Towards mRNA with superior translational activity: synthesis and properties of ARCA tetraphosphates with single phosphorothioate modifications†‡
Malwina
Strenkowska§
a,
Joanna
Kowalska§
a,
Maciej
Lukaszewicz
a,
Joanna
Zuberek
a,
Wei
Su
b,
Robert E.
Rhoads
b,
Edward
Darzynkiewicz
a and
Jacek
Jemielity
*a
aDivision of Biophysics, Institute of Experimental Physics, Faculty of Physics, University of Warsaw, Warsaw, Poland. E-mail: jacekj@biogeo.uw.edu.pl; Fax: +48 22 5540771; Tel: +48 22 5540774
bDepartment of Biochemistry and Molecular Biology, Louisiana State University Health Sciences Center, Shreveport, Louisiana, USA 71130-3932, USA
First published on 10th March 2010
Abstract
We describe the chemical synthesis and preliminary biophysical and biochemical characterization of a series of mRNA 5′-end (cap) analogs designed as reagents for obtaining mRNA molecules with augmented translation efficiency and stability in vivo and as useful tools to study mRNA metabolism. The analogs share three structural features: (i) the 5′,5′-bridge is elongated to a tetraphosphate to increase their affinity to translation initiation factor eIF4E; (ii) a single phosphorothioate modification at either the α, β, γ or δ-position of the tetraphosphate has been made to decrease their susceptibility to enzymatic degradation and/or to modulate their interaction with specific proteins; and (iii) a 2′-O-methyl group has been added to the ribose of 7-methylguanosine (a characteristic of Anti-Reverse Cap Analogs (ARCAs), which are incorporated into mRNA during in vitro transcription exclusively in the correct orientation). The dinucleotides bearing the modified tetraphosphate bridge were synthesized by ZnCl2-mediated coupling between two mononucleotide subunits with isolated yields of 30–65%. The preliminary biochemical results show that mRNAs capped with new analogs are 2.5–4.5 times more efficiently translated in a cell-free system than m7GpppG-capped mRNAs, which makes them promising candidates for RNA-based therapeutic applications such as gene therapy and anticancer vaccines.
Introduction
Dinucleoside 5′,5′-polyphosphates fulfill numerous regulatory, signaling, or energetic functions in biological systems. A particular representative of this group of biologically important molecules is the m7G-containing cap present on the 5′-end of all eukaryotic messenger RNAs. Different types of cap structures exist in nature,1 but they all contain guanosine methylated at the N7-position connected by a 5′,5′-triphosphate bridge to the first nucleotide of the RNA transcript (Fig. 1A).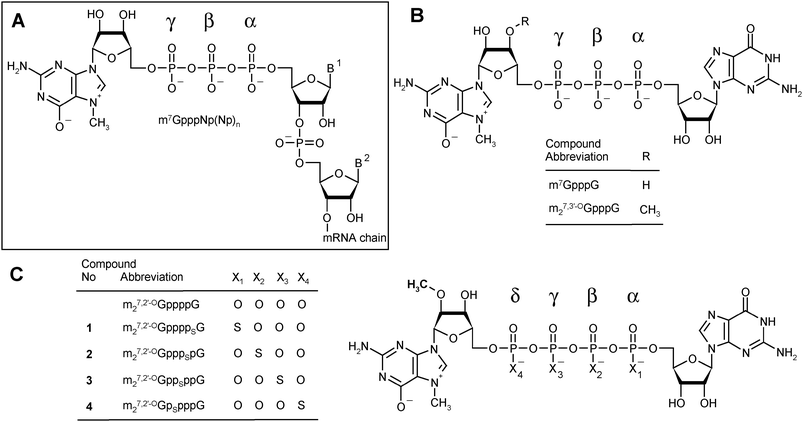 | ||
| Fig. 1 Cap structures. (A) A general depiction of the eukaryotic mRNA 5′-end. (B) Structures of standard triphosphate analogs: m7GpppG and m27,3′-OGpppG (ARCA). (C) Structures of m27,2′-OGppppG and analogs synthesized in this study. | ||
The cap plays a key role in a variety of cellular processes related to mRNA metabolism, including splicing, nucleo-cytoplasmic transport, intracellular targeting, translation, translational repression, and turnover.1,2 Therefore, synthetic analogs of the cap are widely employed as tools for studying these physiological processes as well as being useful in biotechnology. There are also promising potential clinical applications.
One application that has been intensively explored by our groups is the use of cap analogs as reagents for in vitro synthesis of capped mRNAs. Capped mRNAs may be employed for expression of proteins in either in vitro translation systems or in cultured cells. Recently, mRNAs have emerged as an attractive alternative to classical DNA-vector based gene delivery due to several advantages; the use of mRNA avoids the risk of insertional mutagenesis, is effective in non-dividing cells, and does not require introduction into the nucleus because mRNA is translated in the cytoplasm.3
Synthesis of capped mRNA can be achieved by transcribing a DNA template in vitro with either a bacterial4 or bacteriophage5,6 RNA polymerase in the presence of all four NTPs and a dinucleotide cap analog such as m7GpppG. These polymerases normally initiate transcription with nucleophilic attack by the 3′-OH of GTP on the α-phosphate of the next nucleoside triphosphate specified by the DNA template. If a cap analog such as m7GpppG is present in the reaction mixture at a ∼5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio to GTP, the transcription is initiated mainly by attack of 3′-OH of the cap dinucleotide rather than that of GTP, leading to formation of capped transcripts of the form m7GpppGpNp(Np)n. Unfortunately, attack can occur by the 3′-OH of either the Guo or m7Guo moieties of the cap dinucleotide, resulting in one-third to one-half of the transcripts being capped in a reversed orientation, i.e., Gpppm7GpNp(Np)n.7 Such reverse-capped transcripts decreased the overall translational activity of mRNA. This problem was overcome several years ago by the introduction of anti-reverse cap analogs (ARCAs) bearing either 3′-O-methyl, 3′-H or 2′-O-methyl modifications in the m7Guo moiety, ensuring 100% correct orientation.8–10 ARCA-capped mRNAs were shown to have higher translational efficiency than m7GpppG capped mRNAs both in vitro8,10 and in cultured cells.11–13
:1 ratio to GTP, the transcription is initiated mainly by attack of 3′-OH of the cap dinucleotide rather than that of GTP, leading to formation of capped transcripts of the form m7GpppGpNp(Np)n. Unfortunately, attack can occur by the 3′-OH of either the Guo or m7Guo moieties of the cap dinucleotide, resulting in one-third to one-half of the transcripts being capped in a reversed orientation, i.e., Gpppm7GpNp(Np)n.7 Such reverse-capped transcripts decreased the overall translational activity of mRNA. This problem was overcome several years ago by the introduction of anti-reverse cap analogs (ARCAs) bearing either 3′-O-methyl, 3′-H or 2′-O-methyl modifications in the m7Guo moiety, ensuring 100% correct orientation.8–10 ARCA-capped mRNAs were shown to have higher translational efficiency than m7GpppG capped mRNAs both in vitro8,10 and in cultured cells.11–13
One important advantage of the ARCA method over the post-transcriptional enzymatic capping is the option to introduce additional chemical modifications into the mRNA 5′-end that can modify the behavior and properties of the mRNAs. Among dozens of modified synthetic cap analogs examined, several have been identified that augment mRNA translational efficiency, both in vitro and in cultured cells.10,13 Two features of the modified cap structure have emerged that influence the overall yield of protein produced from the mRNA, particularly in cultured cells.11,13 The first one is the cap's intrinsic ability to compete with natural mRNAs for recruitment to the translational machinery, and the second is its susceptibility to enzymatic decapping.
During recruitment of mRNA to the translational machinery, the cap is specifically recognized by eukaryotic initiation factor 4E (eIF4E). mRNA recruitment to form the 48S pre-initiation complex is the rate-limiting step for initiation of translation under normal circumstances (absence of virus infection, cellular stress, etc.) and plays a central role in regulation of translation.14 The presence of the cap greatly increases the rate of translation. Affinity of the cap for eIF4E, and hence for the translational machinery, can be increased by modifying the 5′,5′-triphosphate bridge. One class of compounds with modifications of this type are cap analogs in which the 5′,5′-bridge is extended from triphosphate to tetraphosphate, e.g. m7GppppG, m27,2′-OGppppG or m27,3′-OGppppG.10 Generally, these analogs have binding affinities to eIF4E that are ∼9–10-fold higher than for the corresponding triphosphates and promote translation 20–30% more efficiently when introduced into mRNA. However, it should be mentioned that further increasing the affinity to eIF4E may exert opposite effect on mRNA translational efficiency. Surprisingly, it was found that extending the 5′,5′-bridge to pentaphosphate increases the binding affinity of cap analogs to eIF4E by a factor of ∼3.5–5 compared to tetraphosphates; nonetheless, mRNAs capped with these analogs are translated with efficiency comparable or even lower than for corresponding triphosphates.10
A second important role of the mRNA cap is protecting mRNA against degradation.15 Exonucleolitic mRNA cleavage in the 5′→3′ direction cannot take place until the cap is removed from mRNA 5′-end by a decapping pyrophosphatase termed Dcp1/Dcp2 (a heterodimer consisting of a regulatory and catalytic subunit). The enzyme cleaves the cap, provided it is attached to an RNA of at least 20 nt, between the α and β phosphate moieties to release m7GDP and 5-phosphorylated mRNA chain. This is then degraded by the exonuclease Xrn1.16,17 Both the 5′→3′ and 3′→5′ pathways play major roles in mRNA degradation.15 We have recently demonstrated that mRNAs capped with an ARCA analog containing a phosphorothioate moiety at the β-position of the triphosphate bridge, m27,2′-OGppSpG (D2),18 were resistant to decapping by Dcp2 in vitro, resulting longer half-lives in cultured mouse mammary cells (257 min compared to 155 min for m27,2′-OGpppG-capped mRNA).13 An unexpected further benefit was that m27,2′-OGppSpG (D2)-capped mRNA was 2.4 times more efficiently translated in these cells than m27,2′-OGpppG-capped mRNA.
In the present work, we aimed at obtaining analogs that would combine both features that can enhance the translational yield of mRNA: high affinity for eIF4E and resistance to cleavage by Dcp2, in the expectation that these two features would independently improve the translational properties of mRNA in living cells. We therefore synthesized a series of tetraphosphate ARCA analogs modified with the phosphorothioate group at various positions of the 5′,5′-tetraphosphate bridge (Fig. 1C).
The new tetraphosphate analogs could potentially be useful in other spheres of mRNA metabolism, based on our recent findings with triphosphate phosphorothioate ARCA analogs modified at different positions. Those modified at the γ-position were resistant to another decapping enzyme, DcpS, which acts on the products of 3′→5′ mRNA decay. DcpS cleaves only short capped 5′-oligonucleotides or m7GpppN dinucleotides released after processive degradation of mRNA by the exosome initiated from the 3′-end. Cleavage is between the β- and γ-phosphates of the 5,5-triphosphate bridge, releasing m7GMP and a downstream (oligo)nucleotide.19 Resistance to DcpS is an important factor in design of long-lasting cap analogs that can inhibit translation.
Recently, a cap analog modified at the α-position, m27,2′-OGpppSG (D2), was shown to enhance microRNA-mediated translational repression by a factor of 2 with no change in translation efficiency, providing new insights into the mechanism of this recently discovered but poorly understood process.20
Results
Novel teraphosphate S-ARCA
We designed and prepared a series of four cap analogs 1–4 bearing a single phosphorothioate moiety at either the α-, β-, γ-, or δ-positions of the tetraphosphate chain and 2-O-methyl group (ARCA modification) in the m7Guo moiety. We refer to these as 4P-S-ARCAs (Fig. 1C). The synthetic routes provide these analogs with good yields and can also be applied to the synthesis of a variety of other biologically relevant dinucleoside polyphosphates. Due to the presence of a P-stereogenic center, each 4P-S-ARCA exists as two P-diastereomers, designated D1 and D2 based on their elution order during reverse-phase (RP) HPLC. Two pairs of diastereomers were successfully resolved by RP HPLC, but we could not achieve this for the other two. Subsequently, the new analogs were subjected to biophysical and biochemical characterization. We determined their binding affinities for eIF4E, susceptibility to decapping by DcpS, and translational efficiencies in a rabbit reticulocyte lysate (RRL) system. Other biochemical properties of mRNAs capped with these analogs, including susceptibility to the human decapping enzyme Dcp2, translational efficiency in cultured mammalian cells, and half-life in mammalian cells, are currently under investigation. Preliminary results suggest that mRNA capped with a mixture of diastereomers (D1 plus D2) of m27.2′-OGpppSpG (compound 2) is less susceptible to in vitro decapping by hDcp2 than mRNA capped with m27.2′-OGppSppG (compound 3).21Chemical synthesis of new analogs
The key step in the preparation of each new analog (1–4) was formation of a thio-modified tetraphosphate bridge by ZnCl2-mediated coupling between a nucleoside 5′-O-(thiopolyphosphate) and a second nucleotide activated by conversion into the P-imidazolide. Generally, this approach offers the possibility of synthesizing each compound three ways22 (see Discussion). For each analog we chose the most straightforward synthetic pathway (Scheme 1). Analogs 1 and 4, which are modified at the α- and δ-positions, were obtained by the “3 + 1*” strategy, i.e., by coupling a nucleoside 5′-O-(1-thiotriphosphate) with a nucleoside 5′-monophosphate P-imidazolide. Analogs 2 and 3, which are modified at the β- and γ-positions, were obtained by the “2 + 2*” strategy, i.e., by coupling a nucleoside 5′-(2-thiodiphosphate) with a nucleoside 5′-diphosphate imidazolide. An alternative “3 + 1*” route was also tested for analog 2.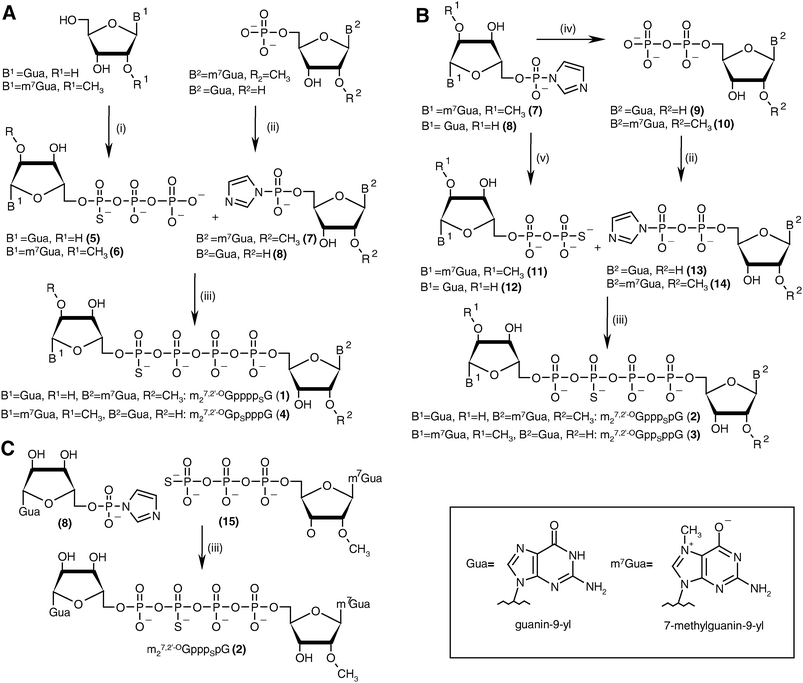 | ||
| Scheme 1 The synthesis of tetraphosphate S-ARCA 1–4. (A) Synthesis of 1 and 4 by the “3 + 1*” strategy. (B) Synthesis of 2 and 3 by the “2 + 2*” strategy. (C) Synthesis of 2 by the “3 + 1*” strategy (the asterisk denotes the activated nucleotide subunit). Reagents: i. (1) PSCl3, trimethylphosphate, 2,6-lutidine, 0 °C, (2) 0.5 M tributylammonium pyrophosphate in DMF; ii. imidazole, 2,2′-dithiodipyridine, PPh3, DMF; iii. ZnCl2, DMF; iv. triethylammonium phosphate, ZnCl2, DMF; v. triethylammonium thiophosphate, ZnCl2, DMF. | ||
The guanosine 5′-O-(1-thiotriphosphate) 5 was obtained by 5′-thiophosphorylation of guanosine in trimethylphosphate in the presence of a non-nucleophilic base, 2,6-lutidine,23 followed by addition of tributylammonium pyrophosphate. As monitored by RP HPLC (see Experimental section), the thiophosphorylation proceeded smoothly, and more than 90% conversion of nucleoside was observed within 5 h. After addition of pyrophosphate solution in DMF and weakly-alkaline hydrolysis (pH 7.5–8.0) a mixture of products was obtained consisting mainly of the desired guanosine 5′-O-(1-thiotriphosphate) (∼60%), guanosine 5′-O-thiophosphate (∼10%) and guanosine 5′-O-(1-thiopentaphosphate) (∼15%). The products were separated by DEAE Sephadex A-25 ion-exchange chromatography, after which 5 was isolated as 7:10 (D1:D2) diastereomeric mixture with 46% yield. The synthesis of 6 was similar to that of 5, but the reaction was less efficient for two reasons: (i) thiophosphorylation proceeded significantly slower, the final conversion (after 24 h) not exceeding 80%, and (ii) the byproduct, 7,2′-O-dimethylguanosine 5′-O-(1-thiopentaphosphate), was formed more efficiently (∼25%). Moreover, 31P NMR analysis unfortunately revealed that the desired product co-eluted with unreacted pyrophosphate, probably due to the partial neutralization of the negative charge on the α-phosphate by the positive charge in the m7Gua moiety. Consequently, additional HPLC purification was necessary before the synthesis scheme could be continued. Compound 6 was finally isolated as a 7:10 (D1:D2) diastereomeric mixture with 30% yield. Both 5 and 6 were used in the subsequent coupling reactions as diastereomeric mixtures.
The final couplings between 5 and 7 or 6 and 8 leading to 1 or 4, respectively, were mediated by ∼16-fold ZnCl2 excess. The P-imidazolide derivatives, 7 and 8, synthesized using a dithiodipyridine/triphenylphosphine activating system,24were used in 1.5–2-fold excess to maximize consumption of the second reactant. Both coupling reactions proceeded smoothly, and essentially complete conversion of 5 or 6 into tetraphosphate-bridged dinucleotides was observed by RP HPLC after several hours (Fig. 2A). The respective HPLC conversions and isolated yields achieved after ion-exchange purification are shown in Table 1.
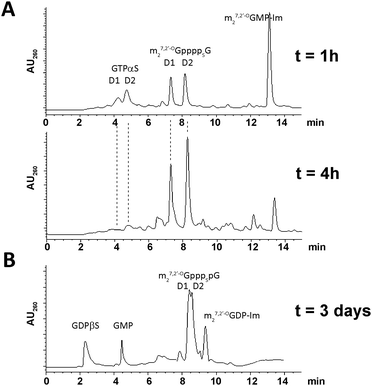 | ||
| Fig. 2 Typical HPLC profiles of reaction mixtures. (A) Progress in synthesis of 1 after 1 h and 4 h. (B) Synthesis of 2 after 3 days. | ||
| Compound | Imidazolide | Nucleophile | Yield (%) | D1:D2 ratiob | ||
|---|---|---|---|---|---|---|
| No. | Abbreviation | HPLC | Isolateda | |||
| a Based on the optical density units at 260 nm of diastereomeric mixtures of products before their HPLC separation. b Based on HPLC and/or 1H NMR. | ||||||
| 1 | m27,2′-OGppppSG | m27,2′-OGMP-Im (7) | GTPαS (5) | >90 | 60 | 7:10 |
| 2 | m27,2′-OGpppSpG | GMP-Im (8) | m27,2′-OGTPγS (15) | 56 | 39 | 9:10 |
| 2 | m27,2′-OGpppSpG | m27,2′-OGDP-Im (14) | GDPβS (12) | 62 | 35 | 8:10 |
| 3 | m27,2′-OGppSppG | GDP-Im (13) | m27,2′-OGDPβS (11) | 68 | 32 | 7:10 |
| 4 | m27,2′-OGpSpppG | GMP-Im (8) | m27,2′-OGTPαS (6) | >90 | 65 | 7:10 |
The synthesis of analogs modified at internal positions of the tetraphosphate bridges, i.e., 2 and 3, involved formation of mixed phosphate–thiophosphate anhydride bonds in the final step (Scheme 1B and C). This required the nucleoside 5′-(2-thiodiphosphates) 11 and 12 or 5′-(3-thiotriphosphate) 15, which were synthesized with 80–85% yields by a procedure reported previously by our group.25 In the case of mixed anhydrides, the formation of dinucleotides proceeded slower than for unmodified ones. The reaction progress usually stopped after 3 d despite some reactants remaining. Partial hydrolysis of 11, 12 or 15 also occurred due to extended reaction times; consequently, HPLC conversions and isolated yields were lower than in the case of 1 and 4 but were still acceptable (Table 1, Fig. 2B). In the case of analog 2, both synthetic pathways proved to be equally efficient.
Analogs 1–4 were isolated after ion-exchange chromatography as diastereomeric mixtures. The diastereomers of 1 and 4, in which the stereogenic P-center is next to the nucleoside moiety, were well separated by either analytical or preparative RP HPLC (see Experimental section). The differences between diastereomers of 2 and 3 were less distinct, perhaps because the P-stereogenic center is located in the more flexible part of tetraphosphate bridge and is more distant from the nucleoside stereogenic centers. Hence, we were unable to find conditions in which these diastereomers could be separated from each other on a preparative basis, although different mobile phases and two types of columns were tested. However, in case of 2 we managed to separate small amounts of compound sufficient to determine some biochemical properties (∼0.5 mg). Thus analogs 1 and 4 were subjected to biological studies as diastereomerically pure samples (1a and 4a = D1, 1b and 4b = D2), but 3 was studied as a diastereomeric mixture. Compound 2 was studied both as a diastereomeric mixture (2) and as pure diastereomers (2a = D1, 2b = D2).26
All newly synthesized compounds were characterized by MS ESI (−), 1H and 31P NMR. Each pair of diastereomers showed some small but significant differences in their 1H chemical shifts, particularly for H8 of the nucleobase and H1′ of the ribose moiety of either guanosine or 7,2′-O-dimethylguanosine. For 3 distinctive differences were also observed for the m27,2′-OGuo H3′ and both methyl groups. In case of all compounds modified with sulfur at a phosphate neighboring a nucleoside (both nucleoside triphosphates 5 and 6 and dinucleoside tetraphosphates 1 and 4) the H8 proton of the diastereomer with shorter RP HPLC retention time (D1) was more up-field shifted than that of D2, with ΔδH chemical shift differences being in the range of 0.2–0.7 ppm (see Experimental section and Table S1). For ATPαS and its analogs, Major et al.27 showed that the faster diastereomer in RP HPLC, with a less shielded H8 proton, has the SP configuration; their model is that the H8 shift depends on its distance from the S atom, which is different for the two diastereomers. The same correlation between the RP HPLC retention time, H8 proton shift and the absolute configuration was subsequently reported for other structurally related compounds, including guanosine nucleotides.28 Some minor differences between diastereomers were also noted in the 31P NMR spectra of 1–6. One observation was that the signal of the phosphorothioate moiety in compounds 1, 4, 5 (GTPαS) and 6 was 0.2–0.3 ppm up-field shifted for D1 compared to D2 (Table S1). The absolute configuration of GTPαS was first reported by Conolly et al.;29 the RP HPLC-faster diastereomer was assinged as SP. Later Ludwig and Eckstein reported that the α-phosphorus signals in GTPαS (and other NTPαSs) were ∼0.2 ppm more up-field for D1 (SP) than for D2 (RP).30 The above-mentioned similarities in the 1H and 31P spectra of compounds 1 and GTPαS (5) suggest that their D1 diastereomers have the same, i.e. SP, absolute configuration. This is further supported by the observation that 1a and 1b are formed in the same diastereomeric ratio as the 5a and 5b ratio before reaction. On the basis of analogous observations made for compounds 4 and 6, one can presume that in their cases the isomer D1 is also of SP configuration. However, this assumption needs to be further confirmed, e.g. by X-ray crystallography.
Binding affinities to eIF4E
The binding affinities of new analogs for murine eIF4E were determined by fluorescence quenching (Fig. 3).31 Experimental curves obtained upon titration of eIF4E with increasing concentrations of cap analogs are depicted in Fig. 3A. The descending fragment of each curve is due to the quenching of intrinsic Trp fluorescence in eIF4E upon cap binding, whereas the increasing fluorescence signal after reaching the plateau originates from fluorescence emission of the unbound cap analog.31 The KAS values and free energies of binding (ΔG°) for the new analogs and reference compounds are presented in Table 2 and Fig. 3B. As expected, all new analogs have binding affinities at least 10-fold higher than the unmodified cap structure, m7GpppG. The comparison to m27,2′-OGppppG reveals that the phosphorothioate moiety generally stabilizes complexes with eIF4E. Modifications in the δ-, γ-, and β-positions (analogs 4a, 4b, 3, 2a, 2b) give additional energetic gains which are similar to those reported for the corresponding modifications in the triphosphate series.32 By contrast, the phosphorothioate substitution in the α-position, which represent the ‘extra’ phosphate moiety (analogs 1a and 1b), has virtually no influence on the binding affinity.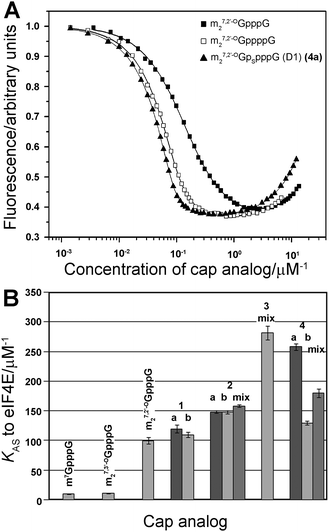 | ||
| Fig. 3 K AS constants for eIF4E–cap analog complexes. (A) Fluorescence quenching curves for titration of eIF4E with selected cap analogs (m27,3′-OGpppG, m27,2′-OGppppG and 4a). (B) Comparison of KAS values of 4P-S-ARCAs (1–4) with standard triphosphate analogs and unmodified tetraphosphate ARCA. The KAS values are means of 3 experiments. | ||
| Cap analoga | K AS cap-eIF4Eb (μM−1) | ΔG° binding energy (kcal mol−1) | Relative translation efficiency of capped mRNAc | Relative cap-dependent mRNA translation efficiencyd |
|---|---|---|---|---|
|
a D1 or D2 designate one of two P-diastereomers; for each pair D1 refers to the one eluted faster from RP HPLC column.
b Values are means of 3 experiments.
c The mean values were calculated from 2–3 assay repetitions for each of two independent mRNA syntheses.
d Calculated by correcting the translation of each mRNA in a particular experiment by subtracting the translation of mRNA capped with ApppG (a nonfunctional cap structure).
e Data from ref. 10.
f The composition of D1/D2 mixtures was as shown in Table 1, except m27.2′-OGpSpppG (mix), which was in a 1:1 ratio.
|
||||
| m7GpppG | 9.4 ± 0.4 | −9.35 ± 0.03 | 1 | 1 |
| ApppG | <0.05 | >−6.20 | 0.21 ± 0.04 | 0 |
| m27.3′-OGpppG | 10.2 ± 0.3e | −9.40 ± 0.02 | 1.62 ± 0.25 | 1.79 ± 0.33 |
| m27.2′-OGppppG | 99.8 ± 6.0e | −10.73 ± 0.04 | 2.01 ± 0.43 | 2.33 ± 0.61 |
| m27.2′-OGppppSG (D1) | 120 ± 7 | −10.83 ± 0.03 | 2.73 ± 0.84 | 3.21 ± 1.02 |
| m27.2′-OGppppSG (D2) | 109 ± 4 | −10.78 ± 0.03 | 2.24 ± 0.45 | 2.58 ± 0.55 |
| m27.2′-OGpppSpG (mix)f | 158 ± 2 | −10.99 ± 0.01 | 2.63 ± 0.40 | 3.03 ± 0.59 |
| m27.2′-OGpppSpG (D1) | 148 ± 2 | −10.95 ± 0.01 | n.d. | n.d. |
| m27.2′-OGpppSpG (D2) | 148 ± 3 | −10.95 ± 0.01 | 3.79 ±1 .12 | 4.57 ± 1.33 |
| m27.2′-OGppSppG (mix)f | 282 ± 12 | −11.33 ± 0.02 | 3.08 ± 0.69 | 3.72 ± 0.90 |
| m27.2′-OGpSpppG (D1) | 258 ± 6 | −11.28 ± 0.01 | 2.15 ± 0.12 | 2.49 ± 0.23 |
| m27.2′-OGpSpppG (D2) | 129 ± 4 | −10.87 ± 0.02 | 2.59 ± 0.53 | 3.04 ± 0.74 |
| m27.2′-OGpSpppG (mix)f | 180 ± 7 | −11.07 ± 0.02 | n.d | n.d |
Susceptibility to enzymatic degradation by specific and nonspecific pyrophosphatases
The susceptibility of new analogs to enzymatic hydrolysis by the cap-specific human DcpS pyrophosphatase was tested by monitoring enzymatic reactions progress by RP HPLC, as described in the Experimental section (Fig. 4). In all experiments m7GpppG was used as a positive control. The amount of DcpS was adjusted to assure complete cleavage of m7GpppG to m7GMP and GDP over 15 min. Under these conditions, analogs bearing modification at the δ-position remained undigested after 24 h of incubation with DcpS, regardless of the P-center absolute configuration, and hence, were considered to be resistant to hydrolysis. Analogs 1–3 were hydrolyzed more than 80% by hDcpS to m27,2′-OGMP and the corresponding nucleoside triphosphate in 2 h, which is comparable to hydrolysis of m27,2′-OGppppG. The identification of hydrolysis product was confirmed by co-injections with authentic samples of m27,2′-OGMP, GTP, and GTPαS SP or RP. The analysis of DcpS degradation products of analogs 1a and 1b adds support to our contention that their absolute configurations around P-stereogenic centers are the same as those of GTPαS (D1) and (D2), respectively, i.e., SP and RP (Fig. 4).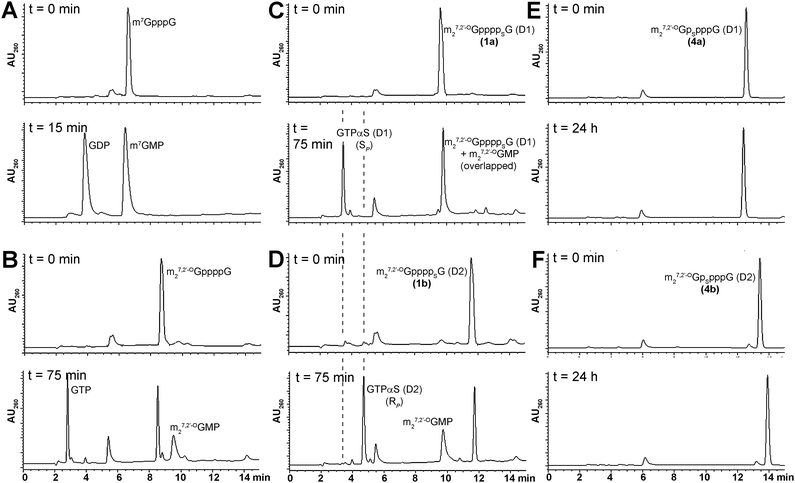 | ||
| Fig. 4 RP HPLC profiles of mRNA cap dinucleotides digestion by hDcpS. A) m7GpppG B) m27,2′-OGppppG C) 1a D) 1b E) 4a F) 4b. Assay conditions are given in the Experimental section. | ||
The new analogs were also subjected to hydrolysis by a non-specific enzyme, Snake Venom Phosphodiesterase I (SVPDE). SVPDE is known to hydrolyze nucleic acids, NTPs, and other structurally related compounds to release nucleoside 5′-monophosphates. The enzyme has been commonly used to determine absolute configurations of phosphorothioate diester bonds and nucleoside-1-thiotriphosphates, as it more readily cleaves phosphorothioate moieties of RP than SP configuration.33,34 By means of this enzyme we hoped to additionally confirm the configurations of analogs modified at the δ-position of tetraphosphate bridge (4a and 4b). We found that SVPDE was capable of hydrolyzing m7GpppG, and surprisingly, the initially (after 15 min) released products were mainly GMP and m7GDP rather than m7GMP and GDP (∼1:3 ratio). During prolonged incubation with SVPDE, the initially formed small amounts of GDP were converted into GMP. Subsequently, a dephosphorylation of GMP was observed, whereas m7GDP remained intact, which suggests it is not a substrate for SVPDE. In the case of m27,2′-OGppppG this regioselectivity of cleavage was even more notable, as initially almost exclusively GMP and m27,2′-OGTP were formed. Prolonged incubation resulted, however, in simultaneous dephosphorylation of GMP and cleavage of m27,2′-OGTP to m27,2′-OGMP. Surprisingly, when analogs modified at the α-position (1a and 1b) were subjected to SVPDE digestion, the regioselectivity (regardless of the configuration of the P-stereogenic center) was opposite to that observed for m7GpppG and m27,2′-OGppppG. The complete cleavage of both 1a and 1b occurred within 1.5 h and the only products observed were m27,2′-OGMP and GTPαS (either D1 or D2). The reaction mixture remained practically unchanged after further incubation with enzyme, so unfortunately, under these conditions, we were not able to observe stereoselectivity of the cleavage of the phosphorothioate moieties. The analogs modified at the δ-position, 4a and 4b, were cleaved to release GMP and m27,2′-OGTPαS (D1) or (D2), respectively. As for 1a and 1b, no further cleavage of NTPαS occurred. Nonetheless, this experiment adds support also to the assumption of the absolute configurations of 4a and 4b, and identifies SVPDE as a new tool for studying modified dinucleotide cap analogs, since its specificity is complementary to that of DcpS.
Translational efficiencies of 4P-S-ARCAs
Finally, the new analogs were investigated for their utility as reagents for increasing the translational activity of mRNA. Transcripts encoding firefly luciferase were synthesized by SP6 RNA polymerase in the presence of a DNA template, four NTPs, and a cap dinucleotide (1a, 1b, 2, 2b, 3, 4a, 4b). The assays included three reference compounds: m7GpppG, m27,3′-OGpppG, and m27,2′-OGppppG. In vitro-synthesized mRNAs were translated in a micrococcal nuclease-treated RRL system and the activity of luciferase per μg of RNA was determined by luminometry after 60 min (Fig. 5A). Cap-dependent translational efficiencies were calculated by correcting the translation of each mRNA in particular experiment by subtracting the translation of mRNA capped with ApppG (a nonfunctional cap structure), which is considered to be cap-independent. The overall translational efficiencies and cap-dependent translational efficiencies (both normalized to the translation of m7GpppG-capped mRNA) are shown in Table 2.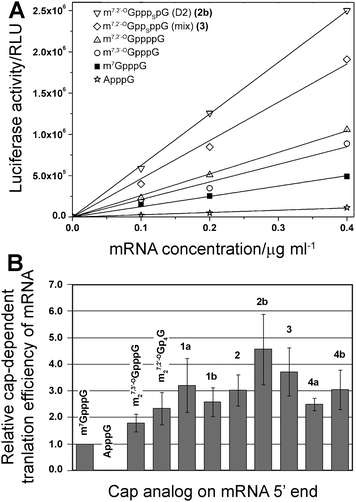 | ||
| Fig. 5 Translational efficiency of mRNA transcripts in vitro. (A) Example experiment of translational efficiency of mRNA transcript encoding firefly luciferase capped with various dinucleotides. (B) Comparison of relative cap-dependent translational efficiency of mRNA transcripts. The mean values were calculated from 2–3 assay repetitions for each of two independent mRNA syntheses. | ||
In our experiment we could reproduce results for cap analogs that were previously tested.10 For instance, mRNA capped with m27,3′-OGpppG and m27,2′-OGppppG were translated 1.79 ± 0.33 and 2.33 ± 0.61 more efficiently than m7GpppG-capped mRNA, respectively, which is similar to the previously reported values of 1.88 ± 0.40 and 2.56 ± 0.18. Importantly, mRNAs capped with all of the new 4P-S-ARCAs were not translated less efficiently than mRNA capped with the parent compound m27,2′-OGppppG. Their relative translation efficiencies ranged from 2.5 to 4.5 compared to m7GpppG (Table 2, Fig. 5B), which makes them attractive candidates for further studies in living cells. mRNAs capped with two analogs, 3 and 2b, were translated more than 3-fold more efficiently than m7GpppG-capped mRNA.
Discussion
Nucleoside and dinucleoside polyphosphates are biologically important molecules. Numerous methods for their synthesis have been developed, several of them providing synthetic routes to dinucleoside tri- or tetraphosphates modified at non-bridging positions in the phosphate group(s).29,35–40 However, most of these methods have drawbacks with regard to synthesis of mRNA cap analogs. These include limited structural variety of possible products available by a particular method (e.g., limitation to symmetrical compounds or those modified at the α-position with respect to nucleoside) or requirement for protected nucleosides as starting materials. Due to the presence of positive charge in 7-methylguanine, m7Guo is unusually polar and thus poorly soluble in organic solvents. It is also susceptible to hydrolysis both in acidic and basic aqueous solutions,41 which are required for removal of the majority of protecting groups. These facts make synthesis of m7Guo-containing nucleotides even more challenging.As first noted by Kadokura et al.42 and then explored by our laboratory,8,10,32,43 and others,44 the formation of pyrophosphate bonds via coupling of nucleotides with nucleotide P-imidazolide derivatives in DMF is greatly accelerated by the presence of excess of anhydrous zinc chloride. The rationale for ZnCl2 catalysis is at least two-fold. First, it increases solubility of nucleotides in DMF, probably through complexing negatively charged phosphates. Due to that, it is possible to obtain completely homogenous DMF solutions of nucleotide triethylammonium salts or even sodium salts in the case of P-imidazolide derivatives. Second, ZnCl2 is thought to act as Lewis acid to activate imidazole as a leaving group.
The synthesis of 4P-S-ARCAs has also been carried out taking advantage of this phenomenon. The formation of dinucleoside tetraphosphates modified at either the α- or β-position with respect to the closest nucleoside can be generally achieved by any of the pathways depicted in Scheme 2. The choice of particular pathway should depend on the availability of the appropriate substrates as well as on the purification method to be applied.
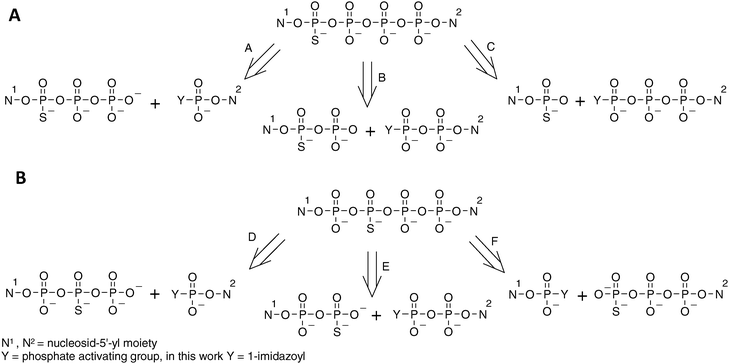 | ||
| Scheme 2 Possible disconnections of tetraphosphate-bridged thio-modified dinucleotides. (A) Tetraphosphates with phosphorothioate modification in the external position. (B) Tetraphosphates with phosphorothioate modification in the internal position. | ||
For the analogs 1 and 4 pathways A, B and C were taken into consideration. Pathway B was excluded, as nucleoside 5′-O-(1-thiodiphsophates) can be obtained by procedures similar to those for 5′-O-(1-thiotriphosphates), albeit with lower yields.28,45 Moreover, nucleoside diphosphates are available from the corresponding activated monophosphates used in pathway A. This would ultimately lengthen the synthesis, giving no obvious advantage over pathway A. Pathway C would involve coupling of nucleoside 5′-monothiophosphates (which are easily available) and imidazolide derivatives of nucleoside triphosphates (which are possible to obtain,10 although – due to decreased solubility in DMF –with lower yields than those for mono- and diphosphates). Moreover, our experience gained during the synthesis of thio-modified dinucleoside triphosphates shows that formation of a mixed phosphate–thiophosphate anhydride bond is much slower than for an unmodified pyrophosphate bond, which would presumably enhance by-product formation, hampering product purification. Hence, the pathway A appeared to us as favorable, since it involves the most available substrates and, additionally, the formation of the tetraphosphate bridge involves coupling between two unmodified phosphate moieties. This route proved to be efficient, as the coupling reactions leading to analogs 1 and 4 were relatively rapid (several hours), with HPLC conversions exceeding 90%.
In regard to the analogs 2 and 3, pathways D, E and F should be considered. Pathway D, although it shares some of the advantages of pathway A, is less favorable since nucleoside 5′-(2-thiotriphosphates) can be obtained only by complicated procedures. Pathways E and F seemed to be both acceptable and both proved to be similarly efficient, with HPLC conversions from 50 to 70%.
It is worth emphasizing that the described synthetic method can be also applied to other biologically important dinucleoside polyphosphates.46 It would be particularly advantageous for the synthesis of unsymmetrical compounds, for instance analogs of uridine adenosine tetraphosphate,47 or for compounds bearing highly polar nucleosides.
The determination of binding affinity to eIF4E by means of fluorescence quenching titration is a relatively simple yet very useful biophysical test that enables one to predict the functionality of any newly synthesized cap analog. A sufficiently high binding affinity for eIF4E is essential for the recognition of mRNA by translational machinery and efficient competing of mRNAs possessing modified caps with natural mRNAs.
In one of our previous studies,11 we identified a triphosphate ARCA analog modified with a methylene group between the α and β phosphates, m27,3′-OGppCH2pG, which protected mRNA against hydrolysis by Dcp2-decapping pyrophosphatase. Due to this, the mRNA had a prolonged half-life when transfected into cultured cells. However, the methylene modification coincidentally decreased the binding affinity to eIF4E from 10.2 ± 0.3 (for m27,3′-OGpppG) to 4.41 ± 0.2 μM−1. This resulted in the mRNA capped with m27,3′-OGppCH2pG (despite being more stable in vivo) being translated even less efficiently than that capped with regular ARCA.11 Another example of the influence of the binding affinity to eIF4E on mRNA translation is the tetraphosphate ARCA analog, m27,2′-OGppppG, which due to the additional phosphate moiety has KAS much higher than the corresponding triphosphate, because of which it was shown to enhance mRNA translation efficiency in vitro.10 It is worth mentioning that the combination of methylene modification with the presence of the additional, fourth, phosphate moiety has been shown recently to compensate for the loss of binding affinity.43
The high binding affinity for eIF4E of new tetraphosphate S-ARCAs constituted thus an important premise that all those analogs should be at least as efficient in promoting translation as m27,2′-OGppppG. The particular KAS values were dependent both on the position of the phosphorothioate modification and on the absolute configuration of the P-stereogenic center. Their comparison to m27,2′-OGppppG and to the S-ARCA triphosphate series reveals that the phosphorothioate moiety in the δ-, γ- and β-positions of tetraphosphate correspond to γ-, β- and α-modifications in the triphosphate bridge.32,48 This implies that eIF4E ‘counts’ the phosphates starting from m7Guo, which is in agreement with the well-established fact that the m7Guo moiety is crucial for mRNA 5′-end recognition by eIF4E. For instance, the determined ΔG° binding energy for 4a was 0.40 and 0.54 kcal mol−1 lower than that of 4b and m27,2′-OGppppG, respectively. In the triphosphate series the ΔΔG° differences between m27,2′-OGpSppG (D1) and either its D2 counterpart or m27,2′-OGpppG were 0.59 and 0.69 kcal mol−1, respectively. This observation indicates that the energetic gains arising from the presence of an additional phosphate and an oxygen-to-sulfur substitution are additive. For analog 3 we were only able to determine the binding affinity for the D1/D2 mixture. In the triphosphate S-ARCA series, modification at the corresponding position (β) resulted in notably different KAS values for D1 and D2 (43.1 ± 1.4 versus 19.3 ± 2.2 μM−1, respectively). From the titration performed on the 1:1 mixture of 4a and 4b, which also differ in their KAS, we found that the KAS value determined for a diastereomeric mixture is roughly a mean of the KAS values for pure diastereomers. Interestingly, the KAS value for analog 3 (282 ± 12 μM−1) is similar to that of pentaphosphate ARCA analog, m27,3′-OGp5G. As mentioned in the Introduction, this analog, despite its high KAS value (299 ± 20 μM−1), was not effective in promoting translation when incorporated into the mRNA 5′-end.10 The proposed explanation was that the extraordinarily high binding affinity interferes with release of the cap at the end of translation initiation, and thereby inhibits further translation events. However, the high translation efficiency of analog 3 suggests that some other factors may contribute to this decreased efficiency of mRNA capped with m27,3′-OGp5G, e.g. low capping percentage during in vitro transcription, or non-specific interactions of highly charged pentaphosphate chain with other proteins.
The results of in vitro translation of mRNAs capped with new 4P-S-ARCAs further support the initial assumption that combining the phosphorothioate modification with elongation of the 5′,5′-bridge to tetraphosphate may produce analogs with beneficial properties in terms of preparation of translationally efficient mRNA. The results suggest that the largest increase in mRNA translation efficiency may be caused by modifications in the internal positions of the tetraphosphate chain (i.e. β and γ). The somehow unexpected difference between translation efficiency of 2 (diastereomeric mixture) and 2b (diastereomer D2) underlines the need for finding a method of separating it into pure diastereomers in order to explore this phenomenon.
This more efficient mRNA translation in vitro may benefit biotechnological in vitro protein production. The determination of translation efficiency in RRL is also a useful preliminary test, enabling selection of most attractive analogs for expensive studies in cultured cells. However, conventionally used rabbit reticulocyte lysates (RRLs), are micrococcal nuclease pre-treated to deplete any endogenous mRNAs present in the lysate. Therefore, this assay does not fully reflect the cellular conditions since: (i) there is no competition of investigated mRNA with natural mRNAs, and (ii) the influence of its stability (half-life) on overall translation yield is rather marginal. Both of these factors have a large impact on the overall protein synthesis in vivo.13 The results from our previous studies suggest that in cellular conditions the differences in translation yield between mRNAs capped with modified and unmodified analogs should be more pronounced.
The ability to produce more stable and more efficiently translated in vivo mRNAs can be advantageous for applications in mRNA-based gene therapy. Several approaches have been exploited, to improve the stability and/or translation efficiency of mRNA vectors in vivo. This includes extending the polyA length, alterations of the mRNA 3′-end, and introducing into mRNA modified nucleosides, which retain coding properties of natural ones.3,12,49,50 Modification of mRNA cap structure is another possibility to facilitate mRNA-based gene delivery. A particularly attractive application of mRNA in therapy is transfection of the patient’s own dendritic cells with mRNAs encoding tumor-associated antigens to evoke auto-immunization against cancer cells.51
We believe that the 4P-S-ARCAs will also enable some new insights into mRNA-related physiological processes. The synthetic cap analogs have been commonly used to investigate mechanism of initiation of protein biosynthesis, and the discovery of the involvement of decapping enzymes in the mRNA degradation pathways expanded further the scope of processes that have been studied by means of cap analogs. One of the still unsolved problems is the detailed mechanism of the Dcp2 decapping enzyme, which belongs to the NuDiX family of pyrophosphatases.16 It is known that the enzyme utilizes cap structures on intact mRNAs and cleaves between the α- and β-phosphates of the triphosphate bridge. The finding that the phosphorothioate group in the β-position protected mRNA from decapping suggests that the β-phosphate is the one directly involved in the catalytic mechanism.13 However, since Dcp2 requires both cap structure and mRNA chain for its activity, it is unclear whether the enzyme recognizes the phosphate groups ‘counting’ them from the m7Guo site or from the mRNA body. The susceptibility to Dcp2 of tetraphosphate S-ARCA modified in the β and γ-positions, which is currently under investigation, may address this issue.
On the other hand, the analogs modified in the δ-position proved to be resistant to the DcpS decapping enzyme, which cleaves cap structures lacking an mRNA chain. DcpS-resistant cap analogs with high affinity for eIF4E are important in the context of developing stable inhibitors of cap-dependent translation which could counteract elevated levels of eIF4E in cancer cells.52 It is assumed that DcpS functions in the cytoplasm to prevent the accumulation of complexes between eIF4E and cap structures that would otherwise accumulate following 3′→5′ mRNA decay,53 which implies that only DcpS-resistant analogs could act as potent eIF4E inhibitors in cellular conditions. Moreover, it was also found that DcpS fulfils also some other biological functions, and constitutes an interesting object for investigation by itself. Its presence has also been detected in the nucleus, where it is implicated in mRNA splicing, and recently, it has been identified as therapeutic target for spinal muscular atrophy.54,55 Non-hydrolysable cap analogs such as m27,2′-OGpSpppG may serve as invaluable tools to investigate these processes, for instance by serving as inhibitors of DcpS activity.
Finally, it has been recently discovered that by modifying the 5′,5′-phosphate bridge in the mRNA cap structure one can observe enhancement in translation repression mediated by microRNAs.20 In the quoted work, two cap analogs have been identified, which in Drosophila melanogaster embryos extract evoke ∼2-fold stronger miRNA-mediated inhibition of translation initiation but are “neutral” towards general cap-dependent translation: the triphosphate ARCA modified at the α-position, m27,2′-OGpppSG (D2), and the analog with the 5′,5′-bridge extended to hexaphosphate, m7Gp6G. Thus a 4P-S-ARCA modified at the α-position, which somehow combines the structural features of these two analogs, may be an interesting tool for studying mechanism of microRNA action.
Conclusions
A general methodology for the synthesis of unsymmetrical dinucleotides bearing a single phosphorothioate modification in either position of the tetraphosphate chain has been developed. Four pairs of tetraphosphate S-ARCAs have been synthesized with good yields. Biophysical and biochemical characterization of new analogs indicates that 4P-S-ARCAs are good candidates for reagents to produce mRNA transcripts with high translational activity. Stabilizing effects of the additional phosphate as well as the phosphorothioate substitutions on the cap–eIF4E complex are additive, which is reflected in KAS values much higher than for triphosphate cap analogs and notably higher than for unmodified tetraphosphate ARCA. This property of 4P-S-ARCAs should be reflected in the kinetics of translation initiation complex formation and make synthetic transcripts more competitive than endogenous mRNA in vivo.It is also shown that phosphorothioate modification in the δ-position of the tetraphosphate bridge protects the dinucleotide cap analog against hydrolysis by DcpS. Similarly, the modification in the appropriate position of the tetraphosphate bridge should decrease the susceptibility of 4P-S-ARCA-containing mRNA transcripts to hydrolysis by Dcp2/Dcp1, which would affect their half-life and give an additional contribution to translational activity in vivo.
The results of studies on translation in vitro show that mRNAs capped with new analogs are 2.5–4.5 times more efficiently translated in a cell-free system than m7GpppG-capped mRNAs. On the basis of our previous studies, one can predict that the effect of these novel caps on translational efficiency should be more pronounced when measured in cultured cells. These observations indicate that 4P-S-ARCAs are promising candidates for studies on therapeutic applications of mRNA, which include mRNA-based gene delivery and antitumor vaccination. In addition to the biotechnological and potential therapeutic applications, 4P-S-ARCAs appear to be useful tools for elucidating mechanisms of various mRNA-related physiological processes.
Experimental section
Chemical syntheses
General procedures
Solvents and other reagents were purchased from Sigma-Aldrich and used without further treatment, unless otherwise stated. Reaction mixtures were analyzed by analytical reverse-phase (RP) HPLC, which was performed on a Agilent Technologies Series 1200 apparatus using a Supelcosil LC-18-T RP column (4.6 × 250 mm, 5 μm, flow rate 1.3 mL/min) developed with a 0–50% linear gradient of methanol in 0.05 M ammonium acetate buffer (pH 5.9) over 30 min, UV-detection at 260 nm and fluorescence detection (excitation at 280 nm and detection at 337 nm). Samples for HPLC analysis were prepared by dissolving 5 μL of a reaction mixture in 100 μL of either water or, in the case of ZnCl2-containing reactions, an aqueous solution of EDTA (10 mg/mL) and NaHCO3 (5 mg/mL).Nucleotides were purified and isolated from reaction mixtures by ion-exchange chromatography on DEAE-Sephadex A-25 (HCO3− form) column using a linear gradient of triethylammonium bicarbonate (TEAB) in deionized water (1.6 L of each) and, after evaporation under reduced pressure with repeated additions of ethanol and drying in a vacuum dessicator over P2O5, isolated as triethylammonium (TEA) salts.
The final products were additionally purified (and separated) by semi-preparative RP HPLC, which was performed on either a Waters 600E Multisolvent Delivery System apparatus using Discovery RP Amide C-16 HPLC column (25 cm × 21.2 mm, 5 μm, flow rate 5.0 mL/min) (or Ascentis C18 HPLC column (25 cm × 21.2 mm, 5 μm, flow rate 5.0 mL/min) developed with linear gradients of methanol in 0.05 M ammonium acetate (AAC) buffer (pH 5.9). UV-detection at 260 nm. After repeated freeze-drying, the products were isolated as ammonium salts. For compound 6, triethylammonium acetate (TEAAC) buffer (pH 7.0) was used in order to isolate compounds as TEA salts.
Yields were preferably calculated based on optical density milliunits at 260 nm (mODU260) of substrates and isolated products, measured in 0.1 M phosphate buffer of pH either 6 (for m7Guo mononucleotides) or pH = 7 (for cap dinucleotides and Guo nucleotides). Optical density miliunits mODU260 are defined as absorption of compound solution in 0.1 M phosphate buffer of appropriate pH at 260 nm multiplied by the volume of the solution in ml. Extinction coefficients taken for calculations were εpH![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 6 = 11400 M−1 cm−1 for m7Guo mononucleotides, εpH7 = 22600 M−1 cm−1 for cap dinucleotides and εpH7 = 12000 M−1 cm−1 for Guo mononucleotides.
6 = 11400 M−1 cm−1 for m7Guo mononucleotides, εpH7 = 22600 M−1 cm−1 for cap dinucleotides and εpH7 = 12000 M−1 cm−1 for Guo mononucleotides.
The structures and homogeneities of synthesized nucleotides were confirmed by chromatography on analytical RP HPLC, mass spectrometry using negative electrospray ionization (MS ESI−) and both 1H and 31P NMR spectroscopy. 1H NMR and 31P NMR spectra were recorded in D2O at 25 °C for samples at 1.5–3.0 mg mL−1 concentrations on a Varian UNITY-plus spectrometer at 399.94 MHz and 161.90 MHz, respectively. 1H NMR chemical shifts in ppm were reported to sodium 3-trimethylsilyl[2,2,3,3-D4]propionate (TSP) in D2O as an internal standard. 31P NMR chemical shifts in ppm were reported to 80% phosphorus acid in D2O as an external standard. J values are given in Hz. Mass spectra were recorded on a Micromass QToF 1 MS spectrometer.
2′-O-Methylguanosine was synthesized as described by Kusmierek and Shugar.56 7,2′-O-Dimethylguanosine was obtained from 2′-O-methylguanosine by methylation with CH3I analogously to that described earlier for 7-methylguanosine.57 Tributylammonium pyrophosphate was prepared according to Ludwig and Eckstein.30 Thiophosphate TEA salt was obtained from its trisodium salt as described earlier.25 Guanosine 5′-monophosphate disodium salt was purchased from Fluka and before use converted into its triethylammonium (TEA) salt by passing through Dowex 50W X8 (100–200 mesh), evaporating collected fractions to dryness and drying in vacuum over P2O5. Nucleotides: guanosine 5′-diphosphate (9), 7,2′-O-dimethylguanosine 5′-monophosphate and 7,2′-O-dimethylguanosine 5′-diphosphate (10) (all as TEA salts) as well as imidazolide derivatives 7, 8, 13 and 14 (all as sodium salts) were synthesized as described previously.10,43
:100 SP:RP (D1:D2, 5a:5b) diastereomeric mixture. ESI MS (−) m/z 537.98 (Calc. for C10H15N5O13P3S: 537.96). 5a: Analytical RP HPLC Rt = 3.5 min δH 8.29 (1H, s, H(8) G); 5.95 (1H, d, J1′,2′ 6.3, H(1′) G); 4.85 (1H, ∼t, J1′,2′ 6.3 J2′,3′ 4.8, H(2′)); 4.60 (1H, dd, J2′,3′ 4.8, J3′,4′ 3.2, H(3′)); 4.39 (1H, m, H(4′)); 4.30 (2H, m, H(5′) H(5′′)). δP 43.12 (1P, d, JP1,P2 27.0, P1), −10.81 (1P, d, JP2,P3 19.6, P3), −23.98 (1P, dd, JP1,P2 27.0, JP2,P3 19.6, P2). 5b: Analytical RP HPLC Rt = 4.1 min δH 8.22 (1H, s, H(8)); 5.95 (1H, d, J1′,2′ 6.3, H(1′)); 4.85 (1H, ∼t, J1′,2′ 6.3 J2′,3′ 4.8, H(2′)); 4.60 (1H, dd, J2′,3′ 4.8, J3′,4′ 3.2, H(3′)); 4.39 (1H, m, H(4′)); 4.30 (2H, m, H(5′) H(5′′)), δP 43.64 (1P, d, JP1,P2 27.0, P1), −10.81 (1P, d, JP2,P3 19.6; P3), −24.02 (1P, dd, JP1,P2 27.0, JP2,P3 19.6; P2).
:10 SP:RP (D1:D2, 6a:6b) diastereomeric mixture. The 31P NMR analysis revealed that the isolated product was contaminated with ∼1.5 eq. of pyrophosphate. Consequently, the product was further purified by semi-preparative RP HPLC using a linear gradient of 0–50% MeOH in TEAAC (pH 7.0) over 45 min and the diastereomers were collected together yielding, after repeated freeze-drying, 6 (1350 mODU260, 120 mg, 0.12 mmol, 30%) as a 7:10 diastereomeric mixture. Additionally, a small portion of 6 was purified using a linear gradient of 0–25% MeOH in AAC (pH 5.9) over 120 min, the diastereomers were collected separately (6aRt = 41 min (SP, D1), 6bRt = 45 min (RP, D2)) and after repeated freeze-drying isolated as NH4+ salts. ESI MS (−) m/z 566.02 (calc. for C12H19N5O13P3S: 565.99). 6a: Analytical RP HPLC Rt = 8.5 min δH 9.33 (1H, s, H(8)); 6.15 (1H, d, J1′,2′ 2.6, H(1′)); 4.70 (1H, ∼t, J2′,3′ = J3′,4′ ∼5.3, H(3′)); 4.42–4.33 (4H, overlapped m, H(2′), H(4′), H(5′), H(5′′)); 4.17 (3H, s, NCH3); δP 43.54 (1P, d, JP1,P2 28.0, P1), −10.32 (1P, d, JP2,P3 20.0; P3), −23.73 (1P, dd, JP1,P2 28.0, JP2,P3 20.0, P2). 6b: Analytical RP HPLC Rt = 9.0 min, δH 9.30 (1H, s, H(8)); 6.14 (1H, d, J1′,2′ 2.1, H(1′)); 4.66 (1H, ∼t, J2′,3′ = J3′,4′ ∼5.3, H(3′)); 4.46–4.27 (4H, overlapped m, H(2′), H(4′), H(5′), H(5′′)); 4.16 (3H, s, NCH3); δP 43.33 (1P, d, JP1,P2 27.6, P1), −10.55 (1P, d, JP2,P3 19.8, P3), −23.73 (1P, dd, JP1,P2 27.6, JP2,P3 19.8, P2).
General procedure for nucleoside 5′-(2-thiodiphosphates) and 5′-(3-thiotriphosphates) – according to ref. 25
An appropriate nucleotide imidazolide derivative (1 eq.) was mixed with thiophosphate TEA salt (3–4 eq.) in DMF followed by immediate addition anhydrous ZnCl2 (at least 8 eq.) and vigorous shaking until a homogenous solution was obtained. The reaction was quenched after 15–25 min by being diluted with ∼20 volumes of aqueous solution of disodium EDTA dihydrate (in 1:1 ratio to ZnCl2) and NaHCO3 (half of the amount of EDTA in mg). If necessary the pH was adjusted to 7–7.5 with an additional portion of NaHCO3. The product was isolated by ion exchange chromatography. Product-containing fractions were poured together and evaporated to dryness with repeated additions of 96% ethanol and, finally, absolute ethanol until a white solid resulted.
:10 SP:RP diastereomeric mixture. The diastereomers were separated using 0–25% gradient of MeOH in AAC (pH = 5.9) over 120 min (1a: Rt = 43 min (SP, D1), 1b: Rt = 49 min (RP, D2)) to yield 484 ODU260 (19.4 mg, 0.02 mmol, 16%) of D1 (1a) and 761 ODU260 (31 mg, 0.033 mmol, 25%) of D2 (1b) as NH4+ salts. ESI MS (−) m/z 911.05 (Calc. for C22H31N10O20P4S1: 911.04). 1a: Analytical RP HPLC Rt = 6.8 min; δH 9.19 (1H, s, H(8)m7G); 8.06 (1H, s, H(8)G); 6.03 (1H, d, J1′,2′ 2.4, H(1′)m7G); 5.83 (1H, d, J1′,2′ 6.0, H(1′)G); 4.72 (1H, ∼t, H(2′)G); 4.61 (1H, ∼t, H(3′)m7G); 4.52 (1H, ∼t, H(3′)G); 4.45-4.42 (7H, overlapped m, H(4′)m7G, H(4′)G, H(2′) m7G, H(5′)G, H(5′′)G, H(5′), H(5′′)m7G); 4.10 (3H, s, N–CH3); 3.58 (3H, s, O–CH3), δP 43.70 (1P, d, JP3,P4 26.0; P4), −11.11 (1P, d, JP1,P2 18.0; P1), −23.80 (1P, ∼t, JP1,P2 18.0, JP2,P3 17.2, P2), −24.90 (1P, dd, JP3,P4 26.0, JP2,P3 17.2; P3). 1b: Analytical RP HPLC Rt = 7.2 min; δH 9.16 (1H, s, H(8)m7G); 8.05 (1H, s, H(8)G); 5.99 (1H, d, J1′,2′ 2.4, H(1′)m7G); 5.80 (1H, d, J1′,2′ 6.0, H(1′)G); 4.75 (1H, ∼t overlapped with HDO, H(2′)G); 4.60–4.45 (2H, overlapped m , H(3′)m7G; H(3′)G); 4.40–4.30 (3H, overlapped m, H(4′)G, H(5′)G, H(5′′)G); 4.30–4.22 (3H, overlapped m, H(4′) m7G, H(5′), H(5′′)m7G); 4.10 (3H, s, N–CH3);3.59 (3H, s, O–CH3), δP 43.47 (1P, d, JP3,P4 26.3, P4), −11.27 (1P, d, JP1,P2 18.0; P1), −23.00 (1P, ∼t, JP1,P2 18.0, JP2,P3 17.0; P2), −24.07 (1P, dd, JP3,P4 26.3, JP2,P3 17.0, P3).
Method I. To a suspension of 14 (Na salt, 102 mg; 1165 mODU260, 0.10 mmol) and 12 (TEA salt, 1730 mODU260, 0.14 mmol) in DMF (3 ml) anhydrous ZnCl2 (250 mg, 1.84 mmol) was added and the mixture was shaken until reagents dissolved (1–2 min). The reaction was maintained at RT for 3 d and then quenched by addition of 685 mg (1.84 mmol) of EDTA in 100 ml of water and adjusted to pH 6 with solid NaHCO3. The product was isolated on DEAE Sephadex using 0–1.3 M gradient of TEAB to yield 795 mODU260 (0.035 mmol, 35%) of 2 (TEA salt). The product was additionally purified by semi-preparative RP HPLC using isocratic 10% MeOH and 90% AAC (pH 5.9) yielding 640 mODU260 (31 mg, 0.031 mmol, 28%) of 2, NH4+ salt) as 8
:10 (D1:D2, 2a:2b) diastereomeric mixture.
Method II. To a suspension of 8 (Na salt, 107 mg; 2570 mODU260, 0.21 mmol) and 15 (TEA salt, 1290 mODU260, 110 mg, 0.11 mmol) in DMF (2 ml) anhydrous ZnCl2 (775 mg, 5.69 mmol) was added and the mixture was shaken until reagents dissolved (1–2 min). The reaction was maintained at RT for 3 days and then quenched by addition of 2.11 g of EDTA (5.69 mmol) in 300 ml of water and neutralized with solid NaHCO3. The purification as in Method I yielded 1010 mODU260 (0.045 mmol, 39%) of 2 (TEA salt) as 9
:10 (2a:2b, D1:D2) diastereomeric mixture, and after additional HPLC purification 820 ODU260 (37 mg, 0.036 mmol, 31%) of 2. ESI MS (−) m/z 911.05 (calc. for C22H31N10O20P4S1: 911.04). 2a: Analytical RP HPLC Rt = 7.5 min, δH 9.04 (1H, s, H(8)m7G); 8.10 (1H, s, H(8)G); 5.97 (1H, d, J1′,2′ 2.9, H(1′)m7G); 5.80 (1H, d, J1′,2′ 5.9, H(1′)G); 4.70 (1H, ∼t, H(2′)G); 4.56 (1H, ∼t, H(3′)m7G); 4.50 (1H, ∼t, H(3′)G); 4.41 (1H, m, H(5′)G); 4.34 (2H, m, overlapped H(4′)m7G, H(4′)G); 4.27 (2H, m overlapped, H(2′)m7G, H(5′′)G); 4.24 (2H, m, H(5′), H(5′′)m7G); 4.08 (3H, s, N–CH3); 3.59 (3H, s, O–CH3). δP 30.49 (1P, t, JP2,P3 = JP3,P4 24.7, P3), −11.14 (1P, d, JP1,P2 17.6, P1), −11.88 (1P, d, JP3,P4 24.7, P4), −23.84 (1P, dd, JP2,P3 24.7, JP1,P2 17.6, P2) 2b: Analytical RP HPLC Rt = 7.6 min, δH 9.00 (1H, s, H(8)m7G); 8.08 (1H, s, H(8)G); 5.96 (1H, d, J1′,2′ 2.9, H(1′) m7G); 5.81 (1H, d, J1′,2′ 5.9, H(1′) G); 4.70 (1H, ∼t, H(2′)G); 4.55(1H, ∼t, H(3′)m7G); 4.48 (1H, ∼t, H(3′)G); 4.40 (2H, overlapped m, H(4′)m7G, H(5′)G); 4.30 (3H, m (overlapped), H(2′) m7G, H(4′)G, H(5′′)G); 4.25 (2H, m, H(5′), H(5′′)m7G); 4.07 (3H, s, N–CH3), 3.62 (3H, s, O–CH3). δP 30.35 (1P, t, JP2,P3 = JP3,P4 24.7, P3), −11.14 (1P, d, JP1,P2 17.6, P1), −11.88 (1P, d, JP3,P4 24.7, P4), −23.86 (1P, dd, JP2,P3 24.7, JP1,P2 17.6, P2).
:10 (3a:3b, D1:D2) diastereomeric mixture. ESI MS (−) m/z 911.04 (calc. for C22H31N10O20P4S1: 911.04) 3a: Analytical RP HPLC Rt = 7.5 min, δH 9.04 (1H, s, H8 m7G); 8.10 (1H, s, H(8)G); 5.97 (1H, d, J 2.9, H(1′)m7G); 5.80 (1H, d, J 5.9, H(1′)G); 4.70 (1H, ∼t, H(2′)G); 4.56 (1H, ∼t, H(3′)m7G); 4.50 (1H, ∼t, H(3′) G); 4.41 (1H, m, H(5′)G); 4.34 (2H, m, overlapped H(4′)m7G, H(4′)G); 4.27 (2H, m overlapped, H(2′)m7G, H(5′′)G); 4.24 (2H, m, H(5′), H(5′′)m7G); 4.08 (3H, s, N–CH3); 3.59 (3H, s, O–CH3), δP 30.21 (1P, t, JP1,P2 = JP2,P3 24.0; P2), −11.12 (1P, d, JP3,P4 18.5, P4), −11.96 (1P, d, JP1,P2 24.0, P1), −23.91 (1P, dd, JP2,P3 24.0, JP3,P4 18.5, P3). 3b: Analytical RP HPLC Rt = 7.6 min, δH 9.00 (1H, s, H(8)m7G), 8.08 (1H, s, H(8)G), 5.96 (1H, d, J 2.9, H(1′)m7G), 5.81 (1H, d, J 5.9, H(1′)G), 4.70 (1H, ∼t, H(2′)G); 4.55 (1H, ∼t, H(3′) m7G), 4.48 (1H, ∼t, H(3′)G), 4.40 (2H, overlapped m, H(4′)m7G, H(5′)G), 4.30 (3H, overlapped m, H(2′)m7G, H(4′)G, H(5′′)G), 4.25 (2H, m, H(5′), H(5′′)m7G), 4.07 (3H, s, N–CH3), 3.62 (3H, s, O–CH,), δP 30.21 (1P, t, JP1,P2 = JP2,P3 24.0, P2), −11.12 (1P, d, JP3,P4 18.5, P4), −11.96 (1P, d, JP1,P2 24.0, P1), −23.91 (1P, dd, JP2,P3 24.0, JP3,P4 18.5, P3).
:10 (SP:RP, D1:D2, 4a:4b) diastereomeric mixture. The diastereomers were separated by RP HPLC using 0–25% gradient of MeOH in AAC (pH = 5.9) over 120 min (4a: Rt = 51 min (SP, D1), 4b: Rt = 52 min (RP, D2)) to yield 196 ODU260 (8.5 mg, 0.0087 mmol, 20%) of 4a and 308 ODU260 (13.4 mg, 0.013 mmol, 31%) of 4b (both as NH4+ salts). ESI MS (−) m/z 911.06 (calc. for C22H31N10O20P4S1: 911.04) 4a: Analytical RP HPLC Rt = 7.5 min, δH 9.04 (1H, s, H(8)m7G), 8.10 (1H, s, H(8)G), 5.97 (1H, d, J 2.9, H(1′) m7G), 5.80 (1H, d, J 5.9, H(1′) G), 4.70 (1H, ∼t, H(2′) G), 4.56 (1H, ∼t, H(3′) m7G), 4.50 (1H, ∼t, H(3′)G), 4.41 (1H, m, H(5′)G), 4.34 (2H, overlapped m, H(4′)m7G, H(4′)G); 4.27 (2H, overlapped m, H(2′)m7G, H(5′′)G), 4.24 (2H, m, H(5′), H(5′′)m7G), 4.08 (3H, s, N–CH3), 3.59 (3H, s, O–CH3). δP 43.92 (1P, d, JP1,P2 23.0, P1), −11.00 (1P, d, JP3,P4 16.6, P4), −22.71 (1P, ∼t, JP3,P4 16.6, JP2,P3 16.5, P3), −23.63 (1P, dd, JP3,P4 23.0, JP2,P3 16.5, P2). 4b: Analytical RP HPLC Rt = 7.8 min: δH 9.00 (1H, s, H(8) m7G); 8.08 (1H, s, H(8)G); 5.96 (1H, d, J 2.9, H(1′)m7G); 5.81 (1H, d, J 5.9, H(1′)G); 4.70 (1H, ∼t, H(2′)G); 4.55 (1H, ∼t, H(3′)m7G); 4.48 (1H, ∼t, H(3′)G); 4.40 (2H, overlapped m, H(4′)m7G, H(5′)G); 4.30 (3H, overlapped m, H(2′)m7G, H(4′)G, H(5′′)G); 4.25 (2H, m, H(5′), H(5′′)m7G); 4.07 (3H, s, N–CH3), 3.62 (3H, s, O–CH3). δP 43.52 (1P, d, JP1,P2 22.5, P4), −11.06 (1P, d, JP3,P4 15.6, P4), −22.89 (1P, ∼t, JP3,P4 15.6, JP2,P3 16.5, P3), −23.74 (1P, dd, JP1,P2 22.5, JP2,P3 16.5, P2).
Enzymatic reactions
Human DcpS was expressed in Escherichia coli according to the procedures described previously.58 The protein of 15 μM concentration was stored at −80 °C in 20 mM Tris buffer, pH 7.5, containing 50 mM KCl, 0.2 mM EDTA, 1 mM DTT, 0.5 mM PMSF, and 20% glycerol. Snake Venom Phosphodiesterase I from Crotalus adamanteus (EC. 3.1.4.1) was purchased from Sigma (0.14 U/mg solid) and before use dissolved at 1 mg/mL concentration in 100 mM Tris buffer, pH 8.0, containing 100 mM NaCl, 14 mM MgCl2 and 50% glycerol, and stored in this form at −20 °C.hDcpS assay: A cap analog at 40 μM concentration was treated with 3.0 μL of DcpS in 500 μl of 50 mM TRIS buffer, pH = 7.9, containing 20 mM MgCl2 and 60 mM (NH4)2SO4 at 30 °C. After 15, 30, 75, 120 min and 24 h a 100 μl sample was collected from the reaction mixture and deactivated by incubation in 90 °C for 3 min.
SVPDE assay: A cap analog at 40 μM concentration was treated with 5.0 μL of SVPDE in 500 μl of 100 mM TRIS buffer, pH = 8.0, containing 14 mM MgCl2 and 100 mM NaCl at 30 °C. After 15, 30, 90, 210, 330 min and 24 h a 50 μl sample was collected from the reaction mixture and deactivated by incubation in 90 °C for 3 min.
All samples were analyzed without further treatment by analytical RP HPLC using a 0–25% linear gradient of methanol in 0.1 M KH2PO4/K2HPO4 buffer (pH = 6.0) over 30 min.
Binding affinities for eIF4E
Murine eukaryotic Initiation Factor eIF4E (residues 28–217) was expressed in BL21(DE3) Escherichia coli strain. The protein was purified from inclusion bodies then guanidinum-solubilized protein was refolded by a one-step dialysis and purified by ion-exchange chromatography on a HiTrapSP column without contact with cap analogs. The concentration of eIF4E was determined spectrophotometrically (ε280 = 53,400 cm−1M−1). Fluorescence titration measurements were carried out on an LS-55 spectrofluorometer (Perkin Elmer Co.) in 50 mM HEPES/ KOH (pH 7.2), 100 mM KCl, 0.5 mM EDTA, 1 mM DTT at 20.0 ± 0.2 °C. 1 μL aliquots of cap analog solutions with increasing concentration were added to 1.4 mL of 0.1 μM protein solutions. Fluorescence intensities (excitation at 280 nm with 2.5 nm bandwidth and detection at 337 nm with 4-nm bandwidth and 290 nm cut off filter) were corrected taking sample dilution and the inner filter effect into account. Equilibrium association constants (KAS) were determined by fitting the theoretical dependence of fluorescence intensity on the total concentration of cap analog to the experimental data points, according to equation described previously.31 The concentration of protein was fitted as a free parameter of equilibrium equation showing amount of “active” protein. The final KAS was calculated as a weighted average of 3–4 independent titrations, with the weights taken as the reciprocals of the numerical standard deviations squared. Numerical nonlinear least-squares regression analysis was performed using ORIGIN 6.0 (Microcal Software Inc.). The Gibbs free energy of binding was calculated from the KAS value according to the standard equation ΔG° = −RTlnKAS.
Synthesis of mRNA transcripts and measuring of translation efficiency in vitro
Capped, polyadenylated luciferase mRNAs were synthesized in vitro on a dsDNA template (amplified by PCR reaction) that contains: SP6 promoter sequence of DNA-dependent RNA polymerase, 5′UTR sequence of rabbit β-globin, the entire firefly luciferase ORF and a string of 31 adenosines. A typical in vitro transcription reaction mixture (40 μL) contained: SP6 transcription buffer (Fermentas), 0.7 μg of DNA template, 1 U/μL RiboLock Ribonuclease Inhibitor (Fermentas), 0.5 mM ATP/CTP/UTP and 0.1 mM GTP and 0.5 mM dinucleotide cap analog (molar ratio cap analog:GTP 5:1). The reaction mixture was preincubated at 37 °C for 5 min before addition of SP6 RNA polymerase (Fermentas) to final concentration of 1 U/μL and reaction was continued for 45 min at 37 °C. After incubation, reaction mixtures were treated with DNase RQ1 (Promega) in transcription buffer, for 20 min at 37 °C at concentration 1 U per μg of template DNA. RNA transcripts were purified using NucAway Spin Columns (Ambion), the integrity of transcripts was checked on a non-denaturating 1% agarose gel and concentrations were determined spectrophotometrically.
A translation reaction in RRL was performed in 10 μL volume for 60 min at 30 °C, under conditions determined as optimal for cap-dependent translation. A typical reaction mixture contained: 40% RRL lysate (Flexi Rabbit Reticulocyte Lysate, Promega), mixture of amino acids (0.01 mM), MgCl2 (0.6 mM), potassium acetate (170 mM) and 5′-capped mRNA. Four different concentrations of each analyzed transcript were tested in an in vitro translation reaction. The activity of synthesized luciferase was measured with a luminometer.
Acknowledgements
We are indebted to Mike Kiledjian (Rutgers University) for providing the hDcpS-encoding plasmid and Zbigniew M. Darzynkiewicz for expressing and purifying the protein, to the Laboratory of Biological NMR (Institute of Biochemistry and Biophysics of the Polish Academy of Sciences, IBB PAS) for access to the NMR apparatus, and to the Laboratory of Mass Spectrometry (IBB PAS) for recording MS spectra. Financial support from the Polish Ministry of Science and Higher Education (No. NN301 243 436), the National Science Support Project 2008–2010 (No. PBZMNiSW-07/I/2007), the Howard Hughes Medical Institute (to E.D.; No. 55005 604) and the National Institute for General Medical Sciences (to R.E.R,; No. GM20818) is gratefully acknowledged.References
- Y. Furuichi and A. J. Shatkin, Adv. Virus Res., 2000, 55, 135 CAS.
- R. E. Rhoads, J. Biol. Chem., 2009, 284, 16711 CrossRef CAS.
- A. Yamamoto, M. Kormann, J. Rosenecker and C. Rudolph, Eur. J. Pharm. Biopharm., 2009, 71, 484 CrossRef CAS.
- R. Contreras, H. Cheroutre, W. Degrave and W. Fiers, Nucleic Acids Res., 1982, 10, 6353 CrossRef CAS.
- M. M. Konarska, R. A. Padgett and P. A. Sharp, Cell, 1984, 38, 731 CrossRef CAS.
- J. K. Yisraeli and D. A. Melton, Methods Enzymol., 1989, 180, 42 Search PubMed.
- A. E. Pasquinelli, J. E. Dahlberg and E. Lund, RNA, 1995, 1, 957 CAS.
- J. Stepinski, C. Waddell, R. Stolarski, E. Darzynkiewicz and R. E. Rhoads, RNA, 2001, 7, 1486 CAS.
- Z. H. Peng, V. Sharma, S. F. Singleton and P. D. Gershon, Org. Lett., 2002, 4, 161 CrossRef CAS.
- J. Jemielity, T. Fowler, J. Zuberek, J. Stepinski, M. Lewdorowicz, A. Niedzwiecka, R. Stolarski, E. Darzynkiewicz and R. E. Rhoads, RNA, 2003, 9, 1108 CrossRef CAS.
- E. Grudzien, M. Kalek, J. Jemielity, E. Darzynkiewicz and R. E. Rhoads, J. Biol. Chem., 2006, 281, 1857 CAS.
- M. Mockey, C. Goncalves, F. P. Dupuy, F. M. Lemoine, C. Pichon and P. Midoux, Biochem. Biophys. Res. Commun., 2005, 340, 1062.
- E. Grudzien-Nogalska, J. Jemielity, J. Kowalska, E. Darzynkiewicz and R. E. Rhoads, RNA, 2007, 13, 1745 CrossRef CAS.
- A. C. Gingras, B. Raught and N. Sonenberg, Annu. Rev. Biochem., 1999, 68, 913 CrossRef CAS.
- J. Coller and R. Parker, Annu. Rev. Biochem., 2004, 73, 861 CrossRef CAS.
- E. van Dijk, N. Cougot, S. Meyer, S. Babajko, E. Wahle and B. Seraphin, EMBO J., 2002, 21, 6915 CrossRef CAS.
- Z. Wang, X. Jiao, A. Carr-Schmid and M. Kiledjian, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12663 CrossRef CAS.
- D2 is a designation for one of the two synthesized P-diastereoisomers of this compound.
- H. Liu, N. D. Rodgers, X. Jiao and M. Kiledjian, EMBO J., 2002, 21, 4699 CrossRef CAS.
- A. Zdanowicz, R. Thermann, J. Kowalska, J. Jemielity, K. Duncan, T. Preiss, E. Darzynkiewicz and M. W. Hentze, Mol. Cell, 2009, 35, 881 CrossRef CAS.
- W. Su and R. E. Rhoads, unpublished work.
- Only the ways in which the unmodified nucleotide is activated are taken into account, since there is practically no efficient method for activation of phosphorothioate nucleotides with an imidazole group.
- J. R. Moran and G. M. Whitesides, J. Org. Chem., 1984, 49, 704 CrossRef CAS.
- T. Mukaiyama and M. Hashimoto, Bull. Chem. Soc. Jpn., 1971, 44, 2284 CrossRef CAS.
- J. Kowalska, M. Lewdorowicz, E. Darzynkiewicz and J. Jemielity, Tetrahedron Lett., 2007, 48, 5475 CrossRef CAS.
- In the case of 2a, we failed to obtain high quality transcripts in the first attempt, and the amount of compound was insufficient to repeat the experiment. Therefore, unfortunately only 2b was tested for translation efficincy in vitro.
- D. T. Major, V. Nahum, Y. Wang, G. Reiser and B. Fischer, J. Med. Chem., 2004, 47, 4405 CrossRef CAS.
- P. Li, Z. H. Xu, H. Y. Liu, C. K. Wennefors, M. I. Dobrikov, J. Ludwig and B. R. Shaw, J. Am. Chem. Soc., 2005, 127, 16782 CrossRef CAS.
- B. A. Connolly, P. J. Romaniuk and F. Eckstein, Biochemistry, 1982, 21, 1983 CrossRef CAS.
- J. Ludwig and F. Eckstein, J. Org. Chem., 1989, 54, 631 CrossRef.
- A. Niedzwiecka, J. Marcotrigiano, J. Stepinski, M. Jankowska-Anyszka, A. Wyslouch-Cieszynska, M. Dadlez, A. C. Gingras, P. Mak, E. Darzynkiewicz, N. Sonenberg, S. K. Burley and R. Stolarski, J. Mol. Biol., 2002, 319, 615 CrossRef CAS.
- J. Kowalska, M. Lewdorowicz, J. Zuberek, E. Grudzien-Nogalska, E. Bojarska, J. Stepinski, R. E. Rhoads, E. Darzynkiewicz, R. E. Davis and J. Jemielity, RNA, 2008, 14, 1119 CrossRef CAS.
- P. M. Burgers, F. Eckstein and D. H. Hunneman, J. Biol. Chem., 1979, 254, 7476 CAS.
- F. R. Bryant and S. J. Benkovic, Biochemistry, 1979, 18, 2825 CrossRef.
- H. T. Ho and P. A. Frey, Biochemistry, 1984, 23, 1978 CrossRef CAS.
- N. Puri, S. Hunsch, C. Sund, I. Ugi and J. Chattopadhyaya, Tetrahedron, 1995, 51, 2991 CrossRef CAS.
- G. M. Blackburn, X. Liu, A. Rosler and C. Brenner, Nucleosides, Nucleotides Nucleic Acids, 1998, 17, 301 CAS.
- Q. Han, S. G. Sarafianos, E. Arnold, M. A. Parniak, B. L. Gaffney and R. A. Jones, Org. Lett., 2007, 9, 5243 CrossRef CAS.
- Q. W. Han, S. G. Sarafianos, E. Arnold, M. A. Parniak, B. L. Gaffney and R. A. Jones, Tetrahedron, 2009, 65, 7915 CrossRef CAS.
- V. Nahum, M. Tulapurkar, S. A. Levesque, J. Sevigny, G. Reiser and B. Fischer, J. Med. Chem., 2006, 49, 1980 CrossRef CAS.
- S. Mikkola, S. Salomaki, Z. Zhang, E. Maki and H. Lonnberg, Curr. Org. Chem., 2005, 9, 999 CrossRef CAS.
- M. Kadokura, T. Wada, C. Urashima and M. Sekine, Tetrahedron Lett., 1997, 38, 8359 CrossRef CAS.
- A. M. Rydzik, M. Lukaszewicz, J. Zuberek, J. Kowalska, Z. M. Darzynkiewicz, E. Darzynkiewicz and J. Jemielity, Org. Biomol. Chem., 2009, 7, 4763 RSC.
- A. R. Kore, M. Shanmugasundaram, I. Charles, A. M. Cheng and T. J. Barta, Bioorg. Med. Chem. Lett., 2007, 17, 5295 CrossRef CAS.
- K. Misiura, D. Szymanowicz and W. J. Stec, Org. Lett., 2005, 7, 2217 CrossRef CAS.
- V. Jankowski, M. van der Giet, H. Mischak, M. Morgan, W. Zidek and J. Jankowski, Br. J. Pharmacol., 2009, 157, 1142 CrossRef CAS.
- V. Jankowski, M. Tolle, R. Vanholder, G. Schonfelder, M. van der Giet, L. Henning, H. Schluter, M. Paul, W. Zidek and J. Jankowski, Nat. Med., 2005, 11, 223 CrossRef CAS.
- The differences in the phosphate designations between tri- and tetraphosphate cap analogs arise from the fact that due to biochemical nomenclature the α-phosphate is always the one attached to 5′-hydroxyl of mRNA transcript after incorporation of the cap analog into mRNA.
- S. Holtkamp, S. Kreiter, A. Selmi, P. Simon, M. Koslowski, C. Huber, O. Tureci and U. Sahin, Blood, 2006, 108, 4009 CrossRef CAS.
- K. Karikó, H. Muramatsu, F. A. Welsh, J. Ludwig, H. Kato, S. Akira and D. Weissman, Mol. Ther., 2008, 16, 1833 CrossRef CAS.
- D. A. Mitchell and S. K. Nair, J. Clin. Invest., 2000, 106, 1065 CrossRef CAS.
- A. de Benedetti and J. R. Graff, Oncogene, 2004, 23, 3189 CrossRef CAS.
- S. W. Liu, X. Jiao, H. Liu, M. Gu, C. D. Lima and M. Kiledjian, RNA, 2004, 10, 1412 CrossRef CAS.
- V. Shen, H. Liu, S. W. Liu, X. Jiao and M. Kiledjian, RNA, 2008, 14, 1132 CrossRef CAS.
- J. Singh, M. Salcius, S. W. Liu, B. L. Staker, R. Mishra, J. Thurmond, G. Michaud, D. R. Mattoon, J. Printen, J. Christensen, J. M. Bjornsson, B. A. Pollok, M. Kiledjian, L. Stewart, J. Jarecki and M. E. Gurney, ACS Chem. Biol., 2008, 3, 711 CrossRef CAS.
- J. Kusmierek and D. Shugar, Nucleic Acids Res, Special Publ. No. 4, 73 Search PubMed.
- J. W. Jones and R. K. Robins, J. Am. Chem. Soc., 1963, 85, 193 CrossRef CAS.
- L. S. Cohen, C. Mikhli, C. Friedman, M. Jankowska-Anyszka, J. Stepinski, E. Darzynkiewicz and R. E. Davis, RNA, 2004, 10, 1609 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Table S1, HPLC profiles, 1H and/or 31P NMR spectra of compounds 1–6. See DOI: 10.1039/b9nj00644c |
| ‡ This article is part of a themed issue on Biophosphates, and is dedicated to Professor Wojciech J. Stec on the occasion of his 70th birthday. |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2010 |