
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyclohexyl-substituted poly(phosphonate)-copolymers with adjustable glass transition temperatures†
Thomas
Wolf
,
Johannes
Naß
and
Frederik R.
Wurm
 *
*
Max Planck-Institut für Polymerforschung, Ackermannweg 10, 55128 Mainz, Germany. E-mail: wurm@mpip-mainz.mpg.de; Fax: +49 6131 370 330; Tel: +49 6131 379 581
First published on 30th March 2016
Abstract
2-Cyclohexyl-2-oxo-1,3,2-dioxaphospholane (cyHexPPn), a new monomer for the anionic ring-opening polymerization to poly(ethylene alkyl phosphonate)s is presented. The organo-catalyzed polymerization produces homopolymers with excellent control over molecular weight and narrow molecular weight distributions. The homopolymer was found to exhibit a glass transition at 15 °C, which is 60 °C higher compared to all previously reported poly(ethylene n-alkyl phosphonate)s. Copolymerization with the water-soluble 2-isopropyl-2-oxo-1,3,2-dioxaphospholane (iPrPPn) resulted in water-soluble, well-defined copolymers. The copolymer composition matched the theoretical value in all cases and Tg showed a linear correlation with the amount of (iPrPPn) incorporated. The copolymer was found to exhibit low cell-toxicity towards sensitive murine macrophage-like cells (RAW264.7).
Poly(phosphonate)s (PPns) are a relatively old polymer class known since the pioneering work of Carraher et al. on the polycondensation of phenyl phosphonic acid dichlorides and aromatic diols.1 Besides their use as flame retardant materials, however, this polymer class has received only little scientific or industrial attention.2–4 Motivated by the properties of poly(phosphate)s as biodegradable polymers for drug delivery, Steinbach et al. developed the first living synthesis of poly(ethylene methyl phosphonate), a water-soluble, biocompatible poly(ethylene alkyl phosphonate), via organocatalytic anionic ring-opening polymerization (AROP).5–7 Following these results, our group continued their research on the elucidation and variation of polymers based on this synthetic platform.8 In contrast to the more prominent poly(phosphate)s with hydrolytically labile phosphorus ester in the main and side chains, in poly(phosphonate)s the labile pendant ester is replaced by a chemically stable P–C bond, which allows the precise adjustment of degradation rates and produces highly water-soluble polymers with low molecular weight dispersity up to full conversion.5,8 One important attribute to modify in order to broaden the potential field of applications of a polymer is its glass transition temperature (Tg). Whereas amorphous low Tg materials may exhibit elastic behavior, the incorporation of a high Tg segment, for example in a block-copolymer, could facilitate phase separation and hence facilitate self-aggregation behavior.9 The glass transition temperature is heavily influenced by the nature and especially the steric demand of the polymer side-chain. The more rotational degrees of freedom available, the lower the glass transition temperature. Rigid cyclic structures in the side-chain are therefore well-suited to increase the Tg of a material. However, not many structures with cyclic aliphatic side-chains have been known to date. Hoogenboom et al. could demonstrate a significant increase in Tg of poly(2-oxazolines) by the incorporation of a cyclopropyl side-chain as well as very recently they presented three novel 2-cycloalkyl-2-oxazoline copolymers which showed high melting temperatures and a complex, non-linear dependence of the Tg with respect to the incorporated linear side-chain monomer.10,11
Here, we present the first synthesis of a new dioxaphospholane derivative, namely 2-cyclohexyl-2-oxo-1,3,2-dioxaphospholane (cyHexPPn, (1)), bearing a cycloalkyl substituent and its well-controlled homo- as well as copolymerization with 2-isopropyl-2-oxo-1,3,2-dioxaphospholane (iPrPPn, (2)) via organocatalytic AROP. The obtained polymers were thoroughly investigated via1H, 31P{H} NMR, SEC and DSC analysis. The Tg of the homopolymer was found to be around 15 °C, which is 60 °C higher than that of the previously reported PPns. A linear correlation between the Tg and the amount of (2) incorporated in the copolymer was found to enable fine tuning. Copolymerization further drastically improved the water-solubility of the polymers. Finally, cell-viability assay showed low cell-toxicity against the sensitive murine macrophage-like cells RAW 264.7.
(1) was synthesized via a two-step reaction (Scheme 1a). In the first step, aluminium chloride, phosphorus(III)chloride and cyclohexyl bromide were converted to cyclohexyl phosphonic acid dichloride according to the literature protocol of Clay et al.12 Ring-closing with ethylene glycol was conducted under high dilution according to a slightly modified literature protocol of Steinbach et al.5 The monomer was received in acceptable yields and high purity as confirmed via1H, 31P{H} and 13C NMR spectroscopy.
 | ||
| Scheme 1 (a) Two-step synthesis of 2-cyclohexyl-2-oxo-1,3,2-dioxaphospholane (1) monomer starting from cyclohexyl bromide; (b) TBD-catalyzed and 2-(benzyloxy)ethanol-initiated anionic ring-opening polymerization of (1) in dichloromethane at 0 °C to afford P(1); (c) TBD catalyzed and 2-(benzyloxy)ethanol initiated anionic ring-opening copolymerization of (1) and (2) in dichloromethane at 0 °C to afford P(1x-co-2y)n. | ||
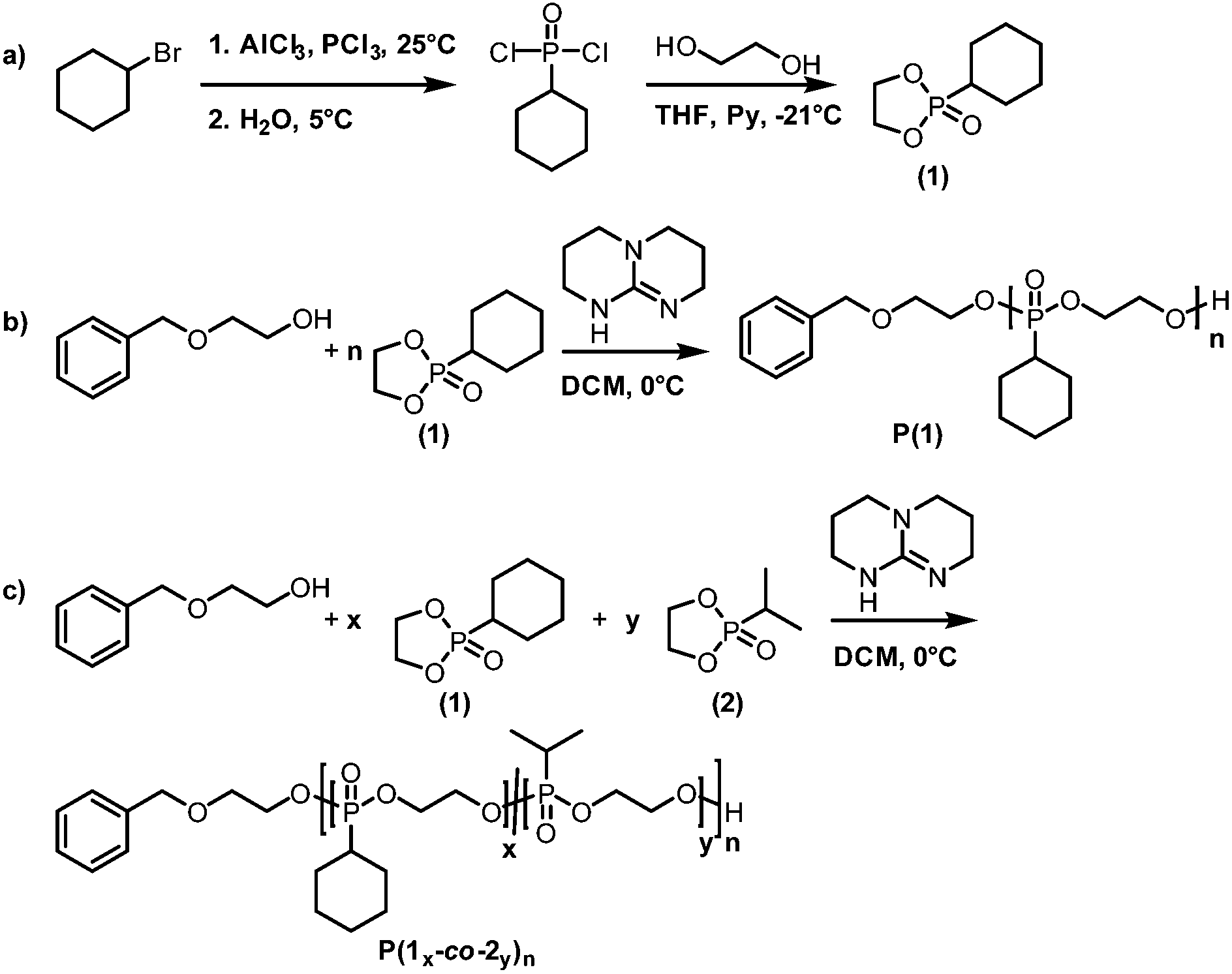
The distinct multiplet of the intracyclic protons in the 1H NMR spectrum between 4.54 and 4.19 ppm as well as the characteristic shift in 31P{H} NMR spectroscopy (47.5 ppm) confirmed the success of the reaction ((Fig. 1a) and Fig. S5†). The protons of the cyclohexane ring showed a complex J-splitting pattern due to the influence of the non-planarity of the cyclohexane ring and JHP coupling (Scheme S1†).
 | ||
| Fig. 1 (a) 1H (500 MHz) NMR spectrum of (1) in CDCl3 at 298 K; (b) 1H (500 MHz) NMR spectrum of P(1) in DMSO-d6 at 298 K; (c) 1H (500 MHz) NMR spectrum of P(10.49-co-20.51)60 in DMSO-d6 at 298 K. | ||
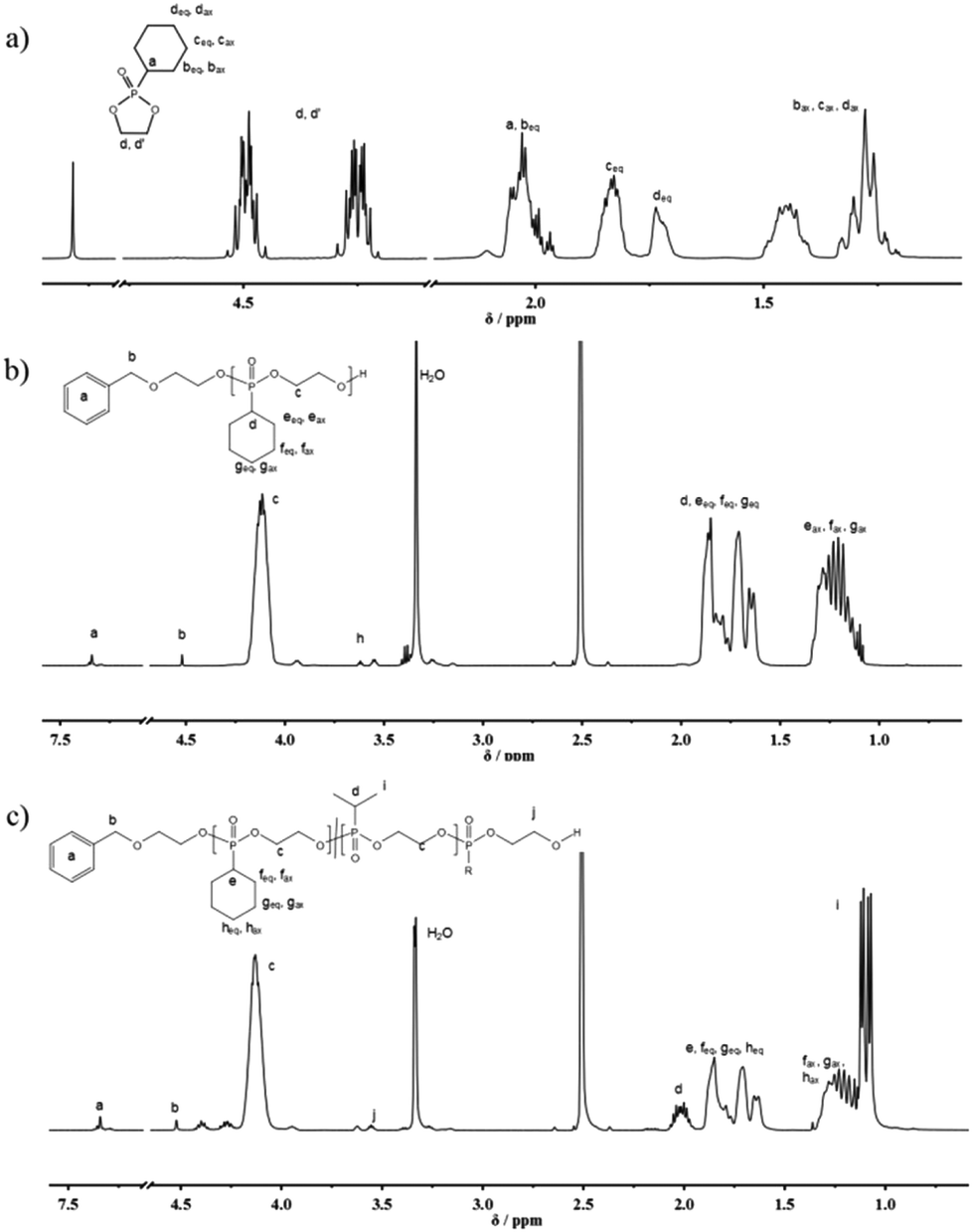
The organocatalytic AROP of the new monomer (1) proceeded in analogy with the previously reported polymerization of dioxaphospholanes with secondary alkyl side-chains.8 1,5,7-Triazabicyclo[4.4.0]dec-5-ene (TBD) was used as a base in the presence of a primary alcohol at 0 °C and proceeded to high conversions (>90%) within 2 h (Scheme 1b). The obtained polymers were thoroughly characterized via1H, 31P{H} and 13C NMR spectroscopy as well as SEC and DSC analysis. The 1H NMR spectrum further showed the disappearance of the characteristic monomer multiplets from 4.54 to 4.19 ppm and the emergence of a broad signal from 4.22 to 4.05 ppm. The accurate determination of Mn was possible via1H NMR assisted end-group analysis through comparison of the integrals of the aromatic initiator (7.26 ppm) with the above mentioned backbone resonance (Fig. 1b). The polymers were synthesized with good control over molecular weights up to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. 31P{H} analysis showed a significant shift from 47.57 ppm of the monomer to a broad signal at 33.12 ppm for the polymer. The end-group phosphorus was visible as a low-intensity signal at 32.96 ppm (Fig. S7†). The obtained polymers were well-defined with a rather narrow molecular weight distribution (Đ) (Fig. 2a)). The glass transition temperature (Tg) of P(1) was found to be around 15 °C, 50–60 °C higher than that of the previously reported poly(ethylene alkyl phosphonate)s.5,8 This comparatively high Tg further enlarges the potential applications of poly(ethylene alkyl phosphonate)s. The complete polymer characterization is summarized in Table 1.
000 g mol−1. 31P{H} analysis showed a significant shift from 47.57 ppm of the monomer to a broad signal at 33.12 ppm for the polymer. The end-group phosphorus was visible as a low-intensity signal at 32.96 ppm (Fig. S7†). The obtained polymers were well-defined with a rather narrow molecular weight distribution (Đ) (Fig. 2a)). The glass transition temperature (Tg) of P(1) was found to be around 15 °C, 50–60 °C higher than that of the previously reported poly(ethylene alkyl phosphonate)s.5,8 This comparatively high Tg further enlarges the potential applications of poly(ethylene alkyl phosphonate)s. The complete polymer characterization is summarized in Table 1.
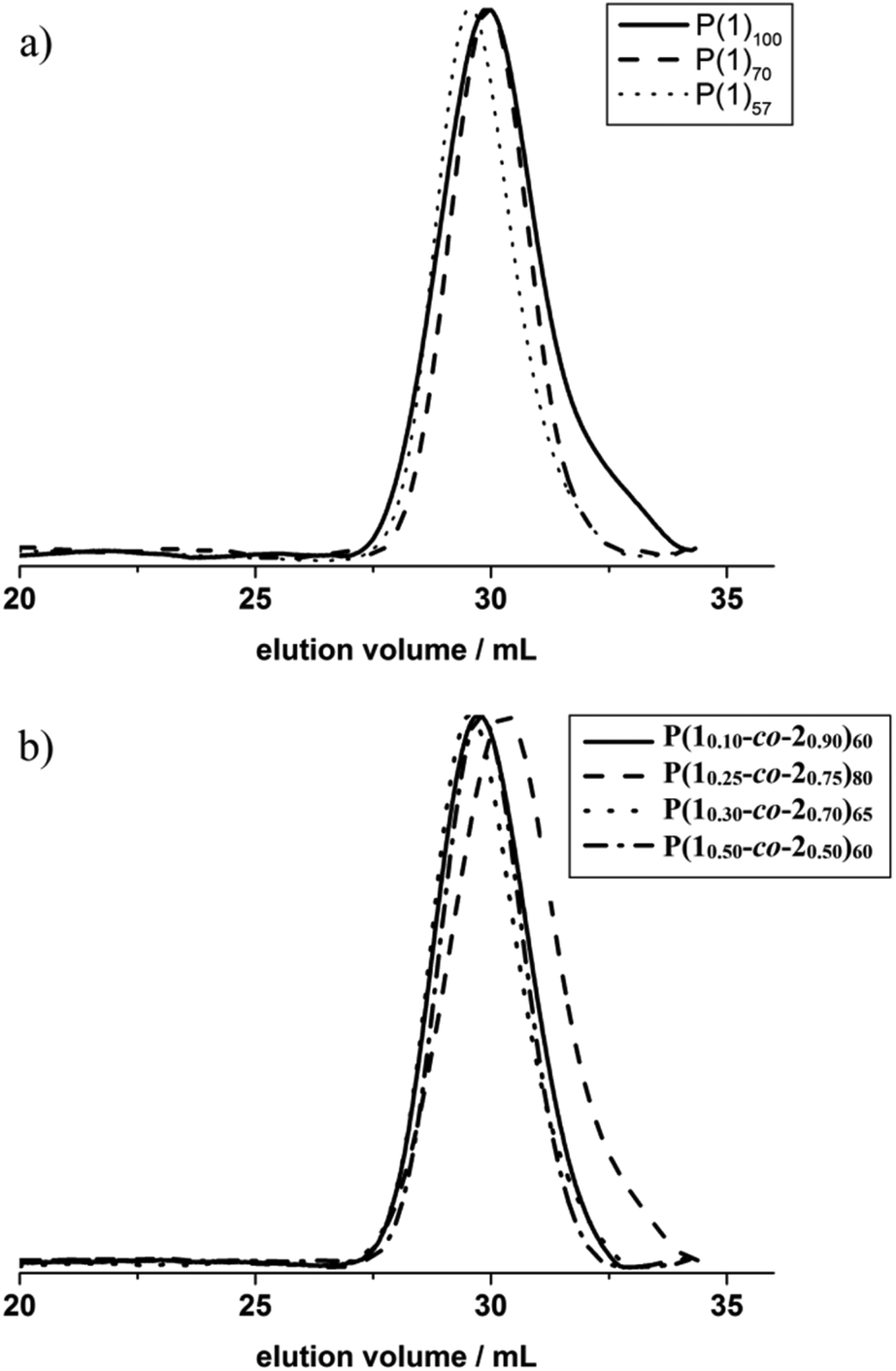 | ||
| Fig. 2 SEC traces (RI detection) of (a) P(1)n and (b) P(1x-co-2y)n in DMF at 333 K (the indices x and y represent the relative copolymer composition whereas n represents the degree of polymerization). | ||
| Sample | Theo. ratio (1)/(2)a |
M
nb/g mol−1 |
Đ |
T
gd/°C |
|---|---|---|---|---|
| a Monomer composition at t = 0, determined via1H NMR spectroscopy in DMSO-d6 at 298 K. b Determined via1H NMR spectroscopy in DMSO-d6 at 298K. c Determined via SEC in DMF (60 °C) (PEG standard). d Determined via DSC at a heating rate of 10 °C min−1. | ||||
| P(1)57 | — | 10800 |
1.28 | 16.1 |
| P(1)70 | — | 13300 |
1.20 | 16.3 |
| P(1)100 | — | 19100 |
1.36 | 15.5 |
| P(10.1-co-20.9)60 | 0.11/0.91 | 9100 | 1.31 | −39.2 |
| P(10.25-co-20.75)80 | 0.25/0.75 | 12700 |
1.49 | −26.4 |
| P(10.30-co-20.70)65 | 0.31/0.69 | 10500 |
1.35 | −26.9 |
| P(10.50-co-20.50)60 | 0.47/0.53 | 10100 |
1.24 | −21.8 |
| P(10.65-co-20.35)67 | 0.65/0.35 | 11800 |
1.26 | −14.1 |
| P(10.66-co-20.34)85 | 0.66/0.34 | 15000 |
1.40 | −15.1 |
As the homopolymers of (1) are hydrophobic materials, copolymers with 2-isopropyl-2-oxo-1,3,2-dioxaphospholane (iPrPPn, (2)) were prepared. The incorporation of these hydrophilic segments produces firstly water-soluble poly(ethylene alkyl phosphonate)s, and secondly allows gradual adjustment of the Tgs. The homopolymer of (2) is known to be highly water-soluble (>10 g L−1), with a Tg at ca. −40 °C and polymerizes under the same conditions as (1), allowing the copolymerization to proceed under the same conditions as the homopolymerization (Scheme 1c).8 (2) was synthesized according to a literature protocol.8 The success of the polymerization was observed by the vanishing of the monomer resonances in the 31P{H} NMR spectra at 47.5 ppm ((1)) and 50.45 ppm ((2)), respectively, and by the emergence of the polymer resonances at 32.96 ppm (P(1)) and 29.88 ppm (P(2)) (Fig. S10†). The broad backbone signal in the 1H NMR spectrum from 4.21 to 4.02 ppm further confirmed the polymerization (Fig. 1c). End-group analysis in 1H NMR spectroscopy again enabled accurate determination of Mn. Both 1H NMR as well as 31P NMR spectroscopy methods were used to calculate the copolymer composition. In 31P{H} NMR comparison of the well-separated signals at 32.96 ppm (P(1)) and 29.88 ppm (P(2)) was used to determine the copolymer composition. In 1H NMR the resonances of the respective side-chains at 1.92 to 1.60 ppm (P(1)) and 2.09 to 1.94 ppm (P(2)), respectively, were used to determine an accurate copolymer composition. Both methods of calculation gave the same composition and the copolymer composition matched the theoretical values in all the cases. 1H DOSY NMR spectroscopy was used to ensure the successful synthesis of a copolymer (Fig. S11†). Both the backbone signals (4.21 to 4.02 ppm) as well as all the signals originating from the respective side-chains and the initiator show the same diffusion coefficient (black box) suggesting the formation of a copolymer. The molecular weight distributions were found to be monomodal and precise control over molecular weight up to 15000 g mol−1 was achieved. (Fig. 2b). The water-solubility of the copolymers was dramatically enhanced by the incorporation of (2). Copolymers with 30 mol% of (2) were soluble at a concentration of 1 g L−1, compared to the insoluble P(1). A further increase of the amount of (2) in the copolymer resulted in gradually increasing water-solubility. These water-soluble copolymers exhibited cloud point temperatures at elevated temperatures. Consequently the cloud point temperature increased with the decreasing amount of the incorporated hydrophobic comonomer (1), indicating a higher hydrophilicity of the copolymer. The turbidity measurements are shown in Fig. S12.† DSC measurements revealed a single Tg in all the cases. This indicated a rather random incorporation of both monomers into the copolymer, as a block-copolymer is expected to show two distinct glass transition temperatures. Additionally a linear correlation between the measured Tg and the amount of (2) incorporated in the polymer was detected (Fig. 3).
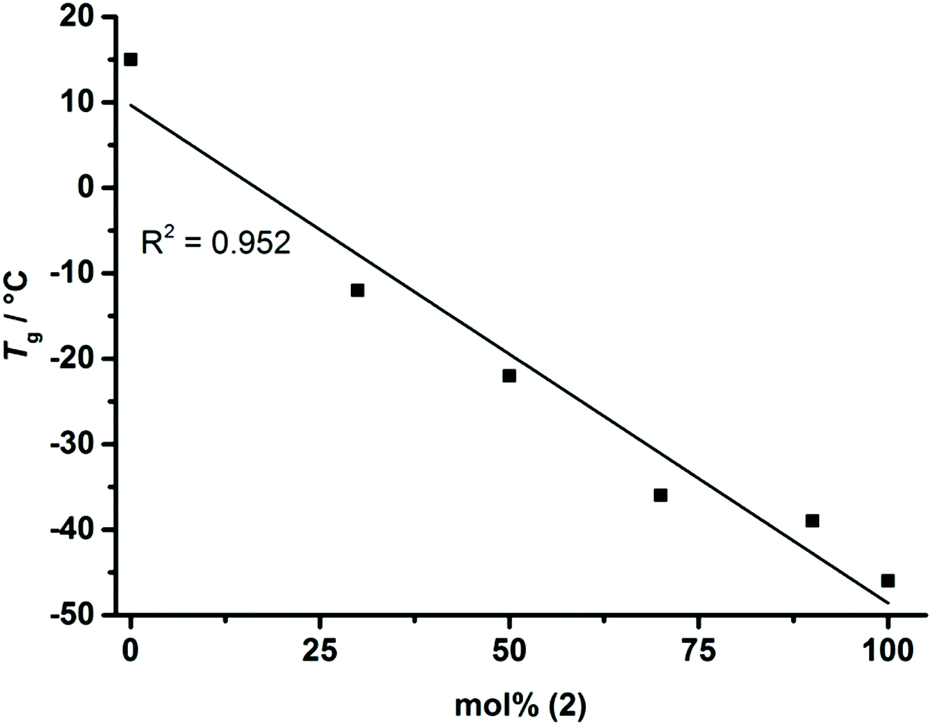 | ||
| Fig. 3 Linear correlation between the measured Tgs (from DSC) and the mol fraction of (2) in the copolymers P(1x-co-2y)n. | ||
Hence, a precise adjustment of the Tg of the copolymers in the range from −45 to 15 °C is possible. (DSC thermograms are shown in Fig. S12†). This makes P(1x-co-2y)n the first known water-soluble poly(ethylene alkyl phosphonate)s with adjustable Tg over a broad temperature range.
Finally, to elucidate the biocompatibility of the newly synthesized materials, a representative in vitro cell-toxicity test of P(10.30-co-20.70)65 was performed with the sensitive murine macrophage-like cells RAW 264.7. The cells were incubated with a solution of P(10.30-co-20.70)65 in medium (+10% FBS) at the respective concentration for 24 h. Afterwards, cell-viability was determined via the commercially available CellTiterGlo® Luminescent Cell-Viability Assay (Promega) according to the delivered protocol. At pharmaceutically relevant concentrations, below 200 μg mL−1, no cytotoxicity was detected, however a significantly reduced viability was found for concentrations exceeding 200 μg mL−1 (Fig. 4).
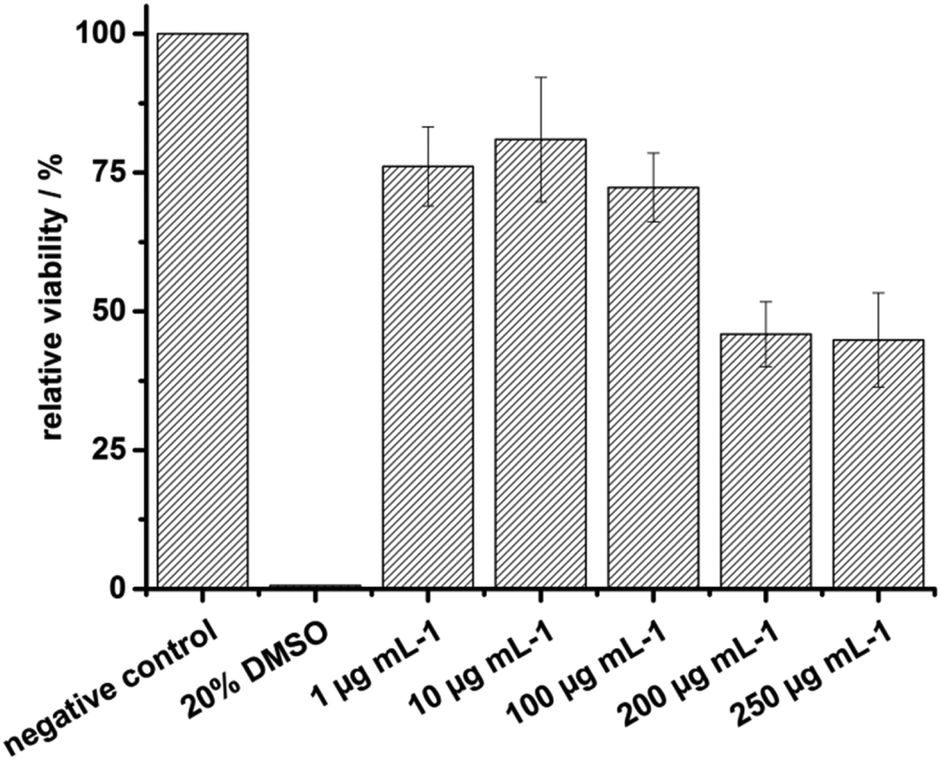 | ||
| Fig. 4 In vitro cell-viability assay against murine macrophage-like cells (RAW 264.7). 24 h incubation of P(10.30-co-20.70)65 at 37 °C with pure medium as negative control and 20% DMSO as positive control. All experiments were performed in triplicate. | ||
To conclude, the successful synthesis of the novel dioxaphospholane monomer 2-cyclohexyl-2-oxo-1,3,2-dioxaphospholane, the first phosphonate monomer for the anionic ring-opening polymerization carrying a cycloalkyl side-chain, is reported. The cyclic monomer polymerized via organocatalytic anionic ring-opening polymerization with TBD and afforded well-defined polymers with good control over molecular weight and Tg of ca. 15 °C, i.e. 60 °C higher compared to the previously reported poly(ethylene n-alkyl phosphonate)s. The Tg of these water-insoluble materials was further adjusted by copolymerization with 2-isopropyl-2-oxo-1,3,2-dioxyphospholane and a linear correlation between the Tg and the incorporated amount of (2) was found. Water-soluble copolymers with low cell-toxicity against macrophage-like cells (RAW 264.7) and Tgs in the range from +15 to −40 °C were obtained. This makes P(1x-co-2y)n the first known water-soluble poly(alkylene alkyl phosphonate)s with adjustable glass transition temperatures. Future studies for the preparation of biodegradable nanocarriers with such systems are in progress and will be reported in due course.
Acknowledgements
The authors thank Prof. Dr K. Landfester for continuous support. The authors acknowledge support from the “Deutsche Forschungsgemeinschaft” (DFG WU 750/6-1). The authors thank Johanna Simon for the cell culture and toxicity assays.References
- F. Millich and C. E. Carraher, Interfacial Syntheses of Polyphosphonate and Polyphosphate Esters. I. Effects of Alkaline Medium, J. Polym. Sci., Part A1, 1969, 7, 2669–2678 CrossRef CAS.
- K.-S. Kim, Phosphorus-Containing polymers. I. Low temperature polycondensation of phenylphosphonic dichloride with bisphenols, J. Appl. Polym. Sci., 1983, 28, 1119–1123 CrossRef CAS.
- T. Ranganathan, J. Zilberman, R. J. Farris, E. B. Coughlin and T. Emrick, Synthesis and characterization of halogen-free antiflammable polyphosphonates containing 4,4 ‘-bishydroxydeoxybenzoin, Macromolecules, 2006, 39, 5974–5975 CrossRef CAS.
- T. Steinbach and F. R. Wurm, Poly(phosphoester)s: A New Platform for Degradable Polymers, Angew. Chem., Int. Ed., 2015, 54, 6098–6108 CrossRef CAS PubMed.
- T. Steinbach, S. Ritz and F. R. Wurm, Water-Soluble Poly(phosphonate)s via Living Ring-Opening Polymerization, ACS Macro Lett., 2014, 3, 244–248 CrossRef CAS.
- Y. H. Lim, K. M. Tiemann, G. S. Heo, P. O. Wagers, Y. H. Rezenom, S. Zhang, F. Zhang, W. J. Youngs, D. A. Hunstad and K. L. Wooley, Preparation and in vitro antimicrobial activity of silver-bearing degradable polymeric nanoparticles of polyphosphoester-block-poly(L-lactide), ACS Nano, 2015, 9, 1995–2008 CrossRef CAS PubMed.
- S. Schottler, G. Becker, S. Winzen, T. Steinbach, K. Mohr, K. Landfester, V. Mailander and F. R. Wurm, Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly(phosphoester)-coated nanocarriers, Nat. Nanotechnol., 2016, 11, 372–377 CrossRef PubMed.
- T. Wolf, T. Steinbach and F. R. Wurm, A Library of Well-Defined and Water-Soluble Poly(alkyl phosphonate)s with Adjustable Hydrolysis, Macromolecules, 2015, 48, 3853–3863 CrossRef CAS.
- C. Nardin, T. Hirt, J. Leukel and W. Meier, Polymerized ABA Triblock Copolymer Vesicles, Langmuir, 2000, 16, 1035–1041 CrossRef CAS.
- M. M. Bloksma, C. Weber, I. Y. Perevyazko, A. Kuse, A. Baumgartel, A. Vollrath, R. Hoogenboom and U. S. Schubert, Poly(2-cyclopropyl-2-oxazoline): From Rate Acceleration by Cyclopropyl to Thermoresponsive Properties, Macromolecules, 2011, 44, 4057–4064 CrossRef CAS.
- V. V. Jerca, K. Lava, B. Verbraeken and R. Hoogenboom, Poly(2-cycloalkyl-2-oxazoline)s: high melting temperature polymers solely based on Debye and Keesom van der Waals interactions, Polym. Chem., 2016, 7, 1309–1322 RSC.
- J. P. Clay, A New Method for the Preparation of Alkane Phosphonyl Dichlorides, J. Org. Chem., 1951, 16, 892–894 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental and analytical data. See DOI: 10.1039/c6py00358c |
| This journal is © The Royal Society of Chemistry 2016 |