
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrafast electron dynamics at water covered alkali adatoms adsorbed on Cu(111)
Michael
Meyer†
ab,
Ishita
Agarwal
c,
Martin
Wolf
ab and
Uwe
Bovensiepen
*ac
aFachbereich Physik, Freie Universität Berlin, Arnimallee 14, 14195 Berlin, Germany
bFritz-Haber-Institut der Max-Planck Gesellschaft, Faradayweg 4–6, 14195 Berlin, Germany
cFakultät für Physik und Zentrum für Nanointegration (CENIDE), Universität Duisburg-Essen, Lotharstr. 1, 47057 Duisburg, Germany. E-mail: uwe.bovensiepen@uni-due.de
First published on 23rd January 2015
Abstract
Here we report on the ultrafast electron dynamics of the alkalis Na, K, and Cs coadsorbed with D2O on Cu(111) surfaces, which we investigated with femtosecond time-resolved two-photon photoemission. The well known transient electronic binding energy stabilization in bare adsorbed alkalis is enhanced by the presence of water which acts as a solvent and increases the transient energy gain. We observe for all adsorbed alkalis a transient binding energy stabilization of 100–300 meV. The stabilization rates range from 1 to 2 eV ps−1. Here the heavier alkali exhibits a slower stabilization which we explain by their weaker static alkali–water interaction observed in thermal desorption spectroscopy. The population dynamics at low water coverage is described by a single exponential. With increasing water coverage the behavior becomes non-exponential suggesting an additional excited state due to electron solvation.
1 Introduction
Optical excitation from a ground state to an excited state potential surface may induce molecular motion and eventually photochemical reactions. In condensed phase the environment of such a photo-excited reaction center can modify the excited state through cooperative processes (like screening or recognition) and, vice versa, the excitation has the potential to modify the environment. In this context, solvation processes are of particular interest as in many cases the optical excitation redistributes charge density which induces interesting and rich solvation dynamics.The interaction of excess electrons at metal–molecule interfaces is of particular interest for complex hybrid systems and moreover, it is highly relevant for technologically important areas like electrochemistry and molecular electronics.1 Alkali covered noble metal surfaces represent well established model interface systems2–10 with an electronic structure characterized by an unoccupied electronic surface resonance. It originates from the single s valence electron of the neutral alkali atoms and is modified by the electronic structure of the substrate. On metal surfaces alkalis are chemisorbed and the valence electron density is redistributed between the metal surface and alkali, which becomes partially ionized. As a result, the work function which is determined by the surface dipole, is reduced considerably and facilitates determination of the alkali coverage.4 Resonant optical excitation populates the normally unoccupied surface resonance transiently for a few 10 femtoseconds (fs). At low temperature (50 K) and in the low coverage regime up to 0.1 monolayers (ML), where alkali atoms are considered not to interact, the life times of the unoccupied alkali resonance were calculated and found to increase from 7 fs for Na to 30 fs for Cs following the polarizability of the alkali series11 in reasonable agreement with experimental observations.9 Since the resonance is associated with a repulsive excited state potential surface, its population weakens the chemisorption bond and induces motion of the ion core along the surface normal.8
The challenge to probe the excited state during its population requires access to femtosecond time scales which is nowadays routinely achieved in combination with surface sensitivity by employing femtosecond time-resolved non-linear photoelectron spectroscopy.12,13 This has been successfully applied to alkali covered metal surfaces to systematically study their resonance lifetimes at the metal surfaces.7–8 Additionally, electron localization14,15 and solvation16,17 were observed at interfaces. These processes represent interactions of the excited electronic state with the molecular environment and result from accommodation of the electronic charge within the surrounding molecular network.
Water as a key solvent molecule has always constituted an active area of interdisciplinary research. Because of the rich structure of liquid, amorphous, and crystalline states in the bulk and the corresponding configurations at interfaces,18 this is demanding, but also offers opportunities to modify the environment and thereby investigate the relation between molecular structure, cooperativity, and its dynamics in response to the excitation. At ice–metal interfaces dynamical processes in ice compete with the manifold of electronic states in the adjacent metal. Electron transfer, localization, and solvation dynamics at metal–ice interfaces have been investigated widely in the last decade.15,17,19,20 The dynamics proceeds mostly on ultrafast time scales up to picoseconds where the electron transfer from the ice adlayer to the metal limits the excess electron residence time in ice. In crystalline layers the electronic excited state is strongly localized at a surface defect which results in electron residence times up to minutes.20 Furthermore, the influence of Na adsorbed on ice surfaces has been studied and a Na+ induced feature exhibiting a considerably longer residence time compared to solvated electrons was identified.21
In this article we go beyond the well studied ultrafast dynamics at alkali–metal interfaces and investigate the influence of a solvation shell and hydration. With this goal we coadsorb D2O on alkali (Na, K, Cs) covered Cu(111) surfaces and analyze the water induced changes in the ultrafast electron dynamics, which we attribute to static and dynamic responses of D2O to the transient electron population in the excited alkali–metal surface resonance. We observe an electronic state which differs from the alkali induced surface resonance and which we assign to a water modified state of the alkali surface resonance. We identify further a dynamical energetic stabilization of this water-induced state which we explain by a dynamical evolution of the population in the excited state. Comparing the three investigated alkalis, Na is found to lead to the fastest stabilization at a rate of more than 2 eV per picosecond (ps) which agrees with the largest alkali–water interaction for the investigates series.
Fig. 1 shows a schematic Marcus type energy diagram as a function of a solvation coordinate q for the ground and excited state of the alkali-induced surface resonance. We considered here Na, but the diagram represents the situation for other alkalis as well. The alkali surface resonance in the ice environment termed 3s Na(aq.) is populated, leading to an excess electron in the water–alkali hybrid state at the Cu(111) surface. Similar to other solvation processes the excited state evolves along a solvation coordinate in order to gain energy by solvent rearrangement. The ground state 2p6 Na+(aq.) is occupied and resides in its minimum energy position before optical excitation. Similar to various photo-induced reactions at surfaces the reaction is mediated by excitations in the underlying metal substrate,22i.e. electrons near the Fermi level are excited by a charge transfer excitation into the 3s Na(aq.), which is a vertical transition along the solvation coordinate axis. Solvent rearrangement will subsequently lead to energy gain of the excited electronic state by energy transfer to solvent degrees of freedom.
 | ||
| Fig. 1 Schematic potential diagram for the Na surface resonance modified by coadsorbed D2O. The ground state 2p6 Na+(aq.) is in its minimum near the equilibrium solvation coordinate q0. The excited state labelled 3s Na(aq.) has its minimum at a displaced coordinate and is considered to be excited by an optical charge transfer excitation by electrons near the Fermi level EF of the metal substrate. The excited state relaxes on a time scale τ. Its transient population is probed in a pump–probe experiment using two fs laser pulses. Photoelectrons emitted in this non-linear photoemission process are detected in an electron time-of-flight spectrometer e-TOF. | ||
2 Experimental details and sample characterization
The employed experimental setup combines a tunable fs laser system based on regenerative amplification and Ti:sapphire technology (Coherent RegA 9050) and an ultrahigh vacuum (UHV) chamber with a base pressure below 10−10 mbar which allows surface science experiments on femtosecond time scales. Details of the experimental setup and the preparation procedure are reported in ref. 21 and 23. For two-photon photoemission spectroscopy UV pulses were generated by frequency doubling of the VIS output of the optical parametric amplifier. We conducted time-resolved two-photon photoemission spectroscopy (tr-2PPE) with a photon energy of hνpump = 3 eV generated by frequency doubling of the RegA fundamental output as pump pulses and hνprobe = 2.2 eV generated by optical parametric amplification as probe pulses, cf.Fig. 1. The kinetic energy of the photoelectrons is measured by a time-of-flight (e-TOF) spectrometer. The energy with respect to the Fermi level of intermediate electronic states in the 2PPE process is calculated by E − EF = Ekin + Φ − hνprobe with Φ = Evac − EF being the sample work function. For time-resolved measurements, 2PPE spectra are recorded as a function of the time delay between pump and probe pulse.Clean and smooth single crystal Cu(111) surfaces are prepared by cycling Ar+ sputtering and thermal annealing. The alkalis Na, K, and Cs are evaporated from commercial getter sources (SAES Getters) on top of the freshly prepared Cu(111) surface at 35 K. The evaporation rate is calibrated by measuring the work function using two-photon photoemission spectroscopy upon alkali adsorption on Cu(111) for a given time and comparing the measured work function with literature results.4,9 The alkali coverage could be determined in a coverage range up to 0.15 ML with a precision of 0.02 ML.21 D2O is dosed onto the surface by expansion of D2O vapour through a 50 μm diameter pinhole into UHV and onto Cu(111). The D2O coverage is determined by means of thermal desorption spectroscopy (TDS) using a Ru(001) substrate with a distinct desorption signature of the first bilayer (BL).23 What remains to be characterized is the specificity of the D2O adsorption to the adsorbed alkali ions. We have used TDS to investigate this aspect.
The alkali coverage was kept below 0.15 ML to avoid interaction among alkali ions and alkali–induced water dissociation, which has been reported for coadsorbed water above a critical alkali coverage depending on the alkali and the metal substrate.18 The water molecules were dosed on to the alkali pre-covered Cu(111) surface at a temperature T = 35 K. The obtained thermal desorption spectra of D2O adsorbed on Na, K, and Cs pre-covered Cu(111) are presented in Fig. 2. The desorption spectrum of water on clean Cu(111) exhibits a single peak between 140 and 165 K which is attributed to the desorption of amorphous ice multilayers.15,18 This water desorption is strongly modified when a sub-monolayer coverage of alkali atoms is pre-adsorbed leading to the appearance of a further desorption peak at higher temperature. This feature is highlighted in the inset of Fig. 2 by arrows for pre-adsorbed Cs, K, and Na. For Cs and K the mass-equivalent water coverage is 0.5 and 0.2 BL, respectively, and the adsorbed water desorbs mostly in the high temperature peak. For higher water coverage, as in the Na case where 4.5 BL of water were dosed, an additional broadening of the high temperature peak towards lower T, which reaches to the water desorption peak, and an additional maximum close to that peak is observed. This broadening is indicated by a hatched area in Fig. 2. The desorption temperature of the water molecules in the high temperature peak increases along the sequence Cs, K, Na from 175 to 210 K. This implies that water molecules are stabilized by an attractive interaction with the alkalis, an effect which was observed previously on various metal substrates.24–28 Our TDS studies indicate that the alkali–water interaction is larger for the lighter alkali following the increase of local charge density. We will show in this contribution that the stronger water–alkali interaction for lighter alkalis is linked to a faster stabilization dynamics. As can be deduced from the appearance of the high temperature peak in the desorption spectra the alkali–water interaction exceeds the water–water interaction and therefore the orientation of the water molecules is governed by the alkalis rather than by adjacent water molecules due to electrostatic or dipole–dipole forces, as discussed extensively in literature.18 For low enough water coverage, water adsorbs preferentially at the alkali sites and no water desorption from the clean Cu(111) surface is observed, see desorption spectra for K and Cs in Fig. 2. In this case, the water molecules form a solvation shell around an alkali due to a hydration like interaction.18 Accordingly, the water molecules in the high temperature desorption state can be attributed to water molecules in a hydration shell around the alkali. The desorption kinetics of the water molecules influenced by the alkalis are complicated. For higher water coverage an additional desorption feature and broadening to lower temperatures is observed, see the Na case in Fig. 2. This may result from water molecules in outer hydration shells where the water molecules are less influenced by alkalis. Nevertheless, only a finite amount of water is affected as still a fraction of the water molecules desorb in the normal ‘clean surface’ peak. The fact that water molecules desorb from alkali-influenced adsorption sites at higher temperatures clearly shows that a preferential binding at these alkali sites occurs.
 | ||
| Fig. 2 Thermal desorption spectra of D2O on clean and alkali pre-covered Cu(111). The inset shows a magnified detail of the high temperature peak associated with the water molecules bound to alkali ions. For details see the text. | ||
Analysis of the water adsorption induced changes of the work function supports this scenario. The relative work function change ΔΦ is depicted in Fig. 3 as a function of D2O coverage on alkali covered Cu(111). The work function was measured by means of 2PPE spectroscopy and analysing the low energy cut off of the spectra (top panel). We find that the water deposition onto a sub-monolayer coverage of alkalis on Cu(111) leads initially to an increase of the work function which decreases after passing a maximum in agreement with previous work.29 This behaviour is considerably different from the work function evolution when water is dosed on the clean Cu(111) surface also shown in Fig. 3. Here, the work function is monotonically decreasing by 0.5 eV and a constant work function of Φ = 3.95 eV is reached when a wetting ice layer is formed.15 We conclude that on bare Cu(111) the water molecules are arranged in a manner which reduces the effective surface dipole. Without any specific interaction between alkalis and water the same decrease in Φ is expected to occur on an alkali covered Cu(111) surface. However, the maximum in the work function change with water coverage indicates that the water molecules orient such upon adsorption that they screen and thus counteract the existing surface dipole. We conclude that the maximum in the measured work function change observed in Fig. 3 indicates a competition of the work function decrease upon water adsorption with a partial compensation of the strongly reduced surface dipole of the alkali covered Cu(111) surface. Hence, in the low water coverage regime formation of alkali–water hybrids on the metal substrate takes place, which is an essential prerequisite for the study reported here.
 | ||
| Fig. 3 Top panel: typical 2PPE spectra for water coadsorbed on Cs/Cu(111). The work function plotted in the bottom panel is determined from the low energy cut off. Bottom panel: relative work function change versus D2O coverage for selected alkali coverages. The water was dosed at 35 K. The lines represent data obtained by deposition of D2O on the bare Cu(111). | ||
3 Experimental results and discussion
Fig. 4 displays a representative overview of fs time-resolved 2PPE spectra for hydrated Na, K, and Cs on Cu(111). For all three investigated alkalis, we observe the three following spectral signatures which are therefore considered to be universal for the electron dynamics of the deposited alkali–water hybrids. (i) An intensity maximum located several 100 meV below the binding energy of bare alkalis on Cu(111);9 (ii) the population in the excited state resonance is observed for few 100 fs which is considerably longer compared to bare adsorbed alkalis;9 and (iii) a pronounced energetic stabilization of the water-induced alkali state expected for energy transfer to molecular reorganization in solvation dynamics. Note that by the term energetic stabilization we refer to a transient binding energy increase, which does not necessarily imply a weakening of the alkali–metal bonding. In the following we will analyse the data in detail to compare the behaviour for the different alkalis and conclude on the fundamental interaction driving the stabilization process.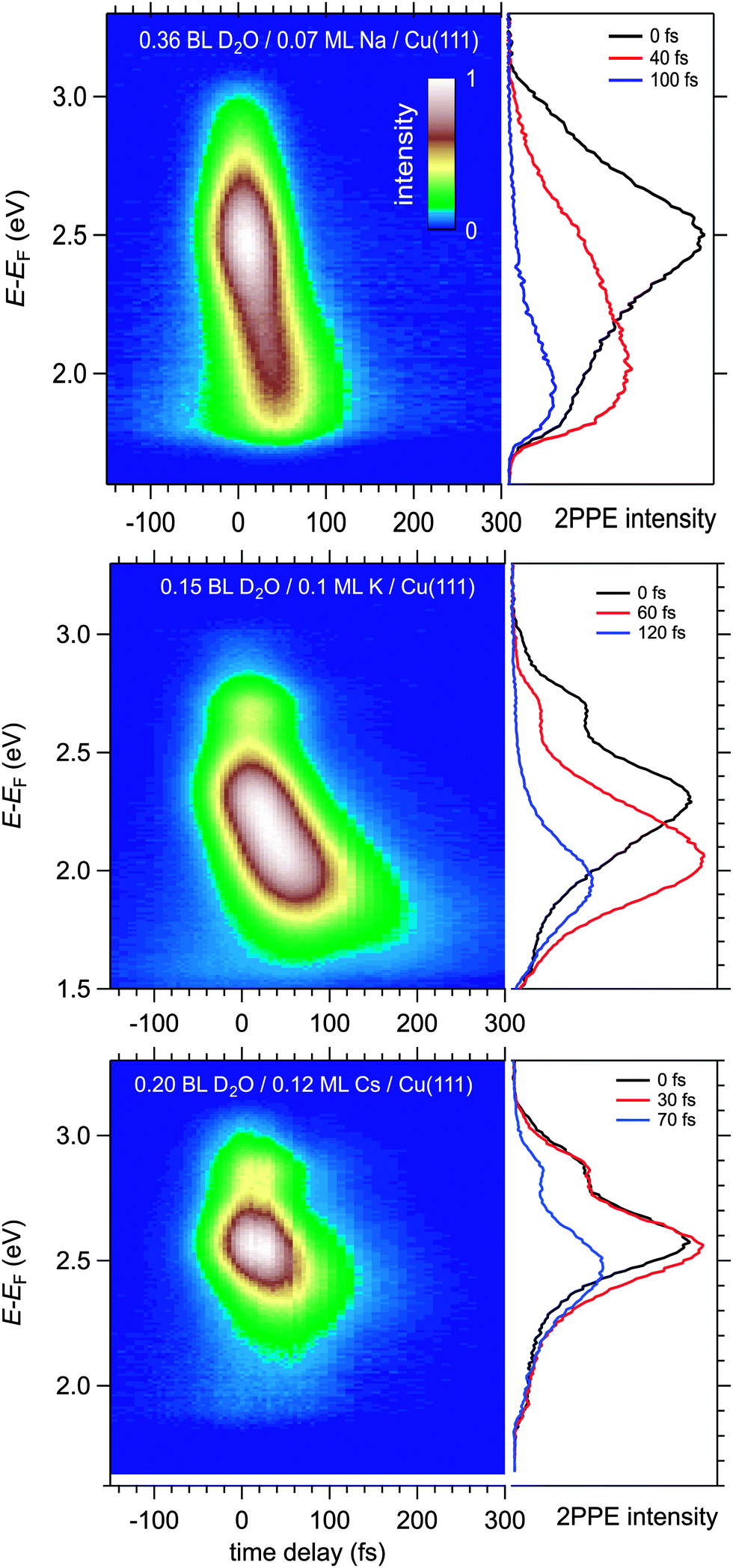 | ||
| Fig. 4 Time-resolved 2PPE data of alkali–water combinations on Cu(111). The 2PPE intensity is given in a false color representation as a function of intermediate state energy (left axis) and pump–probe delay (bottom axis). The spectra for Na, K, and Cs exhibit a short lived state attributed to the alkali resonance and one feature shifting to lower energies with increasing time delay, which is associated with excess electrons at the alkali–water hybrids. Selected spectra are shown in the right panels. | ||
We start by briefly addressing the occurrence of the new water-induced state. The data in Fig. 4 exhibit a clearly visible alkali surface resonance for the K and Cs data with 0.15 and 0.20 BL of D2O coadsorbed. The Na data set was obtained for 0.36 BL D2O where the bare alkali resonance is not resolved. Fig. 5 compares 2PPE spectra for 0.12 ML Cs/Cu(111) for different water coverages. Upon water coadsorption the alkali surface resonance splits in two states. Since increasing water coadsorption quenches the one at higher energy, we assign it to remaining bare alkalis. These contributions exhibit ultrafast relaxation in agreement with literature.9,30 Accordingly, the lower energy feature is attributed to the surface resonance of water–alkali hybrids which appears to originate from the alkali surface resonance but is located at lower binding energy due to the water–alkali interaction. The D2O coverage dependent evolution of the binding energies of the reminiscent alkali resonance and the water-modified feature is complex and beyond the scope of this article. This point will be addressed in detail in a forthcoming publication.
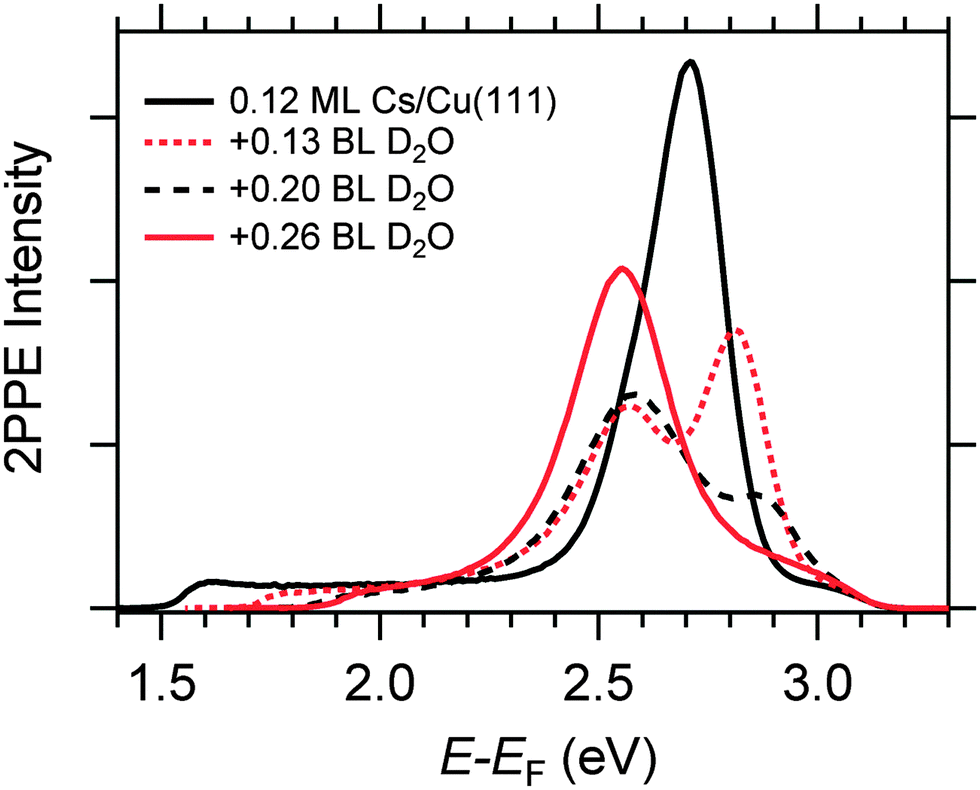 | ||
| Fig. 5 Exemplary 2PPE spectra for Cs/Cu(111) with three different amounts of water coadsorption. | ||
In the following, we focus on the time-dependent changes in population and binding energy. For all investigated alkalis, the energetic position of the alkali–water hybrid state is shifting to lower energies with respect to the Fermi level with increasing time delay, which corresponds to an increase in the binding energy of the state. The time-dependent binding energies were determined by fitting the observed spectral features for the different time delays. The results are depicted in Fig. 6 for Na, K, and Cs. The observed energetic stabilization is pronounced and amounts for 200–300 meV within 200 fs for all three alkalis. Note that, the maximum time delay until which the stabilization can be followed is determined by the population decay and the coupling strength of the excited state to the metal substrate which determines the transfer rate of the excited electron back to the metal substrate. The observed dynamic increase in binding energy is explained by energy transfer to solvent modes, see Fig. 1, i.e. the time-dependent binding energy characterizes the water rearrangement in response to the population of the excited alkali resonance state. Note that no asymptotic limit for the Na(aq.) 3s state, see Fig. 1, can be given because the population decay prevents the stabilization process to continue. Bearing in mind the scenario of Fig. 1, which represents the excited state in a simplified manner as a neutral and the ground state as the solvated state, one might prefer to consider here de-solvation rather than solvation because of the dynamic reduction of dipolar screening of a local charge, compared to increasing screening for solvation. However, in general terms we regard the excited alkali–water–metal resonance as a state with a rearranged charge distribution compared to the ground state. Dynamic evolution on the excited state potential surface along a solvation coordinate q and the measured transient binding energy increase is therefore considered as a response of the solvent to the excitation. In the present case it might be dominated by an evolution towards a more neutral, i.e. less ionic, state compared to the initial one, cf.Fig. 1.
 | ||
| Fig. 6 Top panel: time-dependent change in binding energy of the water modified surface resonance for Na, K, and Cs co-adsorbed with water on Cu(111). The binding energies were obtained by fitting the 2PPE spectra using one or two Gaussian lines dependent on the observation of one or two photoemission lines to account for the water–alkali hybrids state and a bare alkali surface resonance if observed. Bottom panel: stabilization rates obtained from linear fits to the transient change in binding energy in the top panel as a function of D2O coverage. | ||
In our time-resolved experiment we probe the dynamic evolution in the excited state potential surface for three different alkalis. We find that the rate of energy gain is different for Na, K, and Cs. To determine the stabilization rate Σ we fitted a linear function to the time-dependent binding energy. These lines are included in Fig. 6. As apparent for the potassium case, a linear time-dependence of the binding energy describes only a part of the observed dynamics. However, it allows to compare the behavior for Na, K, and Cs in first order. Table 1 summarises the obtained stabilization rates and a clear trend to a slower stabilization rate for heavier alkalis is found.
| Na | K | Cs | |
|---|---|---|---|
| Σ (eV ps−1) | 2.1(2) | 1.7(2) | 1.1(2) |
The obvious question here is, what is the origin of the slower stabilization for heavier alkali adsorbates. Due to the hybrid nature of the D2O–alkali–Cu(111) system different processes might contribute. Note that effects due to weaker electronic interaction or more water accommodated for heavier alkali imply a larger energy gain, which we do not observe. We rather find a slower energy gain for the heavier alkali. To explain this, we consider further below in the text alkali motion away from the surface. Here, at first, we discuss a contribution of the water molecules assuming a fixed position of the alkalis. The observed energy gain is attributed to a (de-)solvation-like response of the water molecules to the optical induced charge transfer and the resulting charge redistribution at the adsorbed alkali. The response time scale of the water molecules in the solvent shell is on the one hand determined by internal solvent molecular modes, which proceeds for water on ultrafast (20 fs) time scales.31 On the other hand, collective and diffusive motion plays an important role in condensed solvents.17 In the latter case the stabilization rate should be determined by the mobility of the solvent molecules in the hybrid system. Under the employed preparation conditions the alkali–water hybrids contain only small amounts of water and, in consequence, the water molecules forming the solvation shell around the excited alkali adsorbate are not fully coordinated in an H-bond network like in bulk ice. We now take into account that the water molecules adjacent to an alkali experience strong electrostatic forces before the optical excitation. Our observations suggest that for adsorbed Cs, where the alkali–water interaction is weakest (Fig. 2), the water molecules adjacent to Cs are more mobile and therefore on average less well aligned than in the case of Na with a stronger interaction. The optical excitation redistributes charge density, which forms a new potential minimum in the excited state potential surface, see Fig. 1, and starts the dynamic evolution along q. A more rigid and aligned structure like around Na will respond and gain energy faster compared to Cs with its weaker water–alkali interaction, in agreement with our observation.
Adding water leads to formation of further solvation shells and an H-bonded network around the alkali. As can be recognized from the bottom panel in Fig. 6, which depicts the changes in stabilization rate with water coverage, the difference for Na and Cs disappears with further water adsorption up to 1 BL. We conclude therefore, that (i) water is decisive for the stabilization dynamics and (ii) additional water tends to quench these differences in the stabilization dynamics.
Now, we address the population relaxation in the adsorbed alkali–water complexes. Fig. 7 shows exemplary data for the time-resolved 2PPE intensity of Na coadsorbed with an increasing amount of water. In the case of adsorption of pure alkalis, the ultrafast population decay is dominated by a single exponential decay.7,9,10 In the case of coadsorption of an increasing amount of water, we find a continuously increasing deviation from a single exponential relaxation behaviour. A similar non-exponential dynamics has been observed previously for solvated electrons in amorphous solid water films.17Fig. 7 shows that least square fits using a double exponential decay work rather well since the experimental data clearly deviate from a single exponential behaviour.
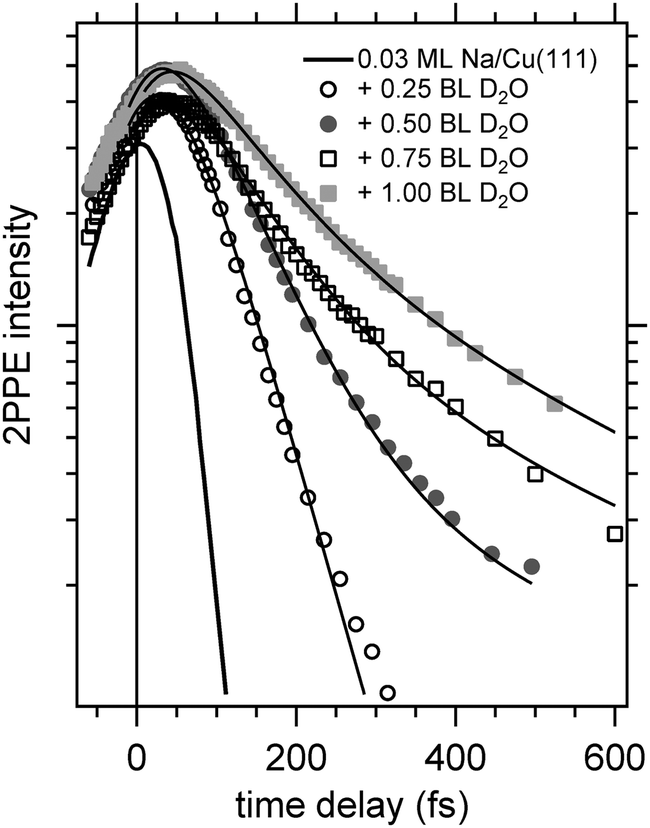 | ||
| Fig. 7 Femtosecond time-resolved 2PPE intensity for Na/Cu(111) with increasing water coverage representing the population dynamics of the alkali–water surface resonance for Na– and Cs–water hybrids adsorbed on Cu(111). Solid lines represent fits to the data using a bi-exponential decay convoluted with the envelope of the laser pulse cross correlation and the data for bare Na adsorption. | ||
Considering previous studies of electron solvation in ice layers the following picture emerges for the observed spectral changes. At low water coverage alkali atoms with binding energies and decay dynamics similar to the bare alkalis coexist with adsorbed alkali–water hybrids, which exhibit different binding energy and dynamics of the unoccupied resonance (see Fig. 5–7). Similar to solvated electrons in thin amorphous ice layers, the decay dynamics for water–alkali hybrids is non-exponential due to the complex dynamics of molecular rearrangements in the solvent shell following excitation. Finally, with an amount of coadsorbed water orders of magnitude above the alkali coverage, electron solvation in a pure water environment is to be expected which changes further the decay dynamics. Since the relaxation of solvated electrons in amorphous ice adsorbed on Cu(111) is considerably slower than for the smallest water coadsorption of 0.25 ML reported in Fig. 7, which is 60 fs,19 the slowing down of the relaxation with increasing water coadsorption can be explained by an onset of a competing, additional state due to electron solvation. This contribution requires a considerable number of adjacent water molecules which explains that it sets in with increasing water coverage. A similar stabilization channel which is competing with the alkali mediated solvation was also identified in an earlier study were alkali atoms were adsorbed on already closed ice layers.21 We assume for the further analysis that this contribution due to electron solvation is not influencing the faster relaxation time scale attributed to the relaxation of the excited water–alkali hybrid state.
Fig. 8 depicts for two different Na coverages the increase in the faster relaxation time as a function of water coverage. Both data sets show a close to linear increase in the relaxation time with water coverage, indeed with similar coverage dependence. What is different is the starting relaxation time at the limit of no water coadsorption. For the smaller Na coverage of 0.03 ML this limit is about 40 fs, for the 0.07 ML coverage, which is about twice as much, this value is just below 20 fs, roughly half of the relaxation time in the low water coverage limit for the 0.03 ML Na data set. This behavior strongly suggests that the decay times are determined by the number of water molecules per adsorbed alkali. We are aware of density functional calculations which indicate an increase of the alkali metal distance with increasing number of water molecules leading to reduced wave function overlap and longer lifetimes,32 in agreement with our observations. Furthermore, half a bilayer of water for the 0.03 ML Na data exhibits within the error bar the same relaxation time than the data for the 0.07 ML Na for 1 BL. This result is supporting our initial conclusions regarding the structure of the water–alkali system on Cu(111) that water molecules are adsorbed adjacent to the alkalis on the Cu surface. The excited electronic alkali–water resonance exhibits ultrafast changes in binding energy for Na, K, and Cs adsorbates due to energy transfer to solvent modes of the hybrid state. The stabilization rate is determined by the local water–alkali interaction. The population dynamics is found to depend on water coverage. Adding water results in a slower and more complex, at least bi-exponential, relaxation. The time scales at which these changes occur are 10–100 fs, see Fig. 8, which we associate with diffusive processes due to nuclear motion. This brings up the question which of the constituents participate in this motion. Fig. 1 implies first of all solvent motion. However, the alkali–surface bond distance is known to be transiently modified by populating the alkali–surface resonance.8 Quantum chemistry calculations were used to study the alkali–ice surface upon impinging of an alkali on the ice surface.33 Since alkali motion is coupled to solvent motion, these results provide some link to our experiment. These results also emphasize that decoupling of the dynamics regarding the involved species is difficult and not necessarily reasonable. On the one hand side the constriction of the excess electron wave function is driven by solvent rearrangement, on the other hand, the most stable state is obtained for a defined position of the alkali within the considered hybrid. Both aspects support our consideration of a coupled solvent-alkali mode, which potentially provides an additional contribution to the transient binding energy increase. Supposing that alkali motion is involved in the energy gain, the slower stabilization dynamics for heavier alkali, Fig. 6, suggests a slower propagation along the alkali–surface coordinate due to inertia. In this view, the longer electronic lifetimes in the hybrid alkali–water–metal systems compared to bare adsorbed alkalis facilitate observation of the stabilization dynamics also for alkalis lighter than Cs. Whether the alkali or solvent motions dominates the dynamics is an interesting, non-trivial question and theoretical calculations for the hybrid system might provide additional insight here. Also, time- and angle-resolved 2PPE experiments potentially provide helpful information, since the spatial extent of localized electrons can be estimated.15 On the other hand side the vanishing difference in the stabilization rates for Na and Cs on Cu(111) at 1 BL water coverage, reported in Fig. 8, indicates that the solvent contribution is decisive.
 | ||
| Fig. 8 Exponential decay times of the faster initial decay component for Na/Cu(111) as a function of water coverage. The solid lines are linear fits to the data. | ||
4 Conclusion
We performed femtosecond time-resolved two-photon photoemission spectroscopy on Na, K, and Cs adatoms which were coadsorbed with D2O on Cu(111). While adsorbed alkalis were already well known to exhibit an ultrafast, transient increase in electron binding energy of the excited surface resonance, coadsorption of water molecules is reported here to enhance this effect up to several 100 meV. This large effect is explained by a solvation like process which stabilizes the excited alkali–water hybrid state in consequence of molecular rearrangement of water molecules. We observe a clear trend of an increasing energetic stabilization rate for lighter alkalis. For Cs coadsorbed with water on Cu(111) the excited water–alkali resonance stabilizes with a rate of 1.1 eV ps−1, while in the case of Na we find 2.1 eV ps−1. This finding is discussed to be related with interaction of the water solvent molecules with the alkali adsorbed on the metal surface, which is larger for Na than for Cs according to our thermal desorption spectroscopy analysis. Also, alkali motion provides a reasonable scenario for an explanation. However, vanishing of the difference in stabilization rates with increasing water coverage up to 1 bilayer favors a mechanism based on water as a solvent. In addition, we find a typical population relaxation time for a rather small water coverage of 0.25 bilayer D2O of 60 fs. With increasing water coverage up to 1.0 bilayer the population relaxation dynamics of the excited state becomes continuously non-exponential which indicates an additional excited state due to electron solvation. In general, we conclude from these experiments that the analysis of the ultrafast response of the water–alkali system coadsorbed on Cu(111) indicates a coupled solvent-alkali mode.Acknowledgements
We gratefully acknowledge fruitful discussions with A. Rubio and funding by the German-Israeli Foundation and by the Deutsche Forschungsgemeinschaft through Sfb 450 and BO 1823/5-1.References
- X.-Y. Zhu, J. Phys. Chem. B, 2004, 108, 8778 CrossRef CAS.
- R. Dudde, K. H. Frank and B. Reihl, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 4897 CrossRef CAS.
- D. Tang, D. McIlroy, X. Shi, C. Su and D. Heskett, Surf. Sci. Lett., 1991, 255, L497 CAS.
- N. Fischer, S. Schuppler, T. Fauster and W. Steinmann, Surf. Sci., 1994, 314, 89 CrossRef CAS.
- Q. Lu, D. O'Connor, B. King and R. MacDonald, Surf. Sci., 1996, 347, L61 CrossRef CAS.
- D. A. Arena, F. G. Curti and R. A. Bartynski, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 15404 CrossRef CAS.
- M. Bauer, S. Pawlik and M. Aeschlimann, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 10040 CrossRef CAS.
- H. Petek, M. J. Weida, H. Nagano and S. Ogawa, Science, 2000, 288, 1402 CrossRef CAS.
- J. P. Gauyacq, A. G. Borisov and M. Bauer, Prog. Surf. Sci., 2007, 82, 244 CrossRef CAS PubMed.
- J. Zhao, N. Pontius, A. Winkelmann, V. Sametoglu, A. Kubo, A. G. Borisov, D. Sánchez-Portal, V. M. Silkin, E. V. Chulkov and P. M. Echenique, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 085419 CrossRef.
- A. G. Borisov, J. P. Gauyacq, E. V. Chulkov, V. M. Silkin and P. M. Echenique, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 235434 CrossRef.
- Dynamics at Solid State Surfaces and Interfaces: Volume 1–Current Developments, ed. U. Bovensiepen, H. Petek and M. Wolf, Wiley–VCH, 2010 Search PubMed.
- U. Bovensiepen and P. S. Kirchmann, Laser Photonics Rev., 2012, 6, 589 CrossRef.
- N.-H. Ge, C. M. Wong, R. L. Lingle Jr., J. D. McNeill, K. J. Gaffney and C. B. Harris, Science, 1998, 279, 202 CrossRef CAS.
- U. Bovensiepen, C. Gahl and M. Wolf, J. Phys. Chem. B, 2003, 107, 8706 CrossRef CAS.
- A. Miller, I. Bezel, K. J. Gaffney, S. Garrett-Roe, S. H. Liu, P. Szymanski and C. B. Harris, Science, 2002, 297, 1163 CrossRef CAS PubMed.
- C. Gahl, U. Bovensiepen, C. Frischkorn and M. Wolf, Phys. Rev. Lett., 2002, 89, 107402 CrossRef CAS.
- M. A. Henderson, Surf. Sci. Rep., 2002, 46, 1 CrossRef CAS.
- J. Stähler, U. Bovensiepen, M. Meyer and M. Wolf, Chem. Soc. Rev., 2008, 37, 2180 RSC.
- U. Bovensiepen, C. Gahl, J. Stähler, M. Bockstedte, M. Meyer, F. Baletto, S. Scand olo, X.-Y. Zhu, A. Rubio and M. Wolf, J. Phys. Chem. C, 2009, 113, 979 CAS.
- M. Meyer, M. Bertin, U. Bovensiepen, D. Wegkamp, M. Krenz and M. Wolf, J. Phys. Chem. C, 2011, 115, 204 CAS.
- C. Frischkorn and M. Wolf, Chem. Rev., 2006, 106, 4207 CrossRef CAS PubMed.
- M. Lisowski, P. Loukakos, U. Bovensiepen, J. Stähler, C. Gahl and M. Wolf, Appl. Phys. A: Mater. Sci. Process., 2004, 78, 165 CrossRef CAS PubMed.
- P. M. Blass, X. L. Zhou and J. M. White, J. Phys. Chem., 1990, 94, 3054 CrossRef CAS.
- J. K. Sass, D. Lackey and J. Schott, Electrochim. Acta, 1991, 36, 1883 CrossRef CAS.
- T. Bornemann, H.-P. Steinrück, W. Huber, K. Eberle, M. Glanz and D. Menzel, Surf. Sci., 1991, 254, 105 CrossRef CAS.
- I. Villegas and M. J. Weaver, Electrochim. Acta, 1996, 41, 661 CrossRef CAS.
- M. J. Weaver and I. Villegas, Langmuir, 1997, 13, 6836 CrossRef CAS.
- H. O. Bonzel, G. Pirug and J. E. Müller, Phys. Rev. Lett., 1987, 58, 2138 CrossRef CAS.
- Using a non-collinear optical parametric amplifier the population relaxation time of Na/Cu(111) at 100 K was determined to be 8 fs.
- M. S. Pshenichnikov, A. Baltuska and D. A. Wiersma, Chem. Phys. Lett., 2004, 389, 171 CrossRef CAS PubMed.
- A. Iacomino, A. Paz and A. Rubio, 2013, private communication.
- T. Vondrak, J. M. C. Plane and S. R. Meech, J. Chem. Phys., 2006, 125, 224702 CrossRef PubMed.
Footnote |
| † Current address: SPECS Surface Nano Analysis, 13355 Berlin, Germany. |
| This journal is © the Owner Societies 2015 |