
p–d Orbital coupling in silicon-based dual-atom catalysts for enhanced CO2 reduction: insight into electron regulation of active center and coordination atoms†

Abstract
Transition metal (TM) dual-atom catalysts (DACs) show promise for carbon dioxide reduction reaction (CO2RR) through d–d orbital cooperative interactions, but their effectiveness is often curtailed by the linear scaling relations between *CO and *CHO on transition metal sites, typically resulting in CO as the predominant product. Specifically, the p–d orbital coupling may exert further influence to regulate the electronic properties and catalytic activity of DACs, which will be of great significance for promoting CO2RR. Herein, we combine density functional theory (DFT) and machine learning (ML) to investigate the potential of heteronuclear DACs with the Si-TM dual-atom active sites in CO2RR and evaluate the influence of the coordination environment. Among 27 SiTMN6 and 336 SiTMN5An (A = B, C, and n represents position) DACs, three SiTMN6 and six SiTMN5An DACs demonstrate high activity and selectivity in converting CO2 to CH4 or CH3OH. The pz band distribution 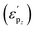 , influenced by both p–d orbital coupling and the coordinating environment, has been elucidated. The optimal
, influenced by both p–d orbital coupling and the coordinating environment, has been elucidated. The optimal 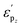 results in superior reaction activity by facilitating optimal adsorption effects for reaction intermediates. This work not only provides comprehensive understanding of reaction mechanisms for CO2 reduction on silicon-based dual-atom catalysts, but also reveals the irreplaceable role of p–d coupling in the performance regulation of DACs. With this knowledge and the aid of machine learning, we establish fundamental principles and descriptors for the accelerated discovery of efficient dual-atom catalysts.
results in superior reaction activity by facilitating optimal adsorption effects for reaction intermediates. This work not only provides comprehensive understanding of reaction mechanisms for CO2 reduction on silicon-based dual-atom catalysts, but also reveals the irreplaceable role of p–d coupling in the performance regulation of DACs. With this knowledge and the aid of machine learning, we establish fundamental principles and descriptors for the accelerated discovery of efficient dual-atom catalysts.

- This article is part of the themed collection: Journal of Materials Chemistry A HOT Papers
 Please wait while we load your content...
Please wait while we load your content...

