
Tailoring magnetic properties of molecular materials through non-covalent interactions
Matteo
Atzori
*a,
Angela
Serpe
a,
Paola
Deplano
a,
John A.
Schlueter
*bc and
Maria
Laura Mercuri
*a
aDipartimento di Scienze Chimiche e Geologiche, Università degli Studi di Cagliari, S.S. 554, Bivio per Sestu, I09042 Monserrato (Cagliari), Italy. E-mail: m.atzori@unica.it; mercuri@unica.it
bMaterials Science Division, Argonne National Laboratory, Lemont, Illinois 60439, USA
cDivision of Materials Research, National Science Foundation, Arlington, Virginia 22230, USA. E-mail: jaSchlueter@anl.gov
First published on 19th December 2014
Abstract
Supramolecular non-covalent interactions, which drive the molecular packing of crystalline functional molecular materials, can be employed as effective tools for mediating magnetic exchange interactions. Specifically, directional hydrogen- and halogen-bonds and π–π interactions can be used to design materials in which their magnetic exchange pathways and strength can increasingly be predicted. Specific examples are presented herein and discussed with the aim of gaining deeper insight into the structure–property relationships that provide a powerful tool to afford new materials with unprecedented physical properties.
Introduction
Interest in molecule-based materials, namely materials built from pre-designed molecular building blocks, has increased in recent years since they are known to exhibit many technologically important properties (e.g., magnetic ordering, electrical conductivity, superconductivity, ferroelectricity etc.) traditionally considered to be solely within the realm of classic atom-based inorganic solids such as metals, alloys or oxides.1 The vast potential of molecular chemistry provides unprecedented possibilities for the design of molecules with the desired size, shape, charge, polarity, and electronic properties. Molecular materials are obtained by arranging such molecules in the solid state through ‘soft’, frequently solution-based routes, involving coordination or supramolecular chemistry.Intermolecular interactions play a key role in assembling these molecular bricks into 1-, 2- and 3-dimensional (D) arrays with the desired structure and functionality. These non-covalent supramolecular interactions include hydrogen- and halogen-bonds, cation–anion electrostatic, π–π, dipole–dipole and van der Waals interactions. A given solid may involve any combination of these with their strength and directionality determining the supramolecular architecture of materials and their physical properties.1 In this context, crystal engineering, the art of designing synthetic crystalline materials through the knowledge of the electronic, steric, topological and intermolecular interactions between the functional groups of their constituent building blocks,2,3 offers a powerful tool for preparing novel molecular materials with appealing physical properties.
In the field of molecular magnets, covalent interactions have been widely employed, mainly by means of bridging ligands between metal centers, to obtain compounds showing cooperative magnetic properties such as ferromagnetism, ferrimagnetism, metamagnetism, and antiferromagnetism.4 Moreover, the spatial arrangement and magnitudes of these couplings can lead to systems such as spin ladders or spin liquids.4
It has been recently established, both from experimental and theoretical studies, that hydrogen and halogen bonds, as strong, specific and highly directional non-covalent interactions, can be, in some cases, as effective as covalent interactions in promoting ferromagnetic (FM) or antiferromagnetic (AFM) interactions.5–23 The use of non-covalent interactions is limited and remains one of the major challenges since they may act both as organizers of the crystal architecture and, in some cases, as mediators of the magnetic exchange coupling between metal centers. Moreover, while covalently linked molecular materials (e.g. n-D coordination polymers, single chain magnets, etc.) show, in most cases, well understood magnetic behaviors, the non-covalently linked systems do not. In fact, they need advanced theoretical models due to the great diversity of structural parameters involved in such interactions, i.e. the nature of the acceptor (A) and donor (D) atoms, the nature of the interacting metal centers, the A⋯D distances and angles, etc.20–24
The aim of the present work is to highlight the capability of these intermolecular interactions in providing efficient magnetic exchange pathways between paramagnetic metal centers and not to review all non-covalently linked molecule-based materials. The crystal engineering of such materials represents a contemporary research challenge and may be considered as the new frontier towards the construction of new molecule-based materials with technological applications. Some of the most relevant examples of crystallographic design of molecular materials where magnetic properties are mediated by non-covalent interactions will be discussed. Ultimately, a deep insight into the structure–property relationships that govern magnetic exchange will provide a powerful way to afford new materials with unprecedented physical properties.
Discussion
Magnetic coupling mediated by hydrogen bonds
H-bonds (hereafter HBs) are ideal noncovalent interactions for rationally constructing supramolecular architectures by molecular self-assembly because they are highly selective and directional.25,26 They are formed when a donor molecule (D) with an available acidic hydrogen atom interacts with an acceptor site (A) on an adjacent molecule carrying an available nonbonding electron lone pair.25,26 Molecular self-assembling offers an attractive tool to obtain supramolecular functional materials with desired physical properties. H-bonding interactions have been widely used to control such molecular assemblies during crystallization thereby engineering both the crystal structures and the physical properties of the resulting molecule-based materials.27 In this respect, several experimental studies have highlighted the importance of HBs in magnetic systems of various dimensions.5–14One of the most powerful strategies to design such materials is based on the use of tectons,2,3 in particular metallotectons which are metal complexes capable of being involved in well identified intermolecular interactions such as HBs. As such, they can serve as building blocks for the rational construction of crystals. There are several advantages of employing such types of metallotectons to construct supramolecular architectures. In particular it is possible to tune both the coordination geometry around the metal center, the bonding capability at the complex's periphery, and the metallotecton shape by varying the metal, its oxidation state, and/or the organic ligands used. When using paramagnetic metallotectons, HBs play an additional structural role that can strongly influence the magnetic properties of the obtained molecular materials since they may force the peripheral donor atoms in close proximity causing their orbital overlap. This can promote a magnetic spin exchange leading, in some cases, to AFM or FM interactions. These interactions range from weak to strong, depending on the nature of the donor and acceptor atoms involved in the D–H⋯A bonds, the bond lengths and angles, the M⋯M distance, etc. Moreover, as thoroughly reported for covalently linked systems,28 the symmetry of the interacting magnetic orbitals plays a crucial role in the magnitude and the sign of the magnetic coupling.9,20–22
Although the role of HBs in mediating magnetic coupling between paramagnetic centers was recognized three decades ago,29 only recently, experimental and theoretical studies have motivated the scientific community to understand, quantify, and rationalize the magnetic properties of such systems.5–14,20–24
In the following, selected examples of molecular materials showing magnetic properties mediated by HBs are reported and discussed with the aim to highlight their peculiarities and potentialities.
The linear chain coordination polymer of formula [CuF2(H2O)2(pyz)] (pyz = pyrazine) was studied by single crystal X-ray diffraction at various temperatures, SQUID magnetometry, pulsed-field magnetization, electron spin resonance (ESR) spectroscopy, and muon-spin relaxation (μSR)30 to investigate its structural, electronic, and magnetic properties.5 In this compound, each Cu2+ ion is located at the center of a distorted CuF2O2N2 octahedron showing axial elongation along the Cu–N bonds due to Jahn−Teller distortion. An extensive network of H-bonding interactions links the structural 1D –Cu–pyz–Cu– chains into a rigid 3D H-bonded supramolecular structure where each fluoride ion forms a HB with two water molecules from two different chains (Fig. 1).5
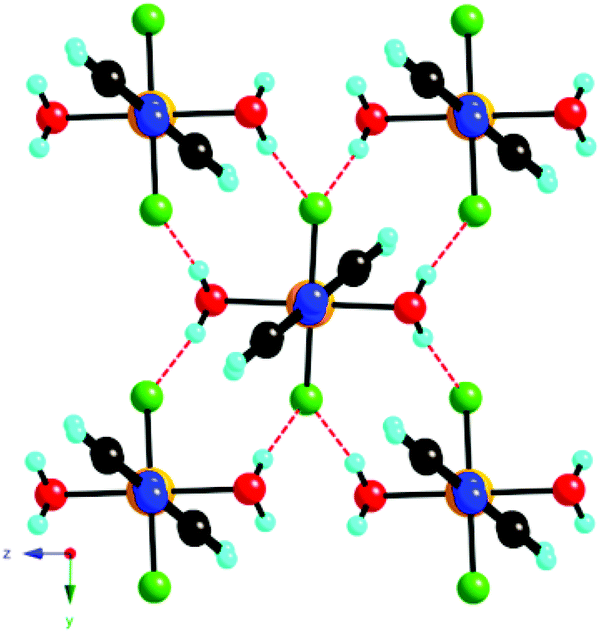 | ||
| Fig. 1 View of the molecular packing of [CuF2(H2O)2(pyz)] along the chain axis showing the O–H⋯F HB interactions between different 1D chains. Color codes: Cu = orange, F = green, O = red, N = blue, C = black and H = cyan. | ||
As the hydrogen atoms of the water molecule are not crystallographically equivalent, these H-bonding interactions are characterized by two slightly different O–H⋯F distances, 2.617(2) and 2.601(2) Å, associated with bond angles of 167.1(4) and 172.7(4)°, respectively, that have been accurately determined from neutron diffraction at 27 K.31 The intrachain Cu⋯Cu separation is 7.625(1) Å, whereas the interchain separations are 7.549(1) and 6.845(1), along the b and c directions, respectively.5
This arrangement gives rise to a quasi-2D rectangular lattice where the dx2−y2 magnetic orbital of the Cu2+ ion lies in the CuF2O2 plane, as confirmed by first principles DFT calculations and ESR measurements, thereby indicating that the nearest-neighbor spin exchange interaction J2D within the bc-plane would lead to a 2D spin lattice (Fig. 2).5 The speculation that the magnetic primary (2D) magnetic exchange occurs through a H-bonded network has been confirmed experimentally through deuterium isotope labelling experiments in which a shift in Tn is observed upon deuteration of the coordinated water molecules, but not upon deuteration of the pyrazine ligands.31,32
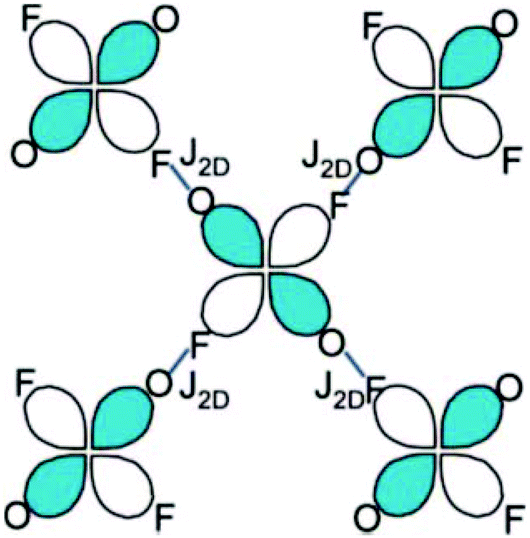 | ||
| Fig. 2 Scheme of the primary superexchange paths, J2D, between H-bonded CuF2O2N2 octahedra. The short blue line represents the F⋯H–O HB interaction. Reprinted with permission from ref. 5. Copyright 2008 American Chemical Society. | ||
The magnetic properties of [CuF2(H2O)2(pyz)] were studied between 2 and 300 K by DC and AC magnetic susceptibility measurements and high-field magnetization. The thermal variation of χ shows a broad maximum at 10.5 K due to a magnetic exchange characterized by a J2D exchange constant, the dominant exchange coupling in the system. Below 10.5 K, the susceptibility decreases smoothly and then increases abruptly near 4.3 K (Fig. 3).5
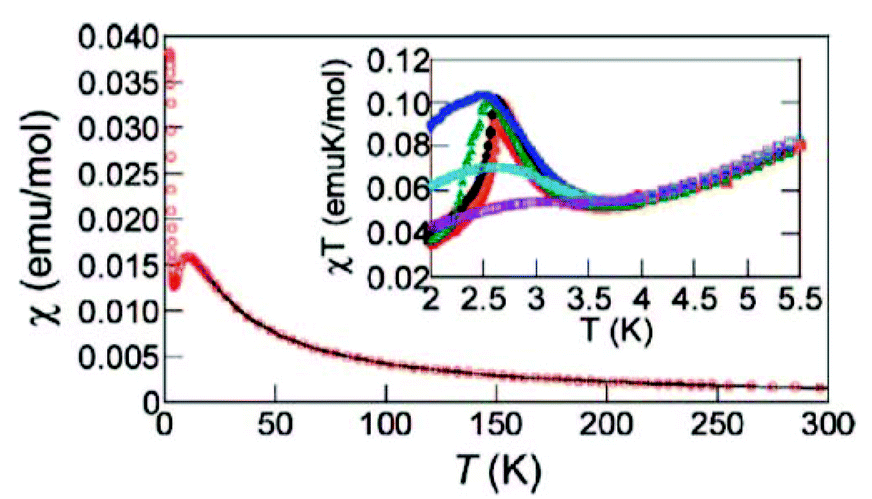 | ||
| Fig. 3 χ vs. T plot for [CuF2(H2O)2(pyz)]. The solid line represents the theoretical fit. Inset shows the χT vs. T plot at different external field values (50 G = red, 100 G = orange, 250 G = black, 500 G = green, 1000 G = blue, 2500 G = cyan, 5000 G = purple). Reprinted with permission from ref. 5. Copyright 2008 American Chemical Society. | ||
A Curie–Weiss fit of χ between 35 and 300 K gave g = 2.28(1) and θ = −15.1(1) K, which indicates the presence of a dominating AFM coupling between the Cu2+ spin centers. Interestingly, the inset in Fig. 3 shows that χT is strongly field-dependent below 3.5 K. This behaviour is most likely due to weak AFM interactions or the onset of a spontaneous magnetic moment as a result of a spin canting.5
The χ vs. T was also fitted with an S = 1/2 2D Heisenberg square lattice model based on the Hamiltonian H = JijΣSi·Sj, and an excellent agreement with the experimental data was obtained between 10 and 300 K for the parameters g = 2.24(1), J2D/kB = −5.58(1) K and zJ⊥/kB = − 0.4(1) K, where J⊥ indicates the intrachain magnetic interactions between Cu2+ ions covalently connected through the pyz ligand. Moreover, μSR measurements indicate that this material is magnetically ordered below 2.62 K, and this long-range ordering is attributed to a spin-canted state that is induced by the strengthening of the O–H⋯F HB network when the temperature is lowered.5 Elastic neutron diffraction measurements indicate a collinear antiferromagnetic structure with moments oriented along the [0.7 0 1] real-space direction and an ordered moment of 0.60 ± 0.03μB/Cu. The spin wave dispersion from the magnetic zone center to the zone boundary points (0.5 1.5 0) and (0.5 0 1.5) can be described by a 2D Heisenberg model with a nearest-neighbor magnetic exchange constant J2D = 0.934 ± 0.0025 meV. The intrachain interaction J⊥ in this compound is less than 1.5% of J2D.33
Perhaps one of the most remarkable properties of this material is that the pressure induces a sequential reorientation of the Jahn–Teller axes. As discussed above, at ambient pressure, the Jahn–Teller axis lies along the Cu–N axis, but near 1 GPa, it reorients along the Cu–O axis,34 resulting in a lowering of the magnetic dimensionality from 2D to 1D. A second reorientation occurs near 3 GPa of pressure, where the Jahn–Teller axis rotates to lie along the Cu–F axis (Fig. 4).
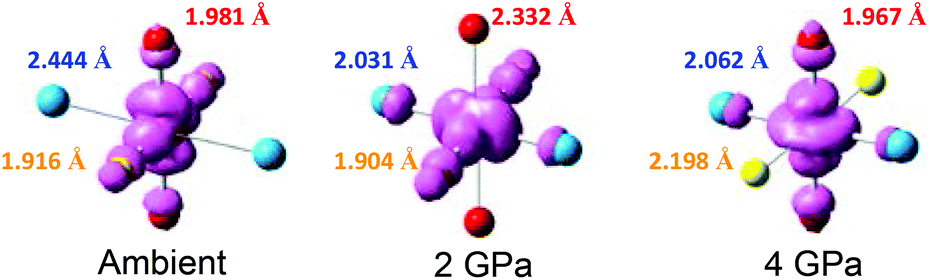 | ||
| Fig. 4 Schematic illustration of the pressure-induced reorientation of the magnetic orbitals in [CuF2(H2O)2(pyz)]. Color codes: F = yellow, O = red, N = cyan. | ||
A further pressure-induced phase has been observed in which one of the water molecules leaves the copper coordination sphere resulting in a proposed ladder-like magnetic structure.35 The reorientation of magnetic orbitals has been confirmed by ESR measurements under pressure,35 while the resulting change in magnetic dimensionality has been confirmed through the use of high-field magnetization and μSR measurements under pressure.36
In a subsequent work, an analogous material, where the Cu–pyz–Cu coordination polymer is replaced by a non-bridging molecular unit, has been prepared and structurally and magnetically characterized. In this system the pyz bridging ligand is replaced by the monodentate 3-chloropyridine (3ClPy) ligand where the interlayer coupling is achieved by π–π interactions.6,7 Also in this case, the molecular units [CuF2(H2O)2(3ClPy)] self-assemble in a H-bonded molecular material that exhibits a long-range magnetic order mediated by the H-bonded network.6,7
As the chemistry of fluoride is markedly different from that of the other halides, new synthetic strategies must be employed to prepare fluoride-based coordination polymers.37 Thus, reports on analogous systems in which H-bonding to fluoride ligands may provide structural stability and magnetic exchange are rare in the literature. Studies in this direction may be of relevant interest in order to better understand the potential to utilize these phenomena to design and control magnetic exchange.
Another interesting example where HBs are responsible for magnetic exchange interactions was reported for the compound K4[Fe(C5O5)(H2O)2](HC5O5)2·4H2O (C5O52− = croconate, dianion of the croconic acid, H2C5O5).8 In this system the paramagnetic FeII metal centers are coordinated by two chelating croconate ligands in the equatorial plane (Fe–O bond distances of 2.138(1) and 2.222(1) Å) and two trans axial water molecules (Fe–O bond distance of 2.079(1) Å). In the crystal structure, this mononuclear building block is arranged in layers connected through an extensive network of moderately-strong H-bonding interactions. The strongest interaction in the structure is related to the O–H⋯O HB between the coordinated water molecule and a peripheral C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O oxygen atom of a ligand of an adjacent unit (O1w–H1A⋯O3 bond distance of 2.771 Å and angle of 174(3)°) (Fig. 5).8
O oxygen atom of a ligand of an adjacent unit (O1w–H1A⋯O3 bond distance of 2.771 Å and angle of 174(3)°) (Fig. 5).8
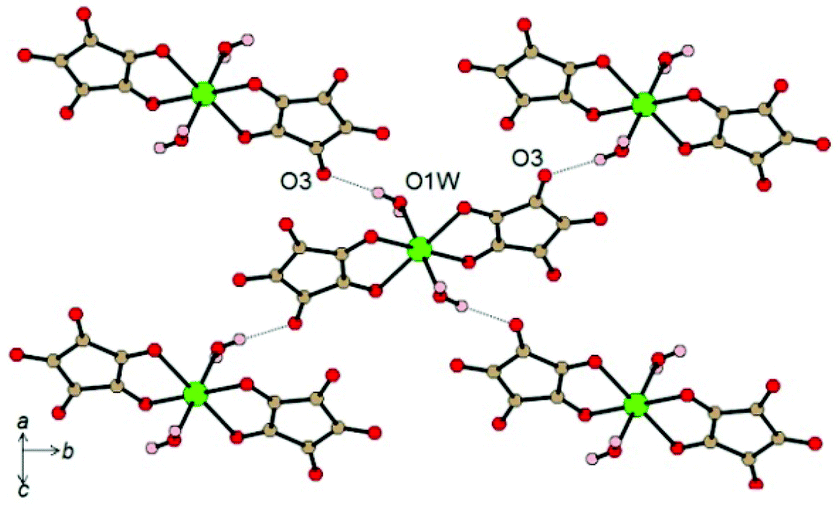 | ||
| Fig. 5 Intralayer H-bonds between discrete [Fe(C5O5)(H2O)2]2− units. Colour codes: C = brown, O = red, H = pink, Fe = green. Reprinted with permission from ref. 8. Copyright 2012 American Chemical Society. | ||
This HB leads to a quadratic regular layer where each [Fe(C5O5)(H2O)2]2− anion is connected to its four neighbors in the plane through four equivalent HBs.
The magnetic properties have been studied in the 2–300 K range by DC magnetization measurements. When the sample is cooled down, the χT value remains constant down to ca. 100 K and, below this temperature, it shows a progressive increase to reach a maximum at ca. 9 K (Fig. 6). Below this temperature the χT shows a sharp decrease until 2 K. This behaviour suggests a predominantly weak FM coupling responsible for the increase of the χT below 100 K.8
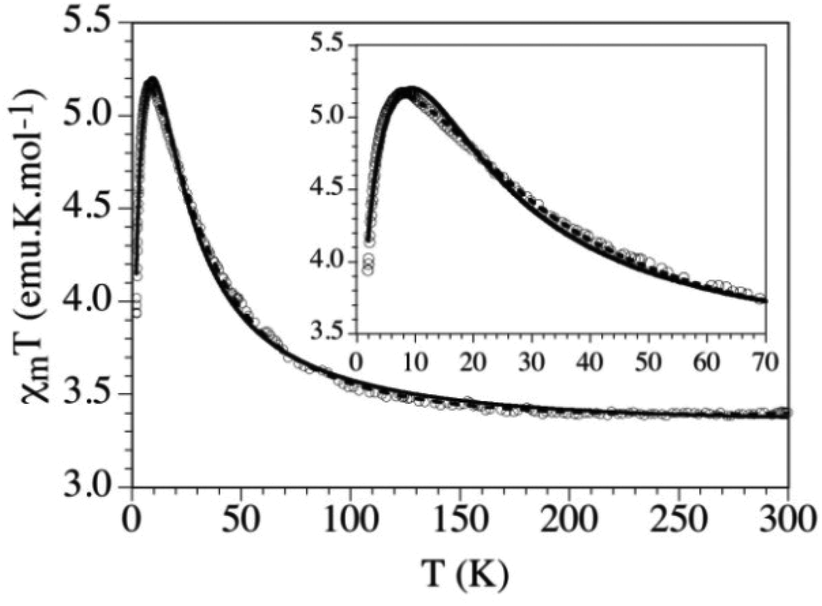 | ||
| Fig. 6 Thermal variation of the χT for K4[Fe(C5O5)(H2O)2](HC5O5)2·4H2O. Solid and dashed lines show the best fit of the employed models. Reprinted with permission from ref. 8. Copyright 2012 American Chemical Society. | ||
Despite the quite long Fe⋯Fe distance (9.016(2) Å) magnetic measurements clearly reveal the presence of FM coupling between metal centers. Since no covalent interactions are present between monomeric units, the pathway for the observed magnetic exchange was attributed to the HBs. The magnetic data were fitted taking into account a perfect S = 2 quadratic layer with only one coupling constant J and only one g value. This led to a Curély model38 that reproduced quite satisfactorily the magnetic data with g = 2.027(2) and J = 4.59(3) cm−1, but only above the maximum (dashed line in Fig. 6). In order to reproduce the decrease observed at temperature lower than 9 K, a weak interplane AFM interaction j was added to this model. This second model reproduces quite well the magnetic data, with more realistic parameters (g = 2.071(7), J = 2.94(7) cm−1, zj = −0.09(1) cm−1, solid line in Fig. 6), and reproduces the magnetic data well at low temperature, although the maximum is not perfectly reproduced (inset in Fig. 6). A possible reason to explain this difference may be the simultaneous presence of both zero-field splitting (ZFS) of the FeII ions and weak AFM interlayer coupling.8
Interestingly, in a previously reported paper, MnIII malonate complexes showing a very similarly layered structure were found to have similar magnetic properties.9 Although the dominant H-bonding mediated FM coupling has similar H-bonding parameters with respect to the described FeII-based material, the magnitude of the coupling constant was lower. This is likely due to the presence of Jahn–Teller distortion on the MnIII center; this leads to a weaker overlap of the magnetic orbitals and, consequently, to a much weaker coupling.9
These examples demonstrate that: (i) the role of water molecules is crucial in self-assembling the metallotectons through HBs in order to afford magnetic supramolecular architectures; (ii) HBs influence both magnetic ordering temperature and the nature of magnetic ordering (FM, AFM, spin-canting); (iii) magnetic coupling, in particular FM interactions, although weak, can be promoted through HBs even between metal centers quite far from each other (9.016(2) Å for the FeII compound and 7.156(1) Å for the MnIII compound); (iv) the magnetic orbital symmetry, due to metal ions of different nature on similar coordination environments, influence the magnetic superexchange. Moreover they demonstrate that strong HB interactions can be the primary magnetic exchange path in the stabilization of long-range ordering. The rational design of such crystal structures by crystal engineering appears to be of fundamental importance for preparing new materials where such properties can be observed and improved.
Magnetic coupling mediated by halogen bonds
Halogen bonding is a strong, specific and highly directional non-covalent interaction characterized by (i) X⋯A distance (X = halogen) shorter than the sum of the van der Waals radii, (ii) linearity of the D–X⋯A bond, higher than in hydrogen bonds, (iii) preferred orientation of the D–X bond in the plane of the lone pair of A with a preference for the lone pair direction within that plane, and (iv) tendency for stronger halogen bonds (XBs) in the order I > Br > Cl.39 The main difference with respect to HBs is that halogen atoms are much larger and polarizable than hydrogen atoms; thus XBs are more sensitive to steric hindrance than HBs. This interaction can be as effective as HB for engineering highly specific crystal packing motifs but, while applications of H-bonding in various research areas such as materials science and synthetic chemistry are abundant,40 the development of relevant halogen-bonded systems is still in its infancy.Although in a very limited number of examples, XB has been demonstrated to be able to promote magnetic exchange interactions due to the orbital overlap of the highly polarizable halogen orbitals.15–18 The strength of this interaction, that normally leads to AFM interactions, appears particularly dependent on the X⋯A distance, the D–X⋯A angle and the electronic nature of the halogen atom. Moreover, the possibility of replacing Cl with Br, or Br with I without altering the crystal packing of a predesigned system, offers a powerful tool to study, in a systematic way, the influence of the nature of the halogen atom in mediating the magnetic exchange.
After the discovery of the strong XB-mediated AFM coupling between 1D coordination polymers of formula [CuBr2(2,5-dmpz)] (2,5-dmpz = 2,5-dimethylpyrazine), where short and highly directional Br⋯Br contacts (3.632 Å, Cu–Br⋯Br–Cu angle = 180.0°) are responsible for a J = −234 K exchange coupling,15 the importance of intermolecular X⋯X contacts on the magnetic properties of molecular systems has been growing up in recent years.
An interesting example where such interactions can be modulated either by varying the nature of the halogen atom or the applied pressure, is represented by the [CuX2(pyzO)(H2O)2] (pyzO = pyrazine-N,N′-oxide; X = Cl, Br) system.16 Their crystal structure consists of 1D chains of Cu2+ ions linked through the bidentate pyzO ligand. These chains are joined together through O–H⋯O HBs between the water ligands and the pzyO oxygen atoms and Cu–X⋯X–Cu contacts (Fig. 7).
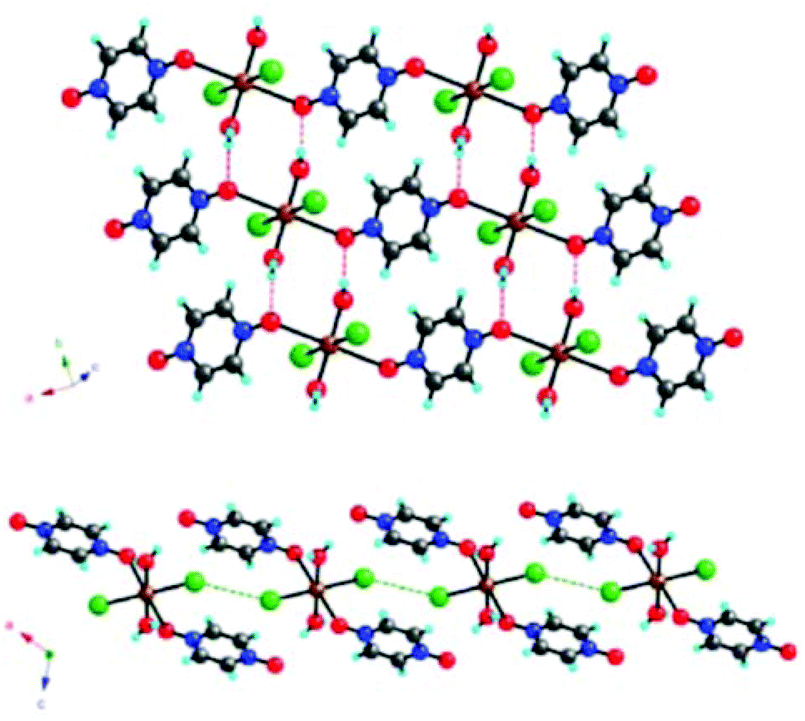 | ||
| Fig. 7 View of the crystal structure of [CuBr2(pyzO)(H2O)2] where the intermolecular interactions between the chains are highlighted (top: H-bonding; down: halogen bonding). Adapted with permission from ref. 16. Copyright 2012 American Chemical Society. | ||
Thus, this is, perhaps, a unique example of a material that contains covalent bonds that link the Cu2+ centers into 1D chains, which are linked into 2D layers through hydrogen bonds, and a 3D structure through halogen bonds.
Bulk magnetic susceptibility measurements at ambient pressure show a broad maximum at 7 and 28 K for X = Cl and Br, respectively, indicating short-range magnetic coupling interactions. The dominant spin-exchange was determined to be the Cu–X⋯X–Cu interaction since the magnetic orbital of the S = 1/2 Cu2+ ion lies in the CuX2(H2O)2 plane, as clearly shown in Fig. 8.16
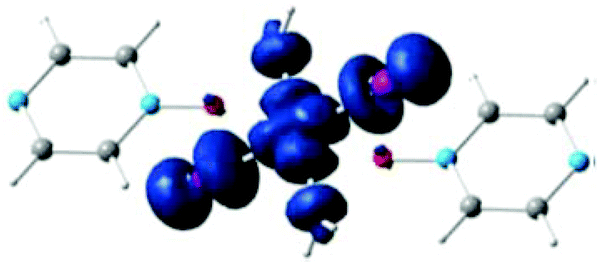 | ||
| Fig. 8 Spin density calculated for an isolated [CuX2(pyzO)(H2O)2], showing that the magnetic orbital of the Cu2+ ion lies in the CuX2(H2O)2 plane. Reprinted with permission from ref. 16. Copyright 2012 American Chemical Society. | ||
Interestingly, the pressure dependence of the magnetic susceptibility for the Br derivatives indicates a gradual increase in the magnitude of the coupling constant (J/kB = −45.9(1) K at ambient pressure) up to −51.2(1) K at 0.84 GPa, suggesting a shortening in the Br⋯Br contacts (Fig. 9), whereas at higher pressures (ca. 3.5 GPa) a structural phase transition occurs.16 μSR measurements indicate that at very low temperatures (1.06 and 0.26 K for Cl and Br respectively) these systems also display a long-range magnetic ordering.16
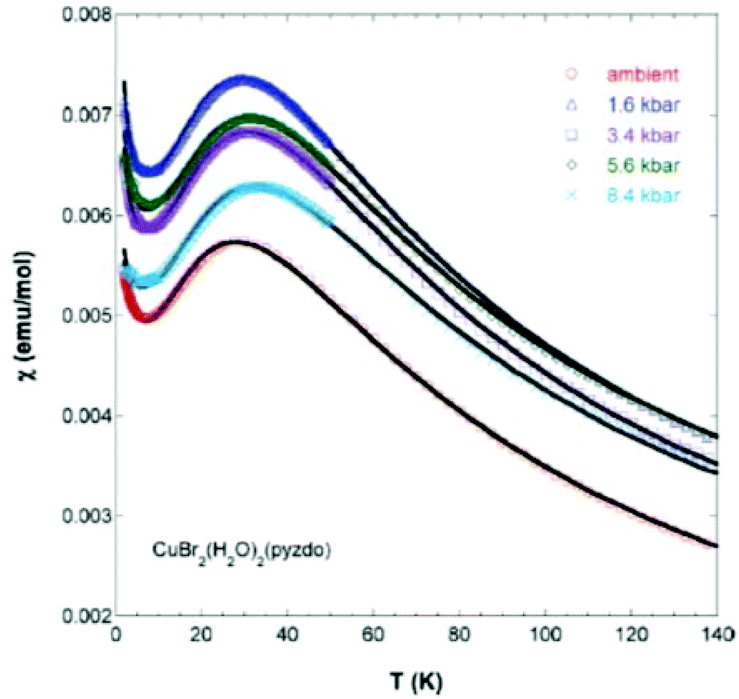 | ||
| Fig. 9 Magnetic susceptibility for [CuBr2(pyzO)(H2O)2] as a function of the applied pressure. Reprinted with permission from ref. 16. Copyright 2012 American Chemical Society. | ||
Halogen-bonded paramagnetic systems where FM or AFM interactions are promoted by XB are very rare in the literature. Among them, the metal complexes [Cu(25dbp)2X2] (25dbp = 2,5-dibromopyridine; X = Cl, Br), where C–Br⋯X–Cu interactions are responsible for weak 1D AFM (Cl) or FM (Br) interactions,17 and the system [Cr(I2An)3]3− (I2An = iodanilate, I2C6O42−) where C–I⋯O interactions are able to promote AFM interactions among paramagnetic Cr(III) centers in halogen-bonded supramolecular dimers.18 In this system the iodine behaves as the XB donor and the oxygen atom as the XB acceptor. This is in agreement with the properties of the electrostatic potential for [Cr(I2An)3]3− that predicts a negative charge accumulation on the peripheral oxygen atoms and a positive charge accumulation on the iodine.18
Despite well-established experimental and theoretical evidence, the possibility to exploit XBs as pathways for mediating magnetic interactions has been investigated only in a limited number of examples. Therefore, the rational design of supramolecular architectures based on metallotectons where suitable functional groups can promote XB interactions strong enough to lead to magnetic exchange, should represent one of the major challenges of crystal engineering in the future. The structure–property correlation in such designed systems will be of fundamental importance for the preparation of novel materials with predetermined and improved physical properties.
Magnetic coupling mediated by π–π interactions
While π–π interactions are common pathways for mediating both conducting and magnetic properties in organic or metallorganic radical-based materials, examples where such interactions have been recognized as responsible for mediation of magnetic exchange interactions between localized magnetic centers (i.e. paramagnetic metal ions) are rare.41 This is primarily due to the competition of such superexchange pathways with more efficient ones. In fact, these interactions are, in most cases, simultaneously present with the above mentioned HB, XB or even covalent bonding interactions, that are more directional and efficient.23,41a–c This leads to a contribution of the π–π stacking that, in some cases, has been evaluated as negligible, as demonstrated for the compounds [Cu2(μ2-CH3COO)2(bpydiol-H)2(H2O)2] (bpydiol-H = monodeprotonated 2,2′-bipyridine-3,3′-diol) and [Cu2(μ2-CH3COO)2(phen)2(H2O)2] (phen = 1,10-phenanthroline) (Fig. 10).23,42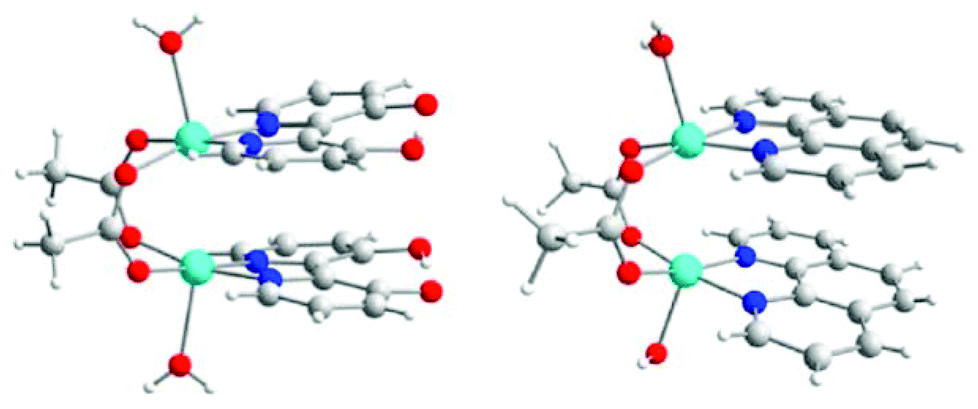 | ||
| Fig. 10 Molecular structures of [Cu2(μ2-CH3COO)2(bpydiol-H)2(H2O)2] (left) and [Cu2(μ2-CH3COO)2(phen)2(H2O)2] (right). Reprinted with permission from ref. 23. Copyright 2012 American Chemical Society. | ||
These complexes are characterized by similar Cu⋯Cu distances (3.01 and 3.06 Å, for the bpydiol-H and phen complexes, respectively) and exhibit moderate AFM interactions between metal centers (J = −59.6 cm−1 and J = −86 cm−1, respectively). The AFM coupling was originally attributed, on the basis of DFT calculations, to a competition between AFM interactions due to the acetate bridges and FM interactions due to the π–π overlap.42a A recent study of the origin of the magnetic properties of the above mentioned complexes has been reported. The analysis of the magnetic exchange coupling based on localized orbitals reveals that π–π interactions do not contribute to the overall antiferromagnetic character of these complexes.23 The main pathways for the magnetic interaction have been recognized, instead, as the direct Cu⋯Cu magnetic overlap, with important superexchange mechanisms through the acetate ligand pathway.23
Molecular materials where π–π interactions can be recognized as the only pathway in mediating magnetic exchange interactions, are usually characterized by small coupling constants associated with both AFM and FM interactions.41 Interestingly, despite the core-like f-type electronic orbitals, these types of magnetic interactions, although weak, have also been observed in lanthanide-based materials.41d
In conclusion, π–π interactions, although in the case of localized magnetic moments commonly give rise to weak magnetic superexchange, are of special interest among the non-covalent interactions since they can be considered as potential magnetic exchange paths also when these interactions are simultaneously present with other non-covalent interactions. An accurate theoretical evaluation of the separate contributions allows for a better understanding of the experimental data.
Concluding remarks
Non-covalent supramolecular interactions, viz. hydrogen bonding, halogen bonding and π–π interactions play a crucial role in determining the crystal packing of crystalline functional molecular materials. While these interactions have been widely used for the engineering of specific supramolecular architectures showing, for example, gas-adsorption and catalytic properties, few examples where such interactions have been thoroughly investigated as tools for mediating magnetic properties have been reported in the literature. We strongly believe that the strategy of combining rationally designed building units (i.e. metallotectons), by following the basic principles of Crystal Engineering for driving specific supramolecular interactions, can lead to novel magnetic supramolecular architectures exhibiting tailored and sometimes unexpected results in the field of non-covalently mediated magnetic properties. The great structural diversity offered by such interactions, i.e. the nature of the A and D atoms, A⋯D distance and angles, M⋯M distance and metal ions’ nature, tunability by external stimuli, and so on, offers a wide range of potentially challenging systems where these properties can be observed and investigated. Moreover, the recent interest in the development of theoretical models capable of rationalizing and predicting their magnetic properties,20,21 along with the improved performance of DFT calculations for a suitable computational evaluation of their magnetic exchange coupling constants,22–24 seems to be the rich soil where this emergent field can grow up.Acknowledgements
M. A. and M. L. M. acknowledge the Regione Autonoma della Sardegna, L.R. 7-8-2007, Bando 2009, CRP-17453 Project “Nano Materiali Multifunzionali per Applicazioni nell'Elettronica Molecolare”, Fondazione Banco di Sardegna and INSTM. J. A. S. acknowledges support from the Independent Research/Development program while serving at the National Science Foundation.Notes and references
- M. L. Mercuri, P. Deplano, L. Pilia, A. Serpe and F. Artizzu, Coord. Chem. Rev., 2010, 254, 1419–1433 CrossRef CAS PubMed.
- (a) J. W. Steed and J. L. Atwood, Supramolecular Chemistry, 2nd edn, John Wiley & Sons, 2009 Search PubMed; (b) J. W. Steed, D. R. Turner and K. J. Wallace, Core concepts in supramolecular chemistry and nanochemistry, John Wiley & Sons, 2007 Search PubMed.
- (a) D. Braga, L. Brammer and N. R. Champness, CrystEngComm, 2005, 7, 1–19 RSC; (b) L. Brammer, Chem. Soc. Rev., 2004, 33, 476–489 RSC.
- See for example: (a) Magnetism: Molecules to Materials, ed. J. S. Miller and M. Drillon, Wiley-VCH, New York, 2001–2005, vol. 1–5 Search PubMed; (b) O. Kahn, Molecular Magnetism, Wiley-VCH, New York, 1993 Search PubMed; (c) H. Tamaki, Z. J. Zhong, N. Matsumoto, S. Kida, M. Koikawa, N. Achiwa, Y. Hashimoto and H. Okawa, J. Am. Chem. Soc., 1992, 114, 6974–6979 CrossRef CAS; (d) C. Mathonière, C. J. Nuttall, S. G. Carling and P. Day, Inorg. Chem., 1996, 35, 1201–1206 CrossRef PubMed; (e) M. Atzori, S. Benmansour, G. Mínguez Espallargas, M. Clemente-León, A. Abhervé, P. Gómez-Claramunt, E. Coronado, F. Artizzu, E. Sessini, P. Deplano, A. Serpe, M. L. Mercuri and C. J. Gómez García, Inorg. Chem., 2013, 52, 10031–10040 CrossRef CAS PubMed.
- J. L. Manson, M. M. Conner, J. A. Schlueter, A. C. McConnell, H. I. Southerland, I. Malfant, T. Lancaster, S. J. Blundell, M. L. Brooks, F. L. Pratt, J. Singleton, R. D. McDonald, C. Lee and M.-H. Whangbo, Chem. Mater., 2008, 20, 7408–7416 CrossRef CAS.
- S. H. Lapidus, J. L. Manson, H. Park, A. J. Clement, S. Ghannadzadeh, P. Goddard, T. Lancaster, J. S. Moller, S. J. Blundell, M. T. F. Telling, J. Kang, M.-H. Whangbo and J. A. Schlueter, Chem. Commun., 2013, 49, 499–501 RSC.
- K. R. O'Neal, T. V. Brinzari, J. B. Wright, C. Ma, S. Giri, J. A. Schlueter, Q. Wang, P. Jena, Z. Liu and J. L. Musfeldt, Sci. Rep., 2014, 4–6 Search PubMed.
- M. Atzori, E. Sessini, F. Artizzu, L. Pilia, A. Serpe, C. J. Gómez-García, C. Giménez-Saiz, P. Deplano and M. L. Mercuri, Inorg. Chem., 2012, 51, 5360–5367 CrossRef CAS PubMed.
- F. S. Delgado, N. Kerbellec, C. Ruiz-Pérez, J. Cano, F. Lloret and M. Julve, Inorg. Chem., 2006, 45, 1012–1020 CrossRef CAS PubMed.
- X. Ren, Y. Chen, C. He and S. Gao, J. Chem. Soc., Dalton Trans., 2002, 3915–3918 RSC.
- Y. Ma, A.-L. Cheng and E.-Q. Gao, Dalton Trans., 2010, 39, 3521–3526 RSC.
- K. Helios, M. Duczmal, A. Pietraszko and D. Michalska, Polyhedron, 2013, 49, 259–268 CrossRef CAS PubMed.
- M. S. Ray, A. Ghosh, R. Bhattacharya, G. Mukhopadhyay, M. G. B. Drew and J. Ribas, Dalton Trans., 2004, 252–259 RSC.
- S. I. Levchenkov, I. N. Shcherbakov, L. D. Popov, V. V. Lukov, V. V. Minin, Z. A. Starikova, E. V. Ivannikova, A. A. Tsaturyan and V. A. Kogan, Inorg. Chim. Acta, 2013, 405, 169–175 CrossRef CAS PubMed.
- R. T. Butcher, J. J. Novoa, J. Ribas-Arino, A. W. Sandvik, M. M. Turnbull, C. P. Landee, B. M. Wells and F. F. Awwadi, Chem. Commun., 2009, 1359–1361 RSC.
- J. A. Schlueter, H. Park, G. J. Halder, W. R. Armand, C. Dunmars, K. W. Chapman, J. L. Manson, J. Singleton, R. McDonald, A. Plonczak, J. Kang, C. Lee, M.-H. Whangbo, T. Lancaster, A. J. Steele, I. Franke, J. D. Wright, S. J. Blundell, F. L. Pratt, J. deGeorge, M. M. Turnbull and C. P. Landee, Inorg. Chem., 2012, 51, 2121–2129 CrossRef CAS PubMed.
- F. F. Awwadi, S. F. Haddad, M. M. Turnbull, C. P. Landee and R. D. Willett, CrystEngComm, 2013, 15, 3111–3118 RSC.
- M. Atzori, F. Artizzu, E. Sessini, L. Marchio, D. Loche, A. Serpe, P. Deplano, G. Concas, F. Pop, N. Avarvari and M. L. Mercuri, Dalton Trans., 2014, 43, 7006–7019 RSC.
- (a) W. Wernsdorfer, N. Aliaga-Alcalde, D. N. Hendrickson and G. Christou, Nature, 2002, 416, 406–409 CrossRef PubMed; (b) S. Hill, R. S. Edwards, N. Aliaga-Alcalde and G. Christou, Science, 2003, 302, 1015–1018 CrossRef CAS PubMed; (c) R. Inglis, G. Papaefstathiou, W. Wernsdorfer and E. K. Brechin, Aust. J. Chem., 2009, 62, 1108–1118 CrossRef CAS.
- C. Desplanches, E. Ruiz, A. Rodríguez-Fortea and S. Alvarez, J. Am. Chem. Soc., 2002, 124, 5197–5205 CrossRef CAS PubMed.
- C. Desplanches, E. Ruiz and S. Alvarez, Chem. Commun., 2002, 2614–2615 RSC.
- N. A. G. Bandeira and B. L. Guennic, J. Phys. Chem. A, 2012, 116, 3465–3473 CrossRef CAS PubMed.
- N. A. G. Bandeira, D. Maynau, V. Robert and B. Le Guennic, Inorg. Chem., 2013, 52, 7980–7986 CrossRef CAS PubMed.
- M. Perić, M. Zlatar, S. Grubišić and M. Gruden-Pavlović, Polyhedron, 2012, 42, 89–94 CrossRef PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS.
- See for example: (a) G. R. Desiraju, J. Chem. Soc., Dalton Trans., 2000, 3745–3751 RSC; (b) G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, Oxford, 1999 Search PubMed; (c) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS PubMed; (d) A. M. Beatty, CrystEngComm, 2001, 51 Search PubMed; (e) M. Atzori, L. Marchiò, R. Clérac, A. Serpe, P. Deplano, N. Avarvari and M. L. Mercuri, Cryst. Growth Des., 2014, 14, 5938–5948 CrossRef CAS.
- J.-P. Launay and M. Verdaguer, Electrons in Molecules, From Basic Principles to Molecular Electronics, Oxford University Press, Oxford, 2013 Search PubMed.
- (a) F. Nepveu, S. Gehring and L. Walz, Chem. Phys. Lett., 1986, 128, 300–304 CrossRef CAS; (b) M. Ardon, A. Bino, K. Michelsen and E. Pedersen, J. Am. Chem. Soc., 1987, 109, 5855–5856 CrossRef CAS.
- (a) S. J. Blundell, Contemp. Phys., 1999, 40, 175–192 CrossRef CAS; (b) T. Lancaster, S. J. Blundell, F. L. Pratt, M. L. Brooks, J. L. Manson, E. K. Brechin, C. Cadiou, D. Low, E. J. L. McInnes and R. E. P. Winpenny, J. Phys.: Condens. Matter, 2004, 16, S4563–S4582 CrossRef CAS; (c) T. Lancaster, S. J. Blundell and F. L. Pratt, Phys. Scr., 2013, 88, 068506 CrossRef.
- J. A. Schlueter, H. Park, J. L. Manson, H. Nakotte and A. J. Schultz, Physica B, 2010, 405, S324–S326 CrossRef CAS PubMed.
- P. A. Goddard, J. Singleton, C. Maitland, S. J. Blundell, T. Lancaster, P. J. Baker, R. D. McDonald, S. Cox, P. Sengupta, J. L. Manson, K. A. Funk and J. A. Schlueter, Phys. Rev. B: Condens. Matter, 2008, 78, 052408 CrossRef.
- C. H. Wang, M. D. Lumsden, R. S. Fishman, G. Ehlers, T. Hong, W. Tian, H. Cao, A. Podlesnyak, C. Dunmars, J. A. Schlueter, J. L. Manson and A. D. Christianson, Phys. Rev. B: Condens. Matter, 2012, 86, 064439 CrossRef.
- G. J. Halder, K. W. Chapman, J. A. Schlueter and J. L. Manson, Angew. Chem., Int. Ed., 2011, 50, 419–421 CrossRef CAS PubMed.
- A. Prescimone, C. Morien, D. Allan, J. A. Schlueter, S. W. Tozer, J. L. Manson, S. Parsons, E. K. Brechin and S. Hill, Angew. Chem., Int. Ed., 2012, 51, 7490–7494 CrossRef CAS PubMed.
- S. Ghannadzadeh, J. S. Moller, P. A. Goddard, T. Lancaster, F. Xiao, S. J. Blundell, A. Maisuradze, R. Khasanov, J. L. Manson, S. W. Tozer, D. Graf and J. A. Schlueter, Phys. Rev. B: Condens. Matter, 2013, 87, 241102 CrossRef.
- S. H. Lapidus, J. L. Manson, J. Liu, M. J. Smith, P. Goddard, J. Bendix, C. V. Topping, J. Singleton, C. Dunmars, J. F. Mitchell and J. A. Schlueter, Chem. Commun., 2013, 49, 3558–3560 RSC.
- (a) J. Curély, Europhys. Lett., 1995, 32, 529 CrossRef; (b) J. Curély, Phys. B, 1998, 245, 263 CrossRef.
- (a) M. Fourmigué, in Halogen Bonding in Conducting or Magnetic Molecular Materials, ed. P. Metrangolo and G. Resnati, Springer-Verlag, Berlin, 2008, vol. 128, 181–207 Search PubMed; (b) M. Fourmigué, Curr. Opin. Solid State Mater. Sci., 2009, 13, 36–45 CrossRef PubMed; (c) P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- (a) D. Braga, L. Maini, M. Polito and F. Grepioni, Struct. Bonding, 2004, 111, 1–32 CrossRef CAS; (b) R. Vilar, Struct. Bonding, 2004, 111, 85–137 CrossRef CAS; (c) V. A. Russell, C. C. Evans, W. Li and M. D. Ward, Science, 1997, 276, 575–579 CrossRef CAS; (d) G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- See for example: (a) Y.-H. Chi, L. Yu, J.-M. Shi, Y.-Q. Zhang, T.-Q. Hu, G.-Q. Zhang, W. Shi and P. Cheng, Dalton Trans., 2011, 40, 1453–1462 RSC; (b) D. Venegas-Yazigi, K. A. Brown, A. Vega, R. Calvo, C. Aliaga, R. C. Santana, R. Cardoso-Gil, R. Kniep, W. Schnelle and E. Spodine, Inorg. Chem., 2011, 50, 11461–11471 CrossRef CAS PubMed; (c) C. Ramirez de Arellano, E. Escriva, C. J. Gomez-Garcia, G. Minguez Espallargas, R. Ballesteros and B. Abarca, CrystEngComm, 2013, 15, 1836–1839 RSC; (d) F. Artizzu, K. Bernot, A. Caneschi, E. Coronado, J. M. Clemente-Juan, L. Marchiò, M. L. Mercuri, L. Pilia, A. Serpe and P. Deplano, Eur. J. Inorg. Chem., 2008, 3820–3826 CrossRef CAS.
- (a) C. Hou, J.-M. Shi, Y.-M. Sun, W. Shi, P. Cheng and L.-D. Liu, Dalton Trans., 2008, 5970–5976 RSC; (b) T. Tokii, N. Watanabe, M. Nakashima, Y. Muto, M. Morooka, S. Ohba and Y. Saito, Bull. Chem. Soc. Jpn., 1990, 63, 364–369 CrossRef CAS.
| This journal is © the Partner Organisations 2015 |