
Syntheses, structures and magnetic properties of a series of mono- and di-nuclear dysprosium(III)-crown-ether complexes: effects of a weak ligand-field and flexible cyclic coordination modes†
You-Song
Ding
,
Tian
Han
,
Yue-Qiao
Hu
,
Minwei
Xu
,
Sen
Yang
and
Yan-Zhen
Zheng
*
Frontier Institute of Science and Technology (FIST), State Key Laboratory for Mechanical Behavior of Materials and MOE Key Laboratory for Nonequilibrium Synthesis and Modulation of Condensed Mater, Xi'an Jiaotong University, Xi'an 710054, China. E-mail: zheng.yanzhen@xjtu.edu.cn
First published on 22nd February 2016
Abstract
Crown ethers, which feature circular and variable coordination sites of oxygen atoms, may be ideal for the symmetry control of the mononuclear lanthanide single-molecule magnets (SMMs) by double-deckering the central metal ions. Herein, a series of Dy(III)-crown-ether complexes have been prepared to test this hypothesis. With a 12-crown-4 ether (12-C-4), a half-sandwich complex Dy(12-C-4)(NO3)3 (1) was obtained, while by changing the counter-anions the double-deck-like complex Dy(12-C-4)2(CH3CN)(ClO4)3 (2) could be prepared. On further adjusting the reaction conditions, a dinuclear complex Dy2(12-C-4)2(ClO4)4(OH)2(H2O)2 (3) was isolated. With the aid of a 15-crown-5 ether (15-C-5), another complex [Dy(12-C-4)(15-C-5)(CH3CN)][Dy(12-C-4)(15-C-5)]2(CH3CN)2(ClO4)9 (4) with a double-decker structure was formed. For the 18-crown-6 ether (18-C-6), two complexes [Dy(18-C-6)(NO3)2]ClO4 (5) and [Dy(18-C-6)(NO3)2]BPh4 (6) have been synthesized using different charge balancing anions. Interestingly, the cationic components in complexes 5 and 6 are isomeric. The only difference is the dihedral angles of the two chelating NO3− groups, namely 90° and 68° for 5 and 6. Systematic magnetic studies reveal that complexes 1–6 show no SMM behaviour in a zero dc field above 2 K, which is probably due to the ligand field provided by crown ethers is too weak to have much impact on the electronic structures of the central Dy(III) ions. However, when the Dy(III) ions are simultaneously coordinated by some counter-anions such as NO3− and OH−, apparent field-induced SMM behaviors can be observed in complexes 1, 3, 5 and 6. Moreover, we found that the axial coordination anions in complexes 5 and 6 have evident impact on the magnetic relaxation behaviours. For the 90° case (complex 5) the energy barrier for magnetisation reversal (ΔE) is 63 K, which is much higher than 43 K for the 68° case (complex 6).
 Yan-Zhen Zheng | Yan-Zhen Zheng was born and raised in Guangdong, China. He received his PhD degree in 2007 under Xiao-Ming Chen and Ming-Liang Tong (Sun Yat-Sen University) for studying low-dimensional magnetic coordination polymers. Yan-Zhen then moved to Europe with the support of Alexander von Humboldt Fellowship (under Annie Powell in Karlsruhe Institute of Technology) and Marie Curie International Incoming Fellowship (under Richard Winpenny in the University of Manchester) for advanced molecular magnetism study. During this period he had a short postdoctoral training for spintronics in Forschungszentrum Jülich (under Paul Kögerler in RWTH Aachen University). In 2012, he was selected as a “National Young 1000-Plan Scholar” and appointed as a professor at Xi'an Jiaotong University, where he started his own adventure in molecular magnetism research. Currently, his research topics mainly include the design and synthesis of low-coordinate mononuclear single-molecule magnets, magnetic refrigerants based on highly-symmetric lanthanide clusters, geometrically spin-frustrated magnets with extended lattices and molecular spintronics comprising of electron-delocalised complexes. |
Introduction
Because of the strong spin–orbital coupling effects, some lanthanide ions (e.g. Tb(III), Dy(III), Ho(III) and Er(III)) can generate huge magnetic anisotropy, and are widely used in the design of high performance single-molecule magnets (SMMs), a kind of nano-sized molecular magnetic material which is believed to have futuristic applications in information storage, quantum computing, and miniature spintronics.1–10 Among these lanthanide ions, the dysprosium(III) ion has particularly attracted a great deal of recent research interest due to the inherent strong spin–orbital coupling effect and hence a very high magnetic anisotropy of the 6H15/2 state.11–23 Although the unpaired 4f electrons of the lanthanide ions are well shielded by the outer 5d and 6s electrons, it is arguably believed that both the coordination geometry and the strength of the ligand field (LF) have great impact on the magnetic performances such as energy barriers for magnetization reversal (ΔE) and the blocking temperatures (TB) of the Dy(III)-SMMs.Specifically, theoretical and experimental studies corroborated that in axial symmetry environments, such as D∞h, S8, D4d, and D5h, some mononuclear lanthanide compounds often show high ΔE.12,24–28 In this regard, Tong et al. have given a good example, where they switched the anisotropy barrier from 469 K to a negligible value by changing the local symmetry for the Dy(III) ion from a pentagonal–bipyramid to an octahedron.28 However, there are also other theories that show ΔE of mononuclear Dy(III) SMMs is mainly determined by the effect of LF,29,30 which is also experimentally evidenced by Winpenny et al. with a Dy(III) bis(methanediide) SMM that shows a large magnetic anisotropy imposed by negatively-charged donor atoms.22
The most successful example that combines the symmetry fixation and a strong LF effect may arguably be the sandwich-type Ln(Pc)2 complexes (Pc = phthalocyanine or substituted phthalocyaninate ligands), because the rigid planar Pc ligands with four strong N donors help fixing the central lanthanide ions with ideal D4d symmetry.4,31 Notable results include a Tb(Pc)2 compound that shows a very high ΔE of 938 K.4 Moreover, other sandwich systems such as polyoxometallate (POM)32,33 and cyclooctatetraene (COT)26,27 also demonstrate the power of cyclic ligands in enhancing the performance of mononuclear SMMs.
Crown ethers, as one of the cyclic ligands with suitable metal binding sites may provide great opportunities for fixing mononuclear lanthanide complexes in axial symmetries, such as C4, C5 and C6 (Scheme 1). In particular, if the large coordination number requirement of the lanthanide ions can be fulfilled, a sandwiched structure may be obtained. Thus, new structures and magnetic properties of coordination complexes based on crown ethers have attracted much attention recently. For instance, the Co(II) ion in the complex [Co(12-C-4)2](I3)2(12-C-4) (12-C-4 = 12-crown-4 ether) is coordinated in a distorted square antiprism geometry and shows hysteresis loops below 1 K at a scan rate of 0.14 T s−1.34 Moreover, two mononuclear Dy(III) crown ether complexes [Dy(15-C-5)(H2O)4](ClO4)3(15-C-5)H2O and [Dy(12-C-4)(H2O)5](ClO4)3 (15-C-5 = 15-crown-5 ether) were also reported with half sandwich or pseudo-capped square antiprismatic geometries and field-induced magnetic relaxation properties, though quantum tunnelling of magnetization (QTM) in these systems is strong.35
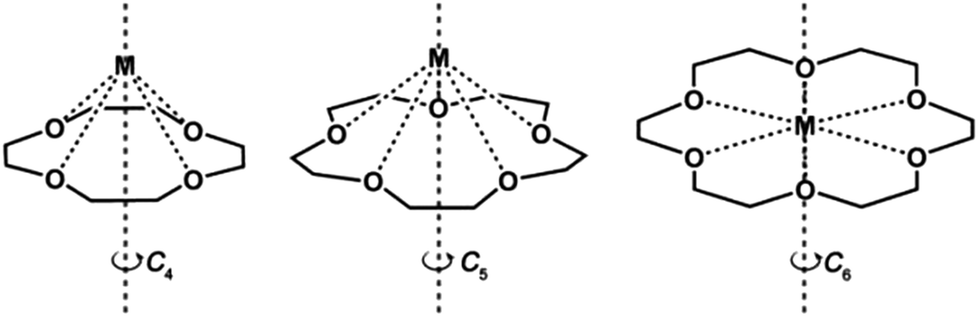 | ||
| Scheme 1 The coordination scheme for crown ethers to lanthanide ions. | ||
Herein, we used three crown ethers (12-C-4 = 12-crown-4 ether, 15-C-5 = 15-crown-5 ether and 18-C-6 = 18-crown-6 ether) to sandwich the Dy(III) ions and isolated six complexes, namely Dy(12-C-4)(NO3)3 (1), Dy(12-C-4)2(CH3CN)(ClO4)3 (2), Dy2(12-C-4)2(ClO4)4(OH)2(H2O)2 (3), [Dy(12-C-4)(15-C-5)(CH3CN)][Dy(12-C-4)(15-C-5)]2(CH3CN)2(ClO4)9 (4), [Dy(18-C-6)(NO3)2]ClO4 (5) and [Dy(18-C-6)(NO3)2]BPh4 (6). Their structures were determined by single-crystal X-ray diffraction, showing various coordination environments for Dy(III) ions. For 1, only one 12-C-4 ligand coordinates and shows a half-sandwich type structure, while for 2, two 12-C-4 and one CH3CN ligands bind, forming a double-deck-like structure. For 3, the central Dy(III) ions are in perfect D4d symmetric coordination environment and are bridged by μ-OH− ligands, forming the only di-nuclear complex. For 4, the central Dy(III) ion is sandwiched between a 15-C-5 and a 12-C-4 ligand, resulting in the only double-decker complex in this series of complexes. Due to the larger size of 18-C-6, the Dy(III) ions in complexes 5 and 6 sit in the centre of the 18-C-6 ring with two apical bidentate nitrate anions. Interestingly, the dihedral angles of the two chelating NO3− groups in complexes 5 and 6 are different, namely 90° and 68° for 5 and 6, respectively. Magnetic studies reveal field-induced magnetic relaxation behaviours in complexes (1, 3, 5 and 6) with coordinated anions such as NO3− and OH−, otherwise the magnetic relaxations are absent regardless of external fields. More importantly, we found that the dihedral angles between two axial coordinating NO3− anions in complexes 5 and 6 have a significant impact on the magnetic relaxation behaviors. For complex 5, the ΔE is 63 K, which is much higher than 43 K for complex 6.
Experimental
Materials and methods
All crown ethers and solvents were commercially available and used without further treatment. Lanthanide salts were prepared by the reaction of rare earth oxides with acid in water, followed by filtration and heating the filtrate to dryness. The FT-IR spectra were recorded from KBr pellets in the range of 400–4000 cm−1 on a Nicolet 6700 FT-IR spectrometer (Thermo Scientific Instrument).| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| Formula | C8H16DyN3O13 | C18H35Cl3DyNO20 | C16H42Cl4Dy2O30 | C60H117Cl9Dy3N3O63 | C12H24ClDyN2O16 | C36H44BDyN2O12 |
| F.w. | 524.74 | 854.32 | 1181.30 | 2695.12 | 650.28 | 870.04 |
| T (K) | 150(2) | 150(2) | 150(2) | 150(2) | 150(2) | 150(2) |
| Space group | P21/c | P43212 | P21/c | P21/c | Pnma | P212121 |
| a (Å) | 12.100(3) | 10.5373(7) | 8.6312(8) | 19.7259(6) | 24.3304(18) | 13.0630(10) |
| b (Å) | 8.5471(19) | 10.5373(7) | 19.9383(18) | 17.2456(6) | 10.8059(8) | 14.8712(11) |
| c (Å) | 15.236(3) | 26.5599(18) | 11.2062(10) | 29.0850(9) | 8.3756(6) | 18.7622(14) |
| α (°) | 90 | 90 | 90 | 90 | 90 | 90 |
| β (°) | 91.661(11) | 90 | 111.5760(10) | 107.1940(10) | 90 | 90 |
| γ (°) | 90 | 90 | 90 | 90 | 90 | 90 |
| V (Å3) | 1575.0(6) | 2949.1(3) | 1793.4(3) | 9452.1(5) | 2202.0(3) | 3644.8(5) |
| Z | 4 | 4 | 2 | 4 | 4 | 4 |
| D c (g cm−3) | 2.213 | 1.924 | 2.188 | 1.894 | 1.961 | 1.586 |
| μ(mm−1) | 4.821 | 2.893 | 4.539 | 2.716 | 3.595 | 2.116 |
| Data collected/unique | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 057/3609 057/3609 |
24207/2893 |
9880/4117 | 79358/21837 |
16108/2289 |
61206/8408 |
| R 1 (>2σ/all data) | 0.0808/0.0844 | 0.0747/0.0827 | 0.0268/0.0290 | 0.0303/0.0385 | 0.0333/0.0356 | 0.0316/0.0365 |
| wR2 (>2σ/all data) | 0.2053/0.2098 | 0.1877/0.1932 | 0.0607/ 0.0616 | 0.0690/ 0.0731 | 0.0760/0.0771 | 0.0696/0.0714 |
| GOF | 1.119 | 1.125 | 1.077 | 1.021 | 1.120 | 1.024 |
| Residues (e Å−3) | 6.180/−2.933 | 2.830/−2.137 | 3.100/−1.566 | 2.099/−1.607 | 0.918/−1.020 | 1.168 /−1.082 |
PXRD measurements
The X-ray powder diffraction (PXRD) measurements for complexes 1–6 were performed on a Rigaku SmartLab (3) X-ray diffractometer at room temperature, which was in good agreement with the results simulated from the single crystal data, indicating the high purity of the synthesized samples (Fig. S1–S6†).Results and discussion
Syntheses and crystal structures
Complex 1 was prepared by dissolving Dy(NO3)3·5H2O and 12-C-4 in acetonitrile and maintaining the mixture at 40 °C to slowly evaporate. Single-crystal X-ray analysis revealed that it is crystallized in the monoclinic space group P21/c. The Dy(III) ion is 10-coordinate, bonded to three bidentate nitrate groups and to the four oxygen atoms of the crown ether, to afford a half-sandwich type structure with a 12-C-4 on one side and three NO3− groups on the other side (Fig. 1a). Similar structures were seen previously.36,37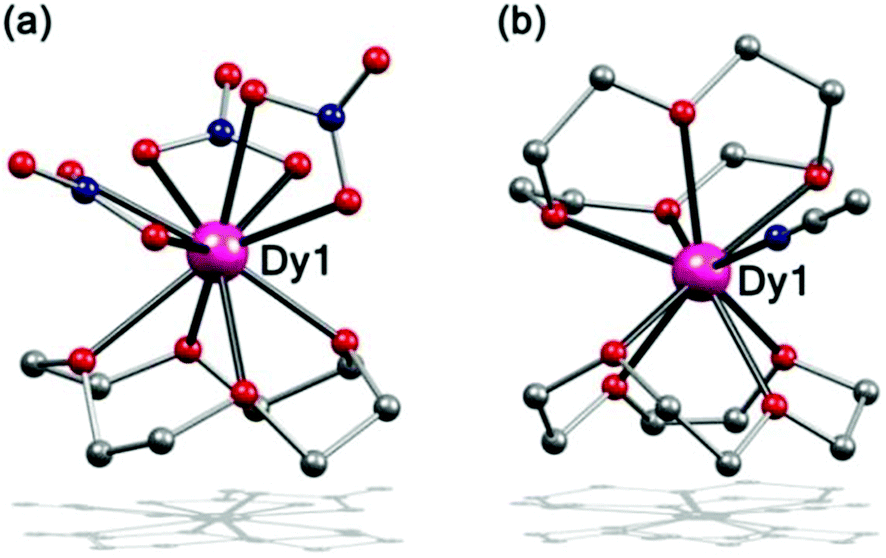 | ||
| Fig. 1 Side view of the molecular structure of complex 1 (a) and 2 (b). H atoms and ClO4− have been omitted for clarity. Colour codes: violet, Dy; red, O; grey, C; blue N. | ||
When the Dy(NO3)3·5H2O was replaced by Dy(ClO4)3·xH2O, complex 2 was obtained under similar reaction conditions, which crystallized in the tetragonal space group P43212. The Dy(III) ion is 9-coordinate, bonded to two 12-C-4 ligands in a non-parallel way and a CH3CN molecule via the N atom, exhibiting a disordered sandwich type structure (Fig. 1b). The crown ether is a kind of flexible ring, so when coordinated to the Dy(III) ion, it is distorted, which means that the four oxygen atoms of the crown ether do not stay in one plane and its C4 conformation is twisted. For both complexes 1 and 2, the crown ethers display large distortions. This is much different than the reported SMMs with a 12-C-4.34,35 Further investigation on the geometry of complexes 1–2 was calculated by continuous shape measures (CSM) (Table S13†).38–40 The result shows that Dy(III) ions in complex 1 are in sphenocorona polyhedra with a CSM value of 2.83 and C2v symmetry. For 2, it is closer to a muffin polyhedron with a CSM value of 1.60 and shows symmetry of Cs.
When a similar reaction for complex 2 was carried out in isopropanol at 130 °C, complex 3 was synthesized, which is a dinuclear complex with Dy(III) ions being bridged by two OH− groups. It crystallized in the monoclinic space group P21/c. The Dy(III) ion is 8-coordinated in a square-antiprismatic coordination environment (Fig. 2). The oxygen atoms from the crown ether almost lie in the same plane and thus, the C4 symmetry is maintained. The oxygen atoms of μ-OH− and H2O ligands comprise the other plane of the square antiprism. As such, the coordination geometry of the Dy(III) ion in 3 shows a perfect D4d symmetry with a CSM value of 0.82 (Table S13†). The Dy–O lengths are 2.42–2.50 Å for O atoms from the crown-ethers, 2.37–2.38 Å for aqua O atoms and 2.20–2.25 Å for OH− groups (Table S5†), indicating that the coordination bond between the crown ether and the lanthanide ion is much weaker than H2O and OH−. Besides, the intramolecular Dy⋯Dy separation is 3.64 Å for complex 3, which is shorter than many reported centrosymmetric Dy2 complexes.20,41–43
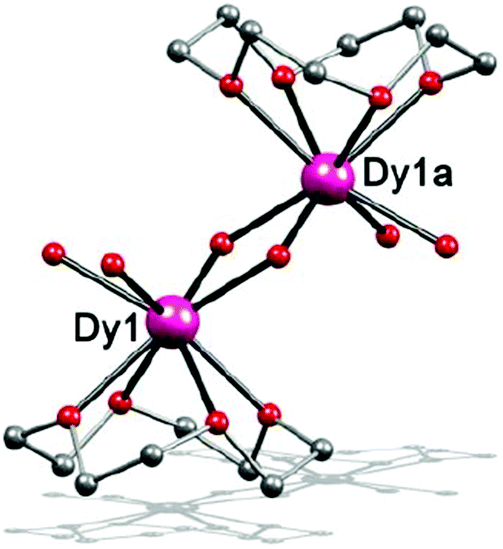 | ||
| Fig. 2 Side view of the molecular structure of complex 3. H atoms and ClO4− have been omitted for clarity. Colours as in Fig. 1. | ||
To prepare complex 4, two equivalents of 12-C-4 for complex 2 were replaced by one equivalent of 15-C-5 and 12-C-4 under the same conditions. Single-crystal X-ray analysis revealed that 4 crystallized in the monoclinic space group P21/c. There are two kinds of coordination models in one unit cell, one is 9-coordinate with a general sandwich type structure (Dy2 and Dy3 in Fig. 3), and the other is 10-coordinate with an additional CH3CN ligand coordinated to the central Dy(III) ion through the cavity of the 15-C-5 ring (Dy1 in Fig. 3). The Dy(III) ion is sandwiched between two crown ether rings with a pseudo-C∞ symmetry, which is similar to the organometallic single-ion magnet (Cp*)Er(COT).25 However, because the crown ethers are flexible, the Dy(III) ions lie in coordination geometries with much lower symmetries. For 9-coordinate Dy(III) ions in complex 4, they locate in the same polyhedra (2.23 for Dy2 and 4.56 for Dy3) as that of 2 with Cs symmetry, while for the 10-coordinated Dy(III) ion, the CSM (1.64 for Dy1) suggests a sphenocorona polyhedron with symmetry of C2v (Table S13†).
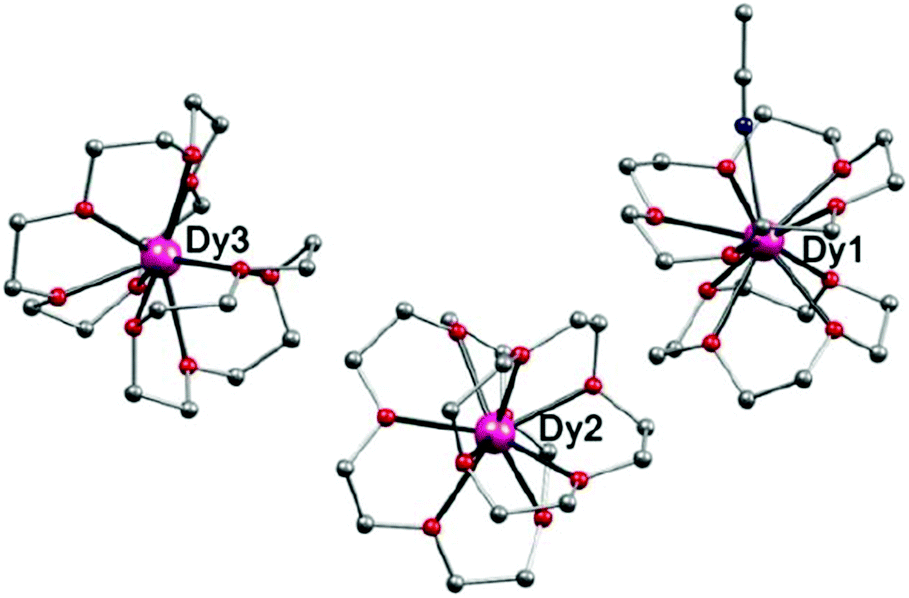 | ||
| Fig. 3 Side view of the molecular structure of complex 4. H atoms, CH3CN and ClO4− have been omitted for clarity. Colours as in Fig. 1. | ||
The slow evaporation of an acetonitrile solution of Dy(NO3)3·5H2O, Dy(ClO4)3·xH2O and 18-C-6 (2:1:3) gave complex 5. Complex 6 was synthesized in a similar way to 5 with the addition of NaBPh4, which gave colorless crystals of 6 in very high yield. 5 and 6 crystallize in the orthorhombic space group Pnma and P212121, respectively. They share an isomeric cation with the common formula of [Dy(18-C-6)(NO3)2]+. Interestingly, the apical positions of the nitrate ligands are different (Fig. 4). For 5, the two nitrate anion groups form a dihedral angle of 90° (Fig. 4b); while for 6, the angle changes to 68°. We suspect that this is caused by the steric-hindrance effect of charge-balancing anions (ClO4− for 5 and BPh4− for 6) in the lattice. Moreover, the Dy–O bond lengths are different. For O atoms from nitrate anions the Dy–O bond lengths are in the range of 2.402(3)–2.439(3) Å, which are shorter than those of O atoms from 18-C-6 ligands (2.450(5)–2.562(3) Å) (Tables S9 and S11†). The shorter Dy–O bonds with the nitrate O atoms are probably due to the central cation which is more attractive to electronegative ligands. On the other hand, the sum of the angles between two adjacent oxygen atoms of 18-C-6 and the DyIII ion is 374° for 5, which is smaller than 380° for 6. This means that the crown-ether ligands in 6 are more distorted than those in 5. As such, the coordination polyhedron is changed from sphenocorona (2.28) for 5 to a bicapped square antiprism (3.10) for 6 (Table S13†).
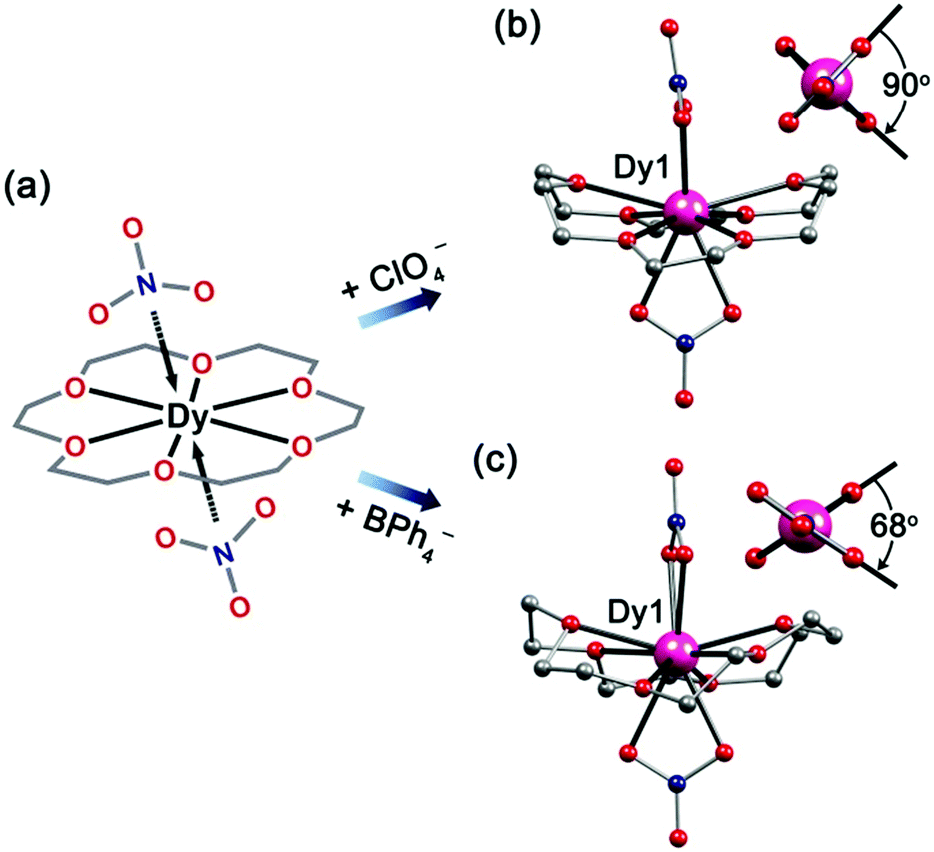 | ||
| Fig. 4 Scheme sketch (a), for side view (b for 5 and c for 6) of Dy(18-C-6)(NO3)2+ cation. Inset: dihedral angles for apical bidentate nitrate anions. H atoms, counter-anions (ClO4− for 5 and BPh4− for 6) have been omitted for clarity. Colours as in Fig. 1. | ||
Magnetic studies
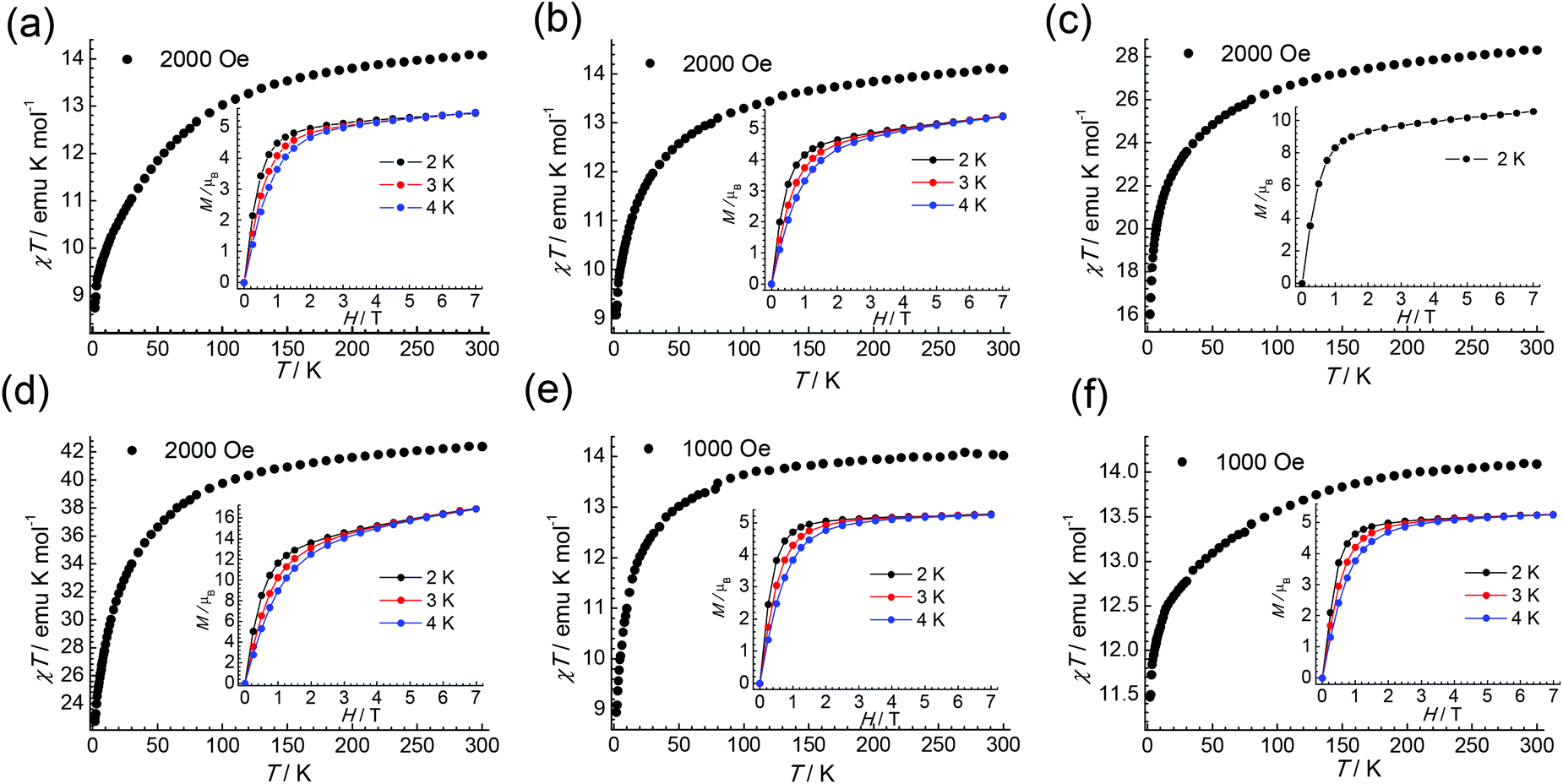 | ||
| Fig. 5 The χT versus T plot of 1–6 (a–f) under indicated dc field. Inset: The field-dependent magnetization plots at indicated temperatures. Lines are visual guides only. | ||
Thus, the dc magnetic field 1000 Oe was applied to suppress the QTM for complexes 1–6. All complexes display some χ′′ signals. But for 2 and 4, only tails of out-of-phase χ′′ ac magnetic susceptibility signals without peaks are observed above 2 K, due to rapid quantum tunnelling of the magnetization (QTM) (Fig. S8 and S11†). While for 1, 3, 5 and 6, as shown in Fig. 6, strong frequency dependent χ’ and χ′′ signals are observed, indicating the presence of slow magnetic relaxation at a low temperature. Complexes 1 and 3 show clear χ′′ signals below 6 K. For complex 5, the peaks are sharp and a maxima signal appears at about 7.0 K for the frequency of 1500 Hz. While for 6, the peaks of χ′′ signals are extremely broad and the maximum of 10 K could be found for the frequency of 1500 Hz, indicating that the relaxation time for 6 is longer than 5 at the same temperature.
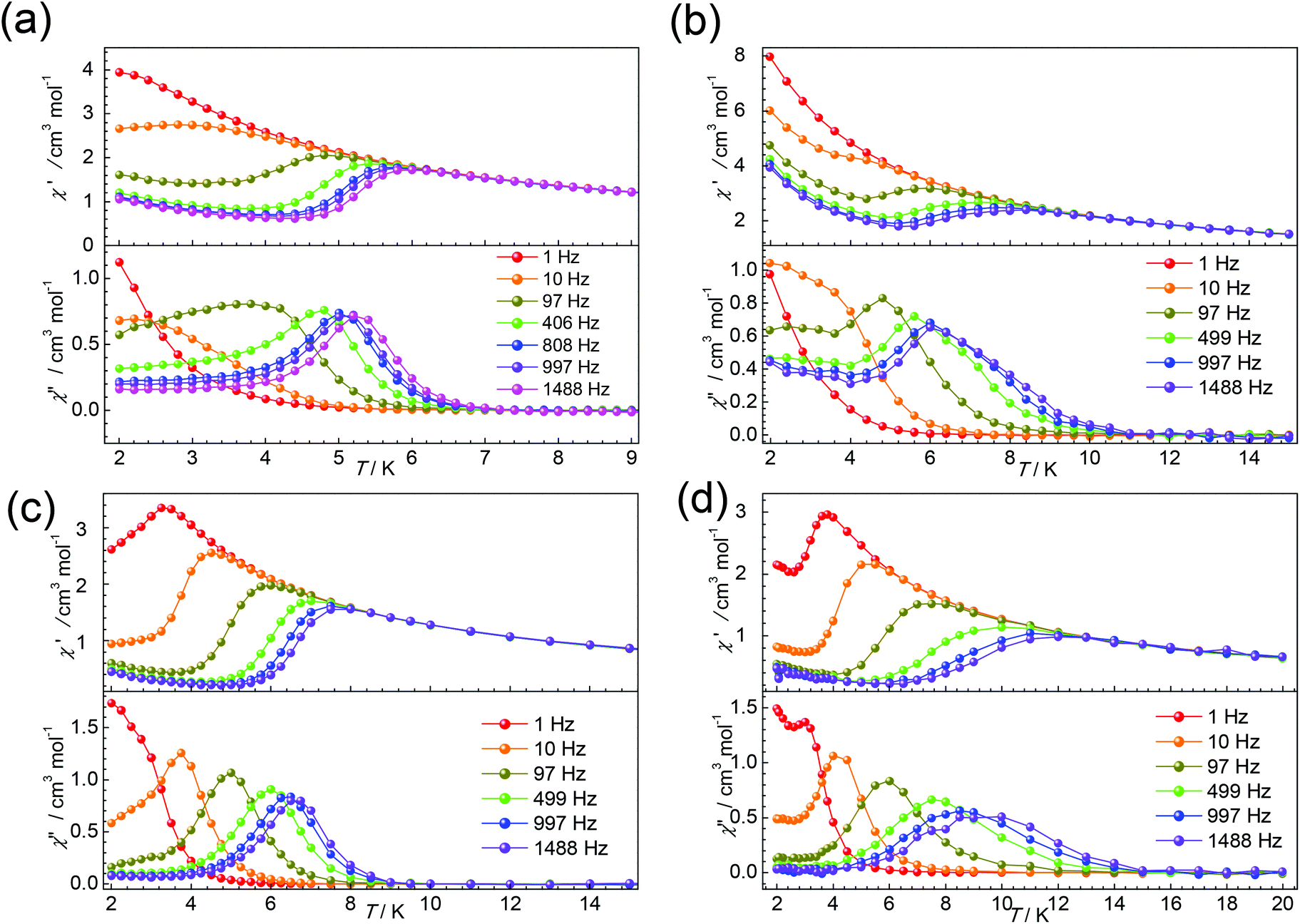 | ||
| Fig. 6 Temperature-dependence of the in-phase (χ′) and out-of-phase (χ′′) ac susceptibility signals under 1000 Oe dc field at the indicated frequencies for complexes 1 (a), 3 (b), 5 (c), and 6 (d). Lines are visual guides only. | ||
Frequency-dependent ac susceptibility data were carried out at the various temperature ranges for 1, 3, 5, and 6 (Fig. 7) under a dc field of 1000 Oe, which were then fitted to a generalized Debye function using the CC-FIT program (Fig. S7, S10, S12 and S13†).46 The obtained α parameters are in the range of 0.05–0.42, 0.15–0.34, 0.03–0.37, and 0.05–0.29 for 1, 3, 5, and 6, respectively (Tables S14–S17†).
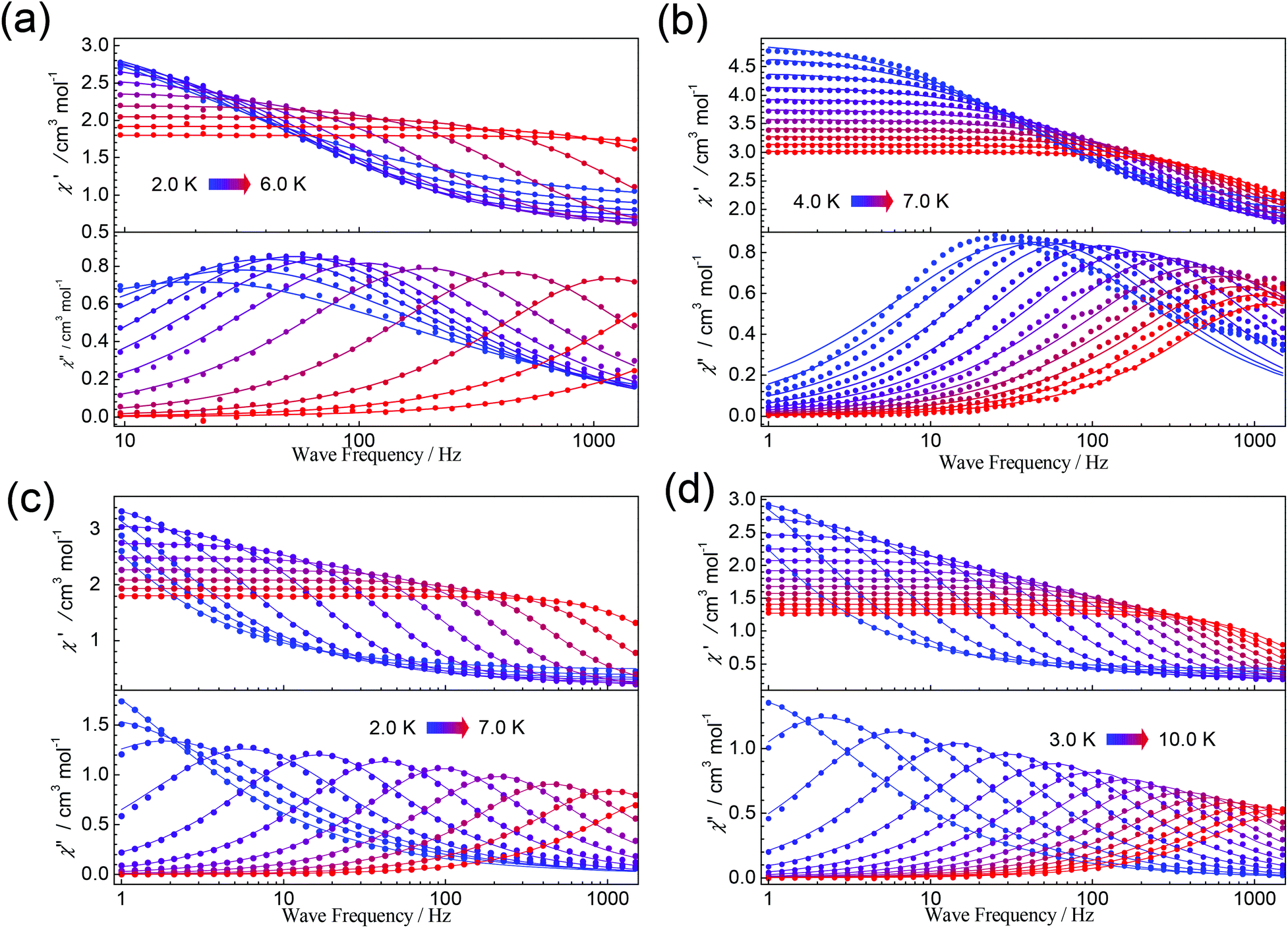 | ||
| Fig. 7 Frequency-dependent ac susceptibility data measured under a dc field of 1000 Oe at indicated temperatures for 1 (a), 3 (b), 5 (c), and 6 (d). Lines are best fits. | ||
It is well known that several processes, including direct, Raman, Orbach relaxation processes etc., may contribute to the magnetic relaxation.47–50 At a high-temperature regime, the Orbach relaxation process is dominant, acting as a temperature-dependent thermally activated regime. The relaxation time τ obtained by the Cole–Cole plots fitting is shown in Fig. 8 (Tables S14–S17†). For 1, the large curvature at low temperature is probably caused by the direct relaxation process. Fitting the high temperature regime with Arrhenius law τ = τ0 exp(ΔE/T) gives ΔE = 68 K and τ0 = 2.07 × 10−10 s. For 3, ΔE = 35 K and τ0 = 8.29 × 10−7 s were obtained. Both complexes 5 and 6 show linear extrapolations (thermally activated regimes) at high temperatures and slight curvatures at low temperatures. The ΔE = 63 K and τ0 = 1.02 × 10−8 s for 5 and the ΔE = 43 K and τ0 = 1.37 × 10−6 s for 6 were obtained by fitting the high temperature regimes using Arrhenius law. Obviously, the relaxation times for 6 are always longer than those of 5 for the same temperatures. Other potential relaxation processes, such as Raman, are also taken into account to simulate the relaxation times, as shown in the ESI.† For complexes 5 and 6, the Raman process may be dominant at low temperature.
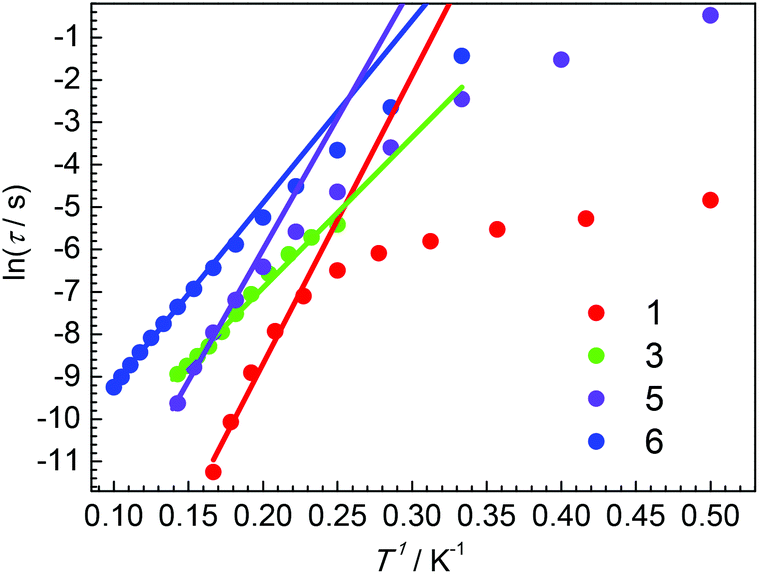 | ||
| Fig. 8 Plots of ln(τ) versus 1/T for 1, 3, 5, and 6. The solid lines are best fits. | ||
Though various coordination environments for Dy(III) ions were obtained, this series of crown-ether-based complexes show no SMM behaviour in a zero dc field above 2 K. This may be due to the weak ligand field provided by crown ethers which have less impact on the structure of the internal 4f electrons. For complexes 1–6, the Dy–Ocrown-ether lengths are in the range of 2.35–2.51 Å, indicating a very weak coordination environment for Dy(III) ions. Compared to the previously reported Dy-SMMs (Table 2), the complex with the highest ΔE for Dy-SMMs is the polymetallic complex, whose Dy(III) ion locates in a distorted octahedral geometry. A key feature of this complex is a strong axial ligand provided by the terminal butoxide.16 The Dy–O length is about 2.10 Å, which is much shorter than the others. For the complex [Zn2DyL2(MeOH)]NO3·3MeOH·H2O with a ΔE of 439 K, a short Dy–O bond of 2.22 Å is observed where the O atom is phenoxyl oxygen.28 For [Dy(L1)2][K(18C6)(THF)2] with ΔE of 813 K and LNCNDyCl2THF2 with ΔE of 335 K, the contribution of short Dy–C bonds to their high ΔE is also mentioned.22,52 On the contrary, the complex Dy(COT)2− held the highest coordination geometry of D8h for all known Dy(III) complexes, but its ΔE is about 11 K, which is much smaller than other Dy-SMMs.26,27 Besides, for the D4d symmetry, which is favourable for Dy-SMMs, the highest ΔE is 187 K.53 It could be concluded that strength of LF is more important to improve Dy-SMMs than the local geometry. This is why Gao et al. in a recent report questioned the effect of symmetry on the performance of Dy-SMMs.51
| Complex | Local symmetry | ΔEb/K | Shortest Dy–X bond/Å | Ref. |
|---|---|---|---|---|
| a X is atom that coordinated to Dy ions. b The number in brackets after some of the ΔE values is the dc field used during the ac measurements. L1 = {C(PPh2NSiMe3)2}2−; LNCN = 2,6-(2,6-C6H3-Et2NCH)2-C6H3; L2 = 2,2′,2′′-(((nitrilotris(ethane-2,1-diyl))tris(azanediyl))tris (methylene))tris-(4-bromophenol); L3 = N,N′-bis(3-methoxysalicylidene)phenylene-1,2-diamine; L4 = dipyrido[3,2-a:20,30-c] phenazine. | ||||
| Dy@[Y4K2O(OtBu)12] | O h | 842 | O−, 2.10 | 16 |
| [Dy(L1)2][K(18C6)(THF)2] | O h | 813 | C2−, 2.10 | 22 |
| [Zn2DyL2(MeOH)]NO3·3MeOH·H2O | Quasi-D5h | 439 | O−, 2.22 | 28 |
| [Zn2(L3)2DyCl3]·2H2O | C 2 | 430 | O−, 2.34 | 51 |
| LNCNDyCl2THF2 | C 2v | 335 | C−, 2.39 | 52 |
| [Dy(L4)(acac)3]·CH3OH | D 4d | 187 | O−, 2.29 | 53 |
| Dy(12-C-4)(NO3)3 | C 2v | 68 (1 kOe) | O−, 2.37 | This work |
| [Dy(acac)3(H2O)2] | D 4d | 64.3 | O−, 2.31 | 12 |
| [Dy(18-C-6)(NO3)2]ClO4 | C 2v | 63 (1 kOe) | O−, 2.42 | This work |
| Na[Dy(DOTA)(H2O)]·4H2O | D 4d | 61 (900 Oe) | O−, 2.32 | 54 |
| [Dy(15-C-5)(H2O)4](ClO4)3 (15-C-5)·H2O | C s | 48.9 (300 Oe) | O, 2.34 | 35 |
| [Dy(18-C-6)(NO3)2]BPh4 | D 4d | 43 (1 kOe) | O−, 2.41 | This work |
| [Pc2Dy]−·TBA+ | D 4d | 40 | N−, 2.44 | 31 |
| [K(18-Crown-6)][Dy(COT)2] | D 8h | 11 | C−, 2.64 (π-System) | 26, 27 |
As such, a rational arrangement of anions coordinated to the Dy(III) ion is very important to improve the magnetic properties of Dy-SMMs. This conclusion is consistent with the field-assisted slow magnetization relaxations only observed in complexes 1, 3, 5 and 6, whose Dy(III) ions are bound to some negatively-charged anions such as NO3− and OH−. This is in accordance with the results summarized in Table 2, where we can see that the strong LF is often provided by anions, such as butoxide, methanediide and phenoxide.
Other than the field-strength of the anions, the coordination geometry also has an unambiguous effect on the slow magnetization relaxation behaviours of the Dy complexes, as evidenced in complexes 5 and 6. The two complexes share a similar coordination environment except for two apical bidentate nitrates, which lead to a significant energy barrier change from 63 K for 5 to 43 K for 6, indicating geometry-perturbed magnetic behavior. As a kind of weak and neutral ligand, the 18-C-6 crown ether may play an important auxiliary role for designing high-performance SMMs with axial negatively-charged donor atoms.
Conclusions
In summary, six Dy(III) complexes based on crown ethers have been synthesized and characterized. Single crystal structure analyses reveal that the coordination of crown ethers is flexible and weak. As such, none of the six Dy(III) complexes behave as SMMs in the absence of a dc field, while obvious field-assisted slow magnetization relaxations were observed if the Dy(III) ions are bound to some negatively-charged anions. Moreover, a subtle geometrical change of the apical coordination anions has a significant effect on the magnetic properties of the Dy(III)-18-C-6 complexes. Therefore, we conclude that both the ligand field and coordination geometry have the effects on the magnetic relaxation behaviours in this series of crown-ether-based Dy-SMMs and the former effect seems more profound.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by “973 projects” (2012CB619401 and 2012CB619402), NSFC (21473129, 21201137, 21103137, 21503155 and IRT13034), “National Young 1000-Plan” program, the open funding of Wuhan National High Magnetic Field Center (2015KF06) and the Fundamental Research Funds for Central Universities. We are grateful to Prof. Dr Zhiping Zheng (University of Arizona) for the recommendation of this work to the inaugural Emerging Investigators themed collection of Inorganic Chemistry Frontiers.Notes and references
- D. Gatteschi, R. Sessoli and J. Villain, Molecular nanomagnets, Oxford University Press, 2006 Search PubMed.
- Molecular Nanomagnets and Related Phenomena, ed. S. Gao, Struct Bond Series 164, Springer, Heidelberg, Germany, 2015 Search PubMed.
- (a) D. N. Woodruff, R. E. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed; (b) H. L. Feltham and S. Brooker, Coord. Chem. Rev., 2014, 276, 1–33 CrossRef CAS; (c) P. Zhang, L. Zhang and J. Tang, Dalton Trans., 2015, 44, 3923–3929 RSC.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC.
- C. R. Ganivet, B. Ballesteros, G. de la Torre, J. M. Clemente-Juan, E. Coronado and T. Torres, Chem. – Eur. J., 2013, 19, 1457–1465 CrossRef CAS PubMed.
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 3, 538–542 CrossRef CAS PubMed.
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14236–14239 CrossRef CAS PubMed.
- F. Luis, A. Repollés, M. Martínez-Pérez, D. Aguilà, O. Roubeau, D. Zueco, P. J. Alonso, M. Evangelisti, A. Camón and J. Sesé, Phys. Rev. Lett., 2011, 107, 117203 CrossRef CAS PubMed.
- M. Urdampilleta, S. Klyatskaya, J.-P. Cleuziou, M. Ruben and W. Wernsdorfer, Nat. Mater., 2011, 10, 502–506 CrossRef CAS PubMed.
- S. Thiele, F. Balestro, R. Ballou, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Science, 2014, 344, 1135–1138 CrossRef CAS PubMed.
- J. Tang, I. Hewitt, N. Madhu, G. Chastanet, W. Wernsdorfer, C. E. Anson, C. Benelli, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 1729–1733 CrossRef CAS PubMed.
- S. D. Jiang, B. W. Wang, G. Su, Z. M. Wang and S. Gao, Angew. Chem., Int. Ed., 2010, 122, 7610–7613 CrossRef.
- R. J. Blagg, C. A. Muryn, E. J. McInnes, F. Tuna and R. E. Winpenny, Angew. Chem., Int. Ed., 2011, 50, 6530–6533 CrossRef CAS PubMed.
- F. Tuna, C. A. Smith, M. Bodensteiner, L. Ungur, L. F. Chibotaru, E. J. McInnes, R. E. Winpenny, D. Collison and R. A. Layfield, Angew. Chem., Int. Ed., 2012, 51, 6976–6980 CrossRef CAS PubMed.
- S. Demir, J. M. Zadrozny, M. Nippe and J. R. Long, J. Am. Chem. Soc., 2012, 134, 18546–18549 CrossRef CAS PubMed.
- R. J. Blagg, L. Ungur, F. Tuna, J. Speak, P. Comar, D. Collison, W. Wernsdorfer, E. J. L. McInnes, L. F. Chibotaru and R. E. P. Winpenny, Nat. Chem., 2013, 5, 673–678 CrossRef CAS PubMed.
- F. Habib, G. Brunet, V. Vieru, I. Korobkov, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2013, 135, 13242–13245 CrossRef CAS PubMed.
- J. L. Liu, J. Y. Wu, Y. C. Chen, V. Mereacre, A. K. Powell, L. Ungur, L. F. Chibotaru, X. M. Chen and M. L. Tong, Angew. Chem., Int. Ed., 2014, 53, 12966–12970 CrossRef CAS PubMed.
- K. Suzuki, R. Sato and N. Mizuno, Chem. Sci., 2013, 4, 596–600 RSC.
- S. Xue, Y. N. Guo, L. Ungur, J. Tang and L. F. Chibotaru, Chem. – Eur. J., 2015, 21, 14099–14106 CrossRef CAS PubMed.
- Y. Dong, P. Yan, X. Zou and G. Li, Inorg. Chem. Front., 2015, 2, 827–836 RSC.
- M. Gregson, N. F. Chilton, A.-M. Ariciu, F. Tuna, I. F. Crowe, W. Lewis, A. J. Blake, D. Collison, E. J. McInnes and R. E. Winpenny, Chem. Sci., 2016, 7, 155–165 RSC.
- W.-M. Wang, S.-Y. Wang, H.-X. Zhang, H.-Y. Shen, J.-Y. Zou, H.-L. Gao, J.-Z. Cui and B. Zhao, Inorg. Chem. Front., 2016, 3, 133–141 RSC.
- C. Görller-Walrand, K. Binnemans, K. A. Gschneidner and L. Eyring, Handbook on the Physics and Chemistry of Rare Earths, 1996, vol. 23 Search PubMed.
- S.-D. Jiang, B.-W. Wang, H.-L. Sun, Z.-M. Wang and S. Gao, J. Am. Chem. Soc., 2011, 133, 4730–4733 CrossRef CAS PubMed.
- K. R. Meihaus and J. R. Long, J. Am. Chem. Soc., 2013, 135, 17952–17957 CrossRef CAS PubMed.
- L. Ungur, J. J. Le Roy, I. Korobkov, M. Murugesu and L. F. Chibotaru, Angew. Chem., Int. Ed., 2014, 53, 4413–4417 CrossRef CAS PubMed.
- J.-L. Liu, Y.-C. Chen, Y.-Z. Zheng, W.-Q. Lin, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M.-L. Tong, Chem. Sci., 2013, 4, 3310–3316 RSC.
- N. F. Chilton, C. A. Goodwin, D. P. Mills and R. E. Winpenny, Chem. Commun., 2015, 51, 101–103 RSC.
- N. F. Chilton, Inorg. Chem., 2015, 54, 2097–2099 CrossRef CAS PubMed.
- N. Ishikawa, M. Sugita, T. Ishikawa, S.-Y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- M. A. AlDamen, J. M. Clemente-Juan, E. Coronado, C. Martí-Gastaldo and A. Gaita-Arino, J. Am. Chem. Soc., 2008, 130, 8874–8875 CrossRef CAS PubMed.
- S. Cardona-Serra, J. Clemente-Juan, E. Coronado, A. Gaita-Ariño, A. Camón, M. Evangelisti, F. Luis, M. Martínez-Pérez and J. Sesé, J. Am. Chem. Soc., 2012, 134, 14982–14990 CrossRef CAS PubMed.
- L. Chen, J. Wang, J. M. Wei, W. Wernsdorfer, X. T. Chen, Y. Q. Zhang, Y. Song and Z. L. Xue, J. Am. Chem. Soc., 2014, 136, 12213–12216 CrossRef CAS PubMed.
- E. L. Gavey, M. Al Hareri, J. Regier, L. D. Carlos, R. A. Ferreira, F. S. Razavi, J. M. Rawson and M. Pilkington, J. Mater. Chem. C, 2015, 3, 7738–7747 RSC.
- E. L. Muetterties and S. Kirschner, Inorganic Syntheses, J. Wiley & Interscience, 1985, vol. 23 Search PubMed.
- J.-C. G. Bünzli and D. Wessner, Inorg. Chim. Acta, 1980, 44, L55–L58 CrossRef.
- D. Casanova, M. Llunell, P. Alemany and S. Alvarez, Chem. – Eur. J., 2005, 11, 1479–1494 CrossRef CAS PubMed.
- A. Ruiz-Martínez, D. Casanova and S. Alvarez, Dalton Trans., 2008, 2583–2591 RSC.
- A. Ruiz-Martínez and S. Alvarez, Chem. – Eur. J., 2009, 15, 7470–7480 CrossRef PubMed.
- J. Long, F. Habib, P.-H. Lin, I. Korobkov, G. Enright, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 5319–5328 CrossRef CAS PubMed.
- J.-D. Leng, J.-L. Liu, Y.-Z. Zheng, L. Ungur, L. F. Chibotaru, F.-S. Guo and M.-L. Tong, Chem. Commun., 2013, 49, 158–160 RSC.
- S. K. Langley, N. F. Chilton, B. Moubaraki and K. S. Murray, Inorg. Chem., 2013, 52, 7183–7192 CrossRef CAS PubMed.
- P. Zhang, L. Zhang, C. Wang, S. Xue, S.-Y. Lin and J. Tang, J. Am. Chem. Soc., 2014, 136, 4484–4487 CrossRef CAS PubMed.
- C. Benelli and D. Gatteschi, Chem. Rev., 2002, 102, 2369–2388 CrossRef CAS PubMed.
- Y.-N. Guo, G.-F. Xu, Y. Guo and J. Tang, Dalton Trans., 2011, 40, 9953–9963 RSC.
- M. Gonidec, F. Luis, À. Vílchez, J. Esquena, D. B. Amabilino and J. Veciana, Angew. Chem., Int. Ed., 2010, 49, 1623–1626 CrossRef CAS PubMed.
- Y.-N. Guo, G.-F. Xu, P. Gamez, L. Zhao, S.-Y. Lin, R. Deng, J. Tang and H.-J. Zhang, J. Am. Chem. Soc., 2010, 132, 8538–8539 CrossRef CAS PubMed.
- K. S. Pedersen, L. Ungur, M. Sigrist, A. Sundt, M. Schau-Magnussen, V. Vieru, H. Mutka, S. Rols, H. Weihe and O. Waldmann, Chem. Sci., 2014, 5, 1650–1660 RSC.
- K. R. Meihaus, S. G. Minasian, W. W. Lukens Jr., S. A. Kozimor, D. K. Shuh, T. Tyliszczak and J. R. Long, J. Am. Chem. Soc., 2014, 136, 6056–6068 CrossRef CAS PubMed.
- W.-B. Sun, P.-F. Yan, S.-D. Jiang, B.-W. Wang, Y.-Q. Zhang, H.-F. Li, P. Chen, Z.-M. Wang and S. Gao, Chem. Sci., 2016, 7, 684–691 RSC.
- Y.-N. Guo, L. Ungur, G. E. Granroth, A. K. Powell, C. Wu, S. E. Nagler, J. Tang, L. F. Chibotaru and D. Cui, Sci. Rep., 2014, 4 Search PubMed , 5471.
- G. J. Chen, Y. N. Guo, J. L. Tian, J. Tang, W. Gu, X. Liu, S. P. Yan, P. Cheng and D. Z. Liao, Chem. – Eur. J., 2012, 18, 2484–2487 CrossRef CAS PubMed.
- P.-E. Car, M. Perfetti, M. Mannini, A. Favre, A. Caneschi and R. Sessoli, Chem. Commun., 2011, 47, 3751–3753 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1441566–1441569, 1044072 and 1044073. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00308c |
| This journal is © the Partner Organisations 2016 |