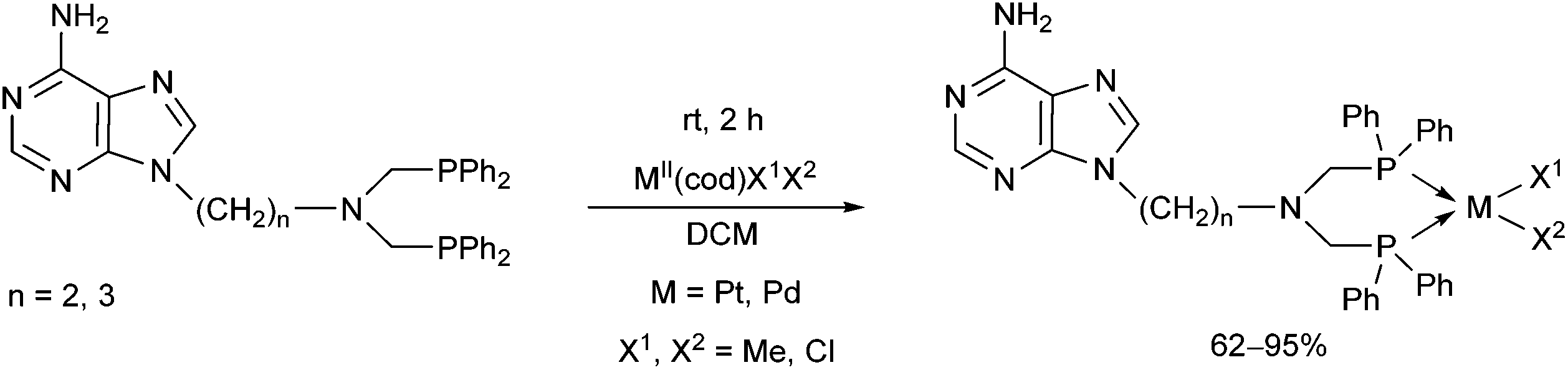 | ||
| Scheme 26 PtII and PdII complexes of bis(α-aminophosphine) ligands containing an adenine moiety. | ||
Crown ether-functionalized PtII and PdII complexes were synthesized by the reaction of bis(diphenylphosphinomethyl)-aminobenzo-15-crown-5 and PtII(cod)Cl2 or PdII(cod)Cl2 at room temperature using toluene–DCM as the solvent in a reaction time of 2 h (Scheme 27).66
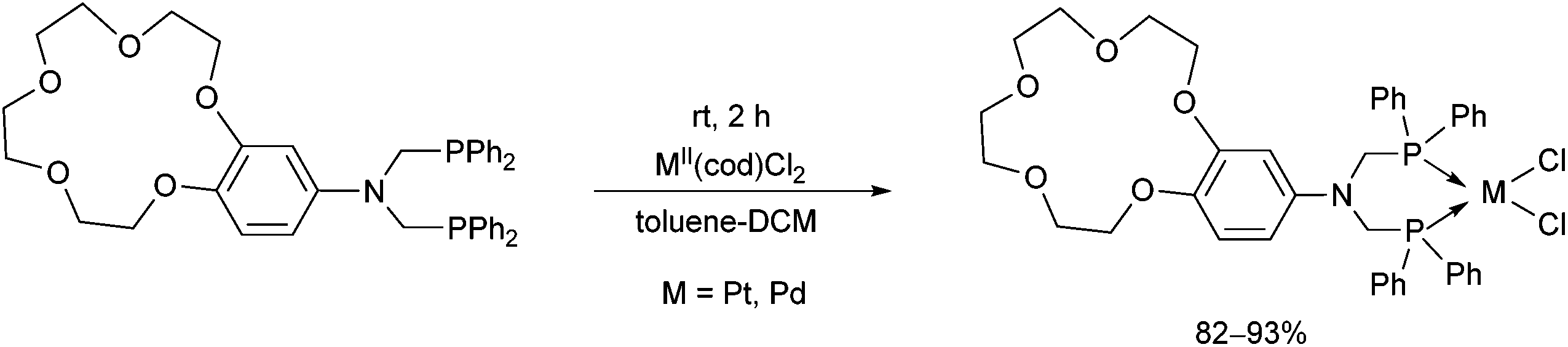 | ||
| Scheme 27 Synthesis of crown ether-functionalized cyclic PtII/PdII complexes. | ||
Water-soluble phosphine ligands incorporating an ethoxylated phosphonate chain were reacted with dihydrogen tetrachloropalladate(II) at the boiling point of butanol for 4 h (Scheme 28).67 The corresponding PdII complexes obtained in yields of 70–78% showed good catalytic activity in the biphasic carbonylation of benzyl chloride.
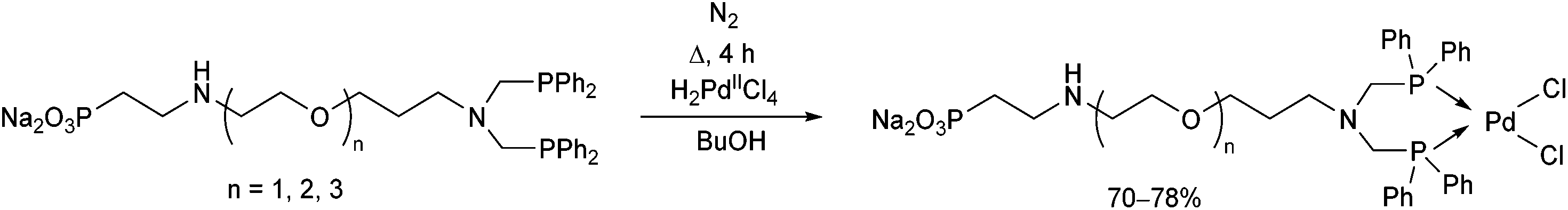 | ||
| Scheme 28 Preparation of PdII complexes incorporating water-soluble bidentate α-aminophosphine ligands. | ||
Complexation of a (3-aminopropyl)triethoxysilane-functionalized bisphosphine ligand with [PdII(η3-allyl)Cl]2 in THF afforded the desired PdII complex, which was co-immobilized with SiO2, as well as with SiO2-supported DABCO (1,4-diazabicyclo[2.2.2]octane) (Scheme 29).68
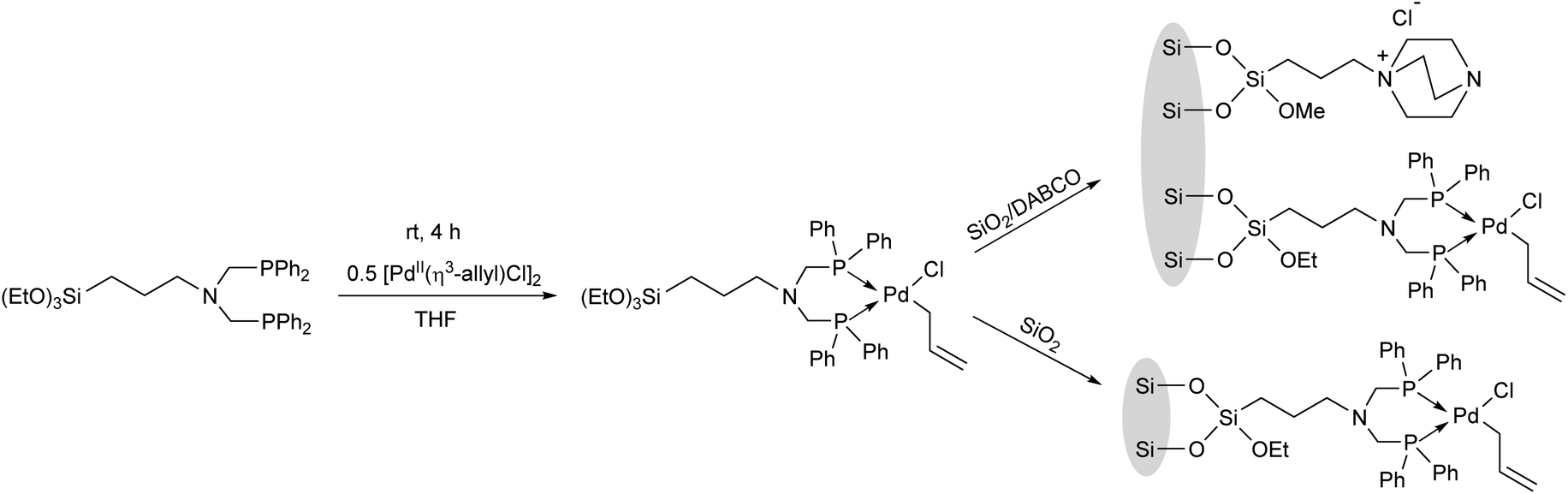 | ||
| Scheme 29 Synthesis of SiO2-supported palladium(II)-bisphosphine complexes. | ||
The complexes prepared were utilized as catalysts in the allylation of ethyl acetoacetate by (allyl)(methyl)carbonate (Table 9).69 The reactions were carried out in the presence of K2CO3 in toluene at 70 °C for 60 min. It was observed that the catalytic activity of the homogeneous PP-PdII complex was similar to the SiO2-supported heterogeneous PP-PdII catalyst (Table 9/entry 1 vs. entry 2). The SiO2/DABCO/PP-PdII catalyst exhibited the highest activity as shown by the complete conversion and the selectivity for the diallylated product (Table 9/entry 3). This allylation reaction was extended to other nucleophiles, such as nitriles, ketoesters, diketones or nitroethane, where the corresponding diallylated products were obtained selectively.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Conversion [%] | Yield (mono:di) [%] |
| 1 | PP-PdII | 97 | 48:42 |
| 2 | SiO2/PP-PdII | 98 | 40:49 |
| 3 | SiO2/DABCO/PP-PdII | 99 | 18:81 |

There are only a few examples of the synthesis of cyclic PdII and PtII complexes bearing alkyl groups on the phosphorus atoms (Table 10). In one case, the complexation of bis(tert-butylaminomethylphosphine) was performed using palladium acetate as the precursor (Table 10/entry 1). The complex synthesized was an efficient catalyst in the Sonogashira cross-coupling of aryl halides with acetylenes. In other instances, PdII(PhCN)2Cl2 or PtII(cod)2X2 (X = Cl, Br) was reacted with the bis(dialkylphosphinomethyl)anilines in THF or in toluene (Table 10/entry 2). In all cases, cis square planar complexes were formed.
The PdII complex of bis(di-tert-butylphosphinomethyl)-benzylamine is a useful catalyst in the Sonogashira cross-coupling of aryl halides and acetylenes (Scheme 30).70 The advantage of this procedure is the possibility of avoiding the use of CuI co-catalysts.
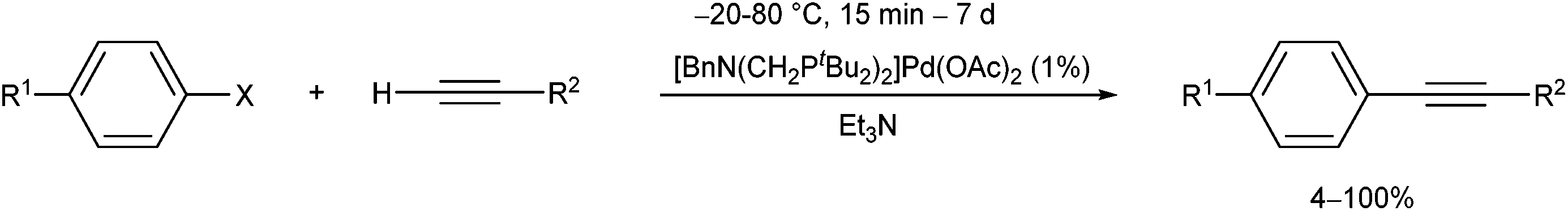 | ||
| Scheme 30 Pd-catalyzed cross-coupling of aryl halides and acetylenes. | ||
The synthesis of zerovalent platinum and palladium complexes was also described, where M0(dba)2 (dba = dibenzylideneacetone) or Pd02(dba)3 was applied as the transition metal precursor (Scheme 31).71 According to X-ray investigations, the transition metal was coordinated to the dba through one dative bond (Fig. 8). The six-membered metallocycle in the complexes is present in a flattened chair conformation.
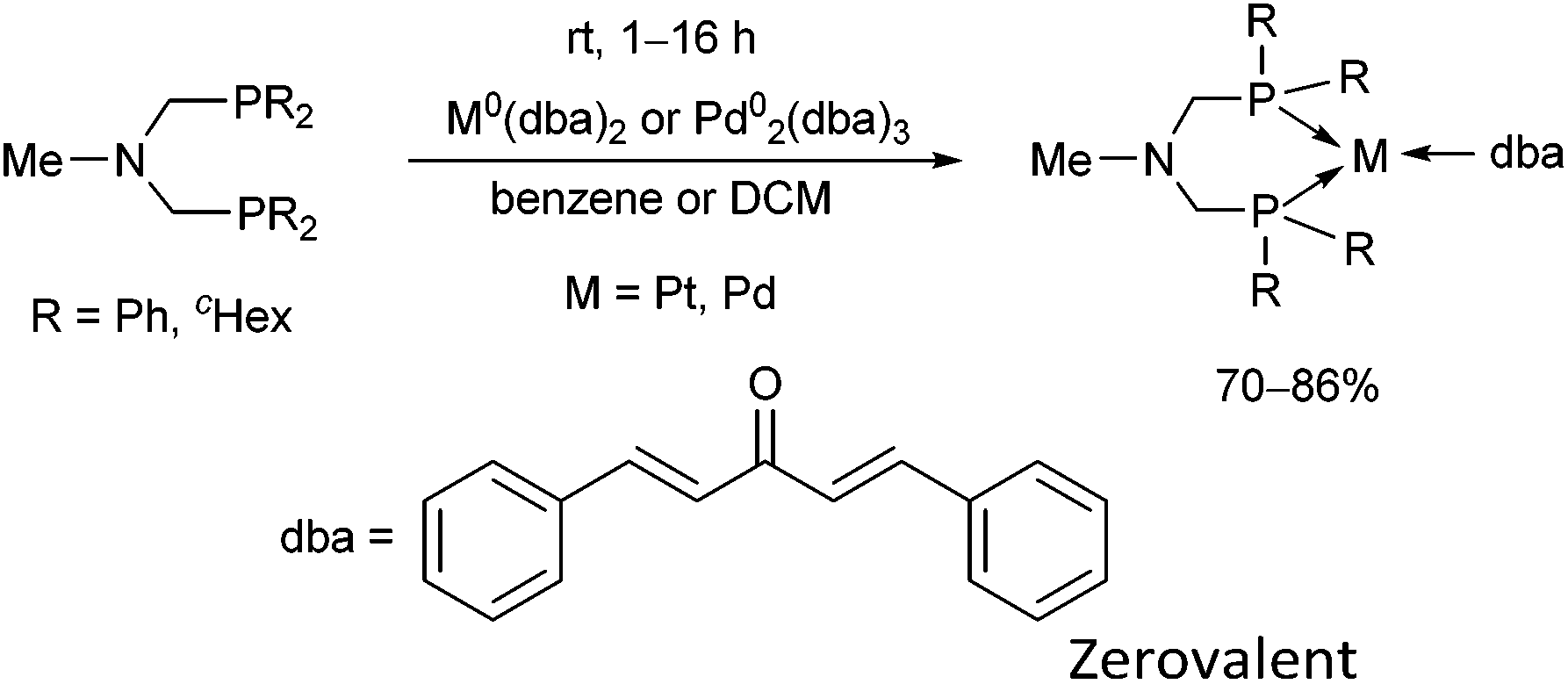 | ||
| Scheme 31 Zerovalent complexes of bis(phosphinomethyl)methylamines. | ||
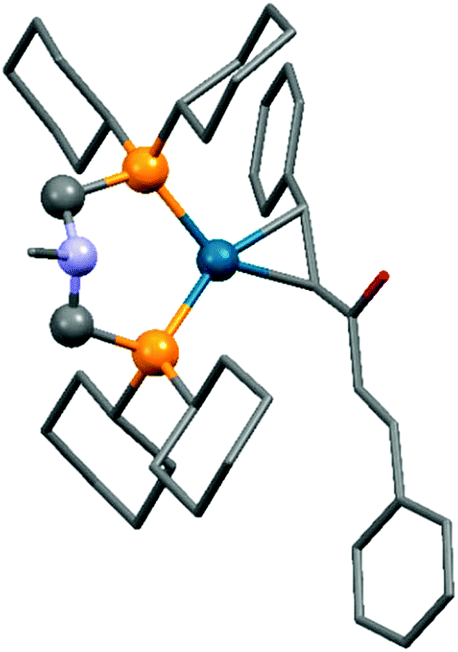 | ||
| Fig. 8 X-Ray structure of Pd(dba)(cHex2PCH2)2NMe [CCDC1304170].71 | ||
A ferrocenyl-substituted ditertiary aminophosphine was applied as a novel ligand in the synthesis of PtII and PdII complexes (Scheme 32).72 By a reaction with MII(cod)Cl2 (M = Pt, Pd), the cis-oriented chelate complexes were obtained in yields of 74–86%, whereas when using PdII(cod)MeCl, a complex with a trans–trans conformation could be prepared. The corresponding structures were proved by single crystal X-ray crystallography.
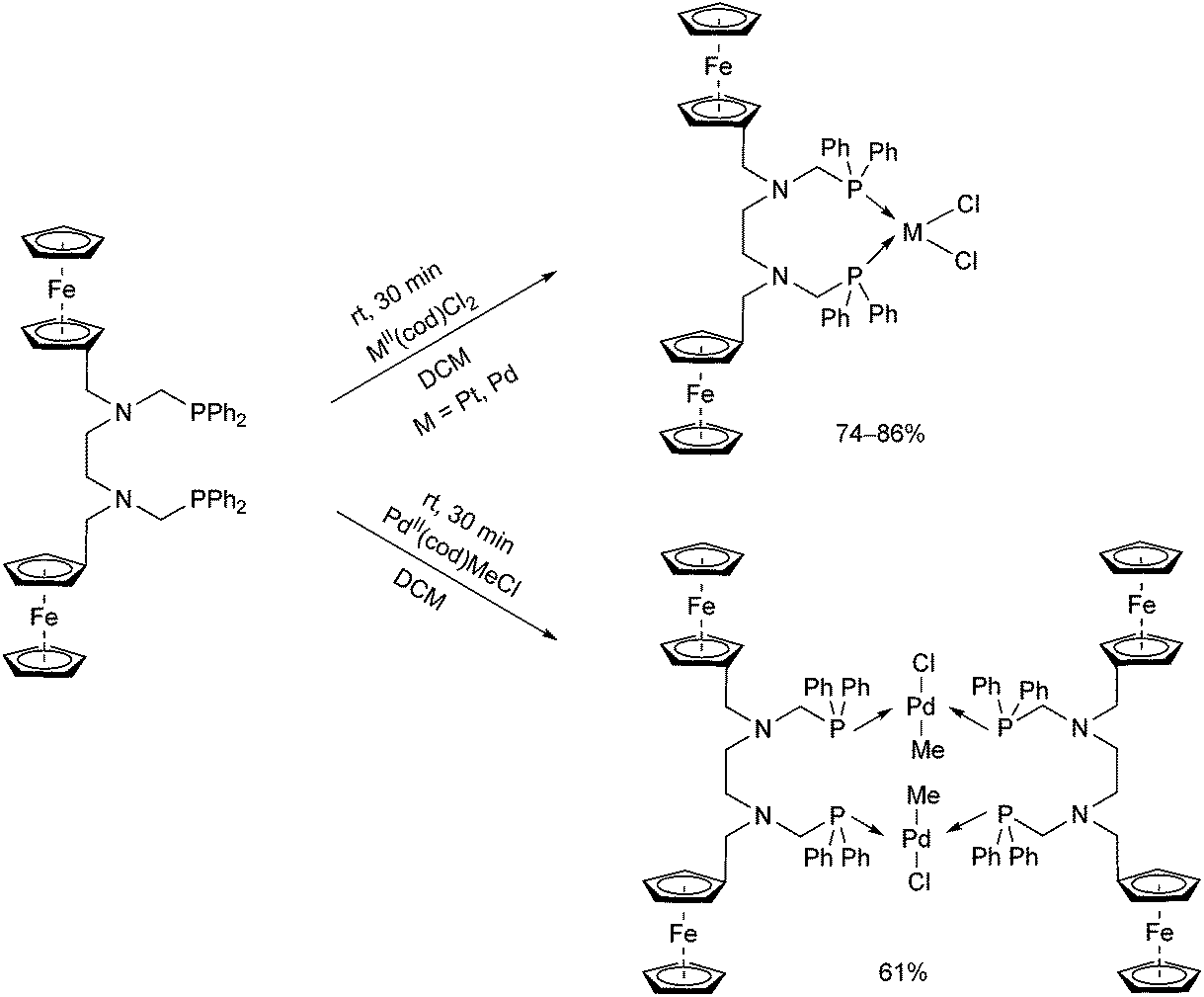 | ||
| Scheme 32 Complexation of an aminophosphine ligand bearing two ferrocenyl groups. | ||
The synthesis of a new hexadentate P2N4 ligand system was also described (Scheme 33).73 The complexation was carried out using MII(cod)Cl2 (M = Pt, Pd) in DCM at ambient temperature for 2 h. According to X-ray analysis, only the P atoms were coordinated to the corresponding transition metal, while the pyridyl groups remained non-bonding. The square planar PtII and PdII centers formed a 5-membered chelate ring with the bisphosphine in both complexes.
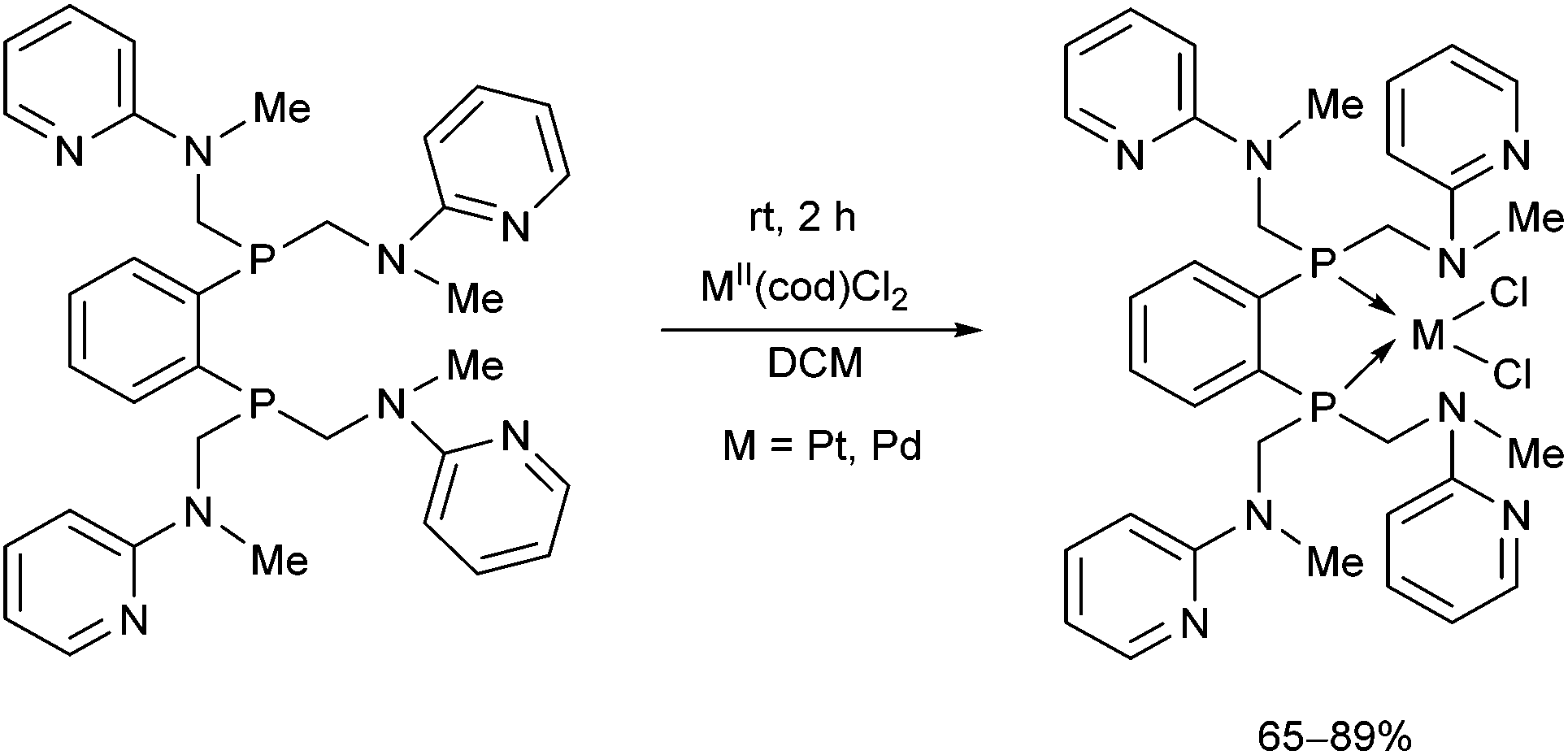 | ||
| Scheme 33 Platinum(II) and palladium(II) complexes of a hexadentate phosphine ligand. | ||
Nonsymmetrical ditertiary phosphines bearing an adamantanate moiety were also applied as efficient ligands (Scheme 34).74 The corresponding PtII and PdII complexes were synthesized in high yields, and their conformation was determined by 31P NMR spectroscopy and single crystal X-ray analysis. Due to the difference in stereoelectronic properties between the two phosphorus atoms, the J(Pt–P) coupling of the –P(Ad) group was twice as much as the coupling on the –PPh unit.
 | ||
| Scheme 34 The complexation of nonsymmetrical bisaminophosphine ligands with MII(cod)Cl2 (M = Pt, Pd). | ||
In the next part, the synthesis of palladium(II) and platinum(II) complexes containing two hetero rings is summarized. Two types of binuclear complexes are known; in one case the phosphine ligand contains a spacer between the donor atoms, which are coordinated to two transition metals. In the other instance, two aminophosphine ligands are coordinated to a single PdII or PtII atom.
A polydentate phosphine ligand including an ethylene spacer was reacted with 2 equivalents of PdII(PhCN)2Cl2 in DCM (Scheme 35).53 The tetrachloro-complex obtained was converted into the corresponding tetraiodo derivative, and its structure was elucidated by X-ray analysis. It was found that the coordination of the two PdII was distorted from planarity, leading to significantly bent trans P–Pd–I angles.
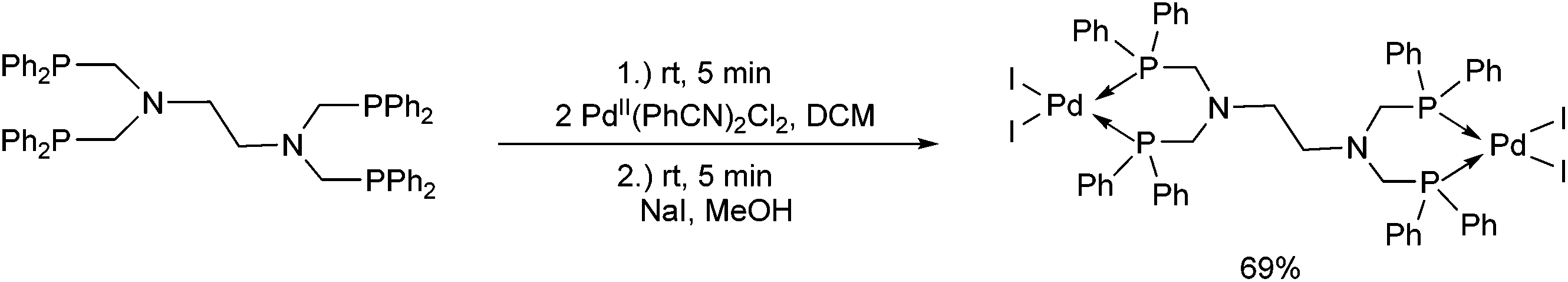 | ||
| Scheme 35 Synthesis of binuclear PdII complexes containing an ethylene spacer. | ||
The reaction of tetra(diphenylphosphinomethyl)diamines with PdII(tab)2Cl2 (tab = 4-trimethylammonio-benzenethiolate) also led to binuclear compounds, but in this case in an ionic form (Scheme 36).75 An X-ray analysis of the complexes revealed a square planar geometry (Fig. 9). Both of the Pd2+ ions were coordinated by two S atoms from the “tab” and two P atoms from the bis(phosphine ligand).
 | ||
| Scheme 36 Binuclear PdII complexes bearing thiolate and aminophosphine ligands. | ||
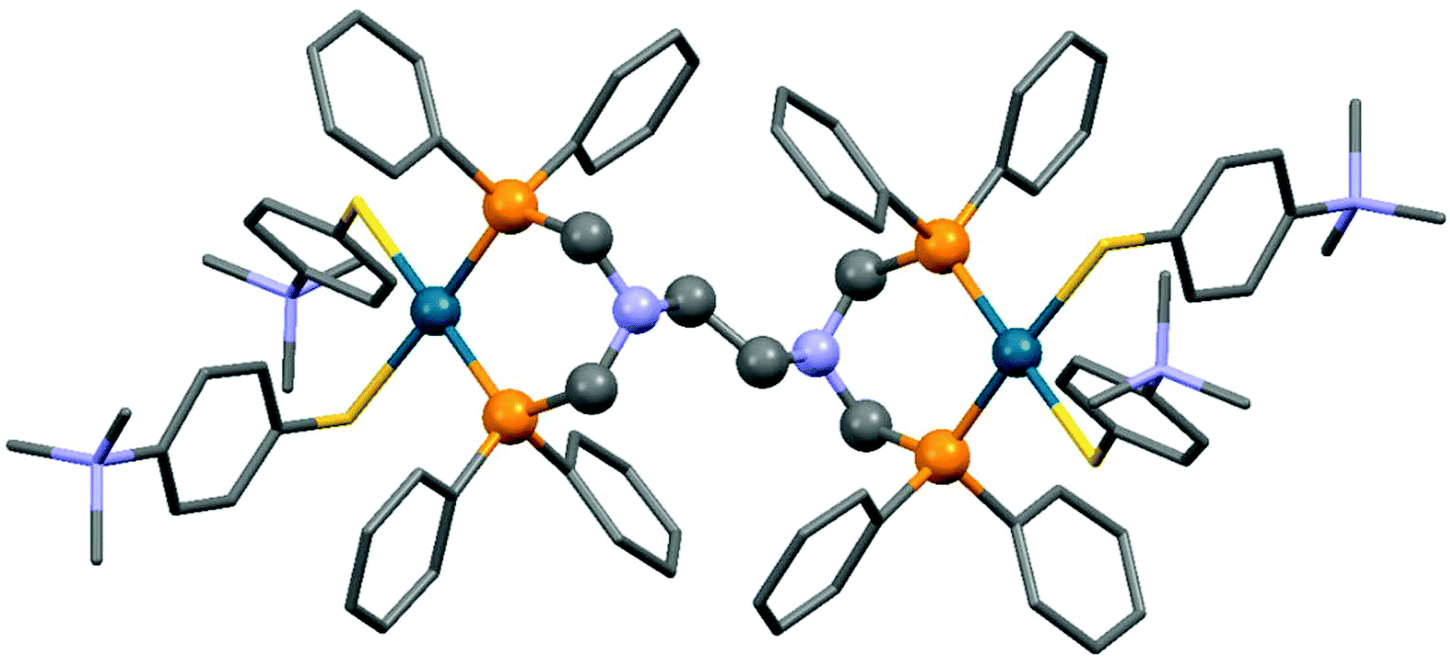 | ||
| Fig. 9 X-Ray structure of {[Pd(tab)2]2(μ-dppeda)}Cl4 [CCDC 800046].75 | ||
Besides the mononuclear PdII complexes of phosphine ligands containing an ethoxylated phosphonate chain (Scheme 28), binuclear-type derivatives were also synthesized by applying 0.5 equivalents of dihydrogen tetrachloropalladate(II) (Scheme 37).67
 | ||
| Scheme 37 Preparation of binuclear PdII complexes with water-soluble ligands. | ||
The synthesis of structurally similar PtII and PdII complexes incorporating ethyl groups on the P atoms was also described (Scheme 38).76 When bis(diethylphosphinomethyl)-methylamine was reacted with PtII(cod)Cl2 in acetonitrile, followed by treatment with ammonium hexafluorophosphate, the corresponding Pt(PNP)2(PF6)2 (PNP = Et2PCH2N(Me)CH2PEt2) complex was obtained in a moderate yield. For the synthesis of the PdII(PNP)2(BF4)2 derivative, [PdII(MeCN)4]BF4 was applied as the metal precursor. The hydride donor ability of the complexes was also investigated, and the PdII derivative proved to be a better reducing agent.
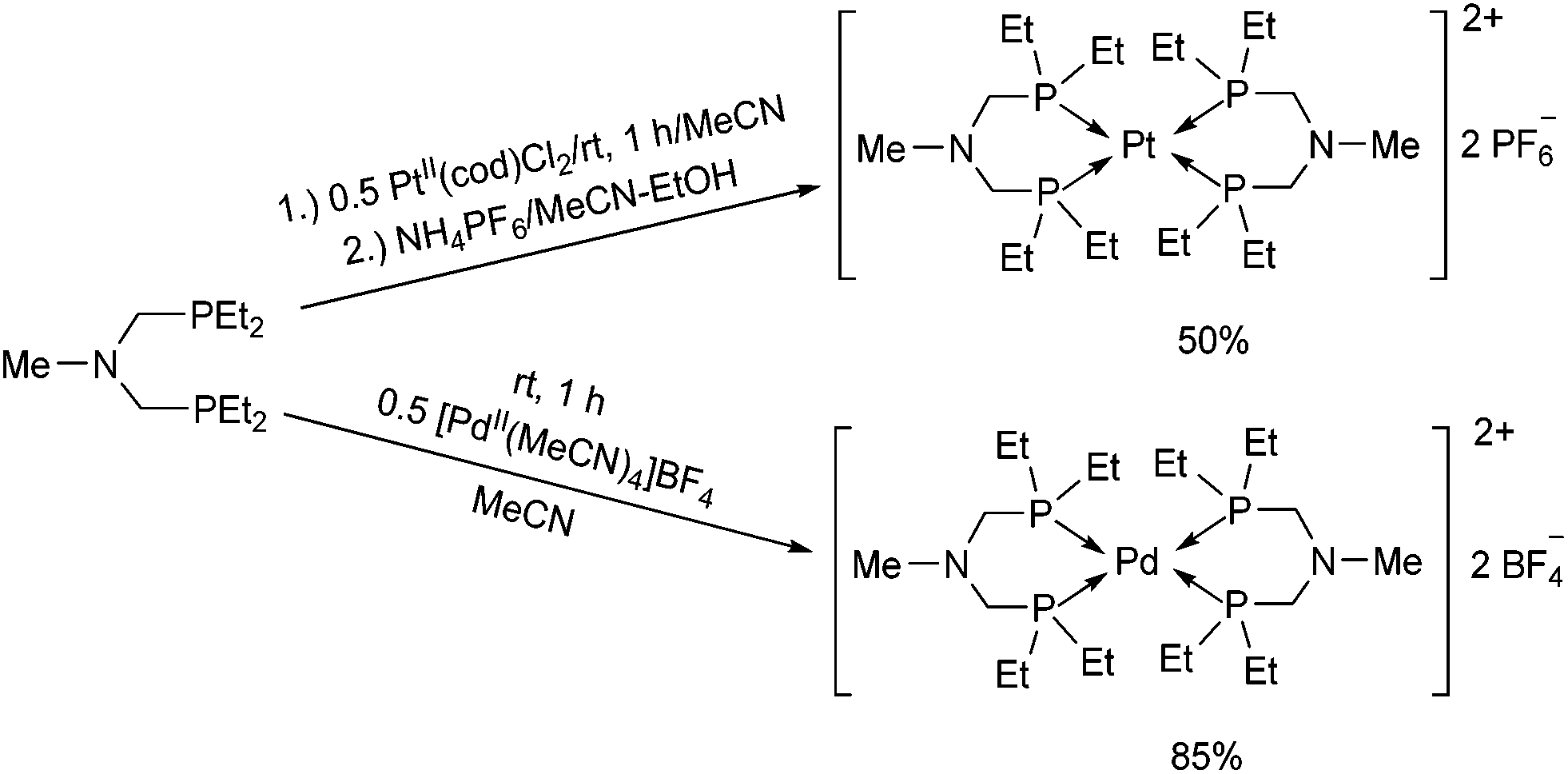 | ||
| Scheme 38 Synthesis of Pt(PNP)2(PF6)2 and Pd(PNP)2(BF4)2 complexes. | ||
3.2. Synthesis of rhodium complexes
Bis(phosphinomethyl)amines were also applied as efficient ligands in the synthesis of bidentate rhodium(I) complexes (Table 11). The complexation of bis(dialkylphosphinomethyl)aniline with chlorocarbonylbis(triphenylphosphine)rhodium(I) in toluene afforded the corresponding ring complexes in yields of 59–84% after 20 min (Table 11/entry 1). The same type of square planar RhI complex was synthesized by the reaction of [RhICl(CO)2Cl]2 with an excess of N,N-bis(diphenylphosphino-methyl)aniline under mild conditions (Table 11/entry 2).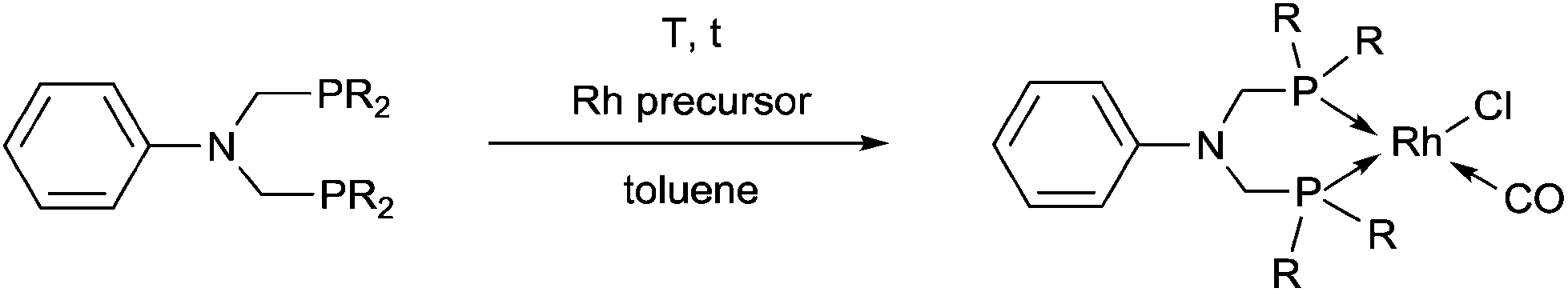
The RhI complex of a bis(phosphinomethyl)amine derivative bearing a hydroxy functionality was synthesized by applying [RhI(cod)Cl]2 as the rhodium precursor (Scheme 39).77 The RhI complex obtained was bonded to the surface of activated carbon, and was tested as catalyst in the hydroformylation of 1-octene, where the formation of the linear aldehydes was predominant. The RhI complexes remained fully active in four consecutive catalytic cycles.
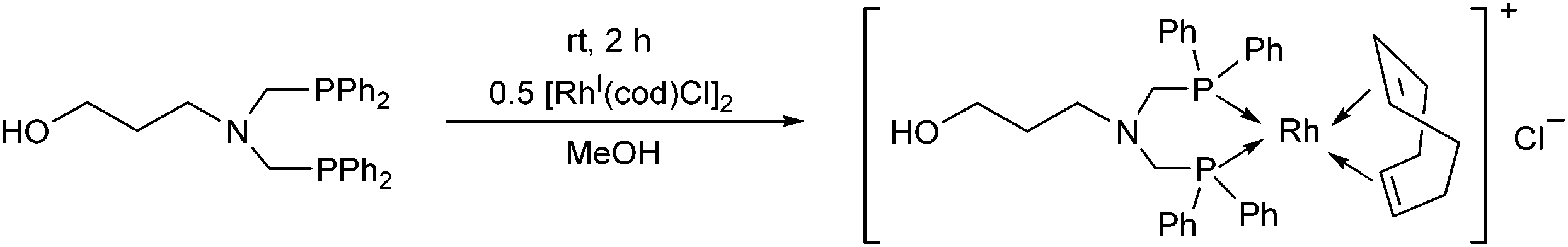 | ||
| Scheme 39 Synthesis of a cyclic RhI complex containing a hydroxyl group. | ||
Bis(diphenylphosphinomethyl)amino acid derivatives were also utilized as bidentate P-ligands in the preparation of RhI complexes (Scheme 40).78 The complexations were carried out with 0.5 equivalents of [RhI(nbd)Cl]2 in methanol. The corresponding complexes obtained in yields of 74–81% were applied as catalysts in the enantioselective hydrogenation of α-acetamidocinnamic acid methyl ester.
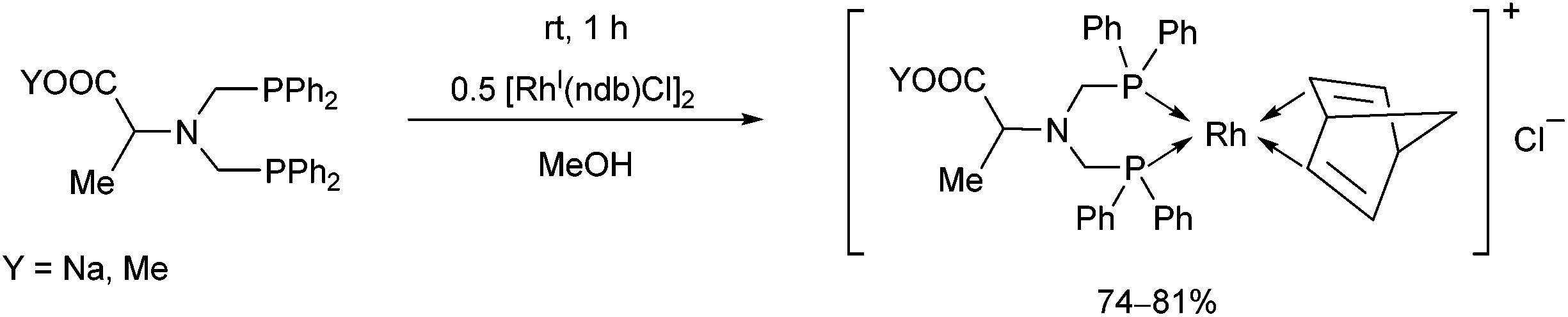 | ||
| Scheme 40 RhI complexes of bis(diphenylphosphinomethyl)amino acid derivatives. | ||
The complexation of the sodium salt of bis(diphenylphosphinomethyl)amino acid was performed using 0.25 equivalents of [RhI(nbd)Cl]2 (Scheme 41).78 In this case, a binuclear RhI complex was obtained in a yield of 65%.
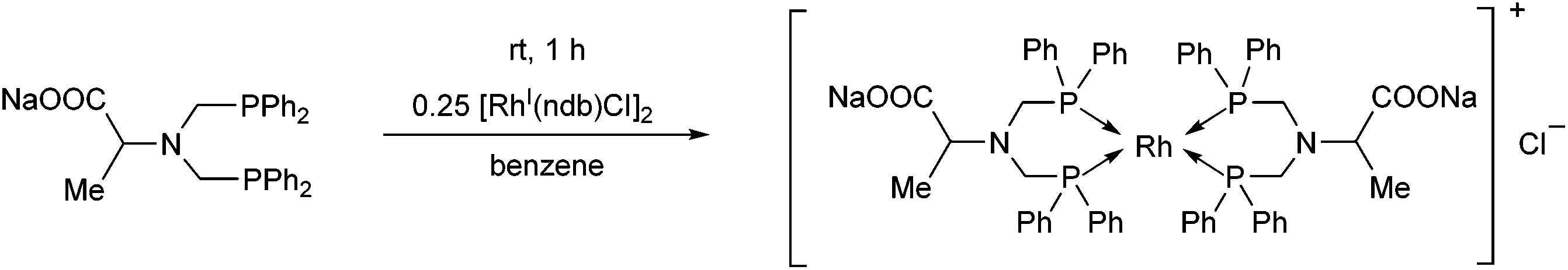 | ||
| Scheme 41 The binuclear RhI complex of the sodium salt of bis(diphenylphosphinomethyl)amino acid. | ||
A ferrocenyl substituted bis(aminophosphine) ligand was utilized in the synthesis of a RhI complex (Scheme 42).72 The complexation was performed with 0.5 equivalents of [RhI(CO)2Cl]2, furnishing the ring product with trans–trans conformation in a yield of 29%. The dimeric structure of the complex was confirmed by X-ray analysis (Fig. 10).
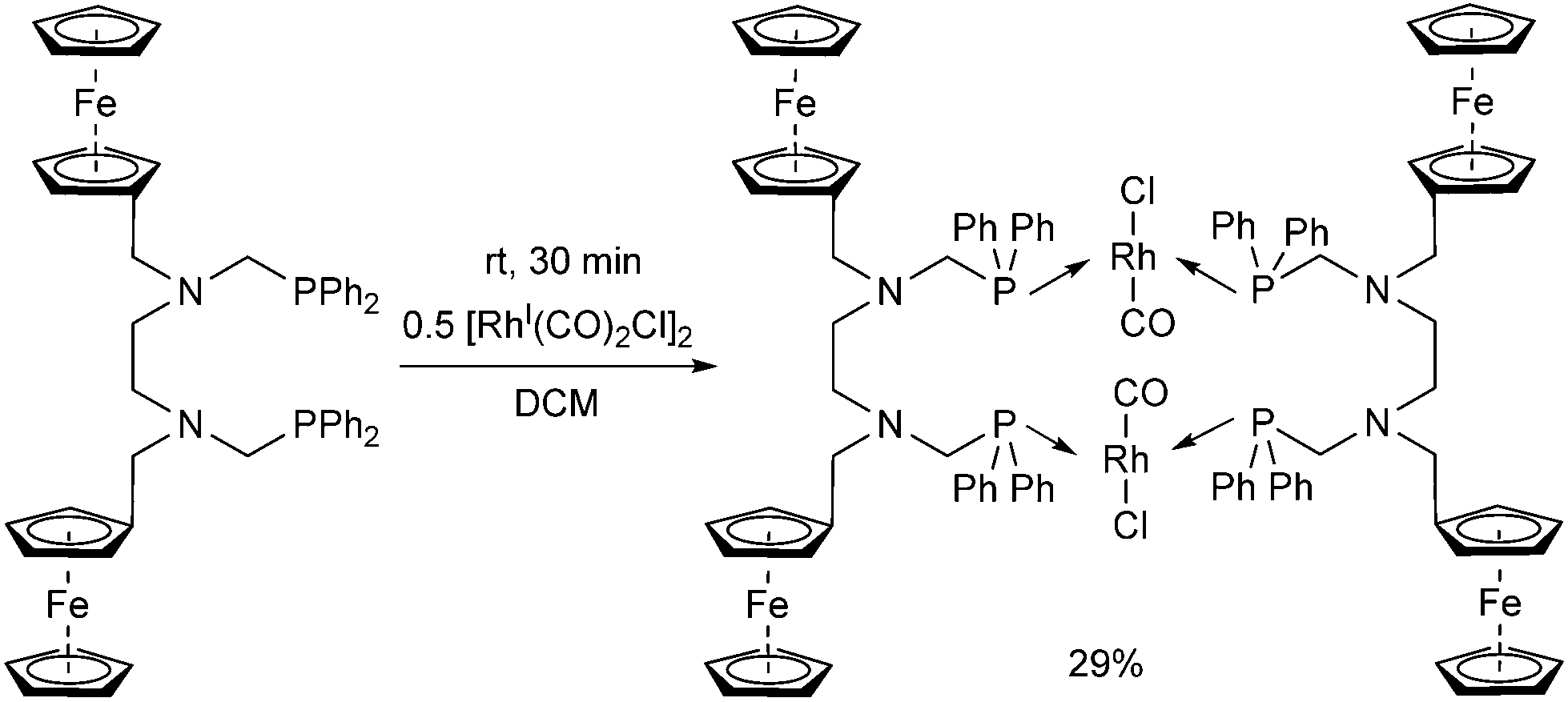 | ||
| Scheme 42 Synthesis of a RhI complex bearing four ferrocenyl groups. | ||
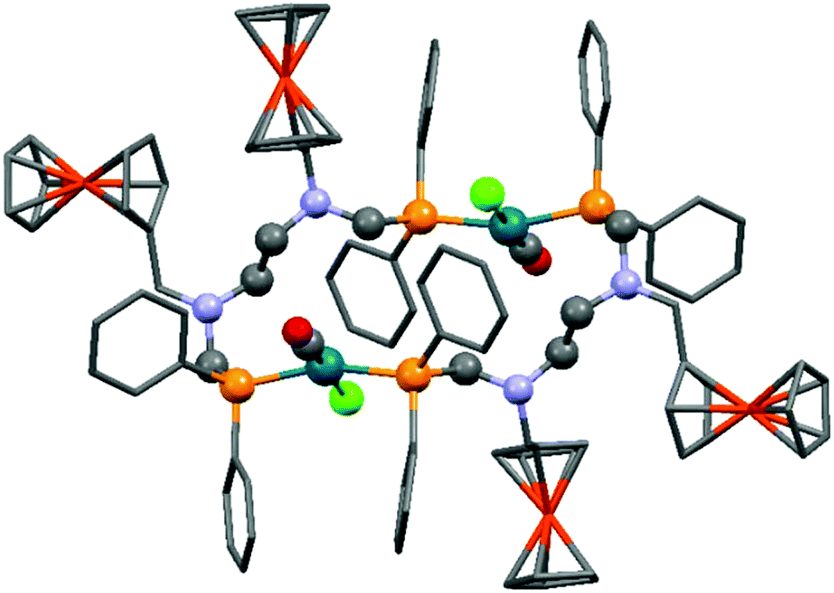 | ||
| Fig. 10 X-Ray structure of [Rh(CO)Cl{FcCH2N(CH2PPh2)CH2}2]2 [CCDC 676683].72 | ||
3.3. General methods for the preparation of complexes incorporating bidentate α-aminophosphines as ligands
Similarly to the previous chapter, general methods for the preparation of complexes incorporating bidentate α-aminophosphines as P-ligands are summarized in Table 12. Pt and Pd complexes containing one bidentate α-aminophosphine ligand (IV) may be synthesized using MII(cod)Cl2 (M = Pt, Pd). In both cases, the products can be obtained in yields of ca. 70–85% using DCM as the solvent. A similar type of Rh complex can be prepared by applying 0.5 equivalents of [RhI(CO)2Cl]2 or 1 equivalent of RhIClCO(PPh3)2 in reaction times of 10 min or 20 h in toluene to afford the products in yields of 59–88%. Towards the synthesis of Pt, Pd and Rh complexes bearing two bidentate α-aminophosphine ligands (V), the starting phosphines should be reacted with 0.5 equivalents of PtII(cod)Cl2 in toluene, 0.5 equivalents of PdII(tab)Cl2 in DCM or 0.25 equivalents of [RhI(ndb)Cl]2 in benzene.| Types of complexes | M | Precursor | t | Solvent | Average yield [%] |
|---|---|---|---|---|---|
|
|
|||||
| a At 80 °C. | |||||
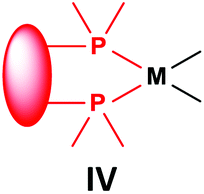
|
Pt | PtII(cod)Cl2 | 0.5–24 h | DCM | 70–85 |
| Pd | PdII(cod)Cl2 | 5 min–4 h | DCM | 68–85 | |
| Rh | 0.5 [RhI(CO)2Cl]2 or RhIClCO(PPh3)2a | 10 min or 20 h | Toluene | 59–88 | |
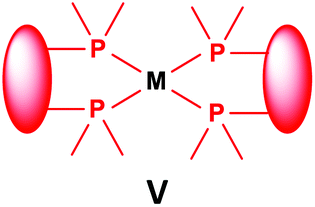
|
Pt | 0.5 PtII(cod)Cl2 | 1 h | MeCN | 50 |
| Pd | 0.5 PdII(tab)Cl2 | 0.5 h | DCM | 86–89 | |
| Rh | 0.25 [RhI(ndb)Cl]2 | 1 h | benzene | 65 | |

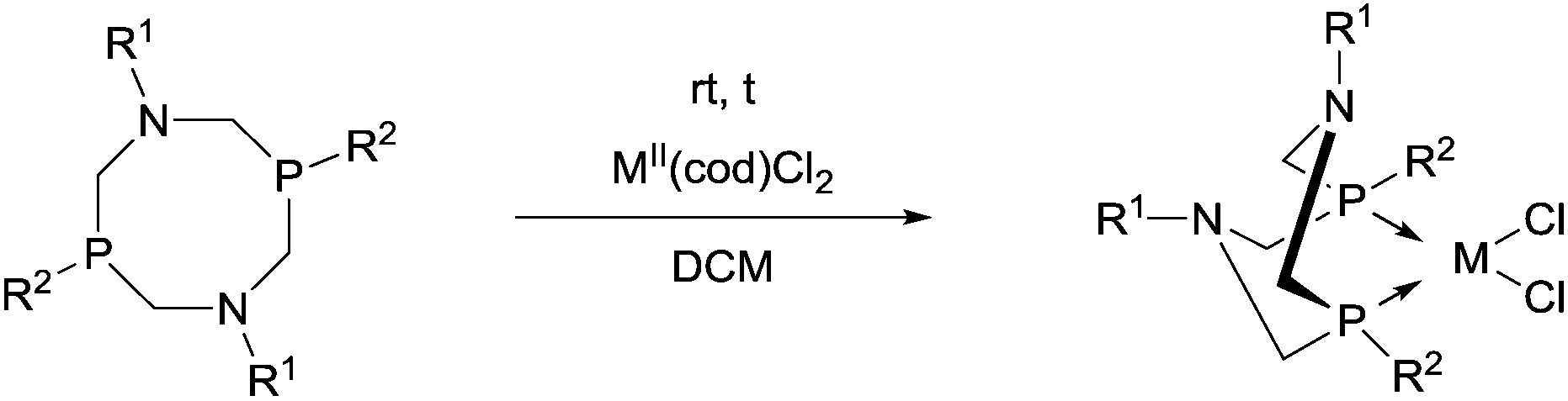
4. Utilization of cyclic α-aminophosphines as ligands
Cyclic α-aminophosphines form another prominent group of commonly used ligands in the synthesis of transition metal complexes. The application of 6-, 7- and 8-membered ring ligands comprising the α-aminophosphine scaffold is discussed in this chapter.4.1. Complexation of 6-membered ring derivatives of α-aminophosphines
In the reaction of PtII(cod)X2 precursors with 1,3-diaza-phosphacyclohexanes incorporating two glycine or glycinate moieties, the corresponding cis complexes were formed (Scheme 43).79 Depending on the functionality (acid or ester), the complexes were soluble in water or in organic solvents, respectively. The structure of the free acid was confirmed by X-ray diffraction analysis.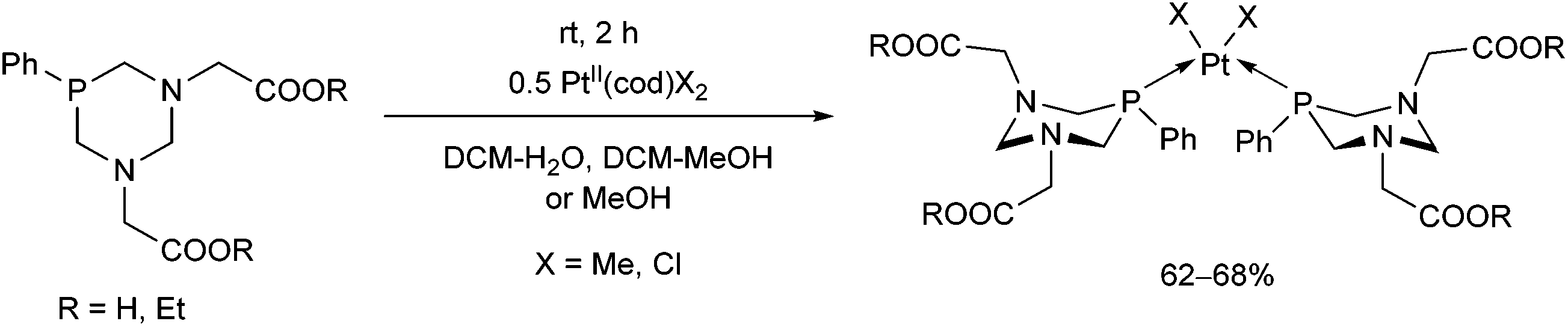 | ||
| Scheme 43 PtII complexes of 1,3-diaza-phosphacyclohexanes comprising amino acid or amino acid ester moieties. | ||
4.2. Complexation of 7-membered ring derivatives of α-aminophosphines
Stereochemistry plays an important role in the chemistry of 7-membered ring derivatives of α-aminophosphines. According to the literature data, derivatives of 1-aza-3,6-diphosphacycloheptanes were applied as ligands in the complexations. Isomers of these heterocycles differ from each other in respect of chelating abilities.80 Only the meso (RS) form contains the two P atoms in the appropriate position suitable for complexation, while in the optically active derivatives (RR and SS), the lone pairs are present at different sides of the 7-membered ring (Scheme 44).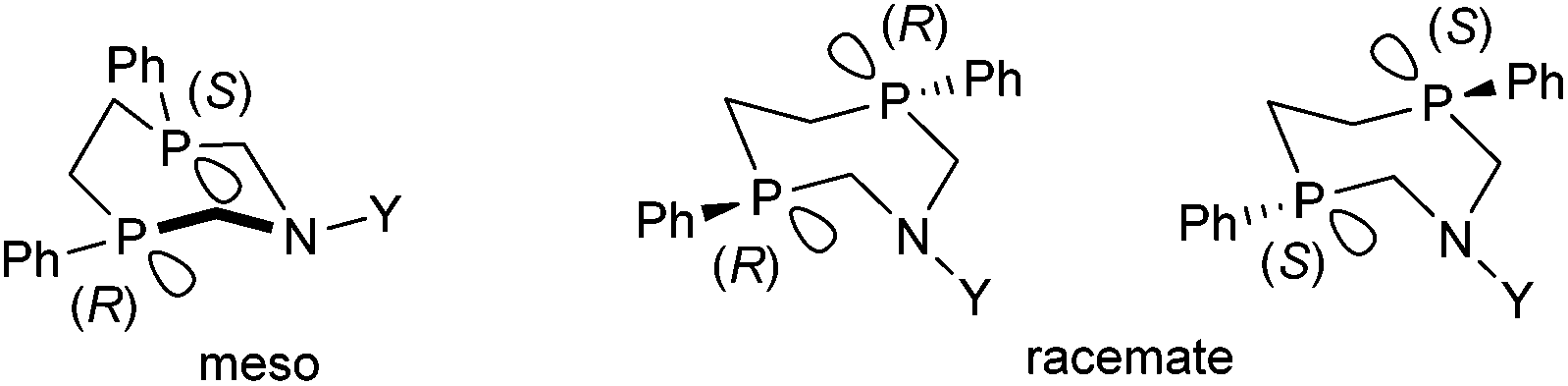 | ||
| Scheme 44 Isomers of 1-aza-3,6-diphosphacycloheptanes. | ||
The meso form of chiral 1-aza-3,6-diphosphacycloheptanes was reacted with PtII(cod)Cl2 at ambient temperature to afford the corresponding cis PtII complexes in yields of 55–63% (Scheme 45).80
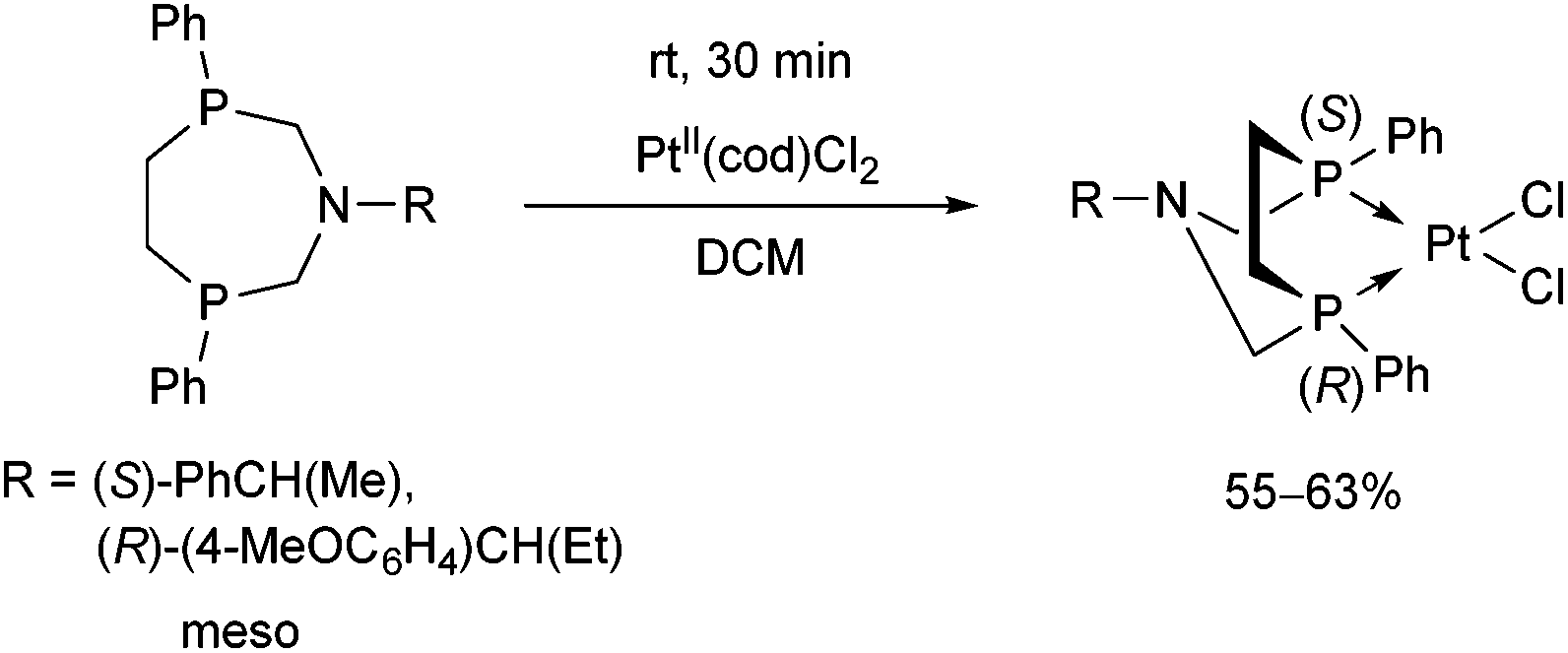 | ||
| Scheme 45 The reaction of meso 1-aza-3,6-diphosphacycloheptanes with PtII(cod)Cl2. | ||
In a similar reaction, by the use of a half equivalent of the PtII precursor, an ionic complex comprising two bidentate cyclic α-aminophosphines was formed (Scheme 46).80 According to the previous explanation, starting from the pure meso isomer, or the mixture of the meso and the racemate, only the complexes incorporating the meso isomer were formed. This method was applicable to separate the racemate from the mixture.
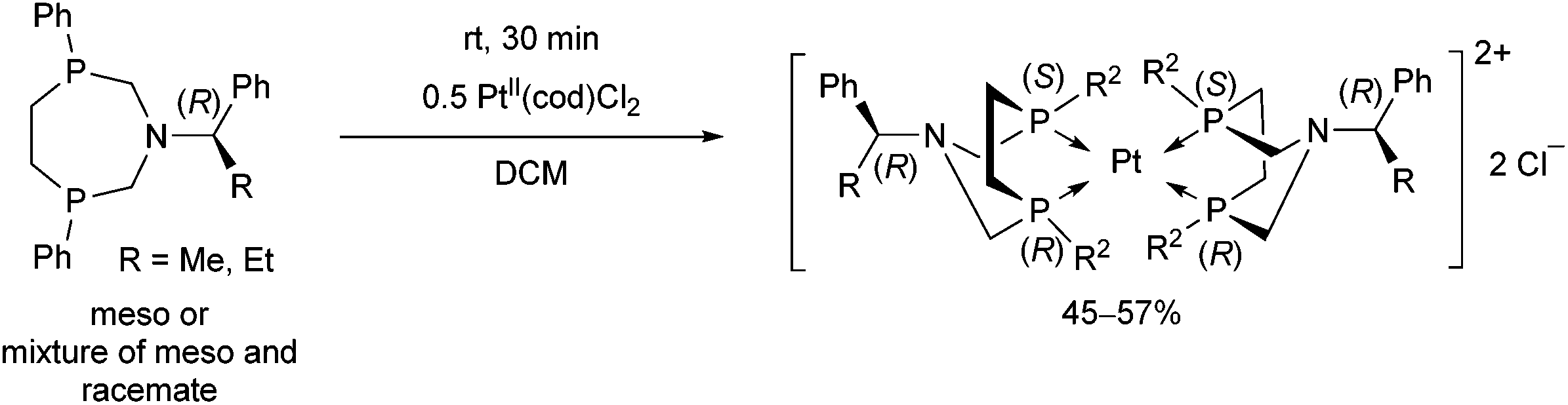 | ||
| Scheme 46 Isolation of the meso isomer by complexation. | ||
A similar aminophosphine containing a benzhydryl substituent on the N atom acted similarly in the complexation (Scheme 47).81 Starting from the mixture of the meso and the racemate forms, only the complexes of the meso form were obtained. The teracoordinated complex was formed in cis and trans forms in a ratio of 78:22.
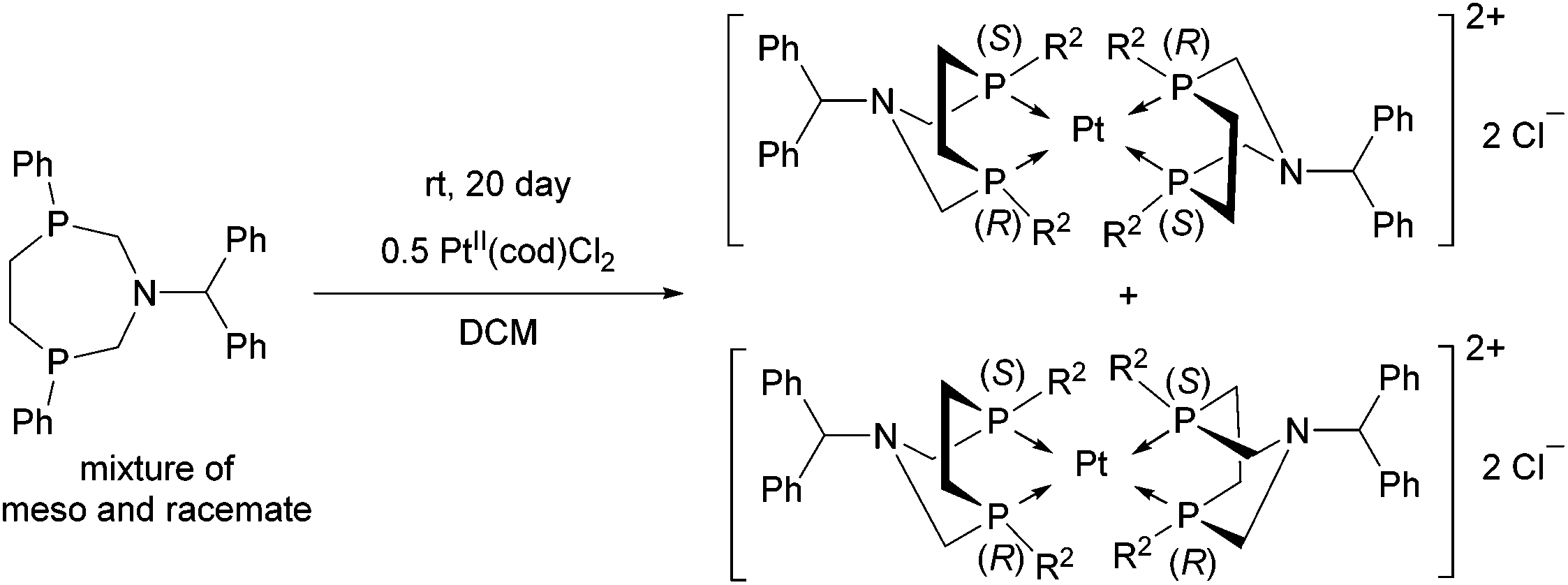 | ||
| Scheme 47 Application of an N-benzhydryl cyclic aminophosphine as a bidentate ligand in PtII complexes. | ||
4.3. Complexation of 8-membered ring derivatives of α-aminophosphines
Novel PtII complexes were prepared by the reaction of 1-aza-3,7-diphosphacyclooctanes with PtII(cod)Cl2 in DMF as the solvent (Scheme 48).82 The structure of the products was confirmed by X-ray analysis.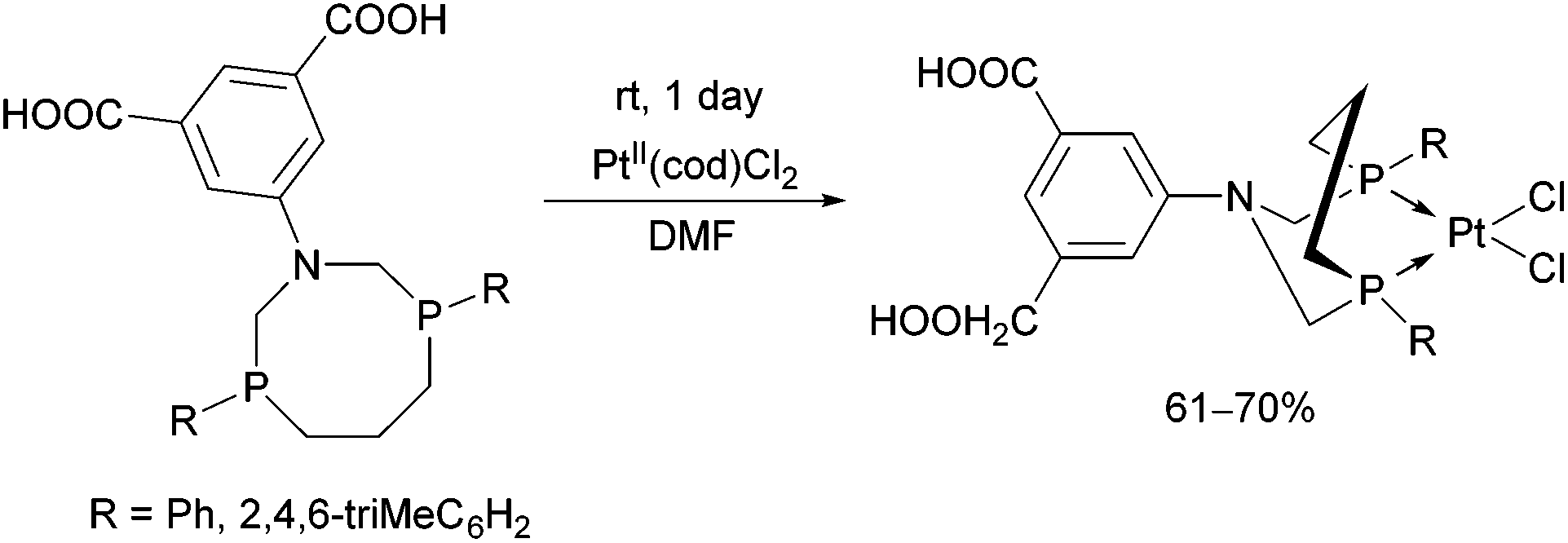 | ||
| Scheme 48 PtII complexes formed from 1-aza-3,7-diphosphacyclooctanes and PtII(cod)Cl2. | ||
Mostly MII(cod)Cl2 (M = Pt, Pd) served as the precursor in the complexation of P- and N-substituted 1,5-diaza-3,7-diphosphacyclooctanes (Table 13). The Ph- and Bn-substituted derivatives were obtained in yields of 79–90% after a reaction time of 4 h (Table 13/entry 1). Both the PtII and PdII complexes of N-4-MeC6H4- and P-menthyl 1,5-diaza-3,7-diphosphacyclooctanes were prepared in yields of 45–46% (Table 13/entry 2). The reaction was also performed starting from an optically active bidentate ligand. The corresponding PdII complex was synthesized in a yield of 50% (Table 13/entry 3). A cyclic aminophosphine bearing a 2-pyridyl substituent on the N atom was also tried out in the complexation (Table 13/entry 4).
Based on X-ray diffraction measurements,84,86 NMR studies83,84 and quantum chemical calculations,83 PtII and PdII complexes of 1,5-diaza-3,7-diphosphacyclooctanes contained two 6-membered metallocycles, one in chair, while the other in boat conformation. As a relevant example, the X-ray structure of [1,5-di(p-tolyl)-3,7-di(2-pyridyl)-1,5-diaza-3,7-diphosphacycylo-octane]PdCl2 (Table 11/entry 4) can be seen in Fig. 11.
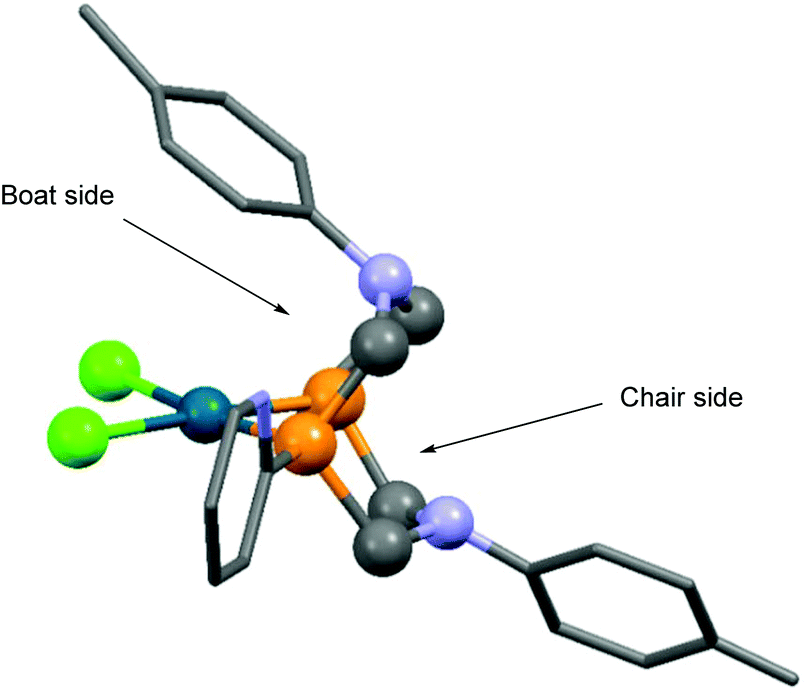 | ||
| Fig. 11 X-Ray structure of [1,5-di(p-tolyl)-3,7-di(2-pyridyl)-1,5-diaza-3,7-diphosphacycylooctane]PdCl2, [CCDC 932996].86 | ||
The 1,5-diphenyl-3,7-dicyclohexyl-1,5-diaza-3,7-diphospha-cyclooctane PdII complex was synthesized by the reaction of the corresponding cyclic α-aminophosphine with Pd(OAc)2 (Scheme 49).87 The in situ prepared PdII complex was proved to be an efficient catalyst in the Suzuki–Miyaura coupling of aryl bromides and arylboronic acids (Scheme 50).
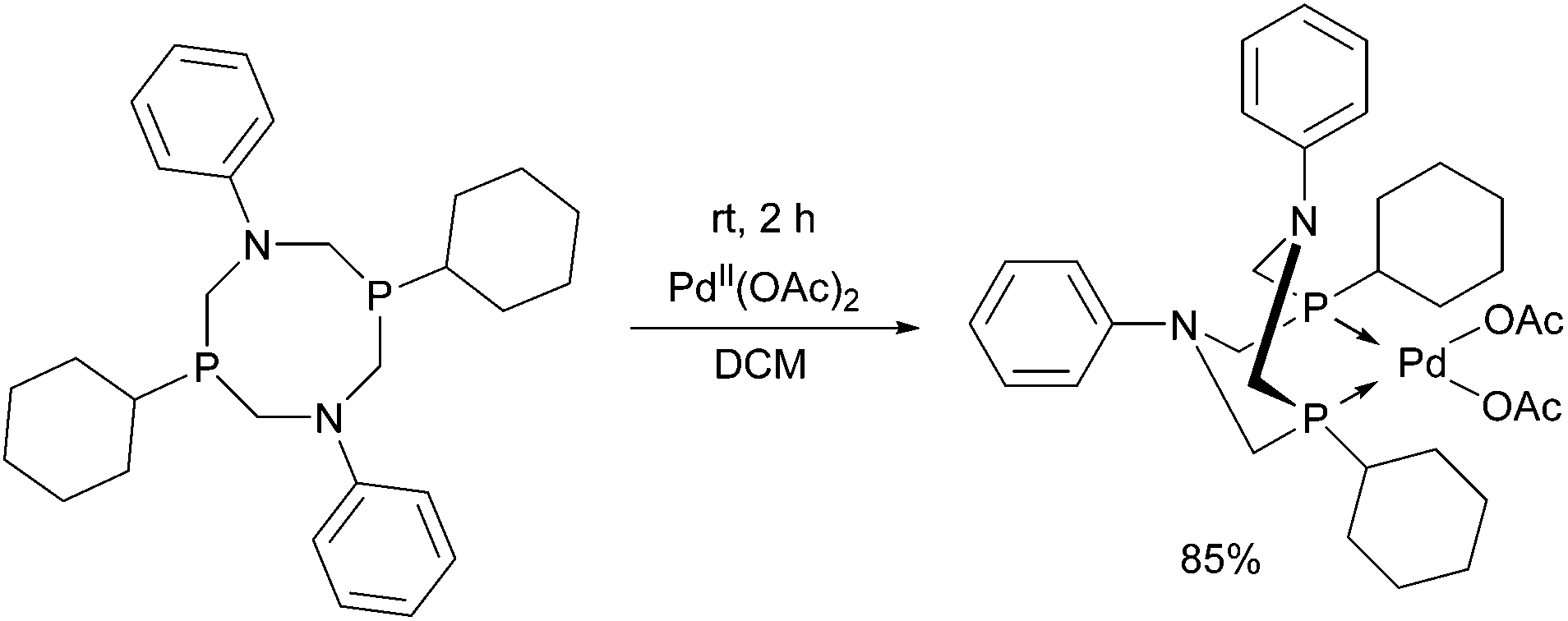 | ||
| Scheme 49 PdII complex of 1,5-diphenyl-3,7-dicyclohexyl-1,5-diaza-3,7-diphosphacycylooctane. | ||
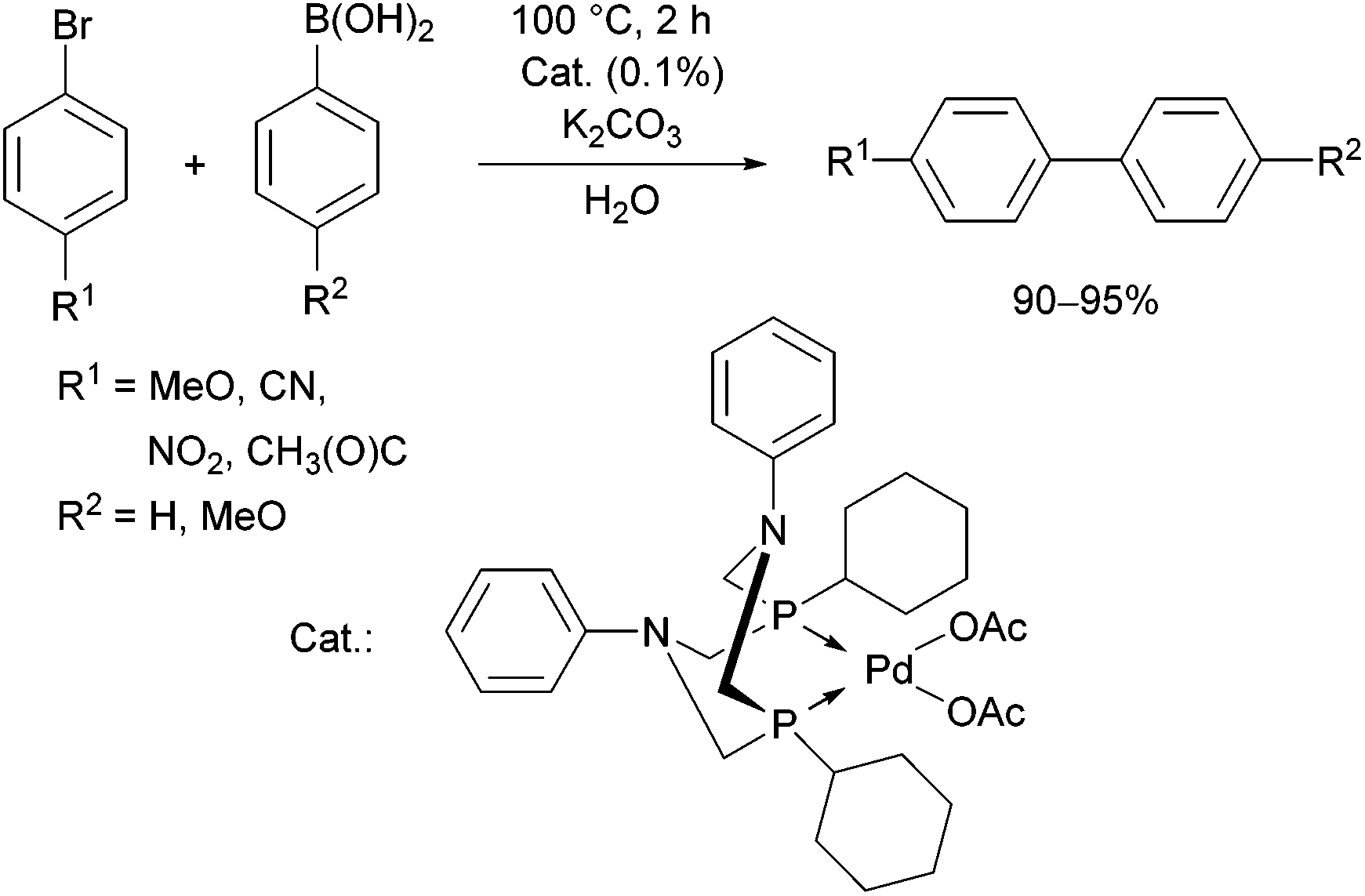 | ||
| Scheme 50 The Suzuki–Miyaura reaction of aryl bromides and arylboronic acids in the presence of the in situ formed complex (1,5-diphenyl-3,7-dicyclohexyl-1,5-diaza-3,7-diphosphacycylo-octane)Pd(OAc)2. | ||
Tetracoordinated ionic PtII and PdII complexes were synthesized by the reaction of 1,5-diaza-3,7-diphosphacyclooctanes with a half equivalent of the metal precursors (Table 14). Bis[(1,3,5,7-tetrabenzyl-1,5-diaza-3,7-diphosphacyclooctane)]PtCl2 was prepared from the 8-membered α-aminophosphine and PtII(cod)Cl2 using DCM as the solvent (Table 14/entry 1). Alkyl- and aryl-substituted cyclic bidentate ligands were reacted with PdII(PhCN)2(BF4)2 in acetonitrile to afford the corresponding complexes in yields of 66–81% (Table 14/entry 2). The products were tested as catalysts in electrochemical proton reduction, but showed lower activity than the similar NiII complexes. Optically active 1,5-diaza-3,7-diphosphacyclooctanes were also utilized as ligands to afford PtII and PdII complexes in good yields (88–91%) (Table 14/entry 3).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | M precursor | Solvent | t | Yield [%] | Ref. |
| 1 | Bn | Bn | PtII(cod)Cl2 | DCM | 12 h | 76 | 83 |
| 2 | Bn, Ph | Me, cHex, Ph | PdII(PhCN)2(BF4)2 | MeCN | 12 h | 66–81 | 88 |
| 3 | (S)-PhCH(Me), (R)-PhCH(Me) | Ph | MII(cod)Cl2 (M = Pt, Pd) | DCM | 1 day | 88–91 | 85 |
| 4 | PhCH(Me), 4-MeC6H4 | 2-Pyridyl | MII(cod)Cl2 (M = Pt, Pd) | DCM | 30 min–1 day | 55–92 | 86 |
Starting from derivatives with 2-pyridyl substituents on the P atom, ionic tetracoordinated complexes comprising PtII or PdII were prepared using MII(cod)Cl2 (M = Pt, Pd) as the precursor. The reaction times were in the range of 30 min to 1 day (Table 14/entry 4). The structure of the products was proved by X-ray measurements.
Rhodium(I) complexes of alkyl- and aryl-substituted 1,5-diaza-3,7-diphosphacyclooctanes were also synthesized by reacting the corresponding bidentate α-aminophosphines with [RhI(cod)BF4]2 (Scheme 51).89 The complexes were tested as catalysts in the reaction of CO2 with hydrogen and showed moderate catalytic activities.
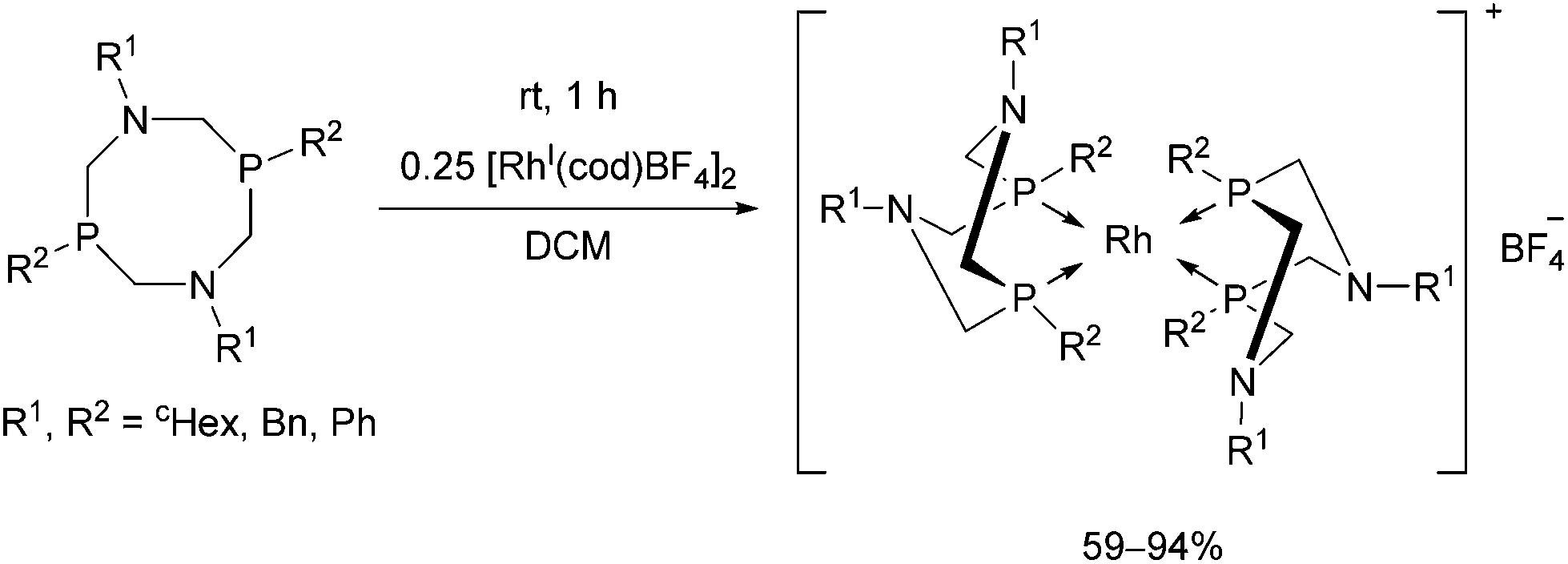 | ||
| Scheme 51 Rhodium(I) complexes of 1,5-diaza-3,7-diphosphacyclooctanes. | ||
4.4. General methods for the preparation of complexes containing cyclic α-aminophosphines as ligands
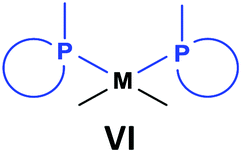
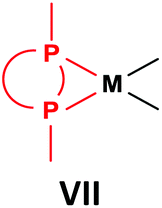
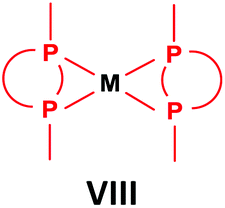
General methods for the preparation of Pt, Pd and Rh complexes incorporating cyclic α-aminophosphines as the ligands are summarized in Table 15. The reactions may be carried out at ambient temperature using DCM as the solvent. Pt complexes of monodentate cyclic α-aminophosphines (VI) may be synthesized using 0.5 equivalents of PtII(cod)X2 (X = Me, Cl) as the precursor. For the synthesis of Pt and Pd complexes of bidentate ligands (VII), MII(cod)Cl2 (M = Pt, Pd) was the most suitable reagent. The products (VII) were obtained in yields of 55–92% after a reaction time of 30 min–1 day. The complexes having two α-aminophosphines as the ligands (VIII) may be synthesized using 0.5 equivalents of MII(cod)Cl2 (M = Pt, Pd) or 0.25 equivalents of [RhI(cod)BF4]2 as the metal precursor in moderate to excellent yields.| Types of complexes | M | Precursor | t | Average yield [%] |
|---|---|---|---|---|
|
|
||||
|
Pt | 0.5 PtII(cod)X2, X = Me, Cl | 2 h | 62–68 |
|
Pt | PtII(cod)Cl2 | 30 min–4 h | 55–90 |
| Pd | PdII(cod)Cl2 | 30 min–1 day | 46–92 | |
|
Pt | 0.5 PtII(cod)Cl2 | 30 min–1 day | 45–92 |
| Pd | 0.5 PdII(cod)Cl2 | 30 min–1 day | 55–88 | |
| Rh | 0.25 [RhI(cod)BF4]2 | 1 h | 59–94 |

5. Conclusions
The utilization of α-aminophosphine ligands in platinum, palladium and rhodium complexes came into the field of vision of organometallic chemists in the 1980s. Since then, this topic has been receiving growing attention; nearly half of the papers have been published in the last five years. To make available α-aminophosphines with different properties, various linear and cyclic ligands were prepared. They were applied as monodentate and bidentate ligands in transition metal complexes. In this review, we have summarized the synthesis of platinum, palladium and rhodium complexes incorporating α-aminophosphines. Furthermore, the structure and utilization of a few relevant derivatives were also discussed. Several complexes described above revealed significant activities as catalysts in hydrogenation, cross-coupling, hydroformylation, allylation or carbonylation. In a few cases, the catalytic activity of α-aminophosphine-based complexes was compared to that of complexes of alkyl and aryl phosphines, where the α-aminophosphine derivatives were found to be more efficient. Besides discussing the literature data, the most commonly applied transition metal precursors and reaction conditions for the synthesis of the different types of α-aminophosphine-M (M = Pt, Pd or Rh) complexes were also summarized. Based on these tendencies, this field of organometallic chemistry will attract further attention in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Hungarian Research Development and Innovation Fund (FK123961 and K119202), and in part (E. B.) by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (BO/00278/17/7). Ádám Tajti thanks Gedeon Richter Talentum Foundation and Pro Progressio Foundation for financial support.Notes and references
- M. M. Pereira, M. J. F. Calvete, R. M. B. Carrilho and A. R. Abreu, Chem. Soc. Rev., 2013, 42, 6990–7020 RSC.
- P. W. N. M. van Leeuwen and P. C. J. Kamer, Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, John Wiley & Sons Ltd, Chichester, UK, 2012 Search PubMed.
- J. Tsuji, Transition Metal Reagents and Catalysts, John Wiley & Sons Ltd, Chichester, UK, 2002 Search PubMed.
- M. Beller and C. Bolm, Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals, John Wiley & Sons Ltd, Weinheim, 2004 Search PubMed.
- L. Kollár and G. Keglevich, Chem. Rev., 2010, 110, 4257–4302 CrossRef PubMed.
- K. Burgess, W. A. van der Donk, C.-H. Jun and Y. J. Park, Chlorotris(triphenylphosphine)-rhodium(I), e-EROS Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons Ltd, Chichester, UK, 2006 Search PubMed.
- P. W. N. M. van Leeuwen, Homogeneous Catalysis, Springer, Dordrecht, Netherlands, 2004 Search PubMed.
- E. A. Sheddon and K. R. Sheddon, The Chemistry of Ruthenium, Elsevier, Amsterdam, Oxford, New York, Tokyo, 1984 Search PubMed.
- W. P. Griffith, Ruthenium Oxidation Complexes, Vol. 34, Springer, Dordrecht, New York, Heidelberg, London, 2011 Search PubMed.
- S. Sabo-Etienne and M. Grellier, Ruthenium: Inorganic & Coordination Chemistry, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons Ltd, 2006 Search PubMed.
- C. Bruneau and P. H. Dixneuf, Ruthenium Catalysts and Fine Chemmistry, Springer, Berlin, Heidelberg, New York, 2004 Search PubMed.
- S. J. Berners-Price and P. J. Sadler, Phosphines and metal phosphine complexes: Relationship of chemistry to anticancer and other biological activity, in Bioinorg. Chem, Springer, Berlin, 1988, vol. 70, pp. 27–102 Search PubMed.
- A. A. Nazarov and P. J. Dyson, in Phosphorus Compounds, vol 37, ed. G. Luca and M. Peruzzini, Springer, Dordrecht, 2011, pp. 445–461 Search PubMed.
- U. Ndagi, N. Mhlongo and M. E. Soliman, Drug Des., Dev. Ther., 2017, 11, 599–616 CrossRef PubMed.
- V. A. Stepanova and I. P. Smoliakova, Curr. Org. Chem., 2012, 16, 2893–2920 CrossRef CAS.
- W. Li and J. Zhang, Chem. Soc. Rev., 2016, 45, 1657–1677 RSC.
- J. Gopalakrishnan, Appl. Organomet. Chem., 2009, 23, 291–318 CrossRef CAS.
- L. A. Labios, C. J. Weiss, J. D. Egbert, S. Lense, R. M. Bullock, W. G. Dougherty, W. S. Kassel and M. T. Mock, Z. Anorg. Allg. Chem., 2015, 641, 105–117 CrossRef CAS.
- N. Priyadarshani, B. Ginovska, J. T. Bays, J. C. Linehan and W. J. Shaw, Dalton Trans., 2015, 44, 14854–14864 RSC.
- J.-F. Zhang, W.-F. Fu, X. Gan and J.-H. Chen, Dalton Trans., 2008, 3093–3100 RSC.
- A. Hazari, J. A. Labiger and J. E. Bercaw, Angew. Chem., Int. Ed., 2012, 51, 8268–8271 CrossRef CAS PubMed.
- R. N. Naumov, E. I. Musina, K. B. Kanunnikov, T. I. Fesenko, D. B. Krivolapov, I. A. Litvinov, P. Lönnecke, E. Hey-Hawkins, A. A. Karasik and O. G. Sinyashin, Dalton Trans., 2014, 43, 12784–12789 RSC.
- R. N. Naumov, A. A. Karasik, O. G. Sinyashin, P. Lönnecke and E. Hey-Hawkins, Dalton Trans., 2004, 357–358 RSC.
- A. Phanopoulos, N. J. Brown, A. J. P. White, N. J. Long and P. W. Miller, Inorg. Chem., 2014, 53, 3742–3752 CrossRef CAS PubMed.
- A. Phanopoulos, A. J. P. White, N. J. Long and P. W. Miller, Dalton Trans., 2016, 45, 5536–5548 RSC.
- C. D. Swor, K. R. Hanson, L. N. Zakharov and D. R. Tyler, Dalton Trans., 2011, 40, 8604–8610 RSC.
- M. Płotek, R. Starosta, U. K. Komarnicka, A. Skórska-Stania, M. Jeżowska-Bojczuk, G. Stochel and A. Kyzioł, Dalton Trans., 2015, 44, 13969–13978 RSC.
- P. Das, M.-H. Ho, M. O'Hagan, W. J. Shaw, R. Morris Bullock, S. Raugei and M. L. Helm, Dalton Trans., 2014, 43, 2744–2754 RSC.
- E. Bálint, E. Fazekas, P. Pongrácz, L. Kollár, L. Drahos, T. Holczbauer, M. Czugler and G. Keglevich, J. Organomet. Chem., 2012, 717, 75–82 CrossRef.
- E. Bálint, A. Tripolszky, E. Jablonkai, K. Karaghiosoff, M. Czugler, Z. Mucsi, L. Kollár, P. Pongrácz and G. Keglevich, J. Organomet. Chem., 2016, 801, 111–121 CrossRef.
- B. B.-N. Ben-Aroya and M. Portnoy, J. Comb. Chem., 2001, 3, 524–527 CrossRef CAS PubMed.
- B. B.-N. Ben-Aroya and M. Portnoy, Tetrahedron, 2002, 58, 5147–5158 CrossRef.
- A. Mansour and M. Portnoy, Tetrahedron Lett., 2003, 44, 2195–2198 CrossRef CAS.
- S. J. Coles, S. E. Durran, M. B. Hursthouse, A. M. Z. Slawin and M. B. Smith, New J. Chem., 2001, 25, 416–422 RSC.
- S. E. Durran, M. B. Smith, A. M. Z. Slawin, T. Gelbrich, M. B. Hursthouse and M. E. Light, Can. J. Chem., 2001, 79, 780–791 CrossRef CAS.
- I. Angurell, E. Puig, O. Rossell, M. Seco, P. Gómez-Sal and A. Martín, J. Organomet. Chem., 2012, 716, 120–128 CrossRef CAS.
- S. E. Durran, M. B. Smith, S. H. Dale, S. J. Coles, M. B. Hursthouse and M. E. Light, Inorg. Chim. Acta, 2006, 359, 2980–2988 CrossRef CAS.
- S. E. Durran, M. B. Smith, A. M. Z. Slawin and J. W. Steed, J. Chem. Soc., Dalton Trans., 2000, 2771–2778 RSC.
- O. Altan, O. Serindag, K. Sayın and D. Karakas, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 993–999 CrossRef CAS.
- M. L. Clarke, D. J. Cole-Hamilton, D. F. Foster, A. M. Z. Slawin and J. D. Woollins, J. Chem. Soc., Dalton Trans., 2002, 1618–1624 RSC.
- D. L. Davies, J. Neild, L. J. S. Prouse and D. R. Russell, Polyhedron, 1993, 12, 2121–2124 CrossRef CAS.
- M. L. Clarke, A. M. Z. Slawin and J. D. Woollins, Polyhedron, 2003, 22, 19–26 CrossRef CAS.
- J. Pfeiffer, G. Kickelbick and U. Schubert, Organometallics, 2000, 19, 62–71 CrossRef CAS.
- Q.-S. Li, F.-B. Xu, D.-J. Cui, K. Yu, X.-S. Zeng, X.-S. Leng, H.-B. Song and Z.-Z. Zhang, Dalton Trans., 2003, 1551–1557 RSC.
- D. A. Clarke, P. W. Miller, N. J. Long and A. J. P. White, Dalton Trans., 2007, 4556–4564 RSC.
- N. W. Waggoner, L. S. Spreer, B. J. Boro, D. L. DuBois and M. L. Helm, Inorg. Chim. Acta, 2012, 380, 14–21 CrossRef CAS.
- R. Starosta, A. Bykowska, M. Barys, A. K. Wieliczko, Z. Staroniewicz and M. Jeżowska-Bojczuk, Polyhedron, 2011, 30, 2914–2921 CrossRef CAS.
- Q. Zhang, S. M. Aucott, A. M. Z. Slawin and J. D. Woollins, Eur. J. Inorg. Chem., 2002, 1635–1646 CrossRef CAS.
- P. Espinet and K. Soulantica, Coord. Chem. Rev., 1999, 193–195, 499–556 CrossRef CAS.
- D. Brackemeyer, C. Schulte to Brinke, F. Roelfes and F. E. Hahn, Dalton Trans., 2017, 46, 4510–4513 RSC.
- E. Payet, A. Auffrant, X. F. Le Goff and P. Le Floch, J. Organomet. Chem., 2010, 695, 1499–1506 CrossRef CAS.
- R. Turpin, P. Dagnac and R. Poilblanc, J. Organomet. Chem., 1987, 319, 247–255 CrossRef CAS.
- A. L. Balch, M. M. Olmstead and S. P. Rowley, Inorg. Chim. Acta, 1990, 168, 255–264 CrossRef CAS.
- H. Han, M. Elsmaili and S. A. Johnson, Inorg. Chem., 2006, 45, 7435–7445 CrossRef CAS PubMed.
- G. Keglevich, A. Szekrényi, Á. Szöllősy and L. Drahos, Synth. Commun., 2011, 41, 2265–2272 CrossRef CAS.
- M. Stickel, C. Maichle-Moessmer, L. Wesemann and H. A. Mayer, Polyhedron, 2013, 52, 1471–1480 CrossRef CAS.
- E. Bálint, Á. Tajti, D. Kalocsai, B. Mátravölgyi, K. Karaghiosoff, M. Czugler and G. Keglevich, Tetrahedron, 2017, 73, 5659–5667 CrossRef.
- M. Keles, Z. Aydin and O. Serindag, J. Organomet. Chem., 2007, 692, 1951–1955 CrossRef CAS.
- M. Keles and M. K. Yilmaz, Heteroat. Chem., 2012, 23, 466–471 CrossRef CAS.
- S. Vigo, R. Andrés, P. Gómez-Sal, J. de la Mata and E. de Jesús, J. Organomet. Chem., 2012, 717, 88–98 CrossRef CAS.
- R.-T. He, J.-F. Wang, H.-F. Wang, Z.-G. Ren and J.-P. Lang, Dalton Trans., 2014, 43, 9786–9794 RSC.
- J.-J. Ning, J.-F. Wang, Z.-G. Ren, D. J. Young and J.-P. Lang, Tetrahedron, 2015, 71, 4000–4006 CrossRef CAS.
- M. R. J. Elsegood, M. B. Smith and P. M. Staniland, Inorg. Chem., 2006, 45, 6761–6770 CrossRef CAS PubMed.
- S. E. Durran, M. R. J. Elsegood and M. B. Smith, New J. Chem., 2002, 26, 1402–1408 RSC.
- Q. Z. Zhang, G. X. Hua, P. Bhattacharyya, A. M. Z. Slawin and J. D. Woollins, Eur. J. Inorg. Chem., 2003, 2426–2437 CrossRef CAS.
- O. Serindag, Synth. React. Inorg. Met.-Org. Chem., 1995, 25, 327–335 CrossRef CAS.
- X. Ma and X. Fu, J. Mol. Catal. A: Chem., 2003, 195, 47–53 CrossRef CAS.
- K. Motokura, K. Saitoh, H. Noda, Y. Uemura, W.-J. Chun, A. Miyaji, S. Yamaguchi and T. Baba, ChemCatChem, 2016, 8, 331–335 CrossRef CAS.
- K. Motokura, K. Saitoh, H. Noda, W.-J. Chun, A. Miyaji, S. Yamaguchi and T. Baba, Catal. Sci. Technol., 2016, 6, 5380–5388 CAS.
- D. Méry, K. Heuzé and D. Astruc, Chem. Commun., 2003, 1934–1935 RSC.
- J. Fawcett, R. D. W. Kemmitt, D. R. Russell and O. Serindag, J. Organomet. Chem., 1995, 486, 171–176 CrossRef CAS.
- M. R. J. Elsegood, A. J. Lake, R. J. Mortimer, M. B. Smith and G. W. Weaver, J. Organomet. Chem., 2008, 693, 2317–2326 CrossRef CAS.
- P. W. Miller, N. J. Long and A. J. P. White, Dalton Trans., 2009, 5284–5286 RSC.
- G. M. Brown, M. R. J. Elsegood, A. J. Lake, N. M. Sanchez-Ballester, M. B. Smith, T. S. Varley and K. Blann, Eur. J. Inorg. Chem., 2007, 1405–1414 CrossRef CAS.
- A.-X. Zheng, Z.-G. Ren, H.-F. Wang, H.-X. Li and J.-P. Lang, Inorg. Chim. Acta, 2012, 382, 43–51 CrossRef CAS.
- C. J. Curtis, A. Miedaner, J. W. Raebiger and D. L. Dubois, Organometallics, 2004, 23, 511–516 CrossRef CAS.
- M. C. Román-Martínez, J. A. Díaz-Auñón, C. Salinas-Martínez de Lecea and H. Alper, J. Mol. Catal. A: Chem., 2004, 213, 177–182 CrossRef.
- K. Kellner, W. Hanke, A. Tzschach, Z. Nagy-Magos and L. Markó, J. Organomet. Chem., 1984, 268, 175–183 CrossRef CAS.
- A. Jain, M. L. Helm, J. C. Linehan, D. L. DuBois and W. J. Shaw, Inorg. Chem. Commun., 2012, 22, 65–67 CrossRef CAS.
- E. I. Musina, A. A. Karasik, A. S. Balueva, I. D. Strelnik, T. I. Fesenko, A. B. Dobrynin, T. P. Gerasimova, S. A. Katsyuba, O. N. Kataeva, P. Lönnecke, E. Hey-Hawkins and O. G. Sinyashin, Eur. J. Inorg. Chem., 2012, 1857–1866 CrossRef CAS.
- A. Plikhta, A. Pöthig, E. Herdtweck and B. Rieger, Inorg. Chem., 2015, 54, 9517–9528 CrossRef CAS PubMed.
- A. A. Karasik, R. N. Naumov, Y. S. Spiridonova, O. G. Sinyashin, P. Lönnecke and E. Hey-Hawkins, Z. Anorg. Allg. Chem., 2007, 633, 205–210 CrossRef CAS.
- S. Latypov, A. Strelnik, A. Balueva, Y. Spiridonova, A. Karasik and O. Sinyashin, Eur. J. Inorg. Chem., 2016, 1068–1084 CrossRef CAS.
- S. N. Ignatieva, A. S. Balueva, A. A. Karasik, S. K. Latypov, A. G. Nikonova, O. E. Naumova, P. Lönnecke, E. Hey-Hawkins and O. G. Sinyashin, Inorg. Chem., 2010, 49, 5407–5412 CrossRef CAS PubMed.
- A. A. Karasik, R. N. Naumov, O. G. Sinyashin, G. P. Belov, H. V. Novikova, P. Lönnecke and E. Hey-Hawkins, Dalton Trans., 2003, 2209–2214 RSC.
- E. I. Musina, V. V. Khrizanforova, I. D. Strelnik, M. I. Valitov, Y. S. Spiridonova, D. M. Krivolapov, I. A. Litvinov, M. K. Kadirov, P. Lönnecke, E. Hey-Hawkins, Y. H. Budnikova, A. A. Karasik and O. G. Sinyashin, Chem. – Eur. J., 2014, 20, 3169–3182 CrossRef CAS PubMed.
- A. Fihri, D. Luart, C. Len, A. Solhy, C. Chevrin and V. Polshettiwar, Dalton Trans., 2011, 40, 3116–3121 RSC.
- C. S. Seu, D. Ung, M. D. Doud, C. E. Moore, A. L. Rheingold and C. P. Kubiak, Organometallics, 2013, 32, 4556–4563 CrossRef CAS.
- A. M. Lilio, M. H. Reineke, C. E. Moore, A. L. Rheingold, M. K. Takase and C. P. Kubiak, J. Am. Chem. Soc., 2015, 137, 8251–8260 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |