
Transforming metal–organic frameworks into functional materials
Ricardo F.
Mendes
and
Filipe A.
Almeida Paz
*
Department of Chemistry, CICECO – Aveiro Institute of Materials, University of Aveiro, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal. E-mail: filipe.paz@ua.pt; Fax: (+351) 234 401470; Tel: (+351) 234 370200 (Ext. 23553)
First published on 16th April 2015
Abstract
This review focuses on a new alternative approach towards MOF preparation through the transformation of known MOF structures into other novel, more active MOFs that, in most cases, could not be obtained by conventional synthetic methods. These transformations are usually of the single-crystal-to-single-crystal type, usually accompanied by a modification of the dimensionality of the MOFs: e.g., 1D → 2D, 2D → 3D, 1D → 3D or even 3D → 3D′ (concerning modification of the connectivity of the primary building units). The literature contains several reports concerning MOF-to-MOF transformations but only in a handful of cases the authors aimed to design new functional compounds, pointing towards applications or the modification (or improvement) of the properties of the materials. This review aims to concisely describe the most significant reports concerning the transformation of MOFs into other more functional and active MOFs. Several types of transformations are possible including solvent removal or insertion, modification of the pH, metal exchange, release of active molecules, among others. These transformations can lead to significant improvements of the properties of MOFs, for example: increase of adsorption of different gases, such as nitrogen and carbon dioxide; creation of sensing centers for different chemical species; pH sensing with, in some cases, a concomitant change in crystal color; improvement of the luminescence properties by the removal of solvent molecules. Other transformations could lead instead to a complete modification of the properties of MOFs such as the appearance of magnetic properties, the creation of storage devices, the design of releasing materials by the incorporation of active molecules or water scavengers.
 Ricardo F. Mendes | Ricardo Mendes born in 1988 completed a first degree in 2009 and an M.A. in 2011, both in chemistry at the University of Porto, Portugal. In 2013 he enrolled in the doctoral programme in materials chemistry under the supervision of Filipe Almeida Paz at CICECO – Aveiro Institute of Materials. His current research is focused on the design, synthesis and transformation of new metal–organic frameworks. |
 Filipe A. Almeida Paz | Filipe Almeida Paz holds a first degree in chemistry from the University of Aveiro (Portugal), and a Ph.D. from the University of Cambridge (U.K.). He returned to Aveiro in 2004 to occupy the position of Auxiliary Researcher at the Associated Laboratory CICECO. His research interests are focused on chemical crystallography and on the design and preparation of novel functional metal–organic frameworks, which may find application as photoluminescent, catalytic and conductive materials. His current research group is composed of three Ph.D. students and five post-doctoral fellows, and it has been financed over the years by numerous MOF R&D projects. Filipe has received numerous awards during his academic career with the most recent one being the Vicente de Seabra Medal from the Portuguese Chemical Society and he was considered an Emerging Young Investigator in Materials Science by RSC in 2014. |
1. Introduction
Metal–Organic Frameworks (MOFs), also known as Coordination Polymers (CPs), are a class of crystalline inorganic–organic hybrid materials that keeps receiving remarkable worldwide interest from all quadrants of the scientific community, particularly those focused on chemistry or materials research. These compounds are in general easy to prepare: starting from naked metal centers (often in solution), or clusters of metals, and appropriate organic molecules bearing coordinating groups in strategically located positions (the so-called Primary Building Units – PBUs), polymeric crystalline compounds can ultimately be isolated. Indeed, in a general sense, four types of compounds can be isolated from the self-assembly of PBUs: “0D”, 1-, 2- or 3-D polymeric structures (Scheme 1). For a better understanding of the MOF versus CP classification the reader is directed to the IUPAC recommendations.1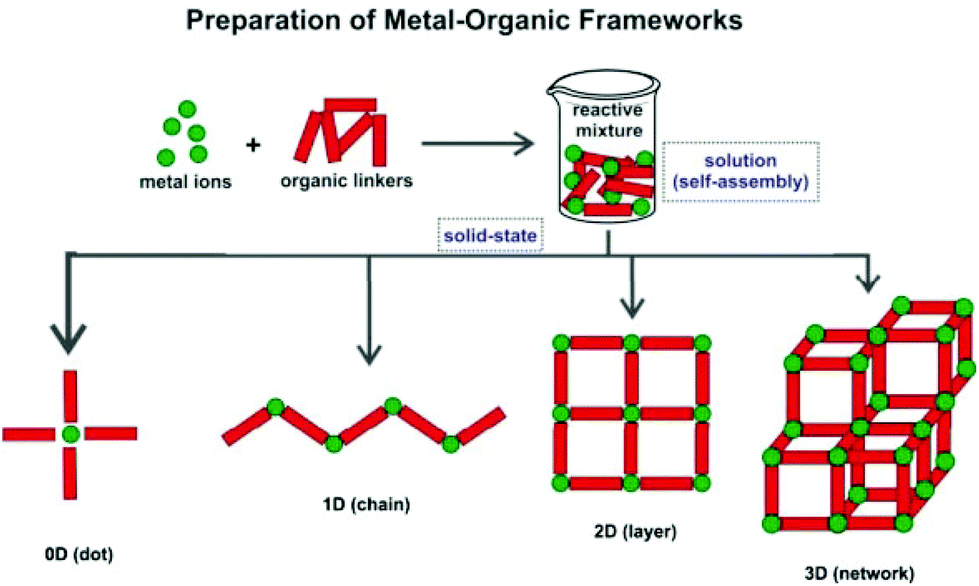 | ||
| Scheme 1 Schematic representation of the formation of MOFs/CPs with different dimensionalities (1D, 2D and 3D) from the same PBUs. | ||
The worldwide remarkable interest in MOFs/CPs remains driven by a number of basic aspects: firstly, there are endless combinations of PBUs, with new compounds being reported on a daily basis; secondly, a close control and design of the employed PBUs (and even of the synthetic conditions), can lead to new remarkable topologies; thirdly, the symbiotic presence in the same network of organic and inorganic components promotes the presence of unusual chemical or physical properties.2 Moreover, because the organic linkers could be chemically modified either before or after the MOF/CP preparation, the properties can be fine-tuned. For example, in porous materials the surface properties, such as hydrophilicity and chirality, could be changed as a result of the controlled incorporation of organic functional groups into the pore walls.3 In this context, it is thus not surprising to see reports of MOFs/CPs with a wide variety of properties and applications such as storage of gases,4–6 selective inclusion of organic molecules and ions,7 use in catalysis,8 as contrast agents,9,10 as drug delivery agents,11–13 and in CO2 uptake,14 among many others. In addition, due to the unprecedented properties of these materials some are already produced on a large scale for commercialization purposes, like the case of Cu-BTC, which has great capacity for gas separation and purification.15
We note that in the last handful of years a number of reviews have been published concerning MOF/CP synthesis and applications.16–19 One of them even tackles structural transformations by using single-crystal X-ray diffraction for a full structural elucidation.18,20 The present review aims not only to show that external stimuli can be used to induce transformations in MOF/CP compounds but also to emphasize how a small change in the structure can drastically improve or even create different properties in these materials.
2. MOF principles
2.1. Low and high dimensional MOFs/CPs
As shown in Scheme 1, and as widely reported in the literature, the polymerization of the PBUs can produce different types of coordination-based compounds. We note that the unit coined as “0D” corresponds, in fact, to the traditional discrete complex based on coordination chemistry principles: even though the organic PBU with bridging capability is present, in some cases discrete complexes are also obtained, hence their inclusion in this systematization.Many researchers refer to MOFs/CPs as solely those frameworks where the self-assembly of the PBUs occurs in the three directions of space, leading to 3D networks (Scheme 1). Others also include in this classification the crystalline compounds obtained when the polymerization is limited in one or two directions, promoting the formation of layered (2D) or chain-type (1D) compounds, respectively. Pursuing low-dimensional MOFs/CPs is not, however, a trivial task: for many combinations of PBUs, stopping the self-assembly process in one or more directions of space requires the use of additional chemical species or the close control of the experimental conditions.
For many chemical systems it is not clear why some experimental conditions lead preferentially to low dimensional MOFs/CPs (e.g., chains) while others to remarkable porous 3D networks. In a handful of cases scientists were able to exactly pinpoint the experimental conditions which control this self-assembly. For example, Liu et al.21 discovered that for the mixed-metal Mo–Cu POM-based system with 1,10-(1,4-butanediyl)bis[2-(4-pyridyl)benzimidazole] (L1), the pH plays a decisive role in the type and dimensionality of the isolated material: while a pH of ca. 3.5 promotes the formation of 1D hybrid chains Cu(HL1)2(β-Mo8O26), adjusting the pH to ca. 2.5 higher dimensional MOFs could be isolated, namely 2D [Cu(H2L1)(β-Mo8O26)(H2O)2]·3H2O and 3D [Cu(H2L1)(γ-Mo8O26)]·3H2O. However, in most experimental cases the reason for the systematic isolation of low-dimensional MOFs remains unknown. One interesting challenge posed to researchers concerns the possibility that if once a low dimensional MOF is formed, can it be transformed into a higher dimension one? For example, 1D-to-2D, 2D-to-3D or 1D-to-3D. A search in the literature reveals that these processes can indeed occur, sometimes with great improvement and fine-tuning of the properties of the materials.
2.2. Changing the dimensionality of MOFs
Chemical modifications on MOFs in order to change their dimensionality have been known for some years.22–33 Only in the last decade or so this process has, however, attracted the interest of scientists, with a handful of reviews on the subject being already available in the literature.20,34 These types of transformations occur through two general pathways: (i) single-crystal-to-single-crystal (SC–SC) transformations, and (ii) transformations via MOF dissociation, usually in solution. In the latter process, the MOF partially or completely dissolves in the solvent employed with the PBUs reorganizing themselves into a new structure. The original MOF can, therefore, be considered as the starting chemical for the formation of a new material. In SC–SC processes the MOF transformation is instead performed in the solid state, generally within the already available crystals, without the dissolution of the MOF PBUs.It is important to emphasize that post-synthetic modification of MOFs is not, usually, included in this group of transformations. Indeed, this relatively new, but highly important and widespread strategy to functionalize MOFs, almost exclusively requires the presence of 3D → 3D′ modifications taking advantage of the porous nature of the network and the maintenance of topological and porosity aspects. This subject has been extensively reviewed in the literature and for more details we direct the reader to the excellent reviews from the research groups of Cohen35 and, more recently, from Doonan.36 We note, however, that in the present review we emphasize a handful of examples where 3D → 3D′ transformations are indeed observed but always accompanied by a boost of the properties because of this change in topological or ligand connectivity.
Transformation of the dimensionality of MOFs can be performed using a wide variety of external stimuli such as temperature, pH variation, and metal exchange.
An interesting example comes from Mobin et al.22 who reported the preparation of three isotypical 1D polymers formulated as [(X)Hg(μ-X)2(L2)] (where X− = Cl−, Br− or I−; L2 = 2-(2-hydroxyethyl)pyridine). Even though the materials are 1D, interactions between the halogen and neighbouring hydrogen atoms lead to the presence of close interactions forming supramolecular layers. On heating up the materials at 383 K for a short period of 90 minutes, the compounds with Br− and I− completely lose their crystallinity. The chloride-based compound undergoes, however, a complete structural transformation as depicted in Fig. 1. This transformation is fully reversible when the material is left under ambient conditions.
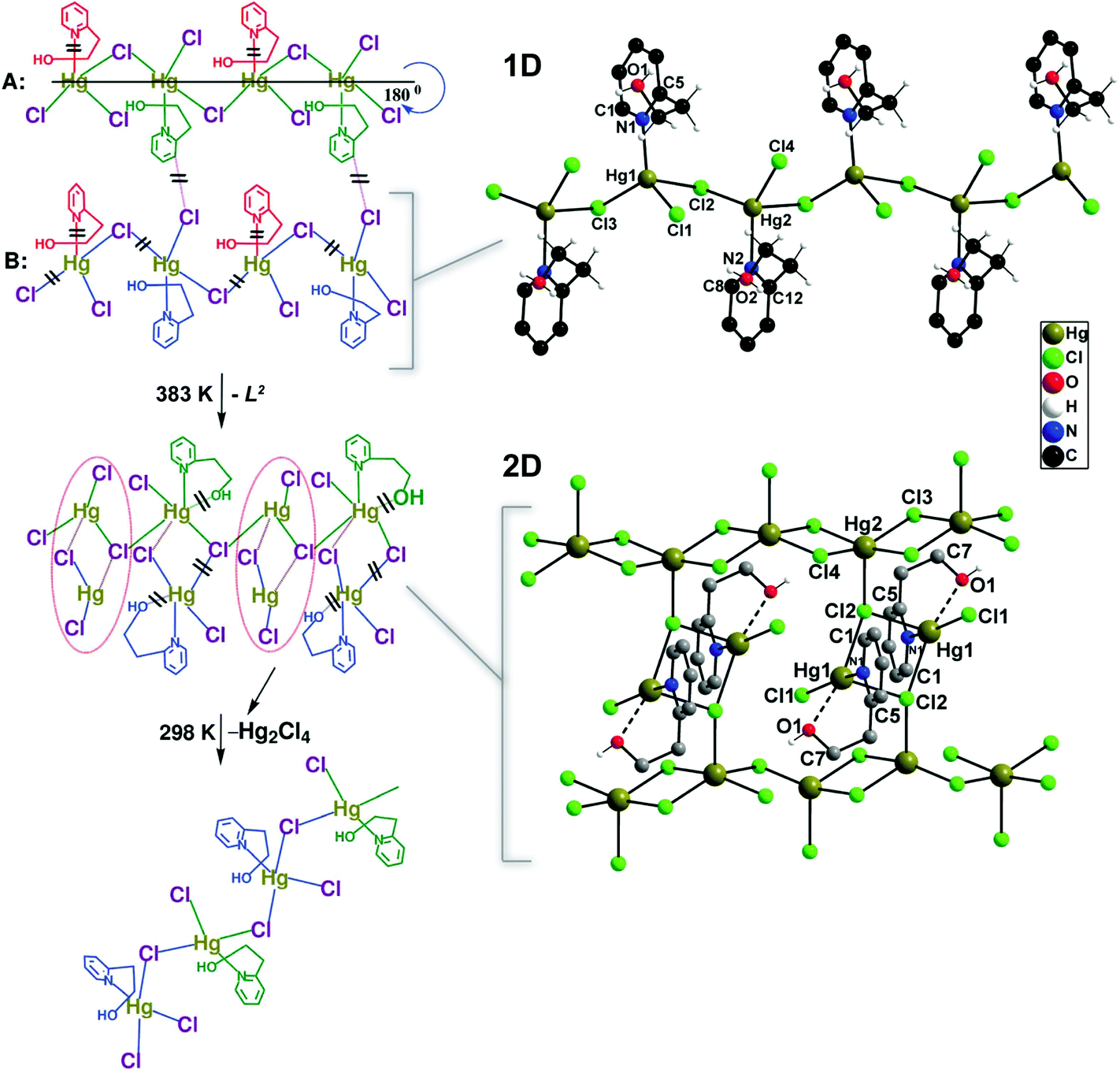 | ||
| Fig. 1 Schematic representation showing the transformation of the 1D polymer [(Cl)Hg(μ-Cl)2(L2)] into a 2D network under the influence of temperature. The proposed pathways by the authors of the original paper are depicted on the left (= bond cleavage; – new bond formation). Reprinted with permission.22 Copyright 2010 Royal Society of Chemistry. | ||
A more extensive structural dynamics was reported by Kondo et al.37 who showed the presence of remarkable stepwise transformations in a Co2+ system (Fig. 2), which could even be accompanied visually by modification of the colour of the compounds. Discrete complexes forming the [Co(bpy)2(CH3CN)2(H2O)2]·2(OTf) compound convert spontaneously into 1D coordination polymers embedded in the [Co(bpy)(OTf)2(H2O)2]·(bpy) crystal structure: acetonitrile solvent molecules and one bpy ligand are released from the coordination sphere of Co2+ being replaced by trifluoromethanesulfonate anions, promoting in this way the polymerization in one direction. Treating the 1D polymer at 423 K for a period of approximately 2 hours under vacuum, a new transformation takes place promoting the formation of a 2D layered material formulated as [Co(bpy)2(OTf)2] (Fig. 2), which exhibited interesting adsorption capabilities. In addition, the discrete complex could even be directly transformed into the layered material by simple immersion in a dried solvent over a period of 10 days. This report by Kondo et al.37 shows that the properties of one given system can be drastically modified by simply changing the dimensionality of the initially isolated compound.
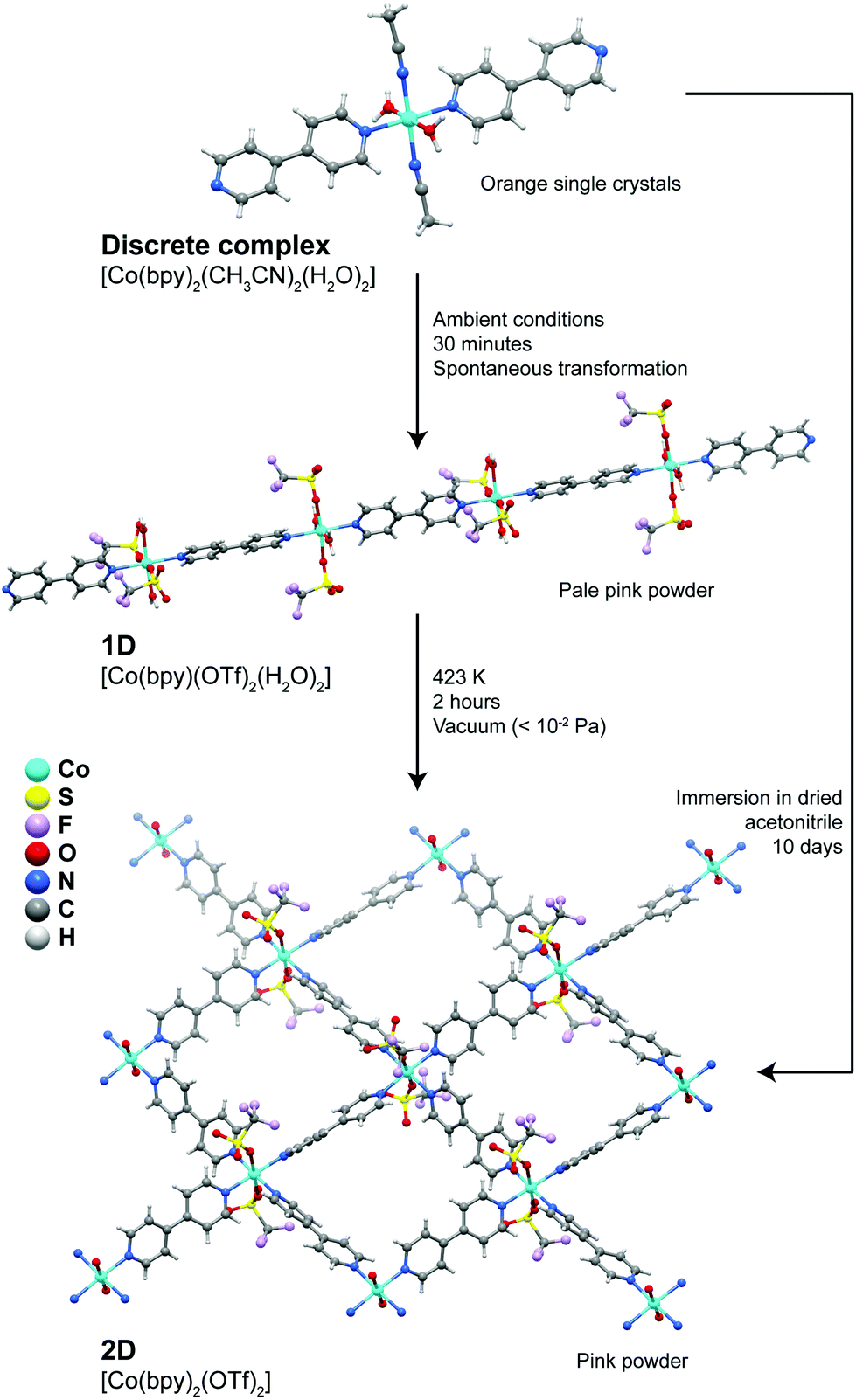 | ||
| Fig. 2 Transformation pathways and experimental conditions for the discrete → 1D, 1D → 2D and discrete → 2D conversions of the [Co(bpy)2(CH3CN)2(H2O)2] (discrete), [Co(bpy)(OTf)2(H2O)2] (1D) and [Co(bpy)2(OTf)2] (2D) compounds. Adapted from a paper by Kondo et al.37 | ||
As highlighted in the previous paragraphs the vast majority of research reports, and the known reviews on the subject, concerning the transformation of MOFs/CPs are mainly focused on purely structural aspects.20,34,39 Taking advantage of these transformations to design new and clever functional materials is not a common aim of the authors, even though in some cases this was indeed achieved and very well explored. In this context, the aim of the present review is to focus on new MOF materials, typically of higher dimensionality, prepared from low dimension ones, with the transformation being concomitantly accompanied by a boost of the observed properties.
3. Transforming MOFs/CPs: designing new functional materials
3.1. Gas adsorption
Transformation of MOFs/CPs through solvent removal or exchange is one of the most common processes reported in the literature, for which usually the dimensionality of the network remains unaltered.40–42 Aiming at potential applications the use of robust 3D networks is, thus, preferred so that the channels can easily be evacuated while the network itself remains crystalline and is able to interact with the guest gases of solvent molecules. If the solvent molecules are attached to the structuring metal centers (i.e., the framework is formed by coordinatively unsaturated metal centers), their removal can in some cases induce an extensive structural modification through the reorganization of the metal coordination spheres. Even though many reports in the literature describe the release (sometimes reversible) of solvent molecules, only a handful of reports explore this to produce functional materials.The manganese MOF [Mn3(μ5-L4)(μ6-L4)(μ-H2O)(H2O)4]·2DMF·H2O (where H3L4 = tris-(3-carboxylphenyl)phosphine oxide) reported by Jeong et al.43 is a 3D MOF with the rare feature of possessing aqua bridges between metal centers. This highly unusual structural feature among crystalline MOFs was explored by the authors so to produce a novel functional material for the adsorption of gases. In a first step, the DMF molecules were exchanged by methanol through immersion at 40 °C over a period of 24 h (please note: the process is fully reversible if the compound is soaked in DMF and water – Fig. 3). In a second step, heating the methanol-based compound at 50 °C under vacuum for 2.5 h leads to a fully desolvated material, [Mn3(L4)2], which besides retaining the main crystalline features of the methanol-based compound, also exhibits interesting adsorption properties. Indeed, the authors found that [Mn3(L4)2] adsorbs CO2 with an uptake in the order of 31.9 cm3 g−1, being approximately ten times more selective for this gas than for hydrogen (not adsorbed), nitrogen (1.8 cm3 g−1) and methane (2.9 cm3 g−1). This report of a 3D → 3D′ transformation through solvent displacement/replacement shows that simple treatments of as-prepared compounds can be used to “activate” the compounds to produce a functional material.
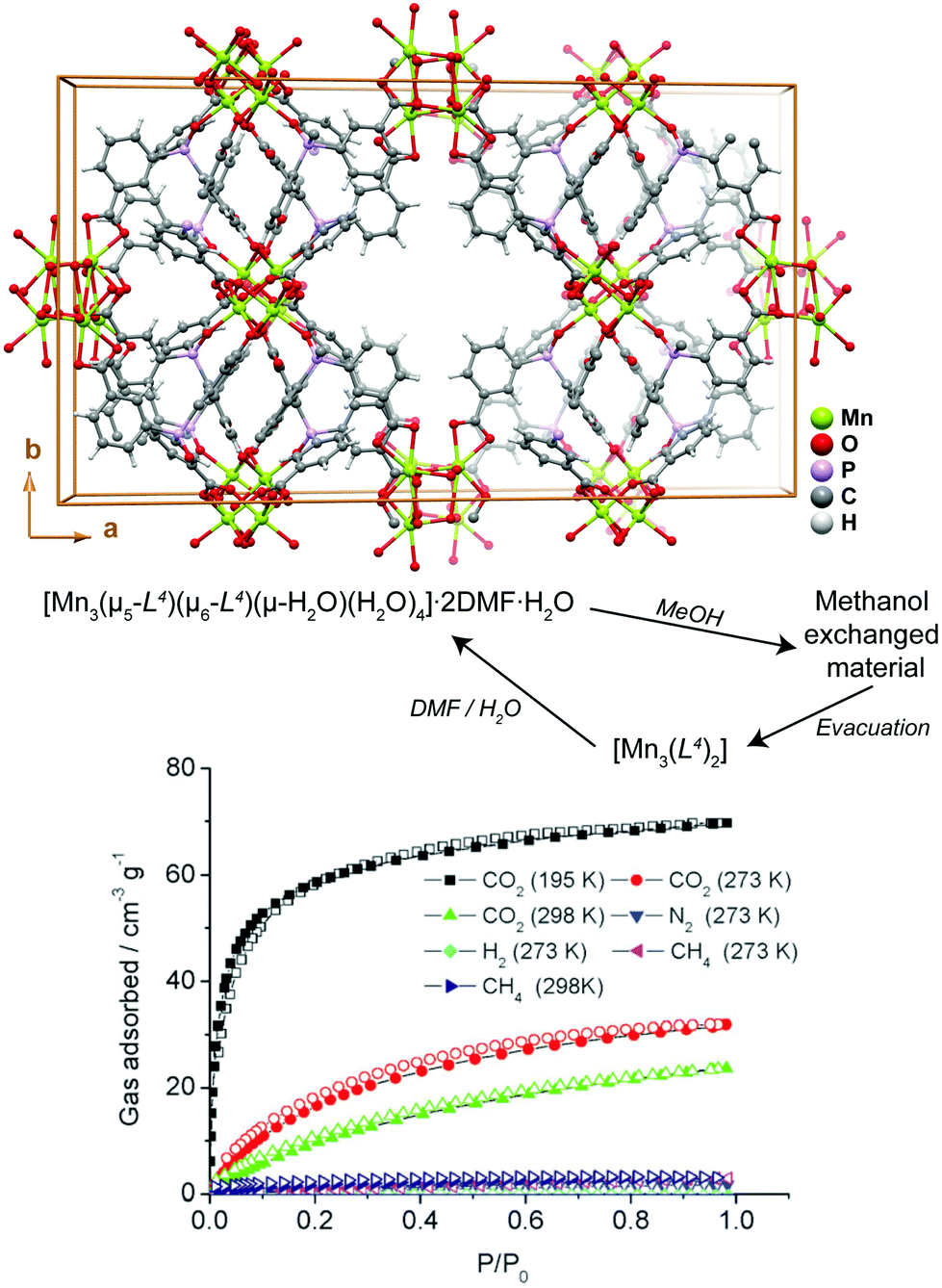 | ||
| Fig. 3 (top) Crystal packing of [Mn3(μ5-L4)(μ6-L4)(μ-H2O)(H2O)4]·2DMF·H2O viewed along the [001] direction of the unit cell. (bottom) Gas uptake isotherms of [Mn3(L4)2]. Adapted and reprinted with permission.43 Copyright 2010 Royal Society of Chemistry. | ||
A “0D” → 2D transformation through solvent replacement and structure re-organization was reported by Zhai et al.44 for Ni2+-based compounds. From hydrothermal synthesis the authors were able to isolate the [Ni3(HL5)6(H2O)6]·9H2O compound (where H2L5 = 4-(1,2,4-triazol-4-yl)phenylphosphonic acid) which is based on a discrete trinuclear Ni2+ complex. This compound was treated under hydrothermal conditions at 180 °C and low pH (ca. 3.0) for 7 hours to yield a new 2D layered CP formulated as [Ni(HL5)2]·7H2O. Indeed, this second step in the overall reaction permitted the displacement of the water molecules composing the Ni2+ coordination spheres, leading to the polymerization of the units into a novel layered material. Because the organic linker is based on phosphonate chelating groups, the resulting compound exhibited a remarkable thermal stability: it could endure refluxing benzene and ethanol for 7 days, acidic media and boiling water. The 2D CP showed a remarkable performance in the adsorption of CO2, with an overall uptake of 80 cm3 g−1, one of the highest reported to date for phosphonate-based MOFs/CPs. Moreover, the authors further showed that the material shows a very high selectivity towards CO2 from a mixture with nitrogen, with a separation ratio of ca. CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N2 of 114:1, much higher than that reported for a number of very stable ZIF materials.
:N2 of 114:1, much higher than that reported for a number of very stable ZIF materials.
The previous cases concern, basically, the removal of solvent molecules, sometimes with the modification of the dimensionality of the material, to produce functional materials. In other cases, the removal of solvent species could permit their replacement by other molecules. A remarkable example of the fabrication of a functional MOF/CP comes from the research group of Lin who prepared a 2D layered CP formulated as [Zn2(L6)(DMF)4]·2DMF·4H2O (where H4L6 = methanetetra(biphenyl-p-carboxylic acid)) using standard solvothermal synthetic conditions (reaction in DMF at 90 °C over a period of 2 days).45 The same structure could also be isolated using Co2+ metal centers. By soaking this 2D CP in dichloromethane at ambient temperature for approximately 8 hours, the authors were able to induce a 2D → 3D SC–SC transformation (the colour and morphology of the crystals remained unaltered – Fig. 4a–c) producing a new crystalline compound formulated as [Zn2(L6)(H2O)2]·xsolv. The 2D → 3D occurs at the level of the metal centers: the departure of the coordinated DMF molecules induces a dimerization, producing a new Secondary Building Unit (SBU) with water molecules in the first coordination sphere of the metal centers. This transformation was accompanied by a boost of the adsorption properties of the materials (Fig. 4d): on the one hand, the BET surface area increases from 177 to 1170 m2 g−1 (a seven-fold enhancement); on the other, the hydrogen uptake capacity (at 77 K and 1 atm) increases from 0.3 to 1.75 wt%, being comparable with the data reported for other well-known porous MOFs.
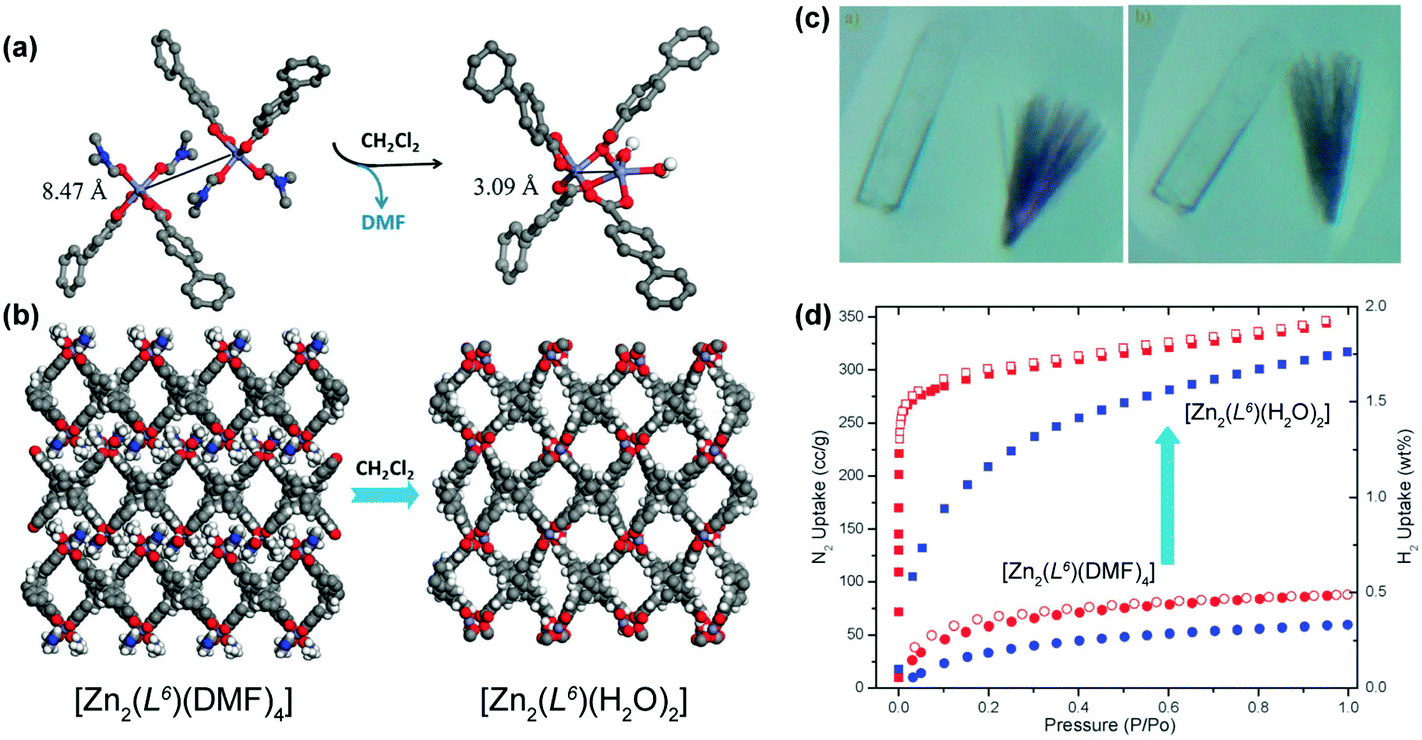 | ||
| Fig. 4 (a) Dimerization of Zn2+ units promoted by the removal of the coordinated DMF molecules. (b) Crystal packing of (left) [Zn2(L6)(DMF)4]·2DMF·4H2O and (right) [Zn2(L6)(H2O)2]·xsolv. (c) Optical photographs of the (left) as-prepared [M2(L6)(DMF)4] (Zn2+ – colourless and Co2+ – purple) compounds and (right) after exposure to dichloromethane for 3 hours. (d) Adsorption isotherms for N2 (in red) and H2 (in blue) at 77 K for [Zn2(L6)(DMF)4] and [Zn2(L6)(H2O)2]. Adapted and reprinted with permission.45 Copyright 2012 Royal Society of Chemistry. | ||
Another striking example of a new material was described by the research group of Oliver who have prepared a 3D Cd2+-based MOF formulated as [Cd(HL7)(L8)]·nDMF.46 The authors showed that this material could undergo a SC–SC transformation to a fully desolvated phase by treating the as-prepared MOF at 300 °C under vacuum. The resulting framework, with empty pores, could accommodate new organic molecules, showing a high selectivity towards benzene, chloroform, 1,4-dioxane and THF (much more than for ethanol or methanol, for example). Besides its solvent selectivity, the authors also showed that the dehydrated MOF could also adsorb gases: for example, 2.5% N2, 4.5% CO2 and 3.4% N2O (in mass).
Boosting gas adsorption on functional MOFs/CPs could also be achieved by acting directly on the chemical species present within the channels. Even though removing solvent can help to increase the available space for the adsorption of gas molecules, the presence of interacting sites to stabilize the gases is also of crucial importance. An example comes from the Long's research group who have chemically modified the charge-balancing ions present in the Mn3[(Mn4Cl)3(L9)8(CH3OH)10]2 (where H3L9 = 1,3,5-benzenetristetrazol) 3D network.47 By immersing the 3D MOF in concentrated solutions of metal chlorides (in freshly distilled methanol) for approximately one month, the charge-balancing Mn3+ cations could be replaced by others such as Li+, Cu+, Fe2+, Co2+, Ni2+, Cu2+ or Zn2+. The authors discovered that even though the different metals in the channels do not lead to significant modifications in the overall H2 adsorption, the adsorption enthalpies are however quite distinct. The most remarkable result concerns the Co2+-exchanged material which exhibits unprecedented initial adsorption enthalpies of 10.5 kJ mol−1, being one of the higher enthalpies observed for H2 adsorption in MOFs. Even though this is a rather straightforward 3D → 3D SC–SC, this remarkable Co2+-based material can only be obtained using this alternative process.
3.2. Luminescence
As exemplified in the previous sub-section, removing or exchanging solvent or guest species can induce important structural modifications that culminate with the improvement of the adsorption of gases or other molecules. The same process can, for example, be used to improve the photoluminescence behaviour of some materials. This is quite an important feature in, for example, lanthanide-based MOFs/CPs. For instance, the [Eu2(L10)2(OH)2]·3H2O (where H2L10 = pyrimidine-4,6-dicarboxylic acid) 3D MOF reported by Sun et al.48 can easily release the uncoordinated solvent molecules with the evacuated material being stable up to ca. 500 °C. In the absence of the quenchers from the crystallization water molecules the authors observed a small increase of the lifetime of the Eu3+ cations by ca. 10%. Lan et al.49 prepared a porous Zn2(L11)0.5(DMSO)(H2O) (abbreviated herein as MOF·DMSO; where H8L11 = 5,5′-(2,2-bis((3,5-dicarboxyphenoxy)methyl)propane-1,3-diyl)bis(oxy)diisophthalic acid) which could easily undergo solvent exchange and replacement (both coordinated and present in the channels) through hydrothermal treatment, leading to MOF·H2O, MOF·DMF, MOF·EtOH and MOF·DMA. The various isolated networks retained the topological features of the parent DMSO-based compound (i.e., a 3D → 3D′ transformation), but the authors observed the presence of small differences concerning the ligand coordination and conformation. Besides the 3D → 3D′ solvent exchange observed for MOF·DMSO, the authors also reported the same transformation concerning the encapsulation of the well-known Alq3 chromophore. The process could be accompanied visually with the colourless crystals of MOF·DMSO changing to yellow after 48 hours for the Alq3@MOF final compound (Fig. 5). Moreover, the 3D → 3D′ transformation of the Alq3@MOF compound permitted a blue shift of the emission spectrum of the chromophore. In this way, the modified host MOF can be envisaged as an alternative way to modulate the luminescence properties of Alq3 and improve the performance of Alq3-based opto-electronic devices. | ||
| Fig. 5 Optical photographs of Zn2(L11)0.5(DMSO)(H2O) (a, d) before and (b, e) after encapsulating Alq3 molecules. (c, f) Optical photographs for Alq3 encapsulated in Zn2(L11)0.5(DMSO)(H2O) irradiated by UV light (λ = 365 nm). Reprinted with permission.49 Copyright 2012 American Chemical Society. | ||
An unusual reversible 1D → 2D transformation was reported by Wang et al.50 who have prepared a 1D ladder-type compound isolated as [Zn(HL7)(L8)0.5(H2O)]·nH2O (where H3L7 = 1,3,5-benzenetricarboxylic acid and L8 = 1,2-bis(4-pyridyl)ethane) (Fig. 6). It is noteworthy that the organic PBUs in this material are identical to those employed by Husain et al.46 (see above) but in this case the resulting polymer has solvent molecules composing the first coordination sphere of the metallic center. Based on the location of these water molecules in relation to the crystal structure (pointing towards crystallographic interplanar spaces) the authors envisaged that if these molecules were removed a structural transformation could be triggered. Moreover, because [Zn(HL7)(L8)0.5(H2O)]·nH2O exhibits a moderate blue emission band centred at ca. 410 nm (lifetime of ca. 1.8 ns), this transformation could also induce different luminescence properties. Based on a combination of various studies, the authors eventually were able to modify the crystal structure by heating the crystals from ambient temperature to ca. 180 °C, promoting the release of all solvent molecules and forming a new layered compound formulated as [Zn(HL7)(L8)0.5]. Remarkably, the fluorescence peak from the original material hypsochromically shifts to ca. 350 nm, with the intensity increasing by a two-fold factor and is also accompanied by an increment of the lifetime to ca. 2.3 ns. This process was shown to be completely reversible in the solid state, indicating a dynamic 1D ⇔ 2D conversion according to the experimental conditions.
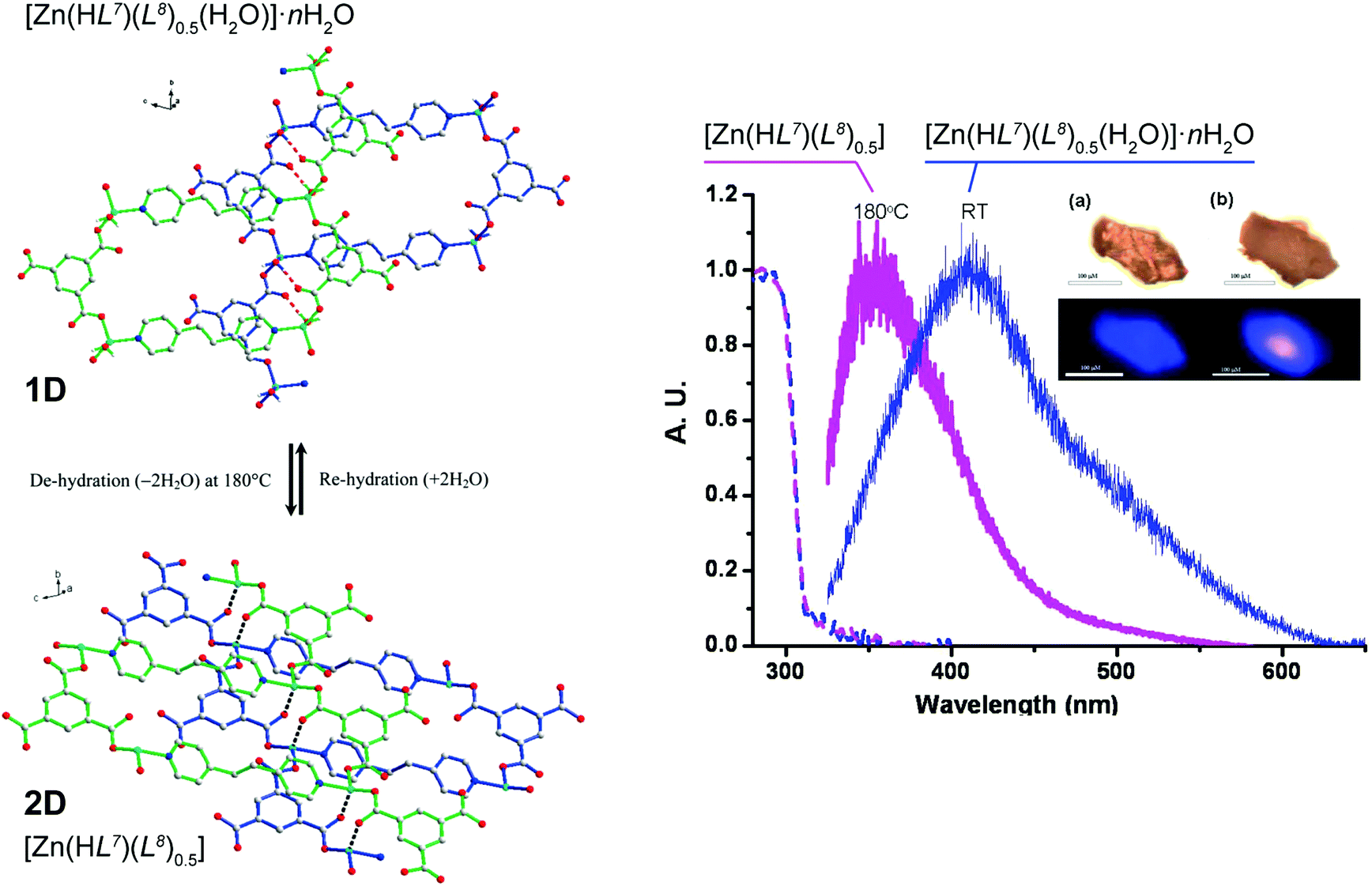 | ||
| Fig. 6 (left) Schematic representation showing the reversible 1D ⇔ 2D conversion between the [Zn(HL7)(L8)0.5(H2O)]·nH2O phase containing the 1D polymer and the fully dehydrated [Zn(HL7)(L8)0.5] 2D phase. (right) Solid state absorption (dashed lines) and emission (solid lines) of [Zn(HL7)(L8)0.5(H2O)]·nH2O at ambient temperature (in blue) and [Zn(HL7)(L8)0.5] at 180 °C (in magenta), λex = 325 nm. Both the absorption and emission spectra have been normalized. Inset: changes of color of (a, upper) [Zn(HL7)(L8)0.5(H2O)]·nH2O at ambient temperature and (b, upper) [Zn(HL7)(L8)0.5] at 180 °C. Luminescence of [Zn(HL7)(L8)0.5(H2O)]·nH2O at ambient temperature (a, lower) and [Zn(HL7)(L8)0.5] (b, lower) at 180 °C upon excitation at 325 nm. Scale bar is 100 μm. Reprinted and adapted with permission.50 Copyright 2011 American Chemical Society. | ||
3.3. Sensors
If MOF transformation is triggered by the presence or absence of a specific chemical stimulus, the system could in principle be employed in the fabrication of a sensing device. The importance of these modifications could be boosted if the modification is, for example, accompanied by a change of a visible aspect of the materials such as colour. A striking example was described by Aslani and Morsali51 who reported a SC–SC transformation of the 1D polymer [Pb2(L12)2(NO3)2(MeOH)] into the 2D layered material [Pb(L12)(NO3)] (where L12 = 8-hydroxyquinoline) (Fig. 7). The authors achieved this transformation by simply heating the as-prepared material up to ca. 165 °C, with the process being accompanied by a modification of the crystal colour from light brown to yellow (Fig. 7 – right). The transformation occurs due to the release of the coordinated methanol molecules. Remarkably, the process is fully reversible, being always of the SC–SC type: when the desolvated material is exposed to methanol it regenerates the original compound. More importantly, the process is also highly selective: in the presence of other common solvents such as ethanol, acetonitrile, dichloromethane, benzene and chloroform, transformation from the layered phase to the chain-type polymer does not occur. In summary, Aslani and Morsali51 described a remarkable CP system in which the reversible 1D ⇔ 2D transformation is solely and highly selectively triggered by the desorption/adsorption of methanol, the process easily accompanied by a drastic change in colour.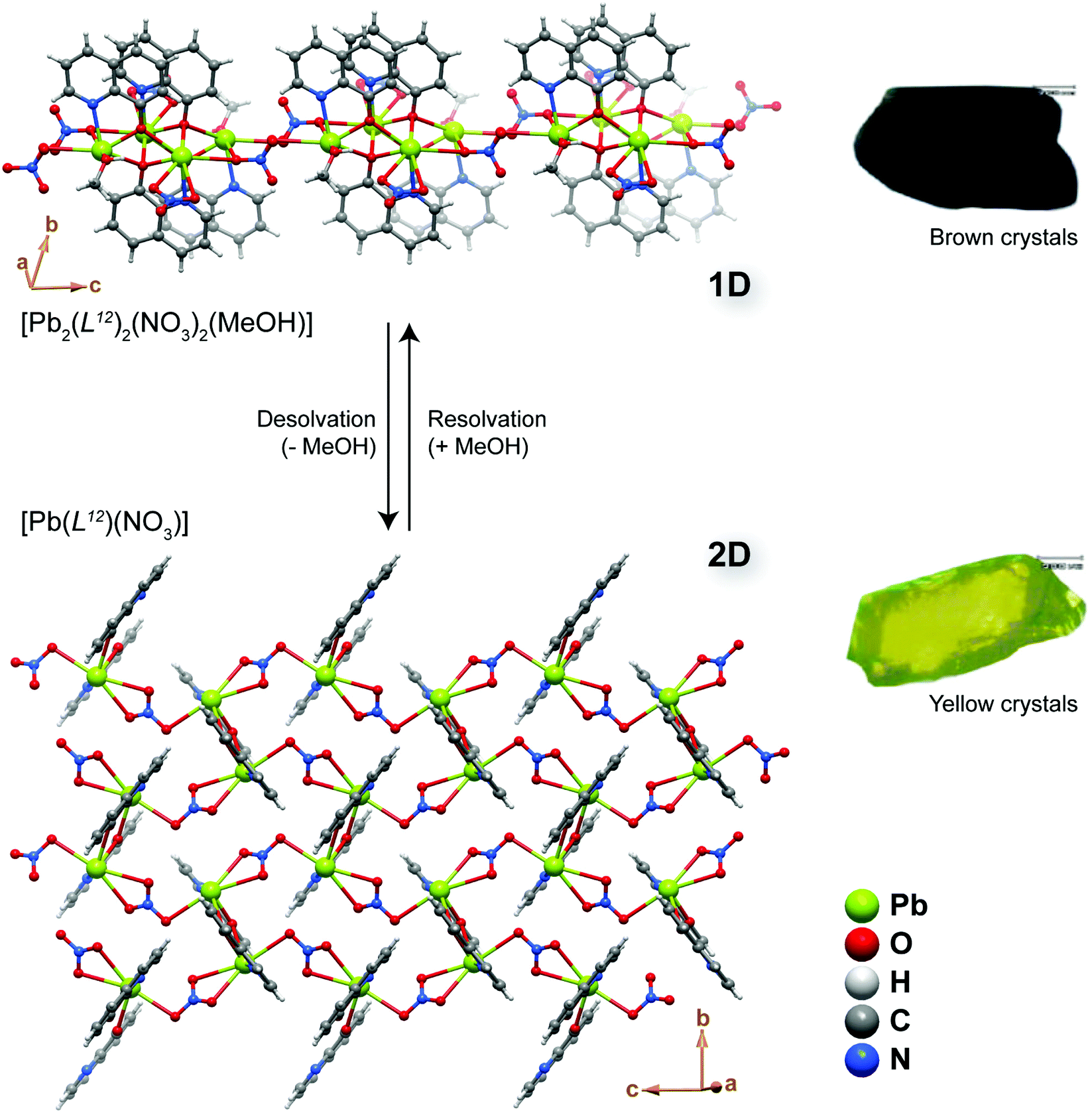 | ||
| Fig. 7 Structural representation of the reversible conversion between the 1D [Pb2(L12)2(NO3)2(MeOH)] compound and the layered 2D [Pb(L12)(NO3)] CP through the removal or inclusion of methanol molecules. During the process the crystals of the materials change colour as depicted on the right. Reprinted and adapted with permission.51 Copyright 2008 Royal Society of Chemistry. | ||
Colour modification, associated with modifications in dimensionality, could also be triggered by the pH of the medium. Yamada et al.52 investigated the transformation of a copper(II)-triazolate system known since the 60s to have both 1D and 3D networks. The authors prepared novel cubic 3D structures, formulated as [Cu3(L13)3(μ3-OH)]X2·6H2O (where HL13 = 1,2,4-triazole and X− = Cl− or Br−), from relatively simple one-pot reactions at ambient temperature between copper(II) salts and the organic PBU. The resulting [Cu3(L13)3(μ3-OH)]X2·6H2O 3D compounds were subjected to external pH stimuli in the solid state. Taking advantage of the insolubility of the 3D MOFs, the authors prepared suspensions of the compounds in water with a pH ranging from ca. 0.1 to 10 (by using HCl/Na2CO3 and HBr/Na2CO3 solutions): for the Cl−-based compound, the colour of the MOF changed from blue to pale green (Fig. 8 – top left); the same was observed for the Br−-based compound but at even more acidic pH the colour changed to brown (Fig. 8 – top right). The investigation was accompanied in detail using powder X-ray diffraction showing that the modification of the pH promoted a drastic structural modification: crystalline 3D [Cu3(L13)3(μ3-OH)]X2·6H2O compounds were gradually converted also into crystalline 1D polymers formulated as [CuX2(HL13)] (where X− = Cl− or Br−) (Fig. 8 – bottom). The authors rationalized this remarkable 3D → 1D transformation through the action of the acid on the bridges of the 3D MOF: basically, the process is triggered by the neutralization of the hydroxyl bridges and protonation of the organic linker, leading to a reorganization in the solid state of the MOF. The inverse 1D → 3D process was attempted by employing NH3 gas, but with no interesting results to be reported.
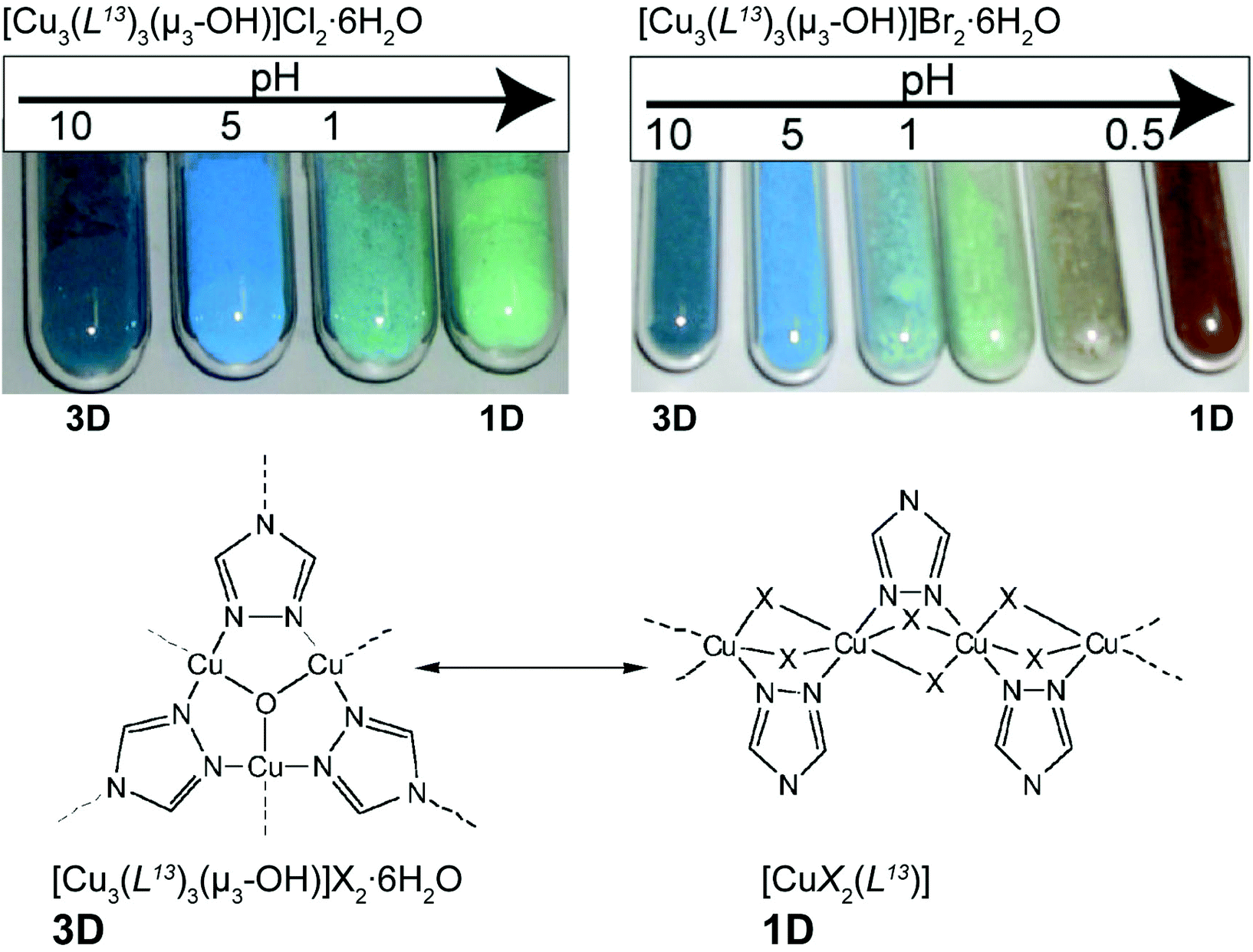 | ||
| Fig. 8 (top) Colour modification of the [Cu3(L13)3(μ3-OH)]X2·6H2O material (where X− = Cl− or Br−) according to the variation of the pH. (bottom) Structural conversion between fragments of the structures of 3D [Cu3(L13)3(μ3-OH)]X2·6H2O and 1D [CuX2(HL13)] MOFs. Reprinted and adapted with permission.52 Copyright 2011 Royal Society of Chemistry. | ||
A rarely encountered feature among crystalline structures, particularly MOFs, concerns the sensing of molecules based on drastic modifications of the resulting crystalline structures. The multidisciplinary research group headed by Lee Brammer recently presented a remarkable example of this feature in MOFs/CPs using a simple two-step process involving the removal, addition or replacement of structural framework organic linkers.53 Vapour diffusion of pyridine (L14) to an ethanolic solution of Zn2+ and (1R,3S)-(+)-camphoric acid (H2L15) produced the 2D MOF compound formulated as [Zn2(L15)2(L14)2]·2EtOH. This layered material possesses the monodentate pyridine ligands and the uncoordinated ethanol solvent molecules pointing towards the interlayer space as shown in Fig. 9. This structural feature facilitated the first step of the structural transformation: heating this parent compound at 150 °C for just 12 hours produces a new crystalline 3D material formulated as [Zn(L15)]. This process allowed the complete removal of both the uncoordinated ethanol molecules and the axially-coordinated pyridine ligands from the paddlewheel SBUs, promoting a rearrangement of the coordination spheres and the interconnection into a 3D network. This was found to be, however, a highly dynamic process (2D ⇔ 3D): reversibility of the transformation was achieved by soaking [Zn(L15)] in an ethanolic solution of pyridine. Because of this feature the authors envisaged that the structural reversibility of the material could permit the inclusion of other molecules. Indeed, two different phases are obtained simply by soaking [Zn(L15)] in solutions of either pyrazine (L16) or 3-bromopyridine (L17), also as crystalline networks (over a period of 7 days – see Fig. 9): 3D [Zn2(L15)2(L16)]·2DMF (i.e., 3D → 3D′) and 2D [Zn2(L15)2(L17)2]·DMF (i.e., 3D → 2D). Even though the authors did not indicate the existence of any visual modification of the crystals (or powders), such as colour, this system constitutes a remarkable example of a two-step dimensionality modification through sensing/replacement of structural organic linkers. As shown in the report, the transformation can be easily followed using powder X-ray diffraction since the crystallinity of the compounds is retained throughout the processes.
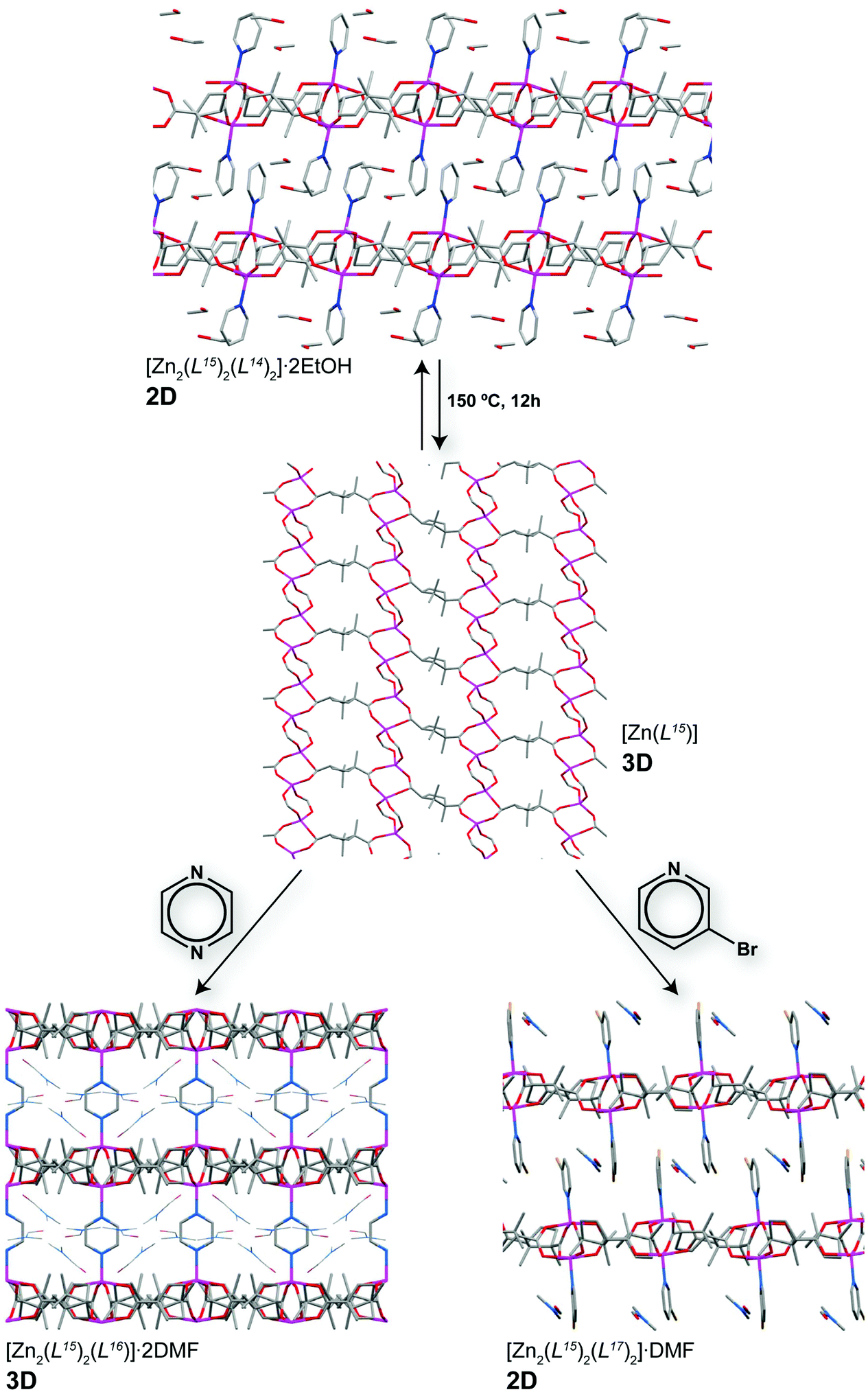 | ||
| Fig. 9 Schematic representation of the various successive framework transformations of the materials based on (1R,3S)-(+)-camphoric acid: non-covalently pillared [Zn2(L15)2(L14)2]·2EtOH undergoes a reversible 2D ⇔ 3D transformation into the 3D non-porous [Zn(L15)] compound, which when immersed in either pyrazine or 3-bromopyridine adsorbs these molecules producing 3D [Zn2(L15)2(L16)]·2DMF (i.e., 3D → 3D′) and 2D [Zn2(L15)2(L17)2]·DMF (i.e., 3D → 2D). Reprinted and adapted with permission.53 Copyright 2013 Wiley-VCH. | ||
3.4. Controlled release of molecules
Once incorporated into the structure of a MOF/CP, active or structurally interesting molecules could be released into the surrounding medium, usually impelled by an external stimulus, leading to a modification of the overall crystalline structure of the compound. An outstanding example was very recently described by He et al.54 based on the 2D Cu2+ CP prepared using standard hydrothermal reaction procedures (130 °C, 3 days) between isonicotinic acid (HL18), iodine and copper(II) nitrate: [Cu(L18)2]·I2. The resulting layered CP, (with (4,4) topology), has the striking feature of possessing molecular iodine occupying the square pores (Fig. 10 – top). The authors discovered that these molecules could be quickly released by heating the compound at 120 °C for just 8 hours under vacuum. During this SC–SC process, the 2D network undergoes a structural transformation into a 3D MOF formulated as [Cu(L18)2], motivated by the modification of the coordination modes of the organic ligand and an increase of the coordination number of Cu2+ (from 4 to 5). The molecular iodine could also be slowly released with time, as demonstrated by soaking [Cu(L18)2]·I2 in dry methanol. It is noteworthy that this process is also accompanied by a change in colour of the crystals (from black to green – Fig. 10 – bottom), which could be used as a quick identification of the presence of a material fully depleted of iodine.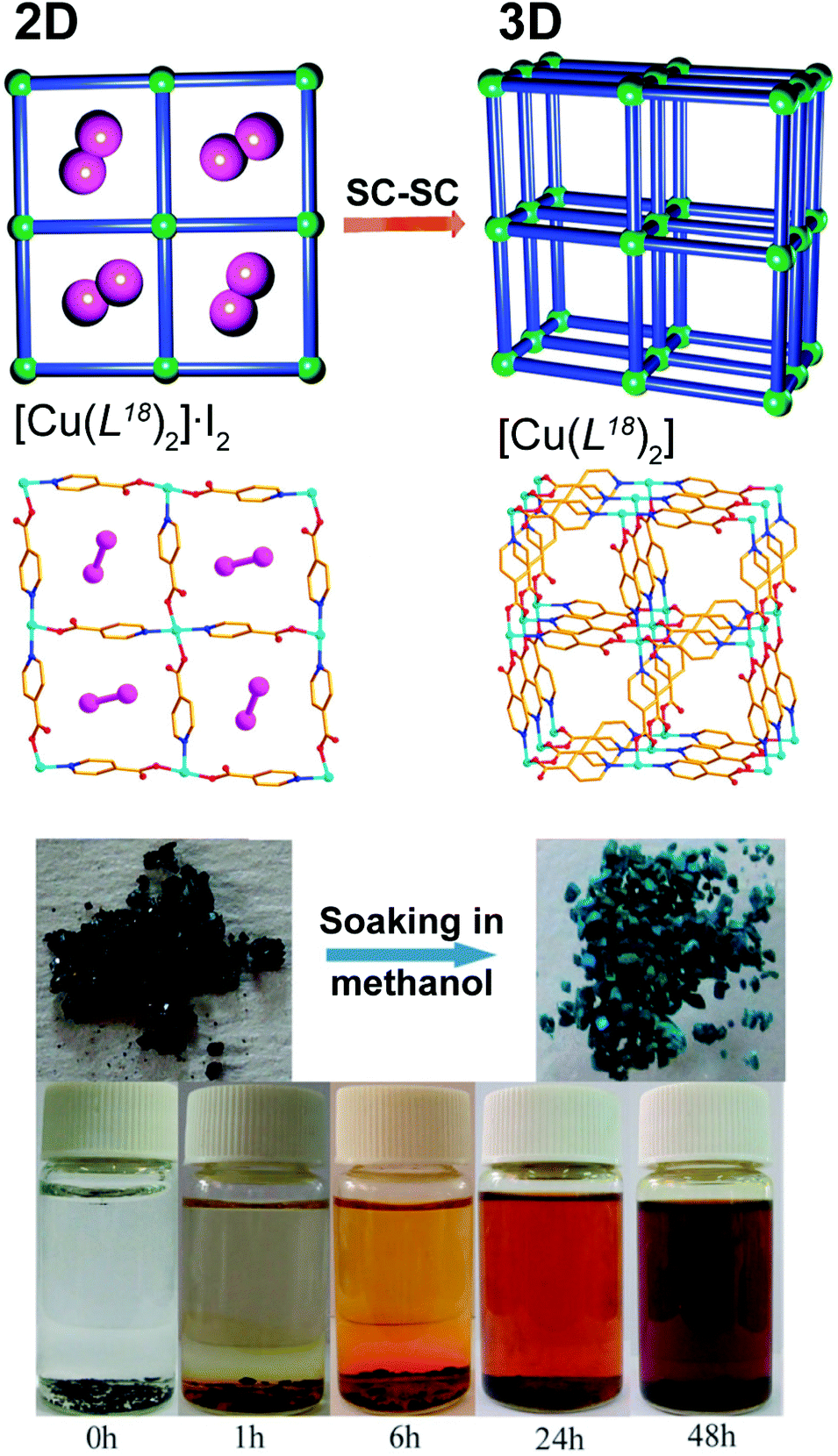 | ||
| Fig. 10 (top) Schematic representation of the 2D → 3D SC–SC transformation of [Cu(L18)2]·I2 into [Cu(L18)2] by the release of the encapsulated iodine molecules. (bottom) Colour change from black to green during the release of iodine (also revealed in the methanol suspension at the bottom). Reprinted and adapted with permission.54 Copyright 2012 Royal Society of Chemistry. | ||
3.5. Water scavengers
In many areas of industrial activity and scientific research, the presence of trace amounts of water hinders applications and activity of materials. Many solutions for this problem are known, such as the use of molecular sieves, heat, adsorption and the use of inorganic agents to promote dehydration. However, for real efficiency, in many systems the use of chemical reactions to remove all traces of water is absolutely necessary such as, for example, jet fuel. Even though MOFs/CPs are typically prepared in water, they can be used as water scavengers from a reaction medium based on structural conversion. The research group of Kitagawa described a series of this type of materials based on the highly flexible organic linker tris(2-carboxyethyl)isocyanuric acid (H3L19). From hydrothermal reactions, this flexible linker in the presence of solutions containing Ce3+ or Pr3+ nitrates and tetramethylammonium chloride (used as a blocking agent) produces a 2D layered network formulated as [Ln(L19)(H2O)2]·2H2O.55 Heating up the as-prepared compound (150 °C for 5 hours) leads to a new water-free 3D material, [Ln(L19)] (Fig. 11 – top). The authors rationalized such transformation based on the presence of coordinated water molecules in the coordination sphere of the lanthanide cations that, upon removal, lead to a rearrangement of the network. Both materials, [Ln(L19)(H2O)2]·2H2O and [Ln(L19)], are very stable in various solvents (e.g., methanol, ethanol, THF, acetone) but once the 3D phase is placed in the presence of small amounts of water it reabsorbs this solvent regenerating the original 2D compound. This remarkable selectivity was even directed towards gases: besides the aforementioned solvent molecules, the 3D network also rejected gases such as nitrogen or CO2 as they are extremely specific for water (Fig. 11 – bottom). In the following year, the same research group reported a similar SC–SC behaviour for a transition metal-based MOF,23 in which the 2D [Cu2(L19)(OH)(H2O)3]·1.5H2O compound could be transformed, reversibly, into 3D [Cu2(L19)(OH)] at an even lower temperature (ca. 80 °C for just 4 hours). This second water scavenger system even has the advantage of the reversible 2D ⇔ 3D transformation being accompanied by a change in colour: from greenish-blue (2D) to deep blue (3D).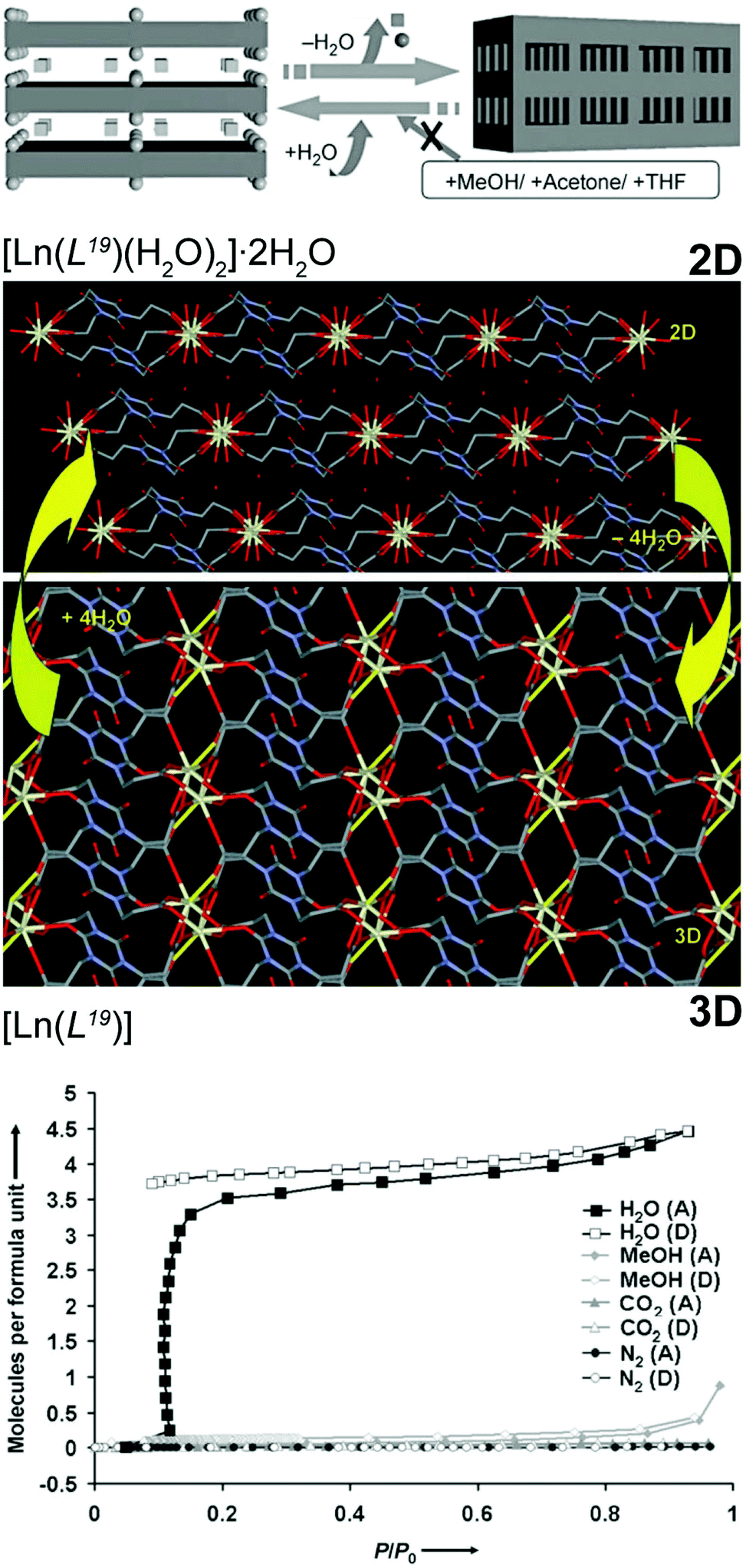 | ||
| Fig. 11 (top) Schematic representation of the reversible 2D ⇔ 3D SC–SC transformation of [Ln(L19)(H2O)2]·2H2O (where Ln3+ = Ce3+ or Pr3+) into [Ln(L19)] by the release of the water molecules. Chemical diagrams prepared from the crystallographic determinations are also provided. (bottom) Adsorption (A) and desorption (D) isotherms of H2O, MeOH, CO2 and N2 of the 3D [Ln(L19)] MOF material. Reprinted and adapted with permission.55 Copyright 2007 Wiley-VCH. | ||
3.6. Magnetic materials
It is feasible to assume that the structural rearrangements associated with the framework transformations described in the present review would also have significant impact on properties intimately associated with the metal centers such as, for example, magnetism. Surprisingly these features have not been explored and reported very much. For example, in the study of the 2D [Cu2(L19)(OH)(H2O)3]·1.5H2O ⇔ 3D [Cu2(L19)(OH)] system reported above,23 the authors indicated that the dimensionality transformation was accompanied by a significant modification of the observed magnetic properties: the increased dimensionality of [Cu2(L19)(OH)] fragmented the magnetic exchange pathways, which ultimately resulted in an overall weakening of the magnetic correlation.Zhang et al.56 described a remarkable system in which discrete (“0D”) complexes could be reversibly converted into 1D polymeric double zigzag chains with strikingly distinct magnetic properties. The achiral hexanuclear cluster [{FeIII(L20)(CN)3}4{FeII(MeCN)(H2O)2}2]·10H2O·2MeCN (“0D”; where L20 = hydrotris(pyrazolyl)borate) is closely packed in the solid state alongside with a tetrameric water cluster which imposes a physical separation between adjacent complexes. This cluster could be, however, removed from the material by heating the crystals at 150 °C under a nitrogen atmosphere, promoting a SC–SC transformation leading to the formation of 1D [FeIII(L20)(CN)3]4FeII(H2O)2FeII chiral polymers. This “0D” → 1D conversion was accompanied by a drastic modification of the magnetic properties: while the discrete cluster has a predominantly antiferromagnetic behaviour, the chiral 1D polymer is ferromagnetic. The authors also discovered that this structural transformation, and the observed properties, are fully reversible (“0D” ⇔ 1D): exposing the 1D [FeIII(L20)(CN)3]4FeII(H2O)2FeII polymer to vapours of water and acetonitrile regenerates quantitatively [{FeIII(L20)(CN)3}4{FeII(MeCN)(H2O)2}2]·10H2O·2MeCN (Fig. 12).
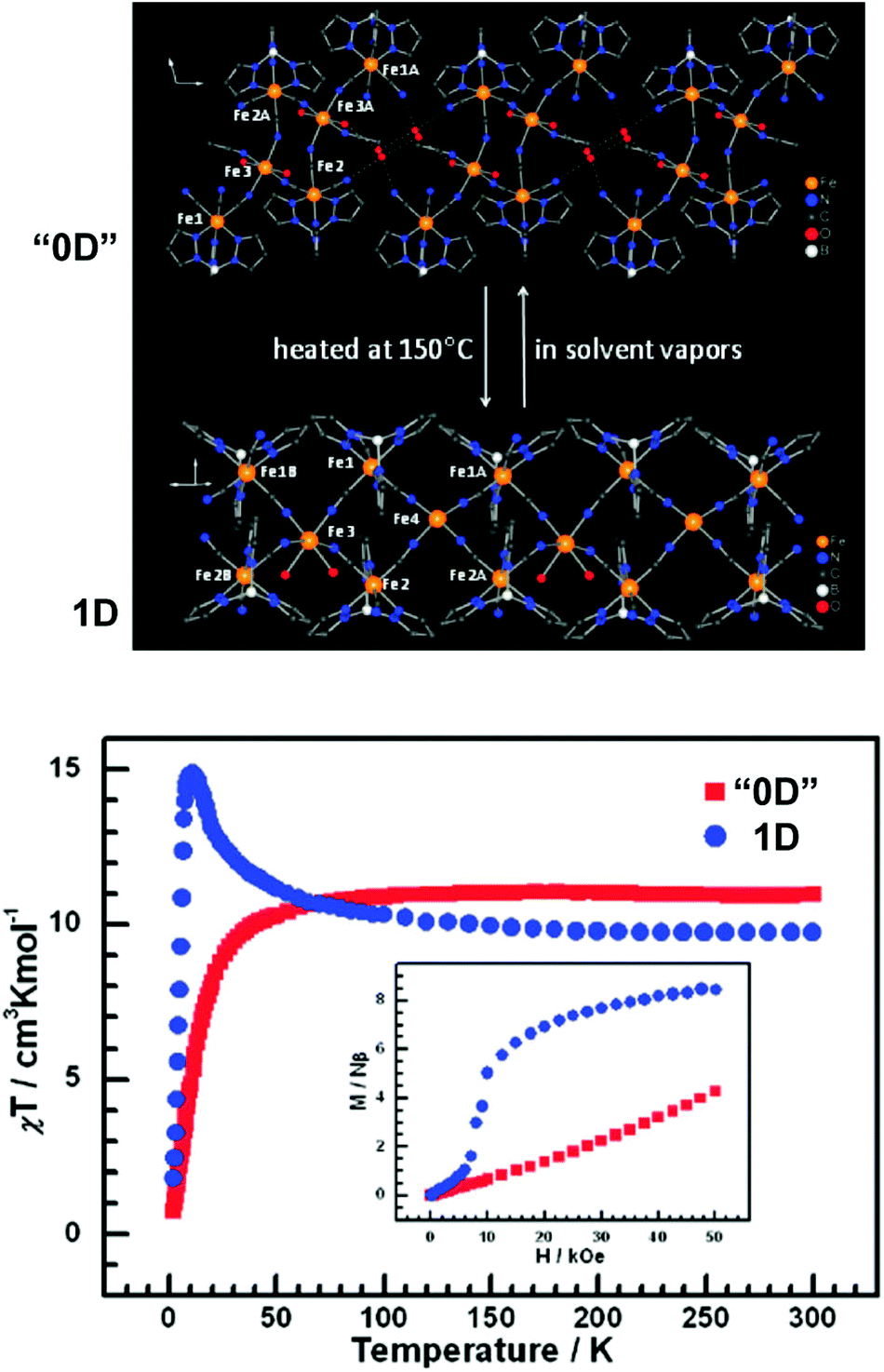 | ||
| Fig. 12 (top) Schematic representation of a portion of the crystal structures of [{FeIII(L20)(CN)3}4{FeII(MeCN)(H2O)2}2]·10H2O·2MeCN (“0D”) and [FeIII(L20)(CN)3]4FeII(H2O)2FeII (1D) showing the presence of discrete complexes and 1D polymers, respectively. (bottom) χT vs. T plots (1 kOe field) and M vs. H plots at 2 K for the “0D” and 1 D compounds. Reprinted and adapted with permission.56 Copyright 2009 American Chemical Society. | ||
4. Conclusions and outlook
The intense research activity in the field of MOFs/CPs is currently giving birth to hundreds of new crystalline structures on a weekly basis. The vast majority of these compounds are only structurally described for their crystalline features with only a handful exhibiting truly interesting properties for potential applications. This short review highlighted the efforts of some research groups who have looked into their new as-prepared MOF compounds, most of them without much applicability, and converted them into new interesting and functional materials, in most of the cases through a modification of the dimensionality of the MOF. This was usually possible through the displacement of solvent molecules composing the coordination sphere of the metallic centers. Indeed, the presence of unsaturated metal centers within these structures seems to be the best indication that some type of transformation of site-centred activity could be indeed envisaged. With this short review we also intend to demonstrate that much work still has to be done in the field of transforming inert, or poorly functional, MOFs/CPs into other new structures, typically of higher dimensionality, which can indeed exhibit interesting applications. It is, therefore, foreseen that this area of research in MOFs/CPs is still in its infancy and we should see an explosion of activity within the next few years.List of abbreviations
| 0D | Discrete complex |
| 1D | One-dimensional |
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| bpy | 4,4′-Bipyridine |
| HL13 | 1,2,4-Triazole |
| HL18 | Isonicotinic acid |
| H2L5 | 4-(1,2,4-Triazol-4-yl)phenylphosphonic acid |
| H2L10 | Pyrimidine-4,6-dicarboxylic acid |
| H2L15 | (1R,3S)-(+)-Camphoric acid |
| H3L4 | Tris-(3-carboxylphenyl)phosphine oxide |
| H3L7 | 1,3,5-Benzenetricarboxylic acid |
| H3L9 | 1,3,5-Benzenetristetrazol |
| H3L19 | Tris(2-carboxyethyl)isocyanuric acid |
| H4L6 | Methanetetra(biphenyl-p-carboxylic acid) |
| H5L3 | 2-{[2-(Dimethyl-amino)ethylimino]methyl}-6-methoxyphenol |
| H8L11 | 5,5′-(2,2-Bis((3,5-dicarboxyphenoxy)methyl)propane-1,3-diyl)bis(oxy)diisophthalic acid |
| L1 | 1,10-(1,4-Butanediyl)bis[2-(4-pyridyl)benzimidazole] |
| L2 | 2-(2-Hydroxyethyl)pyridine |
| L8 | 1,2-Bis(4-pyridyl)ethane |
| L12 | 8-Hydroxyquinoline |
| L14 | Pyridine |
| L16 | Pyrazine |
| L17 | 3-Bromopyridine |
| L20 | Hydrotris(pyrazolyl)borate |
| CP | Coordination Polymer |
| DMF | Dimethylformamide |
| MOF | Metal–Organic Framework |
| OTf | Trifluoromethanesulfonate |
| PBU | Primary Building Unit |
| POM | Polyoxometalate |
| SBU | Secondary Building Unit |
| SC–SC | Single-crystal to single-crystal transformation |
| THF | Tetrahydrofuran |
Acknowledgements
We would like to thank Fundação para a Ciência e a Tecnologia (FCT, Portugal), the European Union, QREN, and FEDER through Programa Operacional Factores de Competitividade (COMPETE). This work was developed in the scope of the project CICECO – Aveiro Institute of Materials (Ref. FCT UID /CTM/50011/2013), financed by national funds through the FCT/MEC and when applicable co-financed by FEDER under the PT2020 Partnership Agreement. We further wish to thank FCT for funding the R&D project FCOMP-01-0124-FEDER-041282 (Ref. FCT EXPL/CTM-NAN/0013/2013) and the Ph.D. scholarship no. SFRH/BD/84231/2012 (to RFM).References
- R. Batten Stuart, R. Champness Neil, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. Paik Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715 Search PubMed.
- M. J. Zaworotko, Nature, 2008, 451, 410–411 CrossRef CAS PubMed.
- R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. V. Belosludov, T. C. Kobayashi, H. Sakamoto, T. Chiba, M. Takata, Y. Kawazoe and Y. Mita, Nature, 2005, 436, 238–241 CrossRef CAS PubMed.
- S. Yang, X. Lin, A. J. Blake, G. S. Walker, P. Hubberstey, N. R. Champness and M. Schröder, Nat. Chem., 2009, 1, 487–493 CrossRef CAS PubMed.
- R. J. Kuppler, D. J. Timmons, Q.-R. Fang, J.-R. Li, T. A. Makal, M. D. Young, D. Yuan, D. Zhao, W. Zhuang and H.-C. Zhou, Coord. Chem. Rev., 2009, 253, 3042–3066 CrossRef CAS.
- Z. Xin, J. Bai, Y. Pan and M. J. Zaworotko, Chem. – Eur. J., 2010, 16, 13049–13052 CrossRef CAS PubMed.
- O. M. Yaghi, G. Li and H. Li, Nature, 1995, 378, 703–706 CrossRef CAS.
- Y.-W. Ren, J.-X. Liang, J.-X. Lu, B.-W. Cai, D.-B. Shi, C.-R. Qi, H.-F. Jiang, J. Chen and D. Zheng, Eur. J. Inorg. Chem., 2011, 4369–4376 CrossRef CAS.
- W. J. Rieter, K. M. L. Taylor, H. An, W. Lin and W. Lin, J. Am. Chem. Soc., 2006, 128, 9024–9025 CrossRef CAS PubMed.
- J. Della Rocca and W. Lin, Eur. J. Inorg. Chem., 2010, 3725–3734 CrossRef CAS.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed.
- W. J. Rieter, K. M. L. Taylor and W. Lin, J. Am. Chem. Soc., 2007, 129, 9852–9853 CrossRef CAS PubMed.
- K. M. L. Taylor-Pashow, J. D. Rocca, Z. Xie, S. Tran and W. Lin, J. Am. Chem. Soc., 2009, 131, 14261–14263 CrossRef CAS PubMed.
- S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 10870–10871 CrossRef CAS PubMed.
- U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastre, J. Mater. Chem., 2006, 16, 626–636 RSC.
- S. Qiu, M. Xue and G. Zhu, Chem. Soc. Rev., 2014, 43, 6116–6140 RSC.
- Z.-J. Lin, J. Lu, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867–5895 RSC.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- J.-P. Zhang, P.-Q. Liao, H.-L. Zhou, R.-B. Lin and X.-M. Chen, Chem. Soc. Rev., 2014, 43, 5789–5814 RSC.
- H.-Y. Liu, H. Wu, J. Yang, Y.-Y. Liu, B. Liu, Y.-Y. Liu and J.-F. Ma, Cryst. Growth Des., 2011, 11, 2920–2927 CAS.
- S. M. Mobin, A. K. Srivastava, P. Mathur and G. K. Lahiri, Dalton Trans., 2010, 39, 8698–8705 RSC.
- B.-C. Tzeng, S.-L. Wei and T.-Y. Chang, Chem. – Eur. J., 2012, 18, 16443–16449 CrossRef CAS PubMed.
- M. Gustafsson, J. Su, H. Yue, Q. Yao and X. Zou, Cryst. Growth Des., 2012, 12, 3243–3249 CAS.
- P. Díaz-Gallifa, O. Fabelo, L. Cañadillas-Delgado, J. Pasán, A. Labrador, F. Lloret, M. Julve and C. Ruiz-Pérez, Cryst. Growth Des., 2013, 13, 4735–4745 Search PubMed.
- M. Mazaj, G. Mali, M. Rangus, E. Žunkovič, V. Kaučič and N. Zabukovec Logar, J. Phys. Chem. C, 2013, 117, 7552–7564 CAS.
- Q. Chen, Z. Chang, W.-C. Song, H. Song, H.-B. Song, T.-L. Hu and X.-H. Bu, Angew. Chem., Int. Ed., 2013, 52, 11550–11553 CrossRef CAS PubMed.
- C.-H. Zhan, F. Wang, Y. Kang and J. Zhang, Inorg. Chem., 2011, 51, 523–530 CrossRef PubMed.
- M. J. Manos, E. J. Kyprianidou, G. S. Papaefstathiou and A. J. Tasiopoulos, Inorg. Chem., 2012, 51, 6308–6314 CrossRef CAS PubMed.
- J. Tian, L. V. Saraf, B. Schwenzer, S. M. Taylor, E. K. Brechin, J. Liu, S. J. Dalgarno and P. K. Thallapally, J. Am. Chem. Soc., 2012, 134, 9581–9584 CrossRef CAS PubMed.
- Z. Zhang, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, J. Am. Chem. Soc., 2013, 135, 5982–5985 CrossRef CAS PubMed.
- K. Davies, S. A. Bourne and C. L. Oliver, Cryst. Growth Des., 2012, 12, 1999–2003 CAS.
- R. Medishetty, L. L. Koh, G. K. Kole and J. J. Vittal, Angew. Chem., Int. Ed., 2011, 50, 10949–10952 CrossRef CAS PubMed.
- J. J. Vittal, Coord. Chem. Rev., 2007, 251, 1781–1795 CrossRef CAS.
- Z. Q. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Soc. Rev., 2014, 43, 5933–5951 RSC.
- A. Kondo, T. Nakagawa, H. Kajiro, A. Chinen, Y. Hattori, F. Okino, T. Ohba, K. Kaneko and H. Kanoh, Inorg. Chem., 2010, 49, 9247–9252 CrossRef CAS PubMed.
- H.-C. Fang, J.-Q. Zhu, L.-J. Zhou, H.-Y. Jia, S.-S. Li, X. Gong, S.-B. Li, Y.-P. Cai, P. K. Thallapally, J. Liu and G. J. Exarhos, Cryst. Growth Des., 2010, 10, 3277–3284 CAS.
- C. K. Brozek and M. Dinca, Chem. Soc. Rev., 2014, 43, 5456–5467 RSC.
- E. Coronado, M. Giménez-Marqués and G. Mínguez Espallargas, Inorg. Chem., 2012, 51, 4403–4410 CrossRef CAS PubMed.
- P. Mahata, C.-M. Draznieks, P. Roy and S. Natarajan, Cryst. Growth Des., 2012, 13, 155–168 Search PubMed.
- H.-L. Jiang, Y. Tatsu, Z.-H. Lu and Q. Xu, J. Am. Chem. Soc., 2010, 132, 5586–5587 CrossRef CAS PubMed.
- E. Jeong, W. R. Lee, D. W. Ryu, Y. Kim, W. J. Phang, E. K. Koh and C. S. Hong, Chem. Commun., 2013, 49, 2329–2331 RSC.
- F. Zhai, Q. Zheng, Z. Chen, Y. Ling, X. Liu, L. Weng and Y. Zhou, CrystEngComm, 2013, 15, 2040–2043 RSC.
- L. Wen, P. Cheng and W. Lin, Chem. Commun., 2012, 48, 2846–2848 RSC.
- A. Husain, M. Ellwart, S. A. Bourne, L. Öhrström and C. L. Oliver, Cryst. Growth Des., 2013, 13, 1526–1534 CAS.
- M. Dincǎ and J. R. Long, J. Am. Chem. Soc., 2007, 129, 11172–11176 CrossRef PubMed.
- H.-L. Sun, D.-D. Yin, Q. Chen and Z. Wang, Inorg. Chem., 2013, 52, 3582–3584 CrossRef CAS PubMed.
- Y.-Q. Lan, H.-L. Jiang, S.-L. Li and Q. Xu, Inorg. Chem., 2012, 51, 7484–7491 CrossRef CAS PubMed.
- C.-C. Wang, C.-C. Yang, C.-T. Yeh, K.-Y. Cheng, P.-C. Chang, M.-L. Ho, G.-H. Lee, W.-J. Shih and H.-S. Sheu, Inorg. Chem., 2010, 50, 597–603 CrossRef PubMed.
- A. Aslani and A. Morsali, Chem. Commun., 2008, 3402–3404 RSC.
- T. Yamada, G. Maruta and S. Takeda, Chem. Commun., 2011, 47, 653–655 RSC.
- P. Smart, C. A. Mason, J. R. Loader, A. J. H. M. Meijer, A. J. Florence, K. Shankland, A. J. Fletcher, S. P. Thompson, M. Brunelli, A. H. Hill and L. Brammer, Chem. – Eur. J., 2013, 19, 3552–3557 CrossRef CAS PubMed.
- Y.-C. He, J. Yang, G.-C. Yang, W.-Q. Kan and J.-F. Ma, Chem. Commun., 2012, 48, 7859–7861 RSC.
- S. K. Ghosh, J.-P. Zhang and S. Kitagawa, Angew. Chem., Int. Ed., 2007, 46, 7965–7968 CrossRef CAS PubMed.
- Y.-J. Zhang, T. Liu, S. Kanegawa and O. Sato, J. Am. Chem. Soc., 2009, 131, 7942–7943 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2015 |